 Na Lyu
Na Lyu Yuqing Feng
Yuqing Feng Yuanlong Pan
Yuanlong Pan Hua Huang3†
Hua Huang3† Yan Liu
Yan Liu Baoli Zhu
Baoli Zhu Yongfei Hu
Yongfei Hu- 1CAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China
- 2State Key Laboratory of Animal Nutrition, College of Animal Science and Technology, China Agricultural University, Beijing, China
- 3Beijing Products Quality Supervision and Inspection Institute, Beijing, China
- 4Beijing Municipal Center for Food Safety Monitoring and Risk Assessment, Beijing, China
Salmonella enterica remains one of the leading causes of foodborne bacterial disease. Retail meat is a major source of human salmonellosis. However, comparative genomic analyses of S. enterica isolates from retail meat from different sources in China are lacking. A total of 341 S. enterica strains were isolated from retail meat in sixteen districts of Beijing, China, at three different time points (January 1st, May 1st, and October 1st) in 2017. Comparative genomics was performed to investigate the genetic diversity, virulence and antimicrobial resistance gene (ARG) profiles of these isolates. The most common serotype was S. Enteritidis (203/341, 59.5%), which dominated among isolates from three different time points during the year. Laboratory retesting confirmed the accuracy of the serotyping results predicted by the Salmonella In Silico Typing Resource (SISTR) (96.5%). The pangenome of the 341 S. enterica isolates contained 13,931 genes, and the core genome contained 3,635 genes. Higher Salmonella phage 118970 sal3 (219/341, 64.2%) and Gifsy-2 (206/341, 60.4%) prevalence contributed to the diversity of the accessory genes, especially those with unknown functions. IncFII(S), IncX1, and IncFIB(S) plasmid replicons were more common in these isolates and were major sources of horizontally acquired foreign genes. The virulence gene profile showed fewer virulence genes associated with type III secretion systems in certain isolates from chicken. A total of 88 different ARGs were found in the 341 isolates. Three beta-lactamases, namely, blaCTX–M–55 (n = 15), blaCTX–M–14 (n = 11), and blaCTX–M–65 (n = 11), were more prevalent in retail meats. The emergence of qnrE1 and blaCTX–M–123 indicated a potential increase in the prevalence of retail meats. After the prohibition of colistin in China, three and four isolates were positive for the colistin resistance genes mcr-1.1 and mcr-9, respectively. Thus, we explored the evolution and genomic features of S. enterica isolates from retail meats in Beijing, China. The diverse ARGs of these isolates compromise food security and are a clinical threat.
Introduction
Foodborne diseases caused by Salmonella are an important public health concern. More than 2600 Salmonella serovars have been classified (Guibourdenche et al., 2010). Human-adapted typhoid serovars (commonly Salmonella enterica serovars Typhi and Paratyphi A) cause the severe systemic disease typhoid fever, while most human non-typhoidal Salmonella (NTS) infections are attributed to broad-host-range serotypes that cause self-limiting gastroenteritis (Coburn et al., 2007).
China has a high incidence of salmonellosis, and retail meat or meat products are recognized as the main reservoirs of human foodborne NTS (Ma et al., 2017; Zhang Z. et al., 2018; Li et al., 2019). Accordingly, high rates of Salmonella contamination in retail meats such as pork, beef, mutton, and duck meat have been reported in several places in China. A high prevalence of Salmonella contamination has been observed in chicken (63.6%), pork (73.1%), and duck meat (41.4%) in Guangdong, China (Zhang L. et al., 2018; Chen et al., 2020). In a study in which a total of 807 retail meat samples were collected from most provincial capitals in China between July 2011 and June 2016, 19.7% of the samples tested positive for Salmonella. The highest contamination rate occurred in pork (37.3%), followed by beef (16.1%) and mutton (10.9%) (Yang et al., 2019).
The pathogenesis of Salmonella is associated with the expression of virulence-associated determinants in Salmonella pathogenicity islands (SPIs) (Groisman and Ochman, 1996). To date, 24 different SPIs have been identified, but not all of them have been experimentally validated (Lerminiaux et al., 2020). Two distinct type III secretion systems (TTSSs) encoded by SPI-1 and SPI-2 are considered central to the pathogenicity of NTS (Suez et al., 2013). SPI-1 genes are responsible for the invasion of host cells and for the regulation of the host immune response, while SPI-2 contributes to intracellular survival and replication (El Ghany et al., 2016; Lou et al., 2019).
In severe cases of salmonellosis, antibiotics are typically prescribed for treatment; however, the emergence of multidrug-resistant (MDR) S. enterica strains has raised global concern, due to an increase in the mortality rate of infected patients (Gong et al., 2019; Zhan et al., 2019). In particular, extended-spectrum beta-lactamase-producing Salmonella strains have been frequently isolated from food animals in many countries (Zhao et al., 2017). High rates of antimicrobial resistance to sulfisozazole (76.1%), tetracycline (75.3%), ampicillin (48.0%), and ofloxacin (44.7%) have been found in Salmonella isolates from retail chicken and pork meat samples in Guangdong, China (Zhang L. et al., 2018). In another study, among 218 Salmonella isolates obtained from retail meat and meat products in China, 181 (83.0%) were resistant to at least one class of antimicrobials, and 128 (58.7%) were resistant to at least three classes (Yang et al., 2019).
Next-generation sequencing technology provides an unparalleled, powerful tool for the characterization of infectious agents such as Salmonella and the investigation of foodborne outbreaks (Mather et al., 2013; den Bakker et al., 2014). Genomic analysis can provide improved and exhaustive data related to pathogen genotypic characteristics and can allow the identification of virulence determinants, antimicrobial resistance genes and serotypes. The classification of Salmonella based on genome-sequencing technologies is especially important for epidemiological investigations and has been implemented at the PulseNet (Nadon et al., 2017).
Beijing is the second largest city in China, with a population of over 20 million people. It has been estimated that the total annual meat consumption in Beijing is more than 1.3 million tons (Shi et al., 2015). However, there is limited available information on the surveillance and large-scale genomics studies of Salmonella in meats from retail markets in Beijing. This study is, to the best of our knowledge, the first comparative genomic study of S. enterica isolates from different sources in Beijing.
Materials and Methods
Experimental Design and Sample Collection
To explore the genomic characteristics of S. enterica isolates from retail meat in Beijing, China, we collected 1,234 raw samples of chicken meat (n = 688), duck meat (n = 108), pork (n = 220), beef (n = 163) and mutton (n = 55) from retail markets located in all sixteen districts of Beijing, China. To study the prevalence of Salmonella over a year in meats, these samples were collected at three different time points (January 1st, May 1st, and October 1st) in 2017.
Salmonella Isolation
The isolation and identification of Salmonella were performed according to procedures described in the National Standards of the People’s Republic of China (GB 4789.1-2016). Briefly, a 25-g raw meat sample was placed in 225 mL of buffered peptone water (Land Bridge Technology Co., Ltd., Beijing, China) and incubated at 37°C for 8 h. One-milliliter aliquots of the pre-enriched cultures were transferred to 10 mL of tetrathionate broth (TTB, Land Bridge Technology Co., Ltd., Beijing, China) or selenite cystine broth (SCB, Land Bridge Technology Co., Ltd., Beijing, China). After incubation at 42°C for 18 h in TTB and SCB, one loopful of each broth was streaked onto bismuth sulfite (BS, Land Bridge Technology Co., Ltd., Beijing, China) or xylose lysine deoxycholate agar (XLD, Land Bridge Technology Co., Ltd., Beijing, China). The BS plates were incubated at 37°C for 40–48 h, and the XLD plates were incubated at 37°C for 18–24 h. After incubation, the plates were examined for morphologically typical Salmonella colonies. Among the positive samples, one presumptive Salmonella isolate was picked and confirmed by sequencing the 16S rRNA gene (Frank et al., 2008).
Serotyping and Antimicrobial Susceptibility Testing
Salmonella isolates were serotyped via the slide agglutination assay according to the instructions provided by the manufacturer of the Salmonella antisera (Statens Serum Institute, Copenhagen, Denmark). The serovars of the isolates were determined using the Kauffmann-White scheme (Grimont and Weill, 2007).
Antimicrobial susceptibility testing of Salmonella isolates was performed using the agar dilution method for ampicillin, ampicillin-sulbactam, piperacillin, piperacillin-tazobactam, cefuroxime, cefotetan, ceftazidime, ceftriaxone, cefepime, aztreonam, imipenem, amikacin, gentamicin, and tobramycin (Sigma, St. Louis, MO, United States). Escherichia coli ATCC 25922 was used as a quality control strain in each run. The breakpoints for determining susceptibility or resistance to antimicrobials were determined by the guidelines of the Clinical Laboratory Standard Institute (CLSI, 2018).
DNA Sequencing, Assembly, and Annotation
For whole-genome sequencing (WGS), the genomic DNA of the isolates was extracted by using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). A DNA library was constructed using a NEXTflex® rapid DNA-Seq Kit according to the manufacturer’s instructions (Illumina, San Diego, United States). In brief, one paired-end library with an insert size of 350 bp for each sample (Tan et al., 2019) was constructed and sequenced with a 150-bp read length from each end on the Illumina HiSeq 2500 platform (Illumina, San Diego, United States). Adaptors were clipped from the Illumina HiSeq-generated sequencing reads with Trimmomatic (Bolger Anthony et al., 2014). The reads were assembled using Unicycler (v0.4.7) with the default parameters (Wick et al., 2017), including short-read assembly via SPAdes (v3.13.0) (Anton et al., 2012) and polishing via Pilon (Walker et al., 2014). The genomes were annotated using Prokka (v1.13.3) (Seemann, 2014), which uses Prodigal (v2.6.3) (Hyatt et al., 2010) to identify open reading frames.
In silico Analysis of Salmonella Genomes
Core genome and pangenome prediction was carried out using Roary software (v3.11.2) (Page et al., 2015), and the core genome was defined by including both hard-core and soft-core genes (i.e., genes present in more than 95.0% of isolates) (Livingstone et al., 2018). The core gene and accessory gene functional annotations of Salmonella were carried out using the eggNOG platform (v4.5) (Huerta-Cepas et al., 2016). To determine clonality or genetic relatedness between the isolates, phylogenetic reconstruction was calculated using kSNP3 (v3.0) with a kmer size of 19 (Gardner et al., 2015). A maximum likelihood (ML) tree was calculated from 277,664 core SNPs using kSNP3. SNP distance based on the 277,664 core SNPs was calculated using the snp-dists1. In addition to serotyping, in silico analysis by MLST was also performed using the Salmonella In Silico Typing Resource (SISTR) on the basis of whole-genome assemblies (Yoshida et al., 2016). We identified putative plasmid sequences with the Basic Local Alignment Search Tool (BLAST, v2.9.0+) by searching PlasmidFinder database replicons against our dataset with BLASTn (Altschul et al., 1990). A sequence identity of 80% was used as a cutoff for calling a specific replicon as present or absent. Prophage predictions were carried out using PHASTER to explore prophages (Arndt et al., 2016). Virulence genes were identified by BLAST searches against the latest version of the Virulence Factor Database (VFDB) (Chen et al., 2016) with cutoff values of ≥ 80% identity and ≥ 50% coverage. The partial plasmid sequences from the draft genomes were predicted using MOB-suite (Robertson and Nash, 2018) and annotated against the VFDB with the same criteria. ARG analysis was performed by using ResFinder v4.0 (Bortolaia et al., 2020). A sequence identity of 80% and ≥ 50% coverage were used as the cutoffs for calling an ARG as present or absent. Mobile genetic elements (MGEs) in mcr-positive isolates were further investigated according to the results of MOB-suite.
Statistical Analysis and Visualization
The Wilcoxon rank-sum test was used to identify differences in the number of ARGs and VFs. The Kruskal-Wallis test was performed when comparing samples from more than two groups. The statistical significance of the categorical variables was assessed using the chi-square test. The phylogenetic tree was edited and exported using the iTOL tool (Letunic and Bork, 2019). All statistical analyses were performed using R software (v3.6.3). The R package ggplot2 was used for data visualization in figures in the present study (Valero-Mora, 2010).
Results
Isolation of Salmonella and Sequence Statistics
In the present study, we isolated 341 Salmonella strains from retail meat samples, including chicken, pork, duck, beef and mutton specimens, collected from sixteen districts of Beijing, China, at three different time points (January 1st, May 1st, and October 1st) in 2017 (Figures 1A,B and Supplementary Table 1). The raw whole-genome sequence data totaled more than ∼800 Mbp for each isolate (Supplementary Table 1). The average lengths of the genomes ranged from 4.50 to 5.15 Mbp, with an overall average length of 4.78 Mbp (Supplementary Table 1).
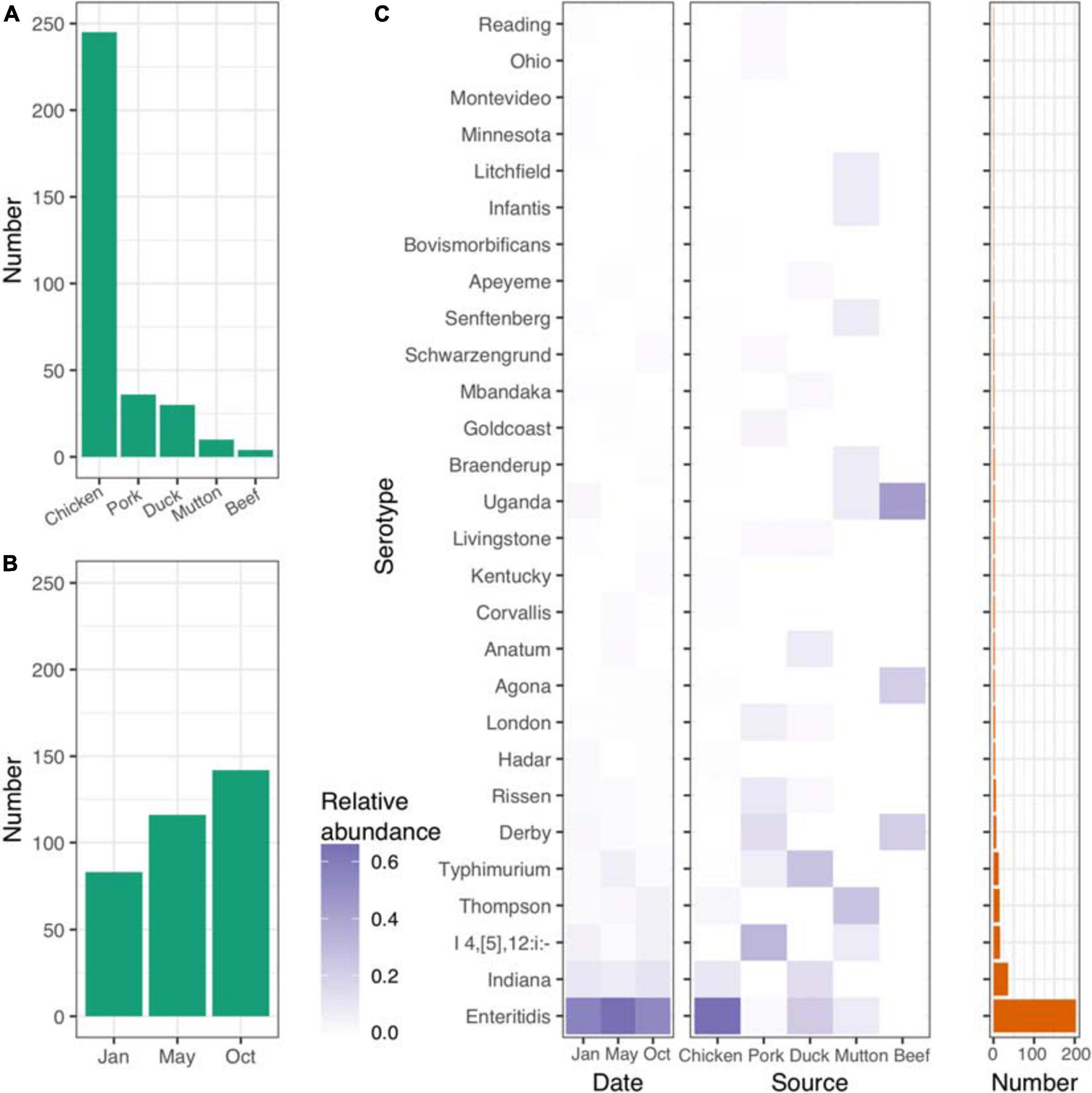
Figure 1. Distribution of isolates and Salmonella serovars. (A) Number of isolates from five different retail meats. (B) Number of isolates from different sampling time points. (C) Distribution of serovars across different time points and retail meats. Left panel, time points; middle panel, retail meats; right panel, number of each serovar.
Prevalence of Serotypes and Sequence Types (STs) Associated With Different Sources and Times
Salmonella serotyping remains the gold-standard tool for the classification of Salmonella isolates. To verify the accuracy of the results from the in silico analysis, we randomly selected 85 isolates for serotyping. SISTR software showed a consistency of 96.5% (82/85). Among the three isolates that were poorly predicted by SISTR, two S. Typhimurium isolates were predicted to belong to the serovar S. I 4,[5],12:i- (Supplementary Table 2). Based on the serovar prediction results from SISTR (Supplementary Table 3), we further explored the connections between the serovars and the corresponding epidemiological information. In total 28 different serovars were identified for the 341 isolates recovered. Most of the isolates were of chicken origin and belonged to S. Enteritidis (Figure 1C and Supplementary Table 4). The frequency of S. Enteritidis was stable even at different time points throughout 2017 (Figure 1C and Supplementary Table 5). However, different sources shared different patterns of serovars. S. Enteritidis, S. I 4,[5],12: i-, S. Typhimurium, S. Thompson and S. Uganda were the dominant serovars in chicken, pork, duck, mutton, and beef, respectively (Supplementary Table 6).
We further explored the STs of the isolates using SISTR. A total of 33 different STs were identified among the 341 isolates recovered. The most frequent ST was ST11 (203/341, 59.5%, Supplementary Table 7), which also belonged to the S. Enteritidis serovar. Some STs, such as ST17 (n = 36), ST34 (n = 17), ST26 (n = 15), ST19 (n = 9), ST40 (n = 7), and ST469 (n = 6), were also common in the isolates. Twenty-four STs (ratio < 1.0%) were rare in the isolates from retail meat (Supplementary Table 8).
Phylogenetic Analysis Revealed Multiple Origins of Salmonella
The results showed that the pangenome of the 341 isolates contained a total of 13,931 genes, while the core genome (3,625 genes) accounted for approximately one-quarter of the pangenome (Figure 2A). On the other hand, most of the Salmonella genome was conserved, considering the total number of genes in each isolate (Supplementary Table 1). The accessory genome harbored more genes associated with the “[S]Function unknown” (Chi-square test, p < 2.2 × 1016), “[L]Replication, recombination and repair” (Chi-square test, p < 2.2 × 1016, p < 2.2 × 1016), and “[K]Transcription” (Chi-square test, p = 0.027) categories (Figure 2B). An ML tree was generated on the basis of the core SNPs (Figure 2C). Considering the prevalence of the serovar from different sources, isolates of the same serovar recovered from different sources were further analyzed on four serovars, i.e., S. Enteritidis, S. Indiana, S. Thompson, and S. Typhimurium. The results showed that the isolates of S. Typhimurium from duck and pork showed longer inter-SNP distance than the intra-SNP distance (p = 7.57 × 109), while the others were not (Supplementary Figure 1). According to the data from the NCBI, 291 out of 341 isolates could be assigned to known SNP clusters (n = 41). Compared with pork (n = 11), duck (n = 8), mutton (n = 8), and beef (n = 2), the number of SNP clusters in chickens was higher (n = 22) (Figure 2D). In addition, there were eight SNP clusters with isolates from multiple sources, among which two SNP clusters (PDS000026869.146 and PDS000004748.44) belonged to S. Enteritidis. Notably, the most common SNP cluster, PDS000026869.146, contained 107 isolates having different sources; 100 were from chicken, 5 from duck, 1 from mutton, and 1 from pork. Additionally, there were two SNP clusters, PDS000004748.44 and PDS000043691.12, contained strains from both chicken and duck. These results strongly suggested the common source of Salmonella contamination in retail meat.
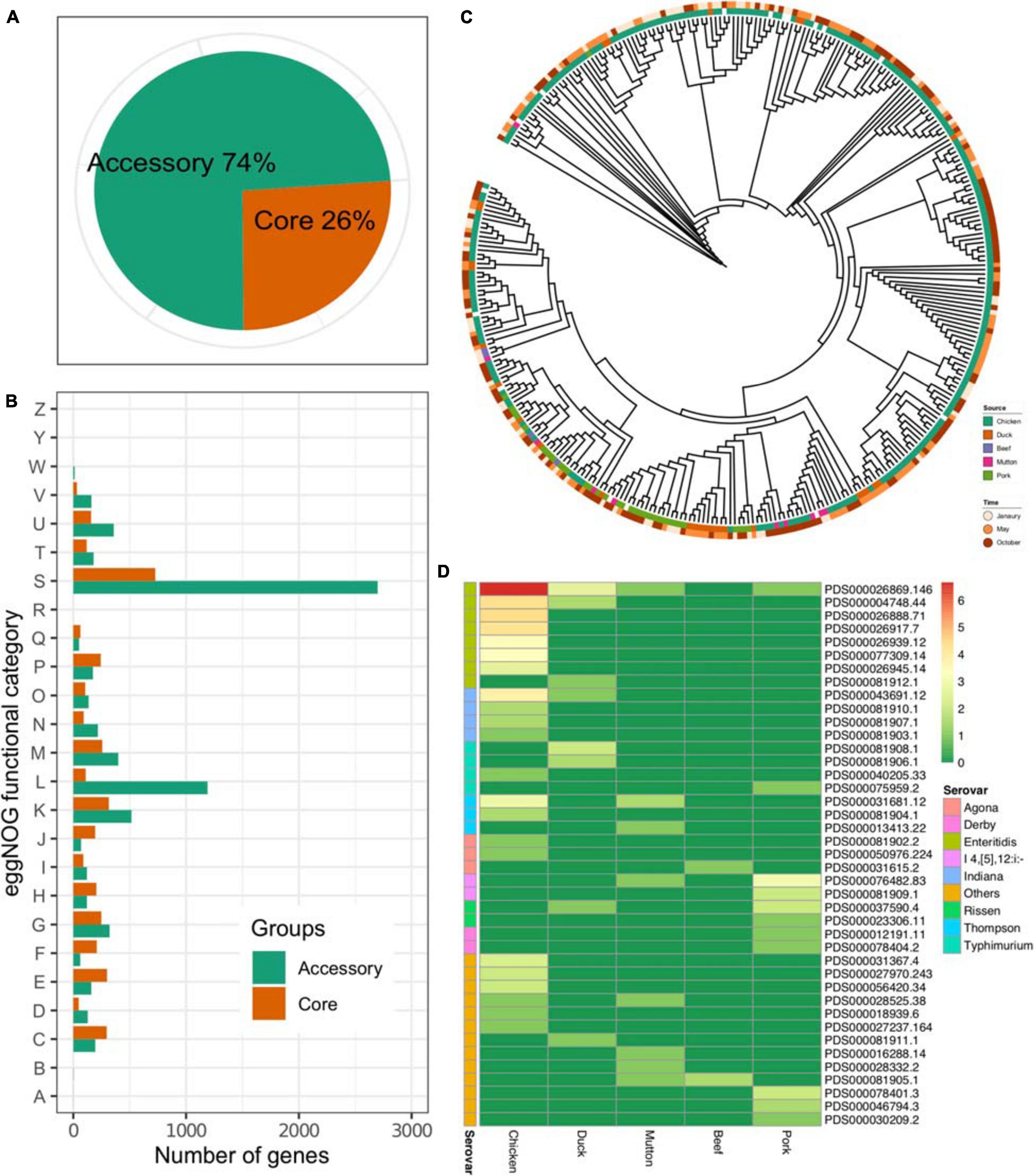
Figure 2. Core genome of Salmonella. (A) Proportions of accessory genes and core genes in the pangenome. (B) Core and accessory genome functional annotation. RNA processing and modification, A; chromatin structure and dynamics, B; energy production and conversion, C; cell cycle control, cell division, chromosome partitioning, D; amino acid transport and metabolism, E; nucleotide transport and metabolism, F; carbohydrate transport and metabolism, G; coenzyme transport and metabolism, H; lipid transport and metabolism, I; translation, ribosomal structure and biogenesis, J; transcription, K; replication, recombination and repair, L; cell wall/membrane/envelope biogenesis, M; cell motility, N; posttranslational modification, protein turnover, chaperones, O; inorganic ion transport and metabolism, P; secondary metabolites biosynthesis, transport and catabolism, Q; general function prediction only, R; function unknown, S; signal transduction mechanisms, T; intracellular trafficking, secretion, and vesicular transport, U; defense mechanisms, V; extracellular structures, W; nuclear structure, Y; cytoskeleton, Z. (C) An ML tree constructed based on the core SNPs (n = 277,664). (D) Prevalence of the SNP clusters from different sources. Color: the number of the isolates (log2-transformed).
Isolates From Retail Meat Presented Different MGEs
Plasmid replicons were widely present among the isolates. Only 11.4% (39/341) of the isolates did not harbor any plasmids in their genomes. A total of 885 plasmid replicons were found among the 341 isolates, with a maximum of ten plasmid replicons observed in a single isolate (Figure 3A). Plasmid replicons of IncFII(S), IncX1, and IncFIB(S) were the three most common types of replicons (n = 181, 177, and 173, respectively; Figure 3B and Supplementary Table 9). The less common types, such as Col(pHAD28), ColRNAI, IncHI2, IncHI2A, IncQ1, and Col4401, were more likely to be present in the isolates from pork upon considering the total number of isolates from chicken (n = 245) and pork (n = 36). Isolates from retail meat showed differences in the prevalence of plasmid replicons at different time points (Supplementary Figure 2A). Specifically, strains isolated in January and May showed a higher prevalence of IncX1. Strains isolated in May and October showed a higher prevalence of IncFII(S) and IncFIB(S). For different serovars, S. Indiana harbored the largest number of plasmid types (n = 21), compared with the other serovars (Supplementary Figure 3). At the same time, isolates from S. Enteritidis were more prone to carry the plasmid replicons of IncFIB(S), IncFII(S) and IncX1.
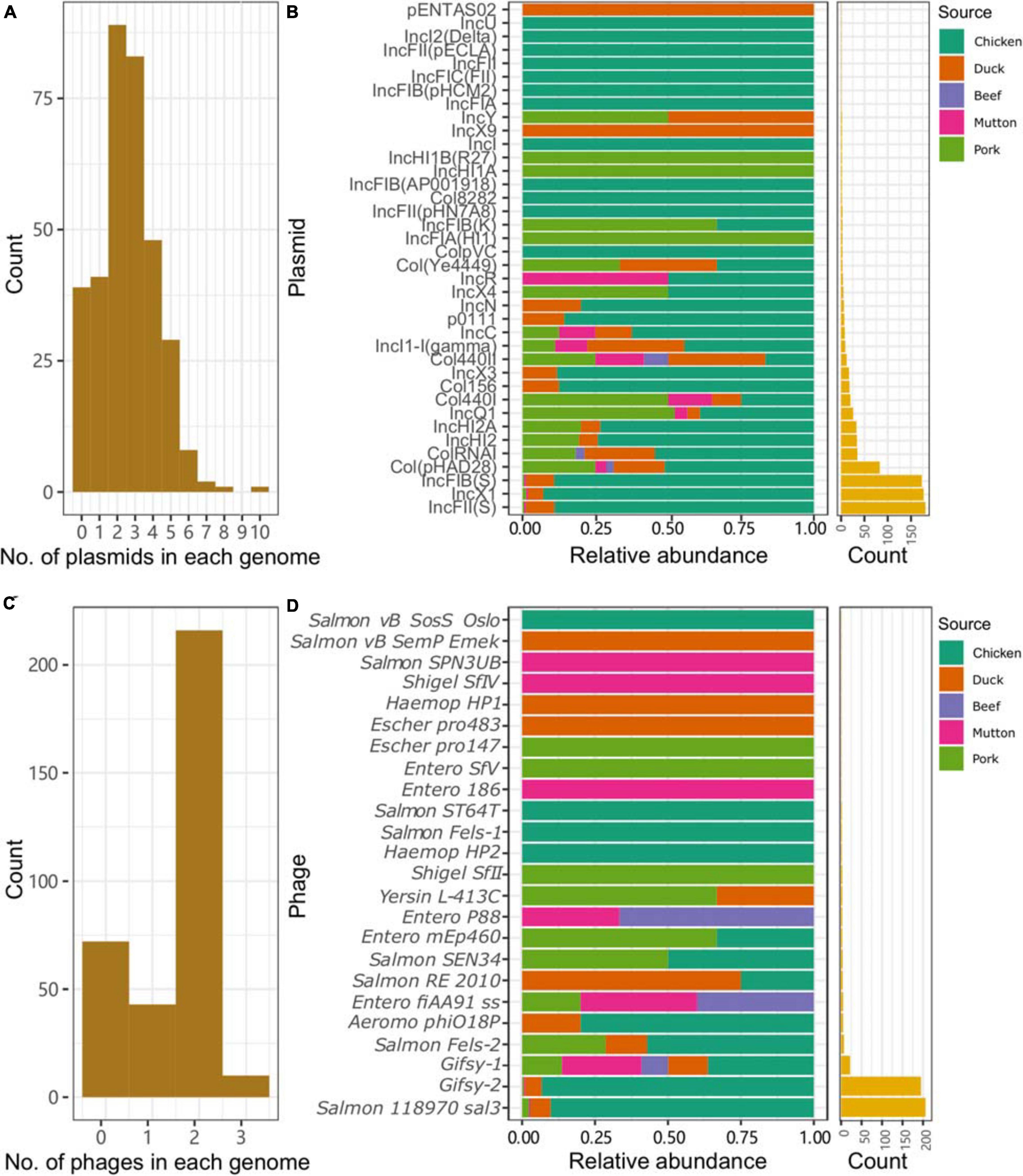
Figure 3. Plasmids and prophages in the Salmonella genome. (A) Number of plasmids in each genome. (B) Distribution of different plasmids in the genomes of Salmonella isolates from different retail meats. (C) Number of prophages in each genome. (D) Distribution of different prophages in the genomes of Salmonella isolates from different retail meats.
Prophages were also highly prevalent among the isolates. Only 72 isolates did not contain any intact prophages in their genomes. A total of 505 prophages were found among the 341 isolates, with a maximum of three observed in a single isolate (Figure 3C). More than half of the isolates (216/341, 63.3%) harbored two intact prophages. Interestingly, 203 isolates harbored both Gifsy-2 and Salmon 118970 sal3 (Figure 3D). The other 22 kinds of prophages were rare in the isolates from retail meat (Supplementary Table 10). The third most common prophage, Gifsy-1, was more likely to be present in isolates from hosts other than chicken. Strains isolated in May and October showed a higher prevalence of Salmon 118970 sal3 and Gifsy-2 prophages (Supplementary Figure 2B). For different serovars, S. I 4,[5],12:i:- harbored the largest number of prophage types (n = 10), compared with the other serovars (Supplementary Figure 4). At the same time, isolates from S. Enteritidis were more prone to contain Gifsy-2 and Salmon 118970 sal3. In addition, the prophages in the isolates were also found to be able carry ARGs (n = 31) and VFs (n = 335) (Supplementary Tables 11, 12).
Isolates From Chicken Showed Fewer Virulence Factors (VFs)
To explore the virulence gene profile of Salmonella, we subjected the genomes to BLAST searches against the full VFDB dataset (Supplementary Table 13). Compared with other sources, isolates from chicken harbored fewer VFs (p < 0.05, Figure 4A). Strains isolated from chicken in October exhibited a lower frequency of VFs than those isolated from chicken at the other two time points (p < 0.05, Figure 4B).
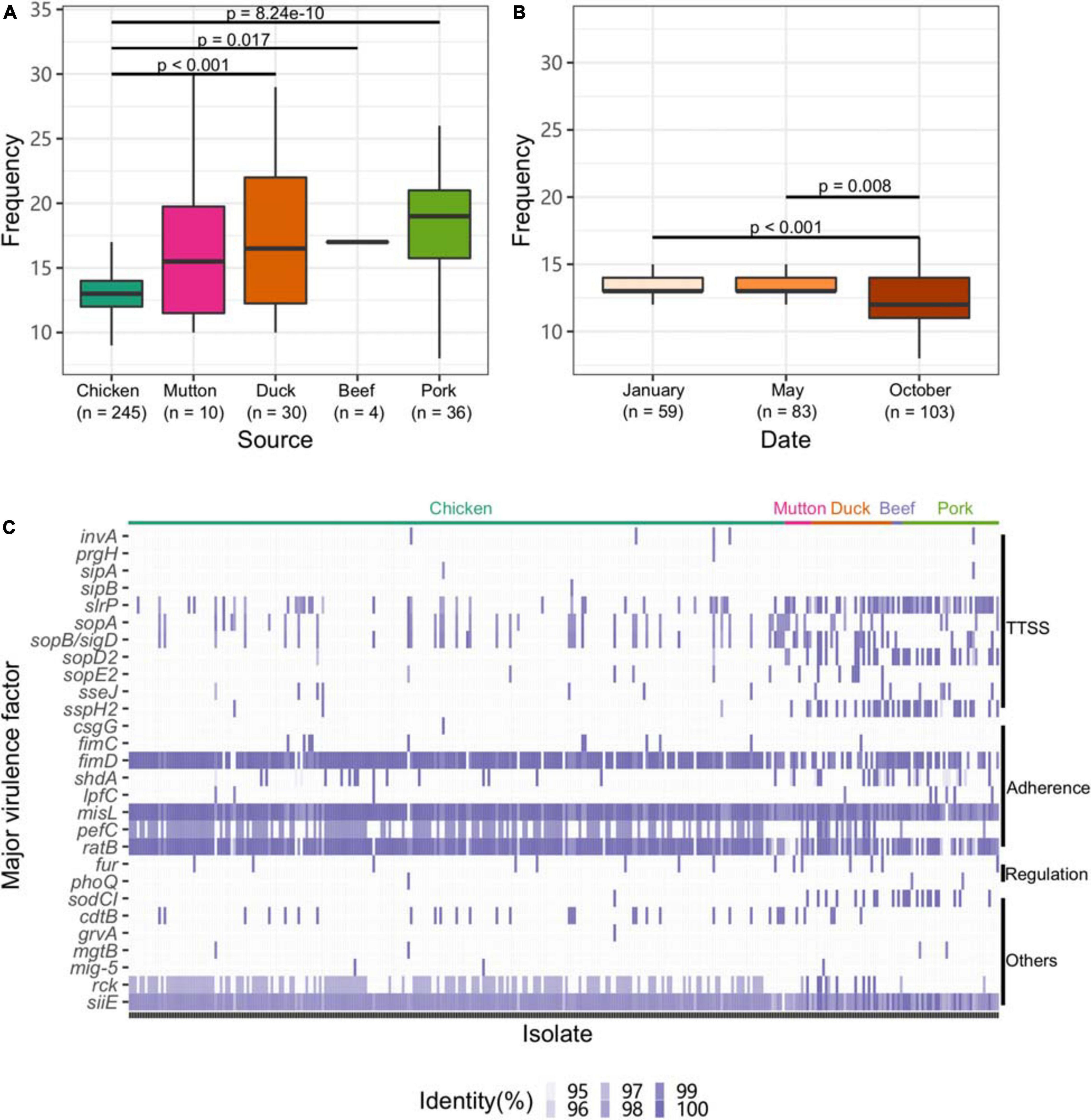
Figure 4. Profile of VFs in the Salmonella genome. (A) Numbers of VFs in the genomes of Salmonella isolates from different retail meats. (B) Numbers of VFs in the genomes of Salmonella isolates from different sampling time points. (C) Major VF profile in the Salmonella genome. TTSS, Type III secretion system.
Eleven genes associated with the TTSS were found among the isolates. Among them, slrP, sopA, and sopB/SopD were common in the isolates. Genes associated with adherence (fimD, misL, and ratB) and siiE were ubiquitously distributed among the isolates. Isolates from chicken harbored fewer genes associated with the TTSS than those from other hosts (Figure 4C). Additionally, the strains isolated from chicken harbored more genes associated with adherence, such as fimD and pefC. We further analyzed the VF categorization based on chromosomes and plasmids. One VF, rck, may be carried by plasmids belonging to the plasmid replicons of IncFII(S), IncX1 and IncFIB(S) (Supplementary Figure 5). Unlike the plasmid and the prophage, the isolates from different serovars almost shared the same patterns of major VFs (Supplementary Figure 6).
ARG Profiles of Salmonella
Antimicrobial susceptibility testing was performed in 117 isolates (Supplementary Table 14). The isolates showed a higher rate of resistance to ampicillin (67.5%, Table 1). All 117 isolates were susceptible to cefotetan, imipenem and piperacillin-tazobactam.
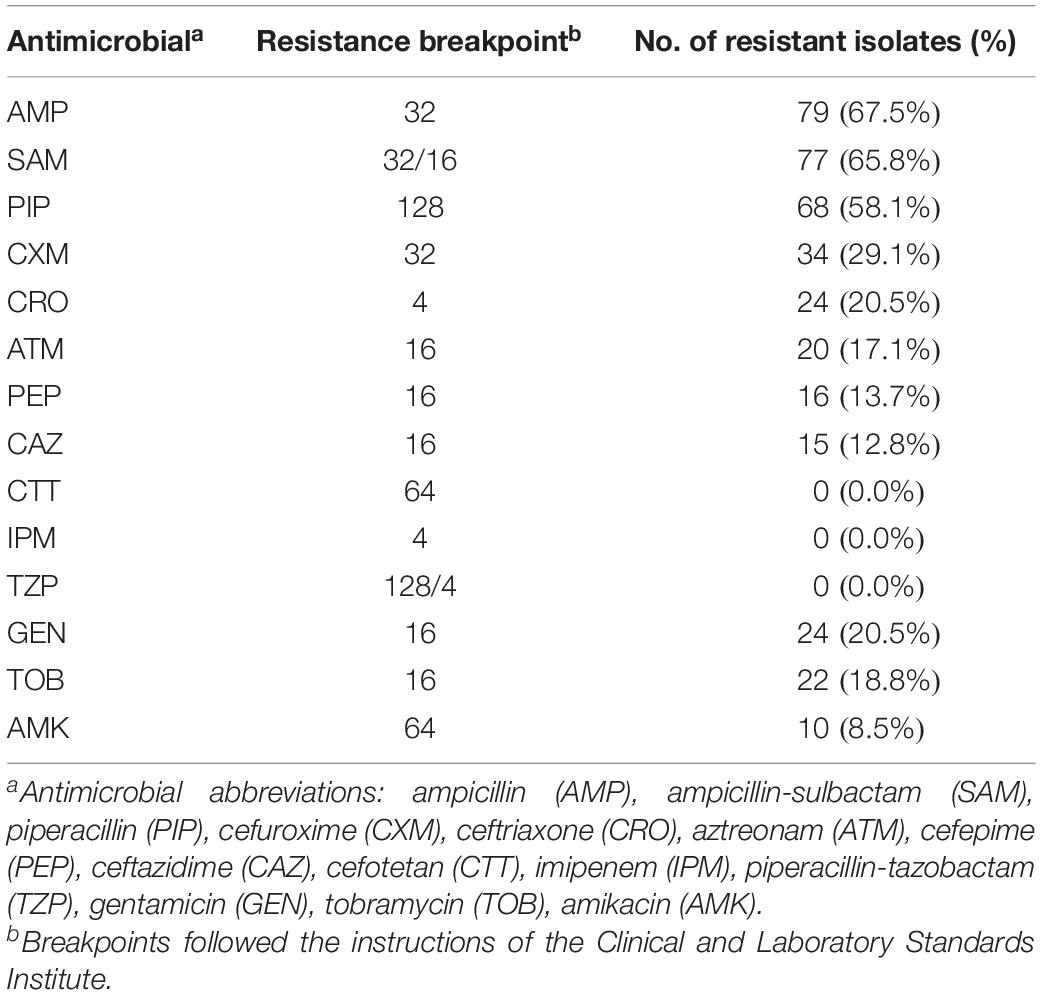
Table 1. Antimicrobial resistance of 117 Salmonella isolates recovered from retail meat.
All sequences of the 341 genomes were screened against the ResFinder database for the presence of resistance genes. A total of 88 different ARGs were found in the 341 isolates (Figure 5A). Most of the ARGs belonged to the categories of beta-lactam resistance (n = 26) and aminoglycoside resistance (n = 25). Among the CTX-M-type beta-lactamases, eight blaCTX–M genes (blaCTX–M–2, blaCTX–M–3, blaCTX–M–14, blaCTX–M–14b, blaCTX–M–15, blaCTX–M–55, blaCTX–M–65, blaCTX–M–123) were identified. The most prevalent genes detected were blaCTX–M–55 (n = 15), blaCTX–M–14 (n = 11), and blaCTX–M–65 (n = 11). Notably, all the isolates were positive for the aminoglycoside resistance gene aac(6′)-Iaa, and half of the isolates possessed a sulfonamide resistance gene (sul2), a beta-lactam resistance gene (blaTEM–1B) and aminoglycoside resistance genes [aph(6)-Id and aph(3″)-Ib] (Supplementary Table 15). Seven kinds of plasmid-mediated quinolone resistance (PMQR) genes were found among the isolates, namely, oqxA (n = 31), oqxB (n = 29), qnrB4 (n = 4), qnrB6 (n = 2), qnrE1 (n = 1), qnrS1 (n = 12), and qnrS2 (n = 2). In addition, three and four isolates were positive for the colistin resistance genes mcr-1.1 and mcr-9, respectively (Table 1). All mcr-9-positive isolates were isolated from chicken. Two of the mcr-1.1-positive isolates were isolated from pork, and the third was isolated from chicken. In two isolates, the IncX4 plasmid replicon was predicted, which was genetically linked with the mcr-1.1 gene on the same contig (Supplementary Table 16). Another mcr-1.1 gene was associated with the IncI2 plasmid replicon. The mcr-9 gene was associated with the IncHI2A/rep_cluster_1088 plasmid replicon. Due to the high prevalence of genes associated with aminoglycoside resistance and beta-lactam resistance, all the isolates harbored at least one ARG in their genomes (Figure 5B). One isolate, SLM309, contained 27 ARGs, which may be involved in resistance to antimicrobials from the beta-lactam, aminoglycoside, phenicol, trimethoprim, macrolide, sulfonamide, rifampicin, and fosfomycin categories (Supplementary Table 17). The strains isolated from pork harbored more ARGs than the others (Figure 5C, p < 0.05). The number of ARGs in the Salmonella genomes from chicken did not change with time (Figure 5D). The isolates in the present study harbored more ARGs and VFs (Supplementary Figure 7) than those of the other four major serovars. The isolates harboring the highest number of ARGs tended to belong to S. Indiana, which may be due to the presence of different kinds of plasmids in the isolates (Table 2).
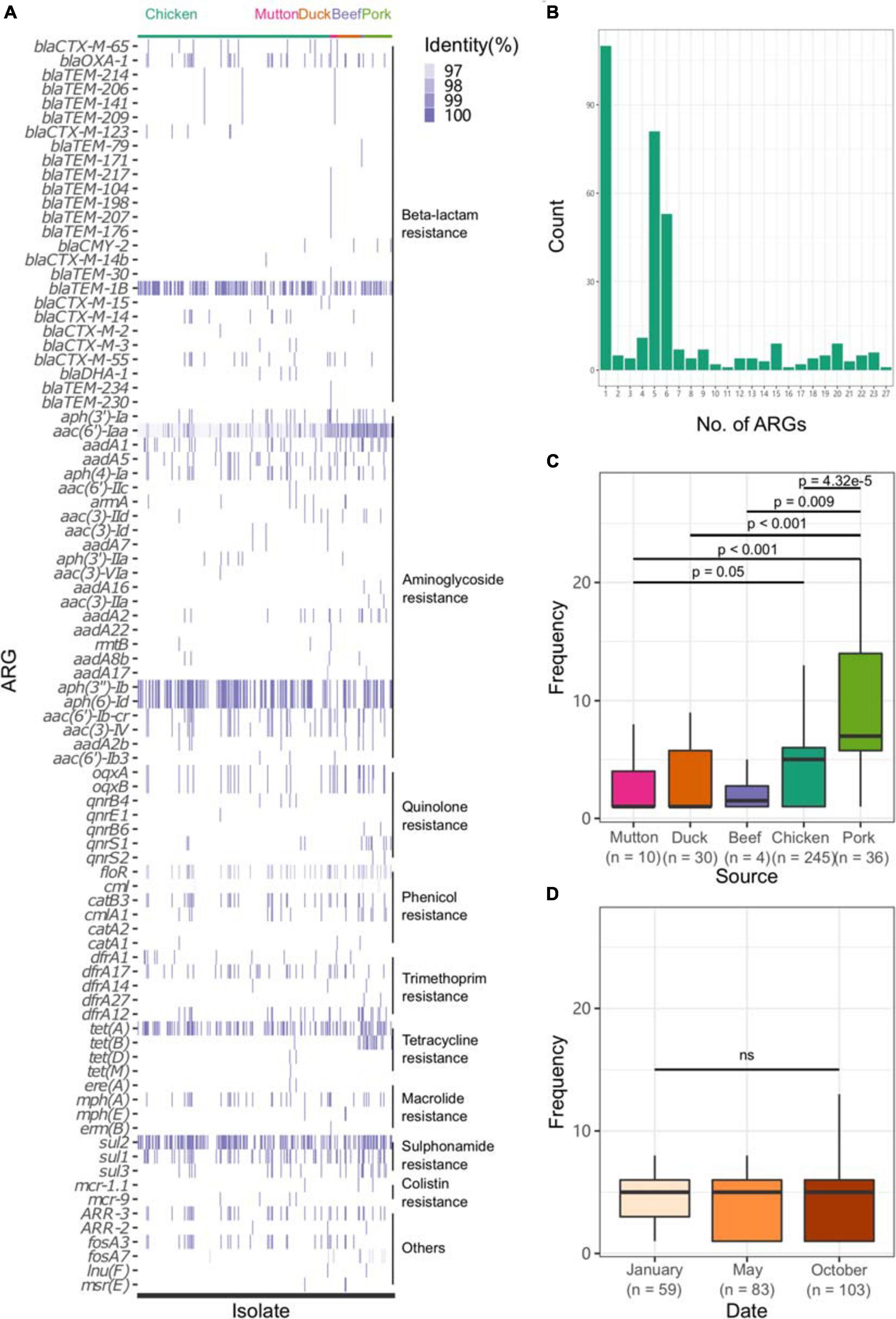
Figure 5. Profile of ARGs in the Salmonella genome. (A) ARG profile in the Salmonella genome. (B) Number of ARGs in each genome. (C) Number of ARGs in the genomes of Salmonella isolates from different retail meats. (D) Number of ARGs in the genomes of Salmonella isolates from different sampling time points.
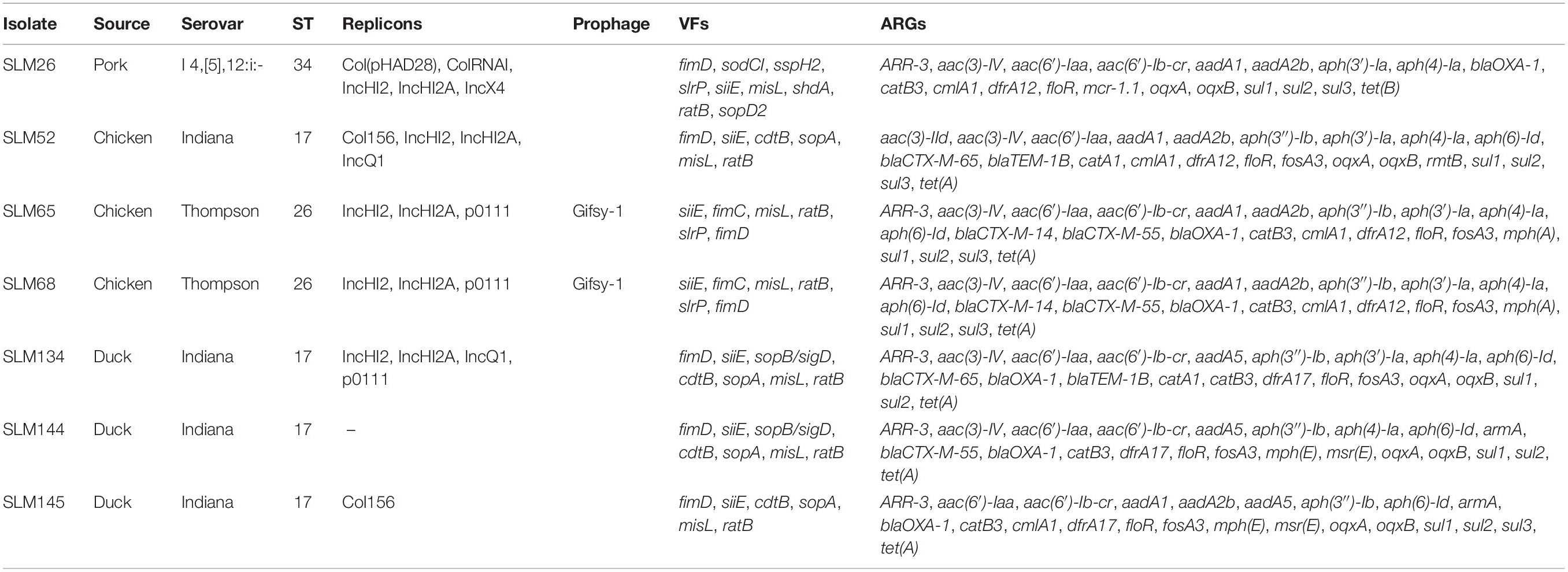
Table 2. Sources and serovar, ARG, VF, and ST profiles of some Salmonella isolates.
Discussion
Salmonella remains the leading cause of reported bacterial foodborne disease in China. Retail meats and meat products are recognized as one of the major sources of human salmonellosis (Yang et al., 2019). To investigate the prevalence of Salmonella in retail meat available on the market throughout the year, we isolated 341 Salmonella strains from Beijing in 2017 and investigated their genomes. To the best of our knowledge, this is the first systematic study reporting the genomic characterization of Salmonella isolates from retail meat in China.
Among the various kinds of retail meat, chicken shows the highest rate of Salmonella contamination (Zhao et al., 2001; Yang et al., 2010). Most of the 341 isolates examined in the present study (71.8%, 245/341) came from chicken. The limited sample sizes obtained for beef (n = 4) and mutton (n = 10) made it difficult to draw a clear conclusion about the other kinds of retail meats, which showed a shortage at the time of the present study.
Most of the retail meat samples from which Salmonella was isolated were imported from at least eight provinces outside of Beijing in China (data not shown). This may explain why the ML tree and the SNP clusters revealed multiple origins of the isolates. Interestingly, the major SNP cluster found in the present study contained many strains from clinical infections in the following years in China, indicating the need for further study on the spread of strains associated with this SNP cluster. The limited sample size may lead to the high one-to-one correspondence between STs and serovars, whereas larger-scale studies (n = 6,887) demonstrated that for a small proportion of STs (four percent of isolates) STs and serotypes were not concordant (Ashton et al., 2016).
MGEs, which include plasmids and bacteriophages, are segments of DNA that encode enzymes and other proteins that mediate the movement of DNA within genomes (Frost et al., 2005). Although the isolates exhibited different backgrounds, including sources, import origins, and sampling time points, they shared 3,625 genes overall. The number of core genes is higher than previous studies on Salmonella (Jacobsen et al., 2011; Nova et al., 2019). Some of the genes with unknown functions may be associated with phage infection (Dutilh et al., 2014). The high frequency of Salmon 118970 sal3 found in the Salmonella genome revealed an emerging phage infection in the strains obtained from retail meat. Salmon 118970 sal3, with a 39,464 bp genome, was first isolated from water buffalo feces in southern Italy and exhibits lytic activity against S. Typhimurium (Paradiso et al., 2016). Salmon 118970 sal3 has a relatively broad host range, which can be also found in E. coli (Li et al., 2020) and Morganella morganii (Minnullina et al., 2019). A broad host range phage could potentially be the equivalent of a broad spectrum antibiotic (Ross et al., 2016).
In addition to phages, plasmids are primary vehicles for horizontal gene transfer (HGT) in bacteria (De la Cruz and Davies, 2000). The plasmid replicon IncX1 was the second most common replicon among the isolates. A previous study indicated that the most common types of plasmid replicons of Salmonella isolated from food animals in the United States were ColE, IncI1, IncF, and IncX (McMillan et al., 2019). The prevalence of plasmid replicons in the Chinese samples was highly consistent with that found in the United States. The IncX1 plasmid observed in Salmonella must remain under surveillance, as many papers have reported the presence of various ARGs, such as tet(X4) (Chen et al., 2019), mcr-1 (He et al., 2020), mcr-3.1 (Sun et al., 2020), mcr-5.1 (Guo et al., 2019), mcr-9 (Cha et al., 2020), and beta-lactamase genes (Zhang X.Z. et al., 2018) in this plasmid. It is clear that infection of humans with Salmonella containing such genes will present a difficult problem in the clinic.
The number of possible VFs of Salmonella is continuing to increase with the ongoing expansion of the knowledge of the molecular mechanism underlying the pathogenicity of Salmonella. For instance, effector proteins involved in survival and replication in so-called Salmonella-containing vacuoles have been characterized. Other VFs (i.e., virulence plasmids, toxins, fimbriae, and flagella) have been the subject of studies for decades and might therefore be annotated as “classic” VFs (Van Asten and Van Dijk, 2005). Pathogenic Salmonella invades non-phagocytic intestinal epithelial cells by delivering a specialized set of effectors though sophisticated machinery comprising the TTSS, which plays a crucial role in the pathogenesis of Salmonella (Que et al., 2013). Although the number of Salmonella isolates obtained from chicken was greater than the numbers obtained from other meat types, they harbored fewer genes associated with the TTSS, such as slrP, sopD2, and sspH2. The lack of genes belonging to SPI-2 may affect the intracellular replication function in the host (Figueira and Holden, 2012). Ubiquitous fimbrial adherence genes indicate that a strain is experiencing strong selective pressure (Worley et al., 2018). This phenomenon accelerates the severity of Salmonella contamination in retail meat. Although the rate of Salmonella contamination was not as high in retail meats such as pork, mutton and beef as it was chicken, the isolates from these other meat types may pose a greater threat to human health.
McDermott and colleagues verified that the correlation of phenotypic resistance with known resistance genes is high (99.0%), except for the resistance to aminoglycosides and beta-lactams (McDermott et al., 2016). More than half of the ARGs (58.0%, 51/88) found in the Salmonella genomes of the 341 isolates were associated with aminoglycoside and beta-lactam resistance. The complex relationship between phenotype and genotype should be further studied regarding the functions of not only known ARGs but also the potential genes whose function is modulated by ARGs.
Antimicrobials are delivered to animals for a variety of reasons, including disease treatment, prevention, and control and the promotion of growth/feed efficiency. The use of antimicrobials causes more resistant bacteria to emerge in farm animals and retail meats. Consistent with previous studies in China (Rao et al., 2014), blaCTX–M–14, blaCTX–M–55, and blaCTX–M–65 were the most dominant CTX-M types. It should be noted that the relatively high prevalence of blaCTX–M–123, which has been identified as a novel hybrid of the blaCTX–M–1 and blaCTX–M–19 β-lactamases recovered from E. coli isolates in China (He et al., 2013). Our findings validated the results indicating the potential spread of the blaCTX–M–123 gene in China, especially in chicken meat samples (Wang W. et al., 2020). Most of PMQR genes could be found in animals from China (Jacoby et al., 2014), except one isolate, YC300, which harbored the qnrE1 gene (Monte et al., 2019). The qnrE1 gene was mainly found in South America (Monte et al., 2019). Colistin, as the last resort for the treatment of infections caused by MDR bacteria, has received extensive attention. In particular, transferable colistin resistance is drawing increasing attention in the study of ARGs (Liu et al., 2016). Our results suggest that the spread of colistin resistance genes, such as the mcr-1.1 and mcr-9 genes in chicken, needs to be under strict surveillance in retail markets. The two plasmids IncX4 and IncI2 may increase the fitness of the host and become the major plasmid types driving the dissemination of the mcr-1 gene (Wu et al., 2018). In the present study, we validated the presence of the mcr-9 gene commonly on large plasmids (Lin et al., 2020). Colistin has been used for decades in veterinary medicine and as a last-resort drug to treat infections in humans (Kempf et al., 2016). Although colistin sulfate premix was prohibited for use as a growth promoter on April 30th, 2017, in China (Wang Y. et al., 2020), retail meat remains an important source of mcr-positive bacteria. In the present study, although not all the genes annotated as aac(6′)-Iaa showed the same identity with the reference sequence in the database, they may still have ability to provide aminoglycoside resistance under low nutrient conditions (Cooper et al., 2020). The emergence of MDR S. Indiana, a dominant Salmonella serovar in China, has raised global awareness because MDR S. Indiana has also recently emerged at a rapid pace in other countries (Gong et al., 2019). The high prevalence of S. Indiana needs to be given more attention in future studies.
Despite some remaining limitations, the comprehensive information provided by WGS will greatly enhance the monitoring of transmission, HGT, VFs and ARGs. The presence of S. enterica carrying virulence and resistance determinants in retail meat compromises food security. The diverse virulence genes and ARGs contained in these isolates may increase the risk of this foodborne pathogen, which warrants constant monitoring. Our data suggested that Salmonella contamination should be monitored in not only chicken meat but also pork and duck meat.
Data Availability Statement
The datasets generated for this study can be found in the online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA675435.
Author Contributions
YH, BZ, and CX conceived and designed the project. NL, HH, and CX isolated and sequenced the isolates. YF analyzed the data. YL and CX performed the serotyping and antimicrobial susceptibility testing. NL, YF, and YP wrote the initial manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Key Research and Development Program of China (No. 2018YFC1603900) and the Starting Grants Programme for Young Talents at China Agricultural University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Yaqiong Guo at the CAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, for assistance with the review of the scripts applied in the present study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.636332/full#supplementary-material
Supplementary Figure 1 | SNP distance among the isolates. (A) SNP distance of isolates of S. Enteritidis from chicken and duck. (B) SNP distance of isolates of S. Indiana from chicken and duck. (C) SNP distance of isolates of S. Thompson from chicken and mutton. (D) SNP distance of isolates of S. Typhimurium from duck and pork. (E) SNP distance of isolates from different sources.
Supplementary Figure 2 | Prevalence of the top 10 plasmid replicons (A) and top 10 prophages (B) at different sampling time points.
Supplementary Figure 3 | Correlations between the serovars and the plasmid replicons.
Supplementary Figure 4 | Correlations between the serovars and the prophages.
Supplementary Figure 5 | Virulence genes in the chromosomes and plasmids. Green: virulence genes in chromosomes; brown: virulence genes in plasmids; purple: plasmid replicons.
Supplementary Figure 6 | Correlations between the serovars and the VFs.
Supplementary Figure 7 | Numbers of ARGs (A) and VFs (B) in the Salmonella genome belonging to different serovars.
Footnotes
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Anton, B., Sergey, N., Dmitry, A., Gurevich Alexey, A., Mikhail, D., Kulikov Alexander, S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Ashton, P. M., Nair, S., Peters, T. M., Bale, J. A., Powell, D. G., Painset, A., et al. (2016). Identification of Salmonella for public health serveillance using whole genome sequencing. PeerJ. 4:e1752. doi: 10.7717/peerj.1752
Bolger Anthony, M., Marc, L., and Bjoern, U. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114. doi: 10.1093/bioinformatics/btu170
Bortolaia, V., Kaas, R. S., Ruppe, E., Roberts, M. C., Schwarz, S., Cattoir, V., et al. (2020). ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 75, 3491–3500. doi: 10.1093/jac/dkaa345
Cha, M.-H., Woo, G.-J., Lee, W., Kim, S.-H., Woo, J.-H., Kim, J., et al. (2020). Emergence of transferable mcr-9 gene-carrying colistin-resistant Salmonella enterica Dessau ST14 isolated from retail chicken meat in Korea. Foodborne Pathog. Dis. 17, 720–727. doi: 10.1089/fpd.2020.2810
Chen, C., Wu, X. T., He, Q., Chen, L., Cui, C. Y., Zhang, Y., et al. (2019). Complete sequence of a tet(X4)-harboring IncX1 plasmid, pYY76-1-2, in Escherichia coli from a cow sample in China. Antimicrob. Agents Chemother. 63:e01528-19. doi: 10.1128/AAC.01528-19
Chen, L., Zheng, D., Liu, B., Yang, J., and Jin, Q. (2016). VFDB 2016: hierarchical and refined dataset for big data analysis-10 years on. Nucleic Acids Res. 44, D694–D697. doi: 10.1093/nar/gkv1239
Chen, Z., Bai, J., Wang, S., Zhang, X., Zhan, Z., Shen, H., et al. (2020). Prevalence, antimicrobial resistance, virulence genes and genetic diversity of Salmonella isolated from retail duck meat in southern China. Microorganisms 8:444. doi: 10.3390/microorganisms8030444
CLSI (2018). Performance Standards for Antimicrobial Susceptibility Testing, 28th Edn. Wayne, PA: CLSI.
Coburn, B., Grassl, G. A., and Finlay, B. B. (2007). Salmonella, the host and disease: a brief review. Immunol. Cell Biol. 85, 112–118. doi: 10.1038/sj.icb.7100007
Cooper, A. L., Low, A. J., Koziol, A. G., Thomas, M. C., Leclair, D., Tamber, S., et al. (2020). Systematic evaluation of whole genome sequence-based predictions of Salmonella serotype and antimicrobial resistance. Front. Microbiol. 11:549. doi: 10.3389/fmicb.2020.00549
De la Cruz, F., and Davies, J. (2000). Horizontal gene transfer and the origin of species: lessons from bacteria. Trends Microbiol. 8, 128–133. doi: 10.1016/S0966-842X(00)01703-0
den Bakker, H. C., Allard, M. W., Bopp, D., Brown, E. W., Fontana, J., Iqbal, Z., et al. (2014). Rapid whole-genome sequencing for surveillance of Salmonella enterica serovar Enteritidis. Emerg. Infect. Dis. 20, 1306–1314. doi: 10.3201/eid2008.131399
Dutilh, B. E., Cassman, N., McNair, K., Sanchez, S. E., Silva, G. G. Z., Boling, L., et al. (2014). A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 5:4498. doi: 10.1038/ncomms5498
El Ghany, M. A., Shi, X., Li, Y., Ansari, H. R., Hill-Cawthorne, G. A., Ho, Y. S., et al. (2016). Genomic and phenotypic analyses reveal the emergence of an atypical Salmonella enterica serovar Senftenberg variant in China. J. Clin. Microbiol. 54, 2014–2022. doi: 10.1128/JCM.00052-16
Figueira, R., and Holden, D. W. (2012). Functions of the Salmonella pathogenicity island 2 (SPI-2) type III secretion system effectors. Microbiology 158, 1147–1161. doi: 10.1099/mic.0.058115-0
Frank, J. A., Reich, C. I., Sharma, S., Weisbaum, J. S., Wilson, B. A., and Olsen, G. J. (2008). Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl. Environ. Microbiol. 74, 2461–2470. doi: 10.1128/AEM.02272-07
Frost, L. S., Leplae, R., Summers, A. O., and Toussaint, A. (2005). Mobile genetic elements: the agents of open source evolution. Nat. Rev. Microbiol. 3, 722–732. doi: 10.1038/nrmicro1235
Gardner, S. N., Slezak, T., and Hall, B. G. (2015). kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 31, 2877–2878. doi: 10.1093/bioinformatics/btv271
Gong, J., Zeng, X., Zhang, P., Zhang, D., Wang, C., and Lin, J. (2019). Characterization of the emerging multidrug-resistant Salmonella enterica serovar Indiana strains in China. Emerg. Microbes Infect. 8, 29–39. doi: 10.1080/22221751.2018.1558961
Grimont, P. A. D., and Weill, F. X. (2007). Antigenic Formulae of the Salmonella Serovars, 9th Edn. Paris: WHO Collaborating Centre.
Groisman, E. A., and Ochman, H. (1996). Pathogenicity islands: bacterial evolution in quantum leaps. Cell 87, 791–794. doi: 10.1016/S0092-8674(00)81985-6
Guibourdenche, M., Roggentin, P., Mikoleit, M., Fields, P. I., Bockemühl, J., Grimont, P. A. D., et al. (2010). Supplement 2003-2007 (No. 47) to the White-Kauffmann-Le Minor scheme. Res. Microbiol. 161, 26–29. doi: 10.1016/j.resmic.2009.10.002
Guo, S., Tay, M. Y. F., Thu, A. K., Seow, K. L. G., Zhong, Y., Ng, L. C., et al. (2019). Conjugative IncX1 plasmid harboring colistin resistance gene mcr-5.1 in Escherichia coli isolated from chicken rice retailed in Singapore. Antimicrob. Agents Chemother. 63:e01043-19. doi: 10.1128/AAC.01043-19
He, D., Partridge, S. R., Shen, J., Zeng, Z., Liu, L., Rao, L., et al. (2013). CTX-M-123, a novel hybrid of the CTX-M-1 and CTX-M-9 group β-lactamases recovered from Escherichia coli isolates in China. Antimicrob. Agents Chemother. 57, 4068–4071. doi: 10.1128/AAC.00541-13
He, T., Wei, R., Li, R., Zhang, L., Sun, L., Bao, H., et al. (2020). Co-existence of tet(X4) and mcr-1 in two porcine Escherichia coli isolates. J. Antimicrob. Chemother. 75, 764–766. doi: 10.1093/jac/dkz510
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2016). EGGNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293. doi: 10.1093/nar/gkv1248
Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Jacobsen, A., Hendriksen, R. S., Aaresturp, F. M., Ussery, D. W., and Friis, C. (2011). The Salmonella enterica pan-genome. Microb. Ecol. 62, 487–504. doi: 10.1080/02724988843000041
Jacoby, G. A., Strahilevitz, J., and Hooper, D. C. (2014). Plasmid-mediated quinolone resistance. Microbiol. Spectr. 2:PLAS-0006–2013. doi: 10.1128/microbiolspec.PLAS-0006-2013
Kempf, I., Jouy, E., and Chauvin, C. (2016). Colistin use and colistin resistance in bacteria from animals. Int. J. Antimicrob. Agents 48, 598–606. doi: 10.1016/j.ijantimicag.2016.09.016
Lerminiaux, N. A., Mackenzie, K. D., and Cameron, A. D. S. (2020). Salmonella pathogenicity island 1 (SPI-1): the evolution and stabilization of a core genomic type three secretion system. Microorganisms 8:576. doi: 10.3390/microorganisms8040576
Letunic, I., and Bork, P. (2019). Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Li, Y., Ma, X., Li, C., Dai, X., and Zhang, L. (2020). Occurrence and genomic characterization of ESBL-producing Escherichia coli ST29 strains from swine with abundant virulence genes. Microb. Pathog. 148:104483. doi: 10.1016/j.micpath.2020.1004483
Li, Y., Pei, X., Zhang, X., Wu, L., Liu, Y., Zhou, H., et al. (2019). A surveillance of microbiological contamination on raw poultry meat at retail markets in China. Food Control 104, 99–104. doi: 10.1016/j.foodcont.2019.04.037
Lin, M., Yang, Y., Yang, Y., Chen, G., He, R., Wu, Y., et al. (2020). Co-occurrence of mcr-9 and blaNDM-1 in Enterobacter cloacae isolated from a patient with bloodstream infection. Infect. Drug Resist. 13, 1397–1402. doi: 10.2147/IDR.S248342
Liu, Y. Y., Wang, Y., Walsh, T. R., Yi, L. X., Zhang, R., Spencer, J., et al. (2016). Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect. Dis. 16, 161–168. doi: 10.1016/S1473-3099(15)00424-7
Livingstone, P. G., Morphew, R. M., and Whitworth, D. E. (2018). Genome sequencing and pan-genome analysis of 23 Corallococcus spp. strains reveal unexpected diversity, with particular plasticity of predatory gene sets. Front. Microbiol. 9:3187. doi: 10.3389/fmicb.2018.03187
Lou, L., Zhang, P., Piao, R., and Wang, Y. (2019). Salmonella pathogenicity island 1 (SPI-1) and its complex regulatory network. Front. Cell. Infect. Microbiol. 9:270. doi: 10.3389/fcimb.2019.00270
Ma, S., Lei, C., Kong, L., Jiang, W., Liu, B., Men, S., et al. (2017). Prevalence, antimicrobial resistance, and relatedness of Salmonella isolated from chickens and pigs on farms, abattoirs, and markets in Sichuan province, China. Foodborne Pathog. Dis. 14, 667–677. doi: 10.1089/fpd.2016.2264
Mather, A. E., Reid, S. W. J., Maskell, D. J., Parkhill, J., Fookes, M. C., Harris, S. R., et al. (2013). Distinguishable epidemics of multidrug-resistant Salmonella Typhimurium DT104 in different hosts. Science 341, 1514–1517. doi: 10.1126/science.1240578
McDermott, P. F., Tyson, G. H., Kabera, C., Chen, Y., Li, C., Folster, J. P., et al. (2016). Whole-genome sequencing for detecting antimicrobial resistance in nontyphoidal Salmonella. Antimicrob. Agents Chemother. 60, 5515–5520. doi: 10.1128/AAC.01030-16
McMillan, E. A., Gupta, S. K., Williams, L. E., Jové, T., Hiott, L. M., Woodley, T. A., et al. (2019). Antimicrobial resistance genes, cassettes, and plasmids present in Salmonella enterica associated with United States food animals. Front. Microbiol. 10:832. doi: 10.3389/fmicb.2019.00832
Minnullina, L., Pudova, D., Shagimardanova, E., Shigapova, L., Sharipova, M., and Mardanova, A. (2019). Comparative genome analysis of uropathogenic Morganella morganii strains. Front. Cell. Infect. Microbiol. 9:167. doi: 10.3389/fcimb.2019.00167
Monte, D. F., Lincopan, N., Cerdeira, L., Fedorka-Cray, P. J., and Landgraf, M. (2019). Early dissemination of qnrE1 in Salmonella enterica serovar Typhimurium from livestock in South America. Antimicrob. Agents Chemother. 63:e00571-19. doi: 10.1128/AAC.00571-19
Nadon, C., Walle, I. V., Gerner-Smidt, P., Campos, J., Chinen, I., Concepcion-Acevedo, J., et al. (2017). PulseNet international: vision for the implementation of whole genome sequencing (WGS) for global food-borne disease serveillance. Euro Surveill. 22:30544. doi: 10.2807/1560-7917.ES.2017.22.23.30544
Nova, M. V., Durimel, L., La, K., Felten, A., Bessieres, P., Mistou, M., et al. (2019). Genetic and metabolic signatures of Salmonella enterica subsp. enterica associated with animal sources at the pangenomic scale. BMC Genomics 20:804. doi: 10.1186/s12864-019-6188-x
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Paradiso, R., Orsini, M., Censi, S. B., Borriello, G., and Galiero, G. (2016). Complete genome sequence of a Myoviridae bacteriophage infecting Salmonella enterica serovar Typhimurium. Genome Announc. 4, e00940-16. doi: 10.1128/genomeA.00940-16
Que, F., Wu, S., and Huang, R. (2013). Salmonella pathogenicity island 1(SPI-1) at work. Curr. Microbiol. 66, 582–587. doi: 10.1007/s00284-013-0307-8
Rao, L., Lv, L., Zeng, Z., Chen, S., He, D., Chen, X., et al. (2014). Increasing prevalence of extended-spectrum cephalosporin-resistant Escherichia coli in food animals and the diversity of CTX-M genotypes during 2003-2012. Vet. Microbiol. 172, 534–541. doi: 10.1016/j.vetmic.2014.06.013
Robertson, J., and Nash, H. E. (2018). MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genom. 4:e000206. doi: 10.1099/mgen.0.000206
Ross, A., Ward, S., and Hyman, P. (2016). More is batter: selecting for broad host range bacteriophages. Front. Microbiol. 7:1352. doi: 10.3389/fmicb.2016.01352
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shi, M., Jun-fei, B., Seale, J., and Wahl, T. (2015). Demographics, societal aging, and meat consumption in China. J. Integr. Agric. 14, 995–1007. doi: 10.1016/S2095-3119(14)60984-9
Suez, J., Porwollik, S., Dagan, A., Marzel, A., Schorr, Y. I., Desai, P. T., et al. (2013). Virulence gene profiling and pathogenicity characterization of non-typhoidal Salmonella accounted for invasive disease in humans. PLoS One 8:e58449. doi: 10.1371/journal.pone.0058449
Sun, D., Jin, S., Wang, J., Wang, Z., Fan, J., Xu, Z., et al. (2020). Multidrug-resistant Escherichia coli strain isolated from swine in China harbors mcr-3.1 on a plasmid of the IncX1 type that cotransfers with mcr-1.1. Foodborne Pathog. Dis. 17, 597–601. doi: 10.1089/fpd.2019.2769
Tan, G., Opitz, L., Schlapbach, R., and Rehrauer, H. (2019). Long fragments achieve lower base quality in Illumina paired-end sequencing. Sci. Rep. 9:2856. doi: 10.1038/s41598-019-39076-7
Valero-Mora, P. M. (2010). ggplot2: Elegant Graphics for Data Analysis. Berlin: Springer. doi: 10.18637/jss.v035.b01
Van Asten, A. J. A. M., and Van Dijk, J. E. (2005). Distribution of “classic” virulence factors among Salmonella spp. FEMS Immunol. Med. Microbiol. 44, 251–259. doi: 10.1016/j.femsim.2005.02.002
Walker, B., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963.t004
Wang, W., Zhao, L., Hu, Y., Dottorini, T., Fanning, S., Fanning, S., et al. (2020). Epidemiological study on prevalence, serovar diversity, multidrug resistance, and CTX-M-type extended-spectrum β-lactamases of Salmonella spp. from patients with diarrhea, food of animal origin, and pets in several provinces of China. Antimicrob. Agents Chemother. 64:e00092-20. doi: 10.1128/AAC.00092-20
Wang, Y., Xu, C., Zhang, R., Chen, Y., Shen, Y., Hu, F., et al. (2020). Changes in colistin resistance and mcr-1 abundance in Escherichia coli of animal and human origins following the ban of colistin-positive additives in China: an epidemiological comparative study. Lancet Infect. Dis. 20, 1161–1171. doi: 10.1016/S1473-3099(20)30149-3
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Worley, J., Meng, J., Allard, M. W., Brown, E. W., and Timme, R. E. (2018). Salmonella enterica phylogeny based on whole-genome sequencing reveals two new clades and novel patterns of horizontally acquired genetic elements. MBio 9, e2303–e2318. doi: 10.1128/mBio.02303-18
Wu, R., Yi, L. X., Yu, L. F., Wang, J., Liu, Y., Chen, X., et al. (2018). Fitness advantage of mcr-1-bearing IncI2 and IncX4 plasmids in vitro. Front. Microbiol. 9:331. doi: 10.3389/fmicb.2018.00331
Yang, B., Qu, D., Zhang, X., Shen, J., Cui, S., Shi, Y., et al. (2010). Prevalence and characterization of Salmonella serovars in retail meats of marketplace in Shaanxi, China. Int. J. Food Microbiol. 141, 63–72. doi: 10.1016/j.ijfoodmicro.2010.04.015
Yang, X., Wu, Q., Zhang, J., Huang, J., Chen, L., Wu, S., et al. (2019). Prevalence, bacterial load, and antimicrobial resistance of Salmonella serovars isolated from retail meat and meat products in China. Front. Microbiol. 10:2121. doi: 10.3389/fmicb.2019.02121
Yoshida, C. E., Kruczkiewicz, P., Laing, C. R., Lingohr, E. J., Gannon, V. P. J., Nash, J. H. E., et al. (2016). The Salmonella in silico typing resource (SISTR): an open web-accessible tool for rapidly typing and subtyping draft Salmonella genome assemblies. PLoS One 11:e0147101. doi: 10.1371/journal.pone.0147101
Zhan, Z., Xu, X., Gu, Z., Meng, J., Wufuer, X., Wang, M., et al. (2019). Molecular epidemiology and antimicrobial resistance of invasive non-typhoidal Salmonella in China, 2007–2016. Infect. Drug Resist. 12, 2885–2897. doi: 10.2147/IDR.S210961
Zhang, L., Fu, Y., Xiong, Z., Ma, Y., Wei, Y., Qu, X., et al. (2018). Highly prevalent multidrug-resistant Salmonella from chicken and pork meat at retail markets in Guangdong, China. Front. Microbiol. 9:2104. doi: 10.3389/fmicb.2018.02104
Zhang, X. Z., Lei, C. W., Zeng, J. X., Chen, Y. P., Kang, Z. Z., Wang, Y. L., et al. (2018). An IncX1 plasmid isolated from Salmonella enterica subsp. enterica serovar Pullorum carrying blaTEM–1B, sul2, arsenic resistant operons. Plasmid 100, 14–21. doi: 10.1016/j.plasmid.2018.09.007
Zhang, Z., Cao, C., Liu, B., Xu, X., Yan, Y., Cui, S., et al. (2018). Comparative study on antibiotic resistance and DNA profiles of Salmonella enterica serovar Typhimurium isolated from humans, retail foods, and the environment in Shanghai, China. Foodborne Pathog. Dis. 15, 481–488. doi: 10.1089/fpd.2017.2414
Zhao, C., Ge, B., De Villena, J., Sudler, R., Yeh, E., Zhao, S., et al. (2001). Prevalence of Campylobacter spp., Escherichia coli, and Salmonella Serovars in retail chicken, turkey, pork, and beef from the greater Washington, D.C., area. Appl. Environ. Microbiol. 67, 5431–5436. doi: 10.1128/AEM.67.12.5431-5436.2001
Keywords: Salmonella enterica, retail meat, next-generation sequencing, serotype, genome plasticity, virulence gene, antimicrobial resistance gene, MLST
Citation: Lyu N, Feng Y, Pan Y, Huang H, Liu Y, Xue C, Zhu B and Hu Y (2021) Genomic Characterization of Salmonella enterica Isolates From Retail Meat in Beijing, China. Front. Microbiol. 12:636332. doi: 10.3389/fmicb.2021.636332
Received: 03 December 2020; Accepted: 16 March 2021;
Published: 07 April 2021.
Edited by:
Catherine D. Carrillo, Canadian Food Inspection Agency, CanadaReviewed by:
Bijay Khajanchi, United States Food and Drug Administration, United StatesJoão Pedro Rueda Furlan, University of São Paulo, Brazil
Copyright © 2021 Lyu, Feng, Pan, Huang, Liu, Xue, Zhu and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongfei Hu, aHV5b25nZmVpQGNhdS5lZHUuY24=
†These authors have contributed equally to this work