 Nikita A. Nikulin
Nikita A. Nikulin Andrei A. Zimin
Andrei A. Zimin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 06 April 2021
Sec. Evolutionary and Genomic Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.632686
The Tevenvirinae viruses are some of the most common viruses on Earth. Representatives of this subfamily have long been used in the molecular biology studies as model organisms – since the emergence of the discipline. Tevenvirinae are promising agents for phage therapy in animals and humans, since their representatives have only lytic life cycle and many of their host bacteria are pathogens. As confirmed experimentally, some Tevenvirinae have non-canonical DNA bases. Non-canonical bases can play an essential role in the diversification of closely related viruses. The article performs a comparative and evolutionary analysis of Tevenvirinae genomes and components of Tevenvirinae genomes. A comparative analysis of these genomes and the genes associated with the synthesis of non-canonical bases allows us to conclude that non-canonical bases have a major influence on the divergence of Tevenvirinae viruses within the same habitats. Supposedly, Tevenvirinae developed a strategy for changing HGT frequency in individual populations, which was based on the accumulation of proteins for the synthesis of non-canonical bases and proteins that used those bases as substrates. Owing to this strategy, ancestors of Tevenvirinae with the highest frequency of HGT acquired genes that allowed them to exist in a certain niche, and ancestors with the lowest HGT frequency preserved the most adaptive of those genes. Given the origin and characteristics of genes associated with the synthesis of non-canonical bases in Tevenvirinae, one can assume that other phages may have similar strategies. The article demonstrates the dependence of genomic diversity of closely related Tevenvirinae on non-canonical bases.
Bacteriophages are some of the most abundant biological objects on Earth (Wommack and Colwell, 2000). Their reproductive cycles, as well as their interactions with the hosts and the environment, are very diverse. The advancement of modern sequencing techniques allows researchers to examine individual genomes, metaviromes, and metagenomes – which, in turn, makes it possible to conduct detailed studies of evolutionary, coevolutionary, and ecological processes in bacteriophages.
One of the first routinely studied bacteriophages were T-even phages: T2, T4, T6 – which played an essential role in the development of molecular genetics (Abedon, 2000). According to ICTV Taxonomy (2019), they belong to the Tequatrovirus genus, Tevenvirinae subfamily, Myoviridae family, Caudovirales order. In addition to these T-even bacteriophages, many representatives of Tevenvirinae (for example, RB phage) were used as objects in the studies of biology of bacterial viruses, (Russell, 1967). These bacteriophages are considered to be in the group of the most promising candidates for phage therapy (Sarker et al., 2012). The development of sequencing technologies has shown a wide distribution of Tevenvirinae and related phages in nature, as well as a great variety of their host bacteria, which include animal and human pathogens (Petrov et al., 2010; Sullivan et al., 2010; Roux et al., 2015).
A wide representation of Tevenvirinae genomes in the online databases has made it possible to analyze their general composition and structural organization, i.e., their pan-genome (Medini et al., 2005; Petrov et al., 2010). The analysis has revealed conserved regions of the Tevenvirinae genome typical of the subfamily members (core-genome), as well as variable zones (quasi-core and plastic genomes) (Comeau et al., 2007; Petrov et al., 2010). Comprehensive knowledge of protein functions of some members of the subfamily (e.g., Escherichia virus T4) has allowed researchers to identify the corresponding protein homologs, whose genes are located in different regions of pan-genome (Petrov et al., 2010). On the basis of these data, studies of the evolutionary divergence of Tevenvirinae have become possible.
One of the experimentally confirmed features of Tevenvirinae is the presence of non-canonical (modified) DNA bases in some of their representatives (Weigele and Raleigh, 2016; Thomas et al., 2018; Hutinet et al., 2019). There are also data on the putative proteins that are highly likely to participate in the biosynthesis of non-canonical bases in Tevenvirinae – however, the functions of these proteins have yet to be experimentally confirmed (Iyer et al., 2013; Hutinet et al., 2019). Some of the non-canonical bases are supposed to serve as a barrier to gene transfer and, therefore, to be a speciation factor among related Tevenvirinae phages (Petrov et al., 2010; Thomas et al., 2018). As shown earlier, transfer of genetic information between phages with different sets of DNA bases can be very difficult (Russell, 1967; Thomas et al., 2018).
To better understand the evolutionary divergence of Tevenvirinae, as well as the influence of non-canonical DNA base systems on this process, we have conducted a comparative genome-wide and pan-genomic study of Tevenvirinae viruses registered in the NCBI database. In the study, we compare data of phylogenetic, phylogenomic, and bipartite network analyses to identify evolutionary changes in the processes of genetic information exchange. In addition, we have performed a comparative analysis of the genetic loci involved in the synthesis of non-canonical bases and have searched for their homologs using BLAST (Madden et al., 1996) – to identify the specifics of inheritance of such genes.
Altogether, the analysis conducted has allowed us to identify specific genetic and genomic changes in the systems of Tevenvirinae responsible for the composition of their sets of DNA bases. This contributes to our understanding of the evolution and divergence of this subfamily of phages and may help to identify similar systems in the phages of other phylogenetic groups.
Two hundred and five complete genomes available in the NCBI database as of July 29, 2019 have been uploaded in the “gbk” format. The genomes met the parameters of (complete genome) and [Taxonomy ID: 1198136 (Organism “Tevenvirinae”)] and were used for further analysis. The GenBank accession numbers, phage names, bacteria used for phage isolation, sources of isolation, and available links are given in Supplementary Table 1 of the Supplementary Section.
The annotations of 205 Tevenvirinae genomes deposited in GenBank were verified according to the following procedure. The annotations were checked for the presence of predicted ORFs and, if they were absent, the genomes were annotated automatically using the Genome Annotation PATRIC service with the parameters (Domain: Viruses) and (Annotation recipe: Phage) (Brettin et al., 2015; Wattam et al., 2018). The homologs of the translated genome frames were clustered using the GET_HOMOLOGUES (get_homologues.pl) script with the parameters of [minimal coverage (-C): 40%], [minimal identity (-S): 50%], [minimal e-value (-E): standard (1e-05)], and [algorithm clustering: COGtriangle] (Kristensen et al., 2010; Contreras-Moreira and Vinuesa, 2013). The minimal values of the coverage and identity parameters were selected on the basis of the best clustering of translated ORFs of core genome of T4-related bacteriophages determined by Petrov et al., 2010. If translated ORFs of core genome were absent in the analyzed genome, its sequence was automatically reannotated using PATRIC and then verified again. If ORF of a core gene was shifted, disrupted or absent, the genome was excluded from further analysis.
The functions of the proteins of core proteome were predicted on the basis of clustering of the corresponding homologs with the translated ORFs of the genome of Escherichia virus T4 (NC_000866.4) (Miller et al., 2003).
To analyze the homolog proteins of Tevenvirinae proteome obtained by the get_homologues.pl script, we used a pan-genomic matrix of presence/absence of clustered sequences, which was generated using the compare_clusters.pl script. The composition of the pan-genome: core-genome (Medini et al., 2005), soft-core-genome (Kaas et al., 2012), shell (Wolf et al., 2012; Vernikos et al., 2015), cloud (Wolf et al., 2012) – was determined using the parse_pangenome_matrix.pl script. A heat map of pan-genome clusters was built using the plot_matrix_heatmap.sh script.
On the basis of the binary matrix of cluster presence/absence, a pan-genomic tree was constructed using the estimate_pangenome_phylogenies.sh GET_PHYLOMARKERS script (Vinuesa et al., 2018) with 100 replications (-r). A phylogenetic tree was constructed from MUSCLE-aligned sequences of the main head protein, which is used as a phylogenetic marker of this group of viruses (Comeau and Krisch, 2008), with the help of W-IQ-TREE (Trifinopoulos et al., 2016). The phylogenomic tree was constructed using VICTOR (Meier-Kolthoff and Göker, 2017) and 100 nucleotide sequences of genomes (tool limitation), which formed separate clades along the pan-genome tree. A cluster-genome bipartite network, which was based on the proteins whose genes were included in the pan-genome shell, was visualized in Cytoscape (Shannon et al., 2003).
The search for the homologs of clusters specific for a certain clade of genomes obtained on the pan-genomic tree and clusters presumably associated with non-canonical bases was carried out using PSI-BLAST (Altschul et al., 1997; Schäffer et al., 2001) and HMMER (Zimmermann et al., 2018).
For the initial search for homologs in PSI-BLAST, we used an amino acid sequence, belonging to one of the viral genomes, from a cluster. Then, a list of homologous proteins was obtained. Sequences of viruses, belonging to Taxonomy ID 1198136 (Tevenvirinae), were selected. The search was repeated (1 iteration). If the cluster was formed by a single protein and after the initial search no other homologous Tevenvirinae sequences were found, then the search was not repeated. The search parameters were as follows: (Max target sequences: 1000), (Word size: 2), [Matrix: BLOSUM45 (in case of CPU exceeding, BLOSUM62)]. To refine the annotations of clusters, in which homologs were found only among hypothetical proteins, HMMER was additionally used. A search was performed with the standard parameters of the nr30 database and a matrix based on the protein sequences aligned with MUSCLE (Edgar, 2004), which is included in the UGENE toolkit (Okonechnikov et al., 2012).
Individual regions of the genomes, containing genes associated with the synthesis of non-canonical bases, were compared using Easyfig (Sullivan et al., 2011).
To further verify the results, a search for Tevenvirinae genomes among uncultured viruses from GenBank and IMG/VR (Roux et al., 2021) databases was conducted.
For GenBank, PSI-BLAST was used with the parameters: (Max target sequences: 1000), (Word size: 2), (Matrix: BLOSUM45), [Organism: Viruses (taxid:10239)]. A PSSM matrix was created based on the amino-acid sequences of the Tevenvirinae (taxid:1198136) proteins that comprise the core. The genomes of the uncultured viruses, whose proteins were found on the first PSI-BLAST iteration, were checked for the genome completeness: only those genomes which were marked as (complete genome) were selected for further analysis.
For IMG/VR, Viral BLAST was used. The search parameters were as follows: (Program: blastn), (Blast Database: Viral Sequence blast database), (E-value: 1e-2). The twenty genomes of the Tevenvirinae viruses, belonging to groups determined by pan-genomic analysis, were used as query sequences. Of the genomes found, only those that met the criterion for high quality genome (Quality: High-quality) were selected for further analysis.
We have also uploaded and included into the dataset the Tevenvirinae genomes deposited within the GenBank database from July 29, 2019 to January 30, 2021.
Clustering and selection of genomes were carried out as described in Genome Clustering and Annotation. Clusters determined by the results of pan-genomic analysis were used as the core genome. Phylogenomic and pan-genomic trees were built as described in the section “Phylogenomic Tree, Phylogenetic Tree, Pan-genomic Tree and Bipartite Network.”
A survey of the annotations of genomes deposited in GenBank showed that Escherichia phages CF2 and HP3 lacked predicted ORFs, so they were reannotated before clustering. In total, the procedure of clustering was carried out three times: (1) clustering of initially annotated genomes (with the exception of CF2 and HP3 genomes); (2) clustering of reannotated genomes that initially lacked core genome components; and (3) clustering of selected genomes. The numbers of pan-genome components in each of the genomes, which were obtained by using the parse_pangenome_matrix.pl script, and the heat map of the cluster presence/absence are given in Supplementary Data Sheet 1 and Supplementary Figure 1.
Clustering of initially annotated genomes (1) showed that 79 of them lacked some core-genome genes. After clustering with reannotated genomes (2), we found that the core protein of the late transcription co-activator (gp33 in the T4 phage) of Vibrio phages, belonging to the genus Schizotequatrovirus, did not form a common homologous cluster with the gp33 co-activator proteins of other Tevenvirinae. Since these clusters of proteins had very low homology with each other, the homologs of the late transcription co-activator of Vibrio phages as a core-gene were not taken into account when genomes were selected. A similar situation was with the genes of tail fibril proteins (gp35, gp36, and gp37). These genes have high mosaicism and are often clustered with non-homologous genes of fibrils of other phages (Petrov et al., 2010).
At the second stage, the genomes Aeromonas phage phiAS5, Shigella phage SHFML-26, Shigella phage Sf25, Vibrio phage nt-1, and Vibrio phage VH7D were not used for the analysis. Due to the shift of the reading frames in a number of genes, they lacked the necessary components of the core genome.
In addition, when examining the soft core of the pan genome, we found that in the genome of Aeromonas phage Ah1 there was a shift in the reading frame in the gene for the head assembly protein (gp40 in phage T4); therefore, this phage was not used for the analysis either.
We clustered the remaining 199 genomes again. As a result, they all contained the necessary set of core genes (with the exception of the genes described above).
At (3) stage 57499 amino acid sequences were used, 4530 homologous clusters were obtained: 61 clusters were included in the core, 76 (taking into account the core-genome clusters) in the soft core, 1966 in the shell, and 2488 in the cloud. As a result, 79 genomes were reannotated, 7 of them were removed and 199 genomes were used for further analysis.
On the basis of the binary matrix of cluster presence/absence, a pan-genomic tree was built (Figure 1). The best model: GTR2 + FO + I + G4. Log-likelihood of the consensus tree: −41953.89217. Figure 2 shows a phylogenetic tree constructed on the basis of homologous sequences of the main head protein. The best model: LG + F + I + G4. The best score found: −15512.735. On the basis of the genome nucleotide sequences, a phylogenomic tree was built (Figure 3). As verified by statistical analysis, the main clades on all trees were identified with high probabilities. Exceptions are some minor clades on the pan-genomic tree, which are represented by a single genome: Pseudomonas phage PspYZU05 [aBayes support (Anisimova et al., 2011) 0.333; ultrafast bootstrap support (Hoang et al., 2018) 15%], Serratia phage PS2 (0.379; 31%, respectively). Groups of viruses which form separate clades are marked by different colors and also by Roman figures (descriptions of groups are given below and in Supplementary Data Sheet 4). Colored circles denote statistical values given in Supplementary Figures 2–4.
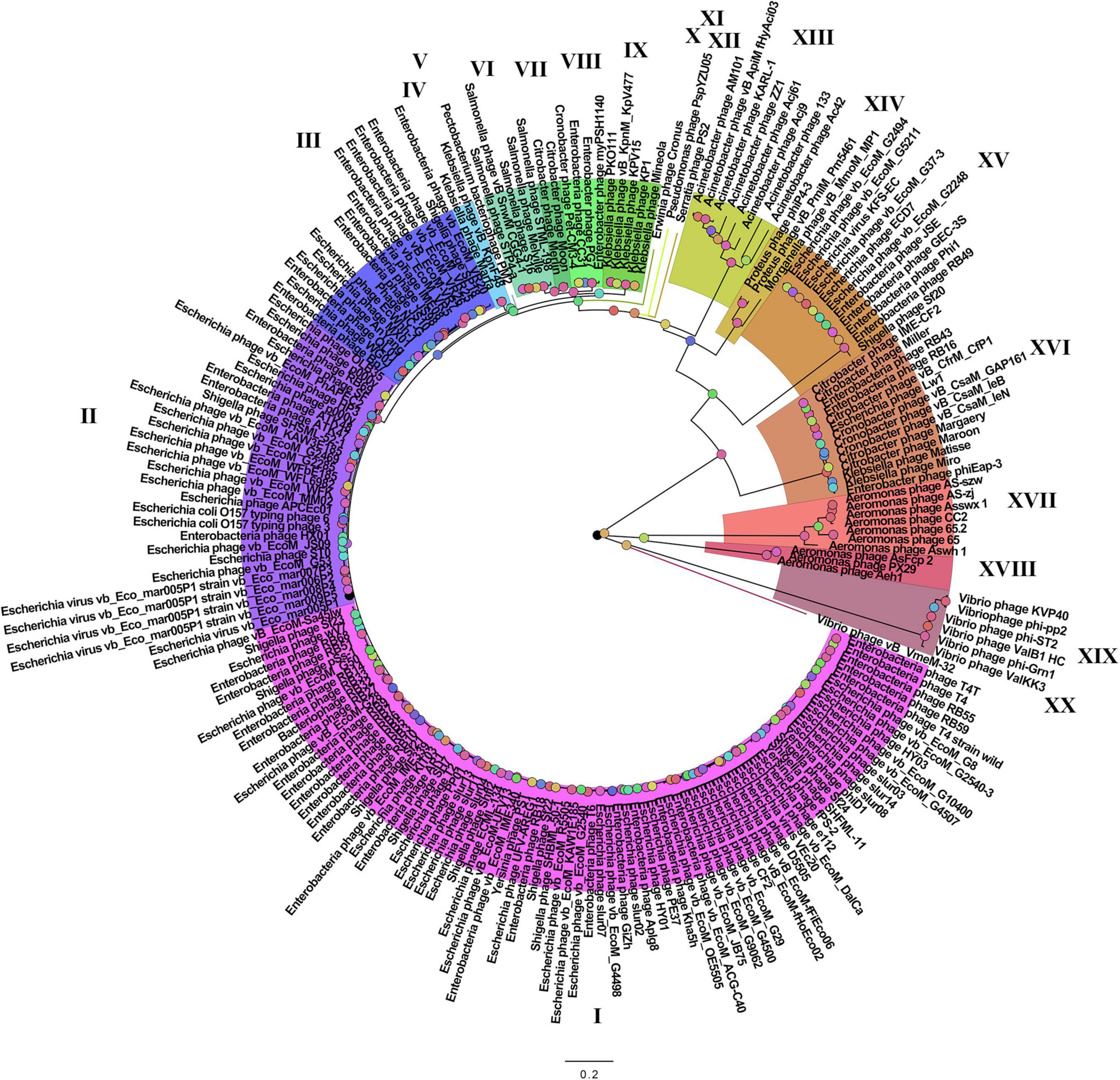
Figure 1. The pan-genomic tree of Tevenvirinae. Selective clades are marked with Roman numerals and colors. The statistical values presented along with the tree in Supplementary Figure 2 are marked with colored circles.
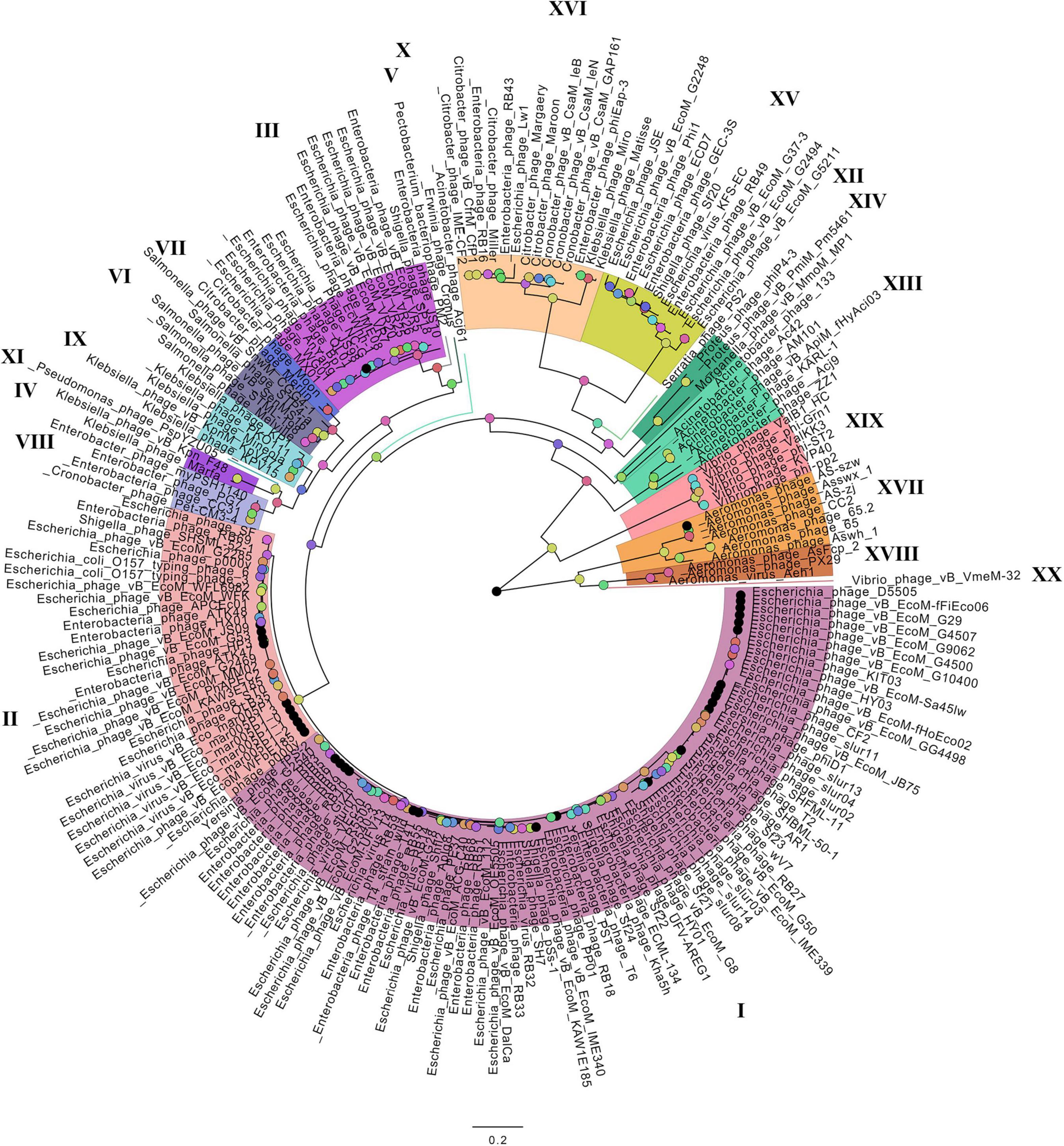
Figure 2. The phylogenetic tree of the main head protein of Tevenvirinae. Selective clades are marked with Roman numerals and colors. The statistical values presented along with the tree in Supplementary Figure 3 are marked with colored circles.
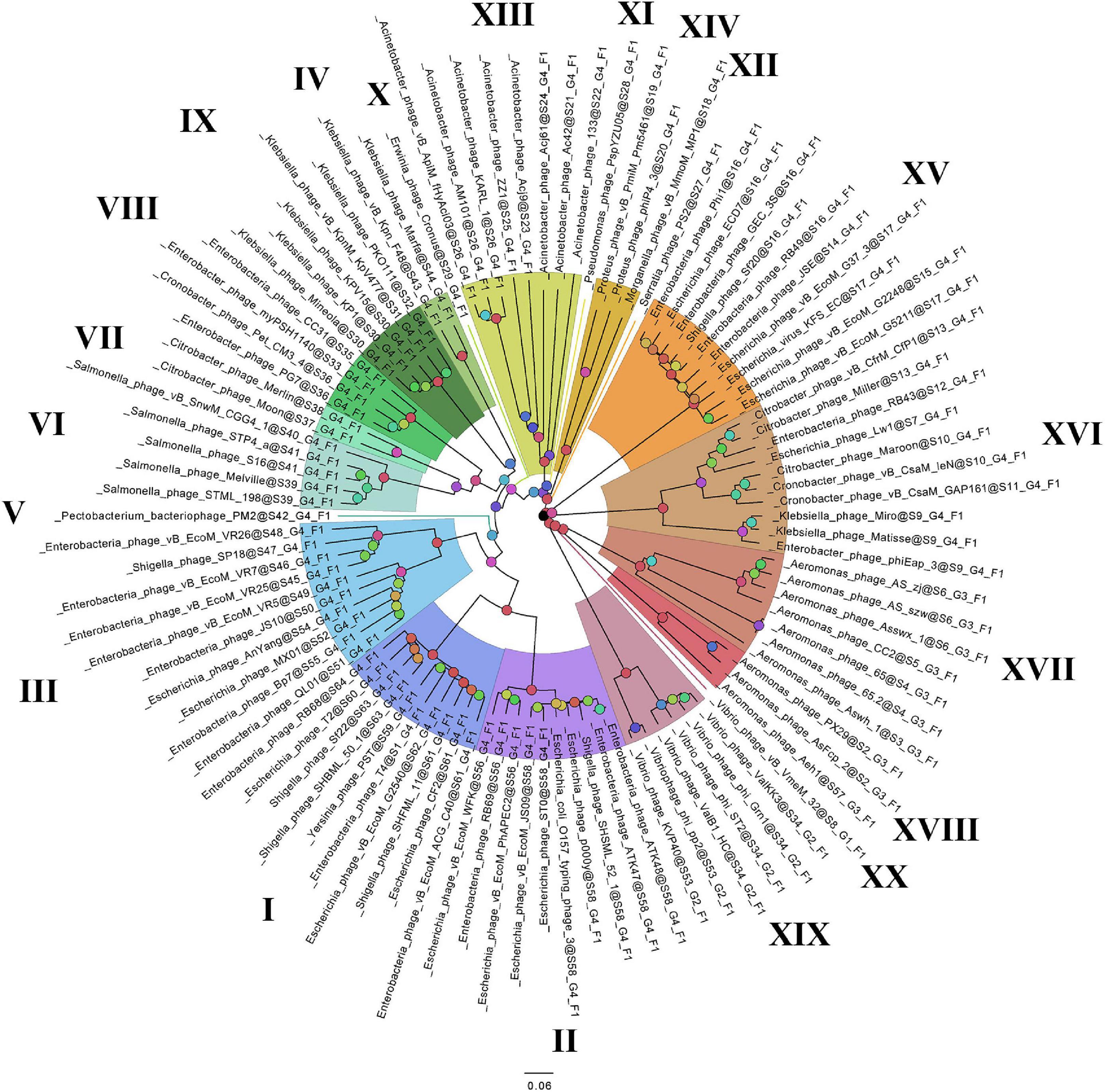
Figure 3. The phylogenomic tree of Tevenvirinae. Selective clades are marked with Roman numerals and colors. The statistical values presented along with the tree in Supplementary Figure 4 are marked with colored circles.
A cluster-genome bipartite network based on the clusters of the pan-genome envelope is shown in Figure 4. Colored nodes are genomes of viruses, color is the group to which the virus belongs. Homologous clusters are represented by the intersections of edges.

Figure 4. A cluster-genome bipartite network of the pan-genome envelope of Tevenvirinae. Genomes that belong to the same cluster (group) are marked with colored circles. The connected nodes represent individual clusters of translated ORF homologs.
Analyzing the pan-genomic tree, we identified 20 Tevenvirinae groups. The following criteria were used to divide genomes into separate groups:
(1) Each group forms a separate clade on the pan-genome tree.
(2) Each group has GSCs (homologous clusters with nucleotide sequences present in all genomes of the group and absent in the genomes of other groups).
(3) NCBI phage taxonomic classification, which can be accessed in the Taxonomy Database, should also be taken into account. Phages, belonging to one genus and forming a separate clade with clearly identifiable GSC, were referred to one group. The only exception is Group 3, which contains members of two phage genera, Dhakavirus and Gaprivervirus. This is because the clade formed by the members of the Dhakavirus genus does not have identifiable GSC.
In the case of phages that have not yet been classified down to the genus level, only the first two criteria were applied.
It should be noted that – as shown by the analysis of the tree and Supplementary Table 1 – phages from the groups represented by more than one genome were isolated from related (at the level of order, family, or genus) host bacteria: Enterobacteriales (I–III, VIII, XV, XVI), Morganellaceae (XIV), Klebsiella (IV, IX), Salmonella (VI), Citrobacter (VII), Acinetobacter (XIII), Aeromonas (XVII, XVIII), Vibrio (XIX).
A comparative analysis of the pan-genomic, phylogenomic, and phylogenetic trees showed that in all cases, the representatives of the identified groups were in the same clades – with the exception of group XIII, a representative of which (Acinetobacter phage Acj61) formed a separate clade on the phylogenetic tree. The main differences between the three trees were in the clade hierarchy and hierarchy of representatives within the clades. These differences may be due to the fact that pan-genomic and phylogenomic trees reflect the exchange of genetic information between Tevenvirinae phages, whereas phylogenetic tree shows the evolution of a single conservative protein, the main protein of the head. Overall, these trees agree with the views on Tevenvirinae bacteriophages presented in other publications (Tétart et al., 2001; Ignacio-Espinoza and Sullivan, 2012).
As noted by Gautreau et al., 2020, the pan-genome shell is of greatest interest in examining the dynamics of the development of bacterial genomes, reflecting their interactions with the ecological niche. This statement may be even more relevant toward bacteriophages, since their evolution is modular (Iranzo et al., 2016). To analyze the envelope, we constructed a bipartite network based on the clusters of this component of pan-genome. One can see that, in general, the cluster network coincides with the pan-genomic tree, yet there is a number of differences. For example, groups I–III of the network are “intermixed” with each other, which is not surprising given the specifics of the ecological conditions of their habitats. In this respect, it is worth to note the genome of Vibrio phage vB_VmeM-32, the only representative of the XX group. It has the smallest number of common clusters with other genomes. However, the majority of these clusters are characteristic of the genomes of most other groups, as indicated by the location of the phage in the network and its connections.
To get more information about the phage groups identified, we analyzed the clusters of homologs specific to each group.
To characterize these clusters (GSCs, group-specific clusters), we searched for homologous sequences. The search revealed (1) GSCs that did not have homologs among the proteins registered in the Non-Redundant Protein Sequences (nr) database; (2) GSCs that had homologs among hypothetical proteins of other organisms; and (3) GSCs that had homologs among proteins of other organisms with a predicted function. The numbers of clusters identified according to these criteria are given in Table 1. Characteristics of clusters of each group are presented in Supplementary Text 1 and Supplementary Table 2.
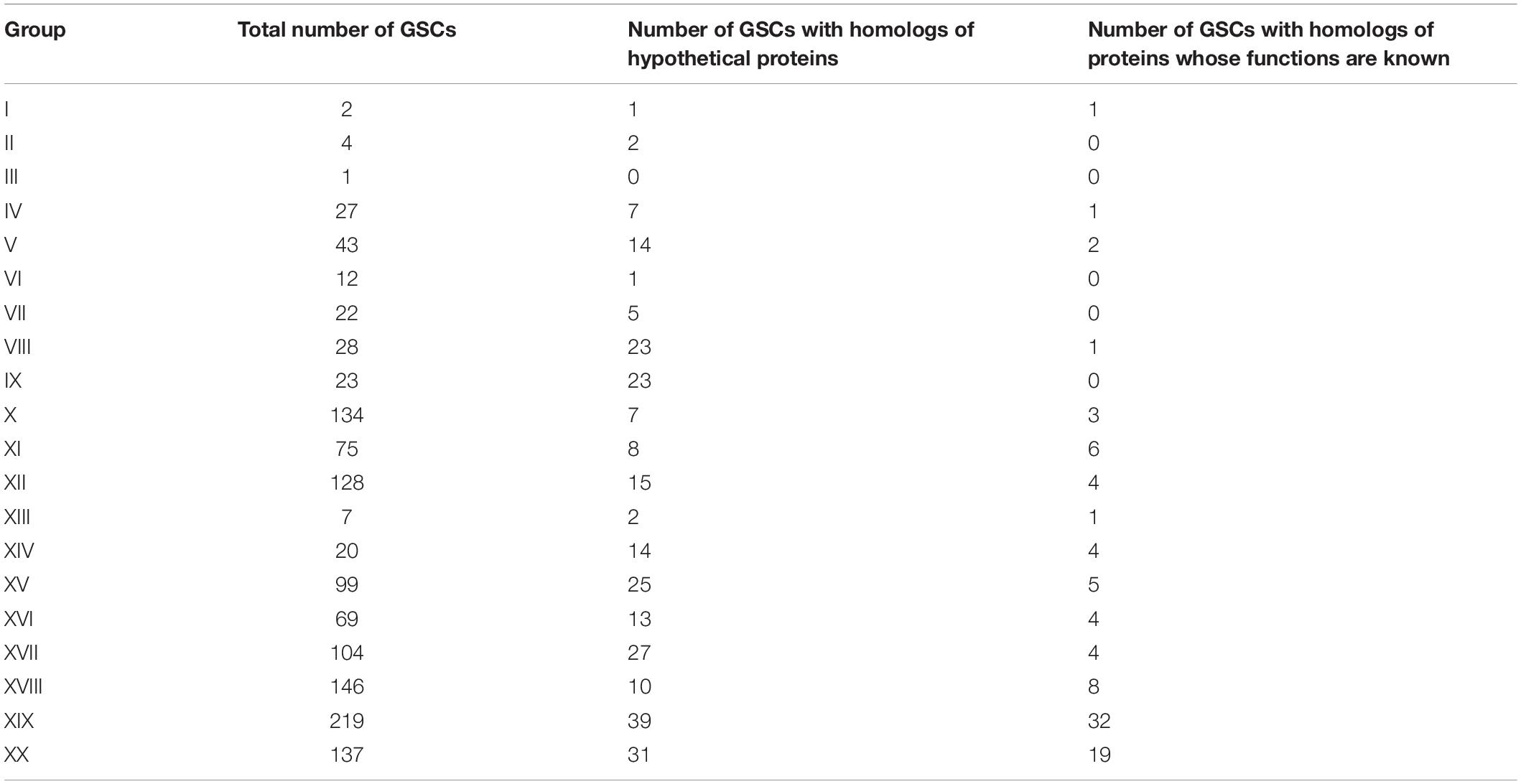
Table 1. The number of group-specific clusters.
As mentioned above, bacteriophages of group XIX have GSCs associated with the synthesis of modifications of 7-deazaguanine. As confirmed experimentally, some Tevenvirinae phages can have other non-canonical bases: glucosylated hmdC (T-even bacteriophages from group I); arabinosylated hmdC (RB69 from group II). Correspondingly, we have searched Tevenvirinae for the protein homologs that are involved – either in vitro or in silico, according to the literature data (Petrov et al., 2010; Iyer et al., 2013; Thomas et al., 2018; Hutinet et al., 2019) – in the synthesis of such bases (or are associated with them in any other way). A list of such proteins and their genes is presented in Table 2.

Table 2. Proteins associated or hypothetically associated with non-canonical bases.
The homologs of these proteins, belonging to the phages considered, are given in Supplementary Table 3. The overlap of group gene sets associated with the synthesis of non-canonical bases is shown in Figure 5.
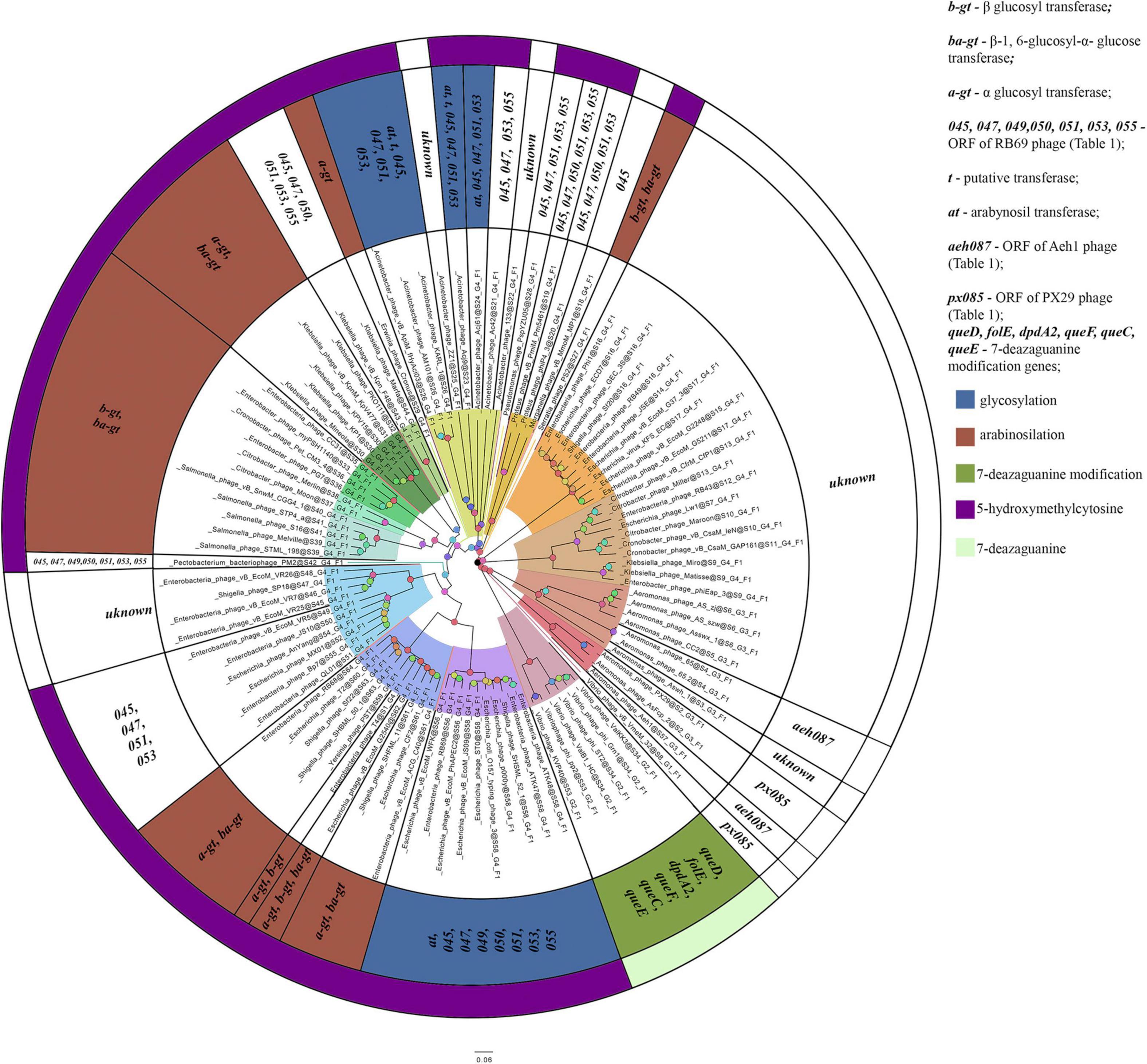
Figure 5. A number of gene sets associated with the synthesis of non-canonical bases. The colors indicate known modifications of non-canonical bases and the non-canonical bases as they are. The genes whose products involved or presumably involved in base modification are additionally marked.
Thomas et al., 2018 showed that most of the genes, whose products were involved or presumably involved in arabinosylation of hmdC in DNA, were located in the region between the genes encoding DNA polymerase and UvsX. The analysis of phages, having proteins associated with the synthesis of hmdC and its modifications, showed that the arrangement of genes encoding those proteins was quite similar. Since the region is surrounded by the genes of head vertex assembly chaperone and DNA polymerase, we have analyzed the genome fragments between them. Figure 6 shows variations of this region revealed in closely related Tevenvirinae phages.
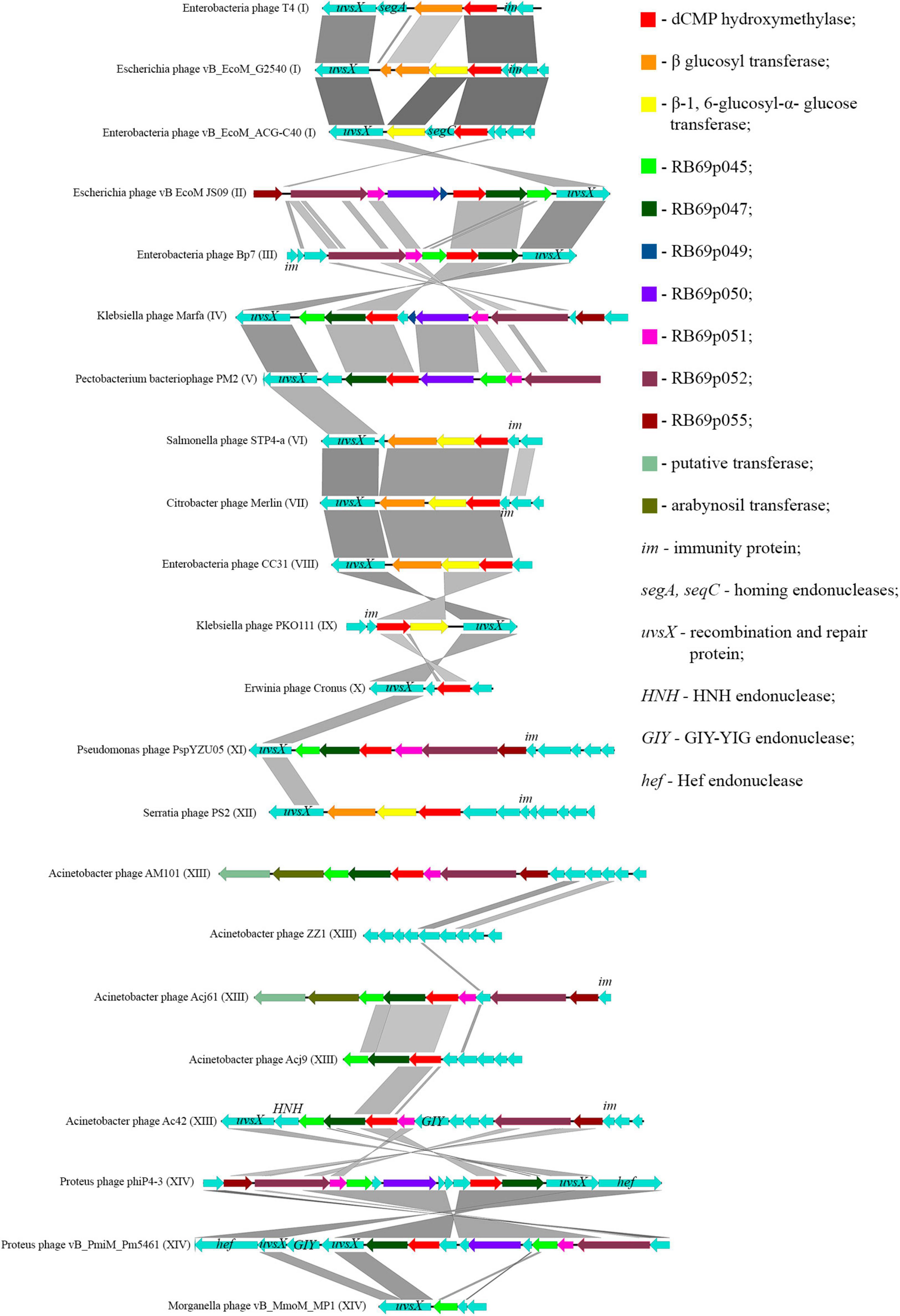
Figure 6. Variations in the region between the genes homologous to the genes of DNA polymerase and head vertex assembly chaperone in closely related Tevenvirinae that have homologs of hmdC-associated proteins. The genes whose products are described in Table 2 are indicated by color; the genes whose products have homologs with known functions are additionally marked.
It should be noted that in Acinetobacter phage Acj9, the genes whose products are supposedly associated with the synthesis of non-canonical bases are located in a wider region of the genome: between the genes of helicase proteins and DNA polymerase (a part of the core genome). The rest of the phages had those genes located in the region considered above.
Interestingly, in the phages closely related to RB69, the gene of arabinosyl transferase is located outside the region considered. However, Acinetobacter phage AM101 and Acinetobacter phage Acj61 have two homologs of the enzyme, and their genes are located within this region.
In the majority of closely related phages, variations in the region are about presence or absence of the genes that presumably encode homing endonucleases. It was previously suggested that homing endonucleases were involved in the horizontal transfer of Tevenvirinae genes (Petrov et al., 2010). Therefore, one could assume that the genes associated with base modification were also involved in this transfer.
It should also be noted that the region conserved for closely related Tevenvirinae.
In this article, we only analyzed the region associated with hmC, since the region associated with the synthesis of 7-deazaguanine modifications was considered in detail in other works (Hutinet et al., 2019). As for the phages with other possible base modifications, only one or two genes are known.
The comparison of the results of GSC and genomic analyses revealed a clear correlation between the presence of proteins associated with non-canonical bases and phylogeny of phages, their position on the pan-genomic tree. After searching of the genomes of uncultured viruses, belonging to Tevenvirinae, only two genomes were selected. Most likely, this was due to the relatively large size of the Tevenvirinae genomes, that makes it difficult to assemble a complete genome. Also, 161 genomes were selected from Tevenvirinae in GenBank. A phage list and accession numbers/Scaffold IDs of these phages are given in Supplementary Table 4. The pan-genome tree and phylogenomic trees with the original and new sets of genomes are given in Supplementary Figures 5,6. As a result, the new set of genomes with the exception of the genome of the Cronobacter phage S13 formed the same groups as the original set did. Cronobacter phage S13 formed a separate clade on the trees and contained 130 unique proteins. The number of GSCs for some groups reduced, most likely due to the appearance of more evolutionary distant viruses in the clades. The total number of GSCs and a list of GSCs with homologs of each group (except for the Cronobacter S13 phage) are given in Supplementary Data Sheet 3. Clusters of known proteins associated with non-canonical bases are given in Supplementary Data Sheet 3 According to the results of additional analysis, the new genome samples contain genes associated with non-canonical bases characteristic of their group.
On the basis of presence/absence of common protein homologs, we identified twenty groups of Tevenvirinae phages. The analysis of data available for the representatives of these groups showed that they were isolated from bacteria inhabiting similar ecological niches. Also, these bacteria belonged to the Gammaproteobacteria. Analysis of genomes in larger number also showed the conservation of the considered groups and the presence of similar characteristics in new genomes.
To consider the exchange of genetic information within Tevenvirinae in more detail, we analyzed connections between the clusters that form the pan-genome cloud, as well as group-specific clusters.
The analysis showed that the cluster-genome bipartite network constructed was, for the most part, similar to the pan-genomic and phylogenomic trees, but there were some differences. The biggest difference was the location of individual representatives of groups I–XIV, which did not have as many connections to the viruses of their own group as they did to the viruses of other groups. Most likely, this can be explained by the specifics of their adaptation to the environment, which is dependent on the genes of the pan-genome cloud and their products. Groups XV and XVI, however, were distant from the groups whose representatives inhabited similar ecological niches. Groups XVII and XVIII, whose phages infect Aeromonas and were isolated from similar sources, were also far apart from each other. Taking into account the characteristics of the pan-genome shell, one can assume that the common ancestors of viruses from groups XVII and XVIII possessed (or acquired in the course of the evolutionary process) an internal (intragenomic) barrier, which blocked active exchange of genes between the phages of the same ecological niche.
To characterize genomes of the identified Tevenvirinae groups, we searched for homologs of their group-specific clusters among the organisms whose amino acid sequences were deposited in the Non-Redundant Sequences database. As it turned out, most GSCs had no homologs among other organisms, with the exception of groups I, II, VIII, and XIV. Leaving aside the groups represented by a single phage (V, X, XI, XII, XX), the largest numbers of GSCs were discovered in the phages of groups XV–XIX. Large numbers of GSCs in the groups represented by a single phage is explained by the fact that they included clusters unique for that single genome representing the group. The survey of the organisms whose proteins had sequences similar to GSCs showed that most of them had similar ecological niches with the phages of one or another group. This may indicate that the ancestors of these organisms shared a common pool of genes.
Thus, comparison of the results of pan-genomic tree, phylogenomic tree, GSC and bipartite network analyses shows that the evolutionary divergence of Tevenvirinae genomes is mostly determined by the characteristics of their ecological niches.
The influence of ecological niches on the evolutionary divergence of viruses was demonstrated earlier for T4-related phages (distinguished in the literature as a superfamily), Far-T4-related phages and other bacterial viruses – on the basis of data obtained from metagenomes, metaviromes and phage genomes from various sources (Desplats and Krisch, 2003; Filée et al., 2005; Bellas and Anesio, 2013; Roux et al., 2015; Braga et al., 2020). However, for some phages, the isolation-related features of their ecological niches are not the only factors that can drive evolutionary divergence in the phage groups.
Analyzing GSCs of group XIV, we found protein homologs associated with the synthesis of modifications of 7-deazaguanine. Taking into account this finding, as well as the presence of other non-canonical bases in Tevenvirinae phages, we hypothesized that the emergence of different bases and their modifications in certain groups of Tevenvirinae ancestors was one of the factors that drive the divergence of genomes in this bacteriophage subfamily. To confirm this, we searched these viruses for the proteins that are associated with non-canonical bases.
The search for the homologs of proteins associated with non-canonical bases (Table 2) revealed that they were present in all the representatives of Tevenvirinae. However, the phages of groups XV and XVI, as well as some representatives of groups III (Gaprivervirus), XIII (Acinetobacter phage ZZ1, Acinetobacter phage 133), and XIV (Morganella phage vB_MmoM_MP1) lacked any known enzymes involved in the synthesis of such bases.
The phages of groups I–XIV (with the exception of the representatives mentioned above and Morganella phage vB_MmoM_MP1) contain homologs of the gene encoding thymidylate synthase, which is involved in the synthesis of hmdCMP. The phages of groups I, VI, VII, VIII, IX, XII additionally contain genes whose products participate in hmdCMP glycosylation, although the composition of these genes varies. Most of the phages of group I have α-glucosyl transferase and β-glucosyl-1,6-α-glucose transferase (bacteriophage T4 and five closely related phages have β-glucosyl transferase instead of β-glucosyl-1,6-α-glucose transferase, and Escherichia phage vB_EcoM_G2540 has genes for all three proteins). The phages of group IX also contain genes encoding α-glucosyl transferase and β-glucosyl-1,6-α-glucose transferase – like most of the phages of group I. The phages of groups VI, VII, VIII, XII have homologs of these two genes. The function of the second protein in these viruses is still unclear, since the enzyme cannot additionally modify bases by any known mechanism in the absence of α-glucosyl hmdC in DNA. Therefore, either this protein modifies DNA in a different way, or its gene remains in the genomes as an atavism.
The phages of group II and XIII (with the exception of Acinetobacter phage ZZ1, Acinetobacter phage 133, and Acinetobacter phage Ac42) have homologs of proteins that are associated with arabinosylation of hmdC in DNA. The phages of groups IV, V, XI and, partly, group XIV (Proteus phage phiP4-3, Proteus phage vB_PmiM_Pm5461) contain proteins that are presumably associated with this process, yet they lack any known arabinosyl transferases. Among the proteins mentioned, two are presumably involved in the synthesis of UDP-arabinose (Thomas et al., 2018) and one is a putative thymidylate kinase.
The bacteriophages of groups XVII (with the exception of Aeromonas phage AS-szw, Aeromonas phage AS-zj and Aeromonas phage Asswx 1), XVIII and XX have homologs of two genes whose products are supposedly associated with base modification. A homolog of one of the genes was common for the phages of group XX and some phages of group XVII (Aeromonas phage AsFcp 2 and Aeromonas phage PX29); and homolog of the second gene was characteristic of the members of group XVIII and some representatives of group XVIII (Aeromonas phage 65.2, Aeromonas phage 65, and Aeromonas phage Aeh1).
As mentioned above, the phages of group XIX have homologs of proteins associated with the synthesis of modifications of 7-diazaguanine. According to Hutinet et al., 2019, Vibrio phage nt-1 has a small number of these non-canonical bases substituting for guanine (in Vibrio phage nt-1 phage, guanine is only partially and rarely replaced by this modifications). No such data are available for the remaining representatives of group XIX. Since they differ from Vibrio phage nt-1 in terms of the genes whose products are involved in the synthesis of 7-diazaguanine modifications, the percentage of substitutions may differ as well.
Some Tevenvirinae phages lack proteins associated with the synthesis of non-canonical bases, yet they have homologs of the genes whose products are dependent on these bases. Namely, these are homologs of DenA, DenB (Kutter et al., 1975, 1994), Alc (Drivdahl and Kutter, 1990), and dCTPase – dUTPase (Kutter et al., 1975). Their functions are listed in Table 2. It should be noted that the presence of DenA and dCTPase – dUTPase homologs is not essential for the synthesis and transfer of DNA with a canonical-nucleotide composition. As shown experimentally, representatives of groups XV and XVI (namely, phages RB43 and RB49) are able to carry out general transduction in the bacteria that contain canonical DNA (Taniashin et al., 2003). At the same time, these phages have genes encoding DenA and dCTPase – dUTPase homologs. The activity of the protein Alc can be inhibited, as shown by Kashlev et al., 1993, by the methylation of CpG sequences in the vicinity of its locus. As for DenB, we did not find any examples that would demonstrate inhibition of its endonuclease activity. Moreover, DenB was shown to play a significant role in the hydrolysis of DNA with a canonical base composition (Kutter et al., 1975, 1994; Petrov et al., 2006). Thus, the presence of genes of DenB and Alc homologs may indicate the presence of non-canonical bases in the viral DNA, whereas the presence of DenA and dCTP – dUTPase homologs only partially indicates this – especially in viruses that are most distant from the phages of group I.
The representatives of group III, forming the genus Gaprivervirus, were found to have homologs of all the four proteins described above. It is possible that these phages have an unknown protein associated with the synthesis of non-canonical bases. The same assumption can be made in respect to Acinetobacter phage ZZ1 and Acinetobacter phage 133 (which have homologs of DenB, Alc, and dCTPase – dUTPase) and for Aeromonas phages AS-szw, AS-zj, and Asswx 1 (these phages have homologs of denA and Alc). The phages of groups XV and XVI, as shown earlier, only have DenA and dCTP – dUTPase homologs. Phages of these two groups do not have any known proteins associated with the synthesis of non-canonical bases – yet the analysis of their GSCs revealed homologs of C-5 cytosine-specific DNA methylase (in the phages of group XV) and DNA-cytosine methyltransferase (in the phages of group XVI). These enzymes can potentially inhibit the activity of DenA and dCTP – dUTPase enzymes. Further investigation of these phages can clarify how they protect themselves from the action of enzymes associated with non-canonical bases (whose genes they carry in their genomes).
A comparative analysis of the sets of proteins associated with non-canonical bases and the relationships between Tevenvirinae phages showed that closely related phages had enzymes involved in the synthesis of the same or chemically similar non-canonical bases. Additional analysis of genomes in larger number also confirmed this. As known, bacteriophage T4 is not capable of general transduction (Wilson et al., 1979); moreover, it is excluded from infection by bacteriophage RB69, which has another modification of hmdC (Russell, 1967). It was also shown in Wilson et al. (1979) that the frequency of transduction changed depending on associated proteins the phage had. Given that some of these proteins affect the host’s DNA, it can be assumed that these proteins will also change the frequency of other HGT events. These facts and our results allow us to suppose that non-canonical bases and the associated proteins formed a barrier for the exchange of genetic information between the related phages, which resulted in their divergence in the process of evolution.
It was also found that genes of the synthesis of non-canonical bases of hmdC and its modifications (Figure 6) as well as genes of the synthesis of 7-deazaguanine modifications are located close to each other (most of them in one genome region). We speculate that genes of the synthesis of hmdC and its modifications was a genome island able to be transferred horizontally. More detailed information on this event is given in Supplementary Data Sheet 5.
Taking into account that non-canonical bases and associated proteins are considered to be one of the mechanisms regulating the frequency of HGT in Tevenvirinae, and based on the results obtained in our study, it is supposed that they play one of the key roles in the genome diversity of these viruses.
Another important function of non-canonical bases is protection of phage DNA from the harm that can be inflicted by the systems of host bacteria restricting the development of phages. Bacteria possess systems of restriction-modification (R-M systems), which, in some cases, cleave canonical, and in other cases, non-canonical DNA. In addition, they have Cas systems, which can be inhibited by non-canonical bases – with the inhibition depending on the composition of the base set (Weigele and Raleigh, 2016; Vlot et al., 2018; Hutinet et al., 2019). In Tevenvirinae, hmdC and its modifications are known to have different protection against the restriction activity of R-M systems, and this is true for Cas systems as well (Bair and Black, 2007; Weigele and Raleigh, 2016; Hutinet et al., 2019). The phages containing 7-deazaguanine have also been demonstrated to have protection against R-M systems (Hutinet et al., 2019). Moreover, representatives of groups I–III possess IpI (Bair et al., 2007) and Arn (Kim et al., 1997) proteins (Table 2 and Supplementary Table 3), which protect them from the actions of type-IV R-M systems (these systems specifically affect DNA with non-canonical bases) (Weigele and Raleigh, 2016). Thus, the existence of multiple protective mechanism against anti-viral host systems erects another barrier to the exchange of genetic information between closely related phages, which increases the divergence of Tevenvirinae.
Summarizing the results of this work and the data on the barrier functions of non-canonical bases and associated proteins, we can envision a possible chain of events that led to the divergence of the genomes of Tevenvirinae ancestors. The first step was infection of various host bacteria: Vibrio, Aeromonas, Enterobacteriales, etc., by closely related phages – and, as a result, similar phages are widely distributed in various types of habitats: marine environments, soil, plants, and animals. Adaptation to the specific conditions of the habitats implied changes in the genomes of phages and host bacteria, which would allow the development of specific metabolic pathways. The ecological niches inhabited by Tevenvirinae phages vary greatly in terms of spatial barriers confining them from the environment – and so do the capabilities of the phages to HGT. In marine bacteriophages, HGT is facilitated; in phages inhabiting the gastrointestinal tract of animals and humans, it is limited. The differences in HGT may be reflected in the sizes of Tevenvirinae genomes (Petrov et al., 2010): the representatives isolated from marine sources are characterized by larger genomes as compared to the phages from other types of habitats – this being associated with the necessity of their ancestors to transfer a larger number of genes. The spatial isolation of bacteriophages and their adaptation to ecological niches played the greatest role in the divergence of ancestral phage genomes.
The second step was the appearance of various non-canonical bases in the DNA of Tevenvirinae ancestors. This event played a key role in the divergence of phage genomes within the same ecological niche. Phages with different sets of non-canonical bases and their modifications became more and more specialized in the infection of closely related bacteria. Next, the ancestors of Tevenvirinae inhabiting the same ecological niches started to diverge in the ways of exchange of genetic information. Some of them retained the ability to exchange genetic information with the organisms whose DNA has a canonical composition of bases. Others began to acquire genes whose products hindered HGT. As a result, the genomes of such phages became more conservative. Furthermore, the emergence of genes whose products protected non-canonical phage DNA from the action of type IV R-M systems expanded the capabilities of those phages to infect bacteria. The differences in the HGT potential also affected the size of the phage genomes. An example are representatives of groups XV and XVI, which have larger genomes as compared to the representatives of group I (Petrov et al., 2010).
As a matter of fact, the use of such a strategy for the exchange of genetic information is one of the reasons for the spread of Tevenvirinae in a variety of ecological niches. On the one hand, it allows them to acquire genes whose products are necessary for the survival in certain environments. On the other hand, it allows them to preserve these genes in their genomes. Under the pressure of natural selection, the populations, in which HGT is less constrained, will have an inherent advantage for propagating their offspring. However, as they became more and more adapted to the specific environmental conditions, the populations, in which HGT is limited, will have an advantage in the conservation of genes that are most critical for survival in a particular ecological niche. Eventually, the populations that manage to conserve the “best” set of survival-related genes will spread within the niche – yet, at the same time, will become less capable of spreading in other niches. In other words, the history of the evolutionary development of Tevenvirinae reflects their gradual specialization to the specific environments, with non-canonical bases and the associated proteins playing their part in the process of specialization.
It is difficult, if not impossible, to observe the full cycle of this evolutionary strategy of Tevenvirinae in every ecological niche, since the first and second steps would occur simultaneously and depend on each other. This is determined by the barrier properties of various habitats. As mentioned above, marine environments have the least number of barriers that would restrict the interaction of bacteria and phages. The total size of genetic pool of marine populations is large, and the flow of genetic information between different organisms is quite intense. Correspondingly, it would take more time for viruses to conserve a set of genes allowing them to excel in those habitats. In the ecological niches with higher barriers, the total size of genetic pool of populations is smaller, and the competition between different organisms is more severe. As a result, the rate of adaptation of viruses to the specific conditions of particular niches increases. We suppose that this strategy of Tevenvirinae, which is based on the restriction of genetic information exchange, may also be found in viruses of other taxonomic groups, inhabiting other niches. Taking into account the fact that the genomic islands associated with the synthesis of DNA bases are quite ancient, the chance to find viruses with such an evolutionary strategy is quite high.
The strategy of Tevenvirinae ancestors, making the exchange of genetic information dependent on the set of non-canonical bases and the associated proteins, was one of the main factors driving the spread of these phages across the environments – as indicated by their frequent occurrence in the metagenomes isolated from various sources.
Also, this strategy correlates with the Constant-Diversity (C-D) model (Mizuno et al., 2014). The model implies the co-existence of competing phage populations in the same biological niche, where each population is comprised by clonal lineages which differ in the presence of certain genes (metaviromic islands). Proteins encoded by such genes may, for example, adjust phage specificity to the host clonal lineages (Mizuno et al., 2014). The co-existence of different bacterial clonal lineages enriches the genetic pool of the bacterial population, and the comparable diversity of phages that prey on those bacteria has a stabilizing effect on it. When applied to our hypothesis, this leads to the conclusion that ancestral Tevenvirinae phages may have used different non-canonical bases to enable their development in the host cells which differed, for example, in genes encoding restriction-modification proteins.
Bacteriophages from other taxonomic groups that have non-canonical DNA bases may also use this strategy. If a sufficient number of phages of certain taxa is registered in the databases, the methods of pan-genomic analysis can be used to reveal the evolutionary strategies of these phages. To be handled efficiently, this task will also require the application of newly developed sequencing techniques. A comprehensive study of metabolism of viruses with non-canonical DNA bases, which would make use, alongside other approaches, the methods of comparative genomics, will contribute to the better understanding of the evolutionary mechanisms underlying the development of such viruses.
Nowadays, Tevenvirinae and a wider group of T4-related phages (Near-T4-related and Far-T4-related) represent an optimal model for studying the influence of non-canonical bases, their hypermodifications and the associated proteins on the evolution and ecology of viruses.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
NN and AZ designed the study, contributed to revision, and read and approved the final manuscript. NN organized the data, performed the data analysis, and drafted the manuscript. AZ conceived the study and critically revised the manuscript. Both authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.632686/full#supplementary-material
Supplementary Figure 1 | Heat map representations of a number of homologous clusters in separate genomes. Horizontal lines represent genomes of viruses, vertical lines are homologous clusters.
Supplementary Figure 2 | The pan-genomic tree of Tevenvirinae (shown in Figure 1) with labeled aBayes support/ultrafast bootstrap support. Black circles indicate the root and the nodes of branches with the same sequences as leaves.
Supplementary Figure 3 | The phylogenetic tree of Tevenvirinae (presented in Figure 2) with labeled SH-aLRT support/aBayes support/ultrafast bootstrap support. Black circles indicate the root and the nodes of branches with the same sequences as leaves.
Supplementary Figure 4 | The phylogenomic tree of Tevenvirinae (shown in Figure 3) with labeled GBDP pseudo-bootstrap support. Black circles indicate the root and the nodes of branches with the same sequences as leaves.
Supplementary Figure 5 | The pan-genomic tree of Tevenvirinae extended genome set with labeled aBayes support/ultrafast bootstrap support. Selective clades are marked with Roman numerals and colors. Black circles indicate the root and the nodes of branches with the same sequences as leaves.
Supplementary Figure 6 | The phylogenomic tree of Tevenvirinae extended genome set with labeled GBDP pseudo-bootstrap support. Selective clades are marked with Roman numerals and colors. Black circles indicate the root and the nodes of branches with the same sequences as leaves.
Supplementary Table 1 | Supplementary data about studied genomes.
Supplementary Table 2 | Supplementary data about studied GSCs.
Supplementary Table 3 | Homologs of proteins associated with non-canonical bases in the studied genomes of Tevenvirinae.
Supplementary Table 4 | GenBank accession numbers and IGM/VR Scaffolds IDs of genomes added for Additional Analysis.
Supplementary Data Sheet 1 | Clustering results (folders “first clusterisation,” “second clusterisation,” and “third clusterisation”): the number of pan-genome components in the individual genome (files “pan genome components of genomes”), the total number of all clusters and components of the pan-genome (files “pan genome components”), a list of clusters of the core genome (files “core list”), a list of clusters of the softcore genome (files “softcore list”). Clusters of the softcore genome (folders “softcore clusters”) are presented in the files which show regions in genomes, protein sequences, names of viruses which have these proteins A description of softcore clusters is also supplied (file “Softcore clusters description”).
Supplementary Data Sheet 2 | The results of the BLAST-assisted (“txt” files) and HMMER-assisted (“xlsx” files) searches for GSC homologs. The clusters containing homologs of proteins from other organisms are listed.
Supplementary Data Sheet 3 | Clusters of the extended set of Tevenvirinae genomes. Lists of GSCs with homologs are presented (folder “GSCs”), which are described in Supplementary Table 2. Also, clusters of protein homologs associated with the synthesis of non-canonical bases are presented (folder “Non-canonical base clusters”), which are described in Table 2.
Supplementary Data Sheet 4 | Characteristics of Tevenvirinae groups.
Supplementary Data Sheet 5 | Characteristics of the region of genes of the synthesis of hmdC and its modifications of Tevenvirinae.
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Anisimova, M., Gil, M., Dufayard, J. F., Dessimoz, C., and Gascuel, O. (2011). Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst. Biol. 60, 685–699. doi: 10.1093/sysbio/syr041
Bair, C. L., and Black, L. W. (2007). A type IV modification dependent restriction nuclease that targets glucosylated hydroxymethyl cytosine modified DNAs. J. Mol. Biol. 366, 768–778. doi: 10.1016/j.jmb.2006.11.051
Bair, C. L., Rifat, D., and Black, L. W. (2007). Exclusion of glucosyl-hydroxymethylcytosine DNA containing bacteriophages is overcome by the injected protein inhibitor IPI∗. J. Mol. Biol. 36, 779–789. doi: 10.1016/j.jmb.2006.11.049
Bellas, C. M., and Anesio, A. M. (2013). High diversity and potential origins of T4-type bacteriophages on the surface of Arctic glaciers. Extremophiles 17, 861–870. doi: 10.1007/s00792-013-0569-x
Braga, L. P. P., Spor, A., Kot, W., Breuil, M. C., Hansen, L. H., Setubal, J. C., et al. (2020). Impact of phages on soil bacterial communities and nitrogen availability under different assembly scenarios. Microbiome 8:52. doi: 10.1186/s40168-020-00822-z
Brettin, T., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Olsen, G. J., et al. (2015). RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5:8365. doi: 10.1038/srep08365
Comeau, A. M., Bertrand, C., Letarov, A., Tétart, F., and Krisch, H. M. (2007). Modular architecture of the T4 phage superfamily: a conserved core genome and a plastic periphery. Virology 362, 384–396. doi: 10.1016/j.virol.2006.12.031
Comeau, A. M., and Krisch, H. M. (2008). The capsid of the T4 phage superfamily: the evolution, diversity, and structure of some of the most prevalent proteins in the biosphere. Mol. Biol. Evol. 25, 1321–1332. doi: 10.1093/molbev/msn080
Contreras-Moreira, B., and Vinuesa, P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701. doi: 10.1128/AEM.02411-13
Desplats, C., and Krisch, H. M. (2003). The diversity and evolution of the T4-type bacteriophages. Res. Microbiol. 154, 259–267. doi: 10.1016/S0923-2508(03)00069-X
Drivdahl, R. H., and Kutter, E. M. (1990). Inhibition of transcription of cytosine-containing DNA in vitro by the alc gene product of bacteriophage T4. J. Bacteriol. 172, 2716–2727. doi: 10.1128/jb.172.5.2716-2727.1990
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Filée, J. L., Tétart, F., Suttle, C. A., and Krisch, H. M. (2005). Marine T4-type bacteriophages, a ubiquitous component of the dark matter of the biosphere. Proc. Natl. Acad. Sci. U.S.A. 102, 12471–12476. doi: 10.1073/pnas.0503404102
Gautreau, G., Bazin, A., Gachet, M., Planel, R., Burlot, L., Dubois, M., et al. (2020). PPanGGOLiN: depicting microbial diversity via a partitioned pangenome graph. PLoS Comput. Biol. 16:e1007732. doi: 10.1371/journal.pcbi.1007732
Gold, L. M., and Schweiger, M. (1969). Synthesis of phage-specific alpha- and beta-glucosyl transferases directed by T-even DNA in vitro. Proc. Natl. Acad. Sci. U.S.A. 62, 892–898. doi: 10.1073/pnas.62.3.892
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q., and Vinh, L. S. (2018). UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281
Hutinet, G., Kot, W., Cui, L., Hillebrand, R., Balamkundu, S., Gnanakalai, S., et al. (2019). 7-Deazaguanine modifications protect phage DNA from host restriction systems. Nat. Commun. 10:5442. doi: 10.1038/s41467-019-13384-y
Ignacio-Espinoza, J. C., and Sullivan, M. B. (2012). Phylogenomics of T4 cyanophages: lateral gene transfer in the ‘core’ and origins of host genes. Environ. Microbiol. 14, 2113–2126. doi: 10.1111/j.1462-2920.2012.02704.x
Iranzo, J., Krupovic, M., and Koonin, E. V. (2016). The double-stranded DNA virosphere as a modular hierarchical network of gene sharing. mBio 7:e0978-16. doi: 10.1128/mBio.00978-16
Iyer, L. M., Zhang, D., Burroughs, A. M., and Aravind, L. (2013). Computational identification of novel biochemical systems involved in oxidation, glycosylation and other complex modifications of bases in DNA. Nucleic Acids Res. 41, 7635–7655. doi: 10.1093/nar/gkt573
Kaas, R. S., Friis, C., Ussery, D. W., and Aarestrup, F. M. (2012). Estimating variation within the genes and inferring the phylogeny of 186 sequenced diverse Escherichia coli genomes. BMC Genom. 13:577. doi: 10.1186/1471-2164-13-577
Kashlev, M., Nudler, E., Goldfarb, A., White, T., and Kutter, E. (1993). Bacteriophage T4 Alc protein: a transcription termination factor sensing local modification of DNA. Cell 75, 147–154. doi: 10.1016/S0092-8674(05)80091-1
Kim, B. C., Kim, K., Park, E. H., and Lim, C. J. (1997). Nucleotide sequence and revised map location of the arn gene from bacteriophage T4. Mol. Cells 7, 694–696.
Kristensen, D. M., Kannan, L., Coleman, M. K., Wolf, Y. I., Sorokin, A., Koonin, E. V., et al. (2010). A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics 26, 1481–1487. doi: 10.1093/bioinformatics/btq229
Kutter, E., Beug, A., Sluss, R., Jensen, L., and Bradley, D. (1975). The production of undegraded cytosine-containing DNA by bacteriophage T4 in the absence of dCTPase and endonucleases II and IV, and its effects on T4-directed protein synthesis. J. Mol. Biol. 99, 591–607. doi: 10.1016/s0022-2836(75)80174-4
Kutter, E., White, T., Kashlev, M., Uzan, M., McKinney, J., and Guttman, B. (1994). “Effects on host genome structure and expression,” in Molecular Biology of Bacteriophage T4, ed. J. Karam (Washington, DC: American Society of Microbiology), 357–368.
Lamm, N., Tomaschewski, J., and Rüger, W. (1987). Nucleotide sequence of the deoxycytidylate hydroxymethylase gene of bacteriophage T4 (g42) and the homology of its gene product with thymidylate synthase of E. coli. Nucleic Acids Res. 15:3920. doi: 10.1093/nar/15.9.3920
Madden, T. L., Tatusov, R. L., and Zhang, J. (1996). Applications of network BLAST server. Methods Enzymol. 266, 131–141. doi: 10.1016/s0076-6879(96)66011-x
Medini, D., Donati, C., Tettelin, H., Masignani, V., and Rappuoli, R. (2005). The microbial pan-genome. Curr. Opin. Genet. Dev. 15, 589–594. doi: 10.1016/j.gde.2005.09.006
Meier-Kolthoff, J. P., and Göker, M. (2017). VICTOR: genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 33, 3396–3404. doi: 10.1093/bioinformatics/btx440
Miller, E. S., Kutter, E., Mosig, G., Arisaka, F., Kunisawa, T., and Rüger, W. (2003). Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 67, 86–156. doi: 10.1128/mmbr.67.1.86-156.2003
Mizuno, C. M., Ghai, R., and Rodriguez-Valera, F. (2014). Evidence for metaviromic islands in marine phages. Front. Microbiol. 5:27. doi: 10.3389/fmicb.2014.00027
Okonechnikov, K., Golosova, O., and Fursov, M. UGENE Team (2012). Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167. doi: 10.1093/bioinformatics/bts091
Petrov, V. M., Nolan, J. M., Bertrand, C., Levy, D., Desplats, C., Krisch, H. M., et al. (2006). Plasticity of the gene functions for DNA replication in the T4-like phages. J. Mol. Biol. 361, 46–68. doi: 10.1016/j.jmb.2006.05.071
Petrov, V. M., Ratnayaka, S., Nolan, J. M., Miller, E. S., and Karam, J. D. (2010). Genomes of the T4-related bacteriophages as windows on microbial genome evolution. Virol. J. 7:292. doi: 10.1186/1743-422X-7-292
Roux, S., Enault, F., Ravet, V., Pereira, O., and Sullivan, M. B. (2015). Genomic characteristics and environmental distributions of the uncultivated Far-T4 phages. Front. Microbiol. 6:199. doi: 10.3389/fmicb.2015.00199
Roux, S., Páez-Espino, D., Chen, I. A., Palaniappan, K., Ratner, A., Chu, K., et al. (2021). IMG/VR v3: an integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Res. 49, D764–D775. doi: 10.1093/nar/gkaa946
Russell, R. L. (1967). Speciation among the T-even Bacteriophages. Ph.D. thesis, California Institute of Technology, Pasadena, CA. doi: 10.7907/SHD0-FM17
Sarker, S. A., McCallin, S., Barretto, C., Berger, B., Pittet, A. C., Sultana, S., et al. (2012). Oral T4-like phage cocktail application to healthy adult volunteers from Bangladesh. Virology 434, 222–232. doi: 10.1016/j.virol.2012.09.002
Schäffer, A. A., Aravind, L., Madden, T. L., Shavirin, S., Spouge, J. L., Wolf, Y. I., et al. (2001). Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 29, 2994–3005. doi: 10.1093/nar/29.14.2994
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Sullivan, M. B., Huang, K. H., Ignacio-Espinoza, J. C., Berlin, A. M., Kelly, L., Weigele, P. R., et al. (2010). Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ. Microbiol. 12, 3035–3056. doi: 10.1111/j.1462-2920.2010.02280.x
Sullivan, M. J., Petty, N. K., and Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. doi: 10.1093/bioinformatics/btr039
Taniashin, V. I., Zimin, A. A., Shliapnikov, M. G., and Boronin, A. M. (2003). Transduction of plasmid antibiotic resistance determinants with pseudo-T-even bacteriophages. Russian J. Genet. 39, 761–772. doi: 10.1023/A:1024748903232
Tétart, F., Desplats, C., Kutateladze, M., Monod, C., Ackermann, H. W., and Krisch, H. M. (2001). Phylogeny of the major head and tail genes of the wide-ranging T4-type bacteriophages. J. Bacteriol. 183, 358–366. doi: 10.1128/JB.183.1.358-366.2001
Thomas, J. A., Orwenyo, J., Wang, L. X., and Black, L. W. (2018). The Odd “RB” phage-identification of arabinosylation as a new epigenetic modification of DNA in T4-Like phage RB69. Viruses 10:313. doi: 10.3390/v10060313
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A., and Minh, B. Q. (2016). W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235. doi: 10.1093/nar/gkw256
Vernikos, G., Medini, D., Riley, D. R., and Tettelin, H. (2015). Ten years of pan-genome analyses. Curr. Opin. Microbiol. 23, 148–154. doi: 10.1016/j.mib.2014.11.016
Vinuesa, P., Ochoa-Sánchez, L. E., and Contreras-Moreira, B. (2018). GET_PHYLOMARKERS, a Software package to select optimal orthologous clusters for phylogenomics and inferring pan-genome phylogenies, used for a critical geno-taxonomic revision of the genus Stenotrophomonas. Front. Microbiol. 9:771. doi: 10.3389/fmicb.2018.00771
Vlot, M., Houkes, J., Lochs, S., Swarts, D. C., Zheng, P., Kunne, T., et al. (2018). Bacteriophage DNA glucosylation impairs target DNA binding by type I and II but not by type V CRISPR-Cas effector complexes. Nucleic Acids Res. 46, 873–885. doi: 10.1093/nar/gkx1264
Wattam, A. R., Brettin, T., Davis, J. J., Gerdes, S., Kenyon, R., Machi, D., et al. (2018). Assembly, annotation, and comparative genomics in PATRIC, the all bacterial bioinformatics resource center. Methods Mol. Biol. 1704, 79–101. doi: 10.1007/978-1-4939-7463-4_4
Weigele, P., and Raleigh, E. A. (2016). Biosynthesis and function of modified bases in bacteria and their viruses. Chem. Rev. 116, 12655–12687. doi: 10.1021/acs.chemrev.6b00114
Wilson, G. G., Young, K. Y., Edlin, G. J., and Konigsberg, W. (1979). High-frequency generalized transduction by bacteriophage T4. Nature 280, 80–82. doi: 10.1038/280080a0
Wolf, Y. I., Makarova, K. S., Yutin, N., and Koonin, E. V. (2012). Updated clusters of orthologous genes for Archaea: a complex ancestor of the Archaea and the byways of horizontal gene transfer. Biol. Direct. 7:46. doi: 10.1186/1745-6150-7-46
Wommack, K. E., and Colwell, R. R. (2000). Virioplankton: viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 64, 69–114. doi: 10.1128/mmbr.64.1.69-114.2000
Keywords: Tevenvirinae, phages, non-canonical bases, modified bases, genome diversity
Citation: Nikulin NA and Zimin AA (2021) Influence of Non-canonical DNA Bases on the Genomic Diversity of Tevenvirinae. Front. Microbiol. 12:632686. doi: 10.3389/fmicb.2021.632686
Received: 23 November 2020; Accepted: 08 March 2021;
Published: 06 April 2021.
Edited by:
Ludmila Chistoserdova, University of Washington, United StatesReviewed by:
Malgorzata Barbara Lobocka, Institute of Biochemistry and Biophysics (PAN), PolandCopyright © 2021 Nikulin and Zimin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nikita A. Nikulin, bi5uaWt1bGluQGlicG0ucnU=; Andrei A. Zimin, YXBvbGxvNjZAcmFtYmxlci5ydQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.