 Amy H. Fitzpatrick
Amy H. Fitzpatrick Agnieszka Rupnik2
Agnieszka Rupnik2 Helen O'Shea
Helen O'Shea Paul Cotter
Paul Cotter
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 22 February 2021
Sec. Virology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.621719
This review aims to assess and recommend approaches for targeted and agnostic High Throughput Sequencing of RNA viruses in a variety of sample matrices. HTS also referred to as deep sequencing, next generation sequencing and third generation sequencing; has much to offer to the field of environmental virology as its increased sequencing depth circumvents issues with cloning environmental isolates for Sanger sequencing. That said however, it is important to consider the challenges and biases that method choice can impart to sequencing results. Here, methodology choices from RNA extraction, reverse transcription to library preparation are compared based on their impact on the detection or characterization of RNA viruses.
Many RNA viruses are of a global health concern from a One Health perspective, which is the intersection of human, animal and environmental health. Environmental transmission of these viruses, whether it be through food, water or recreational activities poses a risk for humans, plants and animals. It is important to adopt One Health principles for the surveillance of RNA viruses as environmental samples can (a) indicate hot spots for viral recombination, (b) serve as an important source of virus transmission, and (c) sequencing these samples allows us to pre-empt new RNA viruses and their variants of potential clinical concern. Viral persistence in the environment increases the opportunity for inter and intra-viral family recombination and increases virus-host exposure, factors that all contribute to the emergence of new viruses; that have the potential to cause large scale outbreaks. Non-enveloped viruses demonstrate remarkable persistence in the environment. Trans-kingdom virus interactions are thought to aid viral persistence in environmental settings, though this has been difficult to investigate, due to a lack of suitable cell culture systems. Furthermore, RNA viruses have high mutation rates as, unlike their DNA counterparts, most do not have a proofreading polymerase, though there are notable exceptions (Smith and Denison, 2013). These mutations can result in non-functional changes but can also enable the virus to evade the host immune system, through changing epitope conformation. Yet emerging RNA viruses are difficult to detect due to (a) lack of cell culture systems and (b) dependence on targeted molecular approaches. Second generation sequencing provided incremental improvements in the monitoring of environmental transmission, persistence and recombination but the costs quickly became prohibitive. This was in part due to the need to isolate viruses using cell culture or clone environmental samples for increased sequencing resolution. In addition, the quantity of input RNA/DNA required to obtain high quality sequences. High throughput sequencing methods (bridge amplification, single molecular real time sequencing, and nanopore-based sequencing) have been widely applied in clinical settings but have had limited success for viral surveillance and aside from Flaviviruses (Zika virus, West Nile virus). There have been important contributions regarding RNA virology from environmental HTS applications (Alberti et al., 2017; Wolf et al., 2020) though HTS investigation of environmental transmission of pathogenic RNA viruses is still in its infancy. Furthermore, the comparatively small size of RNA virus genomes to competing genomic RNA, severely impacts the depth of coverage achievable, due to sequencing saturation. To summarize, RNA viruses are difficult to sequence and characterize using HTS due to (a) their genetic diversity, (b) lack of conserved regions across the genome of viruses and (c) short genome lengths.
In High Throughput Sequencing (HTS), either a targeted or agnostic approach can be taken. Targeted sequencing infers that some level of knowledge is available with respect to the target in question and that the experimental design incorporates this prior knowledge, either through amplicon-based sequencing or probe capture hybridization. An overview of how the different approaches work can be seen in Figure 1.
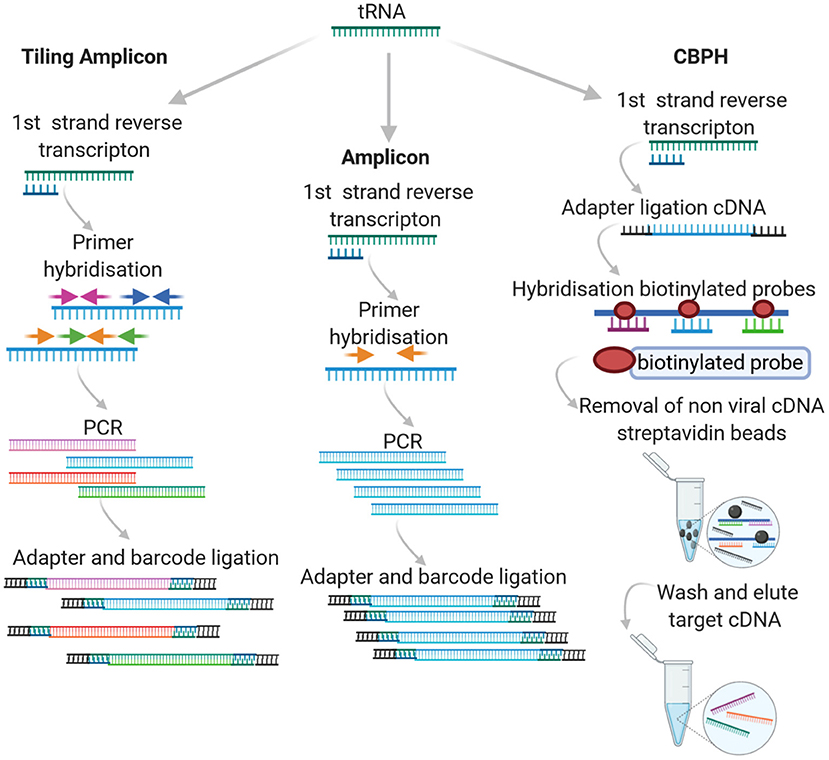
Figure 1. Targeted HTS approaches.
One of the two most common approaches to targeted sequencing is amplicon sequencing. The approach involves amplification of a target genome fragment using specific primers before library preparation and sequencing. It is most often used for the study of diversity and structure of prokaryotic communities in a variety of hosts (human, animal and ecological niches). Often in this case, common PCR-based approaches target highly conserved rRNA genes, such as those encoding the 16S/18S and 28S subunits or the Internal transcribed spacer (ITS) between them. Unlike the 16S rRNA of bacteria, viruses lack universally conserved markers and genome plasticity. In particular, with respect to RNA viruses, this further contributes to the associated challenge, requiring a Family specific priming PCR approach. Amplicon based sequencing approaches, (particularly tiling or “jackhammer”) have been widely applied for the detection of RNA viruses with varying levels of success (Marston et al., 2013; Cruz et al., 2016; Cuevas et al., 2016; Hanke et al., 2016; Imamura et al., 2016b; Boonchan et al., 2017; Johnson et al., 2017; Parra et al., 2017; Quick et al., 2017; Hata et al., 2018; Lun et al., 2018; Suffredini et al., 2018; Wang et al., 2018; Cinek et al., 2019; Di et al., 2019; Fumian et al., 2019; Gradel et al., 2019; Eden et al., 2020; Fauver et al., 2020; Lu et al., 2020; Mancini et al., 2020). Tiling or “jackhammer” approaches involve designing a series of primers that generate short products across the whole target genome and can be Family or genus specific.
The success of amplicon sequencing with respect to RNA viruses is very dependent on the choice of primers. Like traditional Reverse Transcription Polymerase Chain Reaction (RT-PCR) amplification, primers can be designed to anneal to the most conserved sequences of the RNA virus genome(s) in question. In this case, a certain degree of validation is required. Confirmation of the PCR products via Sanger sequencing should be implemented for new primer sets to ensure specificity. There is a high likelihood that degenerate primer sets are required in order to account for virus divergence (Li et al., 2012). This highly targeted approach requires well-characterized viruses for which a number of viral genome sequences are available. In some cases, this may present a technical barrier, especially when developing amplicon sequencing methods to detect emerging infectious diseases, such as at the beginning of recent Ebola virus, Zika, and SARS-CoV-2 outbreaks when, initially, sequencing data was limited.
To circumvent this, Quick et al. (2017) developed a tiling amplicon algorithm called PrimalSeq to facilitate the design of primers that allow short amplicons to be generated across the target genome in a highly multiplexed assay. High quality (non-degenerate) sequences are required in order to design primers that target the entire length of the target genome. For this approach, the detection of recombinant viruses or intra-host nucleotide variants can be accurately detected by applying replicate sequencing, viral input greater than 1000 RNA virus copies and >400x genome coverage (Grubaugh et al., 2019). This approach has been widely applied to obtain whole genome sequences of emerging RNA viruses as it can work with samples with an expected high background host rRNA/MRNA, low concentrations of target viral RNA and limited diversity (Artic Network, 2021).
Capture based probe hybridization (CBPH) requires prior knowledge of the specific sequence variants to be detected. Most capture-based methods use a tiling array approach, where 80 to 120-mer DNA or RNA probes are used to cover the length of the target genome/genomes. The probes typically have 10–50 bp regions between them, adopting a similar approach to the overlapping/jackhammer amplicon approach described above. Target enrichment is based on the biotinylated (or otherwise labeled) probe annealing to complementary sequences in the sample(s). The probes attach to previously fragmented genomic DNA/RNA and the targets are eluted, ligated, and prepared for the specific sequencing platform employed. Amplicon and capture probe hybridization tiling approaches have been widely applied as alternatives for whole genome sequencing of human genomic exons and viruses. In the latter case, this is in large part due to the challenge of sequencing small viral genomes in complex samples containing a high proportion of background host genomic DNA as well as, in some instances, bacteria/archaea.
CBPH was initially implemented to detect Single Nucleotide Variants (SNV) in human genomic exon studies. Various studies have applied this methodology to virus specific studies using widely available commercial kits with custom design options such as SureSelect XT Target Enrichment system, Illumina TruSeq RNA Access, or SeqCap Ez probe design with separate library preparation kit. More recently, the VirCapSeq-VERT and CATCH custom virus oligonucleotide panel have become available as a general tiling array for vertebrate viruses (summarized in Table 1). Metsky et al. (2019), Strubbia et al. (2019b), and Strubbia et al. (2020) are the only studies applying to CBPH to RNA viruses in non-clinical samples.
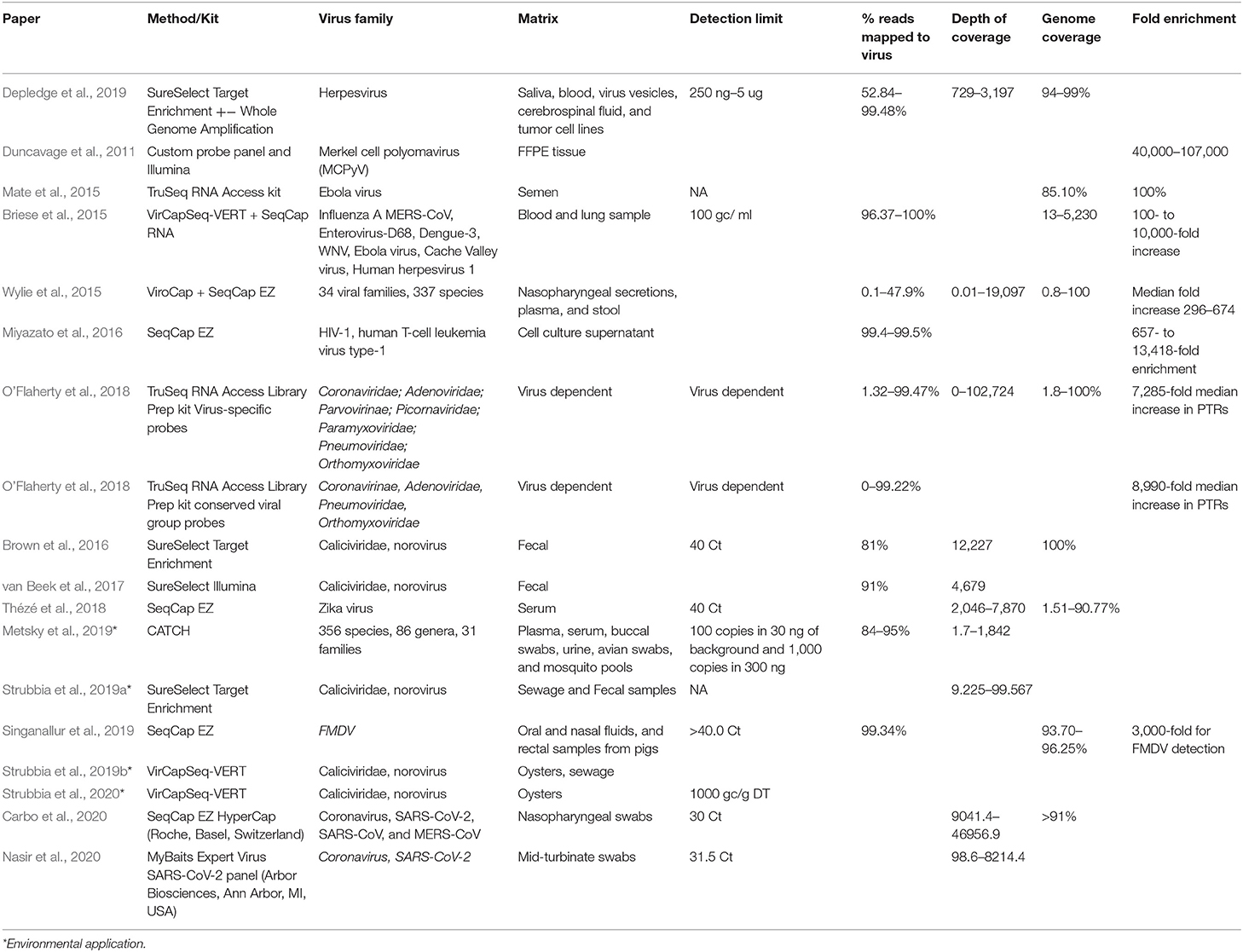
Table 1. Applied CBPH methods used for the characterization of viruses.
There is huge variability across CBPH assays applied for the genetic characterization of viruses, from oligonucleotide bait design approaches (RNA/DNA), matrices and target viruses. Studies attempting to capture a very wide viral diversity have used panels ranging from 300,000 to 2.1 million probes per assay (Duncavage et al., 2011; Wylie et al., 2015; O'Flaherty et al., 2018). There are cost and performance implications for utilizing these large panels, as the number of probes required dictates the cost of probe synthesis (Briese et al., 2015; Wylie et al., 2015; Metsky et al., 2019). These assays tend to use shorter oligonucleotides as each additional nucleotide increases the uniqueness of an oligonucleotide by a factor of four (Hendling and Barišić, 2019). This design difference results in varying genome coverage, as large generic panels have a greater propensity to capture viral diversity but fewer whole genomes, whilst more targeted assays result in improved genome coverage but less viral diversity. When designing or implementing a virus panel, the key point to consider is the research objective (Duncavage et al., 2011; Brown et al., 2016; Thézé et al., 2018) and the limited evidence available suggests that CBPH is a valuable tool for genotypic characterization of RNA viruses in non-clinical samples.
In direct contrast to targeted sequencing, agnostic sequencing requires little prior knowledge of the target genome(s) though hopefully an understanding of the matrix and expected virome in question. For example, if the objective is to characterize human viruses of clinical concern in sewage, concentration and extraction methods must consider that sewage as a matrix will contain PCR inhibitors and that concentration methods may not concentrate both enveloped and non-enveloped RNA viruses of interest. Method validation and the inclusion of appropriate controls is necessary for interpretation of results and for setting quality control thresholds. As agnostic sequencing is not targeted, non-viral RNA can be captured during the library preparation and this can cause downstream issues. Indeed, obtaining sufficient genome coverage of virus RNA genomes against a background of host rRNA and MRNA is a challenge. Various approaches have been developed to enrich samples or to deplete rRNA as outlined below.
Sequence Independent, Single Primer Amplification (SISPA) is a random priming method developed by Reyes and Kim (1991). SISPA involves directional ligation of oligonucleotide(s) to a target population of blunt ended DNA molecules. The common end sequence allows one strand of the double-stranded primer to be used in repeated rounds of annealing, extension and denaturation in the presence of a high-fidelity polymerase. SISPA has been used for the discovery of new viral agents, particularly in the veterinary field (Moser et al., 2016; Chrzastek et al., 2017; Myrmel et al., 2017; Cholleti et al., 2018; Zhao et al., 2018a).
To date, there have been three comparative studies in which SISPA has been compared with other metagenomic methods. Kugelman et al. (2017) compared DNA shotgun metagenomics, RNA template metagenomics using random hexamers and Klenow fragments, amplicon sequencing, SISPA, poly(A) tail enrichment using TruSeq RNA kit and Circular resequencing (CirSeq). Parras-Moltó et al. (2018) compared multiple displacement amplification (MDA) and SISPA to sequence DNA viruses, while Goya et al. (2018) compared the use of various SISPA and random hexamers protocols, with and without rRNA depletion. These comparative studies used clinical samples, cell culture supernatant or plasmid material to assess method efficiency.
Of the numerous approaches they used, Kugelman et al. (2017) determined that SISPA resulted in the highest error increase (9.0-fold) compared to CirSeq or Illumina TruSeq RNA Access kit and SISPA-generated sequences demonstrated an increased number of transition events. The accumulation of these errors could falsely indicate sub-clonal diversity or veil true diversity. Parras-Moltó et al. (2018) found that SISPA-generated viromes displayed uneven coverage profiles, with high coverage peaks in regions with low sequence complexity. Bias induced by random amplification methods had a minor impact, with random hexamers being preferable to SISPA for DNA virus metagenomics. Conversely, when Goya et al. (2018) compared the performance of SISPA with random hexamers, they found that the best performance was achieved with SISPA compared to samples subjected to rRNA depletion prepared with the Nextera XT DNA library kit. The coverage profiles were different for each method, with random hexamers providing a more uniform distribution across the genome, albeit lower coverage. Despite the difference between these three studies, it is apparent that, as currently employed, SISPA is not suitable for the identification of SNVs due to the high number of transition events and uneven coverage of the target genome for both DNA viruses and negative strand RNA viruses studied.
Rolling Circle Amplification (RCA) is an isothermal enzymatic process where a short DNA or RNA primer is amplified to form a long single stranded DNA or RNA using a circular DNA template and specific DNA or RNA polymerases, as can be seen in Figure 2.
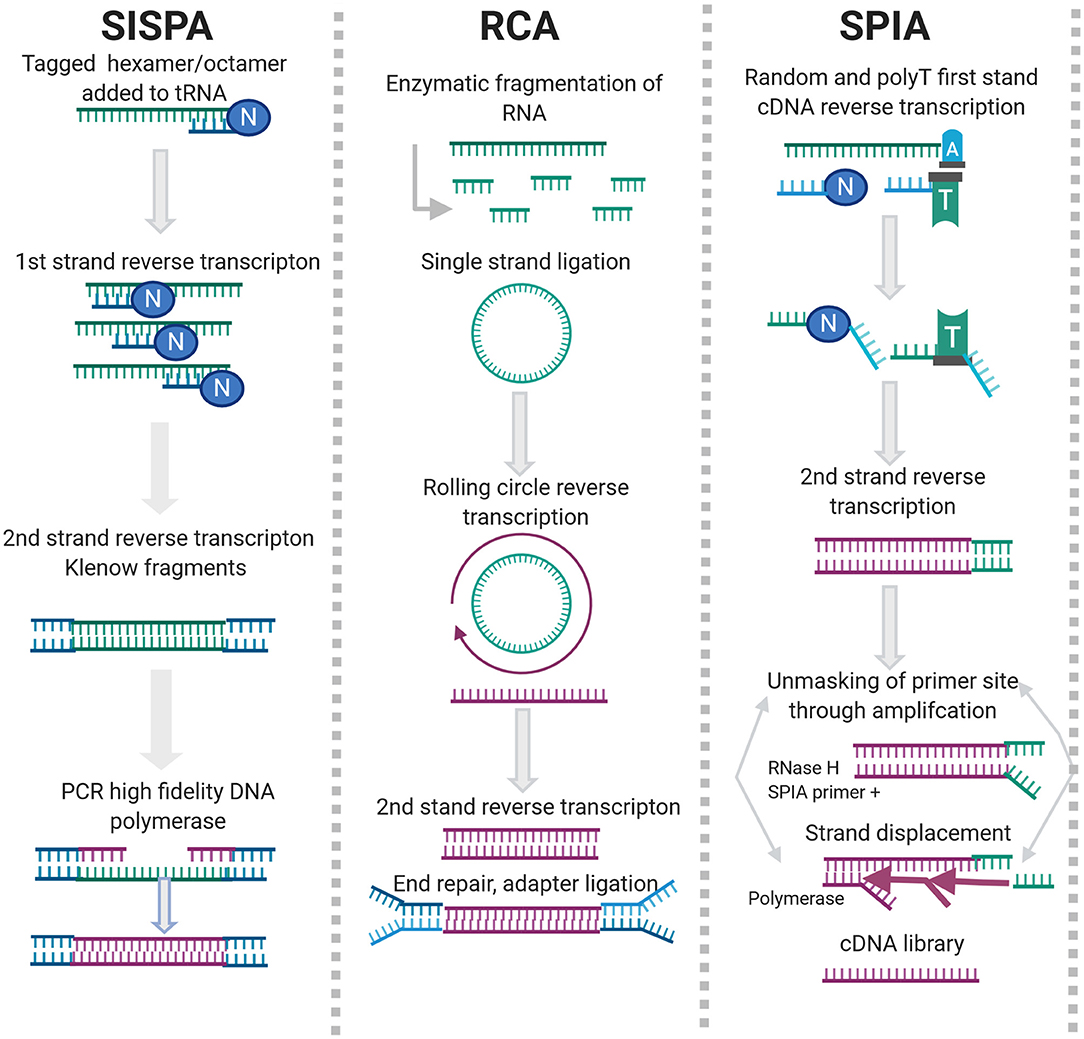
Figure 2. Enrichment methods for agnostic sequencing.
So far only two studies have compared RCA-HTS methods or RCA to other shotgun metagenomics methods for use with DNA/RNA viruses or RNA templates (Kugelman et al., 2017; Sukal et al., 2019). Kugelman et al. (2017) found that CirSeq compared to other target enrichment methods (amplicon, SISPA), was the least error prone (Acevedo et al., 2014). However, Martel et al. (2013) found that the viral load required for CirSeq (1E + 3 IU/ml) is a limiting factor for the application of this method to clinical samples (Hepatitis B virus in serum). Sukal et al. (2019) used variations of RCA to detect and characterize integrated Badnavirus-like sequences in plant host species. Methods included; random-primed RCA primer spiked random-primed RCA, directed RCA and specific-primed RCA. Viral DNA amplified using the optimized directed RCA and specific-primed-RCA protocols showed an 85-fold increase in Badnavirus NGS reads compared with random-primed RCA, showing the benefit of target specific priming strategies.
Ribosomal RNA (rRNA) is the most abundant species of RNA in most cells. For agnostic RNA virus sequencing of complex samples, its presence is problematic as a large number of non-viral reads can be generated, thereby greatly limiting the number of relevant, virus-related, sequences. To increase the number of reads mapping to viral RNA, several methods have been employed to either enrich non-ribosomal RNA or remove unwanted rRNA sequences. Enrichment methods include poly-A selection [TruSeq mRNA (Illumina)], Single Primer Isothermal Amplificaiton (SPIA) (OvationⓇ RNA Amplification System, NuGen) and Not so random (NSR) sequencing (Universal Prokaryotic RNA-Seq, NuGen). For a detailed summary of enrichment methods, refer to Table 2.
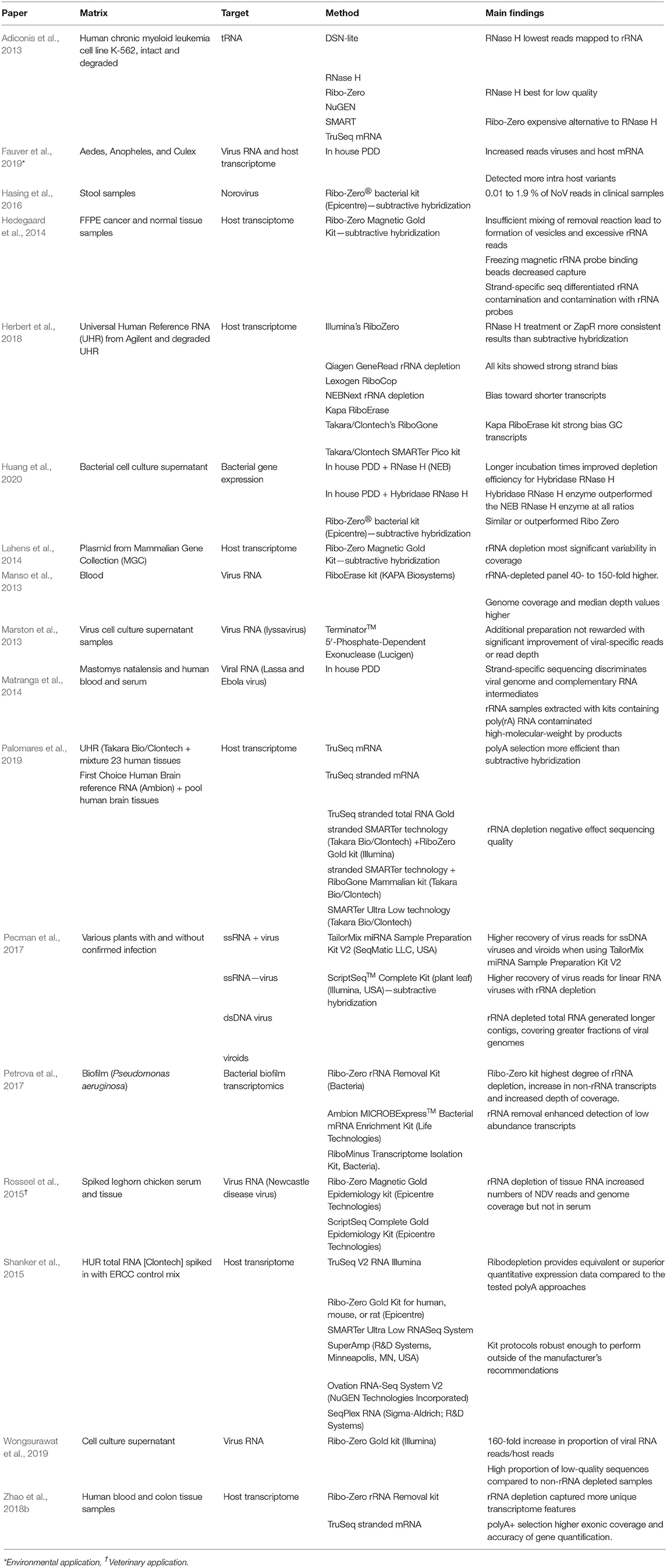
Table 2. Comparative studies of non-rRNA enrichment or rRNA depletion.
During poly-A selection, protein-coding polyadenylated RNA are captured by oligo (dT) primers attached to magnetic beads to isolate RNA. Non-polyadenylated RNA, such as rRNA, are not captured. This approach does result in a strong bias toward the 3' end of RNA targets, though this bias is alleviated by the reduced sequencing depth required to obtain high quality viral reads (Sun et al., 2013; Fonager et al., 2017; Zhao et al., 2018b).
NSR sequencing uses hexamer or heptamer primers that bind to non-rRNA target during reverse transcription (RT) (Figure 2). Several versions of NSR primer panels have been published for various applications (Endoh et al., 2005; Pyrc et al., 2007; de Vries et al., 2011; Manso et al., 2013; Xu et al., 2014; Shanker et al., 2015). NSR sequencing works well with partially degraded or low-input samples but exhibits off-target priming and is species dependent (Armour et al., 2009). During SPIA, a set of reactions occur in which a DNA/RNA chimeric primer binds the complementary sequence and is extended by a DNA polymerase at a constant temperature. Once extension of the primer is complete, the RNA is cleaved and digested by RNase H and the entire process is repeated, producing multiple copies of the amplification product (Figure 2). SPIA requires high input amounts of total RNA, which can be challenging when dealing with clinical samples but has been successfully used for detection of bovine coronavirus (Hrdlickova et al., 2017; Myrmel et al., 2017).
As an alternative to enrichment methods, rRNA can be removed using subtractive hybridization [Ribo-Zero (Illumina)], exonuclease digestion [MICROBExpress (Ambion)], endonuclease digestion (RNase H), or duplex specific nuclease (DSN)/Probe directed degradation (PDD). An overview of the rRNA depletion methods can be viewed in Figure 3 and detailed summary of studies applied rRNA depletion in viral metagenomic studies is included in Table 2.
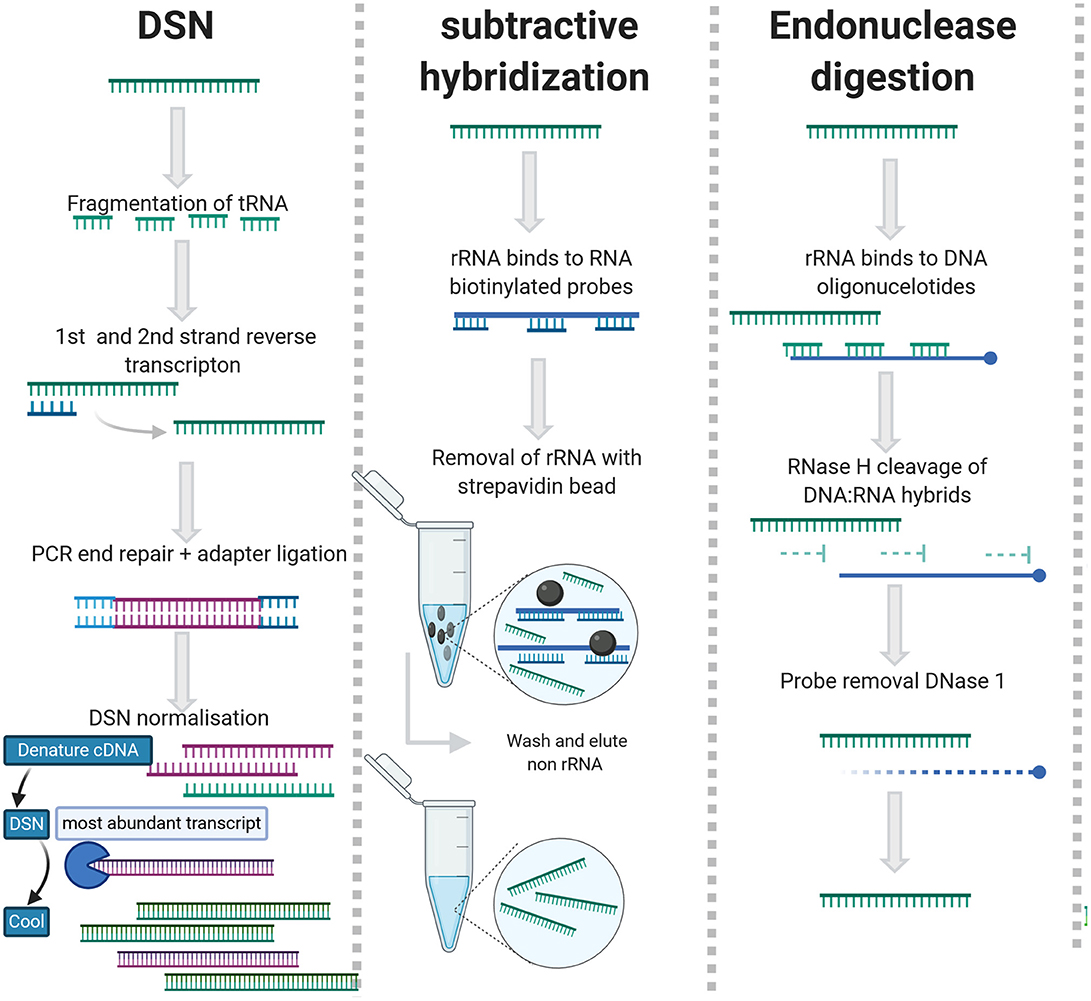
Figure 3. rRNA depletion methods for agnostic sequencing.
DSN generates rRNA depleted libraries by employing the C0t-kinetics-based normalization method to deplete abundant sequences that re-anneal quickly, ergo highly abundant rRNAs and tRNAs (Xiao et al., 2013) (Figure 3). The DSN method works with lower concentrations of RNA and partially degraded rRNA but requires a longer time to prepare libraries (Yi et al., 2011; Qiu et al., 2015). During PDD, rRNAs are targeted by anti-sense DNA oligos and digested by RNase H/DSN (Morlan et al., 2012; Kim et al., 2019) (Figure 3). This requires saturation of rRNA with contiguous oligonucleotide and is slightly less efficient than subtractive hybridization (Archer et al., 2014). The advantage of this approach is that the probes are essentially reverse primers so they are cheap and easy to design (Fauver et al., 2019). DSN has been applied successfully for the sequencing of RNA viruses in complex matrices (Schuh et al., 2020; Zhou et al., 2020). For subtractive hybridization, unwanted rRNAs/cDNAs are hybridized to biotinylated DNA or Locked nucleic acid (LNA) probes and depleted with streptavidin beads (Briese et al., 2015; Culviner et al., 2020).
There are three common methods used for RNA extraction: organic extraction using Acid guanidinium thiocyanate phenol chloroform (AGPC), Silica membrane based spin column technology (SMSC), and silica coated Magnetic beads (MB). Due to the physio-chemical differences between these three extraction methods, the yield, purity, and specificity of the RNA obtained varies and this can have downstream impacts.
AGPC extraction dissolves cell/viral components and maintains the integrity of RNA, due to the denaturing activity of phenol and guanidine thiocyanate with respect to RNases (Le et al., 2018). The addition of chloroform or a chloroform alternative, followed by centrifugation, separates RNA from DNA, proteins, lipids and insoluble matter (Le et al., 2018). However, RNA isolated by this method is often contaminated with protein, cellular materials and organic solvents such as phenol-chloroform, salts and ethanol (Tavares et al., 2011). In addition, the phenol may render the RNA incompatible with downstream applications. SMSC and MB based RNA isolation systems do not require the use of organic solvents, are relatively simple, efficient, low cost, and yield total intact RNA with low levels of contamination from proteins and other cellular materials. However, these methods can often result in significant levels of genomic DNA contamination, an important consideration with respect to sequencing of viral RNA (Tavares et al., 2011).
MB can be coated with silica, oligo(dT) or specific capture probes. Silica coated MB non-selectively bind nucleic acids in the presence of chaotropic salts via electrostatic interactions. The silica-coated beads are most suitable for applications that require nucleic acids other than MRNA while the oligo (dT) beads are best-suited for mRNA targets that are polyadenylated. Specific capture-based systems are best suited for applications that do not tolerate high concentration of non-target nucleic acids (Adams et al., 2015). There is limited information available with respect to the impact of RNA concentration and extraction on sequencing results. Those that are available are summarized in Table 3. Of interest to this review are results from Hjelmsø et al. (2017), comparing the impact of sewage concentration and RNA extraction methods on viral sewage metagenomics. Hjelmsø and colleagues found that (i) highest viral specificity was obtained using Polyethylene glycol (PEG) concentration (ii) Nucleospin RNA XS generated the highest read count for RNA viruses (norovirus, rotavirus and Hepatitis A and E virus) and (iii) viral richness is strongly impacted by extraction method. In a similar fashion, Strubbia et al. (2019a) found that PEG extraction resulted in longer contigs and detection of other viruses in sewage samples. However, this was outperformed by an alternative method which applied sodium pyrophosphate combined with a sonication step prior to PEG concentration. This method successfully generated norovirus reads from both sewage and oyster digestive tissue (Strubbia et al., 2019b). Considering non-viral studies outlined in Table 3, the evidence for improved RNA Integrity number (RIN) values and higher concentrations of RNA using organic extractions is mixed, with no clear trend in either direction.
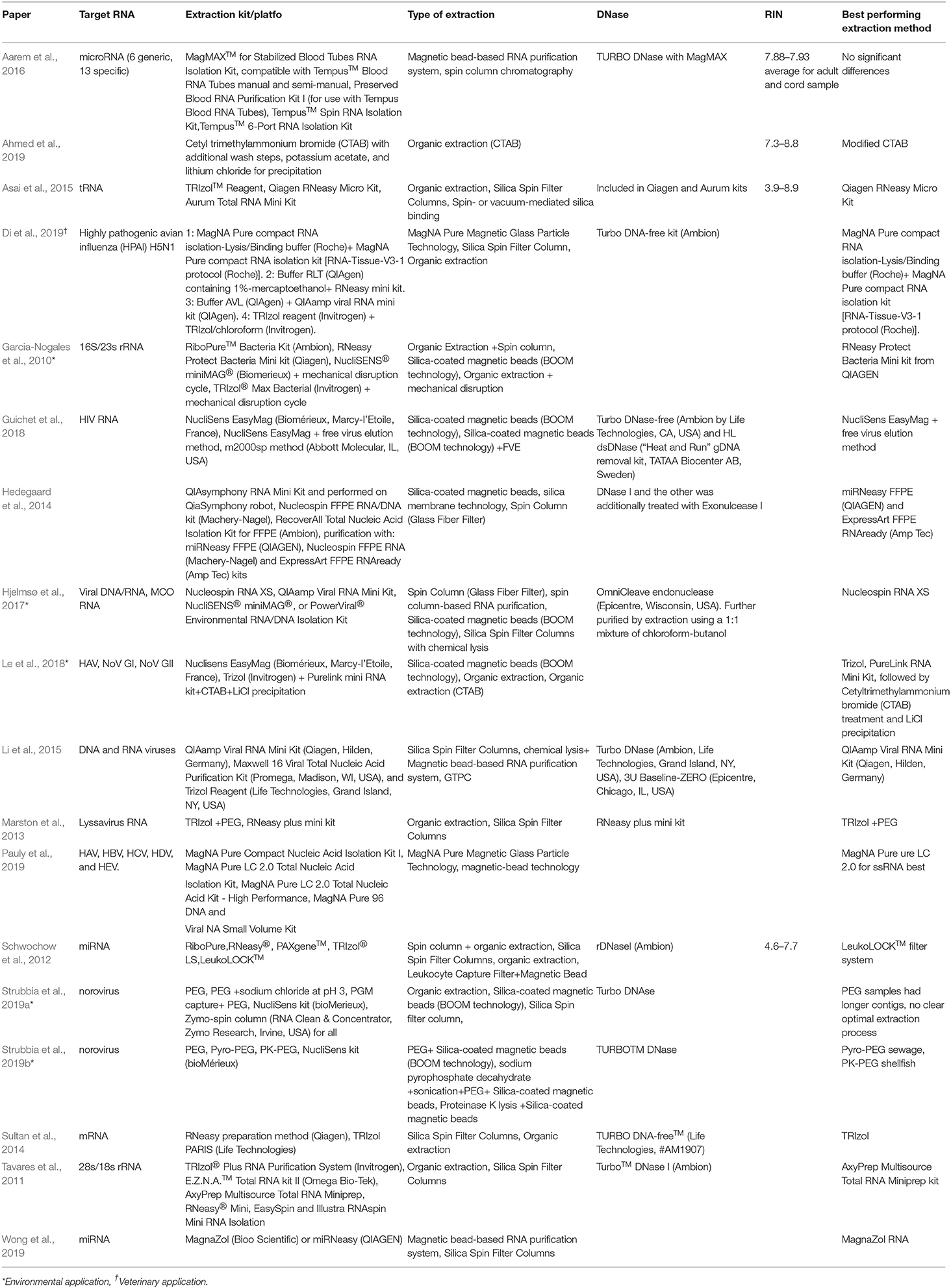
Table 3. Comparison of RNA extraction methods used for HTS.
Both the efficiency of reverse transcription and fidelity is important for the detection of virus quasispecies present at low abundances and the identification of SNV. To date only two studies have investigated reverse transcriptase (RT) fidelity impact on next generation sequencing results (Cholet et al., 2020; Zucha et al., 2020). In order to demonstrate the impact that RT has on the quality of CDNA synthesized, this section includes earlier studies where the focus is on CDNA yield/RT enzyme efficiency for quantitative Reverse Transcription Polymerase Chain Reaction (QRT-PCR) applications (see Table 4). Whilst the focus in these studies is primarily the efficiency in terms of yield, this is still an important consideration for shotgun metagenomics, where relative abundance of viral targets could be interpreted as prevalence, and for the detection of RNA templates present at low concentrations.

Table 4. Literature investigating the impact of RT enzyme on cDNA yield.
Random primers are oligonucleotides with random base sequences widely applied during RT as described in the section on SISPA (2.2.1). As noted above, they are often six nucleotides long and are usually referred to as random hexamers, N6, or dN6. Due to their random binding, they can potentially anneal to any RNA species in the sample. Therefore, these primers may be considered for reverse transcription of RNAs without poly(A) tails, degraded RNA and RNA with known secondary structures. Some random primer sets have been constructed with viral genomes in mind, preferentially priming viral RNA over ribosomal RNA (Endoh et al., 2005; Strubbia et al., 2020).
Oligo(dT) primers consist of a stretch of 12–18 deoxythymidines that anneal to poly(A) tails of eukaryotic mRNAs, which make up only 1–5% of total RNA. Oligo(dT) primers target polyadenylated RNAs, whereas random sequence primers target all RNAs including the abundant rRNA fraction. Mixtures of random hexamers with oligo(dT) are predominantly used in QRT-PCR to maximize yield. Oligo(dT) priming has also been applied as a viral RNA enrichment method as outlined earlier 2.2.3.
Gene-specific primers offer the most specific priming in RT (Miranda and Steward, 2017). These primers are designed based on known sequences of the target RNA, requiring prior knowledge. Since the primers bind to specific RNA sequences, a new set of gene-specific primers is needed for each target RNA. Primers that are specific to a viral genome also efficiently eliminate the influence of ribosomal RNAs.
Strubbia et al. (2020) reviewed three sets of hexamers, those from Endoh et al. (2005), an updated version of this hexamer panel (I-HD), including a probe to reduce host rRNA from oysters, and random hexamers. The I-HD panel resulted in lower read numbers aligning to Mollusc and other Eukaryote genomes. Furthermore, the number of reads targeting virus sequences was higher compared to the random set. Conversely, random hexamers produced more reads aligning to HuNoV than the custom panel and those from Endoh et al. (2005). Random hexamers transcribed HuNoV sequences more efficiently and produced longer contigs, allowing HuNoV genotype identification.
In Table 5 below, the variety of reverse transcriptase enzymes and priming strategies applied in norovirus HTS studies can be seen. SuperScript II, SuperScript III, and High Capacity cDNA RT were commonly used for CDNA synthesis, whilst a balance of random hexamers and oligo(dT) priming strategies were popular. As most publications have not assessed the yield/fidelity post CDNA synthesis, it is not possible to compare these publications based on the RT experimental design. Strubbia et al. (2020) demonstrate that priming strategy for norovirus alters the contig length, which is important for genotypic characterization, but this study did not include oligo(dT)s, or a comparison of RT enzymes.
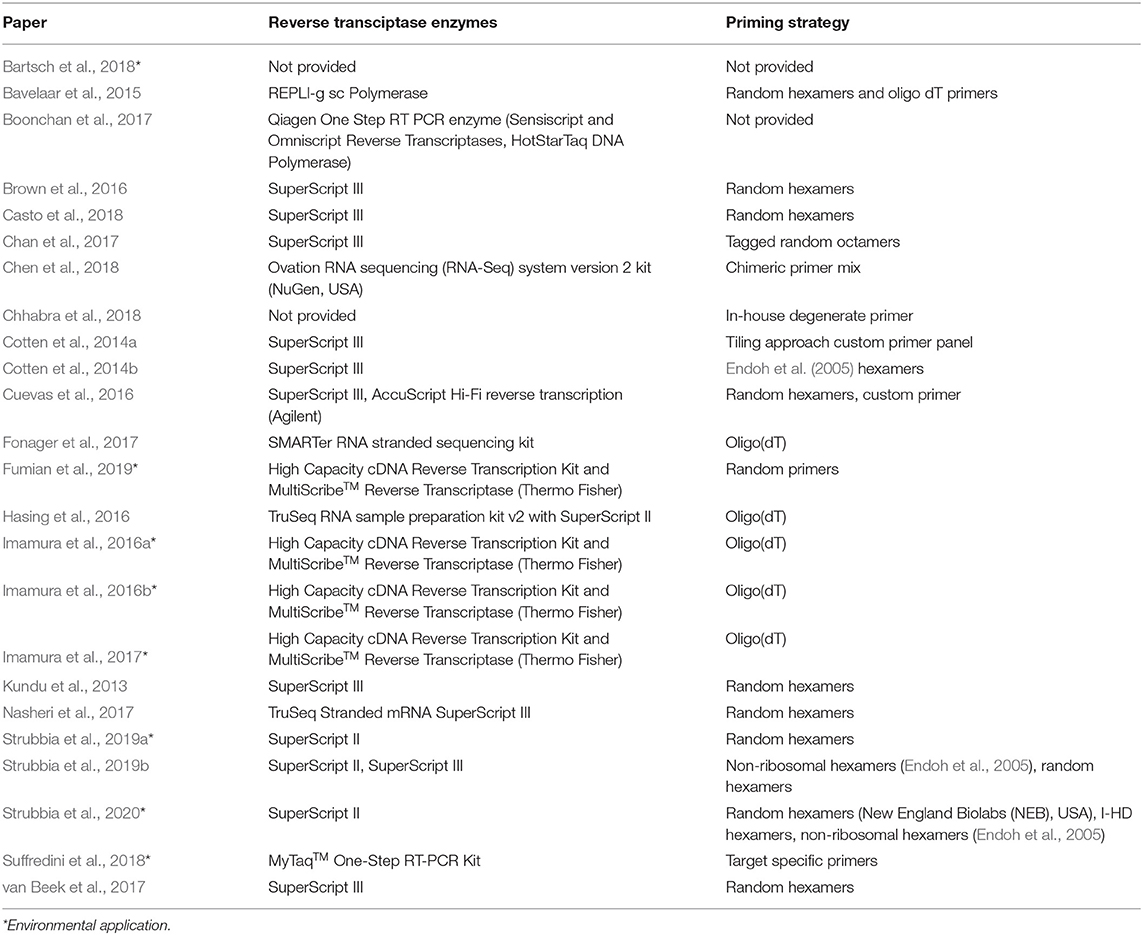
Table 5. RT enzyme and priming strategies used for norovirus NGS studies.
For traditional amplicon sequencing, primers should target a conserved region to allow for reliable detection of the viral target. Primers should be checked against recent sequences of the target question and the PCR conditions (particularly if DNA polymerase enzyme is altered) should be optimized and validated internally. “Jackhammer” PCR allows greater room for error in this aspect, as the primers are targeted across the genome, increasing the probability of successful amplification. That said however, viral RNA is a moving target and “jackhammer” approaches require up to date sequence data to perform consistently. Aside from primer design and method validation, additional considerations given to amplicon generation in HTS protocols is the choice of DNA polymerase and associated PCR cycling conditions and cycle numbers. Amplification errors generated during PCR appear in sequencing data and contribute to false mutations that can ultimately confound genetic analysis (Potapov and Ong, 2017). Several high-fidelity polymerase enzymes are commercially available and have been assessed using a variety of targets for downstream sequencing, see Table 6. Polymerase choice impacts both occurrence and relative abundance estimates and it has been noted that DNA polymerase choice had a greater impact on correct sequence assignment than a reduction in PCR cycles (Quail et al., 2011; Brandariz-Fontes et al., 2015; Nichols et al., 2018). Target characteristics such as prevalence of GC/AT rich regions, as can occur with Hepatitis E, may require an optimized approach for PCR amplification. Additives such as Dimethyl sulphoxide (DMSO) for GC-rich templates or betaine for AT-rich templates can reduce amplification bias for such targets. Betaine may help to keep a GC-rich template single-stranded, but it may also cause premature dissociation of the newly synthesized strand from an AT-rich template, introducing knock on effects for virome analysis (Aird et al., 2011; Nichols et al., 2018). Secondary structures in templates can also bias PCR when molecules with secondary structures, such as hairpin structures common in RNA templates, bind to themselves and inhibit their own amplification. This feature has been utilized in linker-amplification shotgun library second generation sequencing methods (Angly et al., 2006).
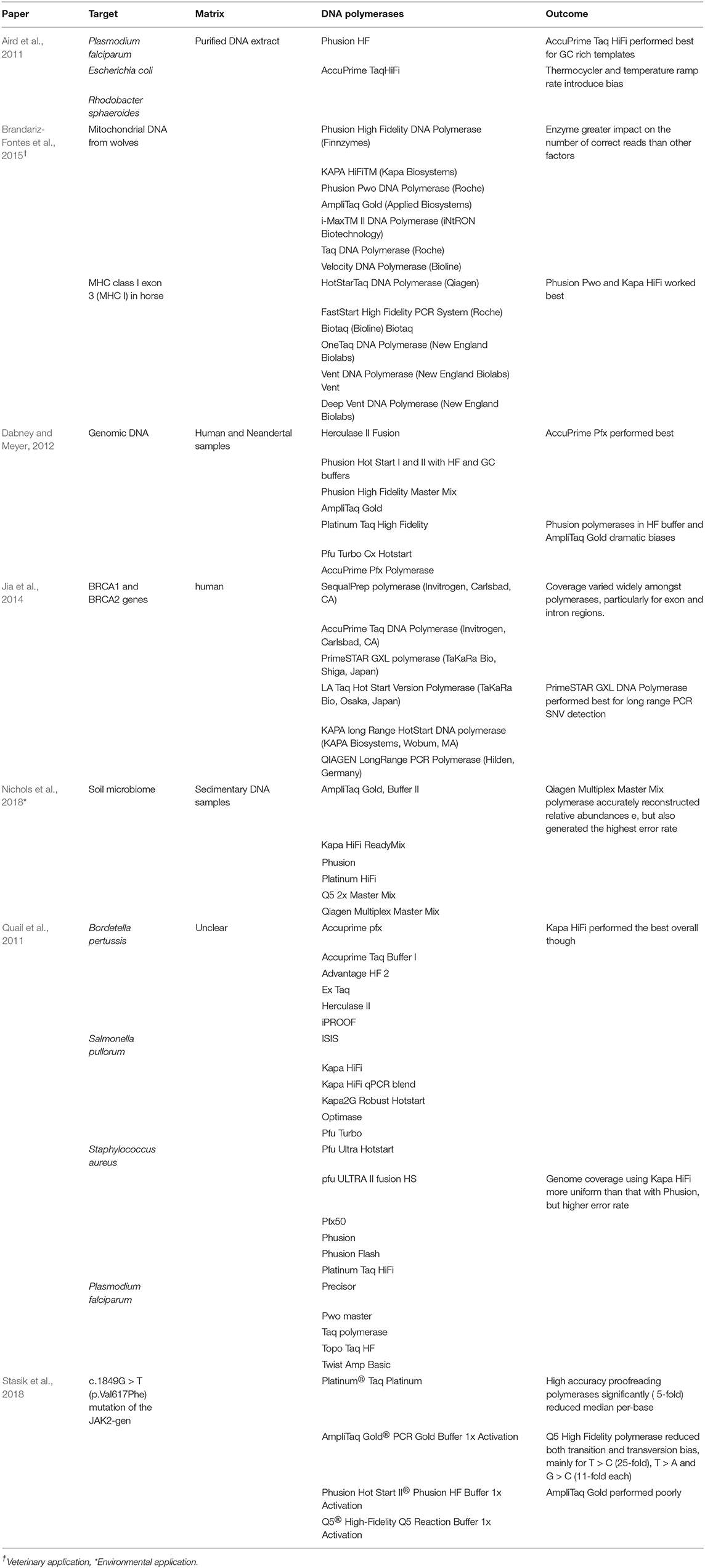
Table 6. Performance of various DNA polymerases enzymes applied during targeted HTS.
Following poly(A) + selection or rRNA depletion, RNA samples are fragmentated to a certain size range, owing to the limitations in the read length of many HTS platforms (Hrdlickova et al., 2017). RNAs can be fragmented with alkaline solutions, solutions with divalent cations, such Mg++, Zn++, or enzymes, such RNase III. Fragmentation with alkaline solutions or divalent cations is typically carried out at an elevated temperature to mitigate the effect of RNA structure on fragmentation (Hrdlickova et al., 2017).
Alternative RNA-Seq library preparations have been suggested to overcome fragmentation bias, including ClickSeq technology and the incorporation of barcoded non-ribosomal hexanucleotide primers during reverse transcription (Routh et al., 2015; Jaworski and Routh, 2017; Wang et al., 2017). In ClickSeq, reverse transcription (RT) reactions are performed with 3'-azido-nucleotides (AzNTPs). AzNTPs are chain-terminators that stochastically terminate CDNA synthesis as determined by AzNTPs:dNTPs. Following chain termination, single-stranded CDNA fragments are generated with an azido-group at their 3' ends. 3'-azido-blocked cDNA molecules can be purified and “click-ligated” to 5' alkyne-modified DNA adaptors via copper-catalysed azide-alkyne cycloaddition (CuAAC). The products of the ClickSeq reaction can be amplified using PCR to generate a CDNA sequencing library. Viral RNAs and MRNA using ClickSeq produced unbiased HTS libraries with low error-rates compared to standard methods (Routh et al., 2015; Jaworski and Routh, 2017).
Alternatively, intact RNAs can be reverse transcribed, and full-length CDNA can be fragmented. DNA is fragmented using either mechanical methods (e.g., nebulization and ultrasonication shearing) or enzymatic digestion. Nebulization involves directing compressed nitrogen or air forces into a DNA sample repeatedly through a small hole, producing mechanically sheared random fragments, leading to a heterogeneous mix of double-stranded DNA molecules containing 3'- or 5' overhangs as well as blunt ends (Knierim et al., 2011). During sonication, DNA is subjected to ultrasonic waves, whose vibrations produce gaseous cavitations in the liquid that shear or break high molecular weight DNA molecules through resonance vibration. Enzymatic digestion of DNA can take many forms, dependent on the library sequencing kit chosen. In general, the fragmented DNA is ligated at both blunt ends of each fragment with specific adaptors, using a transposon-based, tagmentation enzyme. These ligated sites later serve as primer-binding sites for amplification (Poptsova et al., 2014; Hrdlickova et al., 2017). A key issue with fragmentation is that the shear time is difficult to control because DNA or RNA originate from samples with different viral RNA abundance and this treatment may increase the occurrence of artifactual recombination.
Unlike RT-PCR, that is subject to Minimum Information for Publication of Quantiative Real-Time PCR Experiments (MIQE) guidelines, HTS/RNA-Seq has been to slow to include extensive controls, as per other molecular methods (Bustin et al., 2009, 2010). Issues such as contaminant RNA, cross-contamination and human error can be managed by robust experiment design that includes a variety of control samples and quality check points. Human error is unavoidable and 2–3% of samples were estimated to be mis-labeled or mis-pipetted in the Sequencing Quality Contro project (SEQC) (Qing et al., 2013). Given the observation of batch effects across studies; randomization of samples and treatment groups is pivotal and in part helps to circumvent handler bias (Qing et al., 2013; Miller et al., 2016; Eisenhofer et al., 2019).
Whole process negative controls and non-template controls can be included at sample preparation/extraction and library preparation stages. Furthermore, negative controls serve to demonstrate that the method in question does not generate false positives. While there are issues with running blank samples on some HTS platforms, negative samples can be spiked with a unique oligonucleotide to overcome primer-dimer formation issues, similar to internal process controls used in QRT-PCR assays. Cross-contamination can create “batch effects” due to the transfer of sample RNA, barcodes, or amplicons from neighboring wells or tubes. By including negative controls (extraction and library preparation) and comparing controls to biological samples post sequencing, cross-contamination can be identified, thereby aiding the interpretation of sequencing results. Strand specific sequencing can be used to identify the source of contamination during subtractive hybridization or viral genome vs. complementary RNA intermediates (Hedegaard et al., 2014; Matranga et al., 2014). Notably, the use of non-redundant dual indexing prevents index swapping during sequencing, which otherwise can contaminate up to 6% of samples (Costello et al., 2018; Du et al., 2020). Certain sequencing platforms also require maintenance washes between runs to reduce the likelihood of run-to-run cross-contamination.
While a variety of commercial positive sample controls are available, they are not always suitable as external/internal quality controls for HTS of viral RNA. Positive controls available include RNA oligonucleotides, mock virome communities (virus or nucleic acid), Spike in RNA variants (SIRVS) and External RNA Control Consortium (ERCC). RNA oligonucleotides, SIRVs and ERCC samples can be applied as internal controls, spiked into each sample, including the whole process negative control. RNA oligonucleotides and ERCC can be used to assess sample inhibition, which is important to consider in complex matrices, as well as confirm method specificity (Miller et al., 2016; Bal et al., 2018). However, Munro et al. (2014), Qing et al. (2013), and Risso et al. (2014) determined that while ERCC controls could be used as batch controls, they exhibited strong protocol dependent bias and a high degree of variation. SIRVs have been used in previous studies to assess the accuracy of SNV calling in transcriptomic bioinformatic pipelines, though these may not work for SNV detection in RNA viruses. Furthermore, the use of spike in controls assumes that technical effects impact spike-ins and target sequences in the same way. If library preparation steps impact spike-in and target read counts differently, then normalization or inhibition based on the spike-ins may be incorrectly assessed. Mock virome samples or RNA oligonucleotides can be used in a serial dilution to determine limit of detection or false discovery rate and, in the case of mock viromes, demonstrate that a variety of RNA viral families can be sequenced. To date, mock virome controls have not yet been applied in HTS of RNA viruses in environmental samples.
The importance of negative controls in any molecular work, but particularly a method as sensitive as HTS/RNA-Seq, has been emphasized again and again. Multiple studies have been published noting contaminating taxa, likely from reagents (kitome), common environmental taxa introduced through cross-contamination (Salter et al., 2014; Glassing et al., 2016; Bal et al., 2018; Leon et al., 2018) and possible cross contamination (Strubbia et al., 2020). Moreover, the discovery of bacterial reads in cell line data processed using poly-A selection demonstrates that downstream contamination is a source of bacterial reads (Strong et al., 2014). Contamination can also originate from staff, plastic consumables, nucleic acid extraction kits and platforms and laboratory reagents, therefore controls should address these sources as outlined earlier. Negative controls should be compared to biological samples in the final raw sequencing reads. There is much debate as to whether or not contaminating taxa be removed from biological samples but this has been applied in various pipelines (Davis et al., 2018; Leon et al., 2018; Palmer et al., 2018). An additional pre-processing step that has been proposed is to use predictive modeling to identify putative contaminants (Risso et al., 2014; Eisenhofer et al., 2019).
High mutation rate and antigenic drift of most single stranded RNA (SSRNA) viruses, makes it difficult to design reasonably sized CBPH panels that capture species diversity, while also being affordable and technically feasible (Duffy, 2018; Peck and Lauring, 2018). It must be noted though that CBPH resulted in significantly greater genome coverage, % of viral reads and depth of coverage in all studies listed in Table 1 compared to shotgun metagenomics. Whilst some cost comparisons suggest that amplicon “jackhammer” approaches are a similar cost, there is to date only one amplicon vs CBPH comparison study. It was determined that amplicon sequencing had greater on target reads, though CBPH demonstrated a significantly higher standard deviation of genome coverage a more accurate depiction of SNVs (Samorodnitsky et al., 2015). Furthermore, Nasir et al. (2020) noted that CBPH provided an advantage over amplification based protocols such as tiling amplicon approaches due to the absence of amplification artifacts.
Applications of SISPA in the veterinary field has permitted first-time detection or detection of new variants of Newcastle disease virus, Schmallenberg virus, Hantaviruses, and enterovirus C104. However, based on the comparative studies and field work applications of SISPA, it appears that its application is best placed for fieldwork, where speed rather than accuracy is the objective. Follow-up direct sequencing or targeted amplicon sequencing should be used to verify suspected SNVs. There are very few publications applying RCA-HTS to RNA templates and this is likely due to the challenge of working with samples containing abundant background RNA and low target RNA concentrations. While RCA-HTS is the least error prone target amplification approach, it is not suitable in its current format for application to low abundance RNA samples and better suited to studies involving cell culture work.
Various findings from comparative rRNA depletion/enrichment studies found that while rRNA depletion resulted in increased target reads, coverage depth and detection of intra host variants, it also increased the proportion of low-quality reads obtained (Adiconis et al., 2013). PDD incorporating RNaseH provided superior or more consistent results at lower costs, compared to Ribo-Zero/subtractive hybridization (Herbert et al., 2018; Huang et al., 2020). All depletion methods show both strand specific bias as well as a bias toward shorter transcripts (Pecman et al., 2017; Herbert et al., 2018). Pecman et al. (2017) found that rRNA depletion methods worked better for SSRNA viruses than dsDNA viruses. The limitation to these studies is that most focus on commercially available subtractive hybridization kits (see Table 2 for a more in-depth overview of the aforementioned studies). Furthermore, viral RNA is rarely the target, with the host transcriptome more typically the focus.
In terms of recommendations for agnostic sequencing, PDD is more robust and flexible in terms of host rRNA and works better with degraded samples, however there may be issues for low concentration targets, in which cases NSR is a viable alternative. SISPA and SPIA require high input concentrations of RNA and are likely to be unsuitable for samples containing low abundance of specific RNA viral targets. For low concentration targets with a poly-A tail, evidence from transcriptomics indicates that poly-A capture outperforms subtractive hybridization. Choices for targeted sequencing heavily depend on the research question. While CBPH is more expensive, it is a more suitable choice for the detection of SNVs than a “jackhammer” approach. Amplicon sequencing is suitable for well-characterized viruses, with robust PCR assays, where the purpose is genotypic characterization.
Virus-specific approaches increase the chance to detect less abundant species through HTS. The quality of a HTS run has both cost and time implications, and greater viral specificity can reduce the time required for bioinformatics analyses (Hjelmsø et al., 2017). Purification steps during concentration and extraction may not increase viral RNA, but the elimination of background nucleic acids could increase the ratio of viral reads and the quality of contigs obtained (Strubbia et al., 2019b). Therefore, choice of concentration, RNA isolation/extraction and purification steps are influential in determining the quality of RNA obtained and subsequent HTS outputs. In general, AGPC methods result in better quality RNA, however the compromise is often lower concentrations of RNA. While this may not concern studies working with concentrated clinical samples, complex samples such as stool, soil or certain food matrices pose a greater challenge. In these cases, SMSC and MB methods work best for samples, containing low concentrations of viral RNA or in complex samples with high levels of background RNA. Downstream purification (DNase step, spin column purification, ethanol precipitation) of the RNA extracts may be required as SMSC/MB can carry through genomic DNA.
Overall four trends have been observed for RT efficiency, with some conflicting evidence amongst studies as can be seen in Tables 2, 7: (i) background tRNA has a positive impact on RT efficiency, (ii) SuperScript II is more efficient at amplifying low abundance transcripts, (iii) RT efficiency is dependent on template/gene target and (iv) RT enzyme choice contributes more to variation than technical/pipetting variation. RT enzyme plays an important role in generating both accurate and sufficient yields of CDNA, but outcomes are dependent on the target, background RNA, reagent concentrations and priming strategy. Few studies have compared priming strategy during CDNA synthesis, and even fewer have looked at the impact of primer choice on HTS output. Random hexamers tend to produce more variable yields and should be applied at high concentrations (Lekanne Deprez et al., 2002; Stangegaard et al., 2006; Werbrouck et al., 2007; Cholet et al., 2020). Gene specific primers are the most efficient in terms of yield, however they limit HTS output as they require prior knowledge of the target of interest, and do not permit a metagenomic approach (Lekanne Deprez et al., 2002; Miranda and Steward, 2017). In terms of HTS output, random hexamer priming has been shown to conserve the actual proportions of the mock community, however gene specific primers provided better coverage and Operational Taxonomic Unit (OTU) richness of the transcript in question (Schwaber et al., 2019; Cholet et al., 2020; Zucha et al., 2020). This is an important consideration in experimental design and needs to reflect the purpose of the study, i.e., to (a) to assess diversity or (b) characterize a specific target.

Table 7. Literature investigating the impact of RT enzyme on cDNA fidelity.
Optimization of PCR based amplification approaches requires careful consideration of (i) the target(s) in question and (ii) the bias introduced though polymerase choice and cycling conditions. Overall trends from the relevant studies demonstrate that thermostable, high fidelity polymerases outperform the robust alternatives. AmpliTaq Gold has been commonly applied in molecular virology and yet performed poorly in all studies, regardless of target or matrix (Table 6).
Fragmentation of RNA and DNA has been observed to induce bias. The bulk of RNA-Seq studies have investigated the impact of fragmentation on relative gene expression compared to QRT-PCR measurements, rather than the detection of viral quasispecies. Bias observed is dependent on when and what type of fragmentation was applied. For fragmentation of RNA, RNase III-based fragmentation demonstrates a preference for double-stranded RNA sequences. This can result in uneven fragmentation of RNA leading to differential representation of specific regions of RNA (Adiconis et al., 2013). Parekh et al. (2016) found that a large fraction of computationally identified read duplicates were not PCR duplicates and could be explained by sampling and fragmentation bias. Fragmentation bias contributed considerably to computationally identified read duplicates and was stronger for Smart-Seq, i.e., for enzymatic fragmentation, than for TruSeq, i.e., heat fragmentation.
ClickSeq fragmenation (Routh et al., 2015; Jaworski and Routh, 2017; Wang et al., 2017) and a similar method (Wang et al., 2017) were more likely to conserve the relative abundance of the original samples due their robustness against common artifacts of HTS such as chimera formation and artefactual recombination (Routh et al., 2015). This is important as these libraries result in more accurate assessment of polymorphism frequency, species population diversity and accurate de novo genome assembly.
In terms of bias introduced during fragmentation of CDNA, Tn5 and other enzyme-based CDNA fragmentation methods require a precise enzyme:DNA ratio, making method optimization less straightforward than RNA fragmentation (Hrdlickova et al., 2017). When the enzymatic fragmentation is run to completion, the proportion of smaller fragments increases significantly. Furthermore, ultrasound treatment of genomic DNA could induce amplified cleavage of GC-rich areas of genome (Poptsova et al., 2014). As CDNA fragments are sequenced, the number of reads corresponding to each transcript is proportional to the number of CDNA fragments rather than the number of transcripts. Since longer transcripts are generally sheared into more fragments, more reads will be assigned to them than shorter transcripts, dismissing the possibility of relative abundance assessment of viral populations. Indeed, this fragmentation step introduces additional diversity into the starting position of the sequence (Alberti et al., 2014).
Other studies have evaluated mechanical and enzymatic fragmentation of CDNA for virus amplicon-based sequencing though with conflicting results. While Vrancken et al. (2016) determined that the fragmentation had a modest impact on sequencing results, Knierim et al. (2011) observed that while overall sequence quality was similar, enzymatic fragmentation resulted in more insertions/deletions in raw sequence reads yet outperformed mechanical fragmentation when filtering homopolymer errors.
Sequencing platform and bioinformatics pipelines have not been considered in this review, though it is recognized that both impact sequencing results, they are outside the scope of this review and our expertise. Most virus specific pipelines rely on k-mer frequency classification, sometimes with protein alignment based verification (Zhao et al., 2013; Roux et al., 2015; Ren et al., 2017; Alam and Chowdhury, 2020; Nayfach et al., 2020). However as Höper et al. (2020) demonstrated bioinformatic pipelines require further harmonization and standardization for diagnostic application. A comprehensive review on bioinformatic processing of viral sequencing data is required and the current pandemic (COVID-19) has placed our knowledge gaps and ability to interpret sequence data, front, and center. Our current ability to pre-empt RNA viruses of clinical concern detected from sequencing of environmental samples is limited by the need to confirm HTS results in cell culture and animal models.
In this review, the focus has been on how to obtain high quality RNA virus sequences from complex matrices by making careful, informed choices on methodology. This is best described in the decision matrix in Figure 4. For environmental samples, likely containing low concentrations of RNA virus; method choice must be carefully balanced with the objective in mind. Clinical samples with higher viral RNA concentrations could output high quality sequences but if cheaper target amplicon sequencing answers the research questions in mind then it is not necessary. For method steps such as RT and fragmentation, a target specific approach should be taken and current literature surveyed of indications on performance, particularly for RT priming approaches. While agnostic approaches are theoretically preferable, they may not provide sufficient coverage of viral genomes for classification, thus limiting their usefulness as a standard sequencing approach. Therefore, intermediaries such as capture probe hybridization and tiling/jackhammer amplicon approaches should be strongly considered as initial approaches and complemented with long read sequencing.
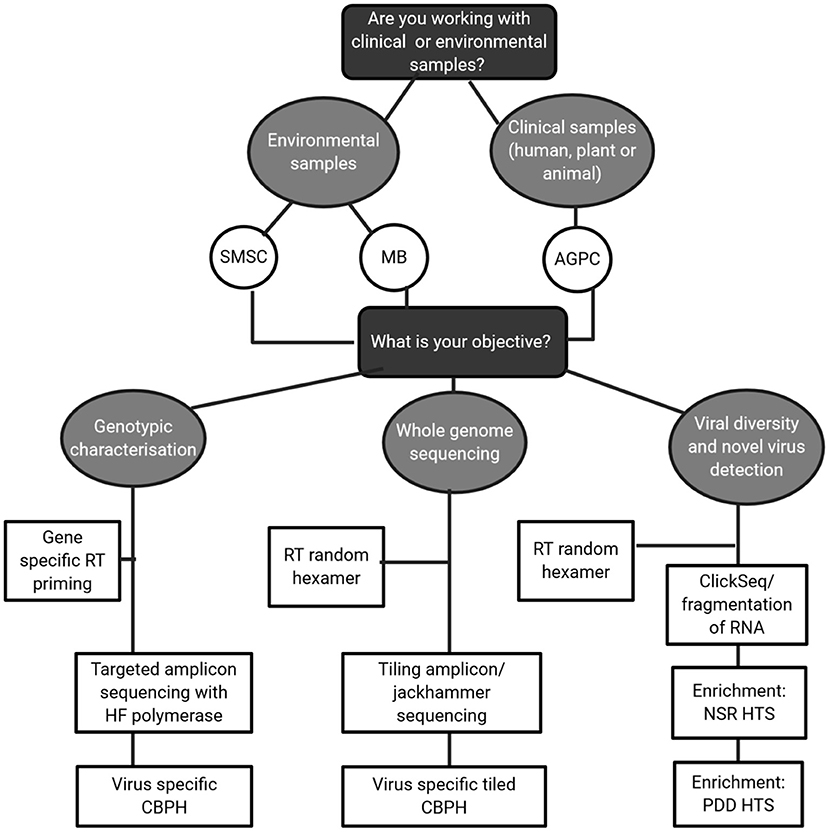
Figure 4. Decision tree guiding method choices for HTS of RNA viruses.
Without controls, results are meaningless. The inclusion of whole process controls, internal process controls such as spike-in DNA and negative controls provide greater certainty on the obtained sequencing reads, particularly in the case of shotgun metagenomics. Novel RNA viruses or variants should be confirmed by PCR and/or Sanger sequencing and relative abundance should be not relied upon as a quantitative measure.
While it is challenging to obtain high quality sequences from environmental samples, the information that could be gleaned is essential for maintaining public health. From developing new PCR/qPCR assays based on recent sequencing data, to monitoring antigenic drift and recombination, identifying new transmission pathways, hosts and viruses or pre-empting RNA viruses and variants of clinical concern in a One Health paradigm, the list of potential benefits goes on. HTS has much to offer to the field of environmental virology but in incorporating it into the arsenal of molecular tools already utilized, it is important to be aware of the challenges and biases and to circumvent these by considering both the matrix and target virus(es) in question.
AF reviewed the literature and compiled the review. AR revised the initial drafts. SK, HO'S, PC, and FC reviewed the final draft. All authors contributed to the article and approved the submitted version.
This work was funded by the Cullen Scholarship Programme which is carried out with the support of the Marine Institute, and funded under the Marine Research Programme by the Irish Government (Funding call: CF/18/01/01).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures 1–4 were created with BioRender.com. The authors would like to thank Teagasc Food Research Centre for supporting this work.
AGPC, Acid guanidinium thiocyanate phenol chloroform; CBPH, Capture based probe hybridization; cDNA, complementary cDNA; DMSO, Dimethyl sulphoxide; DNA, deoxyribonucleic acid; ERCC, External RNA Control Consortium; HTS, High Throughput Sequencing; ITS, Internal transcribed spacer; LNA, Locked nucleic acid; MB, Magnetic beads; MIQE, Minimum Information for Publication of Quantiative Real-Time PCR Experiments; mRNA, messenger RNA; NSR, Not so random; OUT, Operational Taxonomic Unit; PDD, Probe directed degradation; PEG, Polyethylene glycol; qRT-PCR, quantitative Reverse Transcription Polymerase Chain Reaction; RCA, Rolling Circle Amplification; RIN, RNA Integrity number; RNA, ribonucleic acid; RT-PCR, Reverse Transcription Polymerase Chain Reaction; SIRVs, Spike in RNA variants; SISPA, Sequence Independent, Single Primer Amplification; SMSC, Silica membrane based spin column technology; SNV, Single Nucleotide Variants; SPIA, Single Primer Isothermal Amplificaiton; ssRNA, single stranded RNA.
Aarem, J., Brunborg, G., Aas, K. K., Harbak, K., Taipale, M. M., Magnus, P., et al. (2016). Comparison of blood RNA isolation methods from samples stabilized in Tempus tubes and stored at a large human biobank. BMC Res. 9:430. doi: 10.1186/s13104-016-2224-y
Acevedo, A., Brodsky, L., and Andino, R. (2014). Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature 505, 686–690. doi: 10.1038/nature12861
Adams, N. M., Bordelon, H., Wang, K.-K. A., Albert, L. E., Wright, D. W., and Haselton, F. R. (2015). Comparison of three magnetic bead surface functionalities for RNA extraction and detection. ACS Appl. Mater. Interfaces 7, 6062–6069. doi: 10.1021/am506374t
Adiconis, X., Borges-Rivera, D., Satija, R., DeLuca, D. S., Busby, M. A., Berlin, A. M., et al. (2013). Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat. Methods 10, 623–629. doi: 10.1038/nmeth.2483
Ahmed, R., Hossain, M. S., Haque, M. S., Alam, M. M., and Islam, M. S. (2019). Modified protocol for RNA isolation from different parts of field-grown jute plant suitable for NGS data generation and quantitative real-time RT-PCR. Afr. J. Biotechnol. 18, 647–658. doi: 10.5897/AJB2019.16819
Aird, D., Ross, M. G., Chen, W.-S., Danielsson, M., Fennell, T., Russ, C., et al. (2011). Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 12:R18. doi: 10.1186/gb-2011-12-2-r18
Alam, M. N. U., and Chowdhury, U. F. (2020). Short k-mer abundance profiles yield robust machine learning features and accurate classifiers for RNA viruses. PLoS ONE 15:e0239381. doi: 10.1371/journal.pone.0239381
Alberti, A., Belser, C., Engelen, S., Bertrand, L., Orvain, C., Brinas, L., et al. (2014). Comparison of library preparation methods reveals their impact on interpretation of metatranscriptomic data. BMC Genomics 15:912. doi: 10.1186/1471-2164-15-912
Alberti, A., Poulain, J., Engelen, S., Labadie, K., Romac, S., Ferrera, I., et al. (2017). Viral to metazoan marine plankton nucleotide sequences from the Tara Oceans expedition. Sci. Data 4:170093. doi: 10.1038/sdata.2017.93
Angly, F. E., Felts, B., Breitbart, M., Salamon, P., Edwards, R. A., Carlson, C., et al. (2006). The marine viromes of four oceanic regions. PLoS Biol. 4:e368. doi: 10.1371/journal.pbio.0040368
Archer, S. K., Shirokikh, N. E., and Preiss, T. (2014). Selective and flexible depletion of problematic sequences from RNA-seq libraries at the cDNA stage. BMC Genomics 15:401. doi: 10.1186/1471-2164-15-401
Armour, C. D., Castle, J. C., Chen, R., Babak, T., Loerch, P., Jackson, S., et al. (2009). Digital transcriptome profiling using selective hexamer priming for cDNA synthesis. Nat. Methods 6, 647–649. doi: 10.1038/nmeth.1360
Artic Network (2021). Artic Network. Available online at: https://artic.network/ (accessed February 8, 2021).
Asai, S., Ianora, A., Lauritano, C., Lindeque, P. K., and Carotenuto, Y. (2015). High-quality RNA extraction from copepods for next generation sequencing: a comparative study. Mar. Genomics 24, 115–118. doi: 10.1016/j.margen.2014.12.004
Bal, A., Pichon, M., Picard, C., Casalegno, J. S., Valette, M., Schuffenecker, I., et al. (2018). Quality control implementation for universal characterization of DNA and RNA viruses in clinical respiratory samples using single metagenomic next-generation sequencing workflow. BMC Infect. Dis. 18, 1–10. doi: 10.1186/s12879-018-3446-5
Bartsch, C., Hoper, D., Made, D., and Johne, R. (2018). Analysis of frozen strawberries involved in a large norovirus gastroenteritis outbreak using next generation sequencing and digital PCR. Food Microbiol. 76, 390–395. doi: 10.1016/j.fm.2018.06.019
Bavelaar, H. H. J., Rahamat-Langendoen, J., Niesters, H. G. M., Zoll, J., and Melchers, W. J. G. (2015). Whole genome sequencing of fecal samples as a tool for the diagnosis and genetic characterization of norovirus. J. Clin. Virol. 72, 122–125. doi: 10.1016/j.jcv.2015.10.003
Boonchan, M., Motomura, K., Inoue, K., Ode, H., Chu, P. Y., Lin, M., et al. (2017). Distribution of norovirus genotypes and subtypes in river water by ultra-deep sequencing-based analysis. Lett. Appl. Microbiol. 65, 98–104. doi: 10.1111/lam.12750
Brandariz-Fontes, C., Camacho-Sanchez, M., Vile, C., Vega-Pla, J. L., Rico, C., and Leonard, J. A. (2015). Effect of the enzyme and PCR conditions on the quality of high-throughput DNA sequencing results. Sci. Rep. 5:8056. doi: 10.1038/srep08056
Briese, T., Kapoor, A., Mishra, N., Jain, K., Kumar, A., Jabado, O. J., et al. (2015). Virome capture sequencing enables sensitive viral diagnosis and comprehensive virome analysis. mBio 6:e01491–15. doi: 10.1128/mBio.01491-15
Brown, J. R., Roy, S., Ruis, C., Romero, E. Y., Shah, D., Williams, R., et al. (2016). Norovirus whole-genome sequencing by sureselect target enrichment: a robust and sensitive method. J. Clin. Microbiol. 54, 2530–2537. doi: 10.1128/JCM.01052-16
Bustin, S., Beaulieu, J., Huggett, J., Jaggi, R., Kibenge, F., Olsvik, P., et al. (2010). MIQE precis: Practical implementation of minimum standard guidelines for fluorescencebased quantitative real-time PCR experiments. BMC Mol. Biol. 11:74. doi: 10.1186/1471-2199-11-74
Bustin, S., Benes, V., Garson, J., Hellemans, J., Huggett, J., Kubista, M., et al. (2009). The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–22. doi: 10.1373/clinchem.2008.112797
Bustin, S., Dhillon, H. S., Kirvell, S., Greenwood, C., Parker, M., Shipley, G. L., et al. (2015). Variability of the reverse transcription step: practical implications. Clin. Chem. 61, 202–212. doi: 10.1373/clinchem.2014.230615
Carbo, E. C., Sidorov, I. A., Zevenhoven-Dobbe, J. C., Snijder, E. J., Claas, E. C., Laros, J. F. J., et al. (2020). Coronavirus discovery by metagenomic sequencing: a tool for pandemic preparedness. J. Clin. Virol. 131:104594. doi: 10.1016/j.jcv.2020.104594
Casto, A., Adler, A., Makhsous, N., Qin, X., Crawford, K., Jerome, K., et al. (2018). Real-time metagenomic sequencing reveals discrete transmission clusters within a hospital-associated norovirus outbreak. Open Forum Infect. Dis. 5(Suppl 1):S49. doi: 10.1093/ofid/ofy209.117
Chan, M. C. W., Kwok, K., Hung, T.-N., Chan, L.-Y., and Chan, P. K. S. (2017). Complete genome sequence of an emergent recombinant GII.P16-GII.2 norovirus strain associated with an epidemic spread in the winter of 2016-2017 in Hong Kong, China. Genome Announc. 5:e00343-17. doi: 10.1128/genomeA.00343-17
Chen, H., Wang, S., and Wang, W. (2018). Complete genome sequence of a human norovirus strain from the United States classified as genotype GII.P6_gii.6. Genome Announc. 6:e00489-18. doi: 10.1128/genomeA.00489-18
Chhabra, P., Aswath, K., Collins, N., Ahmed, T., Olortegui, M. P., Kosek, M., et al. (2018). Near-complete genome sequences of several new norovirus genogroup II genotypes. Genome Announc. 6:e00007-18. doi: 10.1128/genomeA.00007-18
Cholet, F., Ijaz, U. Z., and Smith, C. J. (2020). Reverse transcriptase enzyme and priming strategy affect quantification and diversity of environmental transcripts. Environ. Microbiol. 22, 2383–2402. doi: 10.1101/2020.03.18.996603
Cholleti, H., Hayer, J., Fafetine, J., Berg, M., and Blomström, A.-L. (2018). Genetic characterization of a novel picorna-like virus in Culex spp. mosquitoes from Mozambique. Virol. J. 15:71. doi: 10.1186/s12985-018-0981-z
Chrzastek, K., Lee, D. H., Smith, D., Sharma, P., Suarez, D. L., Pantin-Jackwood, M., et al. (2017). Use of Sequence-Independent, Single-Primer-Amplification (SISPA) for rapid detection, identification, and characterization of avian RNA viruses. Virology 509, 159–166. doi: 10.1016/j.virol.2017.06.019
Cinek, O., Kramna, L., Mazankova, K., Kunteova, K., Chuda, K., Claas, E. C. J., et al. (2019). Virus genotyping by massive parallel amplicon sequencing: adenovirus and enterovirus in the Norwegian MIDIA study. J. Med. Virol. 91, 606–614. doi: 10.1002/jmv.25361
Costello, M., Fleharty, M., Abreu, J., Farjoun, Y., Ferriera, S., Holmes, L., et al. (2018). Characterization and remediation of sample index swaps by non-redundant dual indexing on massively parallel sequencing platforms. BMC Genomics 19:332. doi: 10.1186/s12864-018-4703-0
Cotten, M., Oude Munnink, B., Canuti, M., Deijs, M., Watson, S. J., Kellam, P., et al. (2014a). Full genome virus detection in fecal samples using sensitive nucleic acid preparation, deep sequencing, and a novel iterative sequence classification algorithm. PLoS ONE 9:e93269. doi: 10.1371/journal.pone.0093269
Cotten, M., Petrova, V., Phan, M. V. T., Rabaa, M. A., Watson, S. J., Ong, S. H., et al. (2014b). Deep sequencing of norovirus genomes defines evolutionary patterns in an urban tropical setting. J. Virol. 88, 11056–11069. doi: 10.1128/JVI.01333-14
Cruz, C. D., Torre, A., Troncos, G., Lambrechts, L., and Leguia, M. (2016). Targeted full-genome amplification and sequencing of dengue virus types 1–4 from South America. J. Virol. Methods 235, 158–167. doi: 10.1016/j.jviromet.2016.06.001
Cuevas, J. M., Combe, M., Torres-Puente, M., Garijo, R., Guix, S., Buesa, J., et al. (2016). Human norovirus hyper-mutation revealed by ultra-deep sequencing. Infect. Genet. Evol. 41, 233–239. doi: 10.1016/j.meegid.2016.04.017
Culviner, P. H., Guegler, C. K., and Laub, M. T. (2020). A simple, cost-effective, and robust method for rRNA depletion in RNA-sequencing studies. mBio 11:e00010–20. doi: 10.1128/mBio.00010-20
Dabney, J., and Meyer, M. (2012). Length and GC-biases during sequencing library amplification: a comparison of various polymerase-buffer systems with ancient and modern DNA sequencing libraries. Biotechniques 52, 87–94. doi: 10.2144/000113809
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A., and Callahan, B. J. (2018). Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6:226. doi: 10.1186/s40168-018-0605-2
de Vries, M., Deijs, M., Canuti, M., van Schaik, B. D. C., Faria, N. R., van de Garde, M. D. B., et al. (2011). A sensitive assay for virus discovery in respiratory clinical samples. PLoS ONE 6:e16118. doi: 10.1371/journal.pone.0016118
Depledge, D. P., Srinivas, K. P., Sadaoka, T., Bready, D., Mori, Y., Placantonakis, D. G., et al. (2019). Direct RNA sequencing on nanopore arrays redefines the transcriptional complexity of a viral pathogen. Nat. Commun. 10:754. doi: 10.1038/s41467-019-08734-9
Di, H., Thor, S., Trujillo, A. A., Stark, T., Marinova-Petkova, A., Jones, J., et al. (2019). Comparison of nucleic acid extraction methods for next-generation sequencing of avian influenza A virus from ferret respiratory samples. J. Virol. Methods. doi: 10.1016/j.jviromet.2019.04.014
Du, M., He, Y., Chen, J., Sun, H., Fu, Y., and Wang, J. (2020). Unique dual indexing PCR reduces chimeric contamination and improves mutation detection in cell-free DNA of pregnant women. Talanta 217:121035. doi: 10.1016/j.talanta.2020.121035
Duffy, S. (2018). Why are RNA virus mutation rates so damn high? PLoS Biol. 16:e3000003. doi: 10.1371/journal.pbio.3000003
Duncavage, E. J., Magrini, V., Becker, N., Armstrong, J. R., Demeter, R. T., Wylie, T., et al. (2011). Hybrid capture and next-generation sequencing identify viral integration sites from formalin-fixed, paraffin-embedded tissue. J. Mol. Diagnost. 13, 325–333. doi: 10.1016/j.jmoldx.2011.01.006
Eden, J.-S., Rockett, R., Carter, I., Rahman, H., de Ligt, J., Hadfield, J., et al. (2020). An emergent clade of SARS-CoV-2 linked to returned travellers from Iran. Virus Evol. 6:veaa027. doi: 10.1093/ve/veaa027
Eisenhofer, R., Minich, J. J., Marotz, C., Cooper, A., Knight, R., and Weyrich, L. S. (2019). Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol. 27, 105–117. doi: 10.1016/j.tim.2018.11.003
Endoh, D., Mizutani, T., Kirisawa, R., Maki, Y., Saito, H., Kon, Y., et al. (2005). Species-independent detection of RNA virus by representational difference analysis using non-ribosomal hexanucleotides for reverse transcription. Nucleic Acids Res. 33:e65. doi: 10.1093/nar/gni064
Fauver, J. R., Akter, S., Morales, A. I. O., Black, W. C., Rodriguez, A. D., Stenglein, M. D., et al. (2019). A reverse-transcription/RNase H based protocol for depletion of mosquito ribosomal RNA facilitates viral intrahost evolution analysis, transcriptomics and pathogen discovery. Virology 528, 181–197. doi: 10.1016/j.virol.2018.12.020
Fauver, J. R., Petrone, M. E., Hodcroft, E. B., Shioda, K., Ehrlich, H. Y., Watts, A. G., et al. (2020). Coast-to-coast spread of SARS-CoV-2 during the early epidemic in the United States. Cell. 181, 990–996.e5. doi: 10.1016/j.cell.2020.04.021
Fonager, J., Stegger, M., Rasmussen, L. D., Poulsen, M. W., Ronn, J., Andersen, P. S., et al. (2017). A universal primer-independent next-generation sequencing approach for investigations of norovirus outbreaks and novel variants. Sci. Rep. 7:813. doi: 10.1038/s41598-017-00926-x
Fumian, T. M., Fioretti, J. M., Lun, J. H., dos Santos, I. A. L., White, P. A., and Miagostovich, M. P. (2019). Detection of norovirus epidemic genotypes in raw sewage using next generation sequencing. Environ. Int. 123, 282–291. doi: 10.1016/j.envint.2018.11.054
Garcia-Nogales, P., Serrano, A., Secchi, S., Gutierrez, S., and Aris, A. (2010). Comparison of commercially-available RNA extraction methods for effective bacterial RNA isolation from milk spiked samples. Electron. J. Biotechnol. 13, 19–20. doi: 10.2225/vol13-issue5-fulltext-10
Glassing, A., Dowd, S. E., Galandiuk, S., Davis, B., and Chiodini, R. J. (2016). Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 8:24. doi: 10.1186/s13099-016-0103-7
Goya, S., Valinotto, L. E., Tittarelli, E., Rojo, G. L., Nabaes Jodar, M. S., Greninger, A. L., et al. (2018). An optimized methodology for whole genome sequencing of RNA respiratory viruses from nasopharyngeal aspirates. PLoS ONE 13:e0199714. doi: 10.1371/journal.pone.0199714
Gradel, C., Terrazos Miani, M. A., Barbani, M. T., Leib, S. L., Suter-Riniker, F., and Ramette, A. (2019). Rapid and cost-efficient enterovirus genotyping from clinical samples using flongle flow cells. Genes 10:659. doi: 10.3390/genes10090659
Grubaugh, N. D., Gangavarapu, K., Quick, J., Matteson, N. L., De Jesus, J. G., Main, B. J., et al. (2019). An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 20:8. doi: 10.1186/s13059-018-1618-7
Guichet, E., Serrano, L., Laurent, C., Eymard-Duvernay, S., Kuaban, C., Vidal, L., et al. (2018). Comparison of different nucleic acid preparation methods to improve specific HIV-1 RNA isolation for viral load testing on dried blood spots. J. Virol. Methods 251, 75–79. doi: 10.1016/j.jviromet.2017.10.014
Hanke, D., Freuling, C. M., Fischer, S., Hueffer, K., Hundertmark, K., Nadin-Davis, S., et al. (2016). Spatio-temporal analysis of the genetic diversity of arctic rabies viruses and their reservoir hosts in Greenland. PLoS Negl. Trop. Dis. 10:e0004779. doi: 10.1371/journal.pntd.0004779
Hasing, M. E. H., Lee, B. E., Preiksaitis, J. K., and Pang, X. L. (2016). A next generation sequencing-based method to study the intra-host genetic diversity of norovirus in patients with acute and chronic infection. BMC Genomics 17:480. doi: 10.1186/s12864-016-2831-y
Hata, A., Kitajima, M., Haramoto, E., Lee, S., Ihara, M., Gerba, C. P., et al. (2018). Next-generation amplicon sequencing identifies genetically diverse human astroviruses, including recombinant strains, in environmental waters. Sci. Rep. 8:11837. doi: 10.1038/s41598-018-30217-y
Hedegaard, J., Thorsen, K., Lund, M. K., Hein, A.-M. K., Hamilton-Dutoit, S. J., Vang, S., et al. (2014). Next-generation sequencing of RNA and DNA isolated from paired fresh-frozen and formalin-fixed paraffin-embedded samples of human cancer and normal tissue. PLoS ONE 9:e98187. doi: 10.1371/journal.pone.0098187
Hendling, M., and Barišić, I. (2019). In-silico design of DNA oligonucleotides: challenges and approaches. Comput. Struct. Biotechnol. J. 17, 1056–1065. doi: 10.1016/j.csbj.2019.07.008
Herbert, Z. T., Kershner, J. P., Butty, V. L., Thimmapuram, J., Choudhari, S., Alekseyev, Y. O., et al. (2018). Cross-site comparison of ribosomal depletion kits for Illumina RNAseq library construction. BMC Genomics 19:199. doi: 10.1186/s12864-018-4585-1
Hjelmsø, M. H., Hellmér, M., Fernandez-Cassi, X., Timoneda, N., Lukjancenko, O., Seidel, M., et al. (2017). Evaluation of methods for the concentration and extraction of viruses from sewage in the context of metagenomic sequencing. PLoS ONE 12:e0170199. doi: 10.1371/journal.pone.0170199
Höper, D., Grützke, J., Brinkmann, A., Mossong, J., Matamoros, S., Ellis, R. J., et al. (2020). Proficiency testing of metagenomics-based detection of food-borne pathogens using a complex artificial sequencing dataset. Front. Microbiol. 11:575377. doi: 10.3389/fmicb.2020.575377
Hrdlickova, R., Toloue, M., and Tian, B. (2017). RNA-seq methods for transcriptome analysis. WIREs RNA 8:e1364. doi: 10.1002/wrna.1364
Huang, Y., Sheth, R. U., Kaufman, A., and Wang, H. H. (2020). Scalable and cost-effective ribonuclease-based rRNA depletion for transcriptomics. Nucleic Acids Res. 48:e20. doi: 10.1093/nar/gkz1169
Imamura, S., Haruna, M., Goshima, T., Kanezashi, H., Okada, T., and Akimoto, K. (2016a). Application of next-generation sequencing to evaluate the profile of noroviruses in pre- and post-depurated oysters. Foodborne Pathog. Dis. 13, 559–565. doi: 10.1089/fpd.2016.2150
Imamura, S., Haruna, M., Goshima, T., Kanezashi, H., Okada, T., and Akimoto, K. (2016b). Application of next-generation sequencing to investigation of norovirus diversity in shellfish collected from two coastal sites in Japan from 2013 to 2014. Jpn. J. Vet. Res. 64, 113–122. doi: 10.14943/jjvr.64.2.113
Imamura, S., Kanezashi, H., Goshima, T., Haruna, M., Okada, T., Inagaki, N., et al. (2017). Next-generation sequencing analysis of the diversity of human noroviruses in Japanese oysters. Foodborne Pathog. Dis. 14, 465–471. doi: 10.1089/fpd.2017.2289
Jaworski, E., and Routh, A. (2017). Parallel ClickSeq and nanopore sequencing elucidates the rapid evolution of defective-interfering RNAs in flock house virus. PLoS Pathog. 13:e1006365. doi: 10.1371/journal.ppat.1006365
Jia, H., Guo, Y., Zhao, W., and Wang, K. (2014). Long-range PCR in next-generation sequencing: comparison of six enzymes and evaluation on the MiSeq sequencer. Sci. Rep. 4:5737. doi: 10.1038/srep05737
Johnson, J. A., Parra, G. I., Levenson, E. A., and Green, K. Y. (2017). A large outbreak of acute gastroenteritis in Shippensburg, Pennsylvania, 1972 revisited: evidence for common source exposure to a recombinant GII.Pg/GII.3 norovirus. Epidemiol. Infect. 145, 1591–1596. doi: 10.1017/S0950268817000498
Kim, I. V., Ross, E. J., Dietrich, S., Doring, K., Alvarado, A. S., and Kuhn, C.-D. (2019). Efficient depletion of ribosomal RNA for RNA sequencing in planarians. BMC Genomics 20:909. doi: 10.1186/s12864-019-6292-y
Knierim, E., Lucke, B., Schwarz, J. M., Schuelke, M., and Seelow, D. (2011). Systematic comparison of three methods for fragmentation of long-range PCR products for next generation sequencing. PLoS ONE 6:e28240. doi: 10.1371/journal.pone.0028240
Kugelman, J. R., Wiley, M. R., Nagle, E. R., Reyes, D., Pfeffer, B. P., Kuhn, J. H., et al. (2017). Error baseline rates of five sample preparation methods used to characterize RNA virus populations. PLoS ONE 12:e0171333. doi: 10.1371/journal.pone.0171333
Kundu, S., Lockwood, J., Depledge, D. P., Chaudhry, Y., Aston, A., Rao, K., et al. (2013). Next-generation whole genome sequencing identifies the direction of norovirus transmission in linked patients. Clin. Infect. Dis. 57, 407–414. doi: 10.1093/cid/cit287
Lahens, N. F., Kavakli, I. H., Zhang, R., Hayer, K., Black, M. B., Dueck, H., et al. (2014). IVT-seq reveals extreme bias in RNA sequencing. Genome Biol. 15:R86. doi: 10.1186/gb-2014-15-6-r86
Le, H. Q., Suffredini, E., Pham, D. T., To, A. K., and Medici, D. D. (2018). Development of a method for direct extraction of viral RNA from bivalve molluscs. Lett. Appl. Microbiol. 67, 426–434. doi: 10.1111/lam.13065
Lekanne Deprez, R. H., Fijnvandraat, A. C., Ruijter, J. M., and Moorman, A. F. M. (2002). Sensitivity and accuracy of quantitative real-time polymerase chain reaction using SYBR green I depends on cDNA synthesis conditions. Anal. Biochem. 307, 63–69. doi: 10.1016/S0003-2697(02)00021-0
Leon, L. J., Doyle, R., Diez-Benavente, E., Clark, T. G., Klein, N., Stanier, P., et al. (2018). Enrichment of clinically relevant organisms in spontaneous preterm-delivered placentas and reagent contamination across all clinical groups in a large pregnancy cohort in the United Kingdom. Appl. Environ. Microbiol. 84:e00483–18. doi: 10.1128/AEM.00483-18
Levesque-Sergerie, J.-P., Duquette, M., Thibault, C., Delbecchi, L., and Bissonnette, N. (2007). Detection limits of several commercial reverse transcriptase enzymes: impact on the low- and high-abundance transcript levels assessed by quantitative RT-PCR. BMC Mol. Biol. 8:93. doi: 10.1186/1471-2199-8-93
Li, K., Shrivastava, S., Brownley, A., Katzel, D., Bera, J., Nguyen, A. T., et al. (2012). Automated degenerate PCR primer design for high-throughput sequencing improves efficiency of viral sequencing. Virol. J. 9:261. doi: 10.1186/1743-422X-9-261
Li, L., Deng, X., Mee, E. T., Collot-Teixeira, S., Anderson, R., Schepelmann, S., et al. (2015). Comparing viral metagenomics methods using a highly multiplexed human viral pathogens reagent. J. Virol. Methods 213, 139–146. doi: 10.1016/j.jviromet.2014.12.002
Lindén, J., Ranta, J., and Pohjanvirta, R. (2012). Bayesian modeling of reproducibility and robustness of RNA reverse transcription and quantitative real-time polymerase chain reaction. Anal. Biochem. 428, 81–91. doi: 10.1016/j.ab.2012.06.010
Lu, J., du Plessis, L., Liu, Z., Hill, V., Kang, M., Lin, H., et al. (2020). Genomic epidemiology of SARS-CoV-2 in Guangdong Province, China. Cell. 181:997–1003. doi: 10.1016/j.cell.2020.04.023
Lun, J. H., Hewitt, J., Sitabkhan, A., Eden, J.-S., Enosi Tuipulotu, D., Netzler, N. E., et al. (2018). Emerging recombinant noroviruses identified by clinical and waste water screening. Emerg. Microb. Infect. 7, 1–14. doi: 10.1038/s41426-018-0047-8
Mancini, P., Bonanno Ferraro, G., Suffredini, E., Veneri, C., Iaconelli, M., Vicenza, T., et al. (2020). Molecular detection of human salivirus in Italy through monitoring of urban sewages. Food Environ. Virol. 12, 68–74. doi: 10.1007/s12560-019-09409-w
Manso, C. F., Torres, E., Bou, G., and Romalde, J. L. (2013). Role of norovirus in acute gastroenteritis in the Northwest of Spain during 2010–2011. J. Med. Virol. 85, 2009–2015. doi: 10.1002/jmv.23680
Marston, D. A., McElhinney, L. M., Ellis, R. J., Horton, D. L., Wise, E. L., Leech, S. L., et al. (2013). Next generation sequencing of viral RNA genomes. BMC Genomics 14:444. doi: 10.1186/1471-2164-14-444
Martel, N., Gomes, S. A., Chemin, I., Trepo, C., and Kay, A. (2013). Improved rolling circle amplification (RCA) of hepatitis B virus (HBV) relaxed-circular serum DNA (RC-DNA). J. Virol. Methods 193, 653–659. doi: 10.1016/j.jviromet.2013.07.045
Mate, S. E., Kugelman, J. R., Nyenswah, T. G., Ladner, J. T., Wiley, M. R., Cordier-Lassalle, T., et al. (2015). Molecular evidence of sexual transmission of Ebola virus. N. Engl. J. Med. 373, 2448–2454. doi: 10.1056/NEJMoa1509773
Matranga, C. B., Andersen, K. G., Winnicki, S., Busby, M., Gladden, A. D., Tewhey, R., et al. (2014). Enhanced methods for unbiased deep sequencing of Lassa and Ebola RNA viruses from clinical and biological samples. Genome Biol. 15:519. doi: 10.1186/s13059-014-0519-7
Metsky, H. C., Siddle, K. J., Gladden-Young, A., Qu, J., Yang, D. K., Brehio, P., et al. (2019). Capturing sequence diversity in metagenomes with comprehensive and scalable probe design. Nat. Biotechnol. 37, 160–168. doi: 10.1038/s41587-018-0006-x
Miller, R. R., Uyaguari-Diaz, M., McCabe, M. N., Montoya, V., Gardy, J. L., Parker, S., et al. (2016). Metagenomic investigation of plasma in individuals with ME/CFS highlights the importance of technical controls to elucidate contamination and batch effects. PLoS ONE 11:e0165691. doi: 10.1371/journal.pone.0165691
Miranda, J. A., and Steward, G. F. (2017). Variables influencing the efficiency and interpretation of reverse transcription quantitative PCR (RT-qPCR): an empirical study using Bacteriophage MS2. J. Virol. Methods 241, 1–10. doi: 10.1016/j.jviromet.2016.12.002
Miyazato, P., Katsuya, H., Fukuda, A., Uchiyama, Y., Matsuo, M., Tokunaga, M., et al. (2016). Application of targeted enrichment to next-generation sequencing of retroviruses integrated into the host human genome. Sci. Rep. 6, 1–10. doi: 10.1038/srep28324
Morlan, J. D., Qu, K., and Sinicropi, D. V. (2012). Selective depletion of rRNA enables whole transcriptome profiling of archival fixed tissue. PLoS ONE 7:e42882. doi: 10.1371/journal.pone.0042882
Moser, L. A., Ramirez-Carvajal, L., Puri, V., Pauszek, S. J., Matthews, K., Dilley, K. A., et al. (2016). A universal next-generation sequencing protocol to generate noninfectious barcoded cDNA libraries from high-containment RNA viruses. mSystems 1:e00039-15. doi: 10.1128/mSystems.00039-15
Munro, S. A., Lund, S. P., Pine, P. S., Binder, H., Clevert, D.-A., Conesa, A., et al. (2014). Assessing technical performance in differential gene expression experiments with external spike-in RNA control ratio mixtures. Nat. Commun. 5:5125. doi: 10.1038/ncomms6125
Myrmel, M., Oma, V., Khatri, M., Hansen, H. H., Stokstad, M., Berg, M., et al. (2017). Single primer isothermal amplification (SPIA) combined with next generation sequencing provides complete bovine coronavirus genome coverage and higher sequence depth compared to sequence-independent single primer amplification (SISPA). PLoS ONE 12:e0187780. doi: 10.1371/journal.pone.0187780
Nasheri, N., Petronella, N., Ronholm, J., Bidawid, S., and Corneau, N. (2017). Characterization of the genomic diversity of norovirus in linked patients using a metagenomic deep sequencing approach. Front. Microbiol. 8:73. doi: 10.3389/fmicb.2017.00073
Nasir, J. A., Kozak, R. A., Aftanas, P., Raphenya, A. R., Smith, K. M., Maguire, F., et al. (2020). A comparison of whole genome sequencing of SARS-CoV-2 using amplicon-based sequencing, random hexamers, and bait capture. Viruses 12:895. doi: 10.3390/v12080895
Nayfach, S., Camargo, A. P., Schulz, F., Eloe-Fadrosh, E., Roux, S., and Kyrpides, N. C. (2020). CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. doi: 10.1038/s41587-020-00774-7
Nichols, R. V., Vollmers, C., Newsom, L. A., Wang, Y., Heintzman, P. D., Leighton, M., et al. (2018). Minimizing polymerase biases in metabarcoding. Mol. Ecol. Resour. 18, 927–939. doi: 10.1111/1755-0998.12895
O'Flaherty, B. M., Li, Y., Tao, Y., Paden, C. R., Queen, K., Zhang, J., et al. (2018). Comprehensive viral enrichment enables sensitive respiratory virus genomic identification and analysis by next generation sequencing. Genome Res. 28, 869–877. doi: 10.1101/gr.226316.117
Okano, H., Baba, M., Hidese, R., Iida, K., Li, T., Kojima, K., et al. (2018). Accurate fidelity analysis of the reverse transcriptase by a modified next-generation sequencing. Enzyme Microb. Technol. 115, 81–85. doi: 10.1016/j.enzmictec.2018.05.001
Palmer, J. M., Jusino, M. A., Banik, M. T., and Lindner, D. L. (2018). Non-biological synthetic spike-in controls and the AMPtk software pipeline improve mycobiome data. PeerJ 6:e4925. doi: 10.7717/peerj.4925
Palomares, M.-A., Dalmasso, C., Bonnet, E., Derbois, C., Brohard-Julien, S., Ambroise, C., et al. (2019). Systematic analysis of TruSeq, SMARTer and SMARTer Ultra-Low RNA-seq kits for standard, low and ultra-low quantity samples. Sci. Rep. 9:7550 doi: 10.1038/s41598-019-43983-0
Parekh, S., Ziegenhain, C., Vieth, B., Enard, W., and Hellmann, I. (2016). The impact of amplification on differential expression analyses by RNA-seq. Sci. Rep. 6, 1–11. doi: 10.1038/srep25533
Parra, G. I., Squires, R. B., Karangwa, C. K., Johnson, J. A., Lepore, C. J., Sosnovtsev, S. V., et al. (2017). Static and evolving norovirus genotypes: implications for epidemiology and immunity. PLoS Pathog. 13:e1006136. doi: 10.1371/journal.ppat.1006136
Parras-Moltó, M., Rodríguez-Galet, A., Suárez-Rodríguez, P., and López-Bueno, A. (2018). Evaluation of bias induced by viral enrichment and random amplification protocols in metagenomic surveys of saliva DNA viruses. Microbiome 6:119. doi: 10.1186/s40168-018-0507-3
Pauly, M. D., Kamili, S., and Hayden, T. M. (2019). Impact of nucleic acid extraction platforms on hepatitis virus genome detection. J. Virol. Methods 113715. doi: 10.1016/j.jviromet.2019.113715
Peck, K. M., and Lauring, A. S. (2018). Complexities of viral mutation rates. J. Virol. 92:e01031–17. doi: 10.1128/JVI.01031-17
Pecman, A., Kutnjak, D., Gutierrez-Aguirre, I., Adams, I., Fox, A., Boonham, N., et al. (2017). Next generation sequencing for detection and discovery of plant viruses and viroids: comparison of two approaches. Front. Microbiol. 8:1998. doi: 10.3389/fmicb.2017.01998
Petrova, O. E., Garcia-Alcalde, F., Zampaloni, C., and Sauer, K. (2017). Comparative evaluation of rRNA depletion procedures for the improved analysis of bacterial biofilm and mixed pathogen culture transcriptomes. Sci. Rep. 7:41114. doi: 10.1038/srep41114
Poptsova, M. S., Il'icheva, I. A., Nechipurenko, D. Y., Panchenko, L. A., Khodikov, M. V., Oparina, N. Y., et al. (2014). Non-random DNA fragmentation in next-generation sequencing. Sci. Rep. 4, 1–6. doi: 10.1038/srep04532
Potapov, V., and Ong, J. L. (2017). Examining sources of error in PCR by single-molecule sequencing. PLOS ONE 12:e0169774. doi: 10.1371/journal.pone.0169774
Pyrc, K., Jebbink, M. F., Berkhout, B., and van der Hoek, L. (2007). Detection of new viruses by VIDISCA. SARS Other Coronaviruses 454, 73–89. doi: 10.1007/978-1-59745-181-9_7
Qing, T., Yu, Y., Du, T., and Shi, L. (2013). mRNA enrichment protocols determine the quantification characteristics of external RNA spike-in controls in RNA-Seq studies. Sci. China Life Sci. 56, 134–142. doi: 10.1007/s11427-013-4437-9
Qiu, X., Zhang, H., Yu, H., Jiang, T., and Luo, Y. (2015). Duplex-specific nuclease-mediated bioanalysis. Trends Biotechnol. 33, 180–188. doi: 10.1016/j.tibtech.2014.12.008
Quail, M. A., Otto, T. D., Gu, Y., Harris, S. R., Skelly, T. F., McQuillan, J. A., et al. (2011). Optimal enzymes for amplifying sequencing libraries. Nat. Methods 9:10. doi: 10.1038/nmeth.1814
Quick, J., Grubaugh, N. D., Pullan, S. T., Claro, I. M., Smith, A. D., Gangavarapu, K., et al. (2017). Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 12, 1261–1276. doi: 10.1038/nprot.2017.066
Ren, J., Ahlgren, N. A., Lu, Y. Y., Fuhrman, J. A., and Sun, F. (2017). VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 5:69. doi: 10.1186/s40168-017-0283-5
Reyes, G. R., and Kim, J. P. (1991). Sequence-independent, single-primer amplification (SISPA) of complex DNA populations. Mol. Cell. Probes 5, 473–481. doi: 10.1016/S0890-8508(05)80020-9
Risso, D., Ngai, J., Speed, T. P., and Dudoit, S. (2014). Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 32, 896–902. doi: 10.1038/nbt.2931
Rosseel, T., Ozhelvaci, O., Freimanis, G., and Van Borm, S. (2015). Evaluation of convenient pretreatment protocols for RNA virus metagenomics in serum and tissue samples. J. Virol. Methods 222, 72–80. doi: 10.1016/j.jviromet.2015.05.010
Routh, A., Head, S. R., Ordoukhanian, P., and Johnson, J. E. (2015). ClickSeq: fragmentation-free next-generation sequencing via click ligation of adaptors to stochastically terminated 3′-azido cDNAs. J. Mol. Biol. 427, 2610–2616. doi: 10.1016/j.jmb.2015.06.011
Roux, S., Enault, F., Hurwitz, B. L., and Sullivan, M. B. (2015). VirSorter: mining viral signal from microbial genomic data. PeerJ 3:e985. doi: 10.7717/peerj.985
Salter, S. J., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffatt, M. F., et al. (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12:87. doi: 10.1186/s12915-014-0087-z
Samorodnitsky, E., Jewell, B. M., Hagopian, R., Miya, J., Wing, M. R., Lyon, E., et al. (2015). Evaluation of hybridization capture versus amplicon-based methods for whole-exome sequencing. Hum. Mutat. 36, 903–914. doi: 10.1002/humu.22825
Schuh, A. J., Amman, B. R., Patel, K., Sealy, T. K., Swanepoel, R., and Towner, J. S. (2020). Human-pathogenic Kasokero virus in field-collected ticks. Emerg. Infect. Dis. 26, 2944–2950. doi: 10.3201/eid2612.202411
Schwaber, J., Andersen, S., and Nielsen, L. (2019). Shedding light: The importance of reverse transcription efficiency standards in data interpretation. Biomol. Detect. Quantif. 17:100077. doi: 10.1016/j.bdq.2018.12.002
Schwochow, D., Serieys, L. E., Wayne, R. K., and Thalmann, O. (2012). Efficient recovery of whole blood RNA - a comparison of commercial RNA extraction protocols for high-throughput applications in wildlife species. BMC Biotechnol. 12:33. doi: 10.1186/1472-6750-12-33
Shanker, S., Paulson, A., Edenberg, H. J., Peak, A., Perera, A., Alekseyev, Y. O., et al. (2015). Evaluation of commercially available RNA amplification kits for RNA sequencing using very low input amounts of total RNA. J. Biomol. Tech. 26, 4–18. doi: 10.7171/jbt.15-2601-001
Singanallur, N. B., Anderson, D. E., Sessions, O. M., Kamaraj, U. S., Bowden, T. R., Horsington, J., et al. (2019). Probe capture enrichment next-generation sequencing of complete foot-and-mouth disease virus genomes in clinical samples. J. Virol. Methods 272:113703. doi: 10.1016/j.jviromet.2019.113703
Smith, E. C., and Denison, M. R. (2013). Coronaviruses as DNA wannabes: a new model for the regulation of RNA virus replication fidelity. PLoS Pathog. 9:e1003760. doi: 10.1371/journal.ppat.1003760
Stangegaard, M., Hogh Dufva, I., and Dufva, M. (2006). Reverse transcription using random pentadecamer primers increases yield and quality of resulting cDNA. Biotechniques 40, 649–657. doi: 10.2144/000112153
Stasik, S., Schuster, C., Ortlepp, C., Platzbecker, U., Bornhauser, M., Schetelig, J., et al. (2018). An optimized targeted next-generation sequencing approach for sensitive detection of single nucleotide variants. Biomol. Detect. Quantif. 15, 6–12. doi: 10.1016/j.bdq.2017.12.001
Strong, M. J., Xu, G., Morici, L., Splinter Bon-Durant, S., Baddoo, M., Lin, Z., et al. (2014). Microbial contamination in next generation sequencing: implications for sequence-based analysis of clinical samples. PLoS Pathog. 10:e1004437. doi: 10.1371/journal.ppat.1004437
Strubbia, S., Phan, M. V. T., Schaeffer, J., Koopmans, M., Cotten, M., and Le Guyader, F. S. (2019a). Characterization of norovirus and other human enteric viruses in sewage and stool samples through next-generation sequencing. Food Environ. Virol. 11, 400–409. doi: 10.1007/s12560-019-09402-3
Strubbia, S., Schaeffer, J., Besnard, A., Wacrenier, C., Le Mennec, C., Garry, P., et al. (2020). Metagenomic to evaluate norovirus genomic diversity in oysters: impact on hexamer selection and targeted capture-based enrichment. Int. J. Food Microbiol. 323:108588. doi: 10.1016/j.ijfoodmicro.2020.108588
Strubbia, S., Schaeffer, J., Oude Munnink, B. B., Besnard, A., Phan, M. V., Nieuwenhuijse, D., et al. (2019b). Metavirome sequencing to evaluate norovirus diversity in sewage and related bioaccumulated oysters. Front. Microbiol. 10:2394. doi: 10.3389/fmicb.2019.02394
Suffredini, E., Iaconelli, M., Equestre, M., Valdazo-Gonzalez, B., Ciccaglione, A. R., Marcantonio, C., et al. (2018). Genetic diversity among genogroup II noroviruses and progressive emergence of GII.17 in wastewaters in Italy (2011-2016) revealed by next-generation and sanger sequencing. Food Environ. Virol. 10, 141–150. doi: 10.1007/s12560-017-9328-y
Sukal, A. C., Kidanemariam, D. B., Dale, J. L., Harding, R. M., and James, A. P. (2019). Assessment and optimization of rolling circle amplification protocols for the detection and characterization of badnaviruses. Virology 529, 73–80. doi: 10.1016/j.virol.2019.01.013
Sultan, M., Amstislavskiy, V., Risch, T., Schuette, M., Dokel, S., Ralser, M., et al. (2014). Influence of RNA extraction methods and library selection schemes on RNA-seq data. BMC Genomics 15:675. doi: 10.1186/1471-2164-15-675
Sun, Z., Asmann, Y. W., Nair, A., Zhang, Y., Wang, L., Kalari, K. R., et al. (2013). Impact of library preparation on downstream analysis and interpretation of RNA-Seq data: comparison between Illumina PolyA and NuGEN ovation protocol. PLoS ONE 8:e71745. doi: 10.1371/journal.pone.0071745
Tavares, L., Alves, P. M., Ferreira, R. B., and Santos, C. N. (2011). Comparison of different methods for DNA-free RNA isolation from SK-N-MC neuroblastoma. BMC Res. 4:3. doi: 10.1186/1756-0500-4-3
Thézé, J., Li, T., du Plessis, L., Bouquet, J., Kraemer, M. U. G., Somasekar, S., et al. (2018). Genomic epidemiology reconstructs the introduction and spread of Zika Virus in Central America and Mexico. Cell Host Microbe 23, 855.e7–864.e7. doi: 10.1016/j.chom.2018.04.017
van Beek, J., de Graaf, M., Smits, S., Schapendonk, C. M. E., Verjans, G. M. G. M., Vennema, H., et al. (2017). Whole-genome next-generation sequencing to study within-host evolution of norovirus (NoV) among immunocompromised patients with chronic NoV infection. J. Infect. Dis. 216, 1513–1524. doi: 10.1093/infdis/jix520
Vrancken, B., Trovao, N. S., Baele, G., Van Wijngaerden, E., Vandamme, A.-M., Van Laethem, K., et al. (2016). Quantifying next generation sequencing sample pre-processing bias in HIV-1 complete genome sequencing. Viruses 8, 12. doi: 10.3390/v8010012
Wang, C. H., Nie, K., Zhang, Y., Wang, J., Zhou, S. F., Li, X. N., et al. (2017). An improved barcoded oligonucleotide primers-based next-generation sequencing approach for direct identification of viral pathogens in clinical specimens. Biomed. Environ. Sci. 30, 22–34. doi: 10.3967/bes2017.003
Wang, H., Sikora, P., Rutgersson, C., Lindh, M., Brodin, T., Bjarlenius, B., et al. (2018). Differential removal of human pathogenic viruses from sewage by conventional and ozone treatments. Int. J. Hyg. Environ. Health 221, 479–488. doi: 10.1016/j.ijheh.2018.01.012
Waugh, C., Cromer, D., Grimm, A., Chopra, A., Mallal, S., Davenport, M., et al. (2015). A general method to eliminate laboratory induced recombinants during massive, parallel sequencing of cDNA library. Virol. J. 12:55. doi: 10.1186/s12985-015-0280-x
Werbrouck, H., Botteldoorn, N., Uyttendaele, M., Herman, L., and Van Coillie, E. (2007). Quantification of gene expression of Listeria monocytogenes by real-time reverse transcription PCR: optimization, evaluation and pitfalls. J. Microbiol. Methods 69, 306–314. doi: 10.1016/j.mimet.2007.01.017
Wolf, Y. I., Silas, S., Wang, Y., Wu, S., Bocek, M., Kazlauskas, D., et al. (2020). Doubling of the known set of RNA viruses by metagenomic analysis of an aquatic virome. Nat. Microbiol. 5, 1262–1270. doi: 10.1038/s41564-020-0755-4
Wong, R. K., MacMahon, M., Woodside, J. V., and Simpson, D. A. (2019). A comparison of RNA extraction and sequencing protocols for detection of small RNAs in plasma. BMC Genomics 20:446. doi: 10.1186/s12864-019-5826-7
Wongsurawat, T., Jenjaroenpun, P., Taylor, M. K., Lee, J., Tolardo, A. L., Parvathareddy, J., et al. (2019). Rapid sequencing of multiple RNA viruses in their native form. Front. Microbiol. 10:260. doi: 10.3389/fmicb.2019.00260
Wylie, T. N., Wylie, K. M., Herter, B. N., and Storch, G. A. (2015). Enhanced virome sequencing using targeted sequence capture. Genome Res. 25, 1910–1920. doi: 10.1101/gr.191049.115
Xiao, Y.-L., Kash, J. C., Beres, S. B., Sheng, Z.-M., Musser, J. M., and Taubenberger, J. K. (2013). High-throughput RNA sequencing of a formalin-fixed, paraffin-embedded autopsy lung tissue sample from the 1918 influenza pandemic. J. Pathol. 229, 535–545. doi: 10.1002/path.4145
Xu, D., Wei, G., Lu, P., Luo, J., Chen, X., Skogerbo, G., et al. (2014). Analysis of the p53/CEP-1 regulated non-coding transcriptome in C. elegans by an NSR-seq strategy. Protein Cell 5, 770–782. doi: 10.1007/s13238-014-0071-y
Yasukawa, K., Iida, K., Okano, H., Hidese, R., Baba, M., Yanagihara, I., et al. (2017). Next-generation sequencing-based analysis of reverse transcriptase fidelity. Biochem. Biophys. Res. Commun. 492, 147–153. doi: 10.1016/j.bbrc.2017.07.169
Yi, H., Cho, Y.-J., Won, S., Lee, J.-E., Jin Yu, H., Kim, S., et al. (2011). Duplex-specific nuclease efficiently removes rRNA for prokaryotic RNA-seq. Nucleic Acids Res. 39:e140. doi: 10.1093/nar/gkr617
Zhao, G., Krishnamurthy, S., Cai, Z., Popov, V. L., da Rosa, A. P. T., Guzman, H., et al. (2013). Identification of novel viruses using virushunter-an automated data analysis pipeline. PLoS ONE 8:e78470. doi: 10.1371/journal.pone.0078470
Zhao, L., Niu, Y., Lu, T., Yin, H., Zhang, Y., Xu, L., et al. (2018a). Metagenomic analysis of the jinding duck fecal virome. Curr. Microbiol. 75, 658–665. doi: 10.1007/s00284-018-1430-3
Zhao, S., Zhang, Y., Gamini, R., Zhang, B., and von Schack, D. (2018b). Evaluation of two main RNA-seq approaches for gene quantification in clinical RNA sequencing: polyA+ selection versus rRNA depletion. Sci. Rep. 8:4781. doi: 10.1038/s41598-018-23226-4
Zhou, H., Chen, X., Hu, T., Li, J., Song, H., Liu, Y., et al. (2020). A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Curr. Biol. 30, 2196.e3–2203.e3. doi: 10.1016/j.cub.2020.05.023
Keywords: high throughput sequencing, RNA viruses, environmental virology, amplicon sequencing, capture based probe hybridization, viral enrichment, RNA depletion
Citation: Fitzpatrick AH, Rupnik A, O'Shea H, Crispie F, Keaveney S and Cotter P (2021) High Throughput Sequencing for the Detection and Characterization of RNA Viruses. Front. Microbiol. 12:621719. doi: 10.3389/fmicb.2021.621719
Received: 26 October 2020; Accepted: 20 January 2021;
Published: 22 February 2021.
Edited by:
Enzo Tramontano, University of Cagliari, ItalyReviewed by:
Alexander Culley, Laval University, CanadaCopyright © 2021 Fitzpatrick, Rupnik, O'Shea, Crispie, Keaveney and Cotter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sinéad Keaveney, c2luZWFkLmtlYXZlbmV5QG1hcmluZS5pZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.