 Xiaoteng Fu
Xiaoteng Fu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 07 May 2021
Sec. Evolutionary and Genomic Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.571212
Background: Members of the Bacillus pumilus group (abbreviated as the Bp group) are quite diverse and ubiquitous in marine environments, but little is known about correlation with their terrestrial counterparts. In this study, 16 marine strains that we had isolated before were sequenced and comparative genome analyses were performed with a total of 52 Bp group strains. The analyses included 20 marine isolates (which included the 16 new strains) and 32 terrestrial isolates, and their evolutionary relationships, differentiation, and environmental adaptation.
Results: Phylogenomic analysis revealed that the marine Bp group strains were grouped into three species: B. pumilus, B. altitudinis and B. safensis. All the three share a common ancestor. However, members of B. altitudinis were observed to cluster independently, separating from the other two, thus diverging from the others. Consistent with the universal nature of genes involved in the functioning of the translational machinery, the genes related to translation were enriched in the core genome. Functional genomic analyses revealed that the marine-derived and the terrestrial strains showed differences in certain hypothetical proteins, transcriptional regulators, K+ transporter (TrK) and ABC transporters. However, species differences showed the precedence of environmental adaptation discrepancies. In each species, land specific genes were found with possible functions that likely facilitate survival in diverse terrestrial niches, while marine bacteria were enriched with genes of unknown functions and those related to transcription, phage defense, DNA recombination and repair.
Conclusion: Our results indicated that the Bp isolates show distinct genomic features even as they share a common core. The marine and land isolates did not evolve independently; the transition between marine and non-marine habitats might have occurred multiple times. The lineage exhibited a priority effect over the niche in driving their dispersal. Certain intra-species niche specific genes could be related to a strain’s adaptation to its respective marine or terrestrial environment(s). In summary, this report describes the systematic evolution of 52 Bp group strains and will facilitate future studies toward understanding their ecological role and adaptation to marine and/or terrestrial environments.
Bacillus are Gram-positive, aerobic or facultative anaerobic, rod-shaped bacteria that can produce highly resistant dormant endospores in response to various environmental stresses (Nicholson et al., 2000; Ravel and Fraser, 2005; Gioia et al., 2007; Earl et al., 2008; Setlow, 2014; Checinska et al., 2015). Benefiting from versatile metabolic capabilities and the ability for spore dispersal, Bacillus is ubiquitous in various natural environments. The genus comprises more than 500 species recognized to date1. Based on 1,172 core proteins, they have been phylogenetically divided into eight clades, including the Cereus, Subtilis, Simplex, Firmus, Jeotgali, Niacini, Fastidiosus, Alcalophilus clades (Patel and Gupta, 2019). Bacillus pumilus and their relatives belong to the Subtilis clade (Priest et al., 1987; Bhandari et al., 2013; Fan et al., 2017; Patel and Gupta, 2019).
The Bacillus pumilus group, abbreviated as the Bp group in this report, is a large group of Bacillus composed of B. pumilus, B. safensis, B. altitudinis, B. xiamenensis, B. zhangzhouensis, and B. australimaris (Liu et al., 2016). The members of this group have attracted wide attention for their applications in agriculture, industry and medicine (Berrada et al., 2012; Khianngam et al., 2014; Kumar et al., 2014; Lateef et al., 2015). At present, most of these industrial Bp group bacteria have been isolated from terrestrial ecosystems. However, the less explored marine Bacillus are even more diverse (Liu et al., 2013, 2017). To date, over one thousand marine Bacillus strains have been deposited in our collections2, approximately one-fifth of which belong to the Bp clade. They are frequently isolated from marine environments ranging from coastal and pelagic water columns to deep sea sediments, and most of these isolates have been preliminarily identified as either one of the three species of B. pumilus, B. safensis, and B. altitudinis based on 16S rRNA gene analysis (unpublished data). Among our collections, approximately 66% of Bp bacteria were isolated from marine sediments, 26% from marine water and 8% from marine animals. Due to the survivability of spores of this group under harsh conditions, we are not certain whether they are indigenous to marine habitats. In a study involving the cultivation of marine Bacillus, B. pumilus was found to be the predominant species in the coastal environment of Cochin, India (Parvathi et al., 2009). However, little is known about the geographic distribution of this group in global oceans and its speciation in various marine natural habitats (Ettoumi et al., 2009; Connor et al., 2010).
16S rRNA gene sequencing is a powerful tool for elucidating the phylogenetic diversity of bacterial families (Fox et al., 1980). However, it may not be applicable for the determination of the relationship of some species (Fox et al., 1992) within the groups of certain genera, such as Bacillus. In studies by other groups, methods such as gyrB gene sequence phylogeny, and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOFMS) protein profiling have been applied to differentiate Bp group species (Dickinson et al., 2004; La Duc et al., 2004; Satomi et al., 2006). Previously, we found that the strains of the Bp group were closely related to each other, sharing over 99.5% 16S rRNA sequence similarity (Liu et al., 2013). To understand the phylogeny and biogeography of bacteria within this group, we performed a multilocus sequence analysis (MLSA) of selected marine Bp isolates. We used seven concatenated housekeeping genes with the order of gyrB-rpoB-pycA-pyrE-mutL-aroE-trpB to construct a phylogenetic tree. The choice of the genes is based on earlier work (Liu et al., 2013). The results showed that the bacteria of this group could be divided into six clades, and the three large clades were represented by B. altitudinis, B. safensis, and B. pumilus, respectively. Furthermore, the gyrB-based phylogenetic analysis of 73 terrestrial and marine isolates clearly showed that marine bacteria tended to cluster together in the three large clades (Liu et al., 2013). Although the MLSA provided clues about the phylogenetic relationship of the Bp strains, it is inefficient for resolving the taxonomic status of the strains of this group, as the housekeeping genes that are used account for only 0.1∼0.2% of the genome (Hall et al., 2010). Moreover, MSLA data is not sufficient to understand bacterial adaptation to different natural habitats. Genome-based analysis can provide essential insights into species differentiation and environmental adaptation, including correlations with significant events in the evolutionary history of microbial species (Yerrapragada et al., 2009; Patané et al., 2018).
In recent years, pan-genomics has been widely applied for the investigation of bacterial species diversity, evolution, adaptability, and population structure (Lawrence and Hendrickson, 2005; Kettler et al., 2007; Muzzi et al., 2007; Tettelin et al., 2008; Vernikos et al., 2015; Tiwary, 2020). The pan-genome includes the “core genome” of genes common to all strains of a species and the “dispensable or accessory genome,” which consists of genes present in at least one but not all strains of a species and strain-unique genes (Medini et al., 2005). The essence of a species in terms of its fundamental biological processes and traits derived from a common ancestor is linked to the core genome. However, genetic traits linked to variations in virulence, adaptation and antibiotic resistance are more often governed by the dispensable or accessory genome (Tettelin et al., 2008; Maistrenko et al., 2020; Naz et al., 2020). Accordingly, it is of interest to estimate the sizes of the pan-genome, core genome, and new genes of a given species as novel genomes are added and to further identify the relative contributions of dispensable genomes and their relationships with specific adaptation to environmental niches (Zhang et al., 2018). Pan-genome analysis has been widely used for studying species evolution and functional gene changes (Wu et al., 2020), including those of several Bacillus species, such as B. amyloliquefaciens, B. anthracis, B. cereus, B. subtilis, and B. thuringiensis (Fang et al., 2011; Bazinet, 2017; Kim et al., 2017; Chun et al., 2019), most of which are terrestrial strains. In a previous report, we gained new insights into the phylogeny and differentiation of the B. cereus group via whole-genome analysis (Liu et al., 2015).
This report aimed at understanding the levels of differences between marine Bp bacteria and their terrestrial counterparts. In order to understand the taxonomy, diversity and environmental adaptation of marine Bp bacteria, we selected and sequenced 16 representative strains from various marine environments, including deep sea, coastal and polar areas. We performed phylogenetic, pan-genomic analyses with 36 other genomes downloaded from the NCBI, which were isolated mainly from soil, plants, animals, food, etc. The results based on these 52 Bp genomes will help elucidate the taxonomy of these strains and gain insights into their evolution and environmental adaptation.
Based on our previous MLSA results (Liu et al., 2013) on the marine Bp bacterial isolates and their positions on different evolutionary branches, we selected 16 marine Bp bacteria for genome sequencing to avoid the bias caused by strain selection. Genomic DNA was extracted using an SBS extraction kit (SBS Genetech Co., Ltd. Shanghai, China) according to the manufacturer’s instructions. In addition, 36 genomes of Bp-group strains with isolation environment records were downloaded from the NCBI ftp site (Bacteria and Bacteria_DRAFT parts in http://ftp.ncbi.nlm.nih.gov/genomes). These strains were isolated from different habitats, such as soils, plants, marine environments, food, insects, air, children, and feces. Since only one genome of each of B. xiamenensis, B. zhangzhouensis and B. australimaris was available in the database, they were excluded from this analysis. The accession numbers of the 52 included strains are listed in Table 1, and information related to the 20 marine isolated strains are listed in Supplementary Table 1.
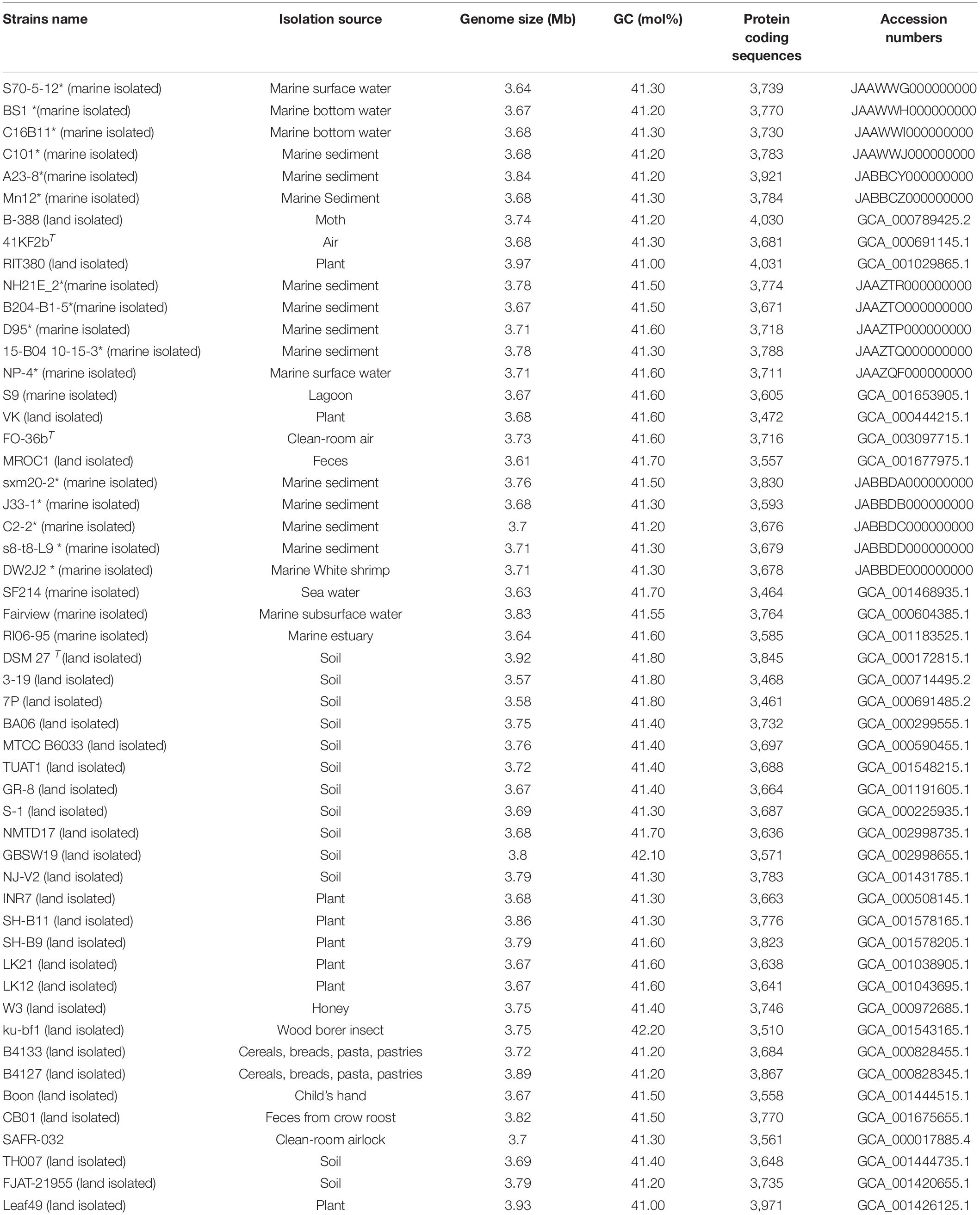
Table 1. Genome information of Bp-group strains (*indicates strains that we isolated and sequenced).
The genome sequencing and assembly of the 16 marine Bp-group strains was conducted on the Illumina HiSeq2000 platform with a sequencing read length of 101 bp by Shanghai Majorbio Bio-pharm Technology Co., Ltd. (Shanghai, China). The high-quality reads were assembled with the SPAdes program (Bankevich et al., 2012). Gene prediction and annotation were done using the Prokka program (Seemann, 2014). The predicted genes were also annotated by performing a BLAST search against the COG database with a cutoff E-value of 1e-5. The assembled genomes and annotated proteins were deposited in the GenBank database. The accession numbers for the 52 strains are listed in Table 1.
All protein sequences of the 52 Bp-group strains and the outgroup of Bacillus subtilis subsp. subtilis 6051-HGW (GenBank accession: CP003329.1) were subjected to ortholog prediction using OrthoMCL (Li et al., 2003), with an E-value of 1e-15, an identity cut-off of 50%, a match cut-off of 50%, and other default settings. Gene clusters that were shared among all strains and contained only single gene copies from each strain were referred to as single-copy core genes. Only single-copy orthologous proteins were chosen for phylogenetic analyses. The single-copy orthologous proteins present in all 53 bacteria were aligned using MAFFT (Katoh and Standley, 2013). The alignment was trimmed using GBlock 0.91b (Castresana, 2000) and used to infer the evolutionary history of the strains with the Randomized Axelerated Maximum Likelihood algorithm (RAxML) and the JTTF model (Jones et al., 1992; Stamatakis, 2014). The reliability of the inferred tree was tested by bootstrapping with 1,000 replicates.
Average nucleotide identity (ANI) values were calculated by using JSpecies software as previously described (Richter and Rosselló-Móra, 2009). A Pearson correlation matrix was generated, and correlation analysis ordered by hierarchical clustering was performed following the procedures of Espariz et al. (2016). In addition, Mash software was chosen for clustering analysis (Ondov et al., 2016) and digital DDH values were determined using the Genome-to-Genome Distance Calculator (GGDC) web server3 using formula 2 as described by Auch et al. (2010) and Meier-Kolthoff et al. (2013).
All the genes from the 52 Bp-group strains were delineated into clusters with putative shared homology via the MultiParanoid (MP) method as implemented in the pan-genome analysis pipeline (PGAP) with a 50% cut-off (Zhao et al., 2011). The pan-genome and core genome profiles were then built. Functional enrichment of orthologous clusters was performed via the PGAP with the parameter “–function” and used for the classification of clusters of orthologous groups (COGs). Twenty genomes of marine isolated strains (including 16 genomes from our collections and four from NCBI) and 32 genomes of strains isolated from terrestrial samples were then utilized to obtain pan-genome profiles via the PGAP. Finally, based on the phylogenetic species assignment results, the gene datasets of each species were used to obtain pan-genome profiles via the PGAP. The significance between COGs was determined with the STAMP (Parks et al., 2014) software using Fisher’s exact test (P < 0.05).
Since dispensable genes in genomes confer selective advantages such as adaptation to different niches and antibiotic resistance (Vernikos et al., 2015), we first extracted the dispensable genomes of the 52 Bp group strains and then used OrthoMCL to cluster orthologous genes shared by all 20 Bp group strains isolated from marine environments. The orthologous genes that have been reported to be related to marine adaptation were considered candidate genes (Penn and Jensen, 2012; Li et al., 2020). COG annotation of candidate genes was then performed through a BLAST search against the downloaded COG database. In addition, we also used R package for core COG hierarchical clustering of 52 Bp group strains. Together with the phylogenetic species assignment results, we compared the different genes in each species between the marine isolated strains and the terrestrial isolated strains. We considered the marine specific genes in each species to be those that were present in at least one-third of the marine strains and were missing in all the terrestrial strains; in each species, terrestrial-specific genes were those that were shared by at least one-third of the terrestrial strains and were missing in all the marine strains.
The analysis of the general features of genomes of the 52 Bp strains showed that the G + C contents ranged from 41.0 to 42.2 mol% and that the genome size ranged from 3.57 to 3.97 Mb (Table 1). Each strain of the Bp group contained 3,705 genes on average, ranging from 3,404 to 4,040 genes per genome, 76–83% of which were assigned biological functions. With respect to their genome sizes, land isolated strains ranged from 3.57 to 3.97 Mb and the marine isolated strains ranged from 3.63 to 3.84 Mb. With respect to their G + C content, the terrestrial strains ranged from 41.0 to 42.2 mol% and the marine strains from 41.2 to 41.7 mol%. Additionally, the terrestrial strains have 3,461–4,031 coding sequences, and the marine strains have 3,464–3,921 coding sequences. Since closely related organisms have similar G (+C contents (Ochman et al., 2005), the similar GC percentages observed for the strains are suggestive of their close relatedness.
Since Bp group bacteria share high identities of the 16S rRNA gene (over 99.5%) (Liu et al., 2013), their phylogenetic relationships need to be resolved using robust higher resolution analyses. In order to do so, we combined core gene phylogenetic analysis, average nucleotide identity correlation and the Mash distance clustering method. B. subtilis subsp. subtilis 6051-HGW was selected as the out-group for the core genes in the phylogenetic analysis. First, 2,039 single copy orthologous proteins shared by 53 genomes, including strain 6051-HGW, were identified and then concatenated to construct an ML (maximum likelihood) phylogenetic tree (Figure 1). As shown in the core gene tree, these Bp strains were clearly clustered into three clades, corresponding to the three species represented by the type strains B. pumilus DSM 27T, B. altitudinis 41KF2bT, and B. safensis FO-36bT. Within each clade, it is our inference that the nomenclature given for the seven strains was incorrect or incomplete. Four of these strains, namely, TUAT1, MTCC B6033, SH-B11, and INR7, have been reported to cluster with other B. altitudinis strains in other studies (Espariz et al., 2016; Tirumalai et al., 2018). Our findings not only corroborate these previous results, but also place the three other strains, namely, TH007, Leaf49 and FJAT-21955, under B. altitudinis. We therefore recommend that all these seven strains be reassigned as B. altitudinis.
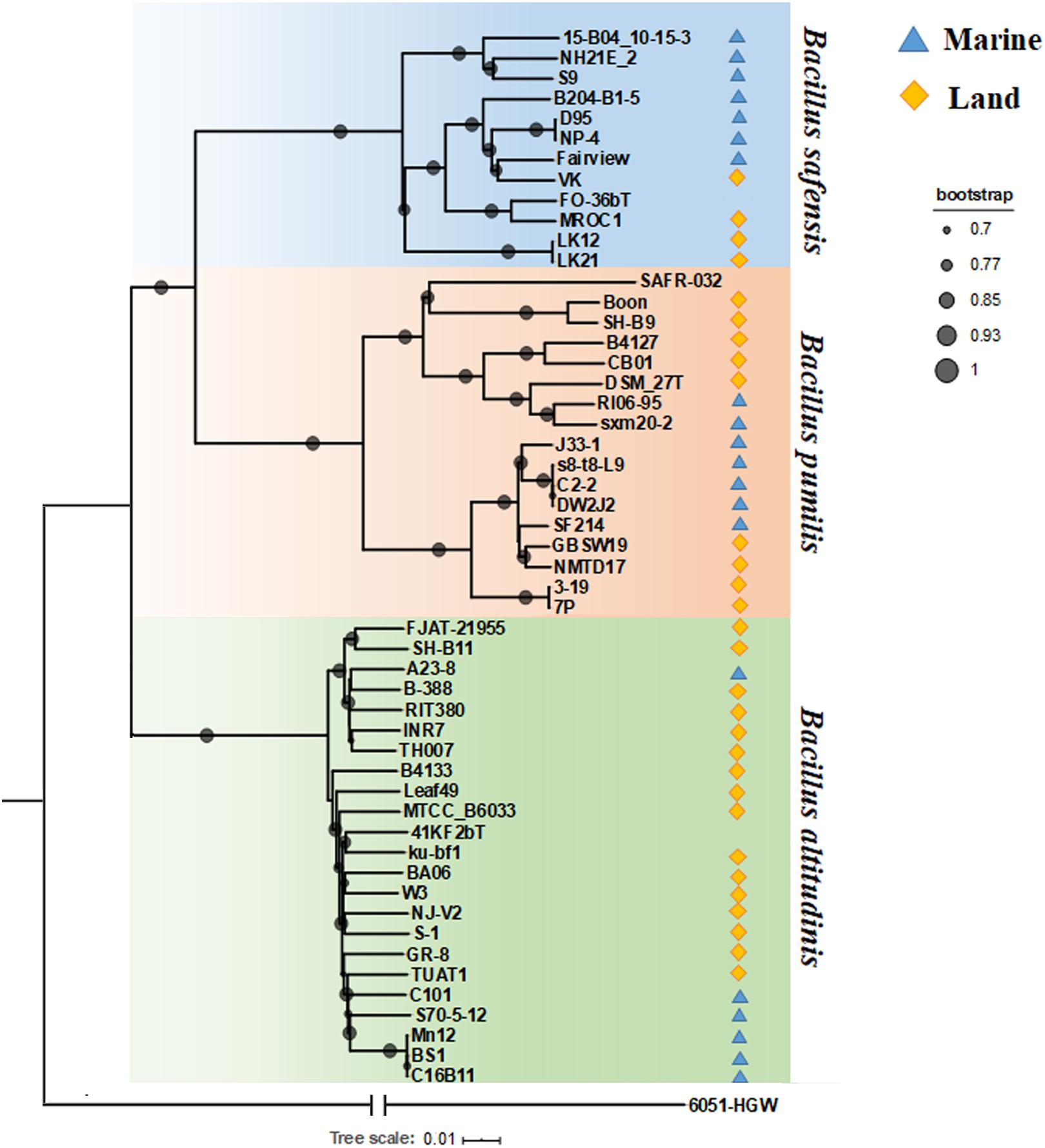
Figure 1. Phylogenetic tree based on total single-copy orthologous. Different colored shading represents the different isolation environments of the strains. Blue represents marine, and yellow represents land environment.
The ML tree based on 2,039 single-copy core genes showed that B. pumilus was more closely related to B. safensis than B. altitudinis (Figure 1), suggesting that B. altitudinis diverged from the other two. Furthermore, B. altitudinis included the majority of the Bp group strains, including six marine strains and 17 terrestrial strains. With the exception of strain A23-8, all of the marine strains of B. altitudinis were grouped together, suggesting that they shared a common ancestor (Figure 1). B. safensis included seven strains isolated from marine environments and five from land environments. Marine strains S9 (Ficarra et al., 2016), NH21E_2 and 15-B04 10-15-3 formed a monophyletic branch; the other branch of B. safensis contained strains from various environments, such as marine environments, plants, feces and air environments. The ancestor of the four marine-derived strains (D95, NP-4, Fairview, and B204-B1-5) might have been transmitted to the marine environment recently, since these strains were closely related to terrestrial strains within this subgroup. The B. pumilus subgroup, contained seven marine strains and ten terrestrial strains, which also clustered into two clades. However, despite occurring in the same subgroup, they differed in their isolation environments. For example, one mixed clade included both soil and marine isolates; four of the marine strains were clustered together, with the exception of SF214; the other clade contained strains of various origins, including samples from marine environments, soil, plants, children, air, food and crow feces, indicating that the bacteria in this clade were widespread in the environment. Overall, the marine bacteria from a given species tended to cluster together, while the terrestrial strains were more diverse and diverged more deeply at the tree nodes (Figure 1). Recently, the phylogeny of 81 bacteria of the genus Bacillus was examined using 196 core genes (Hernández-González et al., 2018). The authors found a clade consisting exclusively of species isolated from aquatic environments and considered some of the aquatic Bacillus species to share a single evolutionary origin. However, we found that the aquatic bacteria tended to group together only within species. We infer that for the Bp group strains, lineage takes priority over the niche effect in driving their divergence.
To confirm the taxonomic identity of these Bp-group strains, pairwise ANI values between the genomes of these strains were calculated, and correlation analysis was conducted to cluster related taxa (Figure 2 and Supplementary Table 2). The 52 strains clustered into three groups, which were identical to the species assignment results based on the core-gene phylogenetic tree. The largest group consisted of B. altitudinis, including 23 strains, sharing more than 98% ANI with each other and 88–90% ANI with the other two species. Accordingly, B. safensis was composed of twelve strains, which shared more than 96% ANI with each other and 90–93% ANI with B. pumilus. B. pumilus consisted of 17 strains, sharing more than 95% ANI with each other, similar to what was observed earlier (Satomi et al., 2006). The ANIs also suggested that B. safensis was more closely related to B. pumilus than B. altitudinis, corroborating previous observations (Tirumalai et al., 2018). In addition, a Mash-based tree was constructed for comparison with the above taxonomic results (Supplementary Figure 1). The Mash approach uses Mash distance to estimate the mutation rate between two sequences and to further cluster species (Ondov et al., 2016). The topology of the tree was similar to that of the core-gene tree in Figure 1 in terms of the species branches. Typically, two genomes belonging to the same species show more than 95% identity using ANI which corresponds to more than 60–70% DDH values (Gupta and Sharma, 2015). When using 60–70% DDH values as the standard cut-off range, the results from each of the following methods, viz., core genes tree, ANI, DDH and Mash distance tree, matched the other very well (Supplementary Table 3). If the cut-off DDH values were set at 70% for species assignment, the DDH results were similar to both the ANI results and the core genes phylogenetic tree, and showed that the B. pumilus strains formed two subclusters. These two subclusters strains shared 60–70% DDH values.
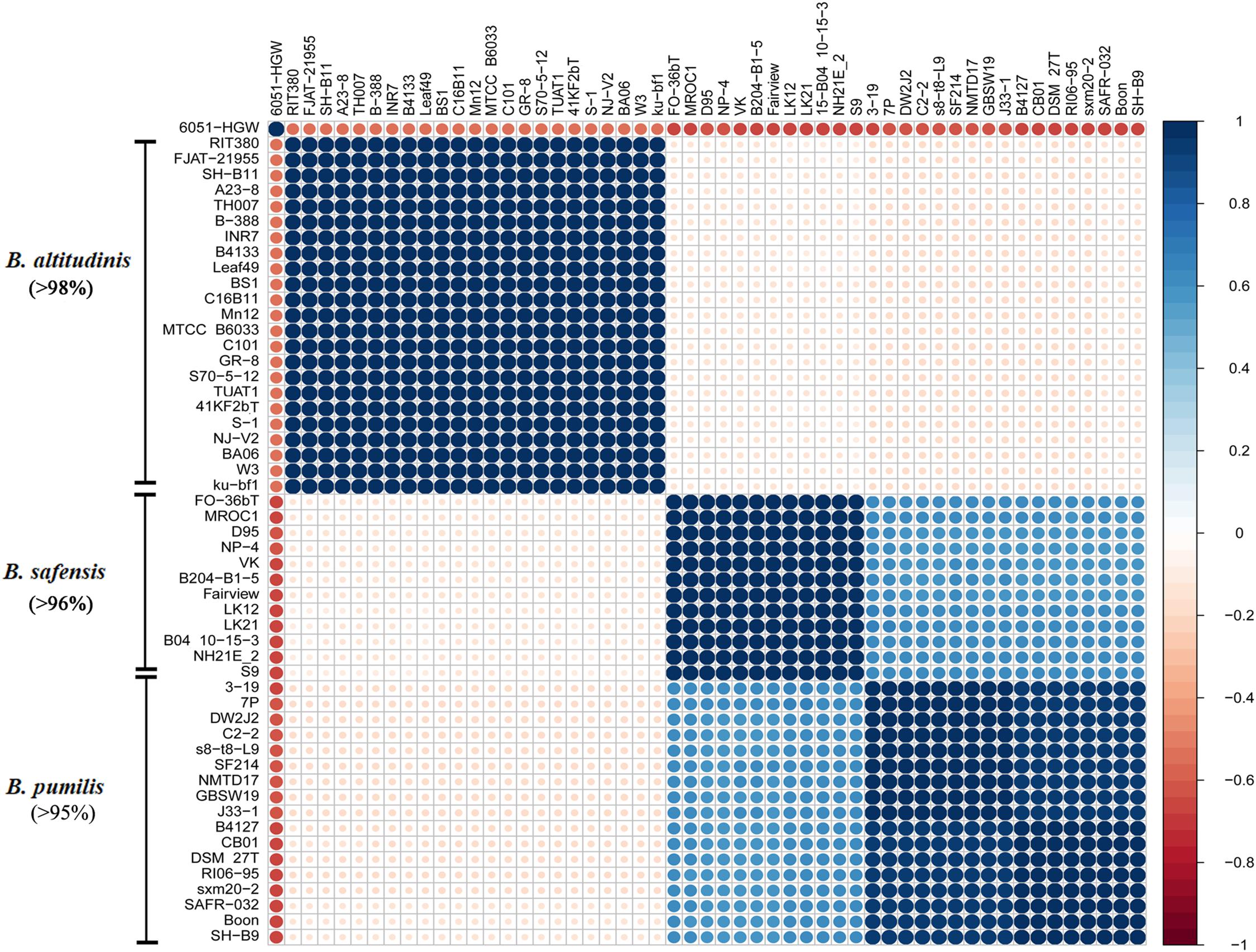
Figure 2. Correlation plot based on strains’ ANI values. ANI values between each strain were calculated using JSpecies software and employed for Pearson correlation matrix construction conducted using R. The plot shows the correlation constructed and ordered by hierarchical clustering using the R package “corrplot.” The minimum percentages of ANI values between the strains of a given cluster are indicated in brackets.
With the combined application of multiple genomic-scale approaches, including core gene analysis, ANI correlation analysis, DDH values and Mash distance analysis, we were able to resolve the taxonomic status of both our marine and terrestrial Bp group strains, by assigning them into three species, and obtain a robust phylogenetic reconstruction. Thus, all these 52 strains were classified as either B. pumilus, or B. altitudinis or B. safensis. The Bp isolates show genomic features that are distinct from each other even as they share a common core, and a common ancestor.
The pan-genome is defined as the entire genomic repertoire of a given phylogenetic clade and encodes all possible lifestyles of the bacteria according to COG (Cluster of Orthologous Genes) analysis (Tettelin et al., 2008). The pan-genome is divided into three categories: the core genome, the dispensable genome and strain-unique genes. Here, (1) we explored the total pan-genome and functional content of Bp group strains; (2) we compared the pangenome of marine isolates with that of terrestrial isolates; and (3) we analyzed the pan-genome of three species according to the phylogenetic tree.
(1) Total Pan-genome Landscape
The pan-genome of the 52 Bp-group strains contained 9,396 genes including 2,370 core genes (Figure 3A). For each strain, the proportion of the core genes was calculated, and the proportion ranged from 58.79 to 68.78%. In comparison, the proportion of the core genes corresponds to 74.01, 30.87, 26.01, and 38.75% of the total gene contents for the 52 B. anthracis strains, the 58 B. cereus strains, the 58 B. subtilis strains, and the 50 B. thuringiensis strains, respectively, as reported earlier (Kim et al., 2017). A total of 3,338 dispensable genes were found; dispensable genes were those that are present in two or more strains, not all strains (Medini et al., 2005; Tettelin et al., 2005; Vernikos et al., 2015), which account for 35.5% of the pan-genome. These dispensable genes are responsible for species diversity, environmental adaptation, and other characteristics of bacteria (Rouli et al., 2015). Strain-unique genes are those that are present in only one strain and are thought to be derived via horizontal gene transfer (Bentley, 2009); these genes accounted for 39.3% of the pan-genome genes among all tested Bp group bacteria, and the number of strain-unique genes varied from 14 to 247 between different strains. Both the dispensable genes and strain-unique genes constitute the non-core genes, representing subsets of the flexible genome. The B. altitudinis RIT380 strain possessed the highest proportion of non-core genes (41.21%), and strain B. pumilus 7P exhibited the lowest proportion (31.52%). This might reflect different levels of gene gains and losses during the evolution of these strains.
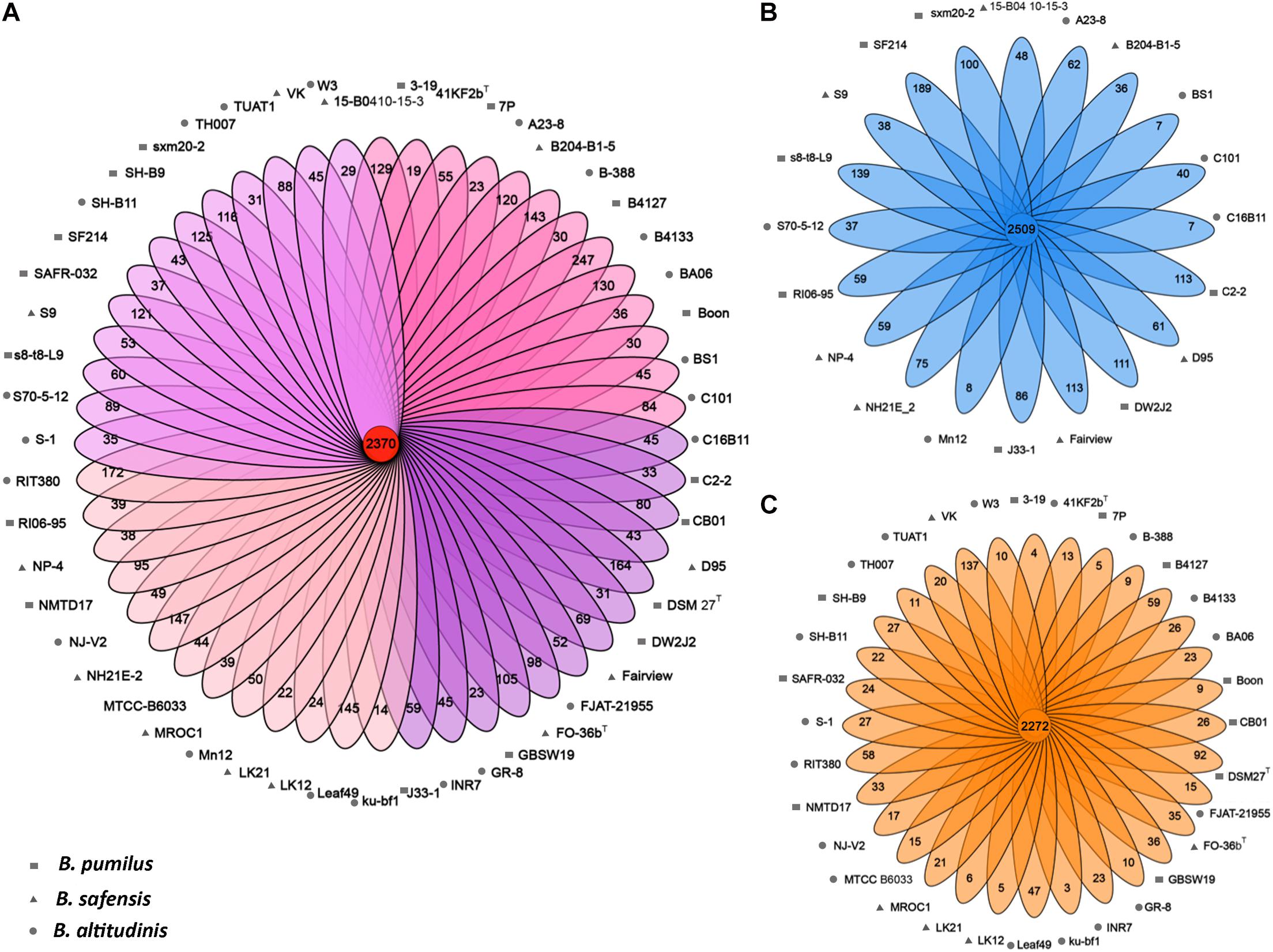
Figure 3. The pan-genomes of Bp group strains. (A) Flower plots showing the core gene number (in the center) and the strain-specific gene numbers (in the petals) in the 52 strains. (B) Flower plots showing the core gene number (in the center) and strain-specific gene number (in the petals) in the marine-isolated subgroups. (C) Flower plots showing the core gene number (in the center) and strain-specific gene number (in the petals) in the land-isolated subgroup.
To understand the relationships between pan-genome size, the core gene number, and the strain numbers of the Bp bacteria, we plotted the fitted curves of the pan-genome profile of the 52 strains. To determine the core genome, the number of conserved genes found upon the sequential addition of each new genome was extrapolated by fitting a decaying function (Figure 4A, red curve), indicating that the average number of core genes converged to a relatively constant number. The core gene number in each genome varied slightly because of the involvement of duplicated genes and prologs in the shared clusters. As shown in Figure 4A, the blue curve increased with the addition of a new strain and was far from saturation, implying that the genetic repertoire of the species was still growing despite suitable adaptation to their diverse ecological niches. Thus, the pan-genome of the Bp group strains is open (Figure 4A, blue curve). This agrees with previous reports showing that environmental samples usually had open pan-genomes (Tettelin et al., 2005; Konstantinidis et al., 2009; Biller et al., 2015). In other words, the Bp group strains are expected to continue to gain genes and evolve in the future. The availability of a large genetic reservoir might be a key survival advantage for Bp group strains when facing environmental challenges. The number of new genes added by the additionally sequenced genomes was also plotted by fitting the decaying exponential in Figure 4B, and these new added genes determined the expansion of the Bp group pan-genome.
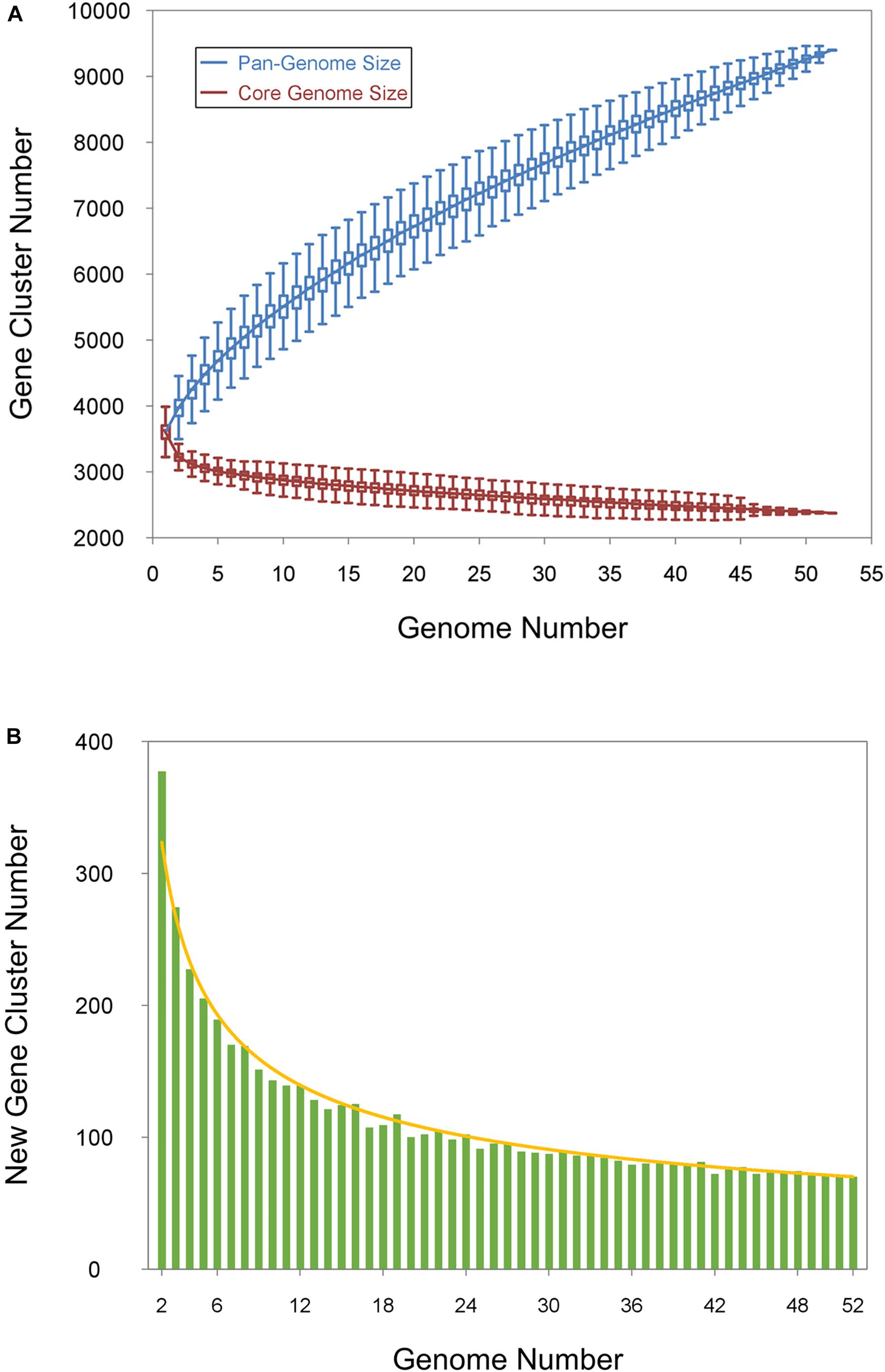
Figure 4. The pangenome curves of Bp group strains. (A) Core and pan-genome calculations for the Bp group. The blue curve shows an open pan-genome; the deduced pan-genome size is y = 726.2n0.552 + 2904.6 (R2 = 0.99998). The red curve represents the core genome, and the deduced core genome size is c = 939.4e– 0.058n + 2379.1 (R2 = 0.94131). (B) New gene size and curve for Bp-group strains.
To predict the functions of the genes that constitute the pan-genome, we took advantage of the COG functional classification. The COG category responsible for the highest proportion were poorly characterized genes (1021 genes), highlighting the important roles of these unknown genes corresponding to the undescribed physiology of Bp strains. The second most abundant category of genes in the pan-genome was general function predicted only including 989 genes and then by genes related to transcription (K) including 803 genes. The over representation of “poorly characterized genes” and “general function predicted only” in the COG categories is similar to what has been observed in other pan-genome studies (Sood et al., 2019; Gaba et al., 2020; Motyka-Pomagruk et al., 2020). Regulation at the transcriptional initiation level is probably the most common scenario for metabolic adaptation in bacteria (Leyn et al., 2013; Arrieta-Ortiz et al., 2015). The abundance of transcriptional regulators was consistent with the existence of complex transcriptional regulatory networks that support morphological and physiological differentiation. The most abundant core genes (40.41%) were associated with metabolism, whereas this category accounted for 26.57% of the strain-specific genes. In a given category of genes, the ratio of the percentage (of genes) in the core-genome, to its percentage in the non-core segment, showed that they could be arranged in a certain order of decreasing ratios. The genes involved in translation, ribosomal structure and biogenesis (J) had a ratio of 7.08:3.18%, while the other gene categories, namely, amino acid transport and metabolism (E), inorganic ion transport and metabolism (P), coenzyme transport and metabolism (H), had their core-genome/pan-genome composition percentages as 9.35/5.52%, 5.19/3.12%, and 5.44/3.47%, respectively. The translational machinery is widely accepted as a very ancient system and universal to life (Fox, 2010), and the translation machinery dominates the set of genes shared as orthologous across the tree of life (Bowman et al., 2020). Thus, given their universality, and essentiality, the genes involved in translation, ribosomal structure and biogenesis contribute to core genome stabilization. In the case of genes involved in repair (L) (3.11/9.69%), defense (V) (2.21/6.24%), transcription (K) (8.07/10.24%), and general function (R) (9.93/13.24%), the reverse was observed since they were better represented in the non-core segment than the core genome. This is suggestive of their possible roles in aiding the strains in their efficient response to different environments (Figure 5A).
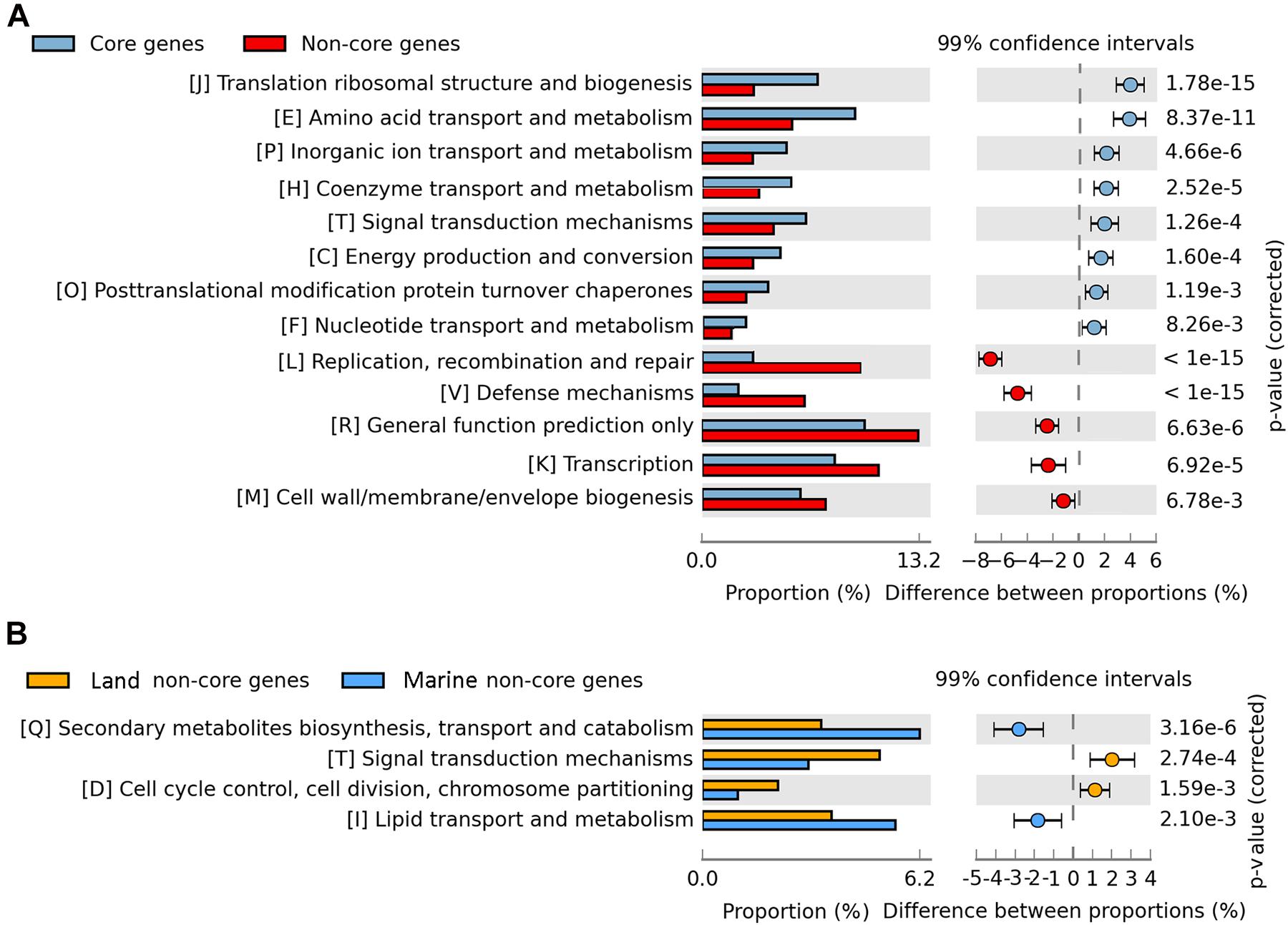
Figure 5. COG categories for total strains and marine vs. land strains. (A) COG categories of core genes and non-core genes. Blue represents core genes; red represents non-core genes. (B) Non-core gene COG categories of marine isolated strains and land isolated strains. Blue represents marine isolated strains; red represents land isolated strains.
(2) Pangenome of Marine and Terrestrial Groups
To detect the niche differentiation of Bp bacteria, they were divided into marine and land groups, and the pan-genome components were compared between marine and terrestrial strains. We found that the two groups exhibited a similar number of core genes, but the marine strains exhibited more strain-specific genes and fewer dispensable genes (Figures 3B,C), suggesting that horizontal gene transfer events have occurred more frequently among these marine strains. No significant difference was observed between the two groups in terms of COG category proportions in the core genome, suggesting that their core genes were highly functionally conserved. In the non-core genome, secondary metabolite biosynthesis, transport, and catabolism (Q) and lipid transport and metabolism (I) seemed to be over-represented in the marine group. The land group was enriched in signal transduction mechanisms (T), cell cycle control, cell division, chromosome partitioning (D) among the COG categories (Figure 5B). It has been suggested that land bacteria might respond quickly to environmental variation (Wan et al., 2019). At the same time, it has also been suggested that having dispensable genes involved in cell wall biogenesis, transport and metabolism of amino acids, carbohydrates, inorganic ions, and secondary metabolites, could be how certain members of Azospirillum adapt to terrestrial niches (Wisniewski-Dyé et al., 2011). However, only twelve strains (refer to Table 1) covered in our study appear to be associated with hosts (plants and animals) on land. Our data is not conclusive enough to make a correlation between the non-core (dispensable) genes and the specific adaptations of the strains to their environment. Further studies are warranted to explore the same.
(3) Pangenomes of the Three Species
The pan-genome of each species included 6,306 orthologous genes in B. altitudinis, 6,284 in B. pumilus, and 5,262 in B. safensis. A total of 2,953 (46.8%), 2,693 (42.9%), and 2,924 (55.5%) core genes were identified in B. altitudinis, B. pumilus and B. safensis, respectively. The numbers of dispensable genes were 1,507 (23.9%), 1,728 (27.5%), and 1,187 (22.6%) in B. altitudinis, B. pumilus and B. safensis, respectively. A total of 1,846 (29.3%), 1,863 (29.6%), and 1,151 (21.9%) strain-unique genes were identified in B. altitudinis, B. pumilus and B. safensis, respectively (Table 2). COG analysis revealed that the core genes showed quite similar distributions in functional categories, reflecting their conservation in the three species. Among the non-core genes, B. altitudinis showed the enrichment of larger number of genes related to DNA recombination and repair (L) (5%) than the other two species (2% in B. pumilus, 1.8% in B. safensis). Such an enrichment could be enabling B. altitudinis strains to better cope with different stresses in land environments, in which they could be continuously exposed to DNA-damaging agents, including UVB, ozone, desiccation, rehydration, salinity, low and high temperature, and air and soil pollutants on land (Fang et al., 2017). B. safensis possessed more genes involved in carbohydrate transport and metabolism (G) (8% in B. safensis, 5.8% in B. pumilus, 5% in B. altitudinis), and secondary metabolite biosynthesis, transport and catabolism (Q) (4.8% in B. safensis, 2.2% in B. pumilus, 2% in B. altitudinis), than the other two species. Overall, B. altitudinis exhibited the largest pan-genome size, and B. safensis possessed the least variation in genetic diversity. B. pumilus exhibited the highest number of non-core genes and strain-specific genes, resulting in high genetic diversity in this species. This has likely facilitated the wide geographic distribution of B. pumilus.
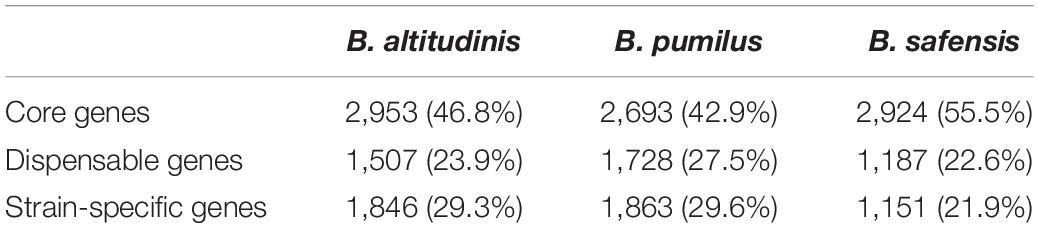
Table 2. Pan-genome size of the three different species.
In our dataset, 20 strains were isolated from marine environments, including 16 strains sequenced in this study and 4 genomes downloaded from NCBI. Since dispensable genome genes confer selective advantages such as adaptation to different niches and antibiotic resistance (Vernikos et al., 2015), we extracted the dispensable genes of the 52 Bp group strains and identified those shared by the 20 marine isolates. In the 20 marine bacterial isolates, 397 dispensable genes were found in total, including 41 hypothetical protein-coding genes. COG functional classification showed that 32 genes were transcription (K) related, which accounted for the highest proportion of the genes, such as transcriptional regulators belonging to the LysR family, AcrR family, GntR family, IclR family, IscR family, LacI/PurR family, MarR family, MerR family, MocR family, MurR/RpiR family, GlxA family, PucR family, etc. Regulatory proteins are thought to aid bacterial responses to specific environmental and cellular signals that modulate transcription, translation, or other events related to gene expression. Such adaptive responses facilitate bacterial survival in unstable environments (Romero-Rodríguez et al., 2015).
Generally, transcriptional regulators function via one- or two-component systems linking a specific type of environmental stimulus to a transcriptional response. The typical two-component systems usually consist of a membrane-bound sensor kinase (SK) and a DNA-binding protein known as a response regulator (RR) (Eswaramoorthy et al., 2010; Romero-Rodríguez et al., 2015). Of particular interest is the gene for the histidine kinase DesK. In B. subtilis, the histidine kinase DesK detects changes in temperature and activates the expression of a desaturase, thereby playing a role in increasing membrane fluidity and cold adaptation (Cybulski et al., 2010; Inda et al., 2014). All the 20 marine strains had the gene for DesK, whereas the homologs of DesK were found in only 7 land strains. The DesK homologs of the marine Bp-group strains showed 70–80% similarity with B. subtilis. Since this gene is found in both marine and terrestrial strains, it is not clear if it’s presence confers any adaptive advantage to either the marine or terrestrial strains.
In addition, we found genes that could possibly play a role in coping with high salinity and oligotrophic conditions in marine bacteria. First, all the marine strains encoded glycine betaine transporters implying that they might use organic compatible solutes to maintain cellular osmotic balance. Glycine betaine is a well-known osmotic regulator that facilitates the accumulation of betaine, carnitine, and choline to maintain the cellular osmotic balance (Kempf and Bremer, 1998; Sleator and Hill, 2002). Compatible solutes such as glycine betaine generally stabilize proteins by preventing the unfolding and denaturation of proteins caused by heating, freezing, and dehydration to maintain cell volume and integrity in response to osmotic stress (Goh et al., 2011). Potassium (K+) ions are crucial for bacteria to regulate solute transport and adapt to changes in osmotic pressure. The K+ transporter (TrK) facilitates the accumulation of K+ in marine-derived strains, which is the primary strategy whereby many extremophiles survive in high-osmolality environments (Sleator and Hill, 2002; Roberts, 2004). There are various mechanisms for the accumulation of K+ in the cell, including TrK, Kdp, Kup and Ktr potassium transport systems (Yaakop et al., 2016). The Kdp system is considered a high-affinity, inducible system; the Ktr system requires Na+; the ability of the Kup system to transport potassium is weak; and the Trk system is a low-affinity but rapid potassium transporter (Cai and He, 2017). The presence of the gene encoding TrK in our marine strains is suggestive of a strategy to survive under relatively high osmolality, similar to what is observed in marine Streptomyces (Tian et al., 2016). Marine bacteria require Na+ for growth, and some use an Na+ circuit for various functions (Oh et al., 1991). Many Na+-dependent transporters were detected in our marine strains, including an Na+: H+ antiporter and Na+ symporters and transporters, which play essential roles in the pH and Na+ homeostasis of cells.
Another major aspect related to habitat adaptation is the availability of nutrients. Multiple sugar ABC transporters were identified in the marine strains, consistent with the report that there was an enrichment of ABC transporter genes in the genomes of deep-sea microorganisms (DeLong et al., 2006). The presence of multiple transporters could facilitate efficient uptake of nutrients under oligotrophic conditions, as reported for marine bacteria (Norris et al., 2020). The identification of sugar hydrolases, including glycosidases, beta-xylosidase, endoglucanase, xylanase/chitin deacetylase, and chitinase, implied that marine Bp group strains could sense and respond to sugar sources and degrade them as carbon and energy sources. Additionally, the existence of amino acid permeases, amino acid transporters, fructose permease, glucose uptake permease and phosphotransferase system proteins likely facilitate the absorbance of amino acids, oligopeptides, and carbohydrates by the marine strains. Most of these enzymes were also reported to enhance adaptation to marine oligotrophic environments in the marine bacterium Zunongwangia profunda SM-A87 (Qin et al., 2010). Drug/metabolite transporters (DMTs) and additional cation/multidrug efflux pump members are involved in drug/metabolite or homoserine lactone removal. Marine Bp group strains employ these transporters to defend against intracellular toxic substances, as a means of survival in the marine environment (Jack et al., 2001). One “cation/multidrug efflux pump,” one “ABC-type dipeptide transport system, periplasmic component,” two “ABC-type sugar transport system, periplasmic component,” one “glycosyltransferase” and one “esterase/lipase” COG were also recently detected among adaptive aquatic Bacillus COGs (Hernández-González et al., 2018), suggesting the existence of specific genes that play roles in niche adaptation.
The marine isolates tended to cluster together in the core-gene phylogenetic tree, reflecting signals of differentiation and niche adaptation embedded in the genome. To make environmental groups more reasonable, COGs of core genes hierarchical clustering analysis for the 52 Bp group strains were performed (Supplementary Figure 2). The result was consistent with the core-gene phylogenetic tree which also exhibited marine origin strains gathered in each species. Therefore, to avoid discrepancies between different species, we compared marine and terrestrial strains within the same species. The presence/absence of genes as niche-specific genes and their COG distribution in the three species are summarized in Supplementary Table 4. Within each species, marine bacteria possessed more specific genes than land bacteria; however, more than half of these genes were of unknown functions.
Marine B. pumilus bacteria possessed more genes related to defense (V) and replication, recombination and repair (L) than those isolated from land (Supplementary Table 5). Additionally, we found that the genomes of our marine B. pumilus strains were enriched in phage and CRISPR elements (Supplementary Table 6), compared with land strains. B. pumilus phages have been isolated and characterized for their roles in horizontal gene transfer (Keggins et al., 1978; Lorenz et al., 2013; Badran et al., 2018), and implicated in acquisition of unique genes (Tirumalai et al., 2018) conferring traits such as spore radiation resistance (Tirumalai and Fox, 2013). Considering the large variety of phages in the marine environment (approximately 5–10 times higher than the number of bacteria), these genes might play roles in antiphage defense strategies (Wommack and Colwell, 2000; Doron et al., 2018). In comparison, land bacteria showed enrichment of genes specifically related to amino acid transport and metabolism (E) and mobile elements (X) and had different carbohydrate metabolism (G) related genes. Intriguingly, seven of the ten terrestrial strains contained a cyanide dehydratase that was not found in any marine B. pumilus strains. This cyanide dehydratase shared 73% amino acid identity with the cyanide dihydratase of B. pumilus C1, which has been proven to catalyze the conversion of cyanide to format and ammonia (Meyers et al., 1993). We could not explain why most of these land bacteria require the ability to perform cyanide detoxification. One possible clue toward answering this question is that cyanide-related compounds are common in terrestrial environments. A previous study indicated that cyanides are ubiquitously present in nature (Kumar et al., 2017), contributed by both anthropogenic activities and higher plant, fungal, and bacterial production (Baxter and Cummings, 2006; Larsen et al., 2004). Soil microbes can degrade cyanide to nitrites or form complexes with trace metals under aerobic conditions (Kjeldsen, 1999), suggesting that the land B. pumilus strains included in our study may utilize cyanide via cyanide dihydratase.
In the case of B. safensis, the marine strains had more specific genes related to transcription (K) and energy production and conversion (C), when compared with the land strains (Supplementary Table 5). The land strains have gained two multidrug transporters and specific genes in functional categories such as inorganic ion transport and metabolism (P), post-translational modification, protein turnover, and chaperones (O), compared with marine strain specific genes to cope with more diverse niches (Supplementary Table 5). All five terrestrial strains have gained the same ATP-binding protein of the ABC transporter, and two harbored the multidrug transporter EmrE, which was absent in the marine strains. These two specific proteins might help terrestrial strains to pump toxic substances outside the cell. Even within the same COG category, the proteins involved in amino acid and carbohydrate utilization were different between the land and marine strains. This reflected specialization in the metabolism of carbohydrates between land and marine strains, respectively. For example, the terrestrial strains have gained galactarate dehydrogenase and 5-dehydro-4-deoxyglucarate dehydratase; the former catalysis the transformation of galactarate to 5-dehydro-4-deoxy-D-glucarate, which is broken down by the latter enzyme (Deacon and Cooper, 1977).
In B. altitudinis, the marine strains contained specific genes involved in defense (V), replication, recombination and repair (L) against bacteriophages and other stresses, such as cold temperatures (Supplementary Table 5). The land strains were enriched in specific genes related to transcription (K), mobile elements (X), carbohydrate transport and metabolism (G), and some spore-related genes, such as the spore germination protein PD, the stage III sporulation protein AF, and the sporulation killing factor system radical SAM maturase. A previous study showed that 52 sporulation and germination genes were absent in the aquatic/halophile Bacillus, including the spore germination protein PD and the stage III sporulation protein AF (Alcaraz et al., 2010), which were also lost in our aquatic B. altitudinis strains. Overall, the large pangenome of B. altitudinis strains is an indicator of their versatility in terms of survival in diverse environments, such as soil, plants, food, feces, and animals.
Our results of the genomic analysis of the Bp group indicate that the transition between marine and non-marine habitats has occurred multiple times during their evolutionary history. Both the land and marine Bp bacteria could be classified into three species. However, core gene based phylogenetic analysis showed that in each species, marine isolates tended to cluster together. The genomic plasticity of B. pumilus group members, and their potential for multi niche adaptation has been reported earlier (Branquinho et al., 2014). Thus, in each species, divergence was prone to occur with niche differentiation by gaining or losing different specific genes to adapt to different environments. With the generation of additional genomes of Bp group strains from a wider range of environments, insights into gene acquisition, deletion, and maintenance will facilitate a better understanding of the evolution of Bp group strains.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article.
ZS and XF planned and managed the projects. YL, QL, and LG conducted the experiments. XF and LG analyzed the data. XF and ZS interpreted the results and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was financially supported by the National Natural Science Foundation of China (No. 41906089), COMRA program (No. DY135-B2-01), the Scientific Research Foundation of Third Institute of Oceanography, MNR (Nos. 2019019 and 2019021) and the Natural Science Foundation of Fujian Province, China (No. 2020J01106).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Prof. Xuewen Gao and his team member Huo Rong in the Department of Plant Pathology, College of Plant Protection, Nanjing Agricultural University, P. R. China, for sharing two soil isolates with us.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.571212/full#supplementary-material
Supplementary Figure 1 | Phylogenetic tree of Bp group strains generated with Mash software.
Supplementary Figure 2 | Hierarchical clustering using core COGs.
Supplementary Table 1 | Information of marine Bp group strains used for analyzing.
Supplementary Table 2 | ANI values between each Bp group strain.
Supplementary Table 3 | DDH values between each Bp group strain.
Supplementary Table 4 | COG distribution of niche-specific genes in the three species.
Supplementary Table 5 | Intra-species niche related specific genes of the three species.
Supplementary Table 6 | B. pumilus strains information containing both phage and CRISPR.
Data Sheet 1 | Alignments of protein (product) sequences of the core genes for phylogenetic tree construction.
Alcaraz, L. D., MorenFo-Hagelsieb, G., Eguiarte, L. E., Souza, V., Herrera-Estrella, L., and Olmedo, G. (2010). Understanding the evolutionary relationships and major traits of Bacillus through comparative genomics. BMC Genom. 11:332. doi: 10.1186/1471-2164-11-332
Arrieta-Ortiz, M. L., Hafemeister, C., Bate, A. R., Chu, T., Greenfield, A., Shuster, B., et al. (2015). An experimentally supported model of the Bacillus subtilis global transcriptional regulatory network. Mol. Syst. Biol. 11:839. doi: 10.15252/msb.20156236
Auch, A. F., Klenk, H. P., and Göker, M. (2010). Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand. Genomic Sci. 2, 142–148. doi: 10.4056/sigs.541628
Badran, S., Morales, N., Schick, P., Jacoby, B., Villella, W., and Lorenz, T. (2018). Complete genome sequence of the Bacillus pumilus Phage Leo2. Genome Announc. 15:e00066-18. doi: 10.1128/genomeA.00066-18
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Baxter, J., and Cummings, S. P. (2006). The current and future applications of microorganism in the bioremediation of cyanide contamination. Antonie Van Leeuwenhoek 90, 1–17. doi: 10.1007/s10482-006-9057-y
Bazinet, A. L. (2017). Pan-genome and phylogeny of Bacillus cereus sensu lato. BMC Evol. Biol. 17:176. doi: 10.1186/s12862-017-1020-1
Bentley, S. (2009). Sequencing the species pan-genome. Nat. Rev. Microbiol. 7, 258–259. doi: 10.1038/nrmicro2123
Berrada, I., Benkhemmar, O., Swings, J., Bendaou, N., and Amar, M. (2012). Selection of halophilic bacteria for biological control of tomato gray mould caused by Botrytis cinerea. Phytopatho. Mediterr. 51, 625–630. doi: 10.2307/43872349
Bhandari, V., Ahmod, N. Z., Shah, H. N., and Gupta, R. S. (2013). Molecular signatures for Bacillus species: demarcation of the Bacillus subtilis and Bacillus cereus clades in molecular terms and proposal to limit the placement of new species into the genus Bacillus. Int. J. Syst. Evol. Microbiol. 63, 2712–2726. doi: 10.1099/ijs.0.048488-0
Biller, S. J., Berube, P. M., Lindell, D., and Chisholm, S. W. (2015). Prochlorococcus: the structure and function of collective diversity. Nat. Rev. Microbiol. 13, 13–27. doi: 10.1038/nrmicro3378
Bowman, J. C., Petrov, A. S., Frenkel-Pinter, M., Penev, P. I., and Williams, L. D. (2020). Root of the tree: the significance, evolution, and origins of the ribosome. Chem. Rev. 120, 4848–4878. doi: 10.1021/acs.chemrev
Branquinho, R., Meirinhos-Soares, L., Carriço, J. A., Pintado, M., and Peixe, L. V. (2014). Phylogenetic and clonality analysis of Bacillus pumilus isolates uncovered a highly heterogeneous population of different closely related species and clones. FEMS Microbiol. Ecol. 90, 689–698. doi: 10.1111/1574-6941.12426
Cai, X., and He, J. (2017). Second messenger c-di-AMP regulates potassium ion transport. Wei Sheng Wu Xue Bao 57, 1434–1442. doi: 10.13343/j.cnki.wsxb.20160479
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Checinska, A., Paszczynski, A., and Burbank, M. (2015). Bacillus and other spore-forming genera: variations in responses and mechanisms for survival. Annu. Rev. Food Sci. Technol. 6, 351–369. doi: 10.1146/annurev-food-030713-092332
Chun, B. H., Kim, K. H., Jeong, S. E., and Jeon, C. O. (2019). Genomic and metabolic features of the Bacillus amyloliquefaciens group-B. amyloliquefaciens, B. velezensis, and B. siamensis–revealed by pan-genome analysis. Food Microbiol. 77, 146–157. doi: 10.1016/j.fm.2018.09.001
Connor, N., Sikorski, J., Rooney, A. P., Kopac, S., and Koeppel, A. F. (2010). Ecology of speciation in the genus Bacillus. Appl. Environ. Microbiol. 76, 1349–1358. doi: 10.1128/aem.01988-09
Cybulski, L. E., Martin, M., Mansilla, M. C., Fernandez, A., and De Mendoza, D. (2010). Membrane thickness cue for cold sensing in a bacterium. Curr. Biol. 20, 1539–1544. doi: 10.1016/j.cub.2010.06.074
Deacon, J., and Cooper, R. A. (1977). D-galactonate utilization by enteric bacteria the catabolic pathway in Escherichia coli. FEBS Lett. 77, 201–205.
DeLong, E. F., Preston, C. M., Mincer, T., Rich, V., Hallam, S. J., Frigaard, N. U., et al. (2006). Community genomics among stratified microbial assemblages in the ocean’s interior. Science 311, 496–503. doi: 10.2307/3843405
Dickinson, D. N., La Duc, M. T., Satomi, M., Winefordner, J. D., Powell, D. H., and Venkateswaran, K. (2004). MALDI-TOFMS compared with other polyphasic taxonomy approaches for the identification and classification of Bacillus pumilus spores. J. Microbiol. Methods 58, 1–12. doi: 10.1016/j.mimet.2004.02.011
Doron, S., Melamed, S., Ofir, G., Leavitt, A., Lopatina, A., Keren, M., et al. (2018). Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359:4120. doi: 10.1126/science.aar4120
Earl, A. M., Losick, R., and Kolter, R. (2008). Ecology and genomics of Bacillus subtilis. Trends Microbiol. 16, 269–275. doi: 10.1016/j.tim.2008.03.004
Espariz, M., Zuljan, F. A., Esteban, L., and Magni, C. (2016). Taxonomic identity resolution of highly phylogenetically related strains and selection of phylogenetic markers by using genome-scale methods: the Bacillus pumilus group case. PLoS One 11:e0163098. doi: 10.1371/journal.pone.0163098
Eswaramoorthy, P., Duan, D., Dinh, J., Dravis, A., Devi, S. N., and Fujita, M. (2010). The threshold level of the sensor histidine kinase KinA governs entry into sporulation in Bacillus subtilis. J. Bacteriol. 192, 3870–3882. doi: 10.1128/JB.00466-10
Ettoumi, B., Raddadi, N., Borin, S., Daffonchio, D., Boudabous, A., and Cherif, A. (2009). Diversity and phylogeny of culturable spore-forming Bacilli isolated from marine sediments. J. Basic Microbiol. 49, S13–S23. doi: 10.1002/jobm.200800306
Fan, B., Blom, J., Klenk, H. P., and Borriss, R. (2017). Bacillus amyloliquefaciens, Bacillus velezensis, and Bacillus siamensis form an “operational group B. amyloliquefaciens” within the B. subtilis species complex. Front. Microbiol. 8:22. doi: 10.3389/fmicb.2017.00022
Fang, H., Huangfu, L., Chen, R., Li, P., Xu, S., Zhang, E., et al. (2017). Ancestor of land plants acquired the DNA-3-methyladenine glycosylase (MAG) gene from bacteria through horizontal gene transfer. Sci. Rep. 24:9324. doi: 10.1038/s41598-017-05066-w
Fang, Y., Li, Z., Liu, J., Shu, C., Wang, X., Zhang, X., et al. (2011). A pangenomic study of Bacillus thuringiensis. J. Genet. Genomics 38, 567–576. doi: 10.1016/j.jgg.2011.11.001
Ficarra, F. A., Santecchia, I., Lagorio, S. H., Alarcón, S., Magni, C., and Espariz, M. (2016). Genome mining of lipolytic exoenzymes from Bacillus safensis S9 and Pseudomonas alcaliphila ED1 isolated from a dairy wastewater lagoon. Arch. Microbiol. 198, 893–904. doi: 10.1007/s00203-016-1250-4
Fox, G. E. (2010). Origin and evolution of the ribosome. Cold Spring Harb. Perspect. Biol. 2:a003483. doi: 10.1101/cshperspect.a003483
Fox, G. E., Stackebrandt, E., Hespell, R. B., Gibson, J., Maniloff, J., Dyer, T. A., et al. (1980). The phylogeny of prokaryotes. Science 209, 457–463. doi: 10.1126/science.6771870
Fox, G. E., Wisotzkey, J. D., and Jurtshuk, P. Jr. (1992). How close is close: 16S rRNA sequence identity may not be sufficient to guarantee species identity. Int. J. Syst. Bacteriol. 42, 166–170. doi: 10.1099/00207713-42-1-166
Gaba, S., Kumari, A., Medema, M., and Kaushik, R. (2020). Pan-genome analysis and ancestral state reconstruction of class halobacteria : probability of a new super-order. Sci. Rep. 10:21205. doi: 10.1038/s41598-020-77723-6
Gioia, J., Yerrapragada, S., Qin, X., Jiang, H., Igboeli, O. C., Muzny, D., et al. (2007). Paradoxical DNA repair and peroxide resistance gene conservation in Bacillus pumilus SAFR-032. PLoS One 2:e928. doi: 10.1371/journal.pone.0000928
Goh, F., Jeon, Y. J., Barrow, K., Neilan, B. A., and Burns, B. P. (2011). Osmoadaptive strategies of the archaeon Halococcus hamelinensis isolated from a hypersaline stromatolite environment. Astrobiology 11, 529–536. doi: 10.1089/ast.2010.0591
Gupta, A., and Sharma, V. K. (2015). Using the taxon-specific genes for the taxonomic classification of bacterial genomes. BMC Genomics 16:396. doi: 10.1186/s12864-015-1542-0
Hall, B. G., Ehrlich, G. D., and Hu, F. Z. (2010). Pan-genome analysis provides much higher strain typing resolution than multi-locus sequence typing. Microbiology 156, 1060–1068. doi: 10.1099/mic.0.035188-0
Hernández-González, I. L., Moreno-Hagelsieb, G., and Olmedo-Álvarez, G. (2018). Environmentally-driven gene content convergence and the Bacillus phylogeny. BMC Evol. Biol. 18:148. doi: 10.1186/s12862-018-1261-7
Inda, M. E., Vandenbranden, M., Fernández, A., Mendoza, D., Ruysschaert, J. M., and Cybulski, L. E. (2014). A lipid-mediated conformational switch modulates the thermosensing activity of DesK. PNAS 111, 3579–3584. doi: 10.1073/pnas.1317147111
Jack, D. L., Yang, N. M., and Saier, M. H. (2001). The drug/metabolite transporter superfamily. Eur. J. Biochem. 268, 3620–3639.
Jones, D. T., Taylor, W. R., and Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282. doi: 10.1093/bioinformatics/8.3.275
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Keggins, K. M., Nauman, R. K., and Lovett, P. S. (1978). Sporulation-converting bacteriophages for Bacillus pumilus. J. Virol. 27, 819–822. doi: 10.1128/JVI.27.3.819-822.1978
Kempf, B., and Bremer, E. (1998). Uptake and synthesis of compatible solutes as microbial stress responses to high-osmolality environments. Arch. Microbiol. 170, 319–330. doi: 10.1007/s002030050649
Kettler, G. C., Martiny, A. C., Huang, K., Zucker, J., Coleman, M. L., Rodrigue, S., et al. (2007). Patterns and implications of gene gain and loss in the evolution of Prochlorococcus. PLoS Genet. 3:e231. doi: 10.1371/journal.pgen.0030231
Khianngam, S., Pootaeng-on, Y., Techakriengkrai, T., and Tanasupawat, S. (2014). Screening and identification of cellulose producing bacteria isolated from oil palm meal. J. Appl. Pharm. Sci. 4, 90–96.
Kim, Y., Koh, I., Lim, M. Y., Chung, W. H., and Rho, M. (2017). Pan-genome analysis of Bacillus for microbiome profiling. Sci. Rep. 7, 1–9. doi: 10.1038/s41598-017-11385-9
Kjeldsen, P. (1999). Behaviour of cyanides in soil and groundwater: a review. Water Air Soil Poll. 115, 279–308. doi: 10.1023/a:1005145324157
Konstantinidis, K. T., Serres, M. H., Romine, M. F., Rodrigues, J. L., Auchtung, J., McCue, L. A., et al. (2009). Comparative systems biology across an evolutionary gradient within the Shewanella genus. Proc. Natl. Acad. Sci. U.S.A. 106, 15909–15914. doi: 10.2307/40484821
Kumar, D., Parshad, R., and Gupta, V. K. (2014). Application of a statistically enhanced, novel, organic solvent stable lipase from Bacillus safensis DVL-43. Int. J. Biol. Macromol. 66, 97–107. doi: 10.1016/j.ijbiomac.2014.02.015
Kumar, R., Saha, S., Dhaka, S., Kurade, M. B., Kang, C. U., Baek, S. H., et al. (2017). Remediation of cyanide-contaminated environments through microbes and plants: a review of current knowledge and future perspectives. Geosyst. Eng. 20, 28–40. doi: 10.1080/12269328.2016.1218303
La Duc, M. T., Satomi, M., Agata, N., and Venkateswaran, K. (2004). gyrB as a phylogenetic discriminator for members of the Bacillus anthracis-cereus-thuringiensis group. J. Microbiol. Methods 56, 383–394. doi: 10.1016/j.mimet.2003.11.004
Larsen, M., Trapp, S., and Pirandello, A. (2004). Removal of cyanide by woody plants. Chemosphere 54, 325–333. doi: 10.1016/S0045-6535(03)00662-3
Lateef, A., Adelere, I. A., Gueguim-Kana, E. B., Asafa, T. B., and Beukes, L. S. (2015). Green synthesis of silver nanoparticles using keratinase obtained from a strain of Bacillus safensis LAU 13. Int. Nano Lett. 5, 29–35. doi: 10.1007/s40089-014-0133-4
Lawrence, J. G., and Hendrickson, H. (2005). Genome evolution in bacteria: order beneath chaos. Curr. Opin. Microbiol. 8, 572–578. doi: 10.1016/j.mib.2005.08.005
Leyn, S. A., Kazanov, M. D., Sernova, N. V., Ermakova, E. O., Novichkov, P. S., and Rodionov, D. A. (2013). Genomic reconstruction of the transcriptional regulatory network in Bacillus subtilis. J. Bacteriol. 195, 2463–2473. doi: 10.1128/jb.00140-13
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Li, Q. M., Zhou, Y. L., Wei, Z. F., and Wang, Y. (2020). Phylogenomic insights into distribution and adaptation of Bdellovibrionota in marine waters. bioRxiv [Preprint]. doi: 10.1101/2020.11.01.364414
Liu, Y., Lai, Q., Dong, C., Sun, F., Wang, L., Li, G., et al. (2013). Phylogenetic diversity of the Bacillus pumilus group and the marine ecotype revealed by multilocus sequence analysis. PLoS One 8:e80097. doi: 10.1371/journal.pone.0080097
Liu, Y., Lai, Q., Du, J., and Shao, Z. (2016). Bacillus zhangzhouensis sp. nov. and Bacillus australimaris sp. nov. Int. J. Syst. Evol. Microbiol. 66, 1193–1199. doi: 10.1099/ijsem.0.000856
Liu, Y., Lai, Q., Du, J., and Shao, Z. (2017). Genetic diversity and population structure of the Bacillus cereus group bacteria from diverse marine environments. Sci. Rep. 7:689. doi: 10.1038/s41598-017-00817-1
Liu, Y., Lai, Q., Göker, M., Meier-Kolthoff, J. P., Wang, M., Sun, Y., et al. (2015). Genomic insights into the taxonomic status of the Bacillus cereus group. Sci. Rep. 5:14082. doi: 10.1038/srep14082
Lorenz, L., Lins, B., Barrett, J., Montgomery, A., Trapani, S., Schindler, A., et al. (2013). Genomic characterization of six novel Bacillus pumilus bacteriophages. Virology 444, 374–383. doi: 10.1016/j.virol.2013.07.004
Maistrenko, O. M., Mende, D. R., Luetge, M., Hildebrand, F., Schmidt, T. S., Li, S. S., et al. (2020). Disentangling the impact of environmental and phylogenetic constraints on prokaryotic within-species diversity. ISME 14, 1247–1259. doi: 10.1038/s41396-020-0600-z
Medini, D., Donati, C., Tettelin, H., Masignani, V., and Rappuoli, R. (2005). The microbial pan-genome. Curr. Opin. Genet. Dev. 15, 589–594. doi: 10.1016/j.gde.2005.09.006
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 14:60. doi: 10.1186/1471-2105-14-60
Meyers, P. R., Rawlings, D. E., Woods, D. R., and Lindsey, G. G. (1993). Isolation and characterization of a cyanide dihydratase from Bacillus pumilus C1. J. Bacteriol. 175, 6105–6112. doi: 10.1128/jb.175.19.6105-6112.1993
Motyka-Pomagruk, A., Zoledowska, S., Misztak, A. E., Sledz, W., Mengoni, A., and Lojkowska, E. (2020). Comparative genomics and pangenome-oriented studies reveal high homogeneity of the agronomically relevant enterobacterial plant pathogen Dickeya solani. BMC Genomics 21:449. doi: 10.1186/s12864-020-06863-w
Muzzi, A., Masignani, V., and Rappuoli, R. (2007). The pan-genome: towards a knowledge-based discovery of novel targets for vaccines and antibacterials. Drug Discov. Today 12, 429–439. doi: 10.1016/j.drudis.2007.04.008
Naz, K., Ullah, N., Zaheer, T., Shehroz, M., Naz, A., and Ali, A. (2020). Pan-Genomics of Model Bacteria and Their Outcomes. In Pan-genomics: Applications, Challenges, and Future Prospects. Cambridge, MA: Academic Press. 189–201.
Nicholson, W. L., Munakata, N., Horneck, G., Melosh, H. J., and Setlow, P. (2000). Resistance of Bacillus endospores to extreme terrestrial and extraterrestrial environments. Microbiol. Mol. Biol. Rev. 64, 548–572. doi: 10.1128/mmbr.64.3.548-572.2000
Norris, N., Levine, N. M., Fernandez, V. I., and Stocker, R. (2020). Mechanistic model of nutrient uptake explains dichotomy between marine oligotrophic and copiotrophic bacteria. bioRxiv [Preprint]. doi: 10.1101/2020.10.08.331785
Ochman, H., Lerat, E., and Daubin, V. (2005). Examining bacterial species under the specter of gene transfer and exchange. Proc. Natl. Acad. Sci. U.S.A. 102, 6595–6599.
Oh, S., Kogure, K., Ohwada, K., and Simidu, U. (1991). Correlation between possession of a respiration-dependent Na+ pump and Na+ requirement for growth of marine bacteria. Appl. Environ. Microbiol. 57, 1844–1846. doi: 10.1128/AEM.57.6.1844-1846.1991
Ondov, B. D., Treangen, T. J., Melsted, P., Mallonee, A. B., Bergman, N. H., Koren, S., et al. (2016). Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17:132. doi: 10.1186/s13059-016-0997-x
Parks, D. H., Tyson, G. W., Hugenholtz, P., and Beiko, R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Parvathi, A., Krishna, K., Jose, J., Joseph, N., and Nair, S. (2009). Biochemical and molecular characterization of Bacillus pumilus isolated from coastal environment in Cochin, India. Braz. J. Microbiol. 40, 269–275. doi: 10.1590/S1517-838220090002000012
Patané, J. S. L., Martins, J. Jr., and Setubal, J. C. (2018). Phylogenomics. Methods Mol. Biol. 1704, 103–187. doi: 10.1007/978-1-4939-7463-4_5
Patel, S., and Gupta, R. S. (2019). A phylogenomic and comparative genomic framework for resolving the polyphyly of the genus Bacillus: proposal for six new genera of Bacillus species, Peribacillus gen. nov., Cytobacillus gen. nov., Mesobacillus gen. nov., Neobacillus gen. nov., Metabacillus gen. nov. and Alkalihalobacillus gen. nov. Int. J. Syst. Evol. Microbiol. 70, 406–438. doi: 10.1099/ijsem.0.003775
Penn, K., and Jensen, P. R. (2012). Comparative genomics reveals evidence of marine adaptation in Salinispora species. BMC Genomics 13:86. doi: 10.1186/1471-2164-13-86
Priest, F. G., Goodfellow, M., Shute, L. A., and Berkeley, R. C. W. (1987). Bacillus amyloliquefaciens sp. nov., nom. rev. Int. J. Syst. Evol. Microbiol. 37, 69–71. doi: 10.1099/00207713-37-1-69
Qin, Q. L., Zhang, X. Y., Wang, X. M., Liu, G. M., and Chen, X. L. (2010). The complete genome of Zunongwangia profunda SM-A87 reveals its adaptation to the deep-sea environment and ecological role in sedimentary organic nitrogen degradation. BMC Genom. 11:247. doi: 10.1186/1471-2164-11-247
Ravel, J., and Fraser, C. M. (2005). Genomics at the genus scale. Trends Microbiol. 13, 95–97. doi: 10.1016/j.tim.2005.01.004
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. PNAS 106, 19126–19131. doi: 10.2307/25593154
Roberts, M. F. (2004). Osmoadaptation and osmoregulation in archaea. Front. Biosci. 9, 1999–2019. doi: 10.2741/1366
Romero-Rodríguez, A., Robledo-Casados, I., and Sánchez, S. (2015). An overview on transcriptional regulators in Streptomyces. BBA Gene Regul. Mech. 1849, 1017–1039. doi: 10.1016/j.bbagrm.2015.06.007
Rouli, L., Merhej, V., Fournier, P. E., and Raoult, D. (2015). The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microb. New Infect. 7, 72–85. doi: 10.1016/j.nmni.2015.06.005
Satomi, M., La Duc, M. T., and Venkateswaran, K. (2006). Bacillus safensis sp. nov., isolated from spacecraft and assembly-facility surfaces. Int. J. Syst. Evol. Microbiol. 56, 1735–1740. doi: 10.1099/ijs.0.64189-0
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Setlow, P. (2014). Spore resistance properties. Microbiol. Spectr. 2:e0003-2012. doi: 10.1128/microbiolspec
Sleator, R. D., and Hill, C. (2002). Bacterial osmoadaptation: the role of osmolytes in bacterial stress and virulence. FEMS Microbiol. Rev. 26, 49–71. doi: 10.1111/j.1574-6976.2002.tb00598.x
Sood, U., Hira, P., Kumar, R., Bajaj, A., and Rao, D. L. N. (2019). Comparative genomic analyses reveal core-genome-wide genes under positive selection and major regulatory hubs in outlier strains of Pseudomonas aeruginosa. Front. Microbiol. 10:53. doi: 10.3389/fmicb.2019.00053
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
Tettelin, H., Riley, D., Cattuto, C., and Medini, D. (2008). Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 12, 472–477. doi: 10.1016/j.mib.2008.09.006
Tian, X., Zhang, Z., Yang, T., Chen, M., Li, J., Chen, F., et al. (2016). Comparative genomics analysis of Streptomyces species reveals their adaptation to the marine environment and their diversity at the genomic level. Front. Microbiol. 7:998. doi: 10.3389/fmicb.2016.00998
Tirumalai, M. R., and Fox, G. E. (2013). An ICEBs1-like element may be associated with the extreme radiation and desiccation resistance of Bacillus pumilus SAFR-032 spores. Extremophiles 17, 767–774. doi: 10.1007/s00792-013-0559-z
Tirumalai, M. R., Stepanov, V. G., Wünsche, A., Montazari, S., Gonzalez, R. O., Venkateswaran, K., et al. (2018). Bacillus safensis FO-36b and Bacillus pumilus SAFR-032: a whole genome comparison of two spacecraft assembly facility isolates. BMC Microbiol. 18:57. doi: 10.1186/s12866-018-1191-y
Tiwary, B. K. (2020). “Evolutionary pan-genomics and applications,” in Pan-genomics: Applications, Challenges, and Future Prospects, eds V. Azevedo, S. Tiwari, and S. C. Soares (Cambridge, MA: Academic Press), 65–80.
Vernikos, G., Medini, D., Riley, D. R., and Tettelin, H. (2015). Ten years of pan-genome analyses. Curr. Opin. Microbiol. 23, 148–154. doi: 10.1016/j.mib.2014.11.016
Wan, S., Liu, Z., Chen, Y., Zhao, J., Ying, Q., and Liu, J. (2019). Effects of lime application and understory removal on soil microbial communities in subtropical eucalyptus L’Hér. Plantations. Forests 2019: 338. doi: 10.3390/f10040338
Wisniewski-Dyé, F., Borziak, K., Khalsa-Moyers, G., Alexandre, G., Sukharnikov, L. O., Wuichet, K., et al. (2011). Azospirillum genomes reveal transition of bacteria from aquatic to terrestrial environments. PLoS Genet. 7:e1002430. doi: 10.1371/journal.pgen.1002430
Wommack, K. E., and Colwell, R. R. (2000). Virioplankton: viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 64, 69–114.
Wu, H., Wang, D., and Gao, F. (2020). Toward a high-quality pan-genome landscape of Bacillus subtilis by removal of confounding strains. Brief. Bioinform. 22, 1951–1971. doi: 10.1093/bib/bbaa013
Yaakop, A. S., Chan, K. G., Ee, R., Lim, Y. L., Lee, S. K., Manan, F. A., et al. (2016). Characterization of the mechanism of prolonged adaptation to osmotic stress of Jeotgalibacillus malaysiensis via genome and transcriptome sequencing analyses. Sci. Rep. 6, 1–14. doi: 10.1038/srep33660
Yerrapragada, S., Siefert, J. L., and Fox, G. E. (2009). Horizontal gene transfer in cyanobacterial signature genes. Methods Mol. Biol. 532, 339–366. doi: 10.1007/978-1-60327-853-9_20
Zhang, X., Liu, X., Yang, F., and Chen, L. (2018). Pan-genome analysis links the hereditary variation of Leptospirillum ferriphilum with its evolutionary adaptation. Front. Microbiol. 9:577. doi: 10.3389/fmicb.2018.00577
Keywords: Bacillus pumilus group, phylogenetics, pan-genome, marine adaptation, differentiation
Citation: Fu X, Gong L, Liu Y, Lai Q, Li G and Shao Z (2021) Bacillus pumilus Group Comparative Genomics: Toward Pangenome Features, Diversity, and Marine Environmental Adaptation. Front. Microbiol. 12:571212. doi: 10.3389/fmicb.2021.571212
Received: 10 June 2020; Accepted: 12 April 2021;
Published: 07 May 2021.
Edited by:
John R. Battista, Louisiana State University, United StatesReviewed by:
Martín Espariz, Consejo Nacional de Investigaciones Cient ficas y T cnicas (CONICET), Instituto de Biología Molecular y Celular de Rosario (IBR), ArgentinaCopyright © 2021 Fu, Gong, Liu, Lai, Li and Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zongze Shao, shaozz@163.com
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.