 Kyle H. Rohde
Kyle H. Rohde Leonardo Sorci
Leonardo Sorci- 1Burnett School of Biomedical Sciences, College of Medicine, University of Central Florida, Orlando, FL, United States
- 2Division of Bioinformatics and Biochemistry, Department of Materials, Environmental Sciences and Urban Planning, Polytechnic University of Marche, Ancona, Italy
Given the upsurge of drug-resistant tuberculosis worldwide, there is much focus on developing novel drug combinations allowing shorter treatment duration and a lower toxicity profile. Nicotinamide adenine dinucleotide (NAD) biosynthesis targeting is acknowledged as a promising strategy to combat drug-susceptible, drug-resistant, and latent tuberculosis (TB) infections. In this review, we describe the potential synergy of NAD biosynthesis inhibitors with several TB-drugs in prospective novel combination therapy. Despite not directly targeting the essential NAD cofactor’s biosynthesis, several TB prodrugs either require a NAD biosynthesis enzyme to be activated or form a toxic chemical adduct with NAD(H) itself. For example, pyrazinamide requires the action of nicotinamidase (PncA), often referred to as pyrazinamidase, to be converted into its active form. PncA is an essential player in NAD salvage and recycling. Since most pyrazinamide-resistant strains are PncA-defective, a combination with downstream NAD-blocking molecules may enhance pyrazinamide activity and possibly overcome the resistance mechanism. Isoniazid, ethionamide, and delamanid form NAD adducts in their active form, partly perturbing the redox cofactor metabolism. Indeed, NAD depletion has been observed in Mycobacterium tuberculosis (Mtb) during isoniazid treatment, and activation of the intracellular NAD phosphorylase MbcT toxin potentiates its effect. Due to the NAD cofactor’s crucial role in cellular energy production, additional synergistic correlations of NAD biosynthesis blockade can be envisioned with bedaquiline and other drugs targeting energy-metabolism in mycobacteria. In conclusion, future strategies targeting NAD metabolism in Mtb should consider its potential synergy with current and other forthcoming TB-drugs.
Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), is the leading infectious cause of mortality, with an estimated 1.2 million deaths and 10 million new cases in 2019, and about a quarter of the world’s population latently infected (WHO, 2020). The shortcomings of currently available TB drugs are a major factor underlying the ongoing TB crisis. The current “short course” regimen involves a cocktail of multiple front-line drugs administered for 6 months. In a sobering finding, Malherbe et al. (2016) reported detection of Mtb RNA within nonresolving granulomas in patients who had been declared cured after standard 6-month treatment using PET-CT imaging. This highlights the inability of current drug regimens to effectively eradicate phenotypically tolerant subpopulations of Mtb persisters sequestered within granulomas, the hallmark lesions of TB disease. Further, exacerbating the challenge of treating TB is the COVID-19 pandemic which is hampering the TB detection and the increasing emergence of drug resistance. According to the latest report on TB epidemic, in 2019, nearly 400,000 people developed multidrug-resistant TB (MDR-TB), which is caused by strains resistant to the two first-line drugs rifampicin (RIF) and isoniazid (INH) with a treatment success rate of 57% globally (WHO, 2020). Even more concerning are extensively drug resistant TB (XDR-TB), resistant to RIF, INH, fluoroquinolones, and at least one of the three classes of injectables (amikacin, kanamycin, and capreomycin), and totally drug resistant TB (TDR-TB), which are not susceptible to known drugs (Udwadia et al., 2012). Thus, there is an urgent need for new multi-drug regimens with potent bactericidal activity against dormant persisters and drug-resistant mycobacteria. Identifying novel antimicrobials with unique mechanisms of action that can synergize with drugs currently in use or at late stages of development is a promising strategy to address this need. Prompted by our efforts to develop inhibitors of nicotinamide adenine dinucleotide (NAD) biosynthesis, this review seeks to highlight scientific evidence that chemical perturbation of this pathway could be an effective component of new TB drug regimens.
Isoniazid and NAD Metabolism
Discovered back in 1952, isoniazid, also known as isonicotinic acid hydrazide (INH), remains today one of the major first-line antituberculosis drugs. INH only kills actively replicating bacteria, with a minimum inhibitory concentration (MIC) against the slowly growing M. tuberculosis of 0.05 μg ml−1. In contrast to the exquisite activity of INH on Mtb, most mycobacteria are only susceptible to INH concentrations over 1 μg/ml (Zhang and Young, 1993). The precise target and mechanism of action of INH have eluded the grasp of science for decades. A combination of biochemical, genetic, and X-ray crystallography studies finally concluded that INH is a prodrug involved in inhibiting the biosynthesis of mycolic acid, an essential cell wall component of Mtb (Winder et al., 1970). This prodrug’s most accepted mechanism of action requires its activation into an acyl radical by the catalase-peroxidase KatG enzyme (Lei et al., 2000). The radical reacts with NAD to form an INH-NAD adduct (Figure 1), which, in turn, inhibits the FASII enoyl-ACP reductase InhA, leading to mycobacterial cell death (Rawat et al., 2003). The elucidation of the INH action mechanism has been controversial, and alternative target pathways, including DNA and lipid biosynthesis (Russe and Barclay, 1955; Ebina et al., 1961; Gangadharam et al., 1963), cell division (Barclay et al., 1953), or altered NAD metabolism (Bekierkunst, 1966), have been proposed in early reports.
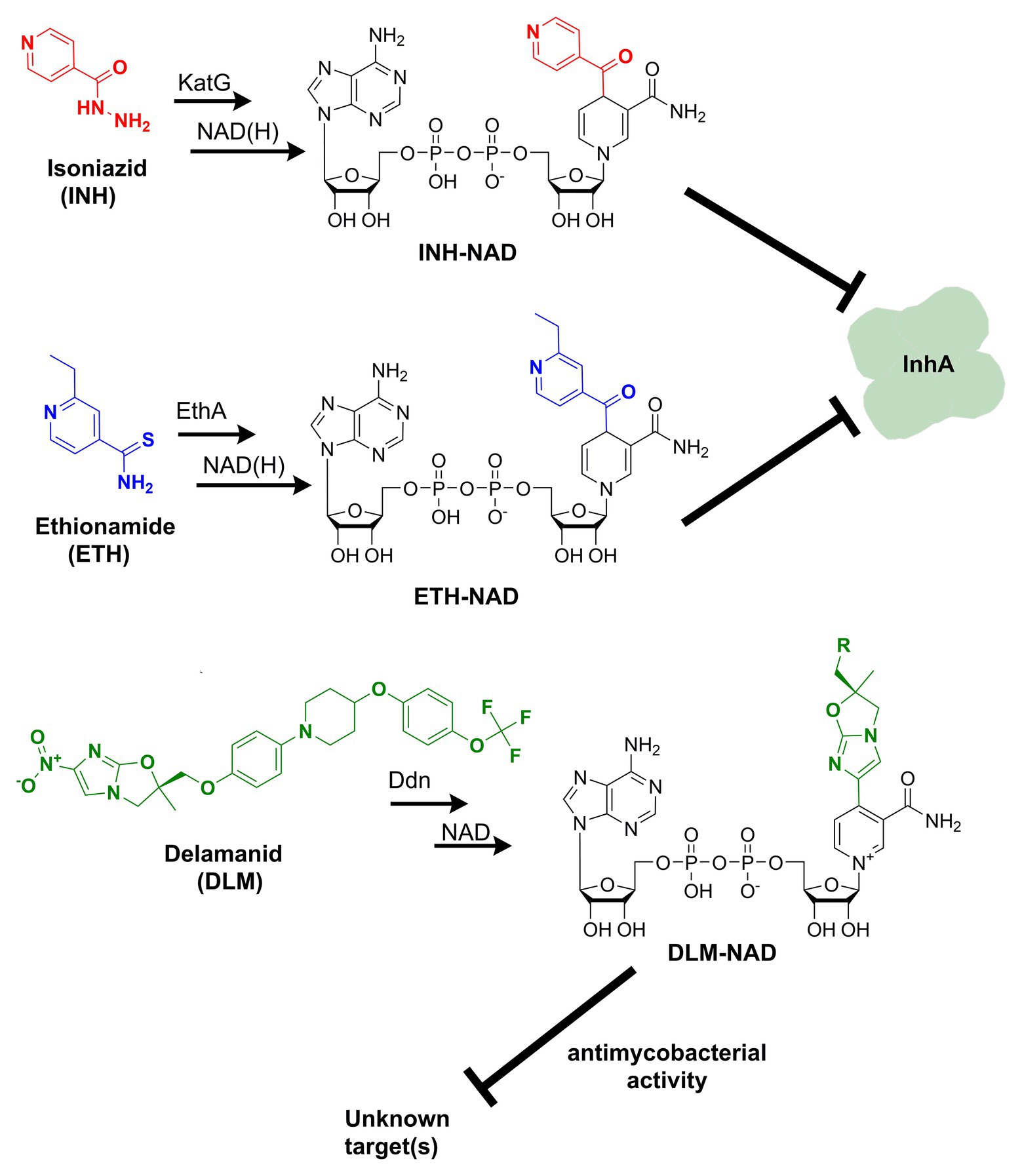
Figure 1. Formation of nicotinamide adenine dinucleotide (NAD) chemical adducts by tuberculosis (TB) drugs. Prothionamide, a close chemical analog of ethionamide, undergoes the same transformation. Pretomanid, belonging to the same class of nitroimidazoles as delamanid, may combine with NAD as well. Their structures have been omitted for clarity.
The emergence of NAD enzyme cofactor biosynthesis as a promising antimycobacterial target pathway warrants a reevaluation of the metabolic interplay between INH and NAD. The perturbation of NAD metabolism by isoniazid was an early finding related to isoniazid action and resistance. In 1966, Bekierkunst reported that in M. tuberculosis H37Rv, a decrease in NAD content could be observed after only 4 h of exposure to isoniazid, rising to 50% after 6 h (Bekierkunst, 1966). Ten years later, Jackett et al. (1977) established that the NAD content declined with increasing drug concentration and increased exposure. These results, though, did not support a direct causal link between NAD depletion and INH’s antibacterial activity, mainly because the NAD drop was marginal at the low bactericidal concentration of INH.
Despite the fact that NAD pool perturbation appears to be an incontrovertible secondary effect of INH action, this link has not been thoroughly elucidated. Several conjectures have been advanced for explaining the mechanism of NAD drop in the presence of INH. These hypotheses have been widely questioned or disproved. For example, it was suggested that NAD loss was as a result of leaks in the cell wall and membrane due to the inhibition of mycolate synthesis, i.e., a post-antibiotic effect (Winder et al., 1971); however, NAD level restored in cells exposed to INH for 24 h, despite the continued inhibition of growth (Jackett et al., 1977). It was also proposed that NAD depletion by INH could be the indirect effect of a NAD glycohydrolase activation (Bekierkunst and Bricker, 1967). Yet, this hypothesis did not explain resistance to isoniazid, as the effects on NAD were equivalent in isoniazid susceptible and resistant strains of M. tuberculosis (Sriprakash and Ramakrishnan, 1970).
More clues into the relationship between NAD and INH had to wait for in depth studies of INH resistance. The primary mechanism of resistance to INH results from mutations that either depress its activator (KatG) activity or overexpress/alter InhA, the target of INH-NAD adduct (Vilcheze and Jacobs, 2007). KatG mutants exhibit decreased or complete loss of catalase and peroxidase activity. Because these enzymatic activities are essential for M. tuberculosis defense against reactive oxygen species (ROS) and virulence in vivo (Pym et al., 2002; Ng et al., 2004), polymorphism in katG may result in a loss of fitness to the bacterium. On the other hand, among a set of overexpressed genes during NAD starvation, KatG came up with the highest upregulation (nearly 7-fold; Vilcheze et al., 2010), pointing to NAD depletion as a trigger to ROS production. Hence, INH and prospective NAD inhibitors may potentiate their effects and reduce the emergence of KatG-mediated resistance mechanisms. In support of this assumption, antagonism was reported between isoniazid and nicotinamide (Jordahl et al., 1961), the primary precursor of mycobacterial NAD salvage pathway.
An alternative mechanism of INH resistance in Mtb is based on defects in the ndh gene (Rv1854c; Lee et al., 2001), which encodes a type II NADH dehydrogenase. This oxidase transfers electrons from NADH to the respiratory chain without proton translocation. The ndh-mediated mechanism of resistance was first described in Mycobacterium smegmatis and M. bovis (Miesel et al., 1998; Vilcheze et al., 2005). As expected, the activity of NdhII mutants was compromised, yielding an increased NADH cellular content and NADH/NAD ratio than wild-type (Vilcheze et al., 2005). In the same study, the authors demonstrated that the accumulation of NADH, the native substrate of InhA, acted as a competitive inhibitor for binding of the INH-NAD adduct to InhA. Despite an impaired NADH to NAD oxidation, the increased NADH/NAD ratio was only achieved by NADH elevation, while NAD remained relatively constant. As a result, the total amount of NAD(H) pyridine nucleotides in ndh mutants grew by 10–50% (Vilcheze et al., 2005). This implies that (i) NAD cofactor homeostasis is tightly regulated in tubercle bacilli and (ii) an increased NAD biosynthesis is necessary to avoid a dramatic NAD drop in ndh mutants.
Consistently, a gene expression analysis of M. tuberculosis exposed to INH showed that nadC (Rv1596) and pntAb (Rv0156) genes are significantly upregulated (Waddell et al., 2004). The gene nadC codes for quinolinate phosphoribosyltransferase (QAPRT), the final enzyme in the de novo branch of NAD biosynthesis (Waddell et al., 2004; Figure 2), while pntAB specifies a component (subunit alpha) of a proton-translocating NAD(P) transhydrogenase transferring reducing equivalents from NADPH to NAD. Supposedly, to counteract INH’s bactericidal action, the two enzymes implement a metabolic response whereby the transhydrogenase elevates the NADH content while NadC replenishes the oxidized NAD cofactor. In this perspective, the blocking of NAD biosynthesis with specific inhibitors could represent not only a novel antimycobacterial strategy but an opportunity to synergize with isoniazid, making the drug more lethal and overcoming some forms of resistance as in ndh mutants.
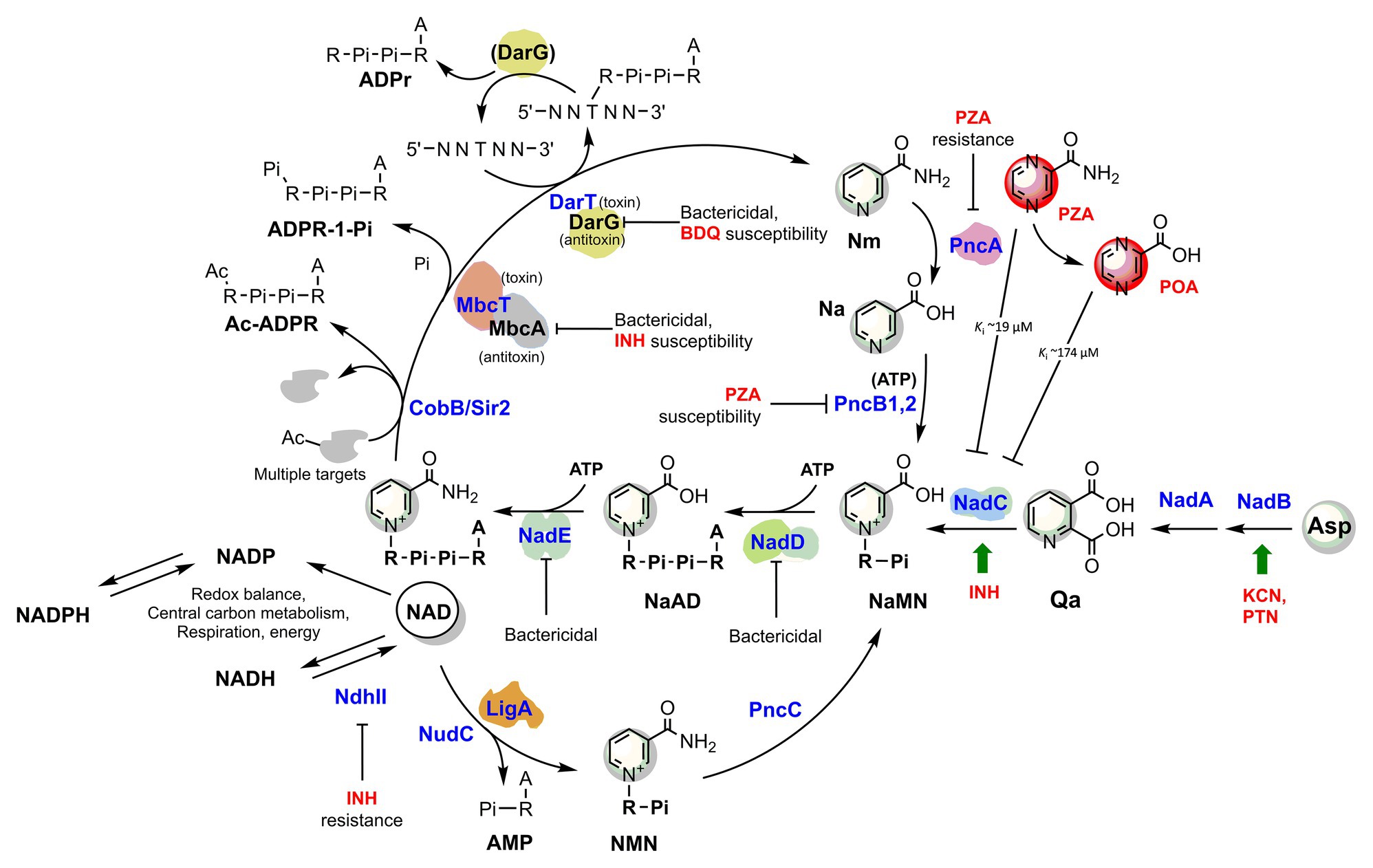
Figure 2. The metabolic interplay between TB drugs and NAD pathways in Mycobacterium tuberculosis. Enzymes (blue) are identified with their gene name, metabolites’ names are in black, and TB drugs or inhibitors are in red. The effects of targeted enzymes’ inhibition/downregulation on bacterial growth or drug efficacy are shown. Green arrows indicate upregulated enzymes upon drug exposure, according to transcriptional analyses. Proteins with available 3D structures are sketched based on their biological quaternary assembly. Enzyme abbreviations: NadB (Rv1595): ASPOX, L-aspartate oxidase; NadA (Rv1594): QSYN, quinolinate synthetase; NadC (Rv1596): QaPRT, quinolinate phosphorybosyltransferase; PncB1 (Rv1330c): NaPRT1, nicotinate phosphoribosyltransferase; PncB2 (Rv0573c): NaPRT1, nicotinate phosphoribosyltransferase; NadD (Rv2421c): NaMNAT, NaMN adenylyltransferase; NadE (Rv2438c): NADS, glutamine-dependent NAD syntetase; PncA (Rv2043c): NMASE, nicotinamide deamidase; PncC (Rv1901): NMND, NMN deamidase; LigA (Rv3014c): NAD-dependent DNA ligase; NudC (Rv3199c): NAD(H) pyrophosphatase; NdhII (Rv1854c); type II NADH:menaquinone oxidoreductase; Sir2/CobB (Rv1151c): NAD-dependent deacetyltransferase. Metabolite abbreviations: NaAD, nicotinic acid adenine dinucleotide; Nam, nicotinamide; Na, nicotinic acid; Qa, quinolinic acid; NaMN, nicotinic acid mononucleotide; NMN, nicotinamide mononucleotide; PRPP, phosphoribosyl diphosphate; PZA, pyrazinamide; POA, pyrazinoic acid; BDQ, bedaquiline; INH, isoniazid; PTN, pretomanid; KCN, potassium cyanide.
It could be argued that NAD depletion would also impair INH activity as less NAD would be available to form the INH-NAD adduct, thus counteracting this effect. Reasonably, this is not the case as INH-NAD is a tight-binding subnanomolar inhibitor of InhA (Ki = 0.75 nM; Rawat et al., 2003). We have demonstrated that a drop of NAD(H) content of only 3-fold becomes bactericidal for Mtb (Rodionova et al., 2014). Thus, given that typical bacterial NAD(H) pool concentration is around 1 mM, the residual NAD would be large enough to drive the INH-NAD adduct formation.
Recently, Freire et al. (2019) demonstrated that the toxin component MbcT of the Toxin-antitoxin MbcTA system is a novel phosphorylase that degrades NAD (Figure 2) and, in the absence of the McbA antidote, reduces mycobacterial survival in macrophages while extending the survival of infected mice. Remarkably, the study revealed a synergistic effect of toxin activity with isoniazid drug in a mouse model of Mtb infection, providing an additional proof-of-concept for INH’s foreseen synergy with NAD synthesis blockers (Figure 2).
Ethionamide/Prothionamide and Delamanid/Pretomanid: Other NAD Adduct-Forming Prodrugs
Ethionamide (ETH) and its close analog prothionamide (PRO) are interchangeable components of second-line drug regimens used for the treatment of drug-resistant TB. Like its structural analog isoniazid, ETH is a prodrug targeting InhA. However, ETH is activated by a different enzyme, the flavin-dependent monooxygenase EthA (Vannelli et al., 2002). The activated ETH reacts with NAD to yield an ETH-NAD adduct (Figure 1), which subsequently inhibits InhA (Wang et al., 2007). Due to the commonalities in the mechanisms of action and cross-resistance of ETH or PRO and isoniazid, similar conclusions regarding the expected interference of ETH or PRO with NAD metabolism can be drawn.
The multi-drug-resistant TB drug delamanid (DLM) is a recently approved drug that also blocks the synthesis of mycolic acids, thereby destabilizing its cell wall (Matsumoto et al., 2006). DLM undergoes activation by the deazaflavin (F420)-dependent nitroreductase (ddn) to yield a reactive intermediate. This activated metabolite is considered to play a crucial role in the drug’s bactericidal effect but whether it represents the final toxic derivative and the exact identification of its molecular target are still unknown (Liu et al., 2018). Strikingly, it has been recently established that after activation, DLM can combine with NAD to form a DLM-NAD adduct (Figure 1), which plays an essential role in the antimycobacterial action of DLM (Hayashi et al., 2020). Hence, delamanid is another TB drug that interferes with the pathogen’s NAD homeostasis and that can potentially synergize with the NAD biosynthesis blockade. Similar conjectures can be drawn for the drug pretomanid (PTN), belonging to the same class of nitroimidazoles, but no experimental evidence that pretomanid can combine with NAD is currently available in the literature.
Pyrazinamide and NAD Metabolism
Pyrazinamide (PZA) drug partners with isoniazid, rifampicin, and ethambutol in the present-day “short-course” TB drug regimen (Mitchison, 1985). Since the discovery of the in vivo sterilizing activity in the animal models and humans (Solotorovsky et al., 1952; Yeager et al., 1952), PZA action mechanism largely remained an enigma. Several modes of action have been proposed in the following decades, though mounting evidence opposing such models was promptly put forward (for in-depth reviews, see Anthony et al., 2018; Lamont et al., 2020). These studies identified a diverse range of potential targets, including fatty acid synthesis (Zimhony et al., 2000), membrane energetics and integrity (Wade and Zhang, 2006), protein translation (Shi et al., 2011), and pantothenate biosynthesis (Zhang et al., 2013a; Dillon et al., 2014). More recently, aspartate decarboxylase PanD, required for Coenzyme A (CoA) biosynthesis, emerged as a convincing target of pyrazinoic acid (PAO), the bioactive form of PZA (Gopal et al., 2019). Remarkably, instead of inhibiting its target protein’s function, as most antibacterials do, PAO functions as a target degrader (Gopal et al., 2020).
Hereafter, we will focus on the link between pyrazinamide and NAD metabolism, with the intent of uncovering foreseeable synergies between PZA and NAD pathway inhibitors. PZA is a prodrug hydrolyzed to the bioactive POA in the mycobacterial cytoplasm by the M. tuberculosis pyrazinamidase/nicotinamidase (PZAse), encoded by pncA (Rv2043c; Scorpio and Zhang, 1996). This amidase hydrolyzes nicotinamide vitamin into nicotinate, subsequently converted by PncB1-2 to nicotinate mononucleotide (NaMN), a shared product with NadABC-mediated de novo biosynthesis (Figure 2). Due to the redundancy of these pathways, PncA is nonessential for survival and virulence of M. tuberculosis (Boshoff et al., 2008; Vilcheze et al., 2010), and loss-of-function mutations represent the predominant mechanism for PZA resistance in clinical isolates (with up to 99.9% frequency; Zhang and Mitchison, 2003). The virulence and fitness of PZA-resistant strains with pncA mutations do not seem to be altered (Cheng et al., 2000). However, PncA resistant mutants may be more vulnerable to antibacterials targeting NAD metabolism, and such molecules could help overcome pncA-mediated resistance. Indeed, pncA-mutants would not efficiently salvage the Nm precursor, which is particularly abundant in the human host, nor recycle it out of NAD-consuming reactions (Figure 2). Mycobacteria encode Nm-releasing enzymes using NAD as a substrate that play vital roles in the dynamic regulation of metabolic functions (Figure 2). For example, in M. tuberculosis, the NAD-dependent Sir2-like family protein CobB (Rv1151c) influences the DNA architecture by deacetylating nucleoid-associated protein HU, a protein essential for growth (Anand et al., 2017; Green et al., 2018). Similarly to other Sir2-family proteins, Rv1151c is inhibited by Nm (Gu et al., 2009), which can accumulate in PZAse-defective Mtb strains. As also discussed in the next paragraph, the Mtb DarTG toxin-antitoxin system consumes NAD to mediate the reversible DNA ADP-ribosylation, a process whose biological significance is still poorly understood (Zaveri et al., 2020). If activated intracellularly, two other toxins, like the abovementioned MbcT and the TNT necrotizing toxin (Sun et al., 2015), may challenge intracellular Mtb NAD homeostasis. An uncharacterized NAD glycohydrolase activity, along with its heat-labile inhibitor, have been identified in Mtb crude extracts in the 1960s (Gopinathan et al., 1964, 1966), which likely correspond to the TNT toxin and its inhibitor IFT (Sun et al., 2015).
Thus, even though Nm salvage or recycling does not significantly affect tubercle bacillus survival or pathogenicity, its absence may impair the bacterial capacity to maintain NAD homeostasis under specific stress conditions that yield a sudden drop in NAD content. Such stress conditions may also be induced by PZA drug itself (Figure 2), which has been reported being an inhibitor of de novo NAD biosynthesis enzyme quinolinate phosphoribosyltransferase (QAPRT; Sharma et al., 1998; Kim et al., 2014). In keeping with this proposition, mutations in NAD pathways and energy production cause increased PZA susceptibility. Of note, mutants in pncB1, involved in NAD synthesis, are 5-fold more susceptible to PZA (Zhang et al., 2013b). Other mutants exhibiting higher PZA susceptibility were defective in energy production and include mutations in NADH dehydrogenase subunits H and N (nuoH and nuoN), nitrate reductase narH, and formate dehydrogenase fdhF (Zhang et al., 2013b). Given these premises, it seems worthwhile to test whether antimycobacterials that target NAD biosynthesis potentiate the effect of PZA and improve the treatment of TB.
The Metabolic Interplay Between NAD and ATP Metabolism: Bright Prospects for Respiratory and NAD Synthesis Inhibitors
It has been assessed that nearly 17% of the central metabolism enzymatic reactions that are essential for the survival of M. tuberculosis require the NAD(H) cofactor and its phosphorylated derivative NADP(H) (Beste et al., 2007). Among these are the key pathways required to produce ATP as NAD(H) is the primary entry electron donor in the respiratory chain and the oxidant driving the glycolysis. Thus, a NAD(H) pool decrease induces a glycolytic slowdown and rapid shutdown of the electron transfer chain, ultimately compromising ATP synthesis from both sources.
In turn, NAD(P) biosynthesis is a highly ATP-dependent pathway (Figure 2). Each of the three universal enzymatic steps converting NaMN precursor to NADP requires an ATP molecule (Sorci et al., 2010b). Moreover, PncB-driven NaMN synthesis from Na and PRPP is strongly activated by the PncB ATPase activity (Vinitsky and Grubmeyer, 1993), and additional ATP molecules are required for recycling the ADP-ribose moiety into the PRPP precursor. The biogenesis and homeostasis of the NAD pool and ATP are undoubtedly interlinked aspects of energy metabolism, and their simultaneous targeting in innovative drug combinations may hold the promise for potential synergy and overcome resistance mechanisms. In line with this concept, Vilcheze et al. (2010) showed that both M. tuberculosis and M. bovis de novo NAD synthesis mutants starved for nicotinamide, i.e., in a metabolic status mimicking NAD synthesis inhibition, strongly upregulate ATP synthetase subunits b, d, and c, the latter being targeted by the recently approved bedaquiline drug (Andries et al., 2005). This implies that electron transport is impaired by limiting NAD and the ATP synthetase upregulation represents an attempt to restore electron transport and oxidative phosphorylation efficiency. Consistently, the transcriptional profiles from M. tuberculosis during exposure with NadE inhibitors resembled those induced by respiratory inhibitors (Boshoff et al., 2008). Conversely, respiratory poisons such as potassium cyanide and pretomanid positively affected the transcription of Mtb nadB (Manjunatha et al., 2009), the first and rate-limiting gene of de novo NAD biosynthesis, regulated by classic feedback inhibition of NAD (Seifert et al., 1990; Tedeschi et al., 1999).
Although they require additional supporting evidence, these observations based on transcriptional profiling emphasize the strict interdependence between ATP and NAD biogenesis and homeostasis in Mtb. Thus, we propose that innovative antimycobacterial strategies disrupting energy metabolism should target both of these two pathways for the best outcome. The latest developments on inhibitors interfering with energy-metabolism in M. tuberculosis are covered in comprehensive reviews (Thompson and Denny, 2019; Appetecchia et al., 2020). Not surprisingly, partial depletion of Mtb DarG (Rv0060), the hydrolase antitoxin reversing DarT-catalyzed DNA ADP-ribosylation (Jankevicius et al., 2016), sensitizes the mycobacterial cell to drugs targeting respiration (i.e., bedaquiline) and DNA metabolism (Zaveri et al., 2020). DarT (Rv0059) consumes NAD as an ADP-ribose donor and interacts with other proteins involved in DNA replication and repair, whereby additional NAD is drained by the activity of the NAD-dependent DNA ligase LigA (Srivastava et al., 2005). Although the Mtb DarTG toxin-antitoxin system’s exact function remains unsolved, the essentiality of DarG and its direct involvement with DNA-repair proteins argues in favor of a significant role in preserving the genome integrity under stress conditions (Ramage et al., 2009). As an obligate human intracellular pathogen, M. tuberculosis has evolved complex defenses to endure the stresses experienced during persistent infection. As exemplified by the DarTG Toxin-Antitoxin system, NAD consumption is required to fuel the Mtb resilience under stress conditions, thus reaffirming bacterial NAD replenishment as an attractive target pathway.
Concluding Remarks and Perspectives
In 1945, nicotinamide was serendipitously discovered to have antituberculosis properties (Chlorine, 1945). This prompted the testing of other pyridine derivatives for their antimycobacterial effects and paved the way for discovering isoniazid and pyrazinamide front-line drugs. Since these agents were much more effective than nicotinamide, further efforts were focused on uncovering their mechanism of action, obscuring the interest in nicotinamide. However, the elucidation of how the vitamin nicotinamide exerts its direct antituberculosis action, which must differ from its renowned derivatives, is of paramount importance as it may disclose novel vulnerable targets tied to the mycobacterial NAD metabolism. For example, Mtb CobB is a NAD-dependent deacetylase member of the SIR2 family of proteins that are notoriously inhibited by nicotinamide (Hu et al., 2014). Of relevance, nicotinamide inhibits PfSir2 activity and intra-erythrocytic growth of Plasmodium falciparum (Prusty et al., 2008). An in vitro antileishmanial activity is also reported for nicotinamide, partially mediated by direct inhibition of the Leishmania Sir2 homolog (Sereno et al., 2005).
Despite the presence of two PncB isozymes, it is well established that Mtb accumulates Na, and its excess is excreted into the culture media, thus allowing it to be detected using the niacin test. Curiously, this metabolic feature may have evolved to respond to high nicotinamide’s toxicity rather than resulting from an intrinsic inability to process niacin.
The recent discoveries of DarTG, MbcTA, and TNT-IFT toxin-antitoxin systems uncovered additional NAD-consuming enzymes lethal to mycobacteria in the absence of functional antitoxins. Indeed, small inhibitors aiming at disrupting such complexes or inactivating the essential “antidotes” are promising antimycobacterial strategies (Williams and Hergenrother, 2012) that will surely mark future TB research. On the other hand, host NAD is a crucial molecule for TB immunity (Singhal and Cheng, 2018). Recent studies have shown that Mtb is capable of counteracting host defenses by subverting immune cells NAD metabolism. In detail, this could be achieved by modulation of human NAD-consuming proteins such as Sirtuins (Cheng et al., 2017; Bhaskar et al., 2020) or by direct depletion of host NAD levels through the TNT toxin (Pajuelo et al., 2020). The emerging roles of NAD and Sirtuin biology in host immune cells could be exploited to develop adjunct host-directed therapies against TB.
This review outlined the metabolic interplay between multiple TB drugs and the NAD cofactor, a crucial molecule for the Mtb growth during acute infections and persistence in a dormant state (Boshoff et al., 2008; Vilcheze et al., 2010; Kim et al., 2013; Rodionova et al., 2014, 2015). The perturbation of NAD homeostasis by these TB drugs is not, alone, bactericidal. However, it represents collateral damage that could be exploited to synergize with prospective drugs that specifically deplete NAD. Indeed, NAD biosynthesis is an established antiinfective target pathway not only in mycobacteria, but it has also been validated in other bacterial pathogens (Sorci et al., 2009, 2010a, 2013; Huang et al., 2010; Orsomando et al., 2016), the protozoan Plasmodium (O’Hara et al., 2014) and Leishmania (Mandal et al., 2019), and the flatworm Schistosoma parasite (Schultz et al., 2020). In mycobacteria, a detailed analysis of the NAD salvage vs. de novo-synthesis pathway concluded that an inhibitor must target either NadD or NadE, which sequentially catalyze the last two shared steps of NAD synthesis (Boshoff et al., 2008). By combining a target-directed and phenotypic screen, our group successfully developed antimycobacterial benzimidazolium compounds targeting MtNadD (Osterman et al., 2019), while other teams optimized biaryl tethered dimers or urea-sulfonamide analogs directed at MtNadE and inhibiting Mtb cell growth in the medium-micromolar range (Boshoff et al., 2008; Wang et al., 2017). A limitation of these studies is that the inhibitors were initially identified for orthologous enzymes from other bacteria (Rodionova et al., 2015; Wang et al., 2017). Thus, computationally-driven, large-scale high-throughput screening on MtNadD and MtNadE may be necessary for identifying more potent mycobacterial NAD synthesis inhibitors that can advance to a drug status.
Author Contributions
LS designed this study and wrote the manuscript. KR critically reviewed the manuscript and contributed to its final writing. Both the authors contributed to the article and approved the submitted version.
Funding
This study was funded by the intramural research grant RSA2018-19 from Polytechnic University of Marche to LS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We want to thank all my lab members for stimulating discussion and Andrei Osterman (SBP Medical Research Institute) for sharing insightful comments.
References
Anand, C., Garg, R., Ghosh, S., and Nagaraja, V. (2017). A Sir2 family protein Rv1151c deacetylates HU to alter its DNA binding mode in Mycobacterium tuberculosis. Biochem. Biophys. Res. Commun. 493, 1204–1209. doi: 10.1016/j.bbrc.2017.09.087
Andries, K., Verhasselt, P., Guillemont, J., Gohlmann, H. W., Neefs, J. M., Winkler, H., et al. (2005). A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307, 223–227. doi: 10.1126/science.1106753
Anthony, R. M., den Hertog, A. L., and van Soolingen, D. (2018). ‘Happy the man, who, studying nature’s laws, thro’ known effects can trace the secret cause.’ Do we have enough pieces to solve the pyrazinamide puzzle? J. Antimicrob. Chemother. 73, 1750–1754. doi: 10.1093/jac/dky060
Appetecchia, F., Consalvi, S., Scarpecci, C., Biava, M., and Poce, G. (2020). SAR analysis of small molecules interfering with energy-metabolism in Mycobacterium tuberculosis. Pharmaceuticals 13:227. doi: 10.3390/ph13090227
Barclay, W. R., Ebert, R. H., and Kochweser, D. (1953). Mode of action of isoniazid. Am. Rev. Tuberc. 67, 490–496. doi: 10.1164/art.1953.67.4.490
Bekierkunst, A. (1966). Nicotinamide-adenine dinucleotide in tubercle bacilli exposed to isoniazid. Science 152, 525–526. doi: 10.1126/science.152.3721.525
Bekierkunst, A., and Bricker, A. (1967). Studies on the mode of action of isoniazid on mycobacteria. Arch. Biochem. Biophys. 122, 385–392. doi: 10.1016/0003-9861(67)90209-3
Beste, D. J., Hooper, T., Stewart, G., Bonde, B., Avignone-Rossa, C., Bushell, M. E., et al. (2007). GSMN-TB: a web-based genome-scale network model of Mycobacterium tuberculosis metabolism. Genome Biol. 8:R89. doi: 10.1186/gb-2007-8-5-r89
Bhaskar, A., Kumar, S., Khan, M. Z., Singh, A., Dwivedi, V. P., and Nandicoori, V. K. (2020). Host sirtuin 2 as an immunotherapeutic target against tuberculosis. eLife 9:e55415. doi: 10.7554/eLife.55415
Boshoff, H. I., Xu, X., Tahlan, K., Dowd, C. S., Pethe, K., Camacho, L. R., et al. (2008). Biosynthesis and recycling of nicotinamide cofactors in Mycobacterium tuberculosis. An essential role for NAD in nonreplicating bacilli. J. Biol. Chem. 283, 19329–19341. doi: 10.1074/jbc.M800694200
Cheng, C. Y., Gutierrez, N. M., Marzuki, M. B., Lu, X., Foreman, T. W., Paleja, B., et al. (2017). Host sirtuin 1 regulates mycobacterial immunopathogenesis and represents a therapeutic target against tuberculosis. Sci. Immunol. 2:eaaj1789. doi: 10.1126/sciimmunol.aaj1789
Cheng, S. J., Thibert, L., Sanchez, T., Heifets, L., and Zhang, Y. (2000). pncA mutations as a major mechanism of pyrazinamide resistance in Mycobacterium tuberculosis: spread of a monoresistant strain in Quebec, Canada. Antimicrob. Agents Chemother. 44, 528–532. doi: 10.1128/AAC.44.3.528-532.2000
Chlorine, V. (1945). Action de l’amide nicotinique sur les bacilles du genre Mycobacterium. C. R. Acad. Sci. 220, 150–151.
Dillon, N. A., Peterson, N. D., Rosen, B. C., and Baughn, A. D. (2014). Pantothenate and pantetheine antagonize the antitubercular activity of pyrazinamide. Antimicrob. Agents Chemother. 58, 7258–7263. doi: 10.1128/AAC.04028-14
Ebina, T., Motomiya, M., Munakata, K., and Kobuya, G. (1961). Effect of isoniazid on fatty acids in Mycobacterium. C. R. Seances Soc. Biol. Fil. 155, 1176–1178.
Freire, D. M., Gutierrez, C., Garza-Garcia, A., Grabowska, A. D., Sala, A. J., Ariyachaokun, K., et al. (2019). An NAD(+) phosphorylase toxin triggers Mycobacterium tuberculosis cell death. Mol. Cell 73, 1282.e1288–1291.e1288. doi: 10.1016/j.molcel.2019.01.028
Gangadharam, P. R., Harold, F. M., and Schaefer, W. B. (1963). Selective inhibition of nucleic acid synthesis in Mycobacterium tuberculosis by isoniazid. Nature 198, 712–714. doi: 10.1038/198712b0
Gopal, P., Gruber, G., Dartois, V., and Dick, T. (2019). Pharmacological and molecular mechanisms behind the sterilizing activity of pyrazinamide. Trends Pharmacol. Sci. 40, 930–940. doi: 10.1016/j.tips.2019.10.005
Gopal, P., Sarathy, J. P., Yee, M., Ragunathan, P., Shin, J., Bhushan, S., et al. (2020). Pyrazinamide triggers degradation of its target aspartate decarboxylase. Nat. Commun. 11:1661. doi: 10.1038/s41467-020-15516-1
Gopinathan, K. P., Ramakrishnan, T., and Vaidyanathan, C. S. (1966). Purification and properties of an inhibitor for nicotinamide-adenine dinucleotidase from Mycobacterium tuberculosis H37Rv. Arch. Biochem. Biophys. 113, 376–382. doi: 10.1016/0003-9861(66)90201-3
Gopinathan, K. P., Sirsi, M., and Vaidyanathan, C. S. (1964). Nicotinamide-adenine dinucleotide glycohydrolase of Mycobacterium tuberculosis H37Rv. Biochem. J. 91, 277–282. doi: 10.1042/bj0910277
Green, K. D., Biswas, T., Pang, A. H., Willby, M. J., Reed, M. S., Stuchlik, O., et al. (2018). Acetylation by Eis and deacetylation by Rv1151c of Mycobacterium tuberculosis HupB: biochemical and structural insight. Biochemistry 57, 781–790. doi: 10.1021/acs.biochem.7b01089
Gu, J., Deng, J. Y., Li, R., Wei, H., Zhang, Z., Zhou, Y., et al. (2009). Cloning and characterization of NAD-dependent protein deacetylase (Rv1151c) from Mycobacterium tuberculosis. Biochemistry 74, 743–748. doi: 10.1134/s0006297909070062
Hayashi, M., Nishiyama, A., Kitamoto, R., Tateishi, Y., Osada-Oka, M., Nishiuchi, Y., et al. (2020). Adduct formation of delamanid with NAD in Mycobacteria. Antimicrob. Agents Chemother. 64, e01755–e01819. doi: 10.1128/AAC.01755-19
Hu, J., Jing, H., and Lin, H. (2014). Sirtuin inhibitors as anticancer agents. Future Med. Chem. 6, 945–966. doi: 10.4155/fmc.14.44
Huang, N., Kolhatkar, R., Eyobo, Y., Sorci, L., Rodionova, I., Osterman, A. L., et al. (2010). Complexes of bacterial nicotinate mononucleotide adenylyltransferase with inhibitors: implication for structure-based drug design and improvement. J. Med. Chem. 53, 5229–5239. doi: 10.1021/jm100377f
Jackett, P. S., Aber, V. R., and Mitchison, D. A. (1977). The relationship between nicotinamide adenine dinucleotide concentration and antibacterial activity of isoniazid in Mycobacterium tuberculosis. Am. Rev. Respir. Dis. 115, 601–607. doi: 10.1164/arrd.1977.115.4.601
Jankevicius, G., Ariza, A., Ahel, M., and Ahel, I. (2016). The toxin-antitoxin system DarTG catalyzes reversible ADP-ribosylation of DNA. Mol. Cell 64, 1109–1116. doi: 10.1016/j.molcel.2016.11.014
Jordahl, C., Prez, R. D., Deuschle, K., Muschenheim, C., and McDermott, W. (1961). Ineffectiveness of nicotinamide and isoniazid in the treatment of pulmonary tuberculosis. Am. Rev. Respir. Dis. 83, 899–900. doi: 10.1164/arrd.1961.83.6.899
Kim, J. H., O’Brien, K. M., Sharma, R., Boshoff, H. I., Rehren, G., Chakraborty, S., et al. (2013). A genetic strategy to identify targets for the development of drugs that prevent bacterial persistence. Proc. Natl. Acad. Sci. U. S. A. 110, 19095–19100. doi: 10.1073/pnas.1315860110
Kim, H., Shibayama, K., Rimbara, E., and Mori, S. (2014). Biochemical characterization of quinolinic acid phosphoribosyltransferase from Mycobacterium tuberculosis H37Rv and inhibition of its activity by pyrazinamide. PLoS One 9:e100062. doi: 10.1371/journal.pone.0100062
Lamont, E. A., Dillon, N. A., and Baughn, A. D. (2020). The bewildering antitubercular action of pyrazinamide. Microbiol. Mol. Biol. Rev. 84, e00070–e00119. doi: 10.1128/MMBR.00070-19
Lee, A. S., Teo, A. S., and Wong, S. Y. (2001). Novel mutations in ndh in isoniazid-resistant Mycobacterium tuberculosis isolates. Antimicrob. Agents Chemother. 45, 2157–2159. doi: 10.1128/AAC.45.7.2157-2159.2001
Lei, B., Wei, C. J., and Tu, S. C. (2000). Action mechanism of antitubercular isoniazid. Activation by Mycobacterium tuberculosis KatG, isolation, and characterization of inha inhibitor. J. Biol. Chem. 275, 2520–2526. doi: 10.1074/jbc.275.4.2520
Liu, Y., Matsumoto, M., Ishida, H., Ohguro, K., Yoshitake, M., Gupta, R., et al. (2018). Delamanid: from discovery to its use for pulmonary multidrug-resistant tuberculosis (MDR-TB). Tuberculosis 111, 20–30. doi: 10.1016/j.tube.2018.04.008
Malherbe, S. T., Shenai, S., Ronacher, K., Loxton, A. G., Dolganov, G., Kriel, M., et al. (2016). Persisting positron emission tomography lesion activity and Mycobacterium tuberculosis mRNA after tuberculosis cure. Nat. Med. 22, 1094–1100. doi: 10.1038/nm.4177
Mandal, H., Vijayakumar, S., Yadav, S., Singh, S. K., and Das, P. (2019). Validation of NAD synthase inhibitors for inhibiting the cell viability of Leishmania donovani: in silico and in vitro approach. J. Biomol. Struct. Dyn. 37, 4481–4493. doi: 10.1080/07391102.2018.1552199
Manjunatha, U., Boshoff, H. I., and Barry, C. E. (2009). The mechanism of action of PA-824: novel insights from transcriptional profiling. Commun. Integr. Biol. 2, 215–218. doi: 10.4161/cib.2.3.7926
Matsumoto, M., Hashizume, H., Tomishige, T., Kawasaki, M., Tsubouchi, H., Sasaki, H., et al. (2006). OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 3:e466. doi: 10.1371/journal.pmed.0030466
Miesel, L., Weisbrod, T. R., Marcinkeviciene, J. A., Bittman, R., and Jacobs, W. R. Jr. (1998). NADH dehydrogenase defects confer isoniazid resistance and conditional lethality in Mycobacterium smegmatis. J. Bacteriol. 180, 2459–2467. doi: 10.1128/JB.180.9.2459-2467.1998
Mitchison, D. A. (1985). The action of antituberculosis drugs in short-course chemotherapy. Tubercle 66, 219–225. doi: 10.1016/0041-3879(85)90040-6
Ng, V. H., Cox, J. S., Sousa, A. O., MacMicking, J. D., and McKinney, J. D. (2004). Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol. Microbiol. 52, 1291–1302. doi: 10.1111/j.1365-2958.2004.04078.x
O’Hara, J. K., Kerwin, L. J., Cobbold, S. A., Tai, J., Bedell, T. A., Reider, P. J., et al. (2014). Targeting NAD+ metabolism in the human malaria parasite Plasmodium falciparum. PLoS One 9:e94061. doi: 10.1371/journal.pone.0094061
Orsomando, G., Agostinelli, S., Bramucci, M., Cappellacci, L., Damiano, S., Lupidi, G., et al. (2016). Mexican sunflower (Tithonia diversifolia, Asteraceae) volatile oil as a selective inhibitor of Staphylococcus aureus nicotinate mononucleotide adenylyltransferase (NadD). Ind. Crop. Prod. 85, 181–189. doi: 10.1016/j.indcrop.2016.03.003
Osterman, A. L., Rodionova, I., Li, X., Sergienko, E., Ma, C. T., Catanzaro, A., et al. (2019). Novel antimycobacterial compounds suppress NAD biogenesis by targeting a unique pocket of NaMN adenylyltransferase. ACS Chem. Biol. 14, 949–958. doi: 10.1021/acschembio.9b00124
Pajuelo, D., Gonzalez-Juarbe, N., and Niederweis, M. (2020). NAD hydrolysis by the tuberculosis necrotizing toxin induces lethal oxidative stress in macrophages. Cell. Microbiol. 22:e13115. doi: 10.1111/cmi.13115
Prusty, D., Mehra, P., Srivastava, S., Shivange, A. V., Gupta, A., Roy, N., et al. (2008). Nicotinamide inhibits Plasmodium falciparum Sir2 activity in vitro and parasite growth. FEMS Microbiol. Lett. 282, 266–272. doi: 10.1111/j.1574-6968.2008.01135.x
Pym, A. S., Saint-Joanis, B., and Cole, S. T. (2002). Effect of katG mutations on the virulence of Mycobacterium tuberculosis and the implication for transmission in humans. Infect. Immun. 70, 4955–4960. doi: 10.1128/IAI.70.9.4955-4960.2002
Ramage, H. R., Connolly, L. E., and Cox, J. S. (2009). Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet. 5:e1000767. doi: 10.1371/journal.pgen.1000767
Rawat, R., Whitty, A., and Tonge, P. J. (2003). The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: adduct affinity and drug resistance. Proc. Natl. Acad. Sci. U. S. A. 100, 13881–13886. doi: 10.1073/pnas.2235848100
Rodionova, I. A., Schuster, B. M., Guinn, K. M., Sorci, L., Scott, D. A., Li, X., et al. (2014). Metabolic and bactericidal effects of targeted suppression of NadD and NadE enzymes in mycobacteria. mBio 5, e00747–e00813. doi: 10.1128/mBio.00747-13
Rodionova, I. A., Zuccola, H. J., Sorci, L., Aleshin, A. E., Kazanov, M. D., Ma, C. T., et al. (2015). Mycobacterial nicotinate mononucleotide adenylyltransferase: structure, mechanism, and implications for drug discovery. J. Biol. Chem. 290, 7693–7706. doi: 10.1074/jbc.M114.628016
Russe, H. P., and Barclay, W. R. (1955). The effect of isoniazid on lipids of the tubercle bacillus. Am. Rev. Tuberc. 72, 713–717. doi: 10.1164/artpd.1955.72.6.713
Schultz, M. D., Dadali, T., Jacques, S. A., Muller-Steffner, H., Foote, J. B., Sorci, L., et al. (2020). Inhibition of the NAD salvage pathway in schistosomes impairs metabolism, reproduction, and parasite survival. PLoS Pathog. 16:e1008539. doi: 10.1371/journal.ppat.1008539
Scorpio, A., and Zhang, Y. (1996). Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat. Med. 2, 662–667. doi: 10.1038/nm0696-662
Seifert, J., Kunz, N., Flachmann, R., Laufer, A., Jany, K. D., and Gassen, H. G. (1990). Expression of the E. coli nadB gene and characterization of the gene product L-aspartate oxidase. Biol. Chem. Hoppe Seyler 371, 239–248.
Sereno, D., Alegre, A. M., Silvestre, R., Vergnes, B., and Ouaissi, A. (2005). In vitro antileishmanial activity of nicotinamide. Antimicrob. Agents Chemother. 49, 808–812. doi: 10.1128/AAC.49.2.808-812.2005
Sharma, V., Grubmeyer, C., and Sacchettini, J. C. (1998). Crystal structure of quinolinic acid phosphoribosyltransferase from Mycobacterium tuberculosis: a potential TB drug target. Structure 6, 1587–1599. doi: 10.1016/S0969-2126(98)00156-7
Shi, W., Zhang, X., Jiang, X., Yuan, H., Lee, J. S., Barry, C. E. 3rd, et al. (2011). Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 333, 1630–1632. doi: 10.1126/science.1208813
Singhal, A., and Cheng, C. Y. (2018). Host NAD+ metabolism and infections: therapeutic implications. Int. Immunol. 31, 59–67. doi: 10.1093/intimm/dxy068
Solotorovsky, M., Gregory, F. J., Ironson, E. J., Bugie, E. J., O’Neill, R. C., and Pfister, K. (1952). Pyrazinoic acid amide—an agent active against experimental murine tuberculosis. Proc. Soc. Exp. Biol. Med. 79, 563–565. doi: 10.3181/00379727-79-19447
Sorci, L., Blaby, I., De Ingeniis, J., Gerdes, S., Raffaelli, N., de Crecy Lagard, V., et al. (2010a). Genomics-driven reconstruction of acinetobacter NAD metabolism: insights for antibacterial target selection. J. Biol. Chem. 285, 39490–39499. doi: 10.1074/jbc.M110.185629
Sorci, L., Blaby, I. K., Rodionova, I. A., De Ingeniis, J., Tkachenko, S., de Crecy-Lagard, V., et al. (2013). Quinolinate salvage and insights for targeting NAD biosynthesis in group A streptococci. J. Bacteriol. 195, 726–732. doi: 10.1128/JB.02002-12
Sorci, L., Kurnasov, O., Rodionov, D. A., and Osterman, A. L. (2010b). “Genomics and enzymology of NAD biosynthesis” in Comprehensive natural products II: Chemistry and biology. eds. L. Mander and H.-W. Lui (Oxford: Elsevier), 213–257.
Sorci, L., Pan, Y., Eyobo, Y., Rodionova, I., Huang, N., Kurnasov, O., et al. (2009). Targeting NAD biosynthesis in bacterial pathogens: structure-based development of inhibitors of nicotinate mononucleotide adenylyltransferase NadD. Chem. Biol. 16, 849–861. doi: 10.1016/j.chembiol.2009.07.006
Sriprakash, K. S., and Ramakrishnan, T. (1970). Isoniazid-resistant mutants of Mycobacterium tuberculosis H37RV: uptake of isoniazid and the properties of NADase inhibitor. J. Gen. Microbiol. 60, 125–132. doi: 10.1099/00221287-60-1-125
Srivastava, S. K., Tripathi, R. P., and Ramachandran, R. (2005). NAD+-dependent DNA ligase (Rv3014c) from Mycobacterium tuberculosis. Crystal structure of the adenylation domain and identification of novel inhibitors. J. Biol. Chem. 280, 30273–30281. doi: 10.1074/jbc.M503780200
Sun, J., Siroy, A., Lokareddy, R. K., Speer, A., Doornbos, K. S., Cingolani, G., et al. (2015). The tuberculosis necrotizing toxin kills macrophages by hydrolyzing NAD. Nat. Struct. Mol. Biol. 22, 672–678. doi: 10.1038/nsmb.3064
Tedeschi, G., Negri, A., Ceciliani, F., Mattevi, A., and Ronchi, S. (1999). Structural characterization of l-aspartate oxidase and identification of an interdomain loop by limited proteolysis. Eur. J. Biochem. 260, 896–903. doi: 10.1046/j.1432-1327.1999.00234.x
Thompson, A. M., and Denny, W. A. (2019). “Inhibitors of enzymes in the electron transport chain of Mycobacterium tuberculosis” in Annual reports in medicinal chemistry. ed. K. Chibale (Cambridge, MA, USA: Elsevier), 97–130.
Udwadia, Z. F., Amale, R. A., Ajbani, K. K., and Rodrigues, C. (2012). Totally drug-resistant tuberculosis in India. Clin. Infect. Dis. 54, 579–581. doi: 10.1093/cid/cir889
Vannelli, T. A., Dykman, A., and Ortiz de Montellano, P. R. (2002). The antituberculosis drug ethionamide is activated by a flavoprotein monooxygenase. J. Biol. Chem. 277, 12824–12829. doi: 10.1074/jbc.M110751200
Vilcheze, C., and Jacobs, W. R. Jr. (2007). The mechanism of isoniazid killing: clarity through the scope of genetics. Annu. Rev. Microbiol. 61, 35–50. doi: 10.1146/annurev.micro.61.111606.122346
Vilcheze, C., Weinrick, B., Wong, K. W., Chen, B., and Jacobs, W. R. Jr. (2010). NAD+ auxotrophy is bacteriocidal for the tubercle bacilli. Mol. Microbiol. 76, 365–377. doi: 10.1111/j.1365-2958.2010.07099.x
Vilcheze, C., Weisbrod, T. R., Chen, B., Kremer, L., Hazbon, M. H., Wang, F., et al. (2005). Altered NADH/NAD+ ratio mediates coresistance to isoniazid and ethionamide in mycobacteria. Antimicrob. Agents Chemother. 49, 708–720. doi: 10.1128/AAC.49.2.708-720.2005
Vinitsky, A., and Grubmeyer, C. (1993). A new paradigm for biochemical energy coupling. Salmonella typhimurium nicotinate phosphoribosyltransferase. J. Biol. Chem. 268, 26004–26010.
Waddell, S. J., Stabler, R. A., Laing, K., Kremer, L., Reynolds, R. C., and Besra, G. S. (2004). The use of microarray analysis to determine the gene expression profiles of Mycobacterium tuberculosis in response to anti-bacterial compounds. Tuberculosis 84, 263–274. doi: 10.1016/j.tube.2003.12.005
Wade, M. M., and Zhang, Y. (2006). Effects of weak acids, UV and proton motive force inhibitors on pyrazinamide activity against Mycobacterium tuberculosis in vitro. J. Antimicrob. Chemother. 58, 936–941. doi: 10.1093/jac/dkl358
Wang, X., Ahn, Y. M., Lentscher, A. G., Lister, J. S., Brothers, R. C., Kneen, M. M., et al. (2017). Design, synthesis, and evaluation of substituted nicotinamide adenine dinucleotide (NAD(+)) synthetase inhibitors as potential antitubercular agents. Bioorg. Med. Chem. Lett. 27, 4426–4430. doi: 10.1016/j.bmcl.2017.08.012
Wang, F., Langley, R., Gulten, G., Dover, L. G., Besra, G. S., Jacobs, W. R. Jr., et al. (2007). Mechanism of thioamide drug action against tuberculosis and leprosy. J. Exp. Med. 204, 73–78. doi: 10.1084/jem.20062100
Williams, J. J., and Hergenrother, P. J. (2012). Artificial activation of toxin–antitoxin systems as an antibacterial strategy. Trends Microbiol. 20, 291–298. doi: 10.1016/j.tim.2012.02.005
Winder, F. G. A., Collins, P., and Rooney, S. A. (1970). Effects of isoniazid on mycolic acid synthesis in Mycobacterium tuberculosis and on its cell envelope. Biochem. J. 117:27P. doi: 10.1042/bj1170027Pa
Winder, F. G., Collins, P. B., and Whelan, D. (1971). Effects of ethionamide and isoxyl on mycolic acid synthesis in Mycobacterium tuberculosis BCG. J. Gen. Microbiol. 66, 379–380. doi: 10.1099/00221287-66-3-379
Yeager, R. L., Munroe, W. G., and Dessau, F. I. (1952). Pyrazinamide (aldinamide*) in the treatment of pulmonary tuberculosis. Trans. Annu. Meet. Nat. Tuberc. Assoc. 48, 178–201.
Zaveri, A., Wang, R., Botella, L., Sharma, R., Zhu, L., Wallach, J. B., et al. (2020). Depletion of the DarG antitoxin in Mycobacterium tuberculosis triggers the DNA-damage response and leads to cell death. Mol. Microbiol. 114, 641–652. doi: 10.1111/mmi.14571
Zhang, S., Chen, J., Shi, W., Liu, W., Zhang, W., and Zhang, Y. (2013a). Mutations in panD encoding aspartate decarboxylase are associated with pyrazinamide resistance in Mycobacterium tuberculosis. Emerg. Microbes Infect. 2:e34. doi: 10.1038/emi.2013.38
Zhang, Y., and Mitchison, D. (2003). The curious characteristics of pyrazinamide: a review. Int. J. Tuberc. Lung Dis. 7, 6–21.
Zhang, Y., Shi, W., Zhang, W., and Mitchison, D. (2013b). Mechanisms of pyrazinamide action and resistance. Microbiol. Spectr. 2, 1–12. doi: 10.1128/microbiolspec.MGM2-0023-2013
Zhang, Y., and Young, D. B. (1993). Molecular mechanisms of isoniazid: a drug at the front line of tuberculosis control. Trends Microbiol. 1, 109–113. doi: 10.1016/0966-842X(93)90117-A
Keywords: isoniazid, pyrazinamide, nicotinamide, ethionamide, delamanid, tuberculosis, toxin-antitoxin system, NAD biosynthesis inhibition
Citation: Rohde KH and Sorci L (2021) The Prospective Synergy of Antitubercular Drugs With NAD Biosynthesis Inhibitors. Front. Microbiol. 11:634640. doi: 10.3389/fmicb.2020.634640
Edited by:
Maria Rosalia Pasca, University of Pavia, ItalyReviewed by:
Satoshi Mitarai, Research Institute of Tuberculosis, Japan Anti-tuberculosis Association, JapanJun Yoshino, Washington University School of Medicine in St. Louis, United States
Copyright © 2021 Rohde and Sorci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leonardo Sorci, bC5zb3JjaUBzdGFmZi51bml2cG0uaXQ=