Motley Crew: Overview of the Currently Available Phage Diversity
 Nikita Zrelovs
Nikita Zrelovs Andris Dislers
Andris Dislers Andris Kazaks
Andris KazaksA Corrigendum on
Motley Crew: Overview of the Currently Available Phage Diversity
by Zrelovs, N., Dislers, A., and Kazaks, A. (2020). Front. Microbiol. 11:579452. doi: 10.3389/fmicb.2020.579452
In the original article, there were some errors.
Several corrections have been made in the “Situation as of today” section,
Second paragraph, second sentence:
Currently, ssDNA bacteriophages (families Microviridae, Inoviridae, Plectroviridae, Finnlakeviridae) are represented with 92 putative species complete genomes (~1.12% of the total genome count), dsDNA phages (Ackermannviridae, Autographiviridae, Chaseviridae, Demerecviridae, Drexlerviridae, Herelleviridae, Myoviridae, Podoviridae, Siphoviridae, Sphaerolipoviridae, Corticoviridae, Tectiviridae, Plasmaviridae) – 7736 genomes (~93.83%), dsRNA phages (Cystoviridae) – 7 genomes (~0.08%), and ssRNA phages (Leviviridae) – 23 genomes (~0.28%), while the putative phage species unclassified at the family level are represented by 387 genomes corresponding to approximately 4.69% of the total genome number (Table 1).
Fifth paragraph, second sentence:
Myo- (long contractile tail) and podo- (short non-contractile tail) phages, on the other hand, are currently found in Myoviridae and Podoviridae, and also in Ackermannviridae, Herelleviridae, Chaseviridae (myophages), and Autographiviridae (podophages) phage families.
Several corrections have been made in the “Discussion” section:
First paragraph, fifth sentence:
Genome physical termini predictions (either by prediction tools or manual inspection of reads mapping onto the putative genome) require a large amount of individual phage reads and may present ambiguous results otherwise (Garneau et al., 2017).
First paragraph, eighth and ninth sentences:
Many of the submissions do not have any manuscript linked to it where the methodology would be stated in-detail, and the submission-associated metadata (that are sometimes very scarce) along with the functional annotation are not always enough to evaluate the plausibility of the “complete genome.” This is raising additional concern for metagenomics acquired phage “complete genomes,” evaluation of which should be handled with particular care, possibly including a brief evidence statement on why the submission authors are confident about the “completeness” of the entry in the sequence metadata (e.g., the “circularity” of the assembly).
Fourth paragraph, second sentence:
Taking the rate of complete phage genome submissions to public sequence repositories into account and addressing concerns about the future usability of entries in such repositories, we, sadly, have to stress the importance of taking the submission process seriously.
Fifth paragraph, first sentence:
First of all, there have been examples of typing errors in the metadata (e.g., “Eschericha” or “Panteoa” instead of the correct Escherichia and Pantoea, “Vibro” instead of Vibrio, and Mycobacterium misspelled in multiple ways).
Sixth paragraph, first sentence:
Secondly, if the sequence of a phage (bacterial virus) is being submitted, submission authors should try to avoid the ambiguous usage of the sequence-related metadata qualifiers (e.g., “/host =” qualifier used for organisms other than bacteria); bacteriophages, being viruses of bacteria, infect and replicate within bacteria, which serve as a natural host.
Table 1, column “Family”:
Replace “Drexlerviridae” for “Drexleviridae”. The corrected Table 1 appears below.
TABLE 1
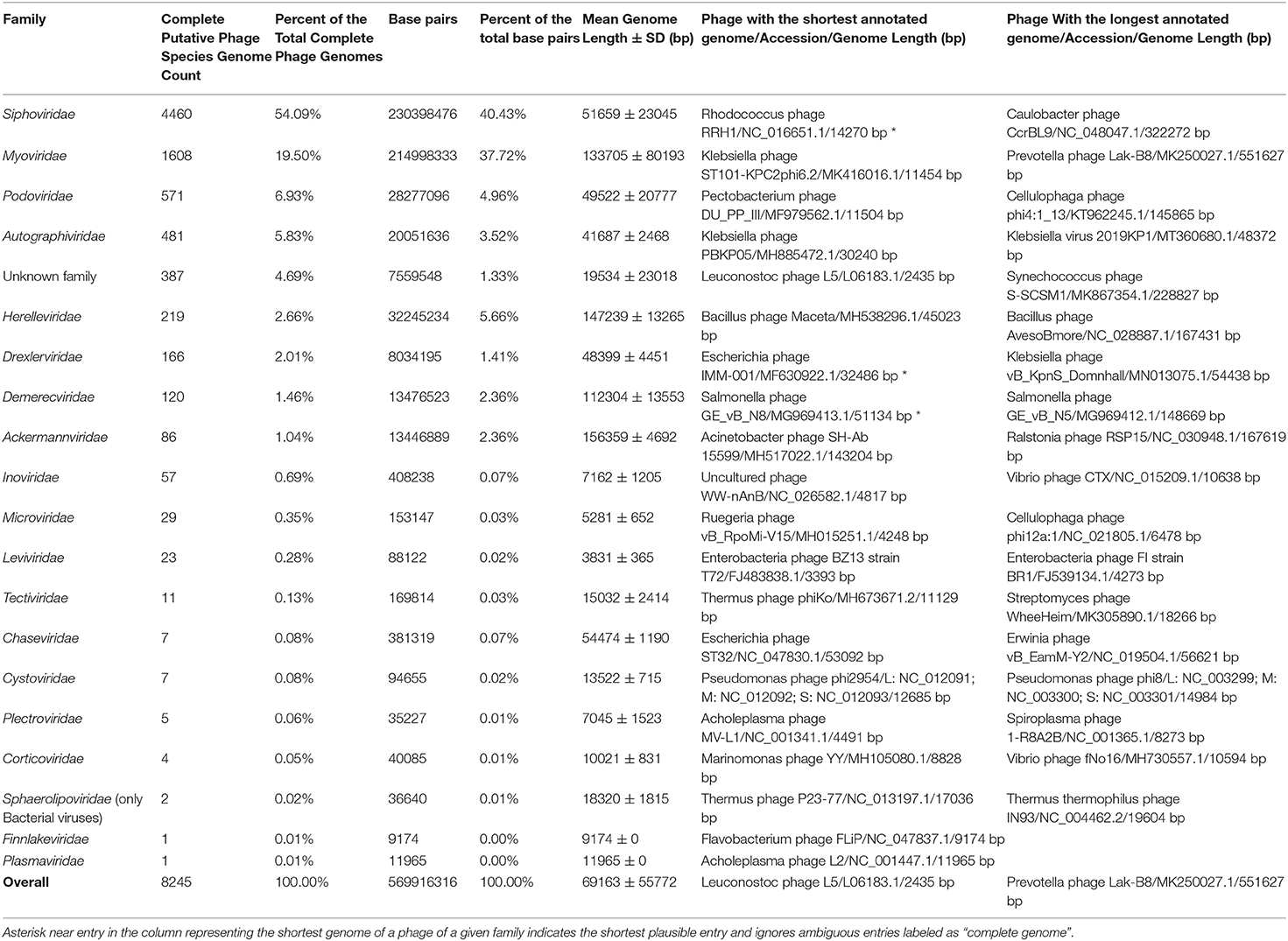
Table 1. Overview of completely sequenced phage genomes.
The authors apologize for all the aforementioned typographic errors and state that they did not change the scientific conclusions of the article in any way.
References
Keywords: complete genome, phage diversity, bioinformatics, genome overview, bacteriophage
Citation: Zrelovs N, Dislers A and Kazaks A (2021) Corrigendum: Motley Crew: Overview of the Currently Available Phage Diversity. Front. Microbiol. 11:626744. doi: 10.3389/fmicb.2020.626744
Received: 06 November 2020; Accepted: 18 November 2020;
Published: 06 January 2021.
Edited and reviewed by: Robert Czajkowski, University of Gdansk, Poland
Copyright © 2021 Zrelovs, Dislers and Kazaks. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andris Kazaks, andris@biomed.lu.lv