 Yu-Shin Nai
Yu-Shin Nai Yu-Chun Huang
Yu-Chun Huang Ming-Ren Yen
Ming-Ren Yen Pao-Yang Chen
Pao-Yang Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol., 22 January 2021
Sec. Evolutionary and Genomic Microbiology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.616922
This article is part of the Research TopicMicrobial Epigenetics and EpigenomicsView all 6 articles
DNA methyltransferases (DNMTs) are a group of proteins that catalyze DNA methylation by transferring a methyl group to DNA. The genetic variation in DNMTs results in differential DNA methylation patterns associated with various biological processes. In fungal species, DNMTs and their DNA methylation profiles were found to be very diverse and have gained many research interests. We reviewed fungal DNMTs in terms of their biological functions, protein domain structures, and their associated epigenetic regulations compared to those known in plant and animal systems. In addition, we summarized recent reports on potential RNA-directed DNA methylation (RdDM) related to DNMT5 in fungi. We surveyed up to 40 fungal species with published genome-wide DNA methylation profiles (methylomes) and presented the associations between the specific patterns of fungal DNA methylation and their DNMTs based on a phylogenetic tree of protein domain structures. For example, the main DNMTs in Basidiomycota, DNMT1 with RFD domain + DNMT5, contributing to CG methylation preference, were distinct from RID + Dim-2 in Ascomycota, resulting in a non-CG methylation preference. Lastly, we revealed that the dynamic methylation involved in fungal life stage changes was particularly low in mycelium and DNA methylation was preferentially located in transposable elements (TEs). This review comprehensively discussed fungal DNMTs and methylomes and their connection with fungal development and taxonomy to present the diverse usages of DNA methylation in fungal genomes.
Epigenetic modifications are external modifications that do not change DNA sequences and play a role in regulating gene expression, among other important biological functions (Holliday, 2006). The major epigenetic mechanisms across the kingdoms of animals, plants, and fungi include DNA methylation, histone modifications, chromatin remodeling, and small non-coding RNAs (Jeltsch, 2010). In this review, we focused on DNA methylation—specifically the 5-methylcytosines (5mC). They are more studied to date comparing with the other types of DNA modifications, such as N4-methylcytosine (4mC) and N6-methyladenine (6mA), and may influence the development of fungal species.
Genome-wide DNA methylation can now be determined at single-base resolution by whole-genome bisulfite sequencing (WGBS) (Su et al., 2011). Treatment of DNA with sodium bisulfite converts unmethylated cytosines (C) to uracils (U); thereafter, PCR amplification will convert these Us to thymines (T), while the methylated cytosines are protected and thus not converted. By using WGBS, researchers have comprehensively compared DNA methylation in living organisms. With the increase in whole-genome sequence data, we now have an unprecedented opportunity for obtaining complete genome-wide DNA methylation maps (DNA methylomes) (Zemach et al., 2010).
Comparison of DNA methylomes across more than 20 eukaryote genomes (including those of animals, plants and fungi) has revealed that most of the genomes undergo DNA methylation, while only a few eukaryotes show zero or little evidence of DNA methylation, i.e., yeast (Saccharomyces cerevisiae), fruit fly (Drosophila melanogaster) and roundworm (Caenorhabditis elegans) (Jeltsch, 2010). DNA methylation was also detected in some model fungal species (Filippovich et al., 2004; Mishra et al., 2011; Montanini et al., 2014). For example, DNA methylation in Neurospora crassa (red bread mold; Ascomycota) is crucial for balancing the formation of sexual and asexual reproductive structures (Filippovich et al., 2004). In the human fungal pathogen Candida albicans, DNA methylation has been recently reported to be primarily located within structural genes and associated with the dimorphic transition between yeast and hyphal forms, switching between white and opaque cells, and iron metabolism (Mishra et al., 2011). In black truffle, Tuber melanosporum, the DNA methylation pattern revealed selective targeting of transposable elements (TEs) rather than gene bodies, and demethylation treatment changed its phenotype (Montanini et al., 2014). Altogether, the current evidence supports the importance of fungal DNA methylation associated with changes in reproductive structures, the dimorphic transition and phenotypes.
In this review, we start by discussing the characteristics and functions of all DNA methyltransferases (DNMTs), which transfer a methyl group to the C-5 position of the cytosine ring and establish DNA methylation, especially fungal-specific DNMT5- and DNMT5-associated potential RNA-directed DNA methylation (RdDM). To explore the correlations between fungal DNMTs and DNA methylation, we list the DNMTs and summarize the DNA methylation data from 40 fungal genomes, mainly belonging to Ascomycota and Basidiomycota, as well as Mucoromycota and Myxomycota (Table 1). In addition, we perform a comprehensive survey of DNMT phylogenies, domain structures, and fungal DNA methylation levels at different life stages. A conserved phylogeny based on the fungal DNMT1 family and Masc1/RID (repeat-induced point mutation defective) is also described in this review. Although the DNA methylation levels in different fungal species were diverse, the dynamics of methylation changed during the fungal life cycle, and a preference for methylated locations in fungi was found. Our review presents a summary of a large survey of fungal DNA methylation levels and the preference for DNMTs in specific groups of fungi.
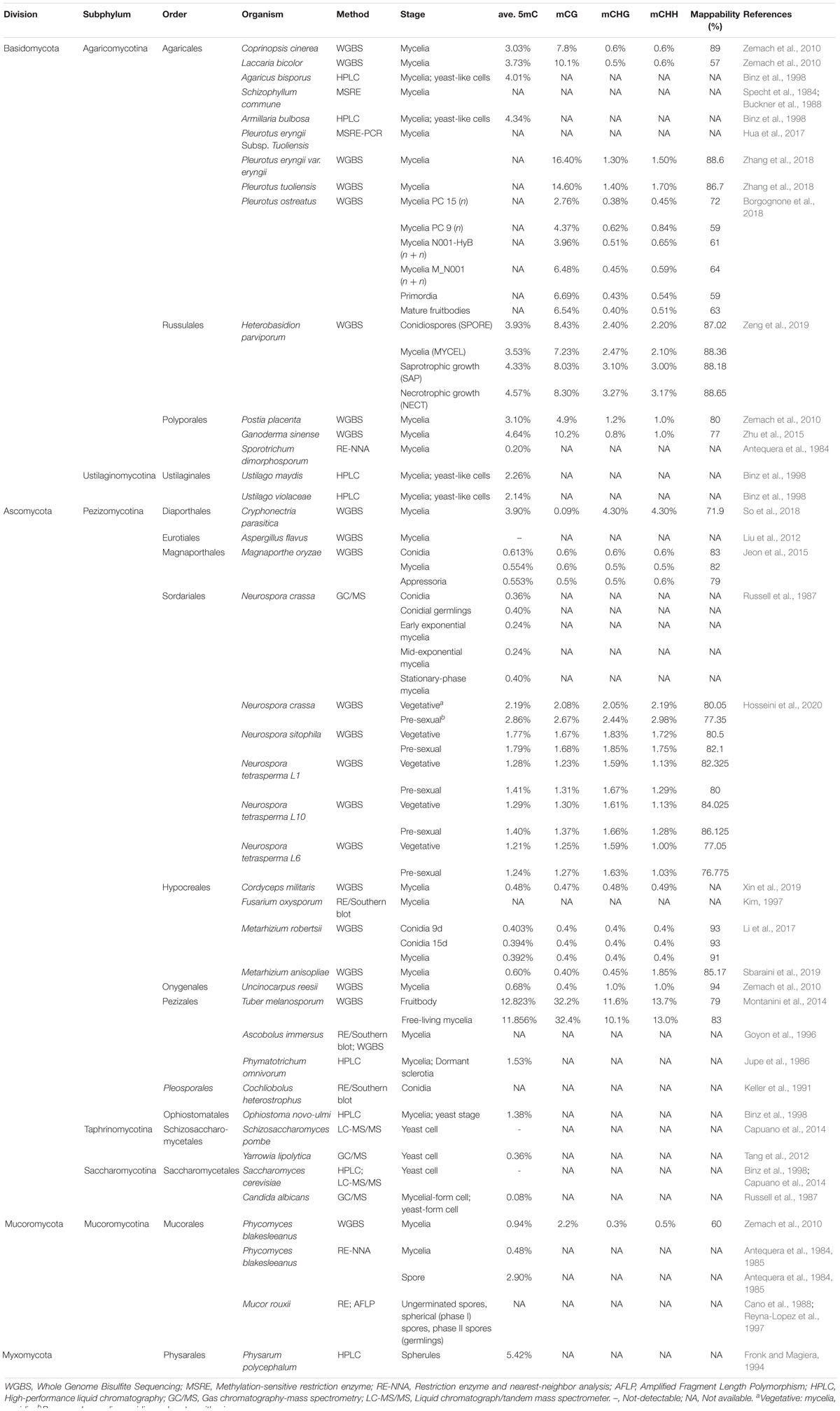
Table 1. List of 40 fungi with the descriptions of methylation data.
DNMTs, containing a DNA methylase domain as the catalytic domain (PF00145), contribute to DNA methylation by transferring the activated methyl group (CH3) from S-adenosyl methionine (SAM) to the 5th position of a cytosine residue (5mC) (Law and Jacobsen, 2010). The DNA methylase domain consists of the catalytic core containing seven beta-sheet and three alpha-helices, and a target recognition domain (TRD) (Martin and McMillan, 2002). During the process of DNA methylation, the catalytic core and TRD are organized into a two-lobe architectural clef to harbor the DNA duplex to form the DNMT-DNA covalent complex in order to complete the enzyme activity (Yang and Xu, 2013).
DNMTs can be categorized into two types: those participating in “maintenance” and “de novo” methylation (Riggs, 1975). Maintenance DNMTs recognize hemimethylated DNA and copy the previous methylation patterns onto the nascent strands after each round of DNA replication, while de novo DNMTs methylate the unmethylated cytosines and establish novel DNA methylation patterns (Chen and Li, 2004). Different DNMTs have been characterized in prokaryotes and eukaryotes. Based on structural and functional similarity, DNMTs can be divided into five classes, including DNMT1, DNMT2, DNMT3/DRM, Masc1/RID, and DNMT5 (Figure 1; Ponger and Li, 2005; de Mendoza et al., 2020).
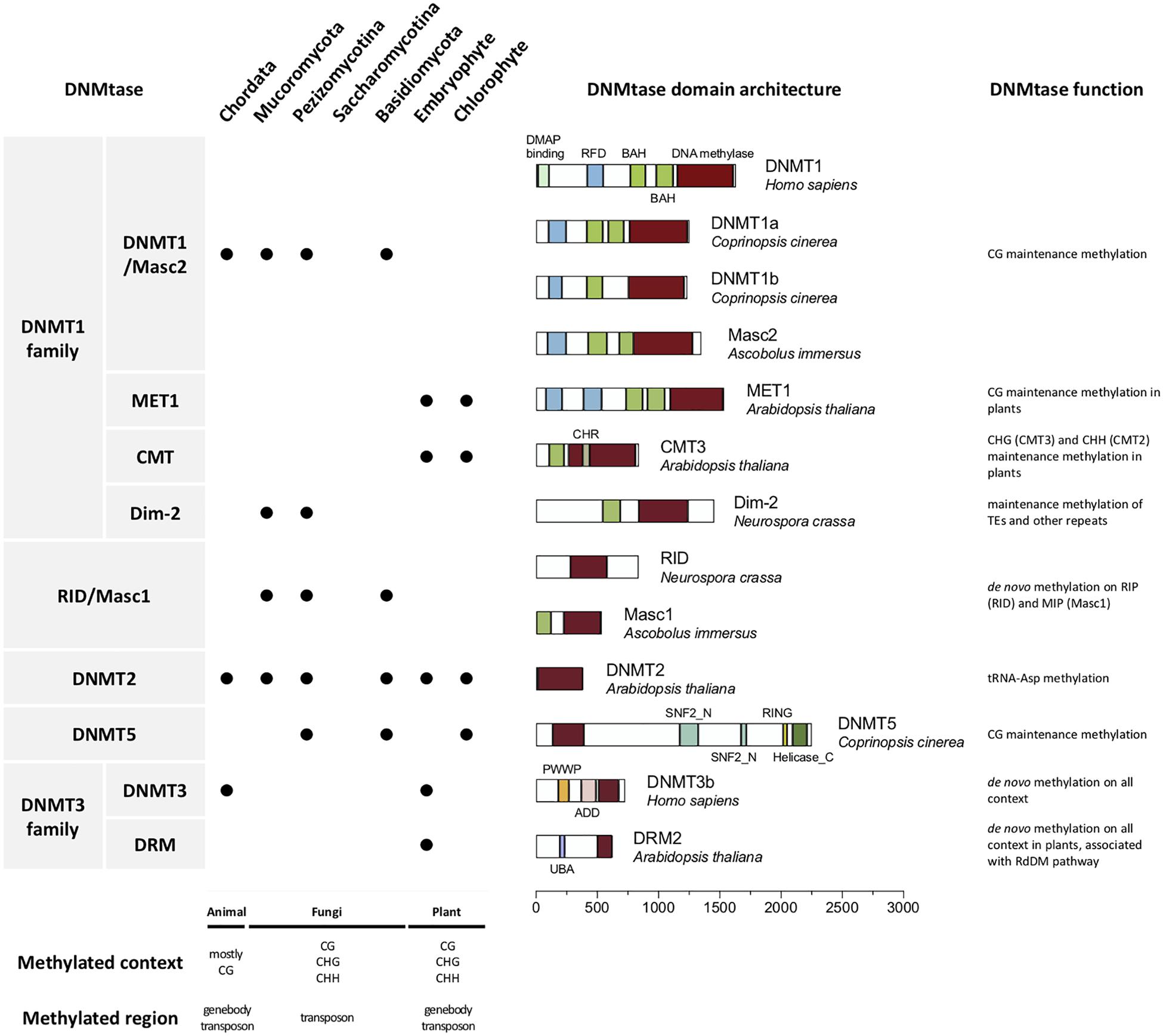
Figure 1. DNA methyltransferases in eukaryotes. The first column shows the names of DNMTs. The second column shows the presence or absence of DNMTs in the animal (Chordata), fungal (Mucoromycota, Pezizomicotina and Saccharomicotina of Ascomycota, and Basidiomycota), and plant (Embryophyte and Chlorophyte) kingdoms. The third column shows the domain architecture of the real DNMTs. The exact species used to generate these DNMT domain architectures are labeled. The fourth column shows function of the DNMTs. The methylation context and region often found in animals, fungi and plants are shown below.
The DNMT1 subfamily, which is generally considered a key factor in maintenance methylation, contains animal and fungal DNMT1, as well as its homologous DNA methyltransferase 1 (MET1) in plants and Masc2 in Ascobolus, Dim-2 in Ascomycota, and plant-specific chromomethylases (CMTs) are also categorized as belonging to this subfamily, owing to having a domain structure and function similar to those of DNMT1 (Ponger and Li, 2005; Huang et al., 2016; de Mendoza et al., 2020).
The DNMT1 subfamily is known to have a bromo-adjacent homology (BAH) domain involved in transcriptional regulation such as gene silencing (Callebaut et al., 1999). The BAH domain is required for DNMT1 to target the replication foci during S phase, and links amongst DNA methylation, replication and transcriptional regulation (Yarychkivska et al., 2018). In plants, CMT3 binds H3K9me2 via both BAH and chromo domains, further suggesting that the BAH domain had the function of histone mark recognition (Du et al., 2012). In DNMT1 and MET1, the replication foci domain (RFD) is also observed, while the RFD is absent in the other two DNMT1 subgroups (Dim-2 and CMT). This RFD is responsible for targeting replication foci to discriminate between unmethylated and hemi-methylated DNA. This allows DNMT1 to methylate the correct residues (Syeda et al., 2011). It was also demonstrated that the RFD of DNMT1 functions as a transcriptional repressor that binds DMAP1 and HDAC2 at the replication foci during the S phase of mitosis (Rountree et al., 2000). Recently, the RFD domain of DNMT1 is referred to as a histone reader of K18/K23 ubiquitylated histone H3, in the assistance of DNMT1 to bind with H3-K18Ub/K23Ub. The module of Dnmt1 binding to two-mono-ubiquitylated H3 is required by Dnmt1 recruitment to the loci for DNA methylation, and the enhancement of its methyltransferase activity (Leonhardt et al., 1992; Ishiyama et al., 2017).
DNMT2 is another class of DNA methylase domain-containing enzymes that is highly conserved in eukaryotes and present in all kingdoms, including those of fungi, animals and plants (Huang et al., 2016). It is responsible for tRNA-Asp methylation and specifically methylates cytosine 38 in the anticodon loop. The function of DNMT2 is also conserved in different organisms, while restoration of human DNMT2 protein facilitated Dnmt2-deficient mouse, fly and Arabidopsis tRNA-Asp methylation in vitro (Goll et al., 2006). Although DNMT2 has zero or very low DNA methyltransferase activity, the DNA fragment in the structural context of a covalent DNA-tRNA hybrid can be more efficiently methylated than the all-ribo tRNAs (Kaiser et al., 2017).
DNMT3 is found in animals, and the homologous in plants are domains of rearranged methyltransferase (DRM) since they both play a role in de novo methylation. In mammals, DNMT3 contributes to the establishment of DNA methylation patterns during embryogenesis. In plants, DRM1/2 participates in the RNA-directed DNA methylation (RdDM) pathway guided by small RNAs (sRNAs) (Ehrlich et al., 1982; Jeltsch et al., 2006; Law and Jacobsen, 2010; Zemach et al., 2010; Matzke and Mosher, 2014). However, DNMT3 homologs are absent in fungal genomes and the genomes of some green algae (i.e., Chlorella sp. NC64A) and animal species (i.e., silkworm) (Zemach and Zilberman, 2010; Huang et al., 2016).
In fungi, the two fungi specific de novo DNMTs occurring on repeat sequences are Masc1 and RID. There is another DNMT, DNMT5, capable of fungal DNA methylation, which is also represented in some chlorophytes, stramenopiles, and haptophytes (Chen and Li, 2004; Huang et al., 2016; Bewick et al., 2019; Schmitz et al., 2019; de Mendoza et al., 2020). The details of fungal-specific DNMTs will be discussed in the following paragraphs.
Fungi are the principal decomposers in ecological systems. Based on their morphological, reproductive traits, and genetic differences, the fungal kingdom may be divided into nine major lineages under three groups of fungi species: (1) Zoosporic fungi (Opisthosporidia, Chytridiomycota, Neocallimastigomycota, Blastocladiomycota); (2) Zygomycetous fungi (Zoopagomycota, Mucoromycota, Glomeromycota); and (3) Dikarya (Ascomycota, Basidiomycota) (Naranjo-Ortiz and Gabaldón, 2019). Dikarya which in general produces dikaryons, is the largest and well-studied group of Fungi, presenting diverse lifestyles and phenotypes (Naranjo-Ortiz and Gabaldón, 2019). In this review we focus on the popular species of Ascomycota and Basidiomycota, and also few Mucoromycota and Myxomycota, depending on the availability of literatures and DNA methylation data.
Recently, the conserved configuration of fungal DNMTs was identified. Based on phylogenetic and domain architecture analyses of the DNMT sequences from fungal species, including Dikarya (Basidiomycota and Ascomycota) and early-diverging fungi (Blastocladiomycota, Chytridiomycota, and Mucoromycota), the fungal DNMTs can be grouped into two monophyletic superclades (Bewick et al., 2019). The first superclade is composed of DNMT1/Masc2, Dim-2, and RID/Masc1. The uncertain de novo DNMTX is also in this clade. The other superclade contains DNMT2 and Rad8/DNMT5. Based on evolutionary analysis (Bewick et al., 2019), Dim-2 existed in the other early-diverging fungi and Ascomycota, but was lost in Basidiomycota after the differentiation between Basidiomycota and Ascomycota in the subkingdom Dikarya. All fungi lack the de novo DNMT3 family, suggesting two independent gains in animals and plants or one single loss in the fungal ancestor that diverged from animals (Huang et al., 2016; Bewick et al., 2019). The distribution of DNMTs exhibited a common characteristic in which each fungus had at least one copy of the DNMT1 family, except for Ustilago maydis, Malassezia globosa, Wallemia sebi of Basidiomycota, and Saccharomyces cerevisiae from Saccharomycotina in Ascomycota (Huang et al., 2016).
Dim-2 is a DNMT1-related methyltransferase. Dim-2 is only found in fungi to mediate methylation of TEs and other repeats. In Neurospora crassa, the relationship between DNA methylation and histone lysine methylation is strong. Lysine 9 methylation of histone H3 (H3 mK9) is needed for DNA methylation mediated by Dim-2 (Goll and Bestor, 2005; Zemach et al., 2010). Moreover, it was proven that the modification state and sequence of DNA can affect the methylation states of accompanying histones in chromatin and vice versa (Goll and Bestor, 2005; Rose and Klose, 2014).
RID and Masc1, which are fungus-specific DNMTs that primarily function in genome defense mechanisms, are closely related to DNMT1 (Gladyshev, 2017). They are required for de novo methylation of TEs and repeats in both CG and non-CG contexts associated with repeat-induced point (RIP) and methylation-induced premeiotic (MIP) mutations, respectively (Chen and Li, 2004; Ponger and Li, 2005; Zemach et al., 2010; Freitag, 2017). The masc1 gene is cloned from Ascobolus immersus and is required for the de novo methylation associated with MIP mutation, which is a process that scans the genome for DNA duplications and subsequently methylates the cytosines in these duplicated sequences (Malagnac et al., 1997). Moreover, the phenotype silencing is reversible during subsequent vegetative growth by the loss of cytosine methylation in MIP mutations (Goyon and Faugeron, 1989). The RIP mutation process in N. crassa is similar to the process of MIP mutation in A. immerses, and both RIP and MIP mutations act at a very precise stage of the sexual cycle (i.e., between fertilization and meiosis) (Selker et al., 1987; Grayburn and Selker, 1989). In contrast, the result of RIP mutation is irreversible; repeated DNA is detected in a pairwise manner and introduced with G-C to A-T transition mutations with a significant bias for 5’−CpA−3’ contexts, and most DNA methylation is found in relics of RIP mutations in N. crassa (Selker, 2002; Selker et al., 2003). In Aspergillus nidulans, DmtA is a DNMT homolog similar to RID mutation and Masc1 that contributes to sexual development, conidiation, sclerotial production, aflatoxin biosynthesis, and virulence (Lee et al., 2008; Yang et al., 2016).
DNMT5 has a unique architecture, including the RING finger with ubiquitination potential following the methyltransferase domain and a long C-terminal region of SNF2 family homology, which contains an ATP-dependent chromatin remodeling domain and helicase domain, suggesting that DNMT5 proteins are multifunctional enzymes (Ponger and Li, 2005; Huff and Zilberman, 2014; Dumesic et al., 2020). The complete CG methylation was lost in Dnmt5 mutant Cryptococcus neoformans. As SNF2 domain ATP hydrolysis is required for driving CG methylation and hemimethylated DNA substrates preferentially activate SNF2 ATPase, DNMT5 is thought to maintain CG methylation with the help of SNF2 domain coupling the recognition and catalysis of hemimethylated DNA (Huff and Zilberman, 2014; Dumesic et al., 2020). DNMT5 is conserved in fungi, haptophytes, stramenopiles, and chlorophytes, indicating that it is ancestral in eukaryotes (de Mendoza et al., 2020). Another protein, the DNA repair protein Rad8, has high similarity to DNMT5 in terms of protein domain architecture. It has been recorded to have conserved RING finger-like motifs and SNF2-family helicase domains but without DNA methylase domains (Doe et al., 1993; Eisen et al., 1995; Ding and Forsburg, 2014); however, Huang and Zhang indicated that the fungal Rad8 subfamily contained DNA methylase domains and acted as DNA methyltransferases (Huang et al., 2016; Zhang et al., 2018). To clarify whether there are any misleading results, we compared the DNMT5 and Rad8 sequences of 20 fungal species reported in Bewick’s and Huang’s research (Huang et al., 2016; Bewick et al., 2019) and found that 18 of them were the same proteins, while the remaining 2 were homologs (blastp e-value ≤1e-30, mismatch <50 within 2,000 bp). Therefore, the Rad8 reported by Huang and Zhang is likely DNMT5, which is actually a DNMT.
In the ancestor of pathogenic Cryptococcus fungal species, Kwoniella, there is a putative DNA methyltransferase DNMTX containing a BAH domain and a DNA methylase domain and acting as a de novo methylase. DNMTX is considered an ancestrally lost DNMT in Cryptococcus neoformans, proven by the accumulation of 5mC when the gene for DNMTX was transferred into a dnmt5 mutant of C. neoformans (Catania et al., 2020).
Helicase-like Snf2 superfamily proteins, such as CLSY1, DRD1 and SNF2-RING-HELICASE–LIKE-1 and -2 (FRG1 and FRG2), are required for RdDM in Arabidopsis (Groth et al., 2014). The chromatin remodeling-associated protein CLSY1 assists in the first step of the RdDM pathway, RNA polymerase IV (Pol IV)-dependent siRNA biogenesis, whereas DRD1 is involved in RNA polymerase V (Pol V)-mediated de novo methylation (Matzke and Mosher, 2014). Although the RdDM pathway is unique to plants, the product of the fungus-specific domain fusion event of the DNA methylase family and Snf2 family is suspected to act as DNMT (Huang et al., 2016). In Pleurotus fungi, the colocation of siRNA abundance and TE methylation also supports that methylation in TE regions is established by siRNA-directed DNA methylation. The genes encoding the proteins taking part in the RdDM pathway, including RNA-dependent RNA polymerase and Dicer-like and Argonaute proteins, are found in Basidiomycota fungi (Zhang et al., 2018). This evidence suggests that there might be an RdDM-like mechanism in the fungal kingdom. As DNMT5 contains the SNF2_N and helicase_C domains of the Snf2 superfamily along with DNA methylase, it is suspected to function in the RdDM-like pathway to achieve DNA methylation in fungi (Groth et al., 2014).
We surveyed the literature to identify fungal species that have both DNA methylation data and DNMT1 family and RID/Masc1 data. There were 40 such fungi, including 15 Basidiomycota species, 22 Ascomycota species, 2 Mucoromycota species, and 1 Myxomycota species (Table 2). The analysis showed that different DNMT families are present in different fungal genomes. Furthermore, there is a distinct DNMT preference between Basidiomycota and Ascomycota. Most fungal genomes in basidiomycetes contain one to three copies of DNMT1, together with only one copy of DNMT5/Rad8 families. DNMT1 and DNMT5 are the main DNMTs in Basidiomycota, which lack Dim-2 and seldom exhibit de novo DNA methyltransferase RID. By contrast, RID and Dim2 are the main DNMTs in Ascomycota, especially in Pezizomycotina (Bewick et al., 2019). DNMT1 is present in most eukaryotes, except for some Dikarya fungi, such as most Ascomycota (Zemach and Zilberman, 2010; Bewick et al., 2019). However, DNMTs were not observed in 5 fungi, namely, Ustilago maydis, Candida albicans, Schizosaccharomyces pombe, Saccharomyces cerevisiae, and Yarrowia lipolytica (Table 2; Ponger and Li, 2005; Huang et al., 2016).

Table 2. Numbers of genes encoding DNMT proteins in the 40 fungal species.
We performed a phylogenetic analysis (see Extended experimental procedures in the Supplementary Material) based on the conserved protein domains of the 54 DNMT1 family and RID/Masc1 proteins from the 26 fungal genomes (Figure 2), including 23 proteins from 10 Basidiomycota, 29 proteins from 15 Ascomycota, and 2 proteins from 1 Mucoromycota. The DNA methylation levels in the CG, CHG, CHH, CA, CC, and CT contexts of each fungal species are also shown in Figure 2. Similar to the report of Huang et al. (2016), we found that the DNMTs in these fungi were clustered into three subgroups: the DNMT1/Masc2 subgroup, Dim-2 subgroup, and RID/Masc1 subgroup. All three subgroups contained the DNA-cytosine methyltransferase (DCM) domain, while the BAH domain was absent from the RID/Masc1 subgroup. Only the DNMT1/Masc2 subgroup and P. blakesleeanus Dim2 contained the RFD domain, which targeted replication foci for discriminating between unmethylated and hemimethylated DNA, resulting in higher CG methylation (>0–30%) than non-CG methylation (0–5%). The DNMT1 proteins of Basidiomycota fungi specifically belonged to the DNMT1/Masc2 subgroup, not Dim-2 (Figure 2). The fungi in the Dim-2 and RID/Masc1 subgroups are from Ascomycota and Mucoromycotina but not Basidiomycota. Without the RFD domain, Dim-2 and RID/Masc1 of Ascomycota usually result in slightly higher non-CG methylation (>0–5%) than CG methylation (>0–0.5%) or equal CG and non-CG methylation (Figure 2). The phylogenetic tree also showed that Dim-2 proteins were absent in Basidiomycota, indicating that Dim-2 was lost after the bifurcation between Ascomycota and Basidiomycota (Huang et al., 2016). In contrast to the findings of Huang et al. (2016), after we blasted the DNMT1 homologs with the Phycomyces blakesleeanus genome, we noticed that P. blakesleeanus Dim2 (132318) in Huang’s research should contain BAH and RFD domains and more likely belonged to the DNMT1/Masc2 subgroup, while P. blakesleeanus DNMT1 (77036) in Huang’s research should belong to the Dim2 subgroup (Figure 2). Therefore, we changed the names of the two DNMTs (Supplementary Table 2).
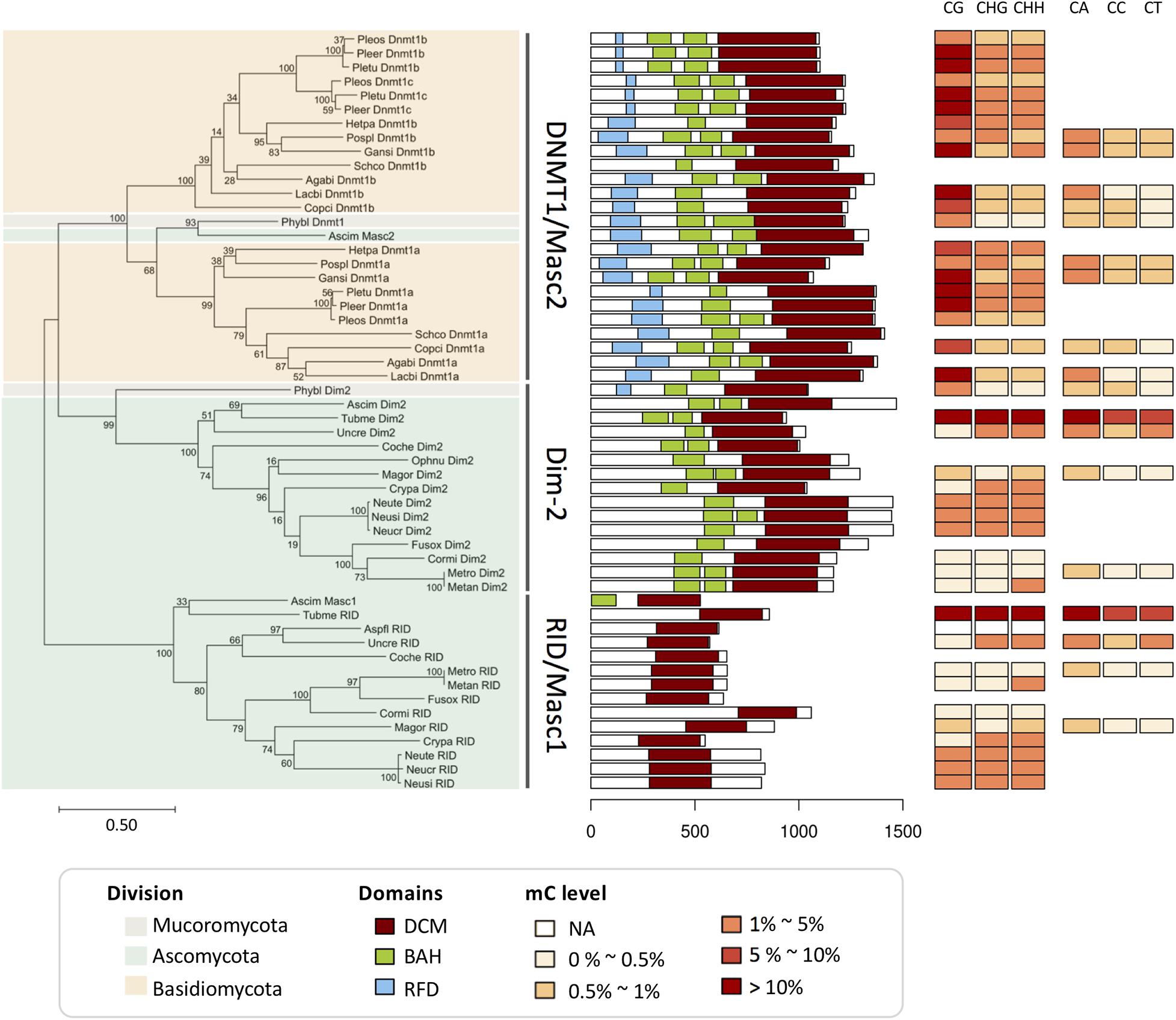
Figure 2. Phylogenetic and domain architecture analyses of the DNMT1 family and RID/Masc1 protein in 26 fungi. The ML tree was constructed with the conserved DCM domains of the 54 predicted proteins from 26 species using the WAG model in MEGA 7.0 with 200 bootstrap replicates. The three groups, namely, DNMT1/Masc2, Dim-2 and Masc1/RID, are indicated.
As shown in Figure 2, the domain architecture indicated that the DNMT1-like protein sequences from different fungi were clustered into the DNMT1/Masc2, Dim-2 and Masc1/RID groups, suggesting a possible functional difference in DNA methylation among these three groups. The biological function of DNMT1 or Masc2 proteins has been demonstrated to act together with or recruit histone deacetylases in fungi (i.e., DNMT1a in Laccaria bicolor and Coprinopsis cinerea) (Fuks et al., 2000). Dim-2 has been shown to be able to catalyze DNA methylation in all sequence contexts in N. crassa (Kouzminova and Selker, 2001). Masc1 and RID are specific to fungi and required for de novo methylation associated with MIP and RIP mutations, respectively (Chen and Li, 2004; Ponger and Li, 2005; Zemach et al., 2010; Freitag, 2017).
To date, several DNA methylation assays have been developed to detect cytosine methylation, including chemical analytic methods, such as high-performance liquid chromatography (HPLC), gas chromatography-mass spectrometry (GC/MS), restriction enzyme digestion of methylated DNA, sequencing and Southern blot hybridization (Table 1). Most early reports on the investigation of DNA methylation performed chemical analytic methods, such as HPLC (Fronk and Magiera, 1994). Instead of HPLC, isotope dilution GC/MS, which has a high sensitivity for analyzing 5mC directly from genomic DNA, was developed and applied in several fungal studies (Russell et al., 1987; Tang et al., 2012). In recent years, more methods have become commonly used for profiling genome-wide methylation levels, including restriction enzyme digestion of methylated DNA followed by hybridization to high-density oligonucleotide arrays or sequencing and Southern blot hybridization, methylated DNA immunoprecipitation (MeDIP) combined with large-scale analysis using microarrays for capturing methylated genomic DNA, methyl-binding domain (MBD) proteins followed by array hybridization or sequencing and WGBS (Lippman et al., 2004; Weber et al., 2005; Zhang et al., 2006; Vaughn et al., 2007). Although these approaches can determine either the status of DNA methylation or genome-wide methylation levels (i.e., HPLC and GC/MS), none of them reached single-base resolution until 2007, when WGBS based on bisulfite sequencing coupled with high-throughput sequencing was developed (Zilberman et al., 2007; Lister and Ecker, 2009; Table 1).
To evaluate the resolution of methylation data generated by different methods, the 40 fungal DNA methylation levels and the assays are listed in Table 1. There were 22 fungi identified by WGBS, 12 by either HPLC or GC/MS, and 8 by restriction enzyme digestion-based methods. The methylation data of the filamentous ascomycete N. crassa were detected by both HPLC and WGBS, and those of Phycomyces blakesleeanus were detected by both restriction enzyme digestion-related methods and WGBS. We compared the DNA methylation levels of the mycelium within three phyla of fungi, namely, Mucoromycota, Ascomycota, and Basidiomycota (Table 1; Zemach et al., 2010; Montanini et al., 2014; Jeon et al., 2015; Zhu et al., 2015; Li et al., 2017). We observed high variation in DNA methylation levels, where the average 5mC% ranged from 0.39 to 12.3% when profiled by WGBS. Among these fungi, five species of Ascomycota, namely, Magnaporthe oryzae, Cordyceps militaris, Metarhizium robertsii, Metarhizium anisopliae, and Uncinocarpus reesii, clearly showed very low levels of DNA methylation (0.55, 0.48, 0.39, 0.60 and 0.68%, respectively). The plant-symbiotic black truffle Tuber melanosporum has the highest DNA methylation (12.3%), followed by the medicinal fungi Gannoderma sinense (4.64%), Cryphonectria parasitica (3.90%), and Laccaria bicolor (3.73%). Unlike the data detected by HPLC, GC/MS, and restriction enzyme digestion-based methods only showing the average 5mC level, the WGBS data contain both methylated CG and non-CG (CHG and CHH) sites. We compared 12 fungi that were profiled by chemical analytic methods (GC/MS and HPLC). Within the 3 profiled by GC/MS, the highest methylation level was found in Y. lipolytica and N. crassa (0.36%), followed by C. albicans (0.08%). The 9 fungi profiled by HPLC also showed variation in methylation ranging from 1.38 to 5.42%. Restriction enzyme digestion-related methods were used to detect the methylation levels of 8 fungi. The results show that methylation seems to occur in all of these fungi. However, more detailed information on the methylation levels was not provided due to the limitations of the assay (Table 1). Of these fungi, Sporotrichum dimorphosporum showed a very low methylation level (∼0.2%) in the mycelium according to restriction and nearest-neighbor analysis (RE-NNA) (Antequera et al., 1984). Fungal methylation data generated by restriction enzyme digestion-based methods should combined with those from other methods to avoid insufficient evidence for data explanation. Both N. crassa and P. blakesleeanus were subjected to two different methylation determination methods. The methylation level of P. blakesleeanus was 2.9% when using RE-NNA, higher than that (0.94%) detected by using WGBS (Antequera et al., 1985). On the other hand, the methylation level of N. crassa stationary-phase mycelia was 0.4% according to GC/MS and lower than 2.19% according to WGBS (Table 1; Hosseini et al., 2020). The results indicated that DNA methylation profiled by distinct methods was different, and it is necessary to compare the methylation data from equal baselines or the same detection method to eliminate the difference. In conclusion, among the four methods that have been reported to be used for fungal DNA methylation assays, WGBS is a better choice for obtaining more details of the fungal methylome. Therefore, in the following discussion, we will only compare fungal methylation data generated by WGBS.
DNA methylation varied among different stages of fungal life cycles. Comparing the spore, yeast stage cell, dormant sclerotia, and conidia with mycelium of 8 fungi, namely, P. blakesleeanus, C. albicans, Ophiostoma novo-ulmi, Phymatotrichum omnivorum, M. oryzae, N. crassa, M. robertsii, and Heterobasidion parviporum, showed that regardless of which method was used for the DNA methylation assay, the lowest methylation levels were mostly found in the mycelium stage, and a higher methylation level was found in the spore or conidia stage of these fungi, except for O. novo-ulmi (Figure 3A). The lower methylation level was also observed in the mycelium stage compared to the fruit body formation stage and the other infection phases, such as protoperithecia of 5 Neurospora species and strains, appressoria of M. oryzae, saprotrophic and necrotrophic growth of H. parviporum, and fruitbodies of T. melanosporum (Figure 3A). In Pleurotus ostreatus, the monokaryotic mycelium stage had the lowest CG methylation level, while the two strains showed differences. In contrast, the non-CG (CHG, CHH) methylation level of mycelium was slightly higher than that of primordia and mature fruitbodies, although non-CG contexts were not predominant in P. ostreatus (Figure 3B; Borgognone et al., 2018). Additionally, the methylation level of the exponentially growing mycelia of N. crassa was lower than that of the stationary-phase mycelia (Figure 3A) (Russell et al., 1987). A lower methylation level may increase genome plasticity and the ability to adapt to environmental changes. Therefore, a lower methylation level in the mycelium stage may allow the genome to undergo evolution and avoid silencing of growth and development processes, especially in monokaryons, the most commonly found developmental stage with high potential in nature (Antequera et al., 1984; Antequera et al., 1985; Russell et al., 1987; Montanini et al., 2014; Borgognone et al., 2018).
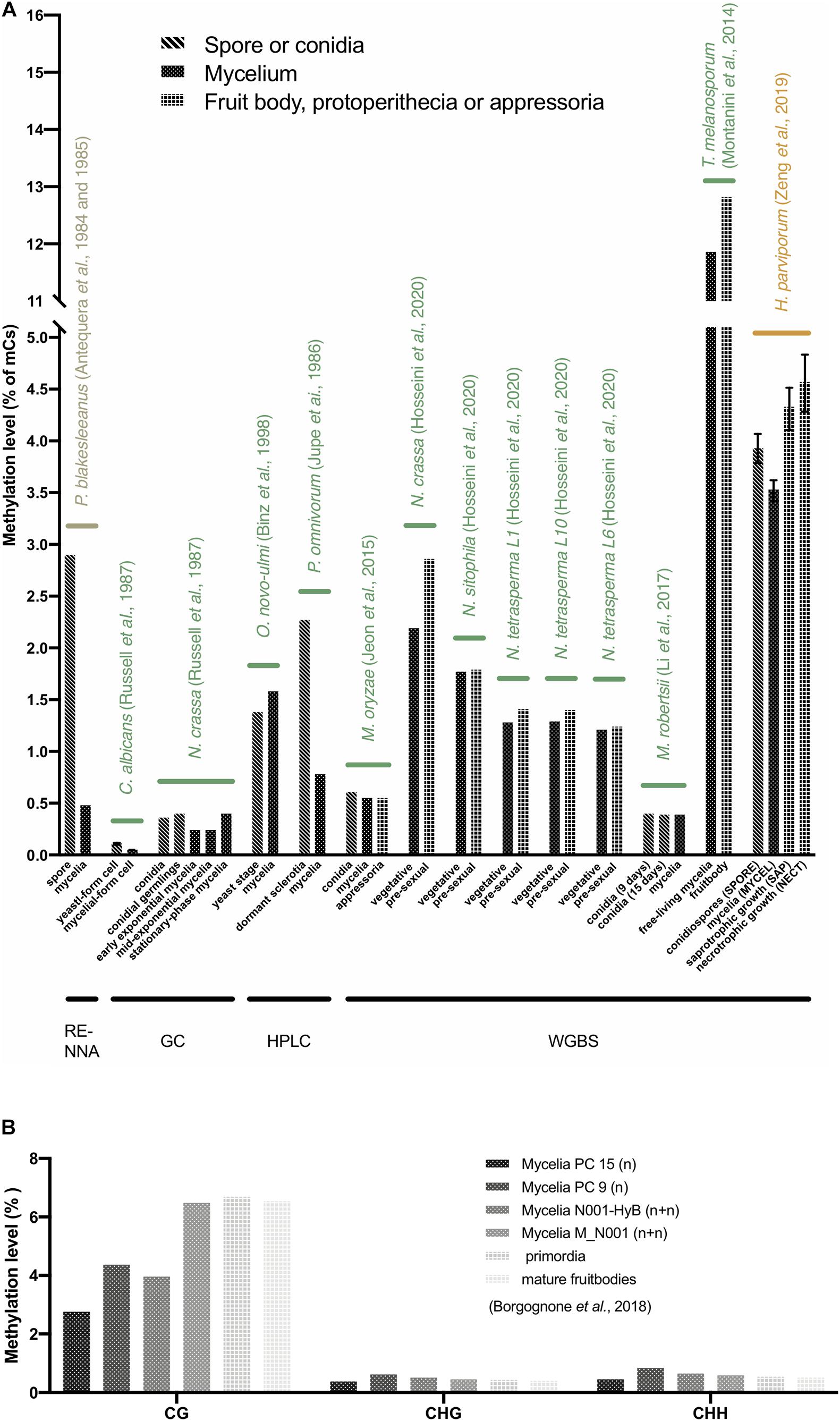
Figure 3. DNA methylation levels at different life stages in fungi. (A) The average 5mC level of different life stages in 14 fungi. The X-axis represents the different life stages of fungi. The Y-axis represents the average 5mC level. The labels with different colors indicate fungi from Mucoromycota (olive), Ascomycota (green), and Basidiomycota (yellow). The methods of DNA methylation assays are shown below. (B) The CG, CHG, and CHH methylation levels of Pleurotus ostreatus (Borgognone et al., 2018). Note that only C. albicans and H. parviporum have replicates, therefore the error bars represent the variances.
DNA methylation can be found in widespread genomic regions, such as gene bodies, promoters, and TEs, causing the DNA methylation pattern to vary across eukaryotes (Jeltsch, 2010; Law and Jacobsen, 2010; Su et al., 2011). For example, both CG and non-CG methylation of plants occurred in the promoters of silent genes and TEs. However, in the transcribed genes of animals and plants, DNA methylation was not found around their promoters but within the gene bodies, except in the TSS-proximal regions. Additionally, TEs are also the targets of methylation in plants and some animals (Taiko et al., 2015; de Mendoza et al., 2020).
In contrast to plants and animals, most fungi lack gene body methylation. In contrast, DNA methylation was enriched in TEs and repeats in the fungal genome (Bewick et al., 2019). As shown in Table 1, the methylation of 12 fungi, namely, Coprinopsis cinerea, L. bicolor, P. placenta, G. sinense, P. eryngii var. eryngii, P. tuoliensis, P. ostreatus, H. parviporum, T. melanosporum, M. oryzae, Neurospora, and P. blakesleeanus, mainly occurred in TE and repeat regions rather than gene bodies (Zemach et al., 2010; Montanini et al., 2014; Jeon et al., 2015; Zhu et al., 2015; Borgognone et al., 2018; Zhang et al., 2018; Zeng et al., 2019; Hosseini et al., 2020). The result of Figure 5 also showed that in all of the three sequence contexts (CG, CHG, and CHH) of the nine fungi, TEs showed the highest methylation level, followed by intergenic regions, comparing to the genebodies and exons that showed less methylation. In the T. melanosporum, TEs were methylated at approximately 80% of CpG sites, whereas genes had almost no methylation, suggesting a non-exhaustive and partly reversible methylation process. This process would lead to transcriptional activation of TEs, thereby potentially playing a role in promoting genome plasticity and the environmental fitness of T. melanosporum (Montanini et al., 2014). In Neurospora, both transcriptional and posttranscriptional gene silencing mechanisms are used for TE silencing, including the RID involved in RIP mutation (Aramayo and Selker, 2013; Hosseini et al., 2020). For the model plant pathogenic fungus M. oryzae, DNA methylation occurs in and around genes as well as TE regions and undergoes global reprogramming during fungal development (Jeon et al., 2015). In contrast to those in previous fungi, the methylation patterns in U. reesii and an entomopathogenic fungus, M. robertsii, showed clear gene body methylation (Li et al., 2017).
DNA methylation of fungi affects gene expression by methylating surrounding TEs instead of directly methylating the gene bodies and their promoters. In Pleurotus ostreatus and Pleurotus tuoliensis, the genes near TEs had higher methylation levels and lower gene expression than those far away from TEs (Borgognone et al., 2018; Zhang et al., 2018). Additionally, the genes located inside TE-rich clusters also had higher methylation levels and lower gene expression (Borgognone et al., 2018). Genes of L. bicolor with TE insertions within a 1 kb upstream and downstream window were repressed. Nevertheless, in S. cerevisiae, a fungus lacking DNA methylation, the genes under TE influence did not show any alteration in expression (Castanera et al., 2016). As a result, TE-associated methylation affected the expression of surrounding genes.
In mammals, CG methylation is predominant. Some of the methylation patterns in plants are similar to those in mammals, except for the two non-CG methylation systems specific to CHG and CHH. These two systems are also present in fungi (Jeltsch, 2010). Moreover, two distinct methylation patterns were found in fungi. One showed a strong preference for CG methylation, and the other showed a preference for CHG and CHH methylation (Figure 4 and Table 1).
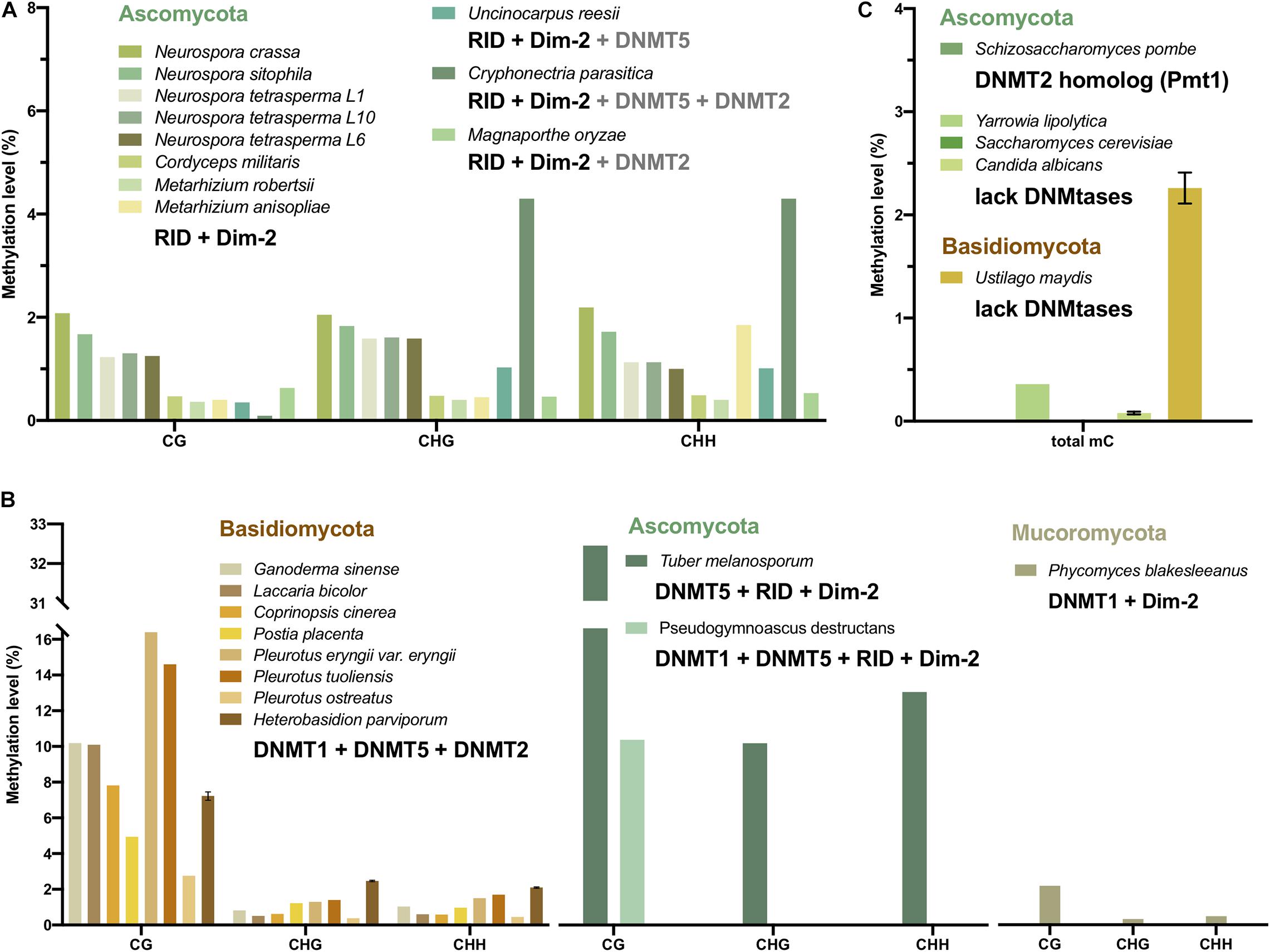
Figure 4. CG, CHG, and CHH methylation levels and the DNMT combination of fungi. The methylation level values are from mycelium, not the other stages. The bars with different colors represent fungi from Mucoromycota (olive), Ascomycota (green), and Basidiomycota (yellow). (A) The DNA methylation level and the DNMT combination of fungi showed a preference for CHG and CHH methylation. (B) The DNA methylation level and the DNMT combination of fungi showed a preference for CG methylation. (C) The DNA methylation level of fungi without DNMT or with only tRNA methyltransferase. Note that only C. albicans, U. maydis, and H. parviporum have replicates, therefore the error bars represent the variances.
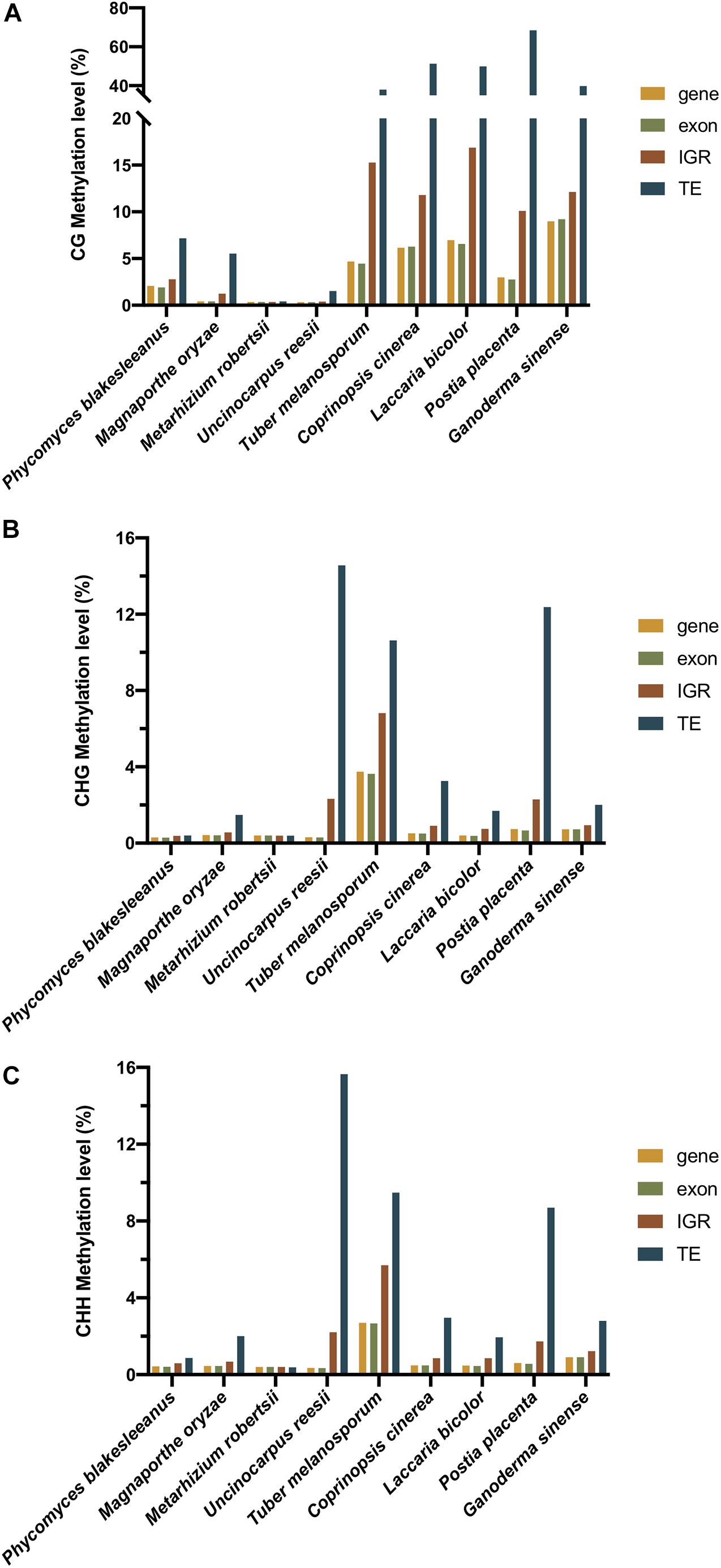
Figure 5. The (A) CG, (B) CHG, and (C) CHH methylation level of different regions of nine fungi (C. cinerea, L. bicolor, P. placenta, U. reesi, G. sinense, M. oryzae, M. robertsii, T. melanosporum, and P. blakesleeanus). The yellow, green, red, and blue bars represent the methylation level of genebody, exon, intergenic region (IGR), and transposable element (TE), respectively.
The DNA methylation of 11 Ascomycota, including 5 Neurospora species and strains, C. militaris, M. robertsii, M. anisopliae, U. reesii, C. parasitica, and M. oryzae, had no preference for CG sites. The methylation level of the non-CG context, which was 0.4–4.3%, was generally similar to and even higher than that of the CG context (0.09–2.08%) (Figure 4A and Table 1). The main DNA methyltransferase combination of these fungi was RID + Dim-2, with some additional DNMT5 (Table 2). CG and non-CG sites might be equally methylated by RID/Masc1 and Dim-2, which are responsible for both symmetric and asymmetric site methylation and are required for de novo DNA methylation and the maintenance of DNA methylation, respectively. In U. reesii, the corresponding increase in CA and TG dinucleotides was consistent with the RID in the genome and involved RIP mutation (Galagan and Selker, 2004).
By contrast, the DNA methylation of 8 Basidiomycota, including G. sinense, L. bicolor, C. cinerea, P. eryngii var. eryngii, P. tuoliensis, P. ostreatus, and H. parviporum, revealed substantial CG methylation (2.76–16.4%) and a small amount of non-CG methylation (0.38–2.47%) (Figure 4B and Table 1). DNMT1 and DNMT5, acting as CG site maintenance enzymes, were the main DNMTs in Basidiomycota (Table 2). In addition, T. melanosporum and Pseudogymnoascus destructans of Ascomycota also showed significantly high CG methylation and low non-CG methylation, while DNMT1 or DNMT5 as well as RID and Dim-2 was found in these two fungi (Figure 4B). The CG methylation of P. blakesleeanus of Mucoromycota with DNMT1 and Dim-2 was also higher than the non-CG methylation (Figure 4B). Therefore, the DNA methylation pattern in fungi was correlated with the DNA methyltransferase combination and associated with the classification of different phyla of fungi.
DNA methylation was not found in several fungal species; more than 20 yeast strains were described as having no detectable DNA methylation levels by GC/MS or LC-MS/MS (Tang et al., 2012; Capuano et al., 2014). Aspergillus flavus, Schizosaccharomyces pombe and Saccharomyces cerevisiae have non-detectable methylation based on WGBS, LC-MS/MS and HPLC methods (Binz et al., 1998; Liu et al., 2012; Capuano et al., 2014). A lack of DNMT homologous in the Saccharomyces cerevisiae genome was reported (Huang et al., 2016; Figure 4C), which can lead to no DNA methylation. S. pombe has a homolog of the DNMT2 family, termed Pombe Methyl Transferase 1 (Pmt1). However, Pmt1 appeared to be specific to tRNA modification and not associated with DNA methylation (Table 2; Wilkinson et al., 1995; Becker et al., 2012). A. flavus has one DNMT1 and one DNMT5, and the methylation level of A. flavus is quite low (similar to that of unmethylated lambda DNA, 0.44∼0.47%); hence, DNA methylation is considered absent in A. flavus, or de novo DNA methylation occurs transiently during the obscure sexual stage (Liu et al., 2012). Nonetheless, there is still low methylation in a few fungi lacking DNMTs, such as Y. lipolytica (0.36%) and Ustilago maydis (2.26%) (Figure 4C).
Variable DNA methylation levels are found in fungi and their life stages, thereby leaving their biological role an open question, especially in their life cycles. Fungal genomic studies that describe DNA methylation levels and methylation preference sites are still limited. To date, more than 80 fungi have been subjected to whole-genome sequencing. Of these, only 40 fungi have methylation data available, and 14 fungi have descriptions of methylation in different life stages (Figure 3A). Different DNA methylation levels were found in different life stages in 14 fungi; lower methylation levels were usually observed in mycelium (Figure 3A).
P. blakesleeanus showed 0.48 and 2.9% methylation levels in mycelium and spores, respectively, suggesting that gene expression may be regulated by DNA methylation and thereby impact the different stages of the life cycle (Figure 3A). Indeed, in P. blakesleeanus, it has been shown that methylation can repress loci transcriptionally; thus, active genes are often unmethylated (Antequera et al., 1985). A higher methylation level was found in the sclerotia of P. omnivorum than in the mycelium (Figure 3A). In P. omnivorum, the sclerotium is the structure for adapting to adverse conditions and allows survival for more than a decade deep in the soil. Therefore, this structure is a potential source of inactive genes and shows a high methylation level (Jupe et al., 1986). The DNA methylation level in gene-flanking regions of the forest pathogen H. parviporum with dual life strategies, saprotrophy on dead wood and necrotrophy on living trees, changes between different stages, suggesting that DNA methylation may contribute to condition-specific gene expression.
At different life stages of fungi, variable methylation may imply complicated epigenetic regulation, possibly associated with changes in morphology upon life stage switches. In T. melanosporum, hypomethylated or unmethylated TEs were found to be transcriptionally active, with higher expression levels in free-living mycelium (FLM) compared to fruitbodies (FBs) (Montanini et al., 2014).
In Basidiomycota, C. cinerea is a model species in studies of photomorphogenesis; it has been revealed that the fruiting mechanism requires specific environmental conditions, such as temperatures between 25°C and 28°C, humidity >85% and a typical day/night rhythm, implying epigenetic coregulation (Kamada et al., 2010). However, the life stage switch does not seem to be conserved among Basidiomycota fungi.
In 2010, keystone research on summarized that genome-wide DNA methylation was an important process in evolutionary adaptation and conservation (Jeltsch, 2010; Zemach et al., 2010; Su et al., 2011). In this review, we provided an overview of fungal DNA methyltransferases, as well as the recent reports on the potential association between fungal DNMT5 and the RdDM pathway. Furthermore, we collected DNA methylation data (reported statistics and raw sequencing data whichever available) and domain features of all the DNA methyltransferases from 40 fungal species to present a comprehensive summary of current fungal DNA methylation, pointing to several directions and gaps for future investigation.
As summarized in our review, the correlation amongst the fungal DNA methylation levels, phylogenies of the fungal DNMT1 family and RID/Masc1 domains, and fungal DNMTs combinations showed that the patterns of DNA methylation are closely associated with the DNMTs. Since the fungal taxonomy has undergone major challenges in the past few decades according to the rapid development in fungal genomics, the highly-diverse features of the species such as morphologies, habitats, lifestyles and epigenomes together with the genetic data may present a comprehensive profile for individual fungal species, to provide an improved description for fungal diversity.
Different fungi often have different types of DNMTs in their genomes, leading to high divergence in their methylation profiles. In truffle, treatment of 5-azacytidine, an inhibitor of DNA methyltransferases, is used to explore the correlation between fungal phenotypes and the potential epigenetic regulation by DNA methylation (Montanini et al., 2014). Still, the epigenetic regulation in fungi is still largely unknown. In addition to DNA methylation, histone modifications and sRNA are also reported to play an important role on diverse aspect of fungal biology (Brosch et al., 2008; Nakayashiki and Nguyen, 2008; Smith et al., 2010; Chang et al., 2012; Jeon et al., 2014; Honda et al., 2016; Dubey and Jeon, 2017; Freitag, 2017; Elías-Villalobos et al., 2019; Chung et al., 2020). There are growing observations describing the profiles of different kinds of epigenetic marks in fungi, yet the studies of integrated fungal epigenetic modifications are limited in only a handful of fungal species, such as N. crassa, S. pombe, and S. cerevisiae. In Neurospora crassa, DNA methylation requires histone deacetylases (HDACs). The mutation of histone deacetylase genes causes global increase in histone acetylation level in correlation with the site-specific loss of DNA methylation (Smith et al., 2010); Trichostatin A (TSA), the inhibitor of HDAC activity, is found to result in the selective loss of demethylation (Selker, 1998). In addition, the histone deacetylase complex HCHC works with the DNMT complex DIM-2–HP1 to establish and maintain heterochromatin, is required for proper DNA methylation of N. crassa (Honda et al., 2012, 2016). Altogether the observations between DNA methylation and HDACs activities suggest a close association between DNA methylation and histone modification in fungal epigenomes.
As fungi tend to have close interactions with the plants, animals and the environments, complicated epigenomic regulations may be involved. By combining the epigenome data of DNA methylation and other epigenetic marks will definitely warrant future fungal research.
Y-SN and Y-CH drafted the manuscript. M-RY performed phylogenetic analysis and methylome reanalysis. P-YC coordinated the study and edited the manuscript. All authors read and approved the final manuscript.
This study was financially supported by grants from the Ministry of Science and Technology in Taiwan (MOST-106-2311-B-001-035-MY3 and MOST 108-2313-B-001-013-MY3).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Chia-Jung Hsieh for collecting the DNA methyltransferase sequences of eukaryotes. We also thank Jo-Wei A. Hsieh, Pei-Yu Lin, Chih-Hung Hsieh, and Ying-Yang Li for editorial assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.616922/full#supplementary-material
Antequera, F., Tamame, M., Vilanueva, J. R., and Santos, T. (1985). Developmental modulation of DNA methylation in the fungus Phycomyces blakesleeanus. Nucleic Acids Res. 13, 6545–6558.
Antequera, F., Tamame, M., Villanueva, J. R., and Santos, T. (1984). DNA methylation in the fungi. J. Biol. Chem. 259, 8033–8036.
Aramayo, R., and Selker, E. U. (2013). Neurospora crassa, a model system for epigenetics research. Cold Spring Harb. Perspect. Biol. 5:a017921. doi: 10.1101/cshperspect.a017921
Becker, M., Muller, S., Nellen, W., Jurkowski, T. P., Jeltsch, A., and Ehrenhofer-Murray, A. E. (2012). Pmt1, a Dnmt2 homolog in Schizosaccharomyces pombe, mediates tRNA methylation in response to nutrient signaling. Nucleic Acids Res. 40, 11648–11658. doi: 10.1093/nar/gks956
Bewick, A. J., Hofmeister, B. T., Powers, R. A., Mondo, S. J., Grigoriev, I. V., James, T. Y., et al. (2019). Diversity of cytosine methylation across the fungal tree of life. Nat. Ecol. Evol. 3, 479–490. doi: 10.1038/s41559-019-0810-9
Binz, T., D’Mello, N., and Horgen, P. A. (1998). A Comparison of DNA methylation levels in selected isolates of higher fungi. Mycologia 90, 785–790. doi: 10.2307/3761319
Borgognone, A., Castanera, R., Morselli, M., López-Varas, L., Rubbi, L., Pisabarro, A. G., et al. (2018). Transposon-associated epigenetic silencing during Pleurotus ostreatus life cycle. DNA Res. 25, 451–464. doi: 10.1093/dnares/dsy016
Brosch, G., Loidl, P., and Graessle, S. (2008). Histone modifications and chromatin dynamics: a focus on filamentous fungi. FEMS Microbiol. Rev. 32, 409–439. doi: 10.1111/j.1574-6976.2007.00100.x
Buckner, B., Novotny, C. P., and Ullrich, R. C. (1988). Organization of the ribosomal RNA genes of Schizophyllum commune. Curr. Genet. 13, 417–424. doi: 10.1007/BF00365663
Callebaut, I., Courvalin, J. C., and Mornon, J. P. (1999). The BAH (bromo-adjacent homology) domain: a link between DNA methylation, replication and transcriptional regulation. FEBS Lett. 446, 189–193. doi: 10.1016/s0014-5793(99)00132-5
Cano, C., Herrera-Estrella, L., and Ruiz-Herrera, J. (1988). DNA methylation and polyamines in regulation of development of the fungus Mucor rouxii. J. Bacteriol. 170, 5946–5948. doi: 10.1128/jb.170.12.5946-5948.1988
Capuano, F., Mulleder, M., Kok, R., Blom, H. J., and Ralser, M. (2014). Cytosine DNA methylation is found in Drosophila melanogaster but absent in Saccharomyces cerevisiae, Schizosaccharomyces pombe, and other yeast species. Anal. Chem. 86, 3697–3702. doi: 10.1021/ac500447w
Castanera, R., Lopez-Varas, L., Borgognone, A., LaButti, K., Lapidus, A., Schmutz, J., et al. (2016). Transposable elements versus the fungal genome: impact on whole-genome architecture and transcriptional profiles. PLoS Genet. 12:e1006108. doi: 10.1371/journal.pgen.1006108
Catania, S., Dumesic, P. A., Pimentel, H., Nasif, A., Stoddard, C. I., Burke, J. E., et al. (2020). Evolutionary persistence of DNA methylation for millions of years after ancient loss of a de novo methyltransferase. Cell 180, 263.–277.
Chang, S.-S., Zhang, Z., and Liu, Y. (2012). RNA interference pathways in fungi: mechanisms and functions. Ann. Rev. Microbiol. 66, 305–323. doi: 10.1146/annurev-micro-092611-150138
Chen, T., and Li, E. (2004). “Structure and function of eukaryotic DNA methyltransferases,” in Current Topics in Developmental Biology, ed. G. Schatten (Amsterdam: Elsevier), 55–89. doi: 10.1016/s0070-2153(04)60003-2
Chung, C.-Y. L. Y.-M., Hunyadi, Y.-C. W. A., and Chang, C. C. W. F.-R. (2020). Natural products development under epigenetic modulation in fungi. Phytochem. Rev. 19, 1323–1340. doi: 10.1007/s11101-020-09684-7
de Mendoza, A., Lister, R., and Bogdanovic, O. (2020). Evolution of DNA methylome diversity in eukaryotes. J. Mol. Biol. 432, 1687–1705. doi: 10.1016/j.jmb.2019.11.003
Ding, L., and Forsburg, S. L. (2014). Essential domains of Schizosaccharomyces pombe Rad8 required for DNA damage response. G3 Genes Genomes Genet. 4, 1373–1384.
Doe, C. L., Murray, J. M., Shayeghi, M., Hoskins, M., Lehmann, A. R., Carr, A. M., et al. (1993). Cloning and characterisation of the Schizosaccharomyces pombe rad8 gene, a member of the SNF2 helicase family. Nucleic Acids Res. 21, 5964–5971. doi: 10.1093/nar/21.25.5964
Du, J., Zhong, X., Bernatavichute, Y. V., Stroud, H., Feng, S., Caro, E., et al. (2012). Dual binding of chromomethylase domains to H3K9me2-containing nucleosomes directs DNA methylation in plants. Cell 151, 167–180. doi: 10.1016/j.cell.2012.07.034
Dubey, A., and Jeon, J. (2017). Epigenetic regulation of development and pathogenesis in fungal plant pathogens. Mol. Plant Pathol. 18, 887–898. doi: 10.1111/mpp.12499
Dumesic, P. A., Stoddard, C. I., Catania, S., Narlikar, G. J., and Madhani, H. D. (2020). ATP Hydrolysis by the SNF2 Domain of Dnmt5 is coupled to both specific recognition and modification of hemimethylated DNA. Mol. Cell 79, 127–139.e4.
Ehrlich, M., Gama-Sosa, M. A., Huang, L. H., Midgett, R. M., Kuo, K. C., McCune, R. A., et al. (1982). Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 10, 2709–2721. doi: 10.1093/nar/10.8.2709
Eisen, J. A., Sweder, K. S., and Hanawalt, P. C. (1995). Evolution of the SNF2 family of proteins: subfamilies with distinct sequences and functions. Nucleic Acids Res. 23, 2715–2723. doi: 10.1093/nar/23.14.2715
Elías-Villalobos, A., Barrales, R. R., and Ibeas, J. I. (2019). Chromatin modification factors in plant pathogenic fungi: insights from Ustilago maydis. Fungal Genet. Biol. 129, 52–64. doi: 10.1016/j.fgb.2019.04.006
Filippovich, S., Bachurina, G. P., and Kritskii, M. S. (2004). [Effect of 5-azacytidine on the light-sensitive formation of sexual and asexual reproductive structures in wc-1 and wc-2 mutants of Neurospora crassa]. Prikl. Biokhim. Mikrobiol. 40, 466–471.
Freitag, M. (2017). Histone methylation by SET domain proteins in fungi. Ann. Rev. Microbiol. 71, 413–439. doi: 10.1146/annurev-micro-102215-095757
Fronk, J., and Magiera, R. (1994). DNA methylation during differentiation of a lower eukaryote, Physarum polycephalum. Biochem. J. 304, 101–104. doi: 10.1042/bj3040101
Fuks, F., Burgers, W. A., Brehm, A., Hughes-Davies, L., and Kouzarides, T. (2000). DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet. 24, 88–91. doi: 10.1038/71750
Galagan, J. E., and Selker, E. U. (2004). RIP: the evolutionary cost of genome defense. Trends Genet. 20, 417–423. doi: 10.1016/j.tig.2004.07.007
Gladyshev, E. (2017). Repeat-induced point mutation and other genome defense mechanisms in fungi. Fungal King. 5, 687–699.
Goll, M. G., and Bestor, T. H. (2005). Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 74, 481–514. doi: 10.1146/annurev.biochem.74.010904.153721
Goll, M. G., Kirpekar, F., Maggert, K. A., Yoder, J. A., Hsieh, C.-L., Zhang, X., et al. (2006). Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311, 395–398. doi: 10.1126/science.1120976
Goyon, C., and Faugeron, G. (1989). Targeted transformation of Ascobolus immersus and de novo methylation of the resulting duplicated DNA sequences. Mol. Cell Biol. 9, 2818–2827. doi: 10.1128/mcb.9.7.2818
Goyon, C., Rossignol, J.-L., and Faugeron, G. (1996). Native DNA repeats and methylation in Ascobolus. Nucleic Acids Res. 24, 3348–3356. doi: 10.1093/nar/24.17.3348
Grayburn, W. S., and Selker, E. U. (1989). A natural case of RIP: degeneration of the DNA sequence in an ancestral tandem duplication. Mol. Cell Biol. 9, 4416–4421. doi: 10.1128/mcb.9.10.4416
Groth, M., Stroud, H., Feng, S., Greenberg, M. V., Vashisht, A. A., Wohlschlegel, J. A., et al. (2014). SNF2 chromatin remodeler-family proteins FRG1 and-2 are required for RNA-directed DNA methylation. Proc. Natl. Acad. Sci. 111, 17666–17671. doi: 10.1073/pnas.1420515111
Holliday, R. (2006). Epigenetics: a historical overview. Epigenetics 1, 76–80. doi: 10.4161/epi.1.2.2762
Honda, S., Bicocca, V. T., Gessaman, J. D., Rountree, M. R., Yokoyama, A., Eun, Y. Y., et al. (2016). Dual chromatin recognition by the histone deacetylase complex HCHC is required for proper DNA methylation in Neurospora crassa. Proc. Natl. Acad. Sci. 113, E6135–E6144.
Honda, S., Lewis, Z. A., Shimada, K., Fischle, W., Sack, R., and Selker, E. U. (2012). Heterochromatin protein 1 forms distinct complexes to direct histone deacetylation and DNA methylation. Nat. Struct. Mol. Biol. 19:471. doi: 10.1038/nsmb.2274
Hosseini, S., Meunier, C., Nguyen, D., Reimegård, J., and Johannesson, H. (2020). Comparative analysis of genome-wide DNA methylation in Neurospora. Epigenetics 15, 972–987. doi: 10.1080/15592294.2020.1741758
Hua, S., Qi, B., Fu, Y.-P., and Li, Y. (2017). DNA Methylation Changes in Pleurotus eryngii Subsp. tuoliensis (Bailinggu) in Response to Low Temperature Stress. Int. J. Agricult. Biol. 19, 328–334. doi: 10.17957/IJAB/15.0286
Huang, R., Ding, Q., Xiang, Y., Gu, T., and Li, Y. (2016). Comparative analysis of DNA methyltransferase gene family in fungi: a focus on basidiomycota. Front. Plant Sci. 7:1556. doi: 10.3389/fpls.2016.01556
Huff, J. T., and Zilberman, D. (2014). Dnmt1-independent CG methylation contributes to nucleosome positioning in diverse eukaryotes. Cell 156, 1286–1297. doi: 10.1016/j.cell.2014.01.029
Ishiyama, S., Nishiyama, A., Saeki, Y., Moritsugu, K., Morimoto, D., Yamaguchi, L., et al. (2017). Structure of the Dnmt1 reader module complexed with a unique two-mono-ubiquitin mark on histone H3 reveals the basis for DNA methylation maintenance. Mol. Cell 68, 350.–360.
Jeltsch, A. (2010). Molecular biology. Phylogeny of methylomes. Science 328, 837–838. doi: 10.1126/science.1190738
Jeltsch, A., Nellen, W., and Lyko, F. (2006). Two substrates are better than one: dual specificities for Dnmt2 methyltransferases. Trends Biochem. Sci. 31, 306–308. doi: 10.1016/j.tibs.2006.04.005
Jeon, J., Choi, J., Lee, G. W., Park, S. Y., Huh, A., Dean, R. A., et al. (2015). Genome-wide profiling of DNA methylation provides insights into epigenetic regulation of fungal development in a plant pathogenic fungus, Magnaporthe oryzae. Sci. Rep. 5:8567. doi: 10.1038/srep08567
Jeon, J., Kwon, S., and Lee, Y.-H. (2014). Histone acetylation in fungal pathogens of plants. Plant Pathol. J. 30, 1–9. doi: 10.5423/ppj.rw.01.2014.0003
Jupe, E. R., Magill, J. M., and Magill, C. W. (1986). Stage-specific DNA methylation in a fungal plant pathogen. J. Bacteriol. 165, 420–423. doi: 10.1128/jb.165.2.420-423.1986
Kaiser, S., Jurkowski, T. P., Kellner, S., Schneider, D., Jeltsch, A., and Helm, M. (2017). The RNA methyltransferase Dnmt2 methylates DNA in the structural context of a tRNA. RNA Biol. 14, 1241–1251. doi: 10.1080/15476286.2016.1236170
Kamada, T., Sano, H., Nakazawa, T., and Nakahori, K. (2010). Regulation of fruiting body photomorphogenesis in Coprinopsis cinerea. Fungal Genet. Biol. 47, 917–921. doi: 10.1016/j.fgb.2010.05.003
Keller, N. P., Bergstrom, G. C., and Yoder, O. (1991). Mitotic stability of transforming DNA is determined by its chromosomal configuration in the fungus Cochliobolus heterostrophus. Curr. Genet. 19, 227–233.
Kim, D. H. (1997). Induced change in DNA methylation of Fusarium oxysporum f. sp. niveum due to successive transfer. BMB Rep. 30, 216–221.
Kouzminova, E., and Selker, E. U. (2001). dim-2 encodes a DNA methyltransferase responsible for all known cytosine methylation in Neurospora. EMBO J. 20, 4309–4323. doi: 10.1093/emboj/20.15.4309
Law, J. A., and Jacobsen, S. E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220. doi: 10.1038/nrg2719
Lee, D. W., Freitag, M., Selker, E. U., and Aramayo, R. (2008). A cytosine methyltransferase homologue is essential for sexual development in Aspergillus nidulans. PLoS One 3:e2531. doi: 10.1371/journal.pone.0002531
Leonhardt, H., Page, A. W., Weier, H.-U., and Bestor, T. H. (1992). A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 71, 865–873. doi: 10.1016/0092-8674(92)90561-p
Li, W., Wang, Y., Zhu, J., Wang, Z., Tang, G., and Huang, B. (2017). Differential DNA methylation may contribute to temporal and spatial regulation of gene expression and the development of mycelia and conidia in entomopathogenic fungus Metarhizium robertsii. Fungal Biol. 121, 293–303. doi: 10.1016/j.funbio.2017.01.002
Lippman, Z., Gendrel, A. V., Black, M., Vaughn, M. W., Dedhia, N., McCombie, W. R., et al. (2004). Role of transposable elements in heterochromatin and epigenetic control. Nature 430, 471–476. doi: 10.1038/nature02651
Lister, R., and Ecker, J. R. (2009). Finding the fifth base: genome-wide sequencing of cytosine methylation. Genome Res. 19, 959–966. doi: 10.1101/gr.083451.108
Liu, S. Y., Lin, J. Q., Wu, H. L., Wang, C. C., Huang, S. J., Luo, Y. F., et al. (2012). Bisulfite sequencing reveals that Aspergillus flavus holds a hollow in DNA methylation. PLoS One 7:e30349. doi: 10.1371/journal.pone.0030349
Malagnac, F., Wendel, B., Goyon, C., Faugeron, G., Zickler, D., Rossignol, J. L., et al. (1997). A gene essential for de novo methylation and development in Ascobolus reveals a novel type of eukaryotic DNA methyltransferase structure. Cell 91, 281–290. doi: 10.1016/s0092-8674(00)80410-9
Martin, J. L., and McMillan, F. M. (2002). SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr. Opin. Struct. Biol. 12, 783–793. doi: 10.1016/s0959-440x(02)00391-3
Matzke, M. A., and Mosher, R. A. (2014). RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat. Rev. Genet. 15, 394–408. doi: 10.1038/nrg3683
Mishra, P. K., Baum, M., and Carbon, J. (2011). DNA methylation regulates phenotype-dependent transcriptional activity in Candida albicans. Proc. Natl. Acad. Sci. U. S. A. 108, 11965–11970. doi: 10.1073/pnas.1109631108
Montanini, B., Chen, P. Y., Morselli, M., Jaroszewicz, A., Lopez, D., Martin, F., et al. (2014). Non-exhaustive DNA methylation-mediated transposon silencing in the black truffle genome, a complex fungal genome with massive repeat element content. Genome Biol. 15:411. doi: 10.1186/s13059-014-0411-5
Nakayashiki, H., and Nguyen, Q. B. (2008). RNA interference: roles in fungal biology. Curr. Opin. Microbiol. 11, 494–502. doi: 10.1016/j.mib.2008.10.001
Naranjo-Ortiz, M. A., and Gabaldón, T. (2019). Fungal evolution: diversity, taxonomy and phylogeny of the Fungi. Biol. Rev. 94, 2101–2137. doi: 10.1111/brv.12550
Ponger, L. C., and Li, W.-H. (2005). Evolutionary diversification of DNA methyltransferases in eukaryotic genomes. Mol. Biol. Evol. 22, 1119–1128. doi: 10.1093/molbev/msi098
Reyna-Lopez, G., Simpson, J., and Ruiz-Herrera, J. (1997). Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms. Mole. Gener. Genet. MGG 253, 703–710. doi: 10.1007/s004380050374
Riggs, A. D. (1975). X inactivation, differentiation, and DNA methylation. Cytogenet. Cell Genet. 14, 9–25. doi: 10.1159/000130315
Rose, N. R., and Klose, R. J. (2014). Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta 1839, 1362–1372. doi: 10.1016/j.bbagrm.2014.02.007
Rountree, M. R., Bachman, K. E., and Baylin, S. B. (2000). DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 25, 269–277. doi: 10.1038/77023
Russell, P., Rodland, K., Rachlin, E., and McCloskey, J. (1987). Differential DNA methylation during the vegetative life cycle of Neurospora crassa. J. Bacteriol. 169, 2902–2905. doi: 10.1128/jb.169.6.2902-2905.1987
Sbaraini, N., Bellini, R., Penteriche, A. B., Guedes, R. L. M., Garcia, A. W. A., Gerber, A. L., et al. (2019). Genome-wide DNA methylation analysis of Metarhizium anisopliae during tick mimicked infection condition. BMC Genom. 20, 1–11. doi: 10.1186/s12864-019-6220-1
Schmitz, R. J., Lewis, Z. A., and Goll, M. G. (2019). DNA methylation: shared and divergent features across eukaryotes. Trends Genet. 35, 818–827. doi: 10.1016/j.tig.2019.07.007
Selker, E. U. (1998). Trichostatin A causes selective loss of DNA methylation in Neurospora. Proc. Natl. Acad. Sci. 95, 9430–9435. doi: 10.1073/pnas.95.16.9430
Selker, E. U. (2002). Repeat-induced gene silencing in fungi. Adv. Genet. 46, 439–450. doi: 10.1016/s0065-2660(02)46016-6
Selker, E. U., Cambareri, E. B., Jensen, B. C., and Haack, K. R. (1987). Rearrangement of duplicated DNA in specialized cells of Neurospora. Cell 51, 741–752. doi: 10.1016/0092-8674(87)90097-3
Selker, E. U., Tountas, N. A., Cross, S. H., Margolin, B. S., Murphy, J. G., Bird, A. P., et al. (2003). The methylated component of the Neurospora crassa genome. Nature 422, 893–897. doi: 10.1038/nature01564
Smith, K. M., Dobosy, J. R., Reifsnyder, J. E., Rountree, M. R., Anderson, D. C., Green, G. R., et al. (2010). H2B-and H3-specific histone deacetylases are required for DNA methylation in Neurospora crassa. Genetics 186, 1207–1216. doi: 10.1534/genetics.110.123315
So, K.-K., Ko, Y.-H., Chun, J., Bal, J., Jeon, J., Kim, J.-M., et al. (2018). Global DNA methylation in the chestnut blight fungus Cryphonectria parasitica and genome-wide changes in DNA methylation accompanied with sectorization. Front. Plant Sci. 9:103. doi: 10.3389/fpls.2018.00103
Specht, C., Novotny, C., and Ullrich, R. (1984). Strain specific differences in ribosomal DNA from the fungus Schizophyllum commune. Curr. Genet. 8, 219–222. doi: 10.1007/BF00417819
Su, Z., Han, L., and Zhao, Z. (2011). Conservation and divergence of DNA methylation in eukaryotes: new insights from single base-resolution DNA methylomes. Epigenetics 6, 134–140. doi: 10.4161/epi.6.2.13875
Syeda, F., Fagan, R. L., Wean, M., Avvakumov, G. V., Walker, J. R., Xue, S., et al. (2011). The replication focus targeting sequence (RFTS) domain is a DNA-competitive inhibitor of Dnmt1. J. Biol. Chem. 286, 15344–15351. doi: 10.1074/jbc.M110.209882
Taiko, K. T., Saze, H., and Kakutani, T. (2015). DNA methylation within transcribed regions. Plant Physiol. 168, 1219–1225. doi: 10.1104/pp.15.00543
Tang, Y., Gao, X. D., Wang, Y., Yuan, B. F., and Feng, Y. Q. (2012). Widespread existence of cytosine methylation in yeast DNA measured by gas chromatography/mass spectrometry. Anal. Chem. 84, 7249–7255. doi: 10.1021/ac301727c
Vaughn, M. W., Tanurdzic, M., Lippman, Z., Jiang, H., Carrasquillo, R., Rabinowicz, P. D., et al. (2007). Epigenetic natural variation in Arabidopsis thaliana. PLoS Biol. 5:e174. doi: 10.1371/journal.pbio.0050174
Weber, M., Davies, J. J., Wittig, D., Oakeley, E. J., Haase, M., Lam, W. L., et al. (2005). Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 37, 853–862. doi: 10.1038/ng1598
Wilkinson, C. R., Bartlett, R., Nurse, P., and Bird, A. P. (1995). The fission yeast gene pmt1+ encodes a DNA methyltransferase homologue. Nucleic Acids Res. 23, 203–210. doi: 10.1093/nar/23.2.203
Xin, X., Yin, J., Zhang, B., Li, Z., Zhao, S., and Gui, Z. (2019). Genome-wide analysis of DNA methylation in subcultured Cordyceps militaris. Archiv. Microbiol. 201, 369–375. doi: 10.1007/s00203-019-01621-3
Yang, K., Liang, L., Ran, F., Liu, Y., Li, Z., Lan, H., et al. (2016). The DmtA methyltransferase contributes to Aspergillus flavus conidiation, sclerotial production, aflatoxin biosynthesis and virulence. Sci. Rep. 6:23259.
Yang, N., and Xu, R. M. (2013). Structure and function of the BAH domain in chromatin biology. Crit. Rev. Biochem. Mol. Biol. 48, 211–221. doi: 10.3109/10409238.2012.742035
Yarychkivska, O., Shahabuddin, Z., Comfort, N., Boulard, M., and Bestor, T. H. (2018). BAH domains and a histone-like motif in DNA methyltransferase 1 (DNMT1) regulate de novo and maintenance methylation in vivo. J. Biol. Chem. 293, 19466–19475. doi: 10.1074/jbc.ra118.004612
Zemach, A., McDaniel, I. E., Silva, P., and Zilberman, D. (2010). Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328, 916–919. doi: 10.1126/science.1186366
Zemach, A., and Zilberman, D. (2010). Evolution of eukaryotic DNA methylation and the pursuit of safer sex. Curr. Biol. 20, R780–R785. doi: 10.1016/j.cub.2010.07.007
Zeng, Z., Wu, J., Kovalchuk, A., Raffaello, T., Wen, Z., Liu, M., et al. (2019). Genome-wide DNA methylation and transcriptomic profiles in the lifestyle strategies and asexual development of the forest fungal pathogen Heterobasidion parviporum. Epigenetics 14, 16–40. doi: 10.1080/15592294.2018.1564426
Zhang, X., Yazaki, J., Sundaresan, A., Cokus, S., Chan, S. W., Chen, H., et al. (2006). Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell 126, 1189–1201. doi: 10.1016/j.cell.2006.08.003
Zhang, Z., Wen, J., Li, J., Ma, X., Yu, Y., Tan, X., et al. (2018). The evolution of genomic and epigenomic features in two Pleurotus fungi. Sci. Rep. 8:8313.
Zhu, Y., Xu, J., Sun, C., Zhou, S., Xu, H., Nelson, D. R., et al. (2015). Chromosome-level genome map provides insights into diverse defense mechanisms in the medicinal fungus Ganoderma sinense. Sci. Rep. 5:11087. doi: 10.1038/srep11087
Keywords: fungi, epigenomics, DNA methyltransferases, DNA methylation, DNMT5, transposable element
Citation: Nai Y-S, Huang Y-C, Yen M-R and Chen P-Y (2021) Diversity of Fungal DNA Methyltransferases and Their Association With DNA Methylation Patterns. Front. Microbiol. 11:616922. doi: 10.3389/fmicb.2020.616922
Received: 13 October 2020; Accepted: 28 December 2020;
Published: 22 January 2021.
Edited by:
Junbiao Dai, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Jiamu Du, Southern University of Science and Technology, ChinaCopyright © 2021 Nai, Huang, Yen and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pao-Yang Chen, cGFveWFuZ0BnYXRlLnNpbmljYS5lZHUudHc=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.