 Sepehr Aghajanian
Sepehr Aghajanian Majid Teymoori-Rad2
Majid Teymoori-Rad2 Ghazale Molaverdi
Ghazale Molaverdi Sayed-Hamidreza Mozhgani
Sayed-Hamidreza Mozhgani- 1Student Research Committee, Alborz University of Medical Sciences, Karaj, Iran
- 2Department of Virology, School of Public Health, Tehran University of Medical Sciences, Tehran, Iran
- 3Non-communicable Diseases Research Center, Alborz University of Medical Sciences, Karaj, Iran
- 4Department of Microbiology, School of Medicine, Alborz University of Medical Sciences, Karaj, Iran
HTLV-1-Associated Myelopathy/Tropical Spastic Paraparesis (HAM/TSP) is a neuropathological disorder in 1–3% of individuals infected with Human T-lymphotropic virus 1 (HTLV-1). This condition is characterized by progressive spastic lower limb weakness and paralysis, lower back pain, bladder incontinence, and mild sensory disturbances resembling spinal forms of multiple sclerosis. This disease also causes chronic disability and is therefore associated with high health burden in areas where HTLV-1 infection is endemic. Despite various efforts in understanding the virus and discovery of novel diagnostic markers, and cellular and viral interactions, HAM/TSP management is still unsatisfactory and mainly focused on symptomatic alleviation, and it hasn’t been explained why only a minority of the virus carriers develop HAM/TSP. This comprehensive review focuses on host and viral factors in association with immunopathology of the disease in hope of providing new insights for drug therapies or other forms of intervention.
Introduction
Human T-lymphotropic virus type 1 (HTLV-1), discovered in 1980, is a retrovirus from the Orthoretrovirinae subfamily of Deltaretrovirus genus. HTLV-1 has seven reported subtypes (subtypes a to g), identified by their related geographic regions and genetic variability (Verdonck et al., 2007; Bangham, 2018; Nozuma and Jacobson, 2019). The virus primarily infects CD4+ T-cells and causes lifelong infections in humans. It propagates mostly, during the chronic phase of infection, through clonal expansion of infected cells and not by de novo infection with cell-free virions, a unique trait even among other genera of retroviruses. The main routes of transmission are through sexual intercourse, breastfeeding, cellular blood products, and to a lesser extent, organ transplantations and perinatal infections (Kawase et al., 1992; Armstrong et al., 2012; Bangham et al., 2015; Rosadas and Taylor, 2019). While most of the infected subjects maintain normal life and are asymptomatic carriers (AC), the virus disrupts the immune system function in some individuals, leading to the development of malignant neoplasms such as Adult T-cell leukemia/leukemia (ATLL) and is associated with immune hypersensitivity conditions like uveitis, arthritis, atherosclerosis, Sjogren’s syndrome, thyroiditis, polymyositis, infective dermatitis, and most notably HTLV-1 associated myelopathy/Tropical spastic paraparesis (HAM/TSP) (Abolbashari et al., 2018; Caswell and Manavi, 2020; Schierhout et al., 2020; Umekita and Okayama, 2020).
HAM/TSP is described as a chronic paraparesis with an insidious onset and a similar clinical picture to Multiple sclerosis (Araujo and Silva, 2006). The disease is characterized by progressive demyelination and neuronal loss in CNS. Lesions occur mainly in posterior, and lateral columns of middle to lower thoracic parts of spinal cord (Iwasaki, 1990; Osame, 2002; Bangham et al., 2015). Spinal cord atrophy and perivascular and parenchymal infiltration of mononuclear cells in CNS have also been observed and are regarded as pathological hallmarks of HAM/TSP (Osame, 2002; Bangham et al., 2015; Nozuma and Jacobson, 2019). Patients present with lower limb paraparesis, lower back pain, dysautonomia, mild sensory disturbances, constipation and cognitive decline (Araujo and Silva, 2006; Araujo and Wedemann, 2019; Tsutsumi et al., 2019). Treatment is primarily focused on control of pain, muscle spasms, and symptomatic treatment of autonomic disturbances, namely, bladder incontinence, constipation, and erectile dysfunction (Bangham et al., 2015). HAM/TSP has a shorter latency than ATLL, ranging from months to decades before the development of symptoms (Olière et al., 2011).
Since the discovery of HTLV-1 as the etiologic agent of HAM/TSP (Poiesz et al., 1980) and establishment of diagnostic criteria for the disease by World Health Organization (World Health Organization. Regional Office for the Western Pacific, 1988), innovative advances in mapping the viral interactions with the host have led to the discovery of new viral mechanisms in the pathogenesis of HTLV-1 diseases and novel insights in diagnosing the disease via pathological markers, but still, the mechanisms underlying tissue damage and disease development are not fully understood. This review characterizes the pathological mechanisms in developing HAM/TSP with emphasis on HTLV-1 genes, structure, and cell-cell and virus-cell interactions throughout the host.
Host Factors in Development of HAM/TSP
Among the host-dependent factors, genotype of major histocompatibility complex/human leukocyte antigen 1 (MHC-I/HLA class I) is one of the factors determining the effectiveness of cell-mediated responses and viral burden which is described as the number of viral copies integrated into the host genome in peripheral blood mononuclear cells (Proviral load; PVL) in individuals. Class-I heterozygosity may also reduce PVL in infected subjects (Jeffery et al., 2000). Certain class II HLAs are also associated with HAM/TSP by increasing CD4+ T-cell activity and subsequent tissue damage (Jeffery et al., 1999; Bangham et al., 2015). Comparatively, protective HLAs are more likely to be detected in ACs and predisposing HLAs are more frequent in patients with HAM/TSP. For example, the protective effects of HLA-A∗02:07 and HLA-C∗08 and the detrimental effects of HLA-B∗54 has been demonstrated in southern Japan (Boelen et al., 2018). HLA-Cw∗08 was similarly seen in both ACs and HAM / TSP patients in a Brazilian study, but was associated with a protective effect in Japan and higher susceptibility to HAM/TSP in Iran (Vallinoto et al., 2019). Class II HLA-DRB1∗0101 has been identified as a predisposing factor to the disease in Japan, Iran, and Spain and it has been shown that the effect is exacerbated in the absence of the protective effect of HLA-A∗02 (Jeffery et al., 1999; Saito, 2019; Vallinoto et al., 2019). Haplotype HLA-B∗0702-Cw∗0702-DRB1∗0101-DQB1∗0501 is also predisposing in the absence of HLA-A∗02 (Catalan-Soares et al., 2009). In addition, the protective effects of HLA-B∗07 in Spain and HLA-A∗0201 and HLA-Cw∗0801 in Japan are documented as well (Vallinoto et al., 2019). The predisposing effects HLA-B∗5401 have been proven, but are not observed in Brazil (Catalan-Soares et al., 2009; Bangham et al., 2015; Enose-Akahata and Jacobson, 2019).
The effect of class 1 HLAs on disease progression in HTLV-1 infection is affected by Inhibitory killer cell immunoglobulin-like receptors (iKIRs), which are expressed on natural killer (NK) cells as well as CD8+ T-cells. iKIR enhance the HLA protective and detrimental effects through binding with their HLA class 1 ligands and increasing the lifespan of CD8+ T-cells (Boelen et al., 2018). The abundance of HLAs involved in determining the course of infection and disease progression indicates that the development of HAM/TSP is reliant on a dynamic interaction between the host and the virus. Polymorphisms and genetic variability in other genes also influence the development of HAM/TSP; some of which are listed in Table 1. It would be an interesting topic to test whether individuals bearing more genetic risk factors would benefit from a more aggressive treatment plan.

Table 1. Predisposing and protective factors for development of HAM/TSP.
Host factors that may precipitate ACs to HAM/TSP may not be limited to genetic factors. Although no concrete study on environmental factors for HAM/TSP exists, it is believed that HAM/TSP is more likely than ATLL to develop as the result infection via blood transfusion, whereas ATLL develops more often in recipients of vertical transmission (Oger, 2007). This observation could be the result of the greater latency period of ATLL (Olière et al., 2011), which would manifest as higher incidence of the disease in individuals infected in the earlier stages of life (Vertical transmission).
HTLV-1 Virology, Gene Expression, and Associated Proteins
HTLV-1 enveloped virions contain the 9-kb positive-sense RNA which serves as a template for the provirus. The RNA is packaged in the viral core with the viral nucleocapsid protein, surrounded by capsid and matrix proteins. The virus relies on cell-to-cell contact of infected host cells to spread, which are usually CD4+ or CD8+ T-cells (Hoshino, 2012). The provirus encodes gag, pro, pol, and env genes observed in other retroviruses. Matrix, Capsid, and Nucleocapsid proteins are products of cleavage of gag encoded p55 by viral protease. The Reverse Transcriptase, RNase H, and Integrase is encoded by the pol gene. Moreover, HTLV-1 virion surface receptor units gp46 (Surface unit) and gp21 (transmembrane unit) are synthesized from cleaving gp61, the precursor protein encoded by the env gene; While HTLV-1 protease is encoded by the pro gene. In addition to these structural proteins, HTLV-1 genome also includes a pX region containing viral regulatory and accessory genes for following proteins with partially overlapping open reading frames: Tax, Rex, p8, p12, p13, p21, p30, and HTLV-1 Basic Zipper Factor (HBZ) (Journo et al., 2009; Giam and Semmes, 2016; Soltani et al., 2019; Figure 1). At each end of the genome is a long terminal repeat (LTR) sequence that regulates various stages in the cycle of infection. The upstream 5′LTR acts as a TATA-box-containing inducible promoter for viral transcription of plus-strand (Kulkarni and Bangham, 2018), whereas HBZ is translated from 3′LTR in minus-strand by a TATA-less, Sp-1 dependent promoter (Yoshida et al., 2008).
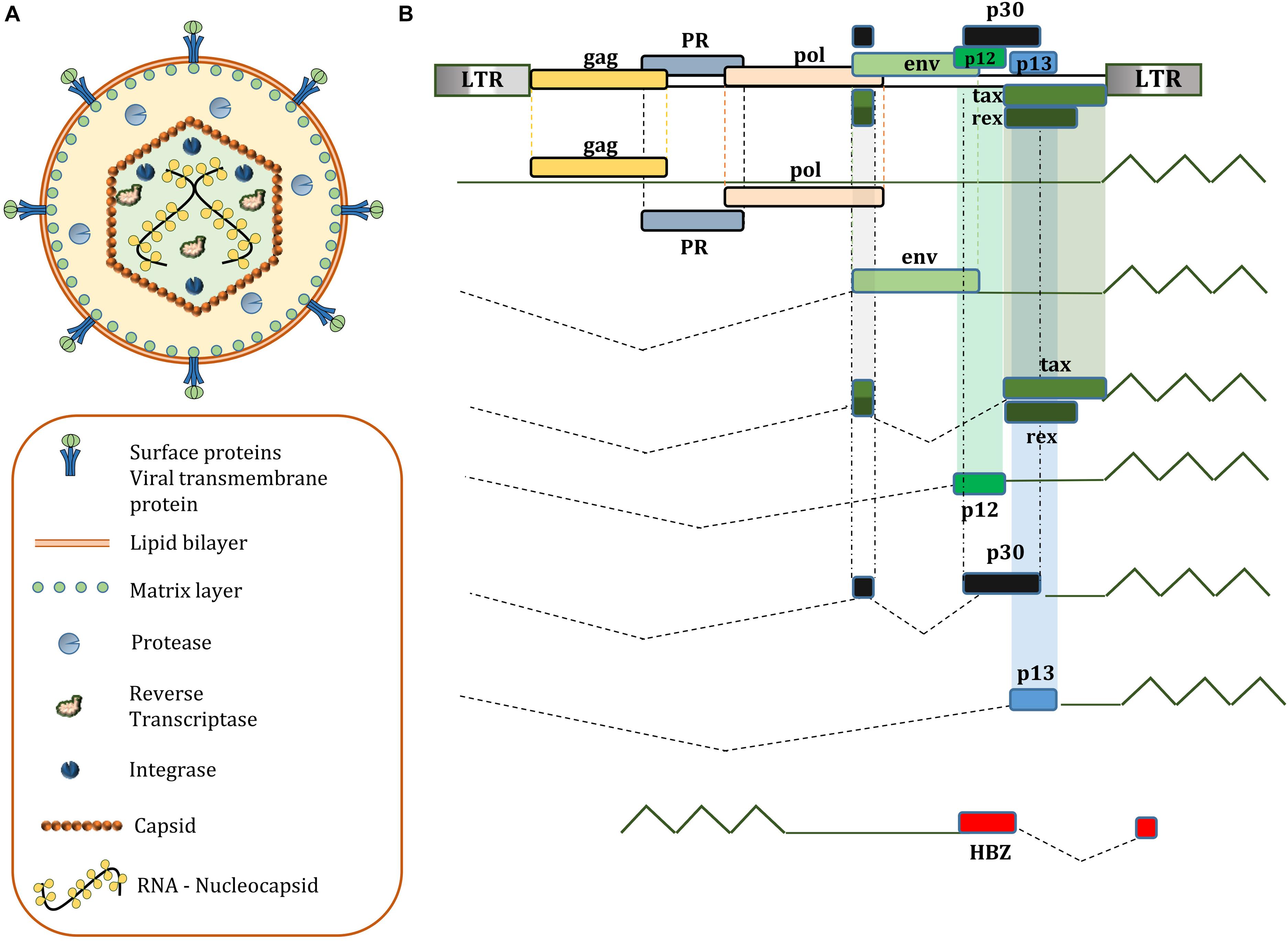
Figure 1. (A) Structure of a mature HTLV-1 virion; (B) genetic composition of HTLV-1; HBZ, p12, p13, p30 accessory proteins are not essential for immortalization of T lymphocytes in vitro, but have a significant role in viral latency and persistent infection in humans.
Tax
Tax as a viral protein affects several cellular pathways, leading to changes in host gene expression, T-cell differentiation, and modification of immune response. The three tandem nucleotide repeats called tax responsive elements (TRE) in the 5′LTR promoter regulate plus-strand expression (Giebler et al., 1997) through binding of proteins of the ATF/CREB family. Plus-strand expression is enhanced by transcription of tax by increasing binding of CREB to TREs through a positive feedback loop, enabling transactivation and spontaneous viral replication (Kulkarni and Bangham, 2018). Furthermore, tax is responsible for modulation of various signaling pathways; including I kappa B kinase, nuclear factor-kappa B (NF-κB), AP-1, JAK1/STAT3, JAK3/STAT5, IRF3, IRF4, and TGF-β signaling (Grant et al., 2008; Olière et al., 2010; Suzuki et al., 2010; Kataoka et al., 2015; Tanaka and Matsuoka, 2018). Tax also acts as an oncoprotein by inhibiting tumor suppressors (p53, Bcl11B, and TP53INP1) and activating cyclin-dependent kinases, both of which lead to increased cell proliferation (Carpentier et al., 2015). Moreover, impaired antiviral signaling in HTLV-1 infections is partially because of Tax-mediated induction of Suppressor of cytokine signaling 1 and reduction of MHC-I in infected cells (Olière et al., 2011). Reduction of interferon type I production via IRF3 phosphorylation by Tax also impairs host viral defense against HTLV-1 (Olière et al., 2011; Bangham, 2018). Furthermore, Tax-mediated expression of proinflammatory mediators and apoptosis is strongly associated with the neuropathology of CNS in HAM/TSP (Banerjee et al., 2007b). Importance of Tax in generating the immune response is emphasized; as while CD4+ T-cells response is predominantly directed toward Env products, the immunodominant antigen recognized by CD8+ T-cells is the Tax protein (Bangham et al., 2015). The reader is referred to review articles for further details regarding Tax interaction with host pathways (Boxus et al., 2008; Matsuoka and Jeang, 2011; Fochi et al., 2018).
HBZ
The functions of HBZ protein are usually in contrast to Tax. HBZ suppresses viral transcription via binding with CREB and selective inhibition of the NF-κB and AP-1 pathways via interacting with p65 and c-Jun, respectively (Tanaka and Matsuoka, 2018; Fochi et al., 2019). HBZ also transactivates the expression of TGF-β, whereas Tax has the opposite effect (Bangham, 2018). These opposing interactions are regarded as advantageous for the virus, because Tax induces a strong immune response in both CD4+ and CD8+ T-cells. The inhibition of transcription of viral structural genes increases viral latency and persistence in infected cells (Bangham et al., 2019). HBZ also downregulates apoptosis and autophagy by reducing FoxO3a-mediated transcription of Bim and mTOR inhibitor, respectively. These regulatory functions contribute to cell proliferation, maintenance of viral reservoirs, and cancer development in ATLL (Mukai and Ohshima, 2014; Vescovo et al., 2020). Although both Tax and HBZ are transcribed in bursts in a single clone (Billman et al., 2017); HBZ is expressed in most of the infected cells (Satou et al., 2006), whereas Tax expression is silenced in various clones through epigenetic and genetic modifications and other viral regulatory genes such as rex, p30, and hbz (Nicot et al., 2004; Takeda et al., 2004; Taniguchi et al., 2005).
Interestingly, only a small fraction of HBZ mRNA is translated into HBZ protein in PBMCs of HAM/TSP subjects (Shiohama et al., 2016). Furthermore, HBZ mRNA levels correlate with PVL and clinical status in HAM/TSP patients (Saito et al., 2009; Leal et al., 2018). This association is partially explained by unique functions of HBZ mRNA distinct from HBZ protein (Shiohama et al., 2016): Untranslated HBZ mRNA (substitution of ATG start codon with TTG) induces T-cell expansion, while HBZ mRNA with silent mutations that interfere with the secondary structure of RNA does not promote proliferation. HBZ RNA likely does this via induction of E2F1 gene expression, which is associated with oncogenesis and increased proliferation (Satou et al., 2006). The low protein level of HBZ can also be explained by strong nuclear retention of HBZ mRNAs, suggesting that the mRNAs might function as non-coding transcripts (Rende et al., 2011). Aberrant proliferation and expression of genes associated with cell cycle in HAM/TSP (Table 2) could also be related to the interplay of HBZ and Tax with host machinery.
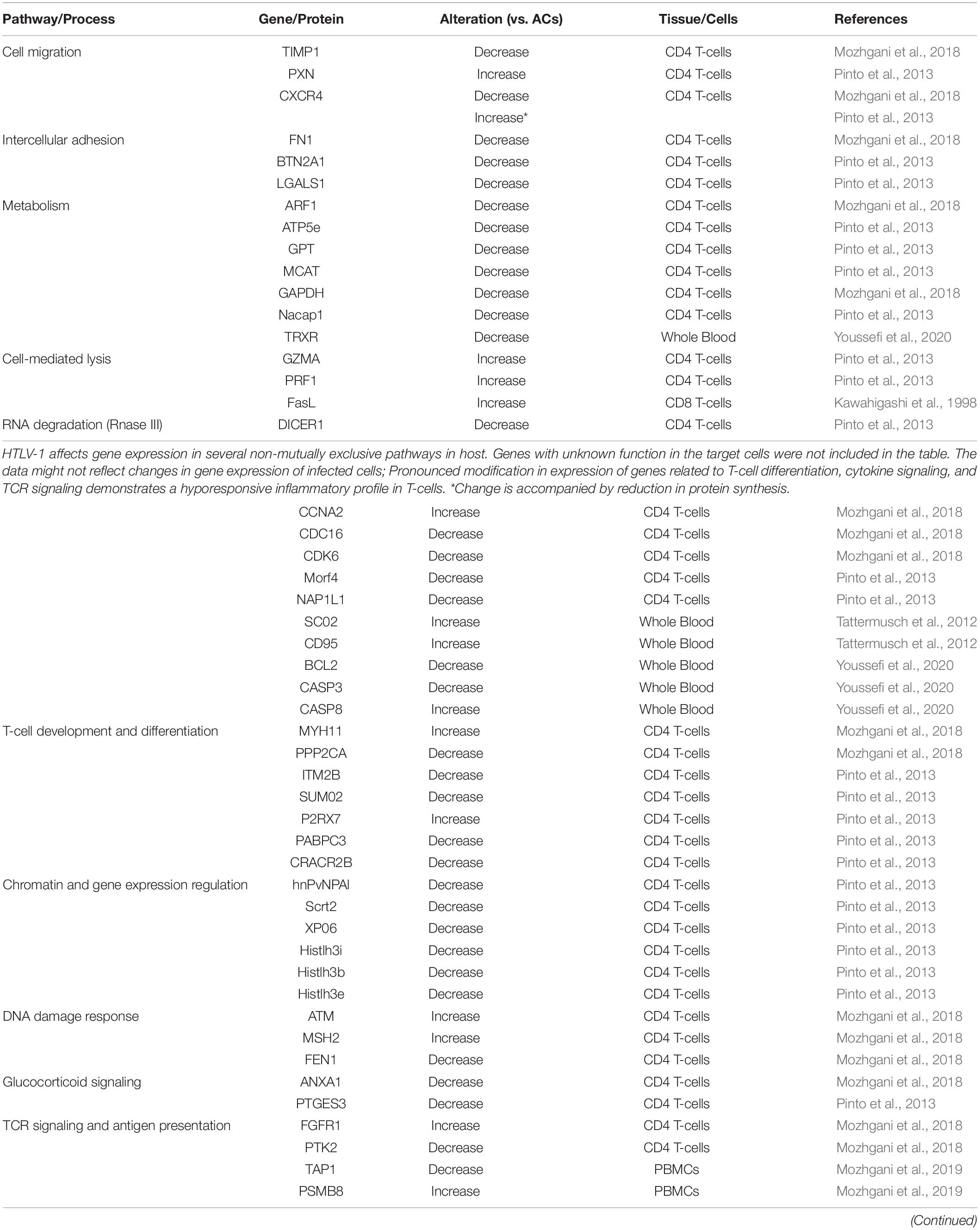
Table 2. Alteration of transcriptomic profile of cellular pathways in HAM/TSP.
Hbz and Tax in Pathogenesis of HTLV-1 Related Diseases
While the significance of PVL in determining the course of HTLV-1 infections and transformation of ACs to ATLL and/or HAM/TSP patients is already determined (Nagai et al., 1998; Iwanaga et al., 2010), the role of HBZ and Tax expression and their balance in the outcome of HTLV-1 infection is also strongly suggested by comparing their levels between ACs, HAM/TSP, and ATLL patients. Relative expression of Tax in ATLL patients is less than ACs and HAM/TSP patients (Furukawa et al., 1995; Yamano et al., 2002; Araya et al., 2011). Conversely, HBZ mRNA expression was found to be significantly lower in HAM/TSP patients compared to ATLL patients. However, a normalized comparison of HBZ mRNA levels (HBZ mRNA/Proviral DNA) did not vary between groups (Saito et al., 2009), suggesting this correlation in peripheral blood cells is based on PVL rather than increased HBZ transcription. Other studies also demonstrate that HBZ protein expression is correlated with PVL and not with host HBZ antibody nor HBZ mRNA levels (Shiohama et al., 2016). All things considered, the conflicting values in different studies may not represent any true correlation since the interdependent expression of Tax and HBZ are modified by various factors both in vitro and in vivo (Kulkarni and Bangham, 2018) that would be transiently affected by certain study circumstances. Moreover, HBZ mRNA levels in peripheral blood of HAM/TSP patients correlated with disease severity and neopterin; a marker of cellular immune response induced by Th1 (Saito et al., 2009). Furthermore, HBZ is primarily visualized in cytoplasm of CD4+ T-cells in HAM/TSP patients, in contrast to ATLL patients in which HBZ is located in the nucleus (Baratella et al., 2017; Forlani et al., 2019). Moreover, transgenic models with HBZ gene demonstrate inflammatory diseases resembling those found in infected individuals (Satou et al., 2011). Finally, higher affinity of HLA class 1 binding to HBZ but not Tax is associated with a lower PVL and more effective viral control and therefore, reduced risk of developing HAM/TSP (Macnamara et al., 2010). The roles of Tax and HBZ in infection and immune response will be discussed further in the section on viral and cellular interactions.
p12 and p8
Open reading frame I of pX region in HTLV-1 viral genome encodes p12 protein which can be cleaved to produce p8 protein (Sarkis et al., 2019). The p12 protein is primarily localized in the Golgi apparatus and endoplasmic reticulum, while p8 interacts with TCR in immunological synapses in plasma membranes, reducing MHC-II expression and antigen presentation, respectively (Sarkis et al., 2019), contributing to HTLV-1 evasion of immune response. p12/p8 also have direct role in activation and proliferation of infected T-cells: Infected T-cells with HTLV-1 show spontaneous proliferation in the absence of IL-2 in vitro due to constitutive activation of JAK-STAT pathway in HTLV-1 infected cells (Migone et al., 1995). However, the role of p12/p8 in this pathway isn’t the direct phosphorylation of JAK/STAT proteins, but rather prevention of transport of IL-2R to the plasma membrane via binding with β and γc chains of the immature IL-2R, lowering IL-2R count on the cell surface (Mulloy et al., 1996; Sarkis et al., 2019). p8 also induces nanotubes in infected cells, facilitating transmission of viral particles and proteins to uninfected cells (Van Prooyen et al., 2010b).
p30 and p13
Both p30 and p13 are encoded by open reading frame II in the pX region (Van Prooyen et al., 2010a). The nucleolus residing protein p30 is reported to be involved in viral latency, gene regulation, and cell cycle progression. p30 contributes to evasion of immune system surveillance by binding to tax/rex mRNA, facilitating its nuclear retention and reduced expression in infected cells. p30 also interferes with the immune response by downregulating TLR4 and disrupting interferon signaling via interacting with transcription factor PU.1 (Moles et al., 2019).
p13 is predominantly localized in the inner membrane of mitochondria of infected cells, where it increases the activity of electron transport chain and production of reactive oxygen species. Increased ROS synthesis in infected cells results in either T-cell activation and proliferation or apoptosis depending on the ROS setpoint in each cell. Nuclear p13 has also been demonstrated to decrease Tax interaction with CREB binding protein and viral transcription (Omsland et al., 2020). Therefore, p13 seems to contribute to viral latency and clonal expansion by inducing apoptosis in transformed cells and favoring proliferation of resting T-cells, respectively.
Cell-To-Cell Viral Dissemination
The entry of virions into host cells and fusion process of viral particles with cell membrane is mediated by the surface unit of HTLV-1 receptor, gp46. The surface unit forms complexes with glucose transporter 1 (GLUT1), Heparan sulfate proteoglycan (HSPG), and neuropilin 1 (NRP-1) on target cells to initiate envelope fusion with the plasma membrane, leading to the release of viral core into cytoplasm (Soltani et al., 2019). The viral RNA is then reverse-transcribed into a double-stranded DNA and is integrated into the host genome.
As mentioned before, cell-free virions have diminished infectivity for host cells aside from dendritic cells (Alais et al., 2015). In addition, even though GLUT1 expression occurs in all tissues (Coskun and Sutton, 2005), the target receptor of HTLV-1 alone cannot adequately describe the viral distribution in host. HTLV-1 infectivity is limited to these cells in vivo with CD4+ T-cells comprising 95% of infected cells: CD4+ or CD8+ T-cells, B lymphocytes, monocytes, macrophages, dendritic cells (DC), glial cells, megakaryocytes, sweat gland epithelia, salivary glands, vascular endothelial cells, and mammary glands (Hoshino, 2012). These facts raise the question about the method of viral spread between host cells, which, as we will see later on, are also critical steps in HTLV-1 related disease pathogenesis.
Virological Synapse and Extracellular Viral Assemblies
The virus utilizes and upregulates cell-cell adhesion molecules for intercellular propagation and heavily relies on intimate cell-cell contact and cytoskeletal remodeling (Igakura et al., 2003). Viral transmission mainly occurs in close cell-to-cell contacts named Virological synapse (VS). VS is the result of tax mediated interaction between intercellular adhesion molecule 1 (ICAM-1) on infected cells with lymphocyte function-associated antigen 1 (LFA-1) on target cells accompanied by the polarization of microtubule organizing center (MTOC) inside the infected cells toward the target cell (Nejmeddine et al., 2009; Gross and Thoma-Kress, 2016). Tax promotes VS formation through upregulation of ICAM-1 on HTLV-1 infected cells, accumulation of Tax in microtubule organizing center, recruitment of fascin bundling protein, and CREB signaling of nuclear Tax (Banerjee et al., 2007a; Gross and Thoma-Kress, 2016; Gross et al., 2016). Cell-cell contact and viral transfer may also be improved by clustering of NRP-1 in VS similar to immunological synapses formed between DCs and T-cells (Tordjman et al., 2002). Upon polarization of MTOC, viral proteins at the cell cortex are released toward the cell contact. Viral components are then visualized inside target cells, demonstrating a successful viral transfer (Figure 2; Pais-Correia et al., 2010).
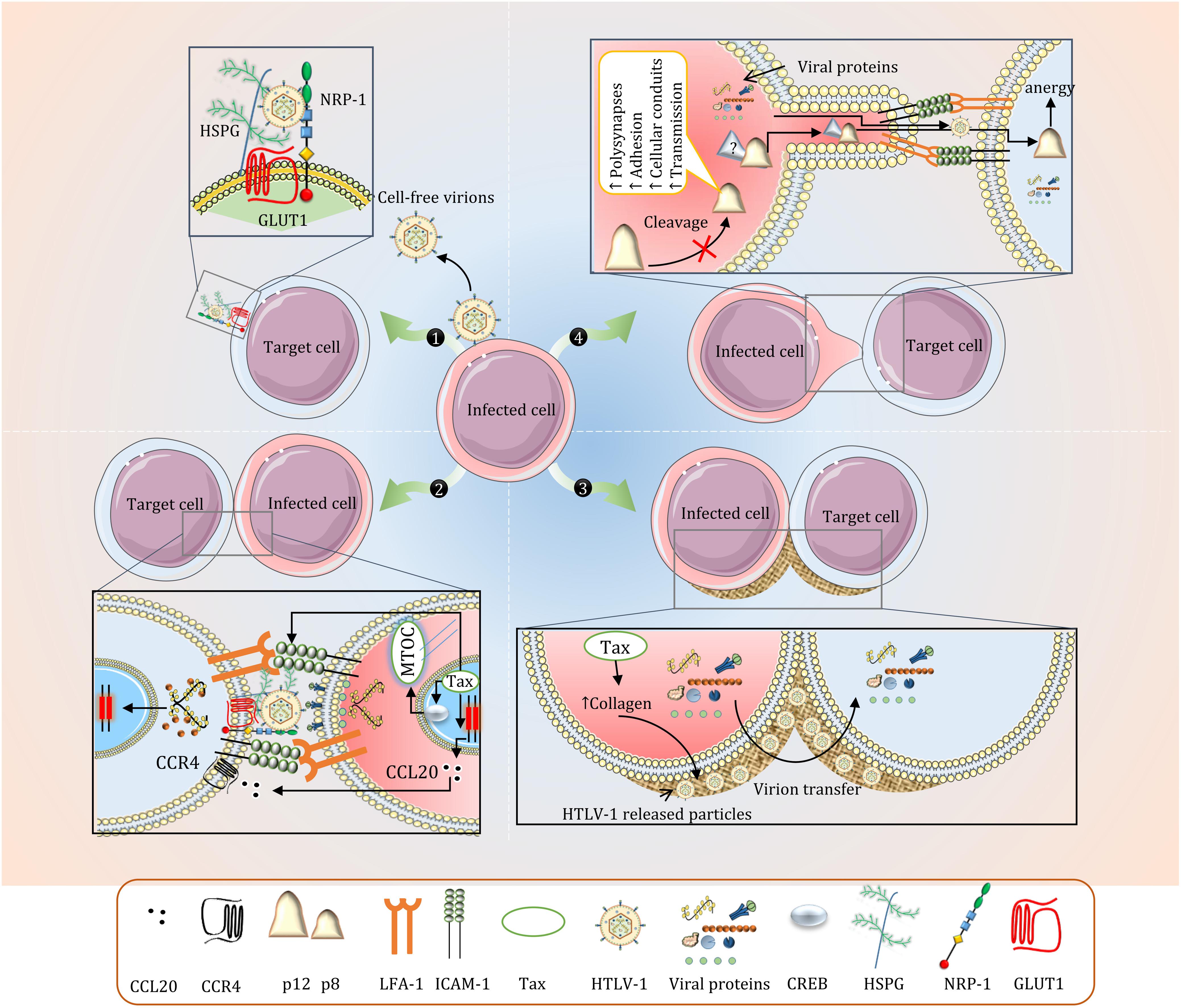
Figure 2. Intercellular Transmission of HTLV-1. (1) Cell-free virions are rarely produced in HTLV-1 infection and these particles are almost non-infectious (only 1 in 105 to 106). The fusion process is mediated by GLUT1, NRP-1, and HSPG. Other cell adhesion molecules including ICAM-3 and VCAM have also been associated with HTLV-1-induced cell fusion. (2) Viral transfer via Virological synapse (VS). Polarization of MTOC and expression of ICAM-1 and CCL22 is enhanced by Tax. While VS formation is primarily mediated by ICAM-1 and LFA-1, blocking CCL22 is shown to significantly reduce VS formation and viral transmission in CD4+ T-cells. (3) Concentrated viral particles in the extracellular assemblies is efficiently transferred to other cells upon contact. (4) The precise mechanisms regarding p8-mediated cellular conduits are not well understood. p8 in recipient CD8+ T-cells induces T-cell anergy which reduces effective CTL response.
The upregulation of adhesion molecules also facilitates concentrated virion transfer in biofilms. These viral assemblies are formed on the cell surface when HTLV-1 released particles are trapped in extracellular matrix components primarily made up of a carbohydrate-rich structure containing sialyl-LewisX and heparan sulfate proteoglycans (Pais-Correia et al., 2010). Collagen was also observed in these assemblies, which is overexpressed in HTLV-1 infected cell lines in a tax-specific fashion (Figure 2; Munoz et al., 1995; Pais-Correia et al., 2010). This form of viral transfer has yet to be reproduced in vivo.
In many aspects, VS is advantageous for HTLV-1 persistence inside the host. The localized release of virions in synaptic cleft increases the efficiency of transmission while avoiding immune system recognition. VS also elucidates preferred tropism of HTLV-1 for CD4+ T-cells (Gross and Thoma-Kress, 2016), as ICAM-1 ligands such as LFA-1 are expressed in CD4+ T-cells. However, It can be speculated that viral spread using VS is limited in later stages of the disease as infected CD4+ T-cells have reduced MHC-I, ICAM-1, and ICAM-2 levels and increased LFA-1, probably due to p12/p8 effect on adhesion molecules (Banerjee et al., 2007a). It can be postulated that this may contribute to the phenomenon regarding the establishment of unique clones early in HTLV-1 infection and dominance of expansion of the existing clones in the chronic stage (Bangham, 2018; Bangham et al., 2019). This observation can also be interpreted as an attempt to escape the NK cell ICAM-mediated lysis of low MHC-I expressing cells (Topham and Hewitt, 2009). The reader is referred to other articles for a list of host cell proteins implicated in HTLV-1 transmission (Gross and Thoma-Kress, 2016).
Cellular Conduits and Extracellular Exosomes
HTLV-1 is also transmitted via p8-induced cellular conduits in vitro. These conduits are thin-membrane extensions of infected cells appearing as an extended VS. While clustering of LFA-1 on the surface of infected cells seems to contribute to the formation of conduits, the precise mechanism for this process is not understood. These cellular conduits may also lead to CTL anergy and viral persistence by transferring p8 to CTLs (Van Prooyen et al., 2010b). The absence of p8 in several viral strains undermines the importance of these conduits in viral transfer.
Unilamellar exosomes released from infected cells during HTLV-1 are generally not infectious, but they may indirectly modulate infectivity by promoting adhesion molecule on recipient cells (Pinto et al., 2019). These extracellular vesicles contribute to the persistence of inflammation in CNS, which is further elaborated in the relevant sections (go to Central Nervous System section).
Viral and Cellular Interactions in the Periphery
For HTLV-1 to enter the host, the virus must first pass through either the intestinal epithelium as in transmission of the virus through breastfeeding or reproductive tract mucosa as in sexual transmission. It is self-evident that acquiring the virus via blood products and organ transplantation bypasses the need to cross mucosal barriers. The virus can then be delivered to bone marrow and lymph nodes, where physiological hypoxia increases plus-strand expression (Kulkarni and Bangham, 2018). This helps to establish numerous clones before an effective anti-viral response could come into play (Figure 3).
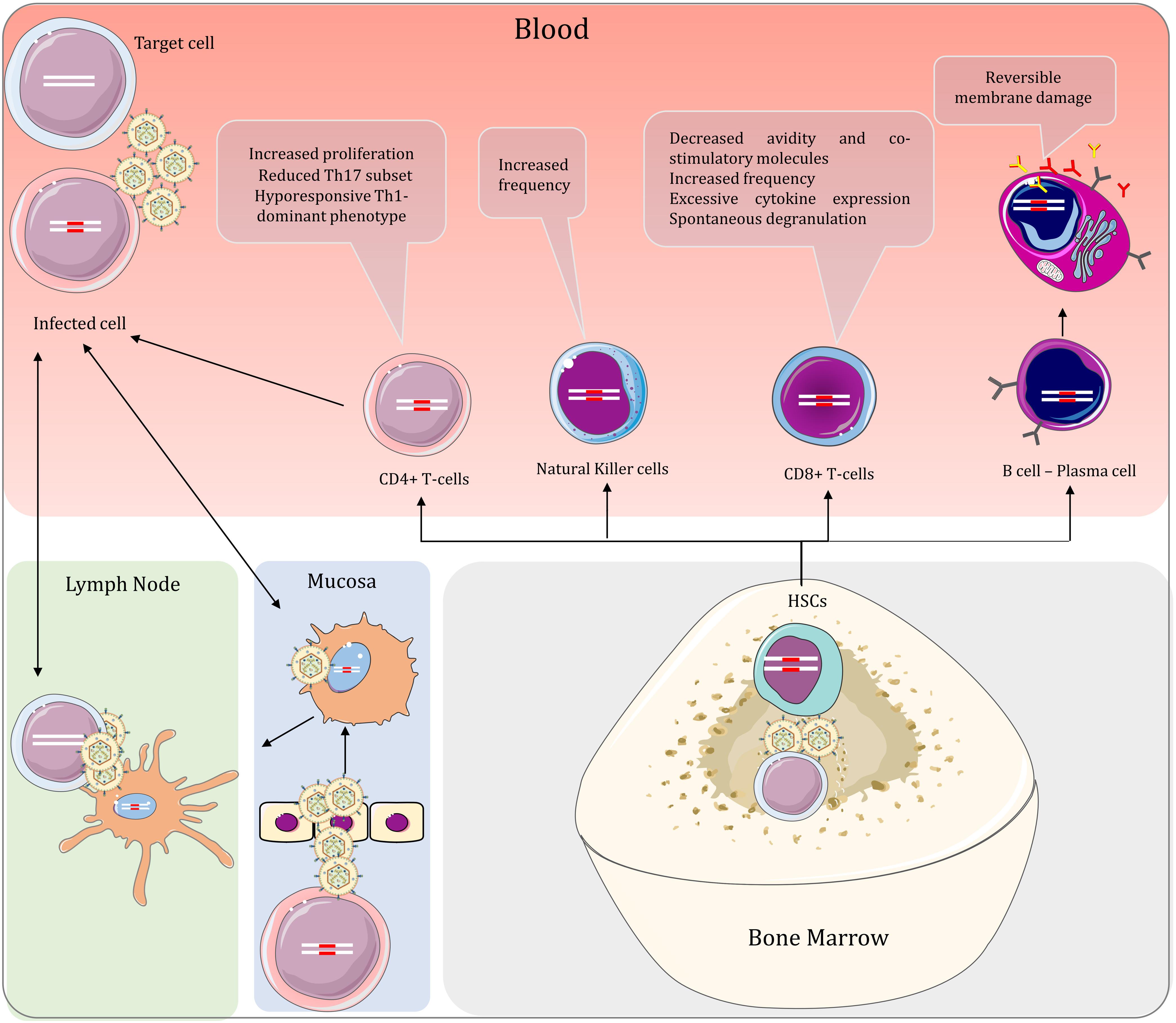
Figure 3. Peripheral interactions of HTLV-1 and the host in HAM/TSP and the effect of the virus on the lymphoid lineage cells. When the virus is acquired via mucosal infection, the dendritic cells spread the virus to immune cells in lymph nodes and the peripheral blood. Consequently, infected T-cells can also infect dendritic cells, spreading the virus bidirectionally between T-cells and dendritic cells in the periphery. HTLV-1 can also be delivered to hematopoietic stem cells in the bone marrow. These cells give rise to all other blood cells, including both myeloid and lymphoid lineages. By virtue of vertical transmission, proviral DNA is found in cells of both lineages, regardless of productive infection. The lymphoid lineage cells are studied more extensively in the context of HTLV-1 infection and are associated with prominent changes in HTLV-1-related diseases.
HTLV-1 can either directly penetrate through a damaged epithelium following injury or utilize cellular-mediated pathways. It is also hypothesized that infected macrophages present in contaminated breast milk or semen can transmigrate through an intact epithelium (Pique and Jones, 2012). Alternatively, free HTLV-1 virions can cross the epithelial barrier via a transcytosis mechanism and infect human DCs beneath the epithelial barrier (Martin-Latil et al., 2012). There are discordant studies regarding the infection of epithelial cells, their role in virus entry, and whether they can be included as viral reservoirs in HTLV-1 infections (Zacharopoulos et al., 1992; Zacharopoulos and Phillips, 1997; Southern and Southern, 1998; Martin-Latil et al., 2012).
Dendritic Cells
Nevertheless, after crossing the barrier, HTLV-1 must either interact with antigen-presenting cells (APCs) such as DCs or directly infect mucosal immune cells. This interaction can also occur without the need to cross the epithelial barrier through direct cell-cell contact of infected cells with patrolling cellular protrusion of DCs in the lumen (Rocamonde et al., 2019). HTLV-1 particles are highly infectious to these cells in vitro (Jones et al., 2008; Jain et al., 2009) and viral DNA is observed in DCs of HAM/TSP patients (Macatonia et al., 1992; de Castro-Amarante et al., 2016). It has also been demonstrated that DC depletion increases gag expression in HTLV-1 infected mouse CD4+ T-cells (Rahman et al., 2010). These findings advocate the critical role of DCs in the pathogenesis of HTLV-1 related diseases.
Dendritic cells are potent APCs that are essential in the immune response against viral infections. While different subtypes of DCs exist in blood (myeloid dendritic cells and plasmacytoid dendritic cells (pDC)) and mucosa (monocyte-derived dendritic cells), they all share their capability in presenting antigens to T-cells in lymphoid organs (Jones et al., 2008; Villadangos and Young, 2008). Infecting DCs is considered favorable for HTLV-1 as viral transmission can occur via viral capture and presentation of virions to resting T-cells in lymph nodes (Jones et al., 2008). Conversely, infected CD4+ T-cells can also infect DCs via VS (albeit to a lesser extent because of the low expression of LFA-1 on DCs compared to activated CD4+ T-cells), establishing a bidirectional viral transmission route (Shimauchi et al., 2019). This process is mediated by HTLV-1 binding to lectin DC-specific ICAM-3-grabbing non-integrin (DC-SIGN) and ICAM-2 and ICAM-3 on target cells (Ceccaldi et al., 2006). In these cells, antigen presentation by MHC-II is also associated with the spontaneous proliferation of lymphocytes (Macatonia et al., 1992). Counterintuitively, HTLV-1 infection reduces monocyte differentiation to DCs which is independent of monocyte infection (Nascimento et al., 2011). It is also reported that DC count is decreased in HAM/TSP (Azakami et al., 2009; Journo and Mahieux, 2011; Nascimento et al., 2011). This reduced differentiation is suspected to be due to increased IL-6 in sera of the patients (Nascimento et al., 2011). The implications of this finding, however, is not understood and requires further studies.
Dendritic cells are also known for their robust antiviral signaling with proinflammatory cytokines. Nevertheless, production of type 1 interferons by DCs provide modest benefit to patients possibly due to interference of tax with the intracellular antiviral signaling cascade (Bangham, 2018), reduced interferon secretion in the majority of DCs (Moles et al., 2019) and reduction in expression and phosphorylation of STAT1 in PBMCs (Mozhgani et al., 2019). Recently, the visualization of DCs in choroid plexus of normal rats has led to the hypothesis that DC access to CNS antigens may play an important role in inflammatory and immune-mediated disorders of CNS (McMenamin, 1999). However, the relevance of this discovery to pathogenesis of CNS inflammation in HAM/TSP remains to be investigated.
Hematopoietic Stem Cells and Myeloid Lineage
All of DC subtypes are derived from a common hematopoietic stem cell (HSC) ancestor. HSCs are self-replicating, multipotent cells, which differentiate to lymphoid and myeloid lineages in bone marrow (Rocamonde et al., 2019). Identical proviral integration site between HSCs, PBMCs and neutrophils in HAM/TSP indicate that these cells are infected in vivo and give rise to HSC derived HTLV-1 infected lymphocytes, monocytes, DCs and other hematopoietic lineages and act as viral reservoirs in persistent infections (Grant et al., 2002; Yasunaga et al., 2016; Furuta et al., 2017). Intriguingly, whereas infected monocytes and pDC are visualized in the blood of HAM/TSP patients, both monocytes and pDC show high resistance to HTLV-1 infections in in vitro studies (Assil et al., 2019; Rocamonde et al., 2019). HTLV-1 infection of monocytes results in abortive infection by two mechanisms: SAMHD1 restriction factor, a dGTP-dependent deoxynucleotide triphosphohydrolase, reduces dNTPs to below the levels required for reverse transcription. Furthermore, SAMHD1 binding to viral intermediates leads to formation of complexes with STING DNA sensor to trigger establishment of an IRF3-Bax complex and subsequent apoptosis of the cell (Sze et al., 2013). SAMHD1 restricting properties are only observed in quiescent T-cells (Descours et al., 2012), explaining why some HTLV-1 proteins aim to activate T-cells. The pDC response against HTLV-1 is mediated by TLR-7 signaling and TRAIL death receptor (Olière et al., 2011; Rocamonde et al., 2019). Studies demonstrate that TLR-7 signaling is impaired by HTLV-1-induced miR146a; Nonetheless, HTLV-1 infected pDCs show a strong TLR7-dependent IFN-a response (Olière et al., 2011). The discrepant observations regarding monocyte infection in different studies and the resistance of activated and mature DCs to HTLV-1, unlike immature DCs, raise the possibility that these cells are primarily infected before differentiation and are not byproducts of de novo infections (Rizkallah et al., 2017; Rocamonde et al., 2019). Notwithstanding, infection of myeloid cell lines is less productive than their lymphoid counterpart (Grant et al., 2002), because Tax transactivation of plus-strand is limited in this cell lineage (Lepoutre et al., 2009).
While neutrophils seem to retain normal functions and cytokine profile in HTLV-1 infections (Bezerra et al., 2011), persistent infection of monocytes has been directly associated with the pathogenesis of HAM/TSP. Infected monocytes and macrophages can migrate to CNS during inflammation-induced blood-brain barrier permeability, delivering the virus to the CNS. HAM/TSP patients also show increased intermediate monocytes (CD14+CD16+) which are also the dominant monocyte subtype in producing Tumor necrosis factor (TNF), a major proinflammatory cytokine in HAM/TSP (Amorim et al., 2014). In addition, non-classical monocytes (CD4lowCD16+) in HAM/TSP patients demonstrate an inflammatory phenotype evidenced by higher levels of CX3CR1 and HLA-DR compared to ACs (Enose-Akahata et al., 2012b). The role of these cells in pathogenesis of CNS is further investigated in the CNS section.
CD4+ T-Cells
It is widely known that HTLV-1 is primarily found in CD4+ T-cells and proliferates by driving the cells to replicate (Asquith et al., 2007). HTLV-1 also modifies the immune response by increasing memory T-cells while reducing naïve T-cells (Yasunaga et al., 2001). This tendency is common between ACs and ATLL and HAM/TSP patients (Furukawa et al., 1992; Takemoto et al., 1994; Bangham, 2018). In HAM/TSP, HTLV-1 also decreases Th17 effector subset, further impairing host immunity (Leal et al., 2013). The imbalance in T helper system in HAM/TSP leads to a Th1 dominant immunophenotype accompanied by proinflammatory cytokines (IFN-γ, TNF-α, and IL-2) (Furuya et al., 1999; da Silva Dias et al., 2016; Nozuma and Jacobson, 2019). The resultant inefficient immune response is rather detrimental than being protective in individuals with HAM/TSP. The mechanism for these changes still remains ambiguous but is visualized by altered expression of genes in processes related to cell differentiation in CD4+ T-cells (Table 2). The function of CD4+ compartment of T-cells is not only dysregulated by the above but also by aberrant expression of other cell markers which is described below.
T-cell factor 1 (TCF 1) and lymphoid-enhancer binding factor 1 (LEF1) both act as transcription factors in Wnt pathway and are primarily expressed in immature T-cells. They antagonize tax transactivation of 5′LTR and may impair viral replication in Thymus and favor viral replication in TCF1/LEF1 low-expressing mature peripheral T-cells observed in HTLV-1 infection (Ma et al., 2015). Apparently, these factors are also reduced in HTLV-1 infection. Considering TCF1 role in preserving effector function of exhausted CD8 T-cells during chronic viral infection (Wang et al., 2019), examining this alteration might provide new insights in treatment of HTLV-1 related diseases. Intriguingly, higher expression of PD-1, another marker associated with T-cell exhaustion, along with loss of CD28, reflecting senescence, is also observed in both CD4+ and CD8+ T-cells in HAM/TSP and ATLL (Shimauchi et al., 2007; Saeed et al., 2020). Surprisingly, it seems that the PD-1 increase is associated with regulatory CD4+ T-cells (Tregs) rather than effector CD4+ T-cells as PD-1 expression in the latter is actually decreased (Leal et al., 2013). The loss of PD-1 on effector T-cells might reflect their active and non-anergic state observed in HAM/TSP.
The reason why HTLV-1 predominantly resides in CD4+ T-cells is not completely understood, yet can be explained by some of the recent discoveries such as the interaction of HBZ with THEMIS. The co-inhibitory receptor T-cell immunoglobulin and ITIM domain (TIGIT) exerts its anti-inflammatory properties via interaction with their intracytoplasmic immunoreceptor tyrosine inhibitory motif (ITIM) domain which subsequently interacts with a complex of SHP-2, Grb2, and THEMIS (Tanaka and Matsuoka, 2018). HBZ interaction with THEMIS inhibits IL-10 production induced by TIGIT that leads to unresponsive proliferation of cell. Knock-down of THEMIS essentially removes cytoplasmic HBZ demonstrating that THEMIS is required for cytoplasmic localization of HBZ (Tanaka and Matsuoka, 2018). Therefore, THEMIS might explain the productive infection of T-cells. CCR4 ligand CCL22 expression by Tax also attracts CCR4+ T-cells and may also contribute to VS formation with CD4+ T-cells (Hieshima et al., 2008). The role of APCs in delivering HTLV-1 to CD4+ T-cells should not be neglected either. Evidence suggests that the predominant presence of HTLV-1 in CD4+ T-cells rather than CD8+ T cells might be a post-infection event due to selective clonal expansion over time (Kannian et al., 2012).
Regulatory CD4+ T-Cells
Regulatory T-cells (Tregs) are a unique subset of T-cells capable of suppressing antigen-specific responses by effector cells (Bangham and Toulza, 2011). Whereas these cells are generally identified as CD4+CD25+Foxp3+, both CD25- and Foxp3- cells with suppressive Treg functions have been demonstrated (Ohkura et al., 2013; Zhao et al., 2017). Furthermore, the transcriptional regulator Foxp3, which is implicated in the development of Treg functions, is expressed in some inflammatory subpopulations of CD4+ T-cells (Ohkura et al., 2013). Moreover, activation of T-cells (as seen with Tax) induces a transient expression of Foxp3 that is not accompanied by Treg functions (Bangham and Toulza, 2011). Therefore, the circumstances have misled the literature to assume that these CD4+CD25+Foxp3+ T-cells, which have been associated a Th1-like overproduction of IFN-γ and induction of proliferation of Tax-specific CD8+ T-cells in HAM/TSP, were originally Tregs (Araya et al., 2011). Nevertheless, CD4+CD25+CCR4+ T-cell population have been noted to be prime viral reservoirs of the virus in both ATLL and HAM/TSP patients (Yamano et al., 2004, 2009).
Tax and HBZ have been associated with the dysregulation of Foxp3 expression. HBZ induces TGF-β/Smad pathway to increase Foxp3+ T-cells in HBZ-transgenic mice (Satou et al., 2011; Zhao and Matsuoka, 2012), but also interact with Foxp3 to interfere with its DNA binding activity (Tanaka and Matsuoka, 2018). Furthermore, it has been shown that induced Tregs are increased in transgenic model with unstable Foxp3 expression (Yamamoto-Taguchi et al., 2013). Although controversial, it seems that the former effect of HBZ in increasing Foxp3 is nullified as Foxp3+ T-cells count is lowered in humans (Yamano et al., 2005; Araya et al., 2011). Moreover, it seems Foxp3+ Tregs are not terminally differentiated and can lose their Foxp3 expression and therefore convert to effector IFN-γ-producing T-cells (Yamamoto-Taguchi et al., 2013). HBZ dysregulation of Foxp3 pathway is confirmed by the fact that immune response of these T-cells in the transgenic model is not directed at HBZ but is attributed to intrinsic properties of HBZ-expressing cells (Yamamoto-Taguchi et al., 2013). Role of Tax in dysregulation of Tregs seems to oppose HBZ; Tax transactivation of OX40 (CD134), a costimulatory molecule from TNF receptor family and its ligand OX40L (gp34), induce proliferation of memory T-cells whereas suppressing the differentiation and activity of Tregs (Higashimura et al., 1996; Enose-Akahata et al., 2017). Tax also inhibits TGF-β pathway and expression of Foxp3, while it increases IFN-γ promoter activity (Sugata et al., 2012; Zhao and Matsuoka, 2012).
In spite of all the evidence, conflict exists whether loss of Foxp3 is associated with reprogramming of Tregs or Foxp3 loss in conventional T-cells (Miyao et al., 2012; Yamamoto-Taguchi et al., 2013) or whether Foxp3 expression is increased or decreased in HAM/TSP (Yamano et al., 2005; Oh et al., 2006; Hayashi et al., 2008; Toulza et al., 2008; Araya et al., 2011; Pinto et al., 2013; Bangham et al., 2015). Nevertheless, there is also the unanswered question whether this pattern is induced by direct infection of Tregs or a dysregulation in differentiation of infected HSCs. These controversies demand much more attention and must be scrutinized.
CD8+ T-Cells and NK Cells
Despite carrying 5% of PVL (Melamed et al., 2015), CD8+ T-cells play a fundamental role in both neuropathology and viral latency in HAM/TSP. It is of note that HTLV-1 specific CTLs are significantly increased in PBMCs of HAM/TSP when compared to ACs. CD8+ T-cell response in peripheral blood of HAM/TSP patients and ACs is normally directed toward the plus-strand protein Tax (Elovaara et al., 1993). The amplitude of the response however, is increased in HAM/TSP. The increased frequency of these cells is associated with higher PVL and might suggest inefficiency in disease prevention (Kubota et al., 2000a,b; Wodarz et al., 2001; Leal et al., 2013). On the other hand, HTLV-1-specific CTL responses with higher efficiency lead to lower PVL and better viral control; This indicates the quality of CTL cell-mediated lysis and not the frequency of these cells to be a major determinant in anti-viral defense vs. HTLV-1 (Bangham and Osame, 2005; Bangham, 2009). This discovery is also evident in the fact that CD8+ T-cells of HAM/TSP patients have reduced co-stimulatory molecules compared to ACs in similar PVLs (Sabouri et al., 2008).
CD8+ T-cells show dysfunctions, some of which are uniquely observed in HAM/TSP (Enose-Akahata et al., 2008). Evidence suggests that aberrant function of these cells in HAM/TSP may be the result of various factors including: high antigenic stimulation, Tax dysregulation of cytokine loops (IL-2/IL-2R and IL-15/IL-15R), and selective expression of surface receptors (CD244) which ultimately results in reduction of T-cell avidity, spontaneous degranulation, excessive cytokine expression (IFN-γ, TNF, and IL-2) and subsequent damage to the host tissues (Kubota et al., 1998; Enose-Akahata et al., 2008, 2017; Sabouri et al., 2008; Kattan et al., 2009; Yasuma-Mitobe and Matsuoka, 2018; Nozuma and Jacobson, 2019). Indeed, PVL is significantly elevated in CSF of HAM/TSP patients compared to their peripheral blood (Puccioni-Sohler et al., 1999). CD14+ cells (Mononuclear phagocytes) may also have a role in dysregulation of CTLs. Spontaneous degranulation and IFN-γ release of CD8+ T-cells which has been linked to higher PVL and Tax mRNA expression is observed when these cells are cocultured with IL-15-expressing-CD14+ cells and not CD4+ T-cells (Enose-Akahata et al., 2008). CTLs role in pathogenesis of HAM/TSP in CNS which will be discussed later.
Similar to CD8+ cytotoxic cells, NK cells also show impairment in their function in form of continuous activation. This had resulted in spontaneous degranulation and hypo-responsiveness as well as increased IFN-γ expression in this cell lineage (Queiroz et al., 2019). These changes apparently reflect malfunction of cell-mediated immunity in HTLV-1 infection. The NK cell (CD56+) frequency is diminished in HTLV-1 infections and HAM/TSP (Azakami et al., 2009; Tattermusch et al., 2012; Amorim et al., 2019) (Although Some studies do not show a reduced count (Tattermusch et al., 2012; Saeed et al., 2020)) and NKT cells (CD56+/CD3+) demonstrate reduced proliferation when exposed to activation molecules (Azakami et al., 2009). NK cell receptors like NKG2D, KIR2DL2, NKp30, and CD137 have been shown to modulate immune response against HTLV-1 and confer anti-viral protection, but these findings can also be attributed to CD8+ T-cells due to overlapping surface receptors and functionalities with NK cells (Bangham and Osame, 2005; Hudspeth et al., 2013; Bangham, 2018; Boelen et al., 2018; Amorim et al., 2019; Wu et al., 2019).
B-Cells and Antibodies in HAM/TSP
The focus of studies regarding B-cells is mainly directed at their capacity to create antibodies as antibody secreting cells (ASCs). Older studies have stated that attempts in infection of B-cells with HTLV-1 had been unsuccessful (Richardson et al., 1990), thus studies regarding the importance of infection of B-cells in HTLV-1 related diseases are scarce (Longo et al., 1984), even though newer studies have demonstrated clonal expansion of HSCs has yielded infected B-cells (Furuta et al., 2017). Be that as it may, B lymphocytes themselves are not primarily infected by HTLV-1; but still, these cells show pathologic changes in HAM/TSP. Both circulating and memory B-cells show reversible membrane damage in which phosphatidylserine residues are exposed at outer membrane leaflet (Furukawa et al., 2000). These cells revert to their normal phenotype over time in vitro. The reason and significance for this disruption is not well understood.
Antibody (Ab) responses exist in viral infections of CNS and HTLV-1 is no exception. Indeed, the laboratory diagnosis of HAM/TSP is based on presence of anti-HTLV-1 Abs in CSF (Yamano and Sato, 2012). Humoral immune responses against Gag and Env are significantly increased in blood of HAM/TSP vs. other HTLV-1 infected individuals; whereas anti-Tax and anti-HBZ Ab levels are similar in HTLV-1 infected subjects (Enose-Akahata et al., 2012a; Shiohama et al., 2016). These Abs appear to provide some levels of protection against HTLV-1 in experimental studies (Tanaka et al., 1994; Saito et al., 2014). Intrathecal antibodies also inversely correspond with a higher PVL and a worse prognosis (Puccioni-Sohler et al., 2007). Despite their protective functions, endogenous antibodies are considered to be ineffective in virus control in humans (Kitze et al., 1996; Cook et al., 2013).
Blood Brain Barrier Dysregulation and Virus Entry to CNS
HAM/TSP shares similarities in many aspects with MS (Puccioni-Sohler et al., 2007; Azodi et al., 2017). In this regard, Inflammatory cells must cross the normally intact blood-brain barrier (BBB) to react with CNS parenchyma. Both diseases show degradation of BBB components and inflammation-mediated increase in its permeability (Umehara et al., 1998; Liebner et al., 2018). BBB dysfunction can occur due to numerous factors HAM/TSP. HTLV-1 methods of access to CNS can be roughly divided into two categories: Direct virus interaction with components of BBB and non-BBB-cell-mediated virus entry to CNS (Figure 4).
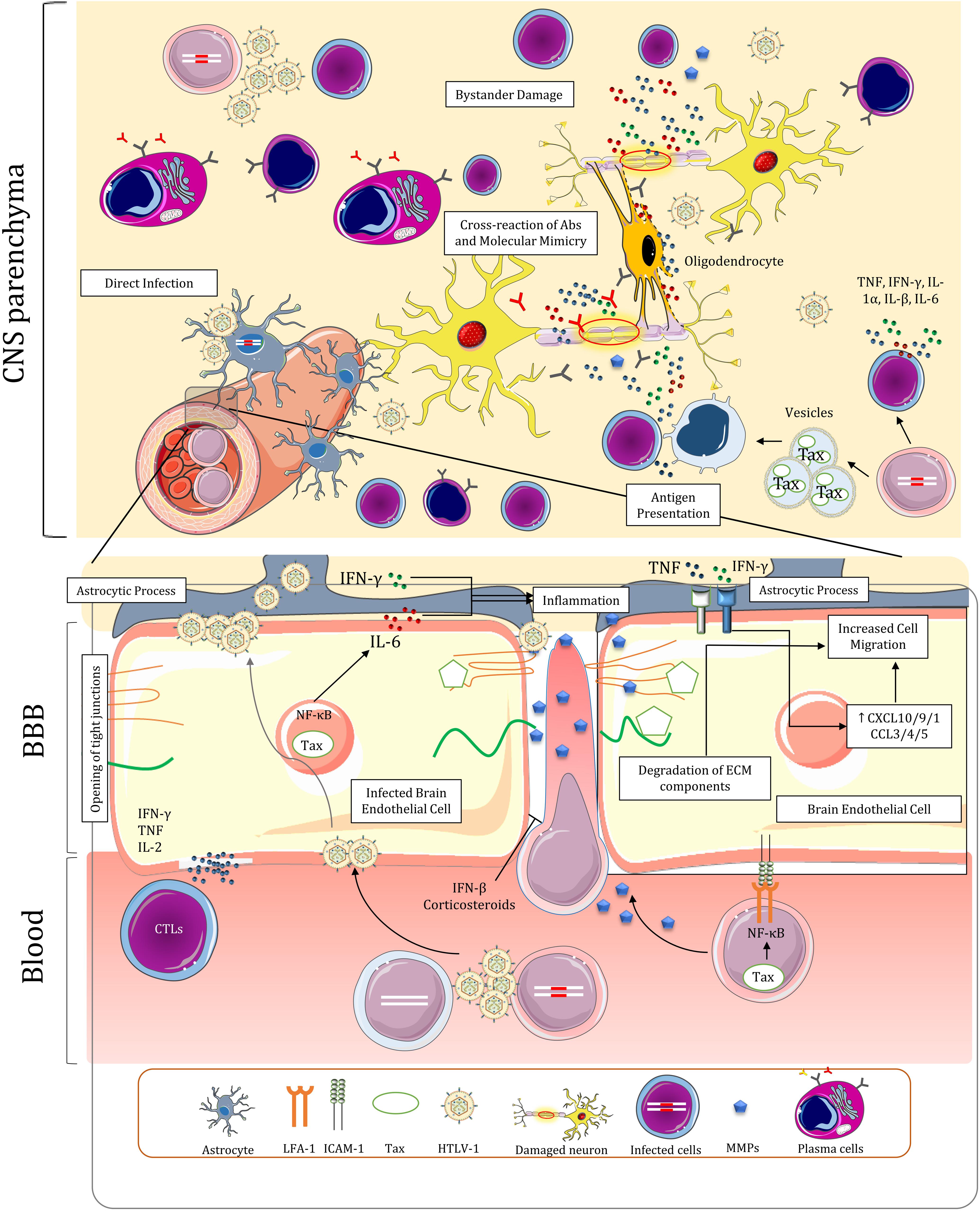
Figure 4. Blood-brain barrier dysregulation, infiltration of immune cells, and proposed mechanisms for the pathogenesis of HAM/TSP in the central nervous system. The BBB is dysregulated due to multiple factors in HAM/TSP, leading to persistent inflammation in the CNS. The inflammation resulting from the presence of the virus and the immune response establishes a positive feedback loop that furthrer disrupts BBB and attracts other immune cells, including infected T-cells.
Brain endothelial cells (BECs) have been shown to be directly involved in BBB dysregulation. It has been demonstrated that HTLV-1 can infect both rat and human (BECs) via virus-induced cell-cell fusion with infected lymphocytes (Uchiyama, 1997; Afonso et al., 2008). Infection of these cells can then lead to release of viral particles at their abluminal surface and infection of astrocyte foot processes which surround brain capillaries, effectively delivering the virus to CNS (Romero et al., 2000). Infected BECs no longer maintain resistance to molecular diffusion and cellular migration in vitro (Afonso et al., 2008). The changes include increased transcytosis and opening of tight junctions between BECs (Romero et al., 2000). These cells can also be targets of CTLs and autoantibodies in vivo resulting in temporary disruption of BBB (Romero et al., 2000; Afonso et al., 2008). The migration of inflammatory cells and their interaction with CNS components also induce a cytokine loop that further disrupts BBB and promotes cell migration. Tax-mediated activation of NF-κB in BECs lead to increased IL-6 secretion (Rott et al., 1993). High IFN-γ expression in the context of viral infection negates IL-6 anti-inflammatory effects via inhibiting STAT3-dependent IL-6 signaling (Bluyssen et al., 2010). This may contribute to cell-mediated entry by inducing focal inflammation and attracting nearby immune cells (Rott et al., 1993). Tax induction of NF-κB and STAT pathway may also increase CD4+ T-cell recruitment via the gateway reflex; however, the thoracic distribution of lesions in spinal cord of patients does not correspond with lumbar entry of cells in this pathway (Kamimura et al., 2019). Depletion of Th17 cells and negligible concentration of CCL20 in HAM/TSP (Ando et al., 2013; Sabharwal et al., 2014), which are both implicated in this reflex, might be responsible for lack of prominent migration in lumbar levels of spinal cord. Lastly, IFN-γ and TNF secretion by immune cells in CNS directly affects the cytokine profile of BECs by upregulating CXCL10, CXCL9, CX3CL1, CCL3, CCL4, and CCL5 expression and redistributing CCL3 to apical surface of these cells all of which ultimately results in further cell migration (Larochelle et al., 2011; Ando et al., 2013).
Upregulation of adhesion molecules by HTLV-1 might facilitate transmigration of immune cells across the BBB. Increased LFA-1 expression on surface of infected lymphocytes may amplify ICAM/LFA-1 interactions with BECs and promote adhesion of HTLV-1 infected cells to these cells (Romero et al., 2000). Endothelial cells in vicinity of spinal cords lesions express higher levels of Vascular cell adhesion molecule 1 (VCAM-1) which may interact with VLA-4 on immune cells to facilitate diapedesis (Umehara et al., 1996, 1998). However, this interaction was described to be of less importance in later studies as VCAM-1 increase is only observed following long-term cytokine stimulation and VLA-4 is decreased in isolated T-cells from HAM/TSP subjects (Romero et al., 2000). Activated Leukocyte Cell Adhesion Molecule (ALCAM) which was reported to be implicated in transmigration of CD4+ T-cells and MPs in MS (Lyck et al., 2017), is increased in HTLV-1 infected Tregs in HAM/TSP (Curis et al., 2016). Finally, OX40 and OX40L have been demonstrated to have a role in adhesion of T-cells to non-brain endothelium in ATLL (Romero et al., 2000). The role of this interaction in HAM/TSP has yet to be determined.
HTLV-1 infection can also modify gene expression in cells that might favor cell migration through the BBB. Gene expression in several pathways related to cell migration and ECM modifications are altered in CD4+ T-cells of HAM/TSP patients (Table 2). Paxillin (PXN) and Cytoskeleton organizer collapsin response mediator protein 2 (CRMP2) which are involved in cell migration pathway are increased in CD4+ T-cells of HAM/TSP patients and HTLV-1 infections, respectively (Varrin-Doyer et al., 2012; Pinto et al., 2013). Furthermore, Matrix metalloproteases (MMPs) which degrade ECM components, have been implicated in the pathogenesis of HAM/TSP by dysregulating BBB function and having a deleterious effect on ECM composition of CNS that leads to neuronal apoptosis (Umehara et al., 1998; Lakhan et al., 2013). MMP expression dysregulation is conspicuous due to observations demonstrating higher expression of MMP gelatinases (MMP-2 and MMP-9) in blood and CSF of HAM/TSP patients and downregulation of Tissue inhibitor of metalloproteinases 1 (TIMP-1) in CD4+ T-cells (Umehara et al., 1998; Mozhgani et al., 2018). Tax-mediated NF-κB activation may be responsible for elevated MMP-9 expression in HTLV-1-infected T-cells (Mori et al., 2002). Inactive chronic lesions of spinal cords of contained fewer MMP-2-positive or MMP-9-positive mononuclear cells than active-chronic lesions (Umehara et al., 1998). In light of the above, MMPs can be regarded as a key factor in bystander damage of CNS resident cells explained later.
IFN-β and corticosteroids, which are used both in MS and HAM/TSP therapy, have restorative effects on BBB function and reduce cell migration (Umehara et al., 1998; Bangham et al., 2015; Sweeney et al., 2019). Glucocorticoid signaling pathway is dysregulated in HAM/TSP which is characterized by downregulation of expression of Annexin A1 and Prostaglandin E synthase 3 in CD4+ T-cells (Mozhgani et al., 2018). The treatments in this regard, however, have shown modest improvement and lose efficacy over the course of disease (Nakagawa et al., 1996; Bangham et al., 2015). Nevertheless, the efficacy of corticosteroids in amelioration of the symptoms outweigh their adverse effects. Transient improvements in treatment with pulsed methylprednisolone and higher long term function with prednisolone has been observed (Araujo et al., 2020). The existing data is insufficient to recommend for other proposed treatment regimens for HAM/TSP such as IFN therapy, blood purification (lymphocytapheresis and plasmapheresis), Vitamin C, antiretroviral drugs, cyclosporine (Bangham et al., 2015), pentosan polysulfate sodium, pentoxifylline, and prosultiamine (Araujo et al., 2020).
Central Nervous System
The neuropathology of HAM/TSP is marked by perivascular infiltrates of PBMCs, chronic demyelination and axonal loss throughout the CNS (Aye et al., 2000; Bangham, 2018; Nozuma and Jacobson, 2019). Lesions are primarily located in white matter of lower thoracic cord of the patients and infiltrating cells mainly comprise T-cells (Iwasaki, 1993; Wu et al., 1993; Matsuoka et al., 1998), where viral and cellular interactions result in spinal manifestations of HAM/TSP. Evidence of neurodegeneration and metabolic changes also exists in brains of both ACs and to a greater extent in HAM/TSP patients (Dimber et al., 2016; Schütze et al., 2017). The inflammation in brain is accompanied by global cognitive impairment and executive dysfunction in HAM/TSP patients (Champs et al., 2019). The exact mechanisms of neurodegeneration and myelin loss in CNS are not understood, but three putative mechanisms have been proposed: (1) direct infection of CNS resident cells; (2) bystander damage to resident cells of CNS during chronic inflammation; and (3) Cross-reaction of Abs and molecular mimicry (Figure 4; Olière et al., 2011; Bangham, 2018; Nozuma and Jacobson, 2019). The validity and contribution of these hypotheses to the pathology of HAM/TSP will be discussed in this section along with major histopathological and functional changes of CNS cells.
Inflammatory Response of Immune Cells in CNS
Unlike monocytes and macrophages (Ginhoux and Prinz, 2015), activated T-cells are recruited to CNS under normal conditions (Hickey et al., 1991; Lehmann, 1998). It can be hypothesized that Infected lymphocytes could cross the barrier in this manner and express HTLV-1 proteins in CNS parenchyma. The presence of HTLV-1 in CNS induces inflammation and cytokine release by the resident CNS cells and migratory immune cells characterized by increased levels of TNF, IFN-γ, IL-1α, IL-β, and IL-6 in CSF and spinal cords of HAM/TSP patients (Umehara et al., 1994; Banerjee et al., 2007b; Champs et al., 2019). The ensuing recruitment of inflammatory cells from peripheral blood induces a vicious cycle of inflammation which is detrimental to CNS resident cells (Umehara et al., 1993; Furuya et al., 1999; Banerjee et al., 2007b). Over the course of inflammation, the number of CD4+ T-cells subsides in lesions (Umehara et al., 1993), probably because they are eliminated by HTLV-1-specific CD8+ T-cells. Indeed, widespread Fas+ T-cells are colocalized with apoptotic T-cells in active lesions (Umehara et al., 2002) while soluble Fas ligand is increased in CSF of HAM/TSP patients (Saito et al., 1999), suggesting increased Fas-mediated apoptosis of CD4+ T-cells in CNS. It is unclear whether the apoptotic T-cells are predominantly the result of immune response or FasL expression by astrocytes and BECs as part of physiological immune tolerance mechanism in CNS (Volpe et al., 2016). Distinct Fas genotypes may be associated with higher PVL and worse disease outcome in HTLV-1 infected carriers (Vallinoto et al., 2017).
Persistent inflammation in the CNS is not limited to the response against virus-infected cells. HTLV-1 infected cells release non-infectious extracellular vesicles containing Tax. It is noteworthy that CSF localization of these vesicles appear to be specific to HAM/TSP patients and is not normally observed in ACs (Anderson et al., 2018). Uptake of these exosomes by uninfected cells upregulates ICAM-1 and increases their susceptibility to VS-mediated infection (Pinto et al., 2019). Acquisition of Tax via these vesicles may also turn the uninfected cells to a potential target of cytotoxic degranulation of CTLs (Anderson et al., 2018). It can be surmised that high cytotoxicity against Tax+ monocytes in vitro (Matsuura et al., 2017) is the result of this interaction; since monocytes are considered resistant to HTLV-1 infection (Rocamonde et al., 2019). Moreover, perivascular macrophages can act as APCs (Hickey and Kimura, 1988; Lepoutre et al., 2009); presenting Tax epitopes to T-cells by scavenging apoptotic bodies of infected cells and the exosomes. It is unclear whether neurons or other CNS resident cells could acquire these vesicles; therefore, the extent of contribution of these exosomes to pathogenesis of HAM/TSP remains obscure (Pinto et al., 2019). One can assume that the antigen presentation, Tax uptake, and immune response of the large populations of infiltrating macrophages and microglia in CNS lead to the low-level inflammation in late-stage, chronic lesions, where infected CD4+ T-cells become sparse (Matsuura et al., 2017). This assumption is questionable owing to the fact that this activity is relative to the amount of HTLV-1 proviral DNA (Abe et al., 1999). These cells also seem to be inactivated during prolonged durations of the illness in chronic lesions (Abe et al., 1999).
Antibody secreting cells in the CSF are mainly involved in anti-Gag intrathecal antibody synthesis. These cells are not observed in ACs indicating a possible role of CSF ASCs in neuropathology of HAM/TSP (Enose-Akahata et al., 2018). Anti-Gag Abs were reported to cross-react with peroxiredoxin-1 (Prx1) and human transaldolase (TAL-H) while anti-Tax Abs react with heterogenous nuclear riboprotein A1 (hnRNPA1) (Banki et al., 1994; Levin et al., 2002; Jernigan et al., 2003; Kalume et al., 2004; Lee et al., 2008). However, the idea of molecular mimicry in CNS damage was later challenged by higher presence of hnRNPA autoantibodies in MS, similar antibody titer in other viral and neurodegenerative diseases, the absence of Tax in some HAM/TSP patients, and the fact that the expression of these genes is not restricted to CNS (Pulford et al., 1995; Ramirez et al., 2003; Yukitake et al., 2008; Fagerberg et al., 2014). It has been demonstrated that HAM/TSP IgG antibodies inhibit neuronal firing of nigral and pyramidal neurons but these immunoglobulins do not alter the viability of neurons during culture (Kalume et al., 2004). Nevertheless, the increase in barrier permeability and presence of antibodies in parenchyma may facilitate complement-mediated cell lysis (Ozden et al., 2002). The scale of protective and pathogenic functions of Abs in HAM/TSP is still largely not understood. In conclusion, there is a positive correlation between ASCs in CSF, Tregs, and PVL in patients (Enose-Akahata et al., 2018), but it is unclear if it translates to a unique immunopathological finding or being a consequence of neuroinflammatory milieu.
HTLV-1 and CNS Resident Cells
HTLV-1 can both directly and indirectly impact the normal function of neurons and glial cells. While HTLV-1 is capable of infecting both neurons and glial cells in vitro (Watabe et al., 1989; Akagi et al., 1992), only astrocytes were shown to be infected by HTLV-1 in patients by in situ hybridization (Lehky et al., 1995; Lepoutre et al., 2009). Numerous studies have elucidated the prominent role of astrocytes in pathology of HAM/TSP (Méndez et al., 1997; Szymocha et al., 2000a,b; Akaoka et al., 2001; Ando et al., 2013). These cells maintain the normal function of neurons throughout the CNS. Dysregulation of this subtype of cells in HAM/TSP disrupts glutamate homeostasis which may lead to neuronal excitotoxicity (Sofroniew and Vinters, 2010). It is demonstrated that HTLV-1 infection may lead to apoptosis of glial cells in vitro, especially oligodendrocytes (Matute et al., 2006). Nonetheless, the impact of direct infection of CNS resident cells is considered to be minimal in neuronal damage (Bangham and Osame, 2005; Bangham, 2018).
The culmination of robust synthesis of proinflammatory cytokines by virtually every cell type present in CNS and the immune response of infiltrating cells during HTLV-1-induced inflammation is the bystander damage of resident cells in CNS (Bangham and Osame, 2005). Tax transduction to astrocytomas and oligodendrogliomas results in significant upregulation of proinflammatory cytokines including TNF, IL-1α, IL-1β, IL-6 (Banerjee et al., 2007b). NT2-N neuron-like cells also express TNF when exposed to Tax (Cowan et al., 1997). High concentration of TNF in active lesions sensitizes oligodendrocytes to apoptosis and induces secretion of MMPs in astrocytes which further disrupts myelin structure and BBB integrity and increases conversion of TNF-precursor to its active form (Giraudon et al., 1997; Banerjee et al., 2007b). High IFN-γ secretion by infected cells also induces CXCL10 expression in astrocytes which in turn recruits more infected cells to the area and prolongs the inflammatory response. Therefore, it appears that astrocyte responses seem to establish a positive feedback loop contributing to chronic CNS inflammation (Ando et al., 2013).
Despite numerous studies revolving around HTLV-1 myelopathy, data regarding neuronal pathogenesis in HAM/TSP is lacking. While the death of glial cells and lack of trophic support coupled by direct damage by free radicals, cytokines, and destructive enzymes are most likely the cause, the precise pathology of axonal degeneration remains unclear. Identifying the mechanism of neuronal damage may provide a major breakthrough in elucidating the course and pathogenesis of not only HAM/TSP but other inflammatory demyelinating diseases as well.
Conclusion
Nearly 40 years of studying on HTLV-1 has yielded a myriad of findings concerning HTLV-1 structure and the effect of the virus on the human host. The study of retroviral clonality in HTLV-1 associated diseases has revealed that the integration site and transcriptional orientation of the provirus in the host genome and the selection force of host immune system against the clones lead to a signature pattern of clonality in malignant infections vs. ACs and patients with HAM/TSP (Gillet et al., 2011). In addition, the unique gene expression profile of PBMCs in HAM/TSP has manifested as disruptions in interferon signaling to the point that this pattern can be employed to distinguish HAM/TSP patients from ACs (Tattermusch et al., 2012) and ATLL patients (Fukutani et al., 2019).
All of these discoveries suggest that HAM/TSP is developed through a dynamic interaction between the host and the virus in all stages of the disease, starting from the proviral integration in active transcription sites (Gillet et al., 2011) and viral modification of host gene expression to cross-communication of infected cells with the immune system and negative selection of active clones to translocation of the virus to CNS. Despite these improvements, further research is required to fully understand the unknown aspects of the disease, which have been highlighted in this article.
Author Contributions
SA performed systematic and data collection and was a major contributor in writing the manuscript. MT-R provided with the illustrations and was a minor contributor to editing the manuscript. GM was a minor contributor in data curation and in writing the manuscript. S-HM conceptualized the review article, had a minor contribution to writing, and provided oversight, critical evaluation, and verification of the manuscript. All authors read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Dr. Charles R. M. Bangham (Division of Infectious Diseases, Faculty of Medicine, Imperial College, London) for providing them with in-depth knowledge on HAM/TSP and his critical evaluation of the manuscript.
References
Abe, M., Umehara, F., Kubota, R., Moritoyo, T., Izumo, S., and Osame, M. (1999). Activation of macrophages/microglia with the calcium-binding proteins MRP14 and MRP8 is related to the lesional activities in the spinal cord of HTLV-I associated myelopathy. J. Neurol. 246, 358–364. doi: 10.1007/s004150050363
Abolbashari, S., Ghayour-Mobarhan, M., Ebrahimi, M., and Meshkat, Z. (2018). The role of human T-lymphotropic virus (HTLV) in cardiovascular diseases: a review of literature. ARYA Atheroscler. 14, 183–187.
Afonso, P. V., Ozden, S., Cumont, M.-C., Seilhean, D., Cartier, L., Rezaie, P., et al. (2008). Alteration of blood-brain barrier integrity by retroviral infection. PLoS Pathog. 4:e1000205. doi: 10.1371/journal.ppat.1000205
Akagi, T., Hoshida, Y., Yoshino, T., Teramoto, N., Kondo, E., Hayashi, K., et al. (1992). Infectivity of human T-lymphotropic virus type I to human nervous tissue cells in vitro. Acta Neuropathol. 84, 147–152. doi: 10.1007/bf00311387
Akaoka, H., Szymocha, R., Beurton-Marduel, P., Bernard, A., Belin, M. F., and Giraudon, P. (2001). Functional changes in astrocytes by human T-lymphotropic virus type-1 T-lymphocytes. Virus Res. 78, 57–66. doi: 10.1016/s0168-1702(01)00284-2
Alais, S., Mahieux, R., and Dutartre, H. (2015). Viral source-independent high susceptibility of dendritic cells to human T-cell leukemia virus type 1 infection compared to that of T lymphocytes. J. Virol. 89, 10580–10590. doi: 10.1128/jvi.01799-15
Amorim, C. F., Carvalho, N. B., Neto, J. A., Santos, S. B., Grassi, M. F. R., Carvalho, L. P., et al. (2019). The role of NK cells in the control of viral infection in HTLV-1 carriers. J. Immunol. Res. 2019:657 4828.
Amorim, C. F., Souza, A. S., Diniz, A. G., Carvalho, N. B., Santos, S. B., and Carvalho, E. M. (2014). Functional activity of monocytes and macrophages in HTLV-1 infected subjects. PLoS Negl. Trop. Dis. 8:e3399. doi: 10.1371/journal.pntd.0003399
Anderson, M. R., Pleet, M. L., Enose-Akahata, Y., Erickson, J., Monaco, M. C., Akpamagbo, Y., et al. (2018). Viral antigens detectable in CSF exosomes from patients with retrovirus associated neurologic disease: functional role of exosomes. Clin. Transl. Med. 7:24.
Ando, H., Sato, T., Tomaru, U., Yoshida, M., Utsunomiya, A., Yamauchi, J., et al. (2013). Positive feedback loop via astrocytes causes chronic inflammation in virus-associated myelopathy. Brain 136, 2876–2887. doi: 10.1093/brain/awt183
Araujo, A., Bangham, C. R. M., Casseb, J., Gotuzzo, E., Jacobson, S., Martin, F., et al. (2020). Management of HAM/TSP. Neurology 1–8. doi: 10.1212/CPJ.0000000000000832
Araujo, A. Q., and Silva, M. T. (2006). The HTLV-1 neurological complex. Lancet Neurol. 5, 1068–1076. doi: 10.1016/s1474-4422(06)70628-7
Araujo, A. Q. C., and Wedemann, D. (2019). HTLV-1 associated neurological complex. what is hidden below the water? AIDS Rev. 21, 211–217.
Araya, N., Sato, T., Yagishita, N., Ando, H., Utsunomiya, A., Jacobson, S., et al. (2011). Human T-lymphotropic virus type 1 (HTLV-1) and regulatory T cells in HTLV-1-associated neuroinflammatory disease. Viruses 3, 1532–1548. doi: 10.3390/v3091532
Armstrong, M. J., Corbett, C., Rowe, I. A., Taylor, G. P., and Neuberger, J. M. (2012). HTLV-1 in solid-organ transplantation: current challenges and future management strategies. Transplantation 94, 1075–1084. doi: 10.1097/tp.0b013e318263ad7a
Asquith, B., Zhang, Y., Mosley, A. J., De Lara, C. M., Wallace, D. L., Worth, A., et al. (2007). In vivo T lymphocyte dynamics in humans and the impact of human T lymphotropic virus 1 infection. Proc. Natl. Acad. Sci. U.S.A. 104, 8035–8040. doi: 10.1073/pnas.0608832104
Assil, S., Futsch, N., Décembre, E., Alais, S., Gessain, A., Cosset, F.-L., et al. (2019). Sensing of cell-associated HTLV by plasmacytoid dendritic cells is regulated by dense β-galactoside glycosylation. PLoS Pathog. 15:e1007589. doi: 10.1371/journal.ppat.1007589
Assone, T., De Souza, F. V., Gaester, K. O., Fonseca, L. A. M., Do Carmo Luiz, O., Malta, F., et al. (2014). IL28B gene polymorphism SNP rs8099917 genotype GG is associated with HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) in HTLV-1 carriers. PLoS Negl. Trop. Dis. 8:e3199. doi: 10.1371/journal.pntd.0003199
Aye, M. M., Matsuoka, E., Moritoyo, T., Umehara, F., Suehara, M., Hokezu, Y., et al. (2000). Histopathological analysis of four autopsy cases of HTLV-I-associated myelopathy/tropical spastic paraparesis: inflammatory changes occur simultaneously in the entire central nervous system. Acta Neuropathol. 100, 245–252. doi: 10.1007/s004019900170
Azakami, K., Sato, T., Araya, N., Utsunomiya, A., Kubota, R., Suzuki, K., et al. (2009). Severe loss of invariant NKT cells exhibiting anti–HTLV-1 activity in patients with HTLV-1–associated disorders. Blood 114, 3208–3215. doi: 10.1182/blood-2009-02-203042
Azodi, S., Nair, G., Enose-Akahata, Y., Charlip, E., Vellucci, A., Cortese, I., et al. (2017). Imaging spinal cord atrophy in progressive myelopathies: HTLV-I-associated neurological disease (HAM/TSP) and multiple sclerosis (MS). Ann. Neurol. 82, 719–728. doi: 10.1002/ana.25072
Banerjee, P., Feuer, G., and Barker, E. (2007a). Human T-cell leukemia virus type 1 (HTLV-1) p12I down-modulates ICAM-1 and-2 and reduces adherence of natural killer cells, thereby protecting HTLV-1-infected primary CD4+ T cells from autologous natural killer cell-mediated cytotoxicity despite the reduction of major histocompatibility complex class I molecules on infected cells. J. Virol. 81, 9707–9717. doi: 10.1128/jvi.00887-07
Banerjee, P., Rochford, R., Antel, J., Canute, G., Wrzesinski, S., Sieburg, M., et al. (2007b). proinflammatory cytokine gene induction by human T-Cell Leukemia Virus Type 1 (HTLV-1) and HTLV-2 Tax in primary human glial cells. J. Virol. 81, 1690–1700.
Bangham, C. R. (2009). CTL quality and the control of human retroviral infections. Eur. J. Immunol. 39, 1700–1712. doi: 10.1002/eji.200939451
Bangham, C. R., Araujo, A., Yamano, Y., and Taylor, G. P. (2015). HTLV-1-associated myelopathy/tropical spastic paraparesis. Nat. Rev. Dis. Primers 1, 1–17.
Bangham, C. R. M. (2018). Human T Cell leukemia virus type 1: persistence and pathogenesis. Annu. Rev. Immunol. 36, 43–71. doi: 10.1146/annurev-immunol-042617-053222
Bangham, C. R. M., Miura, M., Kulkarni, A., and Matsuoka, M. (2019). Regulation of latency in the Human T Cell leukemia virus. HTLV-1. Annu. Rev. Virol. 6, 365–385. doi: 10.1146/annurev-virology-092818-015501
Bangham, C. R. M., and Osame, M. (2005). Cellular immune response to HTLV-1. Oncogene 24, 6035–6046. doi: 10.1038/sj.onc.1208970
Bangham, C. R. M., and Toulza, F. (2011). “Adult T Cell leukemia/lymphoma: FoxP3+ cells and the cell-mediated immune response to HTLV-1,” in Advances in Cancer Research, ed. G. Klein (Cambridge, MA: Academic Press), 163–182. doi: 10.1016/b978-0-12-385524-4.00004-0
Banki, K., Colombo, E., Sia, F., Halladay, D., Mattson, D. H., Tatum, A. H., et al. (1994). Oligodendrocyte-specific expression and autoantigenicity of transaldolase in multiple sclerosis. J. Exp. Med. 180, 1649–1663. doi: 10.1084/jem.180.5.1649
Baratella, M., Forlani, G., Raval, G. U., Tedeschi, A., Gout, O., Gessain, A., et al. (2017). Cytoplasmic localization of HTLV-1 HBZ protein: a biomarker of HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP). PLoS Negl. Trop. Dis. 11:e0005285. doi: 10.1371/journal.pntd.0005285
Bezerra, C. A., Cardoso, T. M., Giudice, A., Porto, A. F., Santos, S. B., Carvalho, E. M., et al. (2011). Evaluation of the microbicidal activity and cytokines/chemokines profile released by neutrophils from HTLV-1-Infected individuals. Scand. J. Immunol. 74, 310–317. doi: 10.1111/j.1365-3083.2011.02579.x
Billman, M. R., Rueda, D., and Bangham, C. R. M. (2017). Single-cell heterogeneity and cell-cycle-related viral gene bursts in the human leukaemia virus HTLV-1. Wellcome Open Res. 2, 87–87. doi: 10.12688/wellcomeopenres.12469.2
Bluyssen, H. A. R., Rastmanesh, M. M., Tilburgs, C., Jie, K., Wesseling, S., Goumans, M.-J., et al. (2010). IFNγ-dependent SOCS3 expression inhibits IL-6-induced STAT3 phosphorylation and differentially affects IL-6 mediated transcriptional responses in endothelial cells. Am. J. Physiol. Cell Physiol. 299, C354–C362.
Boelen, L., Debebe, B., Silveira, M., Salam, A., Makinde, J., Roberts, C. H., et al. (2018). Inhibitory killer cell immunoglobulin-like receptors strengthen CD8(+) T cell-mediated control of HIV-1, HCV, and HTLV-1. Sci. Immunol. 3:eaao2892. doi: 10.1126/sciimmunol.aao2892
Boxus, M., Twizere, J.-C., Legros, S., Dewulf, J.-F., Kettmann, R., and Willems, L. (2008). The HTLV-1 Tax interactome. Retrovirology 5:76. doi: 10.1186/1742-4690-5-76
Carpentier, A., Barez, P.-Y., Hamaidia, M., Gazon, H., De Brogniez, A., Perike, S., et al. (2015). Modes of human T cell leukemia virus type 1 transmission, replication and persistence. Viruses 7, 3603–3624. doi: 10.3390/v7072793
Caswell, R. J., and Manavi, K. (2020). Emerging sexually transmitted viral infections: review of human T-lymphotropic virus-1 disease. Int. J. STD AIDS 31, 92–99. doi: 10.1177/0956462419886940
Catalan-Soares, B., Carneiro-Proietti, A. B. F., Da Fonseca, F., Correa-Oliveira, R., Peralva-Lima, D., Portela, R., et al. (2009). HLA class I alleles in HTLV-1-associated myelopathy and asymptomatic carriers from the Brazilian cohort GIPH. Med. Microbiol. Immunol. 198, 1–3. doi: 10.1007/s00430-008-0096-z
Ceccaldi, P.-E., Delebecque, F., Prevost, M.-C., Moris, A., Abastado, J.-P., Gessain, A., et al. (2006). DC-SIGN facilitates fusion of dendritic cells with human T-cell leukemia virus type 1-infected cells. J. Virol. 80, 4771–4780. doi: 10.1128/jvi.80.10.4771-4780.2006
Champs, A. P. S., De Azeredo Passos, V. M., Carvalho, G., Barreto, S. M., Meirelles, C., and Caramelli, P. (2019). Cognitive impairment in HTLV-1-associated myelopathy, proviral load and inflammatory markers. Int. J. Infect. Dis. 84, 121–126. doi: 10.1016/j.ijid.2019.05.010
Cook, L. B., Elemans, M., Rowan, A. G., and Asquith, B. (2013). HTLV-1: persistence and pathogenesis. Virology 435, 131–140. doi: 10.1016/j.virol.2012.09.028
Coskun, A. K., and Sutton, R. E. (2005). Expression of glucose transporter 1 confers susceptibility to human T-cell leukemia virus envelope-mediated fusion. J. Virol. 79, 4150–4158. doi: 10.1128/jvi.79.7.4150-4158.2005
Cowan, E. P., Alexander, R. K., Daniel, S., Kashanchi, F., and Brady, J. N. (1997). Induction of tumor necrosis factor alpha in human neuronal cells by extracellular human T-cell lymphotropic virus type 1 Tax. J. Virol. 71:6982. doi: 10.1128/jvi.71.9.6982-6989.1997
Curis, C., Percher, F., Jeannin, P., Montange, T., Chevalier, S. A., Seilhean, D., et al. (2016). Human T-Lymphotropic Virus Type 1-Induced overexpression of activated leukocyte cell adhesion molecule (ALCAM) facilitates trafficking of infected lymphocytes through the blood-brain barrier. J. Virol. 90, 7303–7312. doi: 10.1128/jvi.00539-16
da Silva Dias, G. A., Sousa, R. C. M., Gomes, L. F., Caldas, C. A. M., Nassiri, R., Quaresma, J. A. S., et al. (2016). Correlation between clinical symptoms and peripheral immune response in HAM/TSP. Microb. Pathog. 92, 72–75. doi: 10.1016/j.micpath.2015.11.018
de Castro-Amarante, M. F., Pise-Masison, C. A., Mckinnon, K., Parks, R. W., Galli, V., Omsland, M., et al. (2016). Human T cell leukemia virus type 1 infection of the three monocyte subsets contributes to viral burden in humans. J. Virol. 90, 2195–2207. doi: 10.1128/jvi.02735-15
Descours, B., Cribier, A., Chable-Bessia, C., Ayinde, D., Rice, G., Crow, Y., et al. (2012). SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4+ T-cells. Retrovirology 9:87.
Dimber, R., Guo, Q., Bishop, C., Adonis, A., Buckley, A., Kocsis, A., et al. (2016). Evidence of brain inflammation in patients with human T-Lymphotropic Virus Type 1–Associated Myelopathy (HAM): a pilot, multimodal imaging study using 11C-PBR28 PET, MR T1-weighted, and diffusion-weighted imaging. J. Nucl. Med. 57, 1905–1912. doi: 10.2967/jnumed.116.175083
Elovaara, I., Koenig, S., Brewah, A. Y., Woods, R. M., Lehky, T., and Jacobson, S. (1993). High human T cell lymphotropic virus type 1 (HTLV-1)-specific precursor cytotoxic T lymphocyte frequencies in patients with HTLV-1-associated neurological disease. J. Exp. Med. 177, 1567–1573. doi: 10.1084/jem.177.6.1567
Enose-Akahata, Y., Abrams, A., Johnson, K. R., Maloney, E. M., and Jacobson, S. (2012a). Quantitative differences in HTLV-I antibody responses: classification and relative risk assessment for asymptomatic carriers and ATL and HAM/TSP patients from Jamaica. Blood 119, 2829–2836. doi: 10.1182/blood-2011-11-390807
Enose-Akahata, Y., Matsuura, E., Tanaka, Y., Oh, U., and Jacobson, S. (2012b). Minocycline modulates antigen-specific CTL activity through inactivation of mononuclear phagocytes in patients with HTLV-I associated neurologic disease. Retrovirology 9:16. doi: 10.1186/1742-4690-9-16
Enose-Akahata, Y., Azodi, S., Smith, B. R., Billioux, B. J., Vellucci, A., Ngouth, N., et al. (2018). Immunophenotypic characterization of CSF B cells in virus-associated neuroinflammatory diseases. PLoS Pathog. 14:e1007042. doi: 10.1371/journal.ppat.1007042
Enose-Akahata, Y., and Jacobson, S. (2019). Immunovirological markers in HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP). Retrovirology 16, 1–14.
Enose-Akahata, Y., Oh, U., Grant, C., and Jacobson, S. (2008). Retrovirally induced CTL degranulation mediated by IL-15 expression and infection of mononuclear phagocytes in patients with HTLV-I–associated neurologic disease. Blood 112, 2400–2410. doi: 10.1182/blood-2008-02-138529
Enose-Akahata, Y., Vellucci, A., and Jacobson, S. (2017). Role of HTLV-1 Tax and HBZ in the pathogenesis of HAM/TSP. Front. Microbiol. 8:2563. doi: 10.3389/fmicb.2017.02563
Fagerberg, L., Hallström, B. M., Oksvold, P., Kampf, C., Djureinovic, D., Odeberg, J., et al. (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13, 397–406. doi: 10.1074/mcp.m113.035600
Fochi, S., Ciminale, V., Trabetti, E., Bertazzoni, U., D’agostino, D. M., Zipeto, D., et al. (2019). NF-kappaB and MicroRNA deregulation mediated by HTLV-1 Tax and HBZ. Pathogens 8:290. doi: 10.3390/pathogens8040290
Fochi, S., Mutascio, S., Bertazzoni, U., Zipeto, D., and Romanelli, M. G. (2018). HTLV deregulation of the NF-kappaB pathway: an update on tax and antisense proteins role. Front. Microbiol. 9:285. doi: 10.3389/fmicb.2018.00285
Forlani, G., Baratella, M., Tedeschi, A., Pique, C., Jacobson, S., and Accolla, R. S. (2019). HTLV-1 HBZ protein resides exclusively in the cytoplasm of infected cells in asymptomatic carriers and HAM/TSP patients. Front. Microbiol. 10:819. doi: 10.3389/fmicb.2019.00819
Fukutani, E. R., Ramos, P. I. P., Gonçalves, K., Irahe, J., Azevedo, L. G., Rodrigues, M. M. D. S., et al. (2019). Meta-analysis of HTLV-1-infected patients identifies CD40LG and GBP2 as markers of ATLL and HAM/TSP clinical status: two genes beat as one. Front. Genet. 10:1056. doi: 10.3389/fgene.2019.01056
Furukawa, Y., Bangham, C. R. M., Taylor, G. P., Weber, J. N., and Osame, M. (2000). Frequent reversible membrane damage in peripheral blood B cells in human T cell lymphotropic virus type I (HTLV-I)-associated myelopathy/tropical spastic paraparesis (HAM/TSP). Clin. Exp. Immunol. 120, 307–316. doi: 10.1046/j.1365-2249.2000.01211.x
Furukawa, Y., Fujisawa, J., Osame, M., Toita, M., Sonoda, S., Kubota, R., et al. (1992). Frequent clonal proliferation of human T-cell leukemia virus type 1 (HTLV-1)-infected T cells in HTLV-1-associated myelopathy (HAM-TSP). Blood 80, 1012–1016. doi: 10.1182/blood.v80.4.1012.bloodjournal8041012
Furukawa, Y., Osame, M., Kubota, R., Tara, M., and Yoshida, M. (1995). Human T-cell leukemia virus type-1 (HTLV-1) Tax is expressed at the same level in infected cells of HTLV-1-associated myelopathy or tropical spastic paraparesis patients as in asymptomatic carriers but at a lower level in adult T-cell leukemia cells. Blood 85, 1865–1870. doi: 10.1182/blood.v85.7.1865.bloodjournal8571865
Furuta, R., Yasunaga, J.-I., Miura, M., Sugata, K., Saito, A., Akari, H., et al. (2017). Human T-cell leukemia virus type 1 infects multiple lineage hematopoietic cells in vivo. PLoS Pathog. 13:e1006722. doi: 10.1371/journal.ppat.1006722
Furuya, T., Nakamura, T., Fujimoto, T., Nakane, S., Kambara, C., Shirabe, S., et al. (1999). Elevated levels of interleukin-12 and interferon-gamma in patients with human T lymphotropic virus type I-associated myelopathy. J. Neuroimmunol. 95, 185–189.
Giam, C.-Z., and Semmes, O. J. (2016). HTLV-1 infection and adult T-cell leukemia/lymphoma—a tale of two proteins: tax and HBZ. Viruses 8:161. doi: 10.3390/v8060161
Giebler, H. A., Loring, J. E., Van Orden, K., Colgin, M. A., Garrus, J. E., Escudero, K. W., et al. (1997). Anchoring of CREB binding protein to the human T-cell leukemia virus type 1 promoter: a molecular mechanism of Tax transactivation. Mol. Cell. Biol. 17, 5156–5164. doi: 10.1128/mcb.17.9.5156
Gillet, N. A., Malani, N., Melamed, A., Gormley, N., Carter, R., Bentley, D., et al. (2011). The host genomic environment of the provirus determines the abundance of HTLV-1–infected T-cell clones. Blood 117, 3113–3122. doi: 10.1182/blood-2010-10-312926
Ginhoux, F., and Prinz, M. (2015). Origin of microglia: current concepts and past controversies. Cold Spring Harb. Perspect. Biol. 7:a020537. doi: 10.1101/cshperspect.a020537
Giraudon, P., Buart, S., Bernard, A., and Belin, M. F. (1997). Cytokines secreted by glial cells infected with HTLV-I modulate the expression of matrix metalloproteinases (MMPs) and their natural inhibitor (TIMPs): possible involvement in neurodegenerative processes. Mol. Psychiatry 2, 107–110. doi: 10.1038/sj.mp.4000218
Grant, C., Barmak, K., Alefantis, T., Yao, J., Jacobson, S., and Wigdahl, B. (2002). Human T cell leukemia virus type I and neurologic disease: events in bone marrow, peripheral blood, and central nervous system during normal immune surveillance and neuroinflammation. J. Cell. Physiol. 190, 133–159. doi: 10.1002/jcp.10053
Grant, C., Oh, U., Yao, K., Yamano, Y., and Jacobson, S. (2008). Dysregulation of TGF-β signaling and regulatory and effector T-cell function in virus-induced neuroinflammatory disease. Blood 111, 5601–5609. doi: 10.1182/blood-2007-11-123430
Gross, C., and Thoma-Kress, A. K. (2016). Molecular mechanisms of HTLV-1 cell-to-cell transmission. Viruses 8:74. doi: 10.3390/v8030074
Gross, C., Wiesmann, V., Millen, S., Kalmer, M., Wittenberg, T., Gettemans, J., et al. (2016). The tax-inducible actin-bundling protein fascin is crucial for release and cell-to-cell transmission of human T-cell leukemia virus type 1 (HTLV-1). PLoS Pathog. 12:e1005916. doi: 10.1371/journal.ppat.1005916
Hayashi, D., Kubota, R., Takenouchi, N., Tanaka, Y., Hirano, R., Takashima, H., et al. (2008). Reduced Foxp3 expression with increased cytomegalovirus-specific CTL in HTLV-I-associated myelopathy. J. Neuroimmunol. 200, 115–124. doi: 10.1016/j.jneuroim.2008.06.005
Hickey, W. F., Hsu, B. L., and Kimura, H. (1991). T-lymphocyte entry into the central nervous system. J. Neurosci. Res. 28, 254–260. doi: 10.1002/jnr.490280213
Hickey, W. F., and Kimura, H. (1988). Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science 239, 290–292. doi: 10.1126/science.3276004
Hieshima, K., Nagakubo, D., Nakayama, T., Shirakawa, A.-K., Jin, Z., and Yoshie, O. (2008). Tax-inducible production of CC chemokine ligand 22 by human T Cell leukemia virus Type 1 (HTLV-1)-Infected T Cells promotes preferential transmission of HTLV-1 to CCR4-Expressing CD4 T Cells. J. Immunol. 180, 931–939. doi: 10.4049/jimmunol.180.2.931
Higashimura, N., Takasawa, N., Tanaka, Y., Nakamura, M., and Sugamura, K. (1996). Induction of OX40, a Receptor of gp34, on T Cells by trans-acting transcriptional activator, tax, of Human T-Cell Leukemia Virus Type I. Jp. J. Cancer Res. 87, 227–231. doi: 10.1111/j.1349-7006.1996.tb00210.x
Hoshino, H. (2012). Cellular factors involved in HTLV-1 entry and pathogenicit. Front. Microbiol. 3:222. doi: 10.3389/fmicb.2012.00222
Hudspeth, K., Silva-Santos, B., and Mavilio, D. (2013). Natural cytotoxicity receptors: broader expression patterns and functions in innate and adaptive immune cells. Front. Immunol. 4:69. doi: 10.3389/fimmu.2013.00069
Igakura, T., Stinchcombe, J. C., Goon, P. K. C., Taylor, G. P., Weber, J. N., Griffiths, G. M., et al. (2003). Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 299, 1713–1716. doi: 10.1126/science.1080115
Iwanaga, M., Watanabe, T., Utsunomiya, A., Okayama, A., Uchimaru, K., Koh, K.-R., et al. (2010). Human T-cell leukemia virus type I (HTLV-1) proviral load and disease progression in asymptomatic HTLV-1 carriers: a nationwide prospective study in Japan. Blood 116, 1211–1219. doi: 10.1182/blood-2009-12-257410
Iwasaki, Y. (1990). Pathology of chronic myelopathy associated with HTLV-I infection (HAM/TSP). J. Neurol. Sci. 96, 103–123. doi: 10.1016/0022-510x(90)90060-z
Iwasaki, Y. (1993). Human T cell leukemia virus type I infection and chronic myelopathy. Brain Pathol. 3, 1–10. doi: 10.1111/j.1750-3639.1993.tb00719.x
Jain, P., Manuel, S. L., Khan, Z. K., Ahuja, J., Quann, K., and Wigdahl, B. (2009). DC-SIGN mediates cell-free infection and transmission of human T-cell lymphotropic virus type 1 by dendritic cells. J. Virol. 83, 10908–10921. doi: 10.1128/jvi.01054-09
Jeffery, K. J., Siddiqui, A. A., Bunce, M., Lloyd, A. L., Vine, A. M., Witkover, A. D., et al. (2000). The influence of HLA class I alleles and heterozygosity on the outcome of human T cell lymphotropic virus type I infection. J. Immunol. 165, 7278–7284. doi: 10.4049/jimmunol.165.12.7278
Jeffery, K. J., Usuku, K., Hall, S. E., Matsumoto, W., Taylor, G. P., Procter, J., et al. (1999). HLA alleles determine human T-lymphotropic virus-I (HTLV-I) proviral load and the risk of HTLV-I-associated myelopathy. Proc. Natl. Acad. Sci. U.S.A. 96, 3848–3853. doi: 10.1073/pnas.96.7.3848
Jernigan, M., Morcos, Y., Lee, S. M., Dohan, F. C., Raine, C., and Levin, M. C. (2003). IgG in brain correlates with clinicopathological damage in HTLV-1 associated neurologic disease. Neurology 60:1320. doi: 10.1212/01.wnl.0000059866.03880.ba
Jones, K. S., Petrow-Sadowski, C., Huang, Y. K., Bertolette, D. C., and Ruscetti, F. W. (2008). Cell-free HTLV-1 infects dendritic cells leading to transmission and transformation of CD4+ T cells. Nat. Med. 14, 429–436. doi: 10.1038/nm1745
Journo, C., Douceron, E., and Mahieux, R. (2009). HTLV gene regulation: because size matters, transcription is not enough. Fut. Microbiol. 4, 425–440. doi: 10.2217/fmb.09.13
Journo, C., and Mahieux, R. (2011). HTLV-1 and innate immunity. Viruses 3, 1374–1394. doi: 10.3390/v3081374
Kalume, F., Lee, S. M., Morcos, Y., Callaway, J. C., and Levin, M. C. (2004). Molecular mimicry: cross-reactive antibodies from patients with immune-mediated neurologic disease inhibit neuronal firing. J. Neurosci. Res. 77, 82–89. doi: 10.1002/jnr.20137
Kamimura, D., Ohki, T., Arima, Y., Ota, M., and Murakami, M. (2019). Gateway reflex: local neuroimmune interactions that regulate blood vessels. Neurochem. Int. 130:104303. doi: 10.1016/j.neuint.2018.09.011
Kannian, P., Yin, H., Doueiri, R., Lairmore, M. D., Fernandez, S., and Green, P. L. (2012). Distinct transformation tropism exhibited by human T Lymphotropic Virus Type 1 (HTLV-1) and HTLV-2 is the result of postinfection T Cell clonal expansion. J. Virol. 86:3757. doi: 10.1128/jvi.06900-11
Kataoka, K., Nagata, Y., Kitanaka, A., Shiraishi, Y., Shimamura, T., Yasunaga, J.-I., et al. (2015). Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 47:1304.
Kattan, T., Macnamara, A., Rowan, A. G., Nose, H., Mosley, A. J., Tanaka, Y., et al. (2009). The avidity and lytic efficiency of the CTL Response to HTLV-1. J. Immunol. 182, 5723–5729. doi: 10.4049/jimmunol.0900069
Kawahigashi, N., Furukawa, Y., Saito, M., Usuku, K., and Osame, M. (1998). Predominant expression of Fas ligand mRNA in CD8+ T lymphocytes in patients with HTLV-1 associated myelopathy. J. Neuroimmunol. 90, 199–206. doi: 10.1016/s0165-5728(98)00147-7
Kawase, K. I., Katamine, S., Moriuchi, R., Miyamoto, T., Kubota, K., Igarashi, H., et al. (1992). Maternal transmisson of HTLV-1 other than through breast milk: discrepancy between the polymerase chain reaction positivity of cord blood samples for HTLV-1 and the subsequent seropositivity of individuals. Jpn. J. Cancer Res. 83, 968–977. doi: 10.1111/j.1349-7006.1992.tb02009.x
Kitze, B., Usuku, K., Izumo, S., Nakamura, M., Shiraki, H., Ijichi, S., et al. (1996). Diversity of intrathecal antibody synthesis against HTLV-I and its relation to HTLV-I associated myelopathy. J. Neurol. 243, 393–400. doi: 10.1007/bf00868998
Kubota, R., Kawanishi, T., Matsubara, H., Manns, A., and Jacobson, S. (1998). Demonstration of Human T Lymphotropic Virus Type I (HTLV-I) tax-specific CD8 lymphocytes directly in peripheral blood of HTLV-I-associated myelopathy/tropical spastic paraparesis patients by intracellular cytokine detection. J. Immunol. 161, 482–488.
Kubota, R., Kawanishi, T., Matsubara, H., Manns, A., and Jacobson, S. (2000a). HTLV-I specific IFN-γ+ CD8+ lymphocytes correlate with the proviral load in peripheral blood of infected individuals. J. Neuroimmunol. 102, 208–215. doi: 10.1016/s0165-5728(99)00175-7
Kubota, R., Nagai, M., Kawanishi, T., Osame, M., and Jacobson, S. (2000b). Increased HTLV type 1 tax specific CD8+ cells in HTLV type 1-asociated myelopathy/tropical spastic paraparesis: correlation with HTLV type 1 proviral load. AIDS Res. Hum. Retroviruses 16, 1705–1709. doi: 10.1089/08892220050193182
Kulkarni, A., and Bangham, C. R. M. (2018). HTLV-1: regulating the balance between proviral latency and reactivation. Front. Microbiol 9:449. doi: 10.3389/fmicb.2018.00449
Lakhan, S. E., Kirchgessner, A., Tepper, D., and Leonard, A. (2013). Matrix metalloproteinases and blood-brain barrier disruption in acute ischemic stroke. Front. Neurol. 4:32. doi: 10.3389/fneur.2013.00032
Larochelle, C., Alvarez, J. I., and Prat, A. (2011). How do immune cells overcome the blood–brain barrier in multiple sclerosis? FEBS Lett. 585, 3770–3780. doi: 10.1016/j.febslet.2011.04.066
Leal, F. E., Menezes, S. M., Costa, E. A. S., Brailey, P. M., Gama, L., Segurado, A. C., et al. (2018). Comprehensive antiretroviral restriction factor profiling reveals the evolutionary imprint of the ex vivo and in vivo IFN-beta response in HTLV-1-associated neuroinflammation. Front. Microbiol. 9:985. doi: 10.3389/fmicb.2018.00985
Leal, F. E., Ndhlovu, L. C., Hasenkrug, A. M., Bruno, F. R., Carvalho, K. I., Wynn-Williams, H., et al. (2013). Expansion in CD39+ CD4+ immunoregulatory t cells and rarity of Th17 cells in HTLV-1 infected patients is associated with neurological complications. PLoS Negl. Trop. Dis. 7:e2028. doi: 10.1371/journal.pntd.0002028
Lee, S., Shin, Y., Marler, J., and Levin, M. C. (2008). Post-translational glycosylation of target proteins implicate molecular mimicry in the pathogenesis of HTLV-1 associated neurological disease. J. Neuroimmunol. 204, 140–148. doi: 10.1016/j.jneuroim.2008.07.020
Lehky, T. J., Levin, M. C., Flerlage, N., Jacobson, S., Fox, C. H., Koenig, S., et al. (1995). Detection of human T-lymphotropic virus type I (HTLV-I) tax RNA in the central nervous system of HTLV-I–associated myelopathy/tropical spastic paraparesis patients by in situ hybridization. Ann. Neurol. 37, 167–175. doi: 10.1002/ana.410370206
Lehmann, P. V. (1998). The fate of T cells in the brain: veni, vidi, vici and veni, mori. Am. J. Pathol. 153, 677–680.
Lepoutre, V., Jain, P., Quann, K., Wigdahl, B., and Khan, Z. K. (2009). Role of resident CNS cell populations in HTLV-1-associated neuroinflammatory disease. Front. Biosci. 14:1152–1168. doi: 10.2741/3300
Levin, M. C., Lee, S. M., Kalume, F., Morcos, Y., Dohan, F. C., Hasty, K. A., et al. (2002). Autoimmunity due to molecular mimicry as a cause of neurological disease. Nat. Med. 8, 509–513. doi: 10.1038/nm0502-509
Liebner, S., Dijkhuizen, R. M., Reiss, Y., Plate, K. H., Agalliu, D., and Constantin, G. (2018). Functional morphology of the blood–brain barrier in health and disease. Acta Neuropathol. 135, 311–336. doi: 10.1007/s00401-018-1815-1
Longo, D. L., Gelmann, E. P., Cossman, J., Young, R. A., Gallo, R. C., O’brien, S. J., et al. (1984). Isolation of HTLV-transformed B-lymphocyte clone from a patient with HTLV-associated adult T-cell leukaemia. Nature 310, 505–506. doi: 10.1038/310505a0
Lyck, R., Lécuyer, M. A., Abadier, M., Wyss, C. B., Matti, C., Rosito, M., et al. (2017). ALCAM (CD166) is involved in extravasation of monocytes rather than T cells across the blood-brain barrier. J. Cereb. Blood Flow Metab. 37, 2894–2909. doi: 10.1177/0271678x16678639
Ma, G., Yasunaga, J.-I., Akari, H., and Matsuoka, M. (2015). TCF1 and LEF1 act as T-cell intrinsic HTLV-1 antagonists by targeting Tax. Proc. Natl. Acad. Sci. U.S.A. 112, 2216–2221. doi: 10.1073/pnas.1419198112
Macatonia, S. E., Cruickshank, J. K., Rudge, P., and Knight, S. C. (1992). Dendritic cells from patients with tropical spastic paraparesis are infected with HTLV-1 and stimulate autologous lymphocyte proliferation. AIDS Res. Hum. Retroviruses 8, 1699–1706. doi: 10.1089/aid.1992.8.1699
Macnamara, A., Rowan, A., Hilburn, S., Kadolsky, U., Fujiwara, H., Suemori, K., et al. (2010). HLA class I binding of HBZ determines outcome in HTLV-1 infection. PLoS Pathog. 6:e1001117. doi: 10.1371/journal.ppat.1001117
Martin-Latil, S., Gnädig, N. F., Mallet, A., Desdouits, M., Guivel-Benhassine, F., Jeannin, P., et al. (2012). Transcytosis of HTLV-1 across a tight human epithelial barrier and infection of subepithelial dendritic cells. Blood 120, 572–580. doi: 10.1182/blood-2011-08-374637
Matsuoka, E., Takenouchi, N., Hashimoto, K., Kashio, N., Moritoyo, T., Higuchi, I., et al. (1998). Perivascular T cells are infected with HTLV-I in the spinal cord lesions with HTLV-I-associated myelopathy/tropical spastic paraparesis: double staining of immunohistochemistry and polymerase chain reaction in situ hybridization. Acta Neuropathol. 96, 340–346. doi: 10.1007/s004010050903
Matsuoka, M., and Jeang, K. T. (2011). Human T-cell leukemia virus type 1 (HTLV-1) and leukemic transformation: viral infectivity, Tax, HBZ and therapy. Oncogene 30, 1379–1389. doi: 10.1038/onc.2010.537
Matsuura, E., Enose-Akahata, Y., Yao, K., Oh, U., Tanaka, Y., Takashima, H., et al. (2017). Dynamic acquisition of HTLV-1 tax protein by mononuclear phagocytes: role in neurologic disease. J. Neuroimmunol. 304, 43–50. doi: 10.1016/j.jneuroim.2016.09.014
Matute, C., Domercq, M., and Sánchez-Gómez, M. V. (2006). Glutamate-mediated glial injury: mechanisms and clinical importance. Glia 53, 212–224. doi: 10.1002/glia.20275
McMenamin, P. G. (1999). Distribution and phenotype of dendritic cells and resident tissue macrophages in the dura mater, leptomeninges, and choroid plexus of the rat brain as demonstrated in wholemount preparations. J. Comp. Neurol. 405, 553–562. doi: 10.1002/(sici)1096-9861(19990322)405:4<553::aid-cne8>3.0.co;2-6
Melamed, A., Laydon, D. J., Al Khatib, H., Rowan, A. G., Taylor, G. P., and Bangham, C. R. M. (2015). HTLV-1 drives vigorous clonal expansion of infected CD8+ T cells in natural infection. Retrovirology 12:91.
Méndez, E., Kawanishi, T., Clemens, K., Siomi, H., Soldan, S. S., Calabresi, P., et al. (1997). Astrocyte-specific expression of human T-cell lymphotropic virus type 1 (HTLV-1) Tax: induction of tumor necrosis factor alpha and susceptibility to lysis by CD8+ HTLV-1-specific cytotoxic T cells. J. Virol. 71:9143.
Migone, T.-S., Lin, J.-X., Cereseto, A., Mulloy, J. C., O’shea, J. J., Franchini, G., et al. (1995). Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-I. Science 269, 79–81. doi: 10.1126/science.7604283
Miyao, T., Floess, S., Setoguchi, R., Luche, H., Fehling, H. J., Waldmann, H., et al. (2012). Plasticity of Foxp3+ T Cells reflects promiscuous Foxp3 expression in conventional T Cells but not reprogramming of regulatory T Cells. Immunity 36, 262–275. doi: 10.1016/j.immuni.2011.12.012
Moles, R., Sarkis, S., Galli, V., Omsland, M., Purcell, D. F. J., Yurick, D., et al. (2019). p30 protein: a critical regulator of HTLV-1 viral latency and host immunity. Retrovirology 16:42.
Mori, N., Sato, H., Hayashibara, T., Senba, M., Hayashi, T., Yamada, Y., et al. (2002). Human T-cell leukemia virus type I Tax transactivates the matrix metalloproteinase-9 gene: potential role in mediating adult T-cell leukemia invasiveness. Blood 99, 1341–1349. doi: 10.1182/blood.v99.4.1341
Mozhgani, S. H., Piran, M., Zarei-Ghobadi, M., Jafari, M., Jazayeri, S. M., Mokhtari-Azad, T., et al. (2019). An insight to HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) pathogenesis; evidence from high-throughput data integration and meta-analysis. Retrovirology 16:46.
Mozhgani, S. H., Zarei-Ghobadi, M., Teymoori-Rad, M., Mokhtari-Azad, T., Mirzaie, M., Sheikhi, M., et al. (2018). Human T-lymphotropic virus 1 (HTLV-1) pathogenesis: a systems virology study. J. Cell. Biochem. 119, 3968–3979. doi: 10.1002/jcb.26546
Mukai, R., and Ohshima, T. (2014). HTLV-1 HBZ positively regulates the mTOR signaling pathway via inhibition of GADD34 activity in the cytoplasm. Oncogene 33, 2317–2328. doi: 10.1038/onc.2013.181
Mulloy, J. C., Crownley, R., Fullen, J., Leonard, W. J., and Franchini, G. (1996). The human T-cell leukemia/lymphotropic virus type 1 p12I proteins bind the interleukin-2 receptor beta and gammac chains and affects their expression on the cell surface. J. Virol. 70, 3599–3605. doi: 10.1128/jvi.70.6.3599-3605.1996
Munoz, E., Suri, D., Amini, S., Khalili, K., and Jimenez, S. A. (1995). Stimulation of alpha 1 (I) procollagen gene expression in NIH-3T3 cells by the human T cell leukemia virus type 1 (HTLV-1) Tax gene. J. Clin. Invest. 96, 2413–2420. doi: 10.1172/jci118298
Nagai, M., Usuku, K., Matsumoto, W., Kodama, D., Takenouchi, N., Moritoyo, T., et al. (1998). Analysis of HTLV-I proviral load in 202 HAM/TSP patients and 243 asymptomatic HTLV-I carriers: high proviral load strongly predisposes to HAM/TSP. J. Neurovirol. 4, 586–593. doi: 10.3109/13550289809114225
Nakagawa, M., Nakahara, K., Maruyama, Y., Kawabata, M., Higuchi, I., Kubota, H., et al. (1996). Therapeutic trials in 200 patients with HTLV-I-associated myelopathy/tropical spastic paraparesis. J. Neurovirol. 2, 345–355.
Nascimento, C. R., Lima, M. A., De Andrada Serpa, M. J., Espindola, O., Leite, A. C. C., and Echevarria-Lima, J. (2011). Monocytes from HTLV-1–infected patients are unable to fully mature into dendritic cells. Blood 117, 489–499. doi: 10.1182/blood-2010-03-272690
Nejmeddine, M., Negi, V. S., Mukherjee, S., Tanaka, Y., Orth, K., Taylor, G. P., et al. (2009). HTLV-1–Tax and ICAM-1 act on T-cell signal pathways to polarize the microtubule-organizing center at the virological synapse. Blood 114, 1016–1025. doi: 10.1182/blood-2008-03-136770
Nicot, C., Dundr, M., Johnson, J. M., Fullen, J. R., Alonzo, N., Fukumoto, R., et al. (2004). HTLV-1-encoded p30 II is a post-transcriptional negative regulator of viral replication. Nat. Med. 10, 197–201. doi: 10.1038/nm984
Nozuma, S., and Jacobson, S. (2019). Neuroimmunology of human T-Lymphotropic virus Type 1-Associated myelopathy/tropical spastic paraparesis. Front. Microbiol. 10:885. doi: 10.3389/fmicb.2019.00885
Oger, J. (2007). HTLV-1 infection and the viral etiology of multiple sclerosis. J. Neurol. Sci. 262, 100–104. doi: 10.1016/j.jns.2007.06.045
Oh, U., Grant, C., Griffith, C., Fugo, K., Takenouchi, N., and Jacobson, S. (2006). Reduced Foxp3 protein expression is associated with inflammatory disease during human T Lymphotropic virus Type 1 infection. J. Infect. Dis. 193, 1557–1566. doi: 10.1086/503874
Ohkura, N., Kitagawa, Y., and Sakaguchi, S. (2013). Development and maintenance of regulatory T cells. Immunity 38, 414–423. doi: 10.1016/j.immuni.2013.03.002
Olière, S., Douville, R., Sze, A., Belgnaoui, S. M., and Hiscott, J. (2011). Modulation of innate immune responses during human T-cell leukemia virus (HTLV-1) pathogenesis. Cytokine Growth Fact. Rev. 22, 197–210. doi: 10.1016/j.cytogfr.2011.08.002
Olière, S., Hernandez, E., Lézin, A., Arguello, M., Douville, R., Nguyen, T. L.-A., et al. (2010). HTLV-1 evades Type I interferon antiviral signaling by inducing the suppressor of cytokine signaling 1 (SOCS1). PLoS Pathog. 6:e1001177. doi: 10.1371/journal.ppat.1001177
Omsland, M., Silic-Benussi, M., Moles, R., Sarkis, S., Purcell, D. F. J., Yurick, D., et al. (2020). Functional properties and sequence variation of HTLV-1 p13. Retrovirology 17:11.
Osame, M. (2002). Pathological mechanisms of human T-cell lymphotropic virus type I-associated myelopathy (HAM/TSP). J. Neurovirol. 8, 359–364. doi: 10.1080/13550280260422668
Ozden, S., Seilhean, D., Gessain, A., Hauw, J.-J., and Gout, O. (2002). Severe demyelinating myelopathy with low human T Cell lymphotropic virus Type 1 expression after transfusion in an immunosuppressed patient. Clin. Infect. Dis 34, 855–860. doi: 10.1086/338868
Pais-Correia, A.-M., Sachse, M., Guadagnini, S., Robbiati, V., Lasserre, R., Gessain, A., et al. (2010). Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat. Med. 16, 83–89. doi: 10.1038/nm.2065
Pinto, D. O., Demarino, C., Pleet, M. L., Cowen, M., Branscome, H., Al Sharif, S., et al. (2019). HTLV-1 extracellular vesicles promote cell-to-cell contact. Front. Microbiol. 10:2147. doi: 10.3389/fmicb.2019.02147
Pinto, M. T., Malta, T. M., Rodrigues, E. S., Pinheiro, D. G., Panepucci, R. A., Farias, K., et al. (2013). Genes related to antiviral activity, cell migration, and lysis are differentially expressed in CD4+ T cells in HAM/TSP patients. AIDS Res. Hum. Retrovir. 29:130917035218000.
Pique, C., and Jones, K. (2012). Pathways of cell-cell transmission of HTLV-1. Front. Microbiol. 3:378. doi: 10.3389/fmicb.2012.00378
Poiesz, B. J., Ruscetti, F. W., Gazdar, A. F., Bunn, P. A., Minna, J. D., and Gallo, R. C. (1980). Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. U.S.A. 77, 7415–7419. doi: 10.1073/pnas.77.12.7415
Puccioni-Sohler, M., Rios, M., Bianco, C., Zhu, S. W., Oliveira, C., Novis, S. A. P., et al. (1999). An inverse correlation of HTLV-I viral load in CSF and intrathecal synthesis of HTLV-I antibodies in TSP/HAM. Neurology 53, 1335–1339. doi: 10.1212/wnl.53.6.1335
Puccioni-Sohler, M., Yamano, Y., Rios, M., Carvalho, S. M. F., Vasconcelos, C. C. F., Papais-Alvarenga, R., et al. (2007). Differentiation of HAM/TSP from patients with multiple sclerosis infected with HTLV-I. Neurology 68, 206–213. doi: 10.1212/01.wnl.0000251300.24540.c4
Pulford, K., Lecointe, N., Leroy-Viard, K., Jones, M., Mathieu-Mahul, D., and Mason, D. Y. (1995). Expression of TAL-1 proteins in human tissues. Blood 85, 675–684. doi: 10.1182/blood.v85.3.675.bloodjournal853675
Queiroz, G. A. N., Mascarenhas, R. E. M., Vieillard, V., Andrade, R. L., Galvao-Castro, B., and Grassi, M. F. R. (2019). Functional capacity of natural killer cells in HTLV-1 associated myelopathy/tropical spastic paraparesis (HAM/TSP) patients. BMC Infect. Dis. 19:433. doi: 10.1186/s12879-019-4032-1
Rahman, S., Manuel, S. L., Khan, Z. K., Wigdahl, B., Acheampong, E., Tangy, F., et al. (2010). Depletion of dendritic cells enhances susceptibility to cell-free infection of human T cell leukemia virus type 1 in CD11c-diphtheria toxin receptor transgenic mice. J. Immunol. 184, 5553–5561. doi: 10.4049/jimmunol.0903226
Ramirez, E., Fernandez, J., Cartier, L., Villota, C., and Rios, M. (2003). Defective human T-cell lymphotropic virus type I (HTLV-I) provirus in seronegative tropical spastic paraparesis/HTLV-I-associated myelopathy (TSP/HAM) patients. Virus Res. 91, 231–239. doi: 10.1016/s0168-1702(02)00276-9
Rende, F., Cavallari, I., Corradin, A., Silic-Benussi, M., Toulza, F., Toffolo, G. M., et al. (2011). Kinetics and intracellular compartmentalization of HTLV-1 gene expression: nuclear retention of HBZ mRNAs. Blood 117, 4855–4859. doi: 10.1182/blood-2010-11-316463
Richardson, J. H., Edwards, A. J., Cruickshank, J. K., Rudge, P., and Dalgleish, A. G. (1990). In vivo cellular tropism of human T-cell leukemia virus type 1. J. Virol. 64, 5682–5687. doi: 10.1128/jvi.64.11.5682-5687.1990
Rizkallah, G., Alais, S., Futsch, N., Tanaka, Y., Journo, C., Mahieux, R., et al. (2017). Dendritic cell maturation, but not type I interferon exposure, restricts infection by HTLV-1, and viral transmission to T-cells. PLoS Pathog. 13:e1006353. doi: 10.1371/journal.ppat.1006353
Rocamonde, B., Carcone, A., Mahieux, R., and Dutartre, H. (2019). HTLV-1 infection of myeloid cells: from transmission to immune alterations. Retrovirology 16:45.
Romero, I. A., Prevost, M.-C., Perret, E., Adamson, P., Greenwood, J., Couraud, P.-O., et al. (2000). Interactions between brain endothelial cells and human T-cell leukemia virus type 1-infected lymphocytes: mechanisms of viral entry into the central nervous system. J. Virol. 74, 6021–6030. doi: 10.1128/jvi.74.13.6021-6030.2000
Rosadas, C., and Taylor, G. P. (2019). Mother-to-child HTLV-1 transmission: unmet research needs. Front. Microbiol. 10:999. doi: 10.3389/fmicb.2019.00999
Rott, O., Tontsch, U., Fleischer, B., and Cash, E. (1993). Interleukin-6 production in “normal” and HTLV-1 tax-expressing brain-specific endothelial cells. Eur. J. Immunol. 23, 1987–1991. doi: 10.1002/eji.1830230839
Sabharwal, L., Kamimura, D., Meng, J., Bando, H., Ogura, H., Nakayama, C., et al. (2014). The Gateway Reflex, which is mediated by the inflammation amplifier, directs pathogenic immune cells into the CNS. The Journal of Biochemistry 156, 299–304. doi: 10.1093/jb/mvu057
Sabouri, A. H., Usuku, K., Hayashi, D., Izumo, S., Ohara, Y., Osame, M., et al. (2008). Impaired function of human T-lymphotropic virus type 1 (HTLV-1)–specific CD8+ T cells in HTLV-1–associated neurologic disease. Blood 112, 2411–2420. doi: 10.1182/blood-2008-02-140335
Saeed, Z., Rowan, A., Greiller, C., Taylor, G. P., and Pollock, K. M. (2020). Enhanced T-Cell maturation and monocyte aggregation are features of cellular inflammation in human T-Lymphotropic Virus Type 1-Associated myelopathy. Clin. Infect. Dis. 70, 1326–1335.
Saito, M. (2019). Association between HTLV-1 genotypes and risk of HAM/TSP. Front. Microbiol. 10:1101. doi: 10.3389/fmicb.2019.01101
Saito, M., and Bangham, C. R. (2012). Immunopathogenesis of human T-cell leukemia virus type-1-associated myelopathy/tropical spastic paraparesis: recent perspectives. Leuk. Res. Treatment 2012:259045. doi: 10.1155/2012/259045
Saito, M., Eiraku, N., Usuku, K., Nobuhara, Y., Matsumoto, W., Kodama, D., et al. (2005). ApaI polymorphism of vitamin D receptor gene is associated with susceptibility to HTLV-1-associated myelopathy/tropical spastic paraparesis in HTLV-1 infected individuals. J. Neurol. Sci. 232, 29–35. doi: 10.1016/j.jns.2005.01.005
Saito, M., Matsuzaki, T., Satou, Y., Yasunaga, J.-I., Saito, K., Arimura, K., et al. (2009). In vivo expression of the HBZ gene of HTLV-1 correlates with proviral load, inflammatory markers and disease severity in HTLV-1 associated myelopathy/tropical spastic paraparesis (HAM/TSP). Retrovirology 6:19.
Saito, M., Nakamura, N., Nagai, M., Shirakawa, K., Sato, H., Kawahigashi, N., et al. (1999). Increased levels of soluble Fas ligand in CSF of rapidly progressive HTLV-1-associated myelopathy/tropical spastic paraparesis patients. J. Neuroimmunol. 98, 221–226. doi: 10.1016/s0165-5728(99)00095-8
Saito, M., Tanaka, R., Fujii, H., Kodama, A., Takahashi, Y., Matsuzaki, T., et al. (2014). The neutralizing function of the anti-HTLV-1 antibody is essential in preventing in vivo transmission of HTLV-1 to human T cells in NOD-SCID/γcnull (NOG) mice. Retrovirology 11:74. doi: 10.1186/preaccept-6892873451261068
Sarkis, S., Galli, V., Moles, R., Yurick, D., Khoury, G., Purcell, D. F. J., et al. (2019). Role of HTLV-1 orf-I encoded proteins in viral transmission and persistence. Retrovirology 16:43.
Satou, Y., Yasunaga, J.-I., Yoshida, M., and Matsuoka, M. (2006). HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc. Natl. Acad. Sci. U.S.A. 103, 720–725. doi: 10.1073/pnas.0507631103
Satou, Y., Yasunaga, J.-I., Zhao, T., Yoshida, M., Miyazato, P., Takai, K., et al. (2011). HTLV-1 bZIP factor induces T-cell lymphoma and systemic inflammation in vivo. PLoS Pathog. 7:e1001274. doi: 10.1371/journal.ppat.1001274
Schierhout, G., Mcgregor, S., Gessain, A., Einsiedel, L., Martinello, M., and Kaldor, J. (2020). Association between HTLV-1 infection and adverse health outcomes: a systematic review and meta-analysis of epidemiological studies. Lancet Infect. Dis. 20, 133–143. doi: 10.1016/s1473-3099(19)30402-5
Schütze, M., Romanelli, L. C., Rosa, D. V., Carneiro-Proietti, A. B., Nicolato, R., Romano-Silva, M. A., et al. (2017). Brain metabolism changes in patients infected with HTLV-1. Front. Mol. Neurosci. 10:52. doi: 10.3389/fnmol.2017.00052
Shimauchi, T., Caucheteux, S., Finsterbusch, K., Turpin, J., Blanchet, F., Ladell, K., et al. (2019). Dendritic cells promote the spread of human T-cell leukemia virus type 1 via bidirectional interactions with CD4+ T cells. J. Invest. Dermatol. 139, 157–166. doi: 10.1016/j.jid.2018.06.188
Shimauchi, T., Kabashima, K., Nakashima, D., Sugita, K., Yamada, Y., Hino, R., et al. (2007). Augmented expression of programmed death-1 in both neoplastic and non-neoplastic CD4+ T-cells in adult T-cell leukemia/lymphoma. Int. J. Cancer 121, 2585–2590. doi: 10.1002/ijc.23042
Shiohama, Y., Naito, T., Matsuzaki, T., Tanaka, R., Tomoyose, T., Takashima, H., et al. (2016). Absolute quantification of HTLV-1 basic leucine zipper factor (HBZ) protein and its plasma antibody in HTLV-1 infected individuals with different clinical status. Retrovirology 13:29.
Sofroniew, M. V., and Vinters, H. V. (2010). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. doi: 10.1007/s00401-009-0619-8
Soltani, A., Hashemy, S. I., Zahedi Avval, F., Soleimani, A., Rafatpanah, H., Rezaee, S. A., et al. (2019). Molecular targeting for treatment of human T-lymphotropic virus type 1 infection. Biomed. Pharmacother. 109, 770–778. doi: 10.1016/j.biopha.2018.10.139
Southern, ŠO., and Southern, P. J. (1998). Persistent HTLV-I infection of breast luminal epithelial cells: a role in HTLV transmission? Virology 241, 200–214. doi: 10.1006/viro.1997.8978
Sugata, K., Satou, Y., Yasunaga, J.-I., Hara, H., Ohshima, K., Utsunomiya, A., et al. (2012). HTLV-1 bZIP factor impairs cell-mediated immunity by suppressing production of Th1 cytokines. Blood 119, 434–444. doi: 10.1182/blood-2011-05-357459
Suzuki, S., Zhou, Y., Refaat, A., Takasaki, I., Koizumi, K., Yamaoka, S., et al. (2010). Human T cell lymphotropic virus 1 manipulates interferon regulatory signals by controlling the TAK1-IRF3 and IRF4 pathways. J. Biol. Chem. 285, 4441–4446. doi: 10.1074/jbc.m109.031476
Sweeney, M. D., Zhao, Z., Montagne, A., Nelson, A. R., and Zlokovic, B. V. (2019). Blood-brain barrier: from physiology to disease and back. Physiol. Rev. 99, 21–78. doi: 10.1152/physrev.00050.2017
Sze, A., Belgnaoui, S. M., Olagnier, D., Lin, R., Hiscott, J., and Van Grevenynghe, J. (2013). Host restriction factor SAMHD1 limits human T cell leukemia virus type 1 infection of monocytes via STING-mediated apoptosis. Cell Host Microbe 14, 422–434. doi: 10.1016/j.chom.2013.09.009
Szymocha, R., Akaoka, H., Dutuit, M., Malcus, C., Didier-Bazes, M., Belin, M.-F., et al. (2000a). Human T-Cell lymphotropic virus Type 1-Infected T lymphocytes impair catabolism and uptake of glutamate by astrocytes via tax-1 and tumor necrosis factor alpha. J. Virol. 74, 6433–6441. doi: 10.1128/jvi.74.14.6433-6441.2000
Szymocha, R., Brisson, C., Bernard, A., Akaoka, H., Belin, M.-F., and Giraudon, P. (2000b). Long-term effects of HTLV-1 on brain astrocytes: sustained expression of Tax-1 associated with synthesis of inflammatory mediators. J. Neurovirol. 6, 350–357. doi: 10.3109/13550280009030761
Takeda, S., Maeda, M., Morikawa, S., Taniguchi, Y., Yasunaga, J. I., Nosaka, K., et al. (2004). Genetic and epigenetic inactivation of tax gene in adult T-cell leukemia cells. Int. J. Cancer 109, 559–567. doi: 10.1002/ijc.20007
Takemoto, S., Matsuoka, M., Yamaguchi, K., and Takatsuki, K. (1994). A novel diagnostic method of adult T-cell leukemia: monoclonal integration of human T-cell lymphotropic virus type I provirus DNA detected by inverse polymerase chain reaction. Blood 84, 3080–3085. doi: 10.1182/blood.v84.9.3080.bloodjournal8493080
Tanaka, A., and Matsuoka, M. (2018). HTLV-1 Alters T cells for viral persistence and transmission. Front. Microbiol. 9:461. doi: 10.3389/fmicb.2018.00461
Tanaka, Y., Tanaka, R., Terada, E., Koyanagi, Y., Miyano-Kurosaki, N., Yamamoto, N., et al. (1994). Induction of antibody responses that neutralize human T-cell leukemia virus type I infection in vitro and in vivo by peptide immunization. J. Virol. 68, 6323–6331. doi: 10.1128/JVI.68.10.6323-6331.1994
Taniguchi, Y., Nosaka, K., Yasunaga, J.-I., Maeda, M., Mueller, N., Okayama, A., et al. (2005). Silencing of human T-cell leukemia virus type I gene transcription by epigenetic mechanisms. Retrovirology 2:64.
Tattermusch, S., Skinner, J. A., Chaussabel, D., Banchereau, J., Berry, M. P., Mcnab, F. W., et al. (2012). Systems biology approaches reveal a specific interferon-inducible signature in HTLV-1 associated myelopathy. PLoS Pathog. 8:e1002480. doi: 10.1371/journal.ppat.1002480
Topham, N. J., and Hewitt, E. W. (2009). Natural killer cell cytotoxicity: how do they pull the trigger? Immunology 128, 7–15. doi: 10.1111/j.1365-2567.2009.03123.x
Tordjman, R., Lepelletier, Y., Lemarchandel, V., Cambot, M., Gaulard, P., Hermine, O., et al. (2002). A neuronal receptor, neuropilin-1, is essential for the initiation of the primary immune response. Nat. Immunol. 3, 477–482. doi: 10.1038/ni789
Toulza, F., Heaps, A., Tanaka, Y., Taylor, G. P., and Bangham, C. R. M. (2008). High frequency of CD4+FoxP3+ cells in HTLV-1 infection: inverse correlation with HTLV-1–specific CTL response. Blood 111, 5047–5053. doi: 10.1182/blood-2007-10-118539
Tsutsumi, S., Sato, T., Yagishita, N., Yamauchi, J., Araya, N., Hasegawa, D., et al. (2019). Real-world clinical course of HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) in Japan. Orphanet J. Rare Dis. 14:227.
Uchiyama, T. (1997). Human T cell leukemia virus Type I (HTLV-I) and human diseases. Annu. Rev. Immunol. 15, 15–37. doi: 10.1146/annurev.immunol.15.1.15
Umehara, F., Itoh, K., Michizono, K., Abe, M., Izumo, S., and Osame, M. (2002). Involvement of Fas/Fas ligand system in the spinal cords of HTLV-I-associated myelopathy. Acta Neuropathol. 103, 384–390. doi: 10.1007/s00401-001-0478-4
Umehara, F., Izumo, S., Nakagawa, M., Ronquillo, A. T., Takahashi, K., Matsumuro, K., et al. (1993). Immunocytochemical analysis of the cellular infiltrate in the spinal cord lesions in HTLV-I-associated myelopathy. J. Neuropathol. Exp. Neurol. 52, 424–430. doi: 10.1097/00005072-199307000-00010
Umehara, F., Izumo, S., Ronquillo, A. T., Matsumuro, K., Sato, E., and Osame, M. (1994). Cytokine expression in the spinal cord lesions in HTLV-I-associated myelopathy. J. Neuropathol. Exp. Neurol. 53, 72–77. doi: 10.1097/00005072-199401000-00009
Umehara, F., Izumo, S., Takeya, M., Sato, E., Osame, M., Takahasi, K., et al. (1996). Expression of adhesion molecules and monocyte chemoattractant protein-1 (MCP-1) in the spinal cord lesions in HTLV-I-associated myelopathy. Acta Neuropathol. 91, 343–350. doi: 10.1007/s004010050435
Umehara, F., Okada, Y., Fujimoto, N., Abe, M., Izumo, S., and Osame, M. (1998). Expression of matrix metalloproteinases and tissue inhibitors of metalloproteinas HTLV-l-associated myelopathy. J. Neuropathol. Exp. Neurol. 57, 839–849. doi: 10.1097/00005072-199809000-00005
Umekita, K., and Okayama, A. (2020). HTLV-1 infection and rheumatic diseases. Front. Microbiol. 11:152. doi: 10.3389/fmicb.2020.00152
Vallinoto, A. C. R., Cayres-Vallinoto, I., Freitas Queiroz, M. A., Ishak, M. D. O. G., and Ishak, R. (2019). Influence of immunogenetic biomarkers in the clinical outcome of HTLV-1 infected persons. Viruses 11:974. doi: 10.3390/v11110974
Vallinoto, A. C. R., Santana, B. B., Queiroz, M. A. F., Da Silva, A., Cayres-Vallinoto, I. M. V., Da Costa, C. A., et al. (2017). Family aggregation of HTLV-1 infection associated with FAS -670A/G polymorphism: a case report. Front. Microbiol. 8, 2685.
Van Prooyen, N., Andresen, V., Gold, H., Bialuk, I., Pise-Masison, C., and Franchini, G. (2010a). Hijacking the T-cell communication network by the human T-cell leukemia/lymphoma virus type 1 (HTLV-1) p12 and p8 proteins. Mol. Aspects Med. 31, 333–343. doi: 10.1016/j.mam.2010.07.001
Van Prooyen, N., Gold, H., Andresen, V., Schwartz, O., Jones, K., Ruscetti, F., et al. (2010b). Human T-cell leukemia virus type 1 p8 protein increases cellular conduits and virus transmission. Proc. Natl. Acad. Sci. U.S.A. 107, 20738–20743. doi: 10.1073/pnas.1009635107
Varrin-Doyer, M., Nicolle, A., Marignier, R., Cavagna, S., Benetollo, C., Wattel, E., et al. (2012). Human T Lymphotropic Virus Type 1 increases T Lymphocyte migration by recruiting the cytoskeleton organizer CRMP2. J. Immunol. 188, 1222–1233. doi: 10.4049/jimmunol.1101562
Verdonck, K., González, E., Van Dooren, S., Vandamme, A.-M., Vanham, G., and Gotuzzo, E. (2007). Human T-lymphotropic virus 1: recent knowledge about an ancient infection. Lancet Infect. Dis. 7, 266–281. doi: 10.1016/s1473-3099(07)70081-6
Vescovo, T., Pagni, B., Piacentini, M., Fimia, G. M., and Antonioli, M. (2020). Regulation of autophagy in cells infected with oncogenic human viruses and its impact on cancer development. Front. Cell Dev. Biol. 8:47. doi: 10.3389/fcell.2020.00047
Villadangos, J. A., and Young, L. (2008). Antigen-presentation properties of plasmacytoid dendritic cells. Immunity 29, 352–361. doi: 10.1016/j.immuni.2008.09.002
Volpe, E., Sambucci, M., Battistini, L., and Borsellino, G. (2016). Fas–Fas ligand: checkpoint of T Cell functions in multiple sclerosis. Front. Immunol. 7:382. doi: 10.3389/fimmu.2016.00382
Wang, Y., Hu, J., Li, Y., Wang, H., Li, Z., Tang, J., et al. (2019). The transcription factor TCF1 preserves the effector function of exhausted CD8 T cells during chronic viral infection. Front. Immunol. 10:169. doi: 10.3389/fimmu.2019.00169
Watabe, K., Saida, T., and Kim, S. U. (1989). Human and simian glial cells infected by human T-Lymphotropic virus Type I in culture. J. Neuropathol. Exp. Neurol. 48, 610–619. doi: 10.1097/00005072-198911000-00003
Wodarz, D., Hall, S. E., Usuku, K., Osame, M., Ogg, G. S., Mcmichael, A. J., et al. (2001). Cytotoxic T–cell abundance and virus load in human immunodeficiency virus type 1 and human T–cell leukaemia virus type 1. Proc. R. Soc. Lond. Ser. B Biol. Sci. 268, 1215–1221. doi: 10.1098/rspb.2001.1608
World Health Organization. Regional Office for the Western Pacific (1988). Scientific Group on HTLV-I Infections and Associated Diseases, Kagoshima, Japan, 10-15 December 1988 : Report. Manila: WHO Regional Office for the Western Pacific.
Wu, E., Dickson, D. W., Jacobson, S., and Raine, C. S. (1993). Neuroaxonal dystrophy in HTLV-1-associated myelopathy/tropical spastic paraparesis: neuropathologic and neuroimmunologic correlations. Acta Neuropathol. 86, 224–235. doi: 10.1007/bf00304136
Wu, M., Wong, H. Y., Lin, J. L., Moliner, A., and Schwarz, H. (2019). Induction of CD137 expression by viral genes reduces T cell costimulation. J. Cell. Physiol. 234, 21076–21088.
Yamamoto-Taguchi, N., Satou, Y., Miyazato, P., Ohshima, K., Nakagawa, M., Katagiri, K., et al. (2013). HTLV-1 bZIP factor induces inflammation through labile Foxp3 expression. PLoS Pathog. 9:e1003630. doi: 10.1371/journal.ppat.1003630
Yamano, Y., Araya, N., Sato, T., Utsunomiya, A., Azakami, K., Hasegawa, D., et al. (2009). Abnormally high levels of virus-infected IFN-gamma+ CCR4+ CD4+ CD25+ T cells in a retrovirus-associated neuroinflammatory disorder. PLoS One 4:e6517. doi: 10.1371/journal.pone.0006517
Yamano, Y., Cohen, C. J., Takenouchi, N., Yao, K., Tomaru, U., Li, H.-C., et al. (2004). Increased expression of human T Lymphocyte Virus Type I (HTLV-I) Tax11-19 peptide–human histocompatibility leukocyte antigen A∗201 complexes on CD4+ CD25+T Cells detected by peptide-specific, major histocompatibility complex–restricted antibodies in patients with HTLV-I–associated neurologic disease. J. Exp. Med. 199, 1367–1377.
Yamano, Y., Nagai, M., Brennan, M., Mora, C. A., Soldan, S. S., Tomaru, U., et al. (2002). Correlation of human T-cell lymphotropic virus type 1 (HTLV-1) mRNA with proviral DNA load, virus-specific CD8+ T cells, and disease severity in HTLV-1–associated myelopathy (HAM/TSP). Blood 99, 88–94.
Yamano, Y., and Sato, T. (2012). Clinical pathophysiology of human T-lymphotropic virus-type 1-associated myelopathy/tropical spastic paraparesis. Front. Microbiol. 3:389. doi: 10.3389/fmicb.2012.00389
Yamano, Y., Takenouchi, N., Li, H.-C., Tomaru, U., Yao, K., Grant, C. W., et al. (2005). Virus-induced dysfunction of CD4+CD25+ T cells in patients with HTLV-I–associated neuroimmunological disease. J. Clin. Invest. 115, 1361–1368.
Yasuma-Mitobe, K., and Matsuoka, M. (2018). The roles of coinhibitory receptors in pathogenesis of human retroviral infections. Front. Immunol. 9:2755. doi: 10.3389/fimmu.2018.02755
Yasunaga, J.-I., Furuta, R., Miura, M., Sugata, K., Saito, A., Hirofumi, A., et al. (2016). Hematopoietic stem cell infected with HTLV-1 functions as a viral reservoir in vivo. Blood 128:1343.
Yasunaga, J.-I., Sakai, T., Nosaka, K., Etoh, K.-I., Tamiya, S., Koga, S., et al. (2001). Impaired production of naive T lymphocytes in human T-cell leukemia virus type I–infected individuals: its implications in the immunodeficient state. Blood 97, 3177–3183.
Yoshida, M., Satou, Y., Yasunaga, J.-I., Fujisawa, J.-I., and Matsuoka, M. (2008). Transcriptional control of spliced and unspliced human T-cell leukemia virus type 1 bZIP factor (HBZ) gene. J. Virol. 82, 9359–9368.
Youssefi, M., Ghazvini, K., Casseb, J., and Keikha, M. (2020). What is the role of the thioredoxin antioxidant complex in relation to HAM/TSP? Access Microbiol. 2:acmi000090.
Yukitake, M., Sueoka, E., Sueoka-Aragane, N., Sato, A., Ohashi, H., Yakushiji, Y., et al. (2008). Significantly increased antibody response to heterogeneous nuclear ribonucleoproteins in cerebrospinal fluid of multiple sclerosis patients but not in patients with human T-lymphotropic virus type I—associated myelopathy/tropical spastic paraparesis. J. Neurovirol. 14, 130–135.
Zacharopoulos, V., Perotti, M., and Phillips, D. (1992). Lymphocyte-facilitated infection of epithelia by human T-cell lymphotropic virus type I. J. Virol. 66, 4601–4605.
Zacharopoulos, V. R., and Phillips, D. M. (1997). Cell-mediated HTLV-I infection of a cervix-derived epithelial cell line. Microb. Pathog. 23, 225–233.
Zhao, H., Liao, X., and Kang, Y. (2017). Tregs: where we are and what comes next? Front. Immunol. 8:1578. doi: 10.3389/fimmu.2017.01578
Keywords: HAM/TSP, HTLV-1-associated myelopathy, pathogenesis, immunology, human T-lymphotropic virus
Citation: Aghajanian S, Teymoori-Rad M, Molaverdi G and Mozhgani S-H (2020) Immunopathogenesis and Cellular Interactions in Human T-Cell Leukemia Virus Type 1 Associated Myelopathy/Tropical Spastic Paraparesis. Front. Microbiol. 11:614940. doi: 10.3389/fmicb.2020.614940
Received: 07 October 2020; Accepted: 17 November 2020;
Published: 22 December 2020.
Edited by:
Hirofumi Akari, Kyoto University, JapanReviewed by:
Tomoo Sato, St. Marianna University School of Medicine, JapanSteven Jacobson, National Institute of Neurological Disorders and Stroke (NINDS), United States
Copyright © 2020 Aghajanian, Teymoori-Rad, Molaverdi and Mozhgani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sayed-Hamidreza Mozhgani, aGFtaWRyZXphbW96aGdhbmlAZ21haWwuY29t