 Domonkos Sváb1*
Domonkos Sváb1* Linda Falgenhauer2,3
Linda Falgenhauer2,3 Balázs Horváth4
Balázs Horváth4 Gergely Maróti5,6Jane Falgenhauer3,7
Gergely Maróti5,6Jane Falgenhauer3,7 Trinad Chakraborty2,7
Trinad Chakraborty2,7 István Tóth1
István Tóth1- 1Institue for Veterinary Medical Research, Centre for Agricultural Research, Martonvásár, Hungary
- 2Institute of Hygiene and Environmental Medicine, Justus Liebig University Giessen, Giessen, Germany
- 3German Centre for Infection Research, Site Giessen-Marburg-Langen, Giessen, Germany
- 4Seqomics Ltd., Mórahalom, Hungary
- 5Institute of Plant Biology, Biological Research Centre, Szeged, Hungary
- 6Faculty of Water Sciences, University of Public Service, Baja, Hungary
- 7Institute for Medical Microbiology, Justus Liebig University Giessen, Giessen, Germany
Shigella dysenteriae are significant agents of bacillary dysentery, accounting for a considerable number of illnesses with high morbidity worldwide. The Shiga toxin (Stx) encoded by a defective prophage is the key virulence factor of S. dysenteriae type 1 (SD1) strains. Here we present the full genome sequence of an SD1 strain HNCMB 20080 isolated in 1954, compare it to other sequenced SD1 genomes, and assess the diversity of Stx-prophages harbored by previously sequenced SD1 strains. The genome of HNCMB 20080 consists of a chromosome sized 4,393,622 bp containing 5,183 CDSs, as well as two small plasmids. Comparative genomic analysis revealed a high degree of uniformity among SD1 genomes, including the structure of Stx prophage regions, which we found to form two subgroups termed PT-I and PT-II. All PT-I strains are members of the sequence type (ST) 146 or ST260, while the only PT-II harboring strain, Sd1617 proved to be ST untypeable. In accordance with data from previous reports, the Stx1 prophage could not be induced from HNCMB 20080. Our cumulative data do not support the notion that stx-harboring phages in STEC are derived from historical SD1 isolates.
Introduction
Members of the Shigella genus are the leading agents of bacillary dysentery worldwide, especially in developing countries (reviewed by Faherty and Lampel, 2019). Of the four species, Shigella dysenteriae is responsible for historical outbreaks of high morbidity and mortality (reviewed by Anderson et al., 2016).
Shiga toxin (Stx) is the main virulence factor of Shigella dysenateriae type 1 (SD1) strains first discovered in the late nineteenth century by Kiyoshi Shiga (Trofa et al., 1999). Subsequently, Shiga toxin-production was identified in great variety of E. coli (STEC) strains (reviewed by Muniesa and Schmidt, 2014) as well as in other enteral pathogenic bacteria including Enterobacter cloaceae (Probert et al., 2014), as well as S. flexneri (Gray et al., 2014) and recently S. sonnei (Strauch et al., 2001; Carter et al., 2016; Sváb et al., 2017). Two main types of the toxin, Stx1 and Stx2, and several subtypes have been identified in E. coli, with Stx1 being almost identical to the one present in Shigella strains (Scheutz et al., 2012). Importantly, the stx operon is always integrated in the genome of prophages. The Shiga toxin encoding phages constitute a heterogeneous group of temperate lambdoid phages that harbor the stx genes among the late region genes (reviewed by Muniesa and Schmidt, 2014).
The Stx prophages carried by non-SD1 strains frequently proved to be inducible and they can spread horizontally, either because, as is the case of many Stx2 prophages, they retained essential genes for the lysis of the host, or in the case of Stx1 prophages, they undergo recombination with Stx2 prophage sequences (Asadulghani et al., 2009; Muniesa and Schmidt, 2014; Tóth et al., 2016). In contrast to these converting Stx phages, the Stx prophages carried by SD1 strains, are cryptic and cannot be induced.
Earlier there was a comprehensive genome sequencing study of a collection of recent and historical SD1 strains producing hundreds of draft genomes (Njamkepo et al., 2016), which shed a light on the evolutionary history of the pathogen, and established an SNP-based phylogeny consisting of four main lineages numbered I–IV. The study, aiming to outline a global phylogeography of SD1 strains, did not venture into details about the history of individual virulence genes or mobile genetic elements.
In the present study, using genome sequencing and by comparative genomic analysis, the complete genome of an antibiotic-sensitive SD1 strain, HNCMB 20080 isolated in Hungary in 1954, was assembled and analyzed, and the Stx prophage region’s great uniformity across currently available SD1 genome sequences was revealed.
Materials and Methods
Bacterial Strains and Bacteriophages
S. dysenteriae strain HNCMB 20080 was isolated in Hungary from a patient in 1954 and provided by the collection of the Hungarian National Collection of Medical Bacteria (HNCMB), Budapest, Hungary. E. coli K-12 MG1655 was used as phage indicator strain. A rifampicin-resistant mutant of HNCMB 20080 was obtained with the method described in Sváb et al. (2018). Briefly, 200 μl of overnight LB culture of HNCMB 20080 was spread onto LB agar plates supplemented with 150 μg/ml rifampicin and incubated at 37°C for 16 h. Single colonies were selected and further purified by several passages on selective plates. One of the resistant colonies was used for further experiments.
Genome Sequencing, Assembly, and Comparative Analysis
Genomic DNA of SD1 strain HNCMB 20080 was isolated with GenElute Bacterial Genomic DNA Kit (Sigma-Aldrich) according to the manufacturer’s instructions. For genome closure, a combination of short- and long-read sequencing was used. For short-read sequencing, a mate-pair fragment library was generated for the genome of S. dysenteriae strain HNCMB 20080 using the Illumina Nextera Mate-Pair Kit (Cat.Num.: FC-132-1001) with insert sizes ranging between 7 and 11 kb. DNA sequencing was carried out on an Illumina MiSeq machine using V2 sequencing chemistry resulting in 2 × 250 nt reads. Long-read sequencing was performed using the Nanopore technology. For this, the library was prepared using the native barcoding kit (EXP-NBD103, Oxford Nanopore Technologies Ltd., Oxford, United Kingdom) combined with 1D chemistry (SQK-LSK108). Sequencing was performed on a SpotON Flow Cell Mk I R9 Version (FLO−MIN106) on a MinION/MinIT machine. Demultiplexing was performed using Porechop v. 0.2.31. Hybrid assembly of short and long reads was performed using Unicycler v. 0.4.7 (Wick et al., 2017) with default parameters. The assembled chromosome and plasmids of HNCMB 20080 are deposited in GenBank under the accession numbers CP061527-CP061529.
Genomic comparisons and searches were performed with megaBLAST available on the NCBI website. Virulence gene search was conducted with VirulenceFinder 2.0.1 (Joensen et al., 2014).
For comparative purposes, assembly of SD1 isolates sequenced by PacBio technologies (Table 1) was performed using ASA3P (Schwengers et al., 2020). In order to identify chromosomal inversions in the genome, annotation of all genomes in Table 1 was performed with RASTtk (Brettin et al., 2015), and their alignment was conducted using Progressive Mauve (Darling et al., 2010) through the PATRIC platform (Wattam et al., 2017).
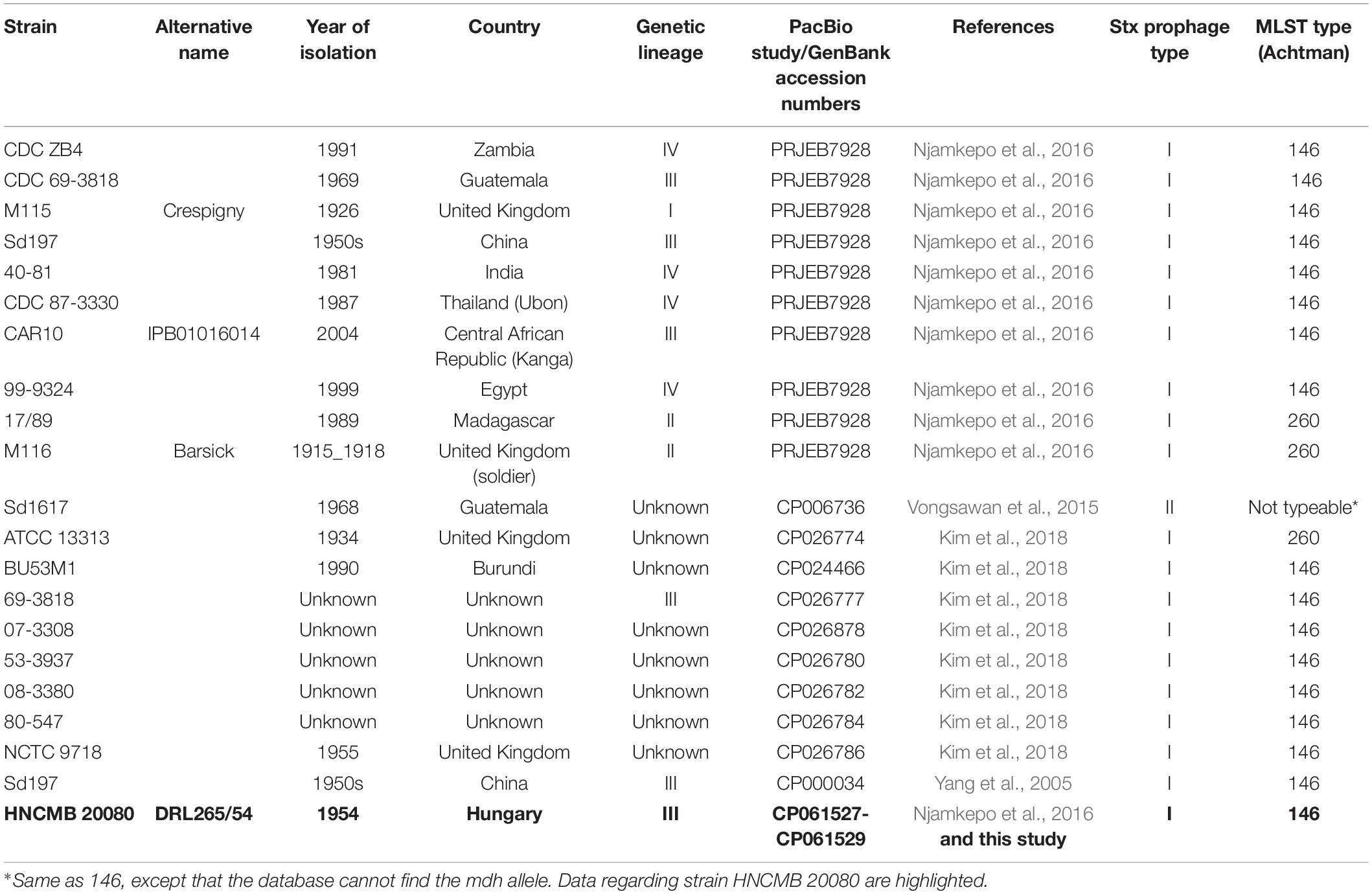
Table 1. List of complete SD1 genomes available in GenBank as well as draft genomes determined by PacBio sequencing used in this study.
For phylogenetic comparison, core-genome SNP analysis was performed using ParSNP using default parameters (Treangen et al., 2014) and HNCMB 20080 as reference. Core genome sequences were extracted. Recombination events were removed using Gubbins (Croucher et al., 2015; 50 iterations, default parameters) and recombination-diminished SNP counts were determined using the “compute pairwise distance” function of Mega 5.2.2 (default parameters) (Tamura et al., 2011). Based on the pairwise SNP analysis, the phylogenetic relationship of HNCMB 20080 to other Sd1 strains was determined. A tree based on the SNPs differences (neighbor-joining method) was visualized and annotated using Mega 5.2.2.
Multi-Locus Sequence Typing
Multi-locus sequence typing (MLST) analysis of HNCMB 200080 as well as complete SD1 genomes shown in Table 1 was performed using according to the E. coli scheme published by Wirth et al. (2006), version 2.0.4 provided by the Centre for Genetic Epidemiology (Larsen et al., 2012).
Prophage and Plasmid Search
Prophages sequences present in the genome of HNCMB 20080 were determined using PHASTER with the “precomputed” mode (Arndt et al., 2016). Plasmids were searched for by PlasmidFinder 2.0 (Carattoli et al., 2014).
Antibiotic Resistance Investigation
Minimum inhibitory concentration (MIC) tests of HNCMB 20080 toward erythromycin were conducted as follows: A liquid culture in LB broth was shaken overnight with 180 rpm at 37°C. Five microliter drops of the culture, containing ~105 cells, were spotted in three parallels on Müller-Hinton agar plates containing concentrations of erythromycin between 1 and 256 μg/ml in halving serial dilution. The lowest concentration of the antibiotic on plates where no growth was observed, was considered the MIC.
Phage Induction Experiments Prophage Induction
We attempted to mobilize prophages carried by HNCMB 20080 with various agents using standard methods as described previously (Sváb et al., 2013). Briefly, an overnight culture of HNCMB 20080 was diluted 50-fold in LB broth and shaken at 180 rpm for 3 h at 37°C. Afterward, the following induction agents were applied in separate batches: mitomycin C in 0.5 μg/ml, norfloxacin in 1.25 μg/ml final concentration were used (both antibiotics were supplied by Sigma-Aldrich, MO, United States). Phage induction was also attempted by using ultraviolet light, according to the protocol described by Hertman and Luria (1967) with some modifications. Briefly, 1 ml of the 3 h culture was spread in a sterile empty Petri-dish and irradiated with a 30 W UV lamp in a distance of 30 cm for 15 s. In all cases, after the addition of the agent or the irradiation, the cultures were incubated with shaking for another 3 h at 37°C. Following centrifugation at 13,000 g for 1 min, and the supernatant was filtered with a 0.22 μm pore-size sterile filter (Acrodisc Supor, Pall Corporation) and spotted on layered soft agar plates of E. coli K-12 derivative strain MG1655. Plates were examined after an overnight incubation at 37°C for the presence of plaques. As control for phage induction, Stx1 and Stx2 phages were induced from non-SD strains (Table 3) by using mitomycin C. E. coli K-12 derivative strain MG1655 was used as indicator and propagating strain.
Attempts for Lysogenization and Phage Transduction
Similar to the phage sensitivity experiments, we attempted to lysogenize S. dysenteriae strain HNCMB 20080 with Stx1 and Stx2-carrying phages or their modified derivates. Ten microliter spots of high titer (>109 PFU/ml) Stx1 and Stx2 phage stocks of non-SD1 strains and their 10-fold dilutions were spread on top of layered soft agar plates containing HNCMB 20080. Surviving colonies in the middle of lysis spots were transferred to new agar plates and after overnight incubation at 37°C, they were checked for the presence of stx genes by PCR. Phages used in these experiments are listed in Table 3. As an alternative method, in the case of MG1655 catGFP and strain 3538/95, 1 ml of 3 h cultures of the respective strains were mixed with an equal amount of 3 h culture of HNCMB 20080 RifR, and mitomycin C was added in the appropriate concentration. After another 3 h without shaking, 1 ml of the mixed culture was centrifuged and spread onto LB agar plates supplemented with 100 μg/ml rifampicin and 25 μg/ml chloramphenicol, as both MG1655 catGFP and strain 3538/95 contain the cat gene encoding chloramphenicol resistance, which trait would also be exhibited by recipient colonies of HNCMB 20080 RifR in case of successful transduction. After overnight incubation at 37°C, the lactose fermenting ability of the surviving colonies was tested on bromophenol-blue plates, to ensure that only lactose-negative colonies, which are most likely of the recipient SD1 strains and not those of the donor E. coli strains, are checked The presence of stx1 and stx2 genes was investigated by PCR with the primers and protocol described by Scheutz et al. (2012).
Results
Genomic Features
The genome of HNCMB 20080 contains a chromosome and two small plasmids. The chromosome of HNCMB 20080 has a size of 4,393,662 bp. The average GC content is 51.3%, and the chromosome contains 5,183 CDSs and 86 tRNA genes. The first plasmid, pHNCMB20080_1 is 8,953 bp long and contains 13 CDSs. Of these, only two were assigned function, one is replication gene, the other is a household gene. The remaining are all encoding “hypothetical proteins.” The second plasmid, pHNCMB20080_2 is 3,069 bp long and has above 98% homology to E. coli strain RHB02-C19 plasmid pRHB02-C19_7 (GenBank CP058069.1). It contains two CDSs, one coding for LPS O-antigen regulator, and the other for an RNA-binding protein. The genomic features of HNCMB 20080 are summarized in Table 2, and the complete list of genes is given in Supplementary Table 1.

Table 2. List of genomic features of Shigella dysenteriae serotype 1 strain HNCMB 20080.
Phylogenetic Relations
MLST analysis showed that, similar to the majority of SD1 strains with complete sequence, HNCMB 20080 belongs to ST-146 according to the Achtman scheme (Table 1).
When comparing the HNCMB 20080 genome to the complete genomes in GenBank as well as with nine further PacBio-sequenced genomes which were successfully assembled to enable Progressive Mauve alignment (Table 1), a total of 42 chromosomal inversions were identified ranging in size from around 700 bp to almost 400 kbp (Supplementary Table 2).
Based on the SNP analysis, the closest relatives to HNCMB 20080 were Sd197, 80–547, and 40–81 (Figure 1 and Supplementary Table 3).
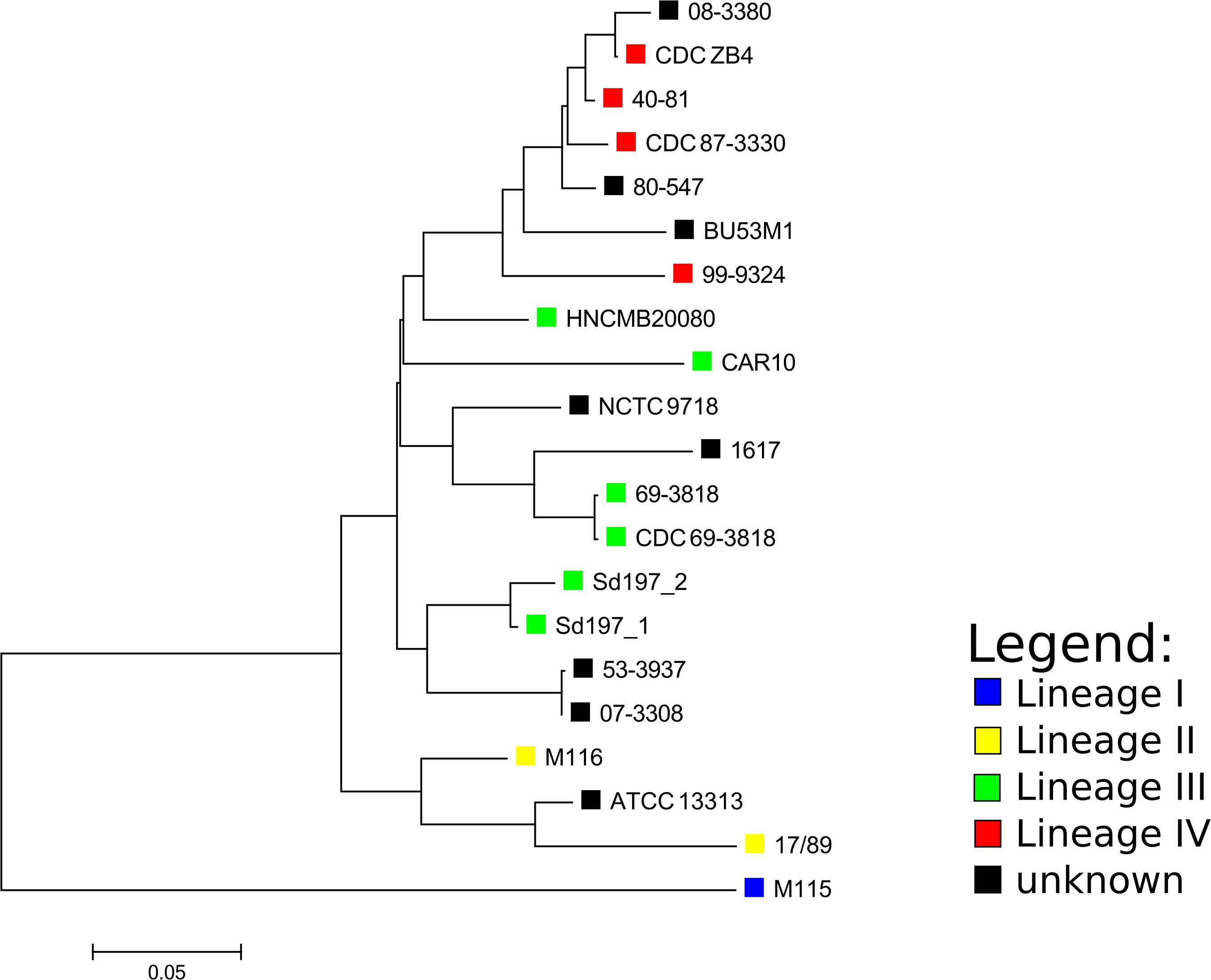
Figure 1. Core SNP-based phylogenetic tree of Shigella dysenteriae complete genomes and Pacbio determined assemblies made with ASA3P. The SNP analysis was performed with ParSNP, followed by removal of SNPs at recombination sites by Gubbins. The tree was visualized with MEGA. The lineages designated for strains by the SNP-based phylogeny made by Njamkepo et al. (2016) are marked with colored squares where applicable.
Virulence Profile
Besides the Stx1-encoding gene cluster, VirulenceFinder only identified the gad gene encoding glutamate-decarboxylase, and the iroN which encodes a tonB-dependent outer membrane siderophore receptor protein. The PlasmidFinder search indicated that the pInv plasmid was missing from the strain.
Prophage Pool
The PHASTER search identified altogether 27 prophage-like regions, with their length between 4.9 and 32 kbp, comprising ~375 kbp of the genome altogether (Supplementary Table 4). Out of these, nine regions were labeled as “intact,” and one of them termed “Region 16” by the program is the Stx1 prophage. The 27 kbp long “Region 15” also contains remnants of a lambdoid prophage, with CDSs encoding a “host killer protein,” CI and cro repressor proteins, and a DNA methylase. The GC content of intact-labeled prophage regions ranged between 48.93 and 52.09%. BLAST searches showed that most of the prophages are present with high coverage and homology in the majority of SD1 strains with complete genomes available, but most of them yielded only low coverage homologies to other enterobacterial phage sequences when BLASTed against viral sequences alone.
Characteristics of the Stx Encoding Prophage
The prophage region containing the genes encoding the stx1 operon in HNCM 20080 comprises 13,682 bp from the start codon of the integrase gene fragment to the stop codon of a mobile element protein, of which the last seven nucleotides overlap with the end of the transcriptional regulator YcjW. This latter gene (part of the multi-gene Ycj putative regulator cluster) flanks the downstream end of the prophage, while upstream from the integrase fragment, the gene encoding peptide transport system permease SapC can be found, with a 294 nt long non-coding region between them. The PHASTER search originally gave a greater length, but it also included the YcjW gene itself, as well as a multi-gene operon encoding a peptide ABC transporter system and two IS110 elements into the prophage sequence.
The prophage region is located in exactly the same region in all SD1 strains with complete genomes available, as well as in the 10 strains sequenced in Njamkepo et al. (2016), where PacBio reads were made (Table 1). The size and genetic content is highly similar in all cases, the sequence identity is above 99% throughout the whole region. The only exception is in strain Sd1617, where orfA of IS600 upstream of the stx genes is truncated. Based on this difference, we termed the two main prophage types “I” and “II” (heretofore referred to as PT-I and PT-II, with PT standing for “prophage type”). The detailed representation of the Stx prophage region with the two main variants is shown on Figure 2. The list of SD1 strains with complete genomes and the type of their Stx prophages is given in Table 1.
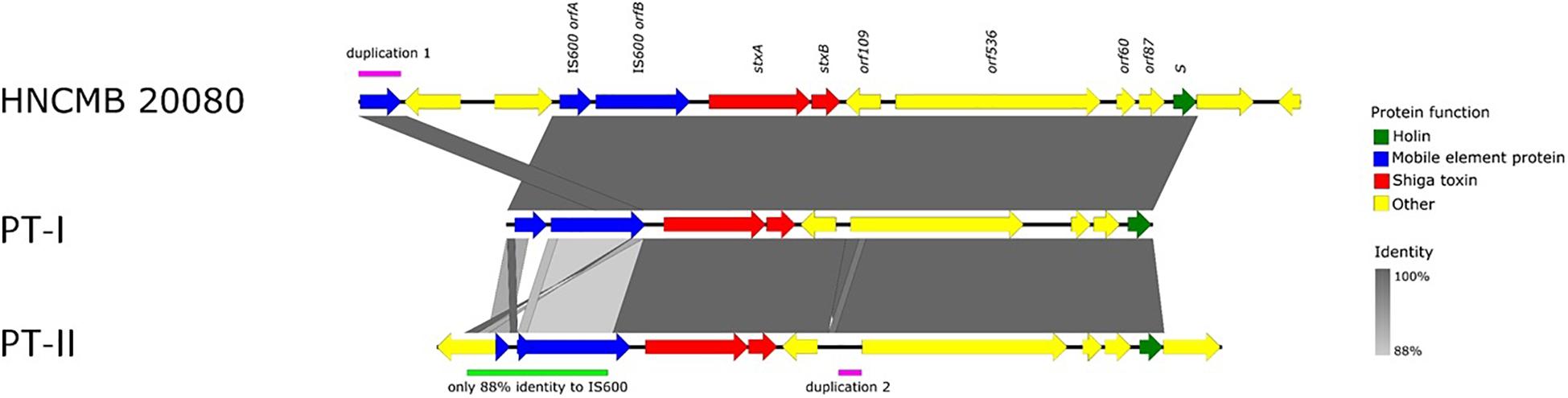
Figure 2. Schematic representation of the two types of Stx1-prophages present in the genomes of SD1 strains.
Antimicrobial Resistance
As the genome sequencing revealed that HNCMB 20080 harbors the gene mdfA associated with macrolide-resistance, we conducted a MIC test with erythromycin. The MIC of HNCMB 20080 against this antibiotic proved to be 64 μg/ml.
Phage Induction
Phage induction of HNCMB 20080 was attempted with various agents (mitomycin C, norfloxacin, UV-light), but none of the experiments resulted in detectable lytic activity on E. coli K-12 derivative strain MG1655.
Lysogenization Experiments
Lysogenization of HNCMB 20080 was attempted with wild type and modified Stx phages listed in Table 3. None of the experiments yielded lysogenic cell lines.

Table 3. Attempts for Lysogenization and Phage Transduction.
Discussion
We sequenced and assembled the complete genome of S. dysenteriae strain HNCMB 20080, a historical strain from the 1950s isolated in Hungary. A draft genome of the strain was published earlier as part of a global S. dysenteriae sequencing project (Njamkepo et al., 2016). Albeit the project included a comprehensive representative collection of samples, complete genome sequences available for this pathogen are still relatively low in number (Table 1), representing only 3% (11/335) of all genome sequences available in GenBank after the study.
In general, the chromosomal structure of S. dysenteriae strains, including HNCMB 20080 is highly conserved, which is also supported by the fact that the complete and PacBio-sequenced genomes of SD1 known up to date are restricted to only two MLST types, except for the untypeable strain Sd1617 (Table 1). However, there were several chromosomal inversions, and all of them associated with insertion sequence (IS) elements. In the first Shigella sequencing project IS elements were highlighted as having a likely role in genomic rearrangements in Shigella genomes, with S. dysenteriae strain Sd197 having the most of these genomic features (Yang et al., 2005).
Phylogenetic relations for the available complete SD1 genomes could not be resolved adequately using MLST, as all strains except for ATCC13313 and 1617 belonged to the same sequence type. The core SNP-based phylogeny created with ParSNP showed that the lineages defined by Njamkepo et al. (2016) using the SNP-based method SMALT2 grouped together, but interestingly, out of the lineage III strains, HNCMB 20080 fell closest to the clusters containing lineage IV strains which harbored strains isolated in the 1980s and 1990s, unlike HNCMB 20080 which was isolated in the 1950s.
HNCMB 20080 was examined in detail because it is a) a historical strain and b) considered sensitive to all major antibiotic classes. The genomic investigation only revealed a macrolide resistance gene mdfA, and the strain showed a 64 μg/ml MIC against erythromycin, which is at the lower end of the reported range between 64 and 1,024 μg/ml for Enterobacteria (Greenberg et al., 1982; Boumghar-Bourtchai et al., 2008).
The genome of HNCMB 20080 contains 27 prophages or prophage-like regions altogether, including the one containing the Stx encoding gene cluster. However, probably due to the passages during storage throughout the decades since its isolation, the strain has most likely lost its pInv plasmid.
For Shiga toxin-producing E. coli (STEC), there have been extensive investigation in the past three decades regarding the genetic properties of Stx encoding genes and its genomic environment. It is generally accepted that it is carried by a lambdoid prophage, but in contrast to STEC, the exact genetic background and origin of Stx in the case of SD1 strains still remains unresolved. Since the work of Unkmeir and Schmidt (2000), there have been no further studies specifically targeting the Stx-carrying prophages of this pathogen.
In light of the above considerations, we examined the genetic structure and relatedness of Stx-encoding prophages found in complete SD1 genomes available in GenBank. We found that the region is remarkably conserved, spanning 13.5 kbp and integrated into the same site in every strain i.e., between the sapC and ychK genes (McDonough and Butterton, 1999). Two main sequence variants, PT-I and PT-II could be distinguished. Currently, the latter is represented by only one strain, Sd1617. In all cases, the prophage is defective, missing most of the key structural genes necessary for it to follow a lytic lifestyle.
The conserved nature of the phage indicated that its integration in the genome most likely precedes the isolation of the first strains, as even in the case of the historical strain M116, which was isolated between 1915 and 1918 (Njamkepo et al., 2016), the region does not differ from those found in the more recently isolated strains. However, as we have noted above, strains with complete genome only represent a small fraction of all the sequenced strains. Completion of further genomes could still reveal new, or possibly more complete variants of the Stx prophage in SD1 strains.
Current evidence also suggests that SD1 had acquired and domesticated the phage earlier than the isolation of the first strains at the turn of the nineteenth and twentieth centuries. This notion strongly suggests that STEC strains obtained Stx1 and Stx2 prophages much later than SD1 strains, as the first STEC were only identified in the 1980s (Riley et al., 1983). Moreover, Stx1-producing S. flexneri and S. sonnei strains were identified as late as in the twenty-first century (Gray et al., 2014; Carter et al., 2016). The notion of their recent acquisition of Stx1 phages is also supported by the inducibility of the phages (Strauch et al., 2001; Tóth et al., 2016). Although sequence data strongly suggests the common origin of the Stx prophages, their diverse states of detoriation and inducibility indicates that they were acquired at different timepoints throughout the past 100 years.
The genomic data of SD1 strains seem to exclude the possibility of the phage being transduced from them to STEC strains, albeit in the case of the prototypic enterohemorrhagic E. coli (EHEC) O157:H7 Sakai strain, the Stx1 gene is harbored on a defective prophage, which in turn can be mobilized through recombination with the Stx2-harboring inducible prophage (Asadulghani et al., 2009; Sváb et al., 2015). Such a recombination event could also have occurred between SD1 Stx1-carrying prophage and a more complete lambdoid phage. However, the observed resistance of HNCMB 20080 to infection with Stx2-carrying phages and its derivates, does not support this possibility. The results of our phage induction experiment seem to exclude the possible recombination, or the presence of satellite or helper phages (reviewed by Penadés and Christie, 2015) between the Stx1 prophage and other prophages in the genome of HNCMB 20080.
In summary, we determined the complete genome sequence of a historical SD1 isolate from Hungary, and the conserved nature of SD1 genomes, together with the presence of multiple chromosomal inversions was confirmed. We have found that the Stx1 prophage also has a conserved sequence, and established the existence of two variants throughout all complete SD1 genomes available so far. The prophage was found uninducible. For further exploration of the evolutionary history of Stx-carrying prophages, as well as that of their carriage in SD1 strains, a larger set of high-quality complete genomes, supplemented with traditional phage induction and infection experiments would be beneficial.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, CP061527-CP061529; https://www.ncbi.nlm.nih.gov/, PRJNA662859.
Author Contributions
IT and TC conceived and designed the study. DS and IT performed phenotypic experiments. BH and GM performed the Illumina sequencing and conducted the assembly. LF and JF conducted the Nanopore sequencing and hybrid assembly, as well as the assembly of PacBio sequences and other bioinformatics work. DS performed the additional comparative bioinformatics work and drafted the manuscript. All authors reviewed and wrote parts of the manuscript.
Funding
This study was funded by the National Research, Development and Innovation Office (NKFIH), grant no. K 124335. DS was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences.
Conflict of Interest
BH was employed by company Seqomics Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Christina Gerstmann for excellent technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.614793/full#supplementary-material
Footnotes
- ^ https://github.com/rrwick/Porechop accessed on August 28th, 2020.
- ^ https://www.sanger.ac.uk/tool/smalt-0/accessed on November 26th, 2020.
References
Anderson, M., Sansonetti, P. J., and Marteyn, B. S. (2016). Shigella diversity and changing landscape: insights for the twenty-first century. Front. Cell. Infect. Microbiol. 6:45. doi: 10.3389/fcimb.2016.00045
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Asadulghani, M., Ogura, Y., Ooka, T., Itoh, T., Sawaguchi, A., Iguchi, A., et al. (2009). The defective prophage pool of Escherichia coli O157: prophage-prophage interactions potentiate horizontal transfer of virulence determinants. PLoS Pathog. 5:e1000408. doi: 10.1371/journal.ppat.1000408
Boumghar-Bourtchai, L., Mariani-Kurkdjian, P., Bingen, E., Filliol, I., Dhalluin, A., Ifrane, S. A., et al. (2008). Macrolide-resistant Shigella sonnei. Emerg. Infect. Dis. 14, 1297–1299. doi: 10.3201/eid1408.080147
Brettin, T., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Olsen, G. J., et al. (2015). RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5:8365. doi: 10.1038/srep08365
Carattoli, A., Zankari, E., García-Fernández, A., Larsen, M. V., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Carter, C. C., Fierer, J., Chiu, W. W., Looney, D. J., Strain, M., and Mehta, S. R. (2016). A novel Shiga toxin 1a-converting bacteriophage of Shigella sonnei with close relationship to Shiga toxin 2-converting pages of Escherichia coli. Open Forum Infect. Dis. 3:ofw079. doi: 10.1093/ofid/ofw079
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43:e15. doi: 10.1093/nar/gku1196
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
Faherty, C. S., and Lampel, K. A. (2019). “Shigella,” in Food Microbiology, eds M. P. Doyle, F. Diez−Gonzalez, and C. Hill (Hoboken, NJ: John Wiley & Sons, Ltd), 317–345. doi: 10.1128/9781555819972.ch12
Gray, M. D., Lampel, K. A., Strockbine, N. A., Fernandez, R. E., Melton-Celsa, A. R., and Maurelli, A. T. (2014). Clinical isolates of Shiga toxin 1a-producing Shigella flexneri with an epidemiological link to recent travel to Hispañiola. Emerg. Infect. Dis. 20, 1669–1677. doi: 10.3201/eid2010.140292
Greenberg, R. N., Pearson, R. D., Innes, D. J., Sauer, K. T., Halterman, L. D., and Guerrant, R. L. (1982). Susceptibility of Shigella species to erythromycin. Antimicrob. Agents Chemother. 21, 347–348. doi: 10.1128/AAC.21.2.347
Hertman, I., and Luria, S. E. (1967). Transduction studies on the role of a rec+ gene in the ultraviolet induction of prophage lambda. J. Mol. Biol. 23, 117–133. doi: 10.1016/s0022-2836(67)80021-4
Imamovic, L., Ballesté, E., Martínez-Castillo, A., García-Aljaro, C., and Muniesa, M. (2016). Heterogeneity in phage induction enables the survival of the lysogenic population. Environ. Microbiol. 18, 957–969. doi: 10.1111/1462-2920.13151
Joensen, K. G., Scheutz, F., Lund, O., Hasman, H., Kaas, R. S., Nielsen, E. M., et al. (2014). Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 52, 1501–1510. doi: 10.1128/JCM.03617-13
Kim, J., Lindsey, R. L., Garcia-Toledo, L., Loparev, V. N., Rowe, L. A., Batra, D., et al. (2018). High-quality whole-genome sequences for 59 historical Shigella strains generated with PacBio sequencing. Genome Announc. 6:e00282-18. doi: 10.1128/genomeA.00282-18
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/JCM.06094-11
McDonough, M. A., and Butterton, J. R. (1999). Spontaneous tandem amplification and deletion of the Shiga toxin operon in Shigella dysenteriae 1. Mol. Microbiol. 34, 1058–1069. doi: 10.1046/j.1365-2958.1999.01669.x
Muniesa, M., and Schmidt, H. (2014). “Shiga toxin-encoding phages: multifunctional gene ferries,” in Pathogenic Escherichia Coli, ed. S. Morabito (Norfolk: Caister Academic Press).
Njamkepo, E., Fawal, N., Tran-Dien, A., Hawkey, J., Strockbine, N., Jenkins, C., et al. (2016). Global phylogeography and evolutionary history of Shigella dysenteriae type 1. Nat. Microbiol. 1:16027. doi: 10.1038/nmicrobiol.2016.27
Penadés, J. R., and Christie, G. E. (2015). The phage-inducible chromosomal islands: a family of highly evolved molecular parasites. Annu. Rev. Virol. 2, 181–201. doi: 10.1146/annurev-virology-031413-085446
Probert, W. S., McQuaid, C., and Schrader, K. (2014). Isolation and identification of an Enterobacter cloacae strain producing a novel subtype of Shiga toxin type 1. J. Clin. Microbiol. 52, 2346–2351. doi: 10.1128/JCM.00338-14
Riley, L. W., Remis, R. S., Helgerson, S. D., McGee, H. B., Wells, J. G., Davis, B. R., et al. (1983). Hemorrhagic colitis associated with a rare Escherichia coli serotype. N. Engl. J. Med. 308, 681–685. doi: 10.1056/NEJM198303243081203
Scheutz, F., Teel, L. D., Beutin, L., Piérard, D., Buvens, G., Karch, H., et al. (2012). Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 50, 2951–2963. doi: 10.1128/JCM.00860-12
Schmidt, H., Bielaszewska, M., and Karch, H. (1999). Transduction of Enteric Escherichia coli Isolates with a Derivative of Shiga Toxin 2-Encoding Bacteriophage φ3538 Isolated from Escherichia coli O157:H7. Appl. Environ. Microbiol. 65, 3855–3861. doi: 10.1128/AEM.65.9.3855-3861.1999
Schwengers, O., Hoek, A., Fritzenwanker, M., Falgenhauer, L., Hain, T., Chakraborty, T., et al. (2020). ASA3P: an automatic and scalable pipeline for the assembly, annotation and higher level analysis of closely related bacterial isolates. PLoS Comput. Biol. 16:e1007134. doi: 10.1371/journal.pcbi.1007134
Strauch, E., Lurz, R., and Beutin, L. (2001). Characterization of a Shiga toxin-encoding temperate bacteriophage of Shigella sonnei. Infect. Immun. 69, 7588–7595. doi: 10.1128/IAI.69.12.7588-7595.2001
Sváb, D., Bálint, B., Maróti, G., and Tóth, I. (2015). A novel transducible chimeric phage from Escherichia coli O157:H7 Sakai strain encoding Stx1 production. Infect. Genet. Evol. 29, 42–47. doi: 10.1016/j.meegid.2014.10.019
Sváb, D., Bálint, B., Vásárhelyi, B., Maróti, G., and Tóth, I. (2017). Comparative genomic and phylogenetic analysis of a Shiga toxin producing Shigella sonnei (STSS) strain. Front. Cell. Infect. Microbiol. 7:229. doi: 10.3389/fcimb.2017.00229
Sváb, D., Falgenhauer, L., Rohde, M., Chakraborty, T., and Tóth, I. (2018). Identification and characterization of new broad host-range rV5-like coliphages C203 and P206 directed against enterobacteria. Infect. Genet. Evol. 64, 254–261. doi: 10.1016/j.meegid.2018.07.004
Sváb, D., Horváth, B., Maróti, G., Dobrindt, U., and Tóth, I. (2013). Sequence variability of P2-like prophage genomes carrying the cytolethal distending toxin V operon in Escherichia coli O157. Appl. Environ. Microbiol. 79, 4958–4964. doi: 10.1128/AEM.01134-13
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Tóth, I., Schmidt, H., Kardos, G., Lancz, Z., Creuzburg, K., Damjanova, I., et al. (2009). Virulence genes and molecular typing of different groups of Escherichia coli O157 strains in cattle. Appl. Environ. Microbiol. 75, 6282–6291. doi: 10.1128/AEM.00873-09
Tóth, I., Sváb, D., Bálint, B., Brown-Jaque, M., and Maróti, G. (2016). Comparative analysis of the Shiga toxin converting bacteriophage first detected in Shigella sonnei. Infect. Genet. Evol. 37, 150–157. doi: 10.1016/j.meegid.2015.11.022
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15:524. doi: 10.1186/s13059-014-0524-x
Trofa, A. F., Ueno-Olsen, H., Oiwa, R., and Yoshikawa, M. (1999). Dr. Kiyoshi Shiga: Discoverer of the Dysentery Bacillus. Clin. Infect. Dis. 29, 1303–1306. doi: 10.1086/313437
Unkmeir, A., and Schmidt, H. (2000). Structural analysis of phage-borne stx genes and their flanking sequences in Shiga toxin-producing Escherichia coli and Shigella dysenteriae type 1 strains. Infect. Immun. 68, 4856–4864. doi: 10.1128/iai.68.9.4856-4864.2000
Vongsawan, A. A., Kapatral, V., Vaisvil, B., Burd, H., Serichantalergs, O., Venkatesan, M. M., et al. (2015). The genome of Shigella dysenteriae strain Sd1617 comparison to representative strains in evaluating pathogenesis. FEMS Microbiol. Lett. 362:fnv011. doi: 10.1093/femsle/fnv011
Wattam, A. R., Davis, J. J., Assaf, R., Boisvert, S., Brettin, T., Bun, C., et al. (2017). Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 45, D535–D542. doi: 10.1093/nar/gkw1017
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Wirth, T., Falush, D., Lan, R., Colles, F., Mensa, P., Wieler, L. H., et al. (2006). Sex and virulence in Escherichia coli: an evolutionary perspective. Mol. Microbiol. 60, 1136–1151. doi: 10.1111/j.1365-2958.2006.05172.x
Keywords: Shigella dysenteriae serotype 1, Stx1 prophage, complete genome, comparative genomics, phylogeny
Citation: Sváb D, Falgenhauer L, Horváth B, Maróti G, Falgenhauer J, Chakraborty T and Tóth I (2021) Genome Analysis of a Historical Shigella dysenteriae Serotype 1 Strain Carrying a Conserved Stx Prophage Region. Front. Microbiol. 11:614793. doi: 10.3389/fmicb.2020.614793
Received: 09 October 2020; Accepted: 09 December 2020;
Published: 08 January 2021.
Edited by:
David W. Ussery, University of Arkansas for Medical Sciences, United StatesReviewed by:
Camilla Sekse, Norwegian Veterinary Institute (NVI), NorwayYanwen Xiong, National Institute for Communicable Disease Control and Prevention (China CDC), China
Copyright © 2021 Sváb, Falgenhauer, Horváth, Maróti, Falgenhauer, Chakraborty and Tóth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Domonkos Sváb, c3ZhYi5kb21vbmtvc0BhdGsuaHU=