 Xiao-Yu Zhu1†
Xiao-Yu Zhu1† Jiwen Liu
Jiwen Liu Jiwei Tian
Jiwei Tian Xiao-Hua Zhang
Xiao-Hua Zhang- 1College of Marine Life Sciences, Ocean University of China, Qingdao, China
- 2Laboratory for Marine Ecology and Environmental Science, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
- 3Institute of Evolution & Marine Biodiversity, Ocean University of China, Qingdao, China
- 4Frontiers Science Center for Deep Ocean Multispheres and Earth System, Ocean University of China, Qingdao, China
Microbial eukaryotes are widespread and play important roles in marine ecosystems. However, their ecological characteristics in the deep sea (>1,000 m), especially hadal trenches, were largely unknown. Here, we investigated the diversity and metabolic potentials of microbial eukaryotes along the whole water column of the Mariana Trench by metagenomics. Our results showed clear depth-related distribution of microbial eukaryotic community and associated metabolic potentials. Surface seawater was dominated by phototrophic/mixotrophic groups (e.g., Dinoflagellata) and genes involved in biosynthesis (photosynthesis and fatty acid biosynthesis), while deep (bathypelagic and/or hadal) seawaters were enriched with heterotrophic groups (e.g., Bicoecea) and genes related to digestion (lysosomal enzymes and V-type ATPase) and carbohydrate metabolism. Co-occurrence analysis revealed high intra-domain connectivity, indicating that microbial eukaryotic composition was more influenced by microbial eukaryotes themselves than bacteria. Increased abundance of genes associated with unsaturated fatty acid biosynthesis likely plays a role in resisting high hydrostatic pressure. Top1 and hupB genes, responsible for the formation and stabilization of DNA structure, were unique and abundant in the hadal zone and thus may be helpful to stabilize DNA structure in the deep sea. Overall, our results provide insights into the distribution and potential adaptability of microbial eukaryotes in the hadal zone.
Introduction
With high morphological and genetic variability, microbial eukaryotes fulfill a variety of crucial ecological roles and are essential participants of marine carbon cycle (Azaml et al., 1983; Worden et al., 2015). Phototrophic microbial eukaryotes are important contributors to global primary productivity (Field et al., 1998; De Vargas et al., 2015), while heterotrophic microbial eukaryotes consume smaller phytoplankton, bacteria, and archaea, and are themselves important prey for larger mesozooplankton, serving as important trophic links to higher trophic levels (Sherr and Sherr, 1994, 2002). In addition, mixotrophic microbial eukaryotes, combining the photosynthesis and the ingestion of living prey, may enhance primary production and biomass transfer to higher trophic levels (Ward and Follows, 2016; Stoecker et al., 2017). Understanding the nutritional strategies of microbial eukaryotes is essential for characterizing their ecological interactions and responses to environmental conditions.
The ocean water column is divided into five depth zones: epipelagic (0–200 m), mesopelagic (200–1,000 m), bathypelagic (1,000–4,000 m), abyssopelagic (4,000–6,000 m), and hadopelagic/hadal (>6,000 m) zones (Arístegui et al., 2009; Liu et al., 2018). The hadal zone (exclusively comprised of trenches) constitutes the deepest 45% of the vertical depth of the global ocean (Jamieson et al., 2010; Jamieson, 2015) and was known to host active and diverse biological communities (Jamieson et al., 2010; Nunoura et al., 2015; Xu et al., 2018; Liu et al., 2019b), indicating the hadal ecosystem is crucial to ocean food web. Previous surveys on the distribution of microbial eukaryotes along the water column on a global scale (Giner et al., 2020) and in specific oceanic regions (Countway et al., 2007; Not et al., 2007; Brown et al., 2009; Hu et al., 2016; Xu et al., 2017) have revealed unexpected high phylogenetic diversity of microbial eukaryotes in deep seas and a clear differentiation between surface photic and deep aphotic ocean communities. However, the maximum depth of these studies was limited to the bathypelagic zone. Jing and Xu recently conducted the first survey of microbial eukaryotes dwelling in the hadal waters (depth up to 8,727 m) of the Mariana Trench (Jing et al., 2018; Xu et al., 2018). They found significant shifts in the community composition of microbial eukaryote with depth. For instance, Alveolata dominated the surface waters whereas Stramenopiles were enriched in the deep waters. Despite these information, little is known about the microbial eukaryotic communities and metabolic potentials in the deeper seawaters (8,727–11,000 m) of the Mariana Trench, where unique microbial populations (e.g., increased amount of SAR406, hydrocarbon-degrading microbes) have been observed (Ichino et al., 2015; Nunoura et al., 2015; Liu et al., 2018, 2019b; Gao et al., 2019). In addition, whether microbial eukaryotes exhibit unique high-pressure adaptation mechanisms as found in bacteria and archaea in our previous studies (Zheng et al., 2020; Zhong et al., 2020) is still unknown. Thus, knowledge of microbial eukaryotes at the bottom of the Mariana Trench, the deepest ocean part on Earth, is necessary for a better understanding of their ecological roles and potential adaptability.
Recently, with technology advances and costs fall, metagenome has shown good performance in resolving microbial eukaryotic diversity from environmental samples (Pernice et al., 2015b; Boeuf et al., 2019; Obiol et al., 2020) free of the potential biases of PCR-dependent methods. However, there are few studies of microbial eukaryotic metabolism using metagenome due to some technical limitations like identifying eukaryotic genes (Levy Karin et al., 2020). Ocean is known to harbor a great number of eukaryotic genes especially in those unexplored zones like hadal zones, half of which however have low similarity with known genes (Carradec et al., 2018). A recent technical progress has made it possible for using metagenome to study in situ community-wide metabolic characteristics of microbial eukaryotes (Levy Karin et al., 2020), which will greatly expand the eukaryotic reference databases. Therefore, it is of great potential for using metagenome to study microbial eukaryotic ecology.
In this study, we extended the information of marine microbial eukaryotes to the bottom waters of the Mariana Trench by analyzing samples at seven water depths ranging from 0 to 10,500 m, a wide span from the surface to near the bottom (∼11,000 m). Metagenome derived ribosomal small subunit (SSU) gene reads were used to reveal the vertical distribution of microbial eukaryotic communities and explore the co-occurrence relationships among different clades of bacteria and microbial eukaryotes. Putative eukaryotic protein-coding genes were used to explore their metabolic potentials across the full water column. As a result, our study showed clear depth-related distribution and metabolic differences of microbial eukaryotes along the water column in the Mariana Trench.
Materials and Methods
Sample Collection, DNA Extraction, and Metagenomic Sequencing
The data used in this study were based on two published datasets from our previous studies (Liu et al., 2019b; Zhong et al., 2020). Those two datasets were from seawater samples of the Challenger Deep at the site 2016 (11.20°N, 142.19°E) and the site 2017 (11.38°N, 142.41°E), respectively (Table 1 and Supplementary Figure 1). The seawater samples were collected by Niskin bottles (36–48 L seawaters per depth) and then were serially filtered through 3- and 0.22-μm polycarbonate membranes. All filters were stored in liquid nitrogen onboard and at −80°C in our laboratory for DNA extraction. Detailed steps for DNA extraction, metagenomic sequencing, and quality control of reads have been described before (Liu et al., 2019b; Zhong et al., 2020). We finally obtained 17 samples from seven different water depths (surface zone: 0 m; bathypelagic zone: 2,000 and 4,000 m; hadal zone: 8,000, 9,600, 10,400, and 10,500 m) as listed in Table 1. Each sample was named according to its fraction size (0.2–3 μm, F; > 3 μm, P), sampling depth and sampling site. For example, P0_2016 represents the >3-μm sample isolated from 0 m at the site 2016. Note that samples from 2,000 and 8,000 m both have two replications and several 0.2–3-μm samples were discarded due to insufficient quantity of DNA for metagenomic sequencing. Water physicochemical parameters from March 2017 cruise were plotted in Supplementary Figure 2.

Table 1. Contexual data of 17 seawater samples from seven depths of the Mariana Trench and diversity index of microbial eukaryotes of each sample.
Taxonomic Profile Based on Extracted 18S rRNA Gene Reads
After quality control, clean reads ranging in size from 11.85 to 16.54 Gb were obtained from each sample (Supplementary Table 1). Metaxa2 version 2.2 (Bengtsson-Palme et al., 2015) was used to extract 18S rRNA gene reads from metagenomic data and then gave them taxonomic information based on SSU sequences in the SILVA database release 128 (Quast et al., 2012) with the default settings (reliability score cutoff = 80). Finally, a raw table of 490 operational taxonomic units (OTUs) was generated by using Metaxa2 (reads belonging to the same species were considered as an OTU) (Supplementary Data 1). As previous studies (Xu et al., 2017; Pasulka et al., 2019; Giner et al., 2020), Metazoa were not included in our study because the high number of 18S rDNA copies in the Metazoan cells may distort the relative abundance of different microbial eukaryotes. All community compositions of microbial eukaryotes in our study were based on the raw OTU table.
Network Analysis
Intra- and inter-domain (bacteria and microbial eukaryotes) co-occurrence networks were constructed using the igraph, Hmisc, and qvalue libraries in R. The bacterial OTU table (Supplementary Data 2) was obtained using the Metaxa2 version 2.2 (Bengtsson-Palme et al., 2015). For both bacteria and microbial eukaryotes, only OTUs with a total proportion above 0.3% in all samples and occurring in more than three samples were retained. The pairwise Spearman’s correlations between OTUs were calculated, with a correlation coefficient > |0.7| and a p < 0.01 (Benjamini and Hochberg adjusted) being considered as a valid relationship. The network was visualized in Gephi.1
Functional Profile Based on Putative Eukaryotic Genes
Clean reads of each sample were assembled independently using the MegaHit version 1.1.2 (Li et al., 2016). Assembled contigs of at least 1 kbp in length were used to identify potential eukaryotic contigs by EukRep with the default settings (West et al., 2018). A total of 43,406 eukaryotic contigs were obtained (Supplementary Data 3). Then, potential eukaryotic contigs were used to discover eukaryotic protein-coding genes by the new developed MetaEuk (Levy Karin et al., 2020) based on a provided database composed of three datasets: Uniclust50 (Mirdita et al., 2017), marine eukaryotic metatranscriptomes (MERC) (Steinegger et al., 2019), and Marine Microbial Eukaryotic Transcriptome Sequencing Project (MMETSP; Keeling et al., 2014; Johnson et al., 2019). This is a comprehensive and largest database focused on eukaryotes to date, and could be a valuable resource for discovering protein-coding genes from environmental samples. Note that some of predictions generated with this method are partial, because some protein-coding genes span more than one contig. Subsequently, taxonomic annotation of predicted eukaryotic genes was also performed with MetaEuk against an integrated taxonomic database composed of Uniclust90 dataset (Mirdita et al., 2017) and MMETSP (Keeling et al., 2014; Johnson et al., 2019) dataset. In this step, non-eukaryotic genes were still detected and were then deleted.
Nucleotide sequences of all predicted eukaryotic protein-coding genes from all samples were clustered at 97% similarity to generate non-redundant gene set using CD-Hit (Fu et al., 2012). Then, estimated non-redundant gene abundances (CPM – copies per million reads) based on paired end reads were determined using the Salmon version 1.2.1 (Patro et al., 2017). The Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation for each gene was done using the eggNOG-mapper (Huerta-Cepas et al., 2015) with an e-value cutoff of 1e–5 against the eggNOG database. Referring to a previous study, several pathways/enzymes (Supplementary Table 2) were chosen to represent the core biogeochemical functions relevant to known metabolisms (Hu et al., 2018). Genes that appeared in multiple KEGG pathways were counted multiple times. Abundance, KEGG assignment, and taxonomic assignment of a total of 70,291 non-redundant genes were summarized in Supplementary Data 4. To understand the carbohydrate metabolism in different layers, the dbCAN2 web server (Zhang et al., 2018) was used for identifying carbohydrate-active genes with three methods: HMMER, DIAMOND, and Hotpep. Only the carbohydrate-active genes identified by two or three methods were retained.
Statistical Analyses and Graphic Plotting
Both alpha and beta diversity analyses were performed with the Vegan package (Oksanen et al., 2016) in R. To equalize sequencing depth, each sample was rarefied to 709 reads (the lowest sequence number across all samples) and a total of 314 OTUs were reserved (Supplementary Data 5). Richness (observed OTUs) and Shannon indices were calculated based on rarified OTU table and the differences between different predefined groups were tested with the Wilcoxon test. For beta diversity, the non-metric multidimensional scaling analysis (NMDS) and permutational multivariate analysis of variance (ANOSIM) were performed based on Bray-Curtis dissimilarities. The heatmap was generated by the TBtools software v0.6662 (Chen et al., 2020). The upset plots and the ternary plot were generated by the UpSetR (Conway et al., 2017) and ggtern package (Hamilton and Ferry, 2018) in R, respectively.
Access to Data
Metagenomic data of two published datasets are available in NCBI under BioProject numbers of PRJNA412741 and PRJNA541485, respectively. The nucleotide acid and amino acid sequences of each predicted eukaryotic non-redundant gene are available at zenodo (DOI: 10.5281/zenodo.3971257).
Results
Environmental Conditions Along the Water Column
Salinity was constant throughout the water column (34.0−34.8), while temperature and pH generally decreased from the surface (29°C; 8.2) to the trench bottom (∼1°C; 7.8) (Supplementary Figure 2). The temperature and salinity profiles were similar to previous observations (Taira et al., 2005; Nunoura et al., 2015; Xu et al., 2018). The concentration of dissolved oxygen (DO) at the sea surface was 193.6 μM and decreased to 84.9 μM at 1,000 m. Then, DO increased to 157.7 μM at 4,000 m and presented at relatively constant concentrations between 156.0 and 174.8 μM below 4,000 m. Uniform concentrations of ammonia (17.5–26.7 nM) and nitrite (0.01−0.11 μM) were observed over the depth. Silicate, phosphate, and NOx– concentrations increased from the surface to 2,000 m and then remained stable at depths greater than 2,000 m.
Alpha and Beta Diversity of Microbial Eukaryotes
The diversity (richness and Shannon) of microbial eukaryotes generally decreased with depth, with a significant difference (Wilcoxon test; p < 0.05) between surface and hadal waters (Table 1 and Supplementary Figures 3A,B). The NMDS analysis revealed significant dissimilarity of microbial eukaryotic communities between surface and deep (bathypelagic and/or hadal) communities (Figure 1A and Supplementary Table 3; ANOSIM: R2 > 0.4, p < 0.05). This OTU-based grouping pattern was similar to that based on the abundance profile of functional genes (Figure 4A and Supplementary Table 4; ANOSIM: R2 > 0.5, p < 0.01), indicating taxonomy-related metabolic differentiations among different depths.
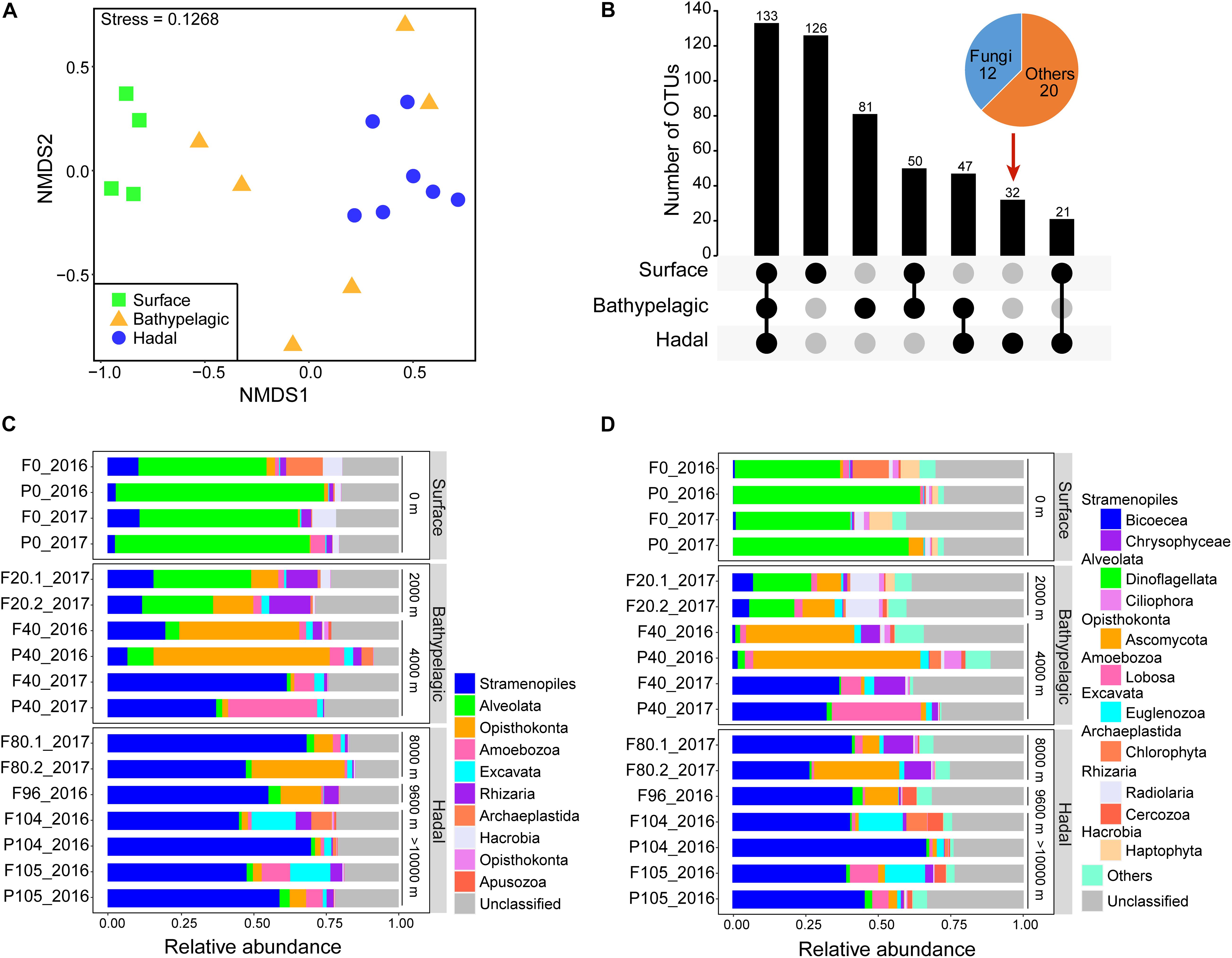
Figure 1. Community characteristics of microbial eukaryotes in the Mariana Trench. (A) Comparison of community structure based on a non-metric multidimensional scaling analysis (NMDS). Samples were grouped according to three layers: surface, bathypelagic, and hadal layers. (B) Core and differential operational taxonomic units (OTUs) among the surface, bathypelagic, and hadal layers. Layers included in a set were represented by filled dots in the matrix below each bar. Community composition of microbial eukaryotes from the surface to the bottom of the Mariana Trench at superphylum (C) and phylum/class levels (D).
Taxonomic Distribution Along the Water Column
Of the total 490 OTUs, 133 were shared among the surface, bathypelagic, and hadal layers. The surface layer had the most unique OTUs while the hadal layer possessed the least unique OTUs (mainly fungi) (Figure 1B). Throughout the water column, Stramenopiles, Alveolata, and Opisthokonta were the top three superphyla (Figure 1C and Supplementary Data 1), in consistent with the taxonomic results of functional genes (Supplementary Figures 4A,B). On average, about 20% of eukaryotic OTUs and 56% eukaryotic genes were unclassified in all samples (Figure 1C and Supplementary Figure 4B). Distinct microbial eukaryotic communities were observed at different water depths of the Mariana Trench. Alveolata represented by Dinoflagellata (36.2–64.2%) dominated the surface water, while Stramenopiles represented by Bicoecea (26.5–66.6%) dominated the deeper waters (≥4,000 m). The relative abundance of Opisthokonta (mainly Ascomycota) was higher at depths ranging from 2,000 to 9,600 m, especially at 4,000 m. The most abundant genus across all samples was Caecitellus (belonging to Bicoecea), comprising 18.3–58.8% of the hadal community (Supplementary Figure 5).
To reveal the distribution of major taxonomic groups with different trophic modes, we compared their relative abundances at different layers (Figure 2). As expected, most phototrophic/mixotrophic (e.g., Dinoflagellata, Haptophyta, Chlorophyta) phytoplankton showed higher relative abundances at the surface, except for phototrophic Diatom and mixotrophic Chrysophyceae and Synurophyceae, which displayed higher relative abundances at the bathypelagic and/or hadal zones. All heterotrophic groups were enriched in the deep sea. Among these heterotrophic groups, Bicoecea, Euglenozoa, and Cercozoa showed the highest relative abundances at the hadal zone, indicating their potential ecological roles in the hadal trench.
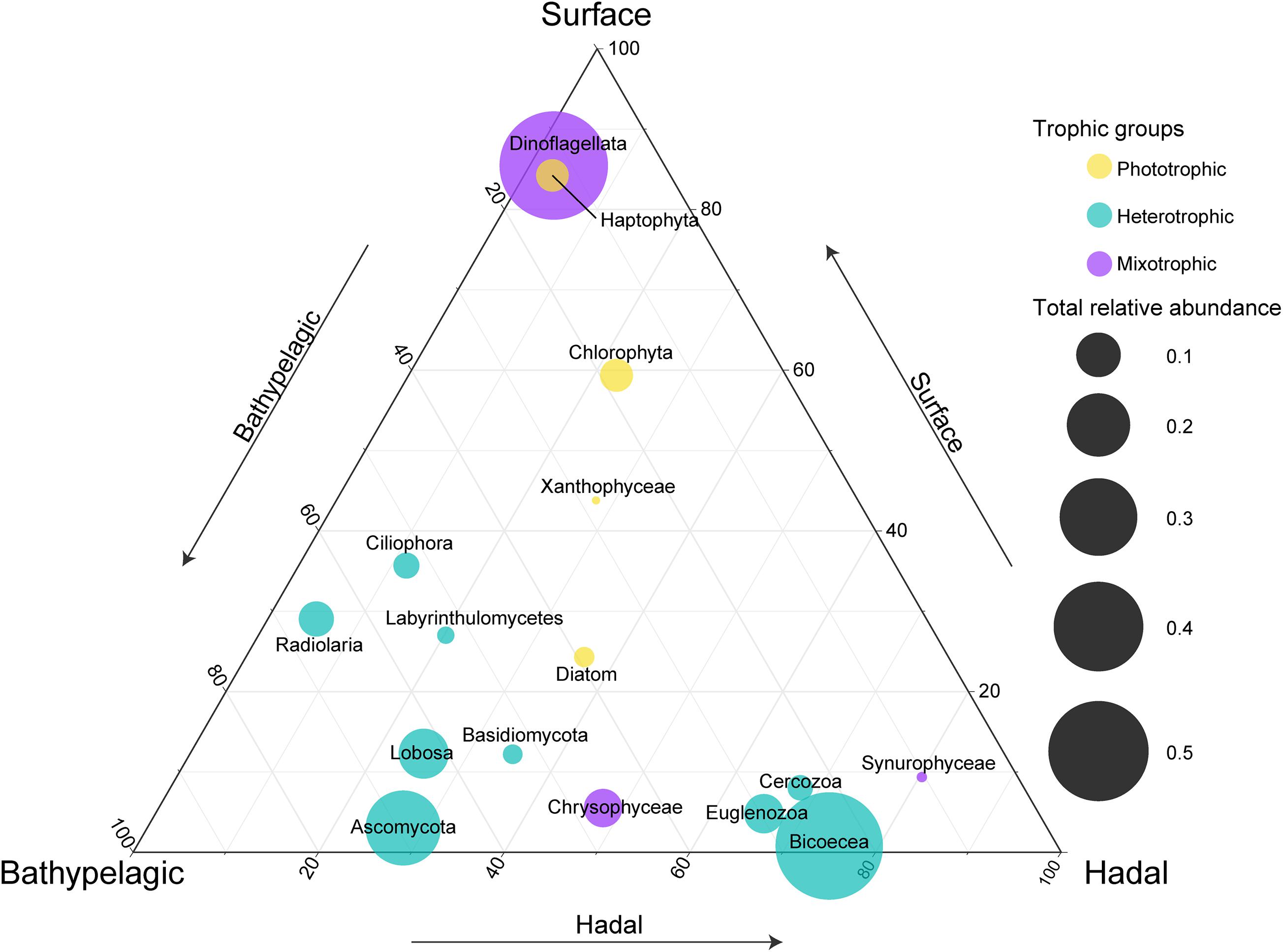
Figure 2. Comparisons of relative abundances for each major taxonomic group at the surface, bathypelagic, and hadal layers. Circle placement represents the relative size of relative abundances among the three layers (one layer per axis, anticlockwise with increasing relative size). Circle size represents the sum of relative abundances for each taxonomic group at three layers. Note that the taxonomic groups were listed according to their main trophic modes (mixotrophic, heterotrophic, or phototrophic).
Co-occurrence Networks Between Microbial Eukaryotes and Bacteria
To explore the ecological linkages among microbial eukaryotes and between microbial eukaryotes and bacteria, a correlation-based network analysis was conducted (Figures 3A,B). The network was composed of 291 nodes (101 eukaryotic OTUs and 190 bacteria OTUs) and 558 edges (correlations). Correlations within eukaryotic taxa were stronger than those between bacteria and eukaryotes (Figure 3A). Modularity analysis revealed eight major modules (subunits with highly inter-connected nodes) (Figure 3B). Most of these modules were comprised of a group of OTUs that were phylogenetically close or even belonged to the same clade. For example, module 3 and 5 were predominated by Proteobacteria OTUs and module 2 was dominated by Alveolata OTUs (Figures 3A,B). Thus, taxonomic relatedness plays a key role in determining the network modular structure. Clades with similar trophic modes also tended to co-occur. For example, module 6 consisted of Alveolata (mainly phototrophic/mixotrophic Dinoflagellata) and Hacrobia (mainly phototrophic Haptophyte) OTUs, and module 4 comprised metatrophic Opisthokonta (totally fungi) and Actinobacteria OTUs. Additionally, connections between ecologically linked clades were observed. For example, Stramenopiles (mostly flagellates) OTUs were dispersedly distributed in the network and connected with some bacterial phyla.
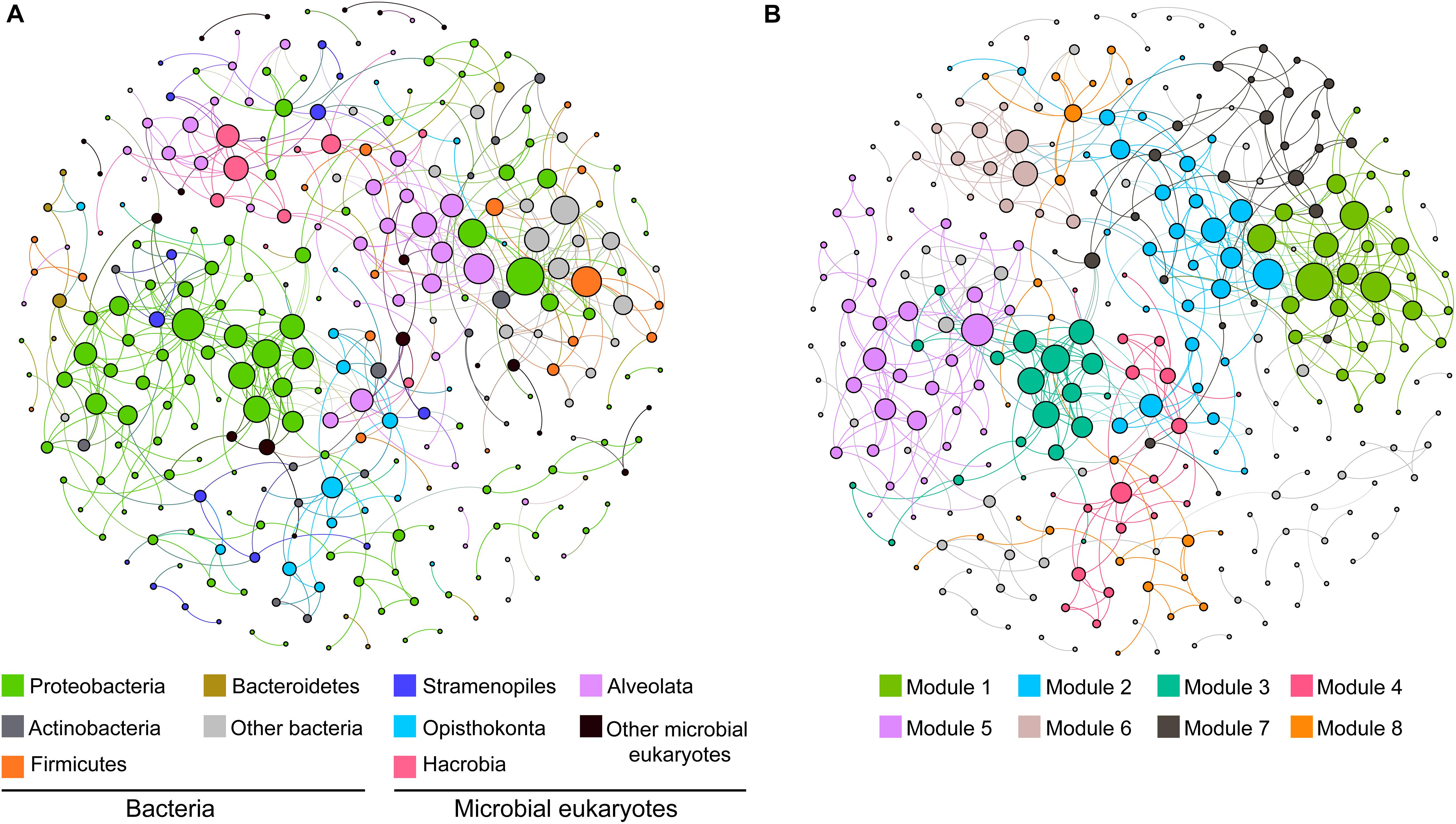
Figure 3. Co-occurrence networks of the dominant clades of microbial eukaryotes and bacteria based on pairwise Spearman’s correlations between OTUs. Only connection with a correlation coefficient > | 0.7| and a p < 0.01 was shown. The size of each node is proportional to the number of connections. The two panels show the network of all samples with OTUs colored by taxonomy (A) and modularity (B).
Community-Wide Metabolic Potentials Across the Whole Water Column of the Mariana Trench
Of the total 70,291 eukaryotic non-redundant genes (56.3% had KEGG identities), most were from the bathypelagic and hadal waters (Figure 4B). Only 7,760 genes were shared within the surface, bathypelagic, and hadal layers, mainly belonging to Stramenopiles and Opisthokonta (Figure 4B and Supplementary Figure 6). The bathypelagic and hadal layers had the most common genes (22,004, 56.8% had KEGG identities). Bathypelagic layer had the most unique genes, over 70% of which were taxonomically unclassified. Compared to the surface (40.6%), the hadal zone had a higher percentage of genes (66%) that can be assigned to KEGG identities. There were 3,872 genes unique for the hadal layer, mainly belonging to Stramenopiles (Figure 4B and Supplementary Figure 6). Among the top five abundant hadal-unique genes, two were involved in DNA formation and stabilization [K03163 (top1; DNA topoisomerase I) and K03530 (hupB; DNA-binding protein HU-beta)], and three were related to alcohol dehydrogenation [K13979 (yahK; uncharacterized zinc-type alcohol dehydrogenase-like protein), K00002 (adh; alcohol dehydrogenase), and K13953 (adhP; alcohol dehydrogenase, propanol-preferring)] (Supplementary Figure 7 and Data 6).
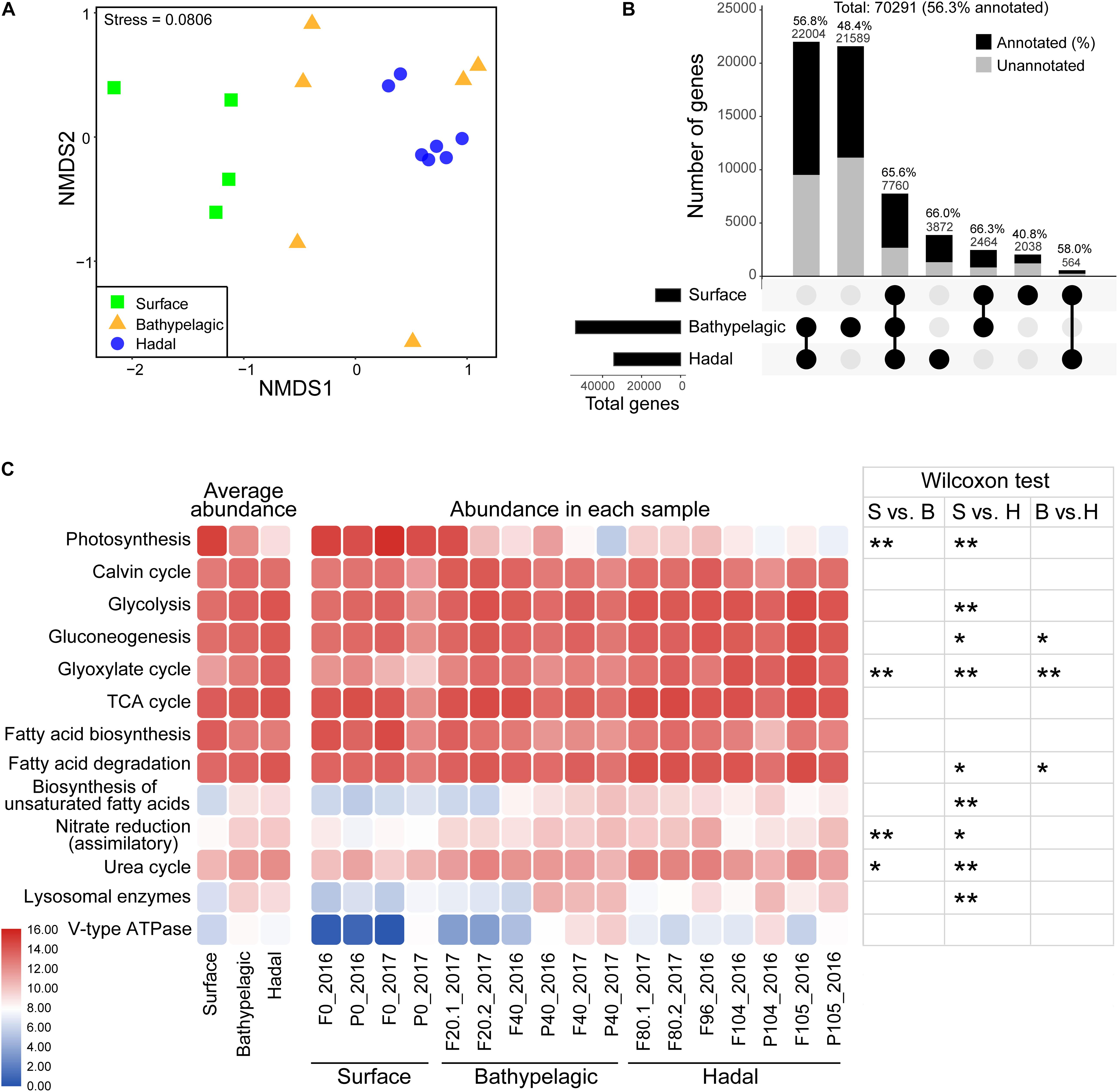
Figure 4. Metabolic characteristics of microbial eukaryotes in the Mariana Trench. (A) Differential analysis of functional structure of microbial eukaryotes based on a NMDS analysis. Samples were grouped according to three layers: surface, bathypelagic, and hadal layers. (B) Shared and unique non-redundant genes between epipelagic, bathypelagic, and hadal zones. Layers included in a set were represented by filled dots in the matrix below each bar. The number of genes that had KEGG identities was represented by black (also given as percentages). (C) Comparisons of key pathways at different samples. Heatmaps show the average abundances (CPM: copies per million reads) of KEGG pathways in the epipelagic, bathypelagic, and hadal zones (left) and their abundances in all the samples (medium). Abundance values generating this heatmap were log2 scaled. Wilcoxon test was used for significance test (right); *p < 0.05, **p < 0.01. S, surface; B, bathypelagic; H, hadal.
Most pathways/enzymes had significant differences (p < 0.05) in abundance between the surface and the hadal zones (Figure 4C and Supplementary Tables 5, 6), which may be due to the dramatically different environments. Genes associated with biosynthetic pathways like photosynthesis (p < 0.01) and fatty acid biosynthesis were higher in abundance at the surface compared to deeper depths, except for the Calvin cycle which showed similar abundance among all depths. Genes involved in several carbon metabolism pathways (Glycolysis, Gluconeogenesis, Glyoxylate cycle, fatty acid degradation, and biosynthesis of unsaturated fatty acid), nitrogen metabolism pathways (assimilatory nitrate reduction and urea cycle), and digestion (lysosomal enzymes and V-type ATPase) were higher in abundance in the deep sea (bathypelagic and/or hadal zones).
To understand carbohydrate metabolism characteristics in different layers, genes were assigned to different enzyme families and only 309 (0.44% of all 70,291 genes) genes were annotated (Supplementary Table 7). The total abundance of genes involved in six enzyme families was higher in the deep sea (Figure 5). The main enzyme families increasing in the deep seawater (≥2,000 m) were glycosyltransferases (GTs, catalyzing the formation of the glycosidic linkage to form a glycoside) and glycoside hydrolases (GHs, catalyzing the hydrolysis of O–, N–, and S–linked glycosides), which are crucial to carbohydrate biosynthesis and degradation, respectively.
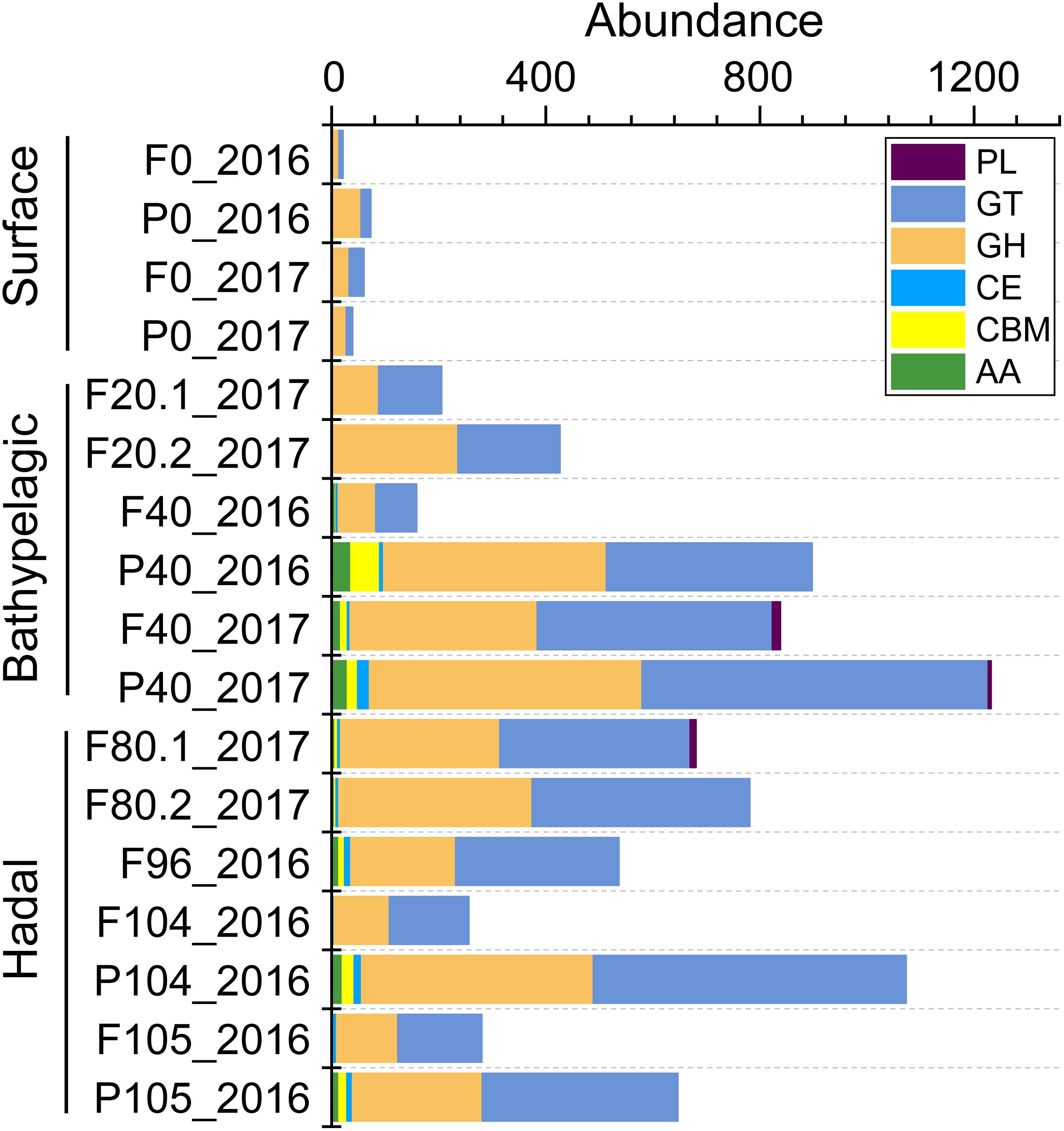
Figure 5. The abundance (CPM) of each enzyme family involved in carbohydrate metabolism across all the depths. AAs, auxiliary activities; CBMs, carbohydrate-binding modules; CEs, carbohydrate esterases; GHs, glycoside hydrolases; GTs, glycosyltransferases; PLs, polysaccharide lyases.
Discussion
Stratified Community Structure of Microbial Eukaryotes
The vertical changes in community composition and metabolic potentials of microbial eukaryotes across the full water column of the Mariana Trench (0–10,500 m) were investigated through metagenomic sequencing in this study. Similar to previous reports in the Mariana Trench (Jing et al., 2018; Xu et al., 2018) and other oceanic regions (Countway et al., 2007; Brown et al., 2009; Xu et al., 2017; Giner et al., 2020), decreased diversity with depth and depth-related community separation were observed. Abiotic factors like depth and depth-related environmental conditions (e.g., temperature, hydrostatic pressure, nutrient availability) have been recognized as the important factors in shaping the distribution of microbial eukaryotic communities on a regional scale as seen in the above references.
Other than abiotic factors, complex microbial interactions (e.g., host–parasite, predator–prey, symbiotic relationships) were also crucial for community assembly (Steele et al., 2011; Faust and Raes, 2012; Lima-Mendez et al., 2015) and may be partially revealed by co-occurrence networks (Faust and Raes, 2012; Liu et al., 2019a). Our results showed that interactions mainly occurred among phylogenetically close taxa, which might due to similar lifestyles and metabolic or physiological characteristics. Consistent with a previous study (Gilbert et al., 2012), associations within domains (bacteria or microbial eukaryotes) were stronger than those between them. Despite this, several connections between bacteria and microbial eukaryotes were observed, indicating their potential ecological linkages. For example, the most connected eukaryotic clade with bacteria was Stramenopiles (Figure 3), which was likely due to their role as important predators of prokaryotes (Burki, 2014). Here, we only discussed the interactions between microbial eukaryotes and bacteria. Interactions between microbial eukaryotes, and other taxa like bigger megafauna [e.g., amphipods and snailfish; (Jamieson et al., 2010)], viruses and archaea may be also important for community assembly of microbial eukaryotes and still need further study.
Enriched Groups at the Bathypelagic and/or Hadal Zones
As expected, the surface waters were dominated by phototrophic/mixotrophic phytoplankton accompanied by a high abundance of genes involved in biosynthesis (photosynthesis and fatty acid synthesis) (Figures 2, 4C), which was also observed in a metatranscriptomic study at the San Pedro Ocean (Hu et al., 2018). Specifically, the relative abundances of phototrophic Diatom and mixotrophic Chrysophyceae were higher in the bathypelagic and hadal zones of the Mariana Trench (Figure 2). Indeed, the presence of living phytoplankton like diatoms, cyanobacteria, and dinoflagellates in the dark ocean has been reported, and fast-sinking or changing nutritional strategies may be the possible explanations (Jiao et al., 2014; Agusti et al., 2015; Xu et al., 2017; Giner et al., 2020). As for Chrysophyceae, they are common bacteriovores and were most abundant phytoplankton frequently detected in the water column at depths of 3,000–4,000 m worldwide (Pernice et al., 2015b).
Fungi perform vital functions as decomposers, driving nutrient cycles in detritus environments, and as parasites and symbionts in the ecosystems (Webster and Weber, 2007). Similar to previous studies (Xu et al., 2018; Wang et al., 2019), the relative abundance of fungi (mainly branched with Ascomycete) was highest in the bathypelagic zone (Figure 2). In addition, a fungi-like group labyrinthulomycetes was also enriched in the bathypelagic zone (Figure 2). Marine snow, aggregation of organisms, and organic matter like fecal pellets and phytodetritus, is important to the biological pump, transporting particulate carbon to greater depths (Turner, 2015). Fungi and labyrinthulomycetes have been reported to be the dominant eukaryotes of bathypelagic marine snow and have the potential to significantly contribute to the degradation of organic matter in the deep sea (Bochdansky et al., 2016). In addition, fungi prefer utilizing organic matter with high C:N ratio (Raghukumar, 2012), which was reported to increase with depth (Hopkinson and Vallino, 2005). Therefore, these saprotrophic organisms have the potential to significantly complement bacteria and archaea in utilizing and recycling of nutrients in deep seas.
Our study complemented the information of microbial eukaryotes in the bottom waters (≥9,600 m) of the Mariana Trench compared to previous studies (Jing et al., 2018; Xu et al., 2018). We found Bicoecea, Euglenozoa, and Lobosa were the dominant heterotrophic protists in the bottom waters (Figure 1D), indicating their potential high-pressure adaptability. Moreover, Bicoecea was the most dominant group in the shallower hadal zone, which was only found in one of two previous studies (Jing et al., 2018; Xu et al., 2018). Considering different primers applied to recover microbial eukaryotes in their studies, primer bias may be a main factor resulting in this differentiation (Stoeck et al., 2006). In addition, significant increase in relative abundances of Bicoecea along the water column was also reported on a global scale (Giner et al., 2020). The dominant genus of Bicoecea is Caecitellus (Supplementary Figure 5), which is a group of small flagellates exerting high grazing activity on prokaryotes and other eukaryotes (Boenigk and Arndt, 2002) and showing general tolerance to high pressure (up to 550 bar under experimental conditions) (Atkins et al., 1998; Živaljić et al., 2018), indicating their important role in deep seas. Small heterotrophic protists are considered as the first consumers of prokaryotic biomass and main consumers of suspended organic matter, promoting organic matter remineralization (Boenigk and Arndt, 2002). Recent studies showed higher-than-expected grazing activity by protists down to mesopelagic depths (Pernice et al., 2015a). Abundant heterotrophic bacteria like SAR406 and hydrocarbon-degrading microbes have been reported in the hadal zone of the Mariana Trench (Nunoura et al., 2015; Gao et al., 2019; Liu et al., 2019b), probably breeding microbial eukaryotes therein. Thus, heterotrophic protists may have capabilities of regenerating nutrients and inorganic molecules in the hadal zone.
Metabolic Changes in the Hadal Zone Compared to the Surface
In the hadal zone, we observed high abundances of genes involved in digestion (lysosomal enzymes and V-type ATPase) and carbohydrate metabolism (glycosyltransferases and glycoside hydrolases) (Figures 2, 4C), all of which are important in the heterotrophic process. For example, lysosomal enzymes are involved in many cellular processes like the internal breakdown and recycling of biomolecules (Eskelinen and Saftig, 2009; Settembre et al., 2013) and are highly expressed in heterotrophic flagellates (Labarre et al., 2020). V-type ATPases play a role in digestive processes by lowering the pH in phagosomes (Finbow and Harrison, 1997). In view of abundant heterotrophic clades found in the hadal zone, it is reasonable that genes associated with heterotrophic processes were enriched therein.
High hydrostatic pressures reduce the fluidity of lipid bilayers and the reversibility of their phase transitions, ultimately leading to the denaturation and functional disorder of membrane-associated proteins (Chong et al., 1983; Kato et al., 2002). Many heterotrophic protists have been reported to be able to survive exposure to high hydrostatic pressure, indicating their potential genetic adaptation (Živaljić et al., 2018). However, their adaptation mechanism is still unknown. In our study, we found increased abundance of genes responsible for biosynthesis of unsaturated fatty acid at depths exceeding 4,000 m (Figure 4C). Likewise, several biochemical studies have suggested that membranes of deep-sea-adapted organisms contain a higher weight percentage of unsaturated fatty acids than the equivalent membranes of shallow-sea species (Behan et al., 1992; Fang et al., 2000). It has been acknowledged that an increase in fluidity of membranes by incorporation of unsaturated fatty acids plays a role in the bacterial survival at high hydrostatic pressures (DeLong and Yayanos, 1985; Allen et al., 1999) and low temperatures (DeLong and Yayanos, 1986). Therefore, we speculated unsaturated fatty acid may be also effective in resisting high hydrostatic pressures for microbial eukaryotes through improving the fluidity of membrane.
The hadal zone had a relatively higher percentage of annotated genes than the surface zone (Figure 4B). The reason might be the higher diversity of both prokaryotes and eukaryotes in the shallow ocean compared to the deeper ocean (Nunoura et al., 2015; Jing et al., 2018; Tian et al., 2018; Xu et al., 2018), as many organisms cannot adapt to the extreme deep-sea environments. Of the five most abundant genes unique in the hadal zone, two (Top1 and hupB) were involved in DNA related processes like replication and transcription (Champoux, 2001; Gupta et al., 2014). Indeed, high pressure treatment was found to affect DNA structure in bacteria (Bartlett, 2002). Thus, these two unique genes might play a role in stabilizing DNA structure in microbial eukaryotes in the hadal zone, which needs further study.
Conclusion
This study investigated the distribution of microbial eukaryotes along the water column of the deepest known ocean. Stratification in community structure and shift of dominant taxonomies with depth were observed, indicating potential influence of depth-related environmental conditions on community composition. In addition, co-occurrence analysis showed that microbial interactions were also important for community composition. Intra-domain connectivity was higher than that between domains (bacteria and microbial eukaryotes), and clades with close phylogenetic relationships or similar trophic modes tend to coexist. Metagenome was firstly used for exploring the community-wide metabolic potentials of microbial eukaryotes in environmental samples, revealing the coordinating variations between metabolic characteristics and community composition. Metabolic changes like increased abundance of genes involved in the biosynthesis of unsaturated fatty acid, and abundant unique genes related to the formation and stabilization of DNA structure in hadal seawaters may help microbial eukaryotes adapt to high hydrostatic pressure. Our study is only a snapshot of microbial eukaryotic metabolism in the Mariana Trench. More efforts are needed to help improve our understanding of how microbial eukaryotes survive in the deep sea and respond to extreme conditions at the transcriptional level. Furthermore, this study indicated that with comprehensive databases metagenomics can be an important approach to studying microbial eukaryotic diversity and has potentials for exploring community-wide metabolic characteristics.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
X-HZ designed the experiments. X-YZ performed the experiments, analyzed the data, and wrote the draft. JL, C-XX, and JT provided great help on bioinformatics analyses and manuscript writing. All authors read and approved the final manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (91751202, 41730530, 41976101, and 92051115), National Key Research and Development Program of China (2018YFE0124100 and 2016YFA0601303), and National Program on Global Change and Air-Sea Interaction (GASI-02-PAC-ST-MSwin).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all of the scientists and crews on the R/V Dong Fang Hong 2 for their assistance with sampling during the cruise. We would also like to thank Hongan Long from Ocean University of China, Lingfeng Huang and Xin Guo from Xiamen University, Wuchang Zhang from Institute of Oceanology, Chinese Academy of Sciences and Jianming Chen from Minjiang University for their constructive comments that greatly contribute to the improvement of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.603692/full#supplementary-material
Footnotes
References
Agusti, S., Gonzalez-Gordillo, J. I., Vaque, D., Estrada, M., Cerezo, M. I., Salazar, G., et al. (2015). Ubiquitous healthy diatoms in the deep sea confirm deep carbon injection by the biological pump. Nat. commun. 6:7608. doi: 10.1038/ncomms8608
Allen, E. E., Facciotti, D., and Bartlett, D. H. (1999). Monounsaturated but not polyunsaturated fatty acids are required for growth of the deep-sea bacterium photobacterium profundum SS9 at high pressure and low temperature. Appl. Environ. Microbiol. 65, 1710–1720. doi: 10.1128/aem.65.4.1710-1720.1999
Arístegui, J., Gasol, J. M., Duarte, C. M., and Herndld, G. J. (2009). Microbial oceanography of the dark ocean’s pelagic realm. Limnol. Oceanogr. 54, 1501–1529. doi: 10.4319/lo.2009.54.5.1501
Atkins, M. S., Anderson, O. R., and Wirsen, C. O. (1998). Effect of hydrostatic pressure on the growth rates and encystment of flagellated protozoa isolated from a deep-sea hydrothermal vent and a deep shelf region. Mar. Ecol. Prog. Ser. 171, 85–95. doi: 10.3354/meps171085
Azaml, F., Fenche, T., Field, J., Gra, J., Meyer-Rei, L., and Thingstad, F. (1983). The ecological role of water-column microbes in the sea. Mar. Ecol. Prog. Ser. 10, 257–263. doi: 10.3354/meps010257
Bartlett, D. H. (2002). Pressure effects on in vivo microbial processes. Biochim. Biophys. Acta 1595, 367–381. doi: 10.1016/S0167-4838(01)00357-0
Behan, M. K., Macdonald, A. G., Jones, G. R., and Cossins, A. R. (1992). Homeoviscous adaptation under pressure: the pressure dependence of membrane order in brain myelin membranes of deep-sea fish. Biochim. Biophys. Acta 1103, 317–323. doi: 10.1016/0005-2736(92)90102-r
Bengtsson-Palme, J., Hartmann, M., Eriksson, K. M., Pal, C., Thorell, K., Larsson, D. G., et al. (2015). METAXA2: improved identification and taxonomic classification of small and large subunit rRNA in metagenomic data. Mol. Ecol. Resour. 15, 1403–1414. doi: 10.1111/1755-0998.12399
Bochdansky, A. B., Clouse, M. A., and Herndl, G. J. (2016). Eukaryotic microbes, principally fungi and labyrinthulomycetes, dominate biomass on bathypelagic marine snow. ISME J. 11, 362–373. doi: 10.1038/ismej.2016.113
Boenigk, J., and Arndt, H. (2002). Bacterivory by heterotrophic flagellates: community structure and feeding strategies. Antonie Van Leeuwenhoek 81, 465–480. doi: 10.1023/A:1020509305868
Boeuf, D., Edwards, B. R., Eppley, J. M., Hu, S. K., Poff, K. E., Romano, A. E., et al. (2019). Biological composition and microbial dynamics of sinking particulate organic matter at abyssal depths in the oligotrophic open ocean. Proc. Natl. Acad. Sci. U.S.A. 116, 11824–11832. doi: 10.1073/pnas.1903080116
Brown, M. V., Philip, G. K., Bunge, J. A., Smith, M. C., Bissett, A., Lauro, F. M., et al. (2009). Microbial community structure in the North Pacific ocean. ISME J. 3, 1374–1386. doi: 10.1038/ismej.2009.86
Burki, F. (2014). The eukaryotic tree of life from a global phylogenomic perspective. Cold Spring Harb. Perspect. 6:a016147. doi: 10.1101/cshperspect.a016147
Carradec, Q., Pelletier, E., Da Silva, C., Alberti, A., Seeleuthner, Y., Blanc-Mathieu, R., et al. (2018). A global ocean atlas of eukaryotic genes. Nat. commun. 9:373. doi: 10.1038/s41467-017-02342-1
Champoux, J. J. (2001). DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 70, 369–413. doi: 10.1146/annurev.biochem.70.1.369
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chong, P. L. G., Cossins, A. R., and Weber, G. (1983). A differential polarized phase fluorometric study of the effects of high hydrostatic pressure upon the fluidity of cellular membranes. Biochemistry 22, 409–415. doi: 10.1021/bi00271a026
Conway, J. R., Lex, A., and Gehlenborg, N. (2017). UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940. doi: 10.1093/bioinformatics/btx364
Countway, P. D., Gast, R. J., Dennett, M. R., Savai, P., Rose, J. M., and Caron, D. A. (2007). Distinct protistan assemblages characterize the euphotic zone and deep sea (2500 m) of the western North Atlantic (Sargasso Sea and Gulf Stream). Environ. Microbiol. 9, 1219–1232. doi: 10.1111/j.1462-2920.2007.01243.x
De Vargas, C., Audic, S., Henry, N., Decelle, J., Mahé, F., Logares, R., et al. (2015). Eukaryotic plankton diversity in the sunlit ocean. Science 348:1261605. doi: 10.1126/science.1261605I
DeLong, E. F., and Yayanos, A. A. (1985). Adaptation of the membrane lipids of a deep-sea bacterium to changes in hydrostatic pressure. Science 228, 1101–1103. doi: 10.1126/science.3992247
DeLong, E. F., and Yayanos, A. A. (1986). Biochemical function and ecological significance of novel bacterial lipids in deep-sea procaryotes. Appl. Environ. Microbiol. 51, 730–737. doi: 10.1128/AEM.51.4.730-737.1986
Eskelinen, E.-L., and Saftig, P. (2009). Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta 1793, 664–673. doi: 10.1016/j.bbamcr.2008.07.014
Fang, J., Barcelona, M. J., Nogi, Y., and Kato, C. (2000). Biochemical implications and geochemical significance of novel phospholipids of the extremely barophilic bacteria from the Marianas Trench at 11,000m. Deep Sea Res. I. 47, 1173–1182. doi: 10.1016/S0967-0637(99)00080-1
Faust, K., and Raes, J. (2012). Microbial interactions: from networks to models. Nat. Rev. Microbiol. 10:538. doi: 10.1038/nrmicro2832
Field, C. B., Behrenfeld, M. J., Randerson, J. T., and Falkowski, P. (1998). Primary production of the biosphere: integrating terrestrial and oceanic components. Science 281, 237–240. doi: 10.1126/science.281.5374.237
Finbow, M. E., and Harrison, M. A. (1997). The vacuolar H+-ATPase: a universal proton pump of eukaryotes. Biochem. J. 324, 697–712. doi: 10.1042/bj3240697
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gao, Z. M., Huang, J. M., Cui, G. J., Li, W. L., Li, J., Wei, Z. F., et al. (2019). In situ meta-omic insights into the community compositions and ecological roles of hadal microbes in the Mariana Trench. Environ. Microbiol. 21, 4092–4108. doi: 10.1111/1462-2920.14759
Gilbert, J. A., Steele, J. A., Caporaso, J. G., Steinbruck, L., Reeder, J., Temperton, B., et al. (2012). Defining seasonal marine microbial community dynamics. ISME J. 6, 298–308. doi: 10.1038/ismej.2011.107
Giner, C. R., Pernice, M. C., Balagué, V., Duarte, C. M., Gasol, J. M., Logares, R., et al. (2020). Marked changes in diversity and relative activity of picoeukaryotes with depth in the world ocean. ISME J. 14, 437–449. doi: 10.1038/s41396-019-0506-9
Gupta, M., Sajid, A., Sharma, K., Ghosh, S., Arora, G., Singh, R., et al. (2014). HupB, a nucleoid-associated protein of Mycobacterium tuberculosis, is modified by serine/threonine protein kinases in vivo. J. Bacteriol. 196, 2646–2657. doi: 10.1128/jb.01625-14
Hamilton, N. E., and Ferry, M. (2018). ggtern: ternary diagrams using ggplot2. J. Stat. Softw. 87, 1–17. doi: 10.18637/jss.v087.c03
Hopkinson, C. S., and Vallino, J. J. (2005). Efficient export of carbon to the deep ocean through dissolved organic matter. Nature 433, 142–145. doi: 10.1038/nature03191
Hu, S. K., Campbell, V., Connell, P., Gellene, A. G., Liu, Z., Terrado, R., et al. (2016). Protistan diversity and activity inferred from RNA and DNA at a coastal ocean site in the eastern North Pacific. FEMS Microbiol. Ecol. 92:fiw050. doi: 10.1093/femsec/fiw050
Hu, S. K., Liu, Z., Alexander, H., Campbell, V., Connell, P. E., Dyhrman, S. T., et al. (2018). Shifting metabolic priorities among key protistan taxa within and below the euphotic zone. Environ. Microbiol. 20, 2865–2879. doi: 10.1111/1462-2920.14259
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2015). eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293. doi: 10.1093/nar/gkv1248
Ichino, M. C., Clark, M. R., Drazen, J. C., Jamieson, A., Jones, D. O. B., Martin, A. P., et al. (2015). The distribution of benthic biomass in hadal trenches: a modelling approach to investigate the effect of vertical and lateral organic matter transport to the seafloor. Deep Sea Res. I. 100, 21–33. doi: 10.1016/j.dsr.2015.01.010
Jamieson, A. J. (2015). The Hadal Zone: Life in The Deepest Oceans. Cambridge: Cambridge University Press.
Jamieson, A. J., Fujii, T., Mayor, D. J., Solan, M., and Priede, I. G. (2010). Hadal trenches: the ecology of the deepest places on Earth. Trends Ecol. Evol. 25, 190–197. doi: 10.1016/j.tree.2009.09.009
Jiao, N., Luo, T., Zhang, R., Yan, W., Lin, Y., Johnson, Z., et al. (2014). Presence of Prochlorococcus in the aphotic waters of the western Pacific Ocean. Biogeosciences 11, 2391–2400. doi: 10.5194/bgd-10-9345-2013
Jing, H., Zhang, Y., Li, Y., Zhu, W., and Liu, H. (2018). Spatial variability of picoeukaryotic communities in the Mariana Trench. Sci. Rep. 8:15357. doi: 10.1038/s41598-018-33790-4
Johnson, L. K., Alexander, H., and Brown, C. T. (2019). Re-assembly, quality evaluation, and annotation of 678 microbial eukaryotic reference transcriptomes. GigaScience 8:giy158. doi: 10.1093/gigascience/giy158
Kato, M., Hayashi, R., Tsuda, T., and Taniguchi, K. (2002). High pressure-induced changes of biological membrane. Study on the membrane-bound Na(+)/K(+)-ATPase as a model system. Eur. J. Biochem. 269, 110–118. doi: 10.1046/j.0014-2956.2002.02621.x
Keeling, P. J., Burki, F., Wilcox, H. M., Allam, B., Allen, E. E., Amaral-Zettler, L. A., et al. (2014). The marine microbial eukaryote transcriptome sequencing project (MMETSP): illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PLoS Biol. 12:e1001889. doi: 10.1371/journal.pbio.1001889
Labarre, A., Obiol, A., Wilken, S., Forn, I., and Massana, R. (2020). Expression of genes involved in phagocytosis in uncultured heterotrophic flagellates. Limnol. Oceanogr. 65, S149–S160. doi: 10.1002/lno.11379
Levy Karin, E., Mirdita, M., and Söding, J. (2020). MetaEuk—sensitive, high-throughput gene discovery, and annotation for large-scale eukaryotic metagenomics. Microbiome 8:48. doi: 10.1186/s40168-020-00808-x
Li, D., Luo, R., Liu, C., Leung, C., Ting, H., Sadakane, K., et al. (2016). MEGAHIT v1. 0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11. doi: 10.1016/j.ymeth.2016.02.020
Lima-Mendez, G., Faust, K., Henry, N., Decelle, J., Colin, S., Carcillo, F., et al. (2015). Determinants of community structure in the global plankton interactome. Science 348:1262073. doi: 10.1126/science.1262073
Liu, J., Meng, Z., Liu, X., and Zhang, X. H. (2019a). Microbial assembly, interaction, functioning, activity and diversification: a review derived from community compositional data. Mar. Life Sci. Technol. 1, 112–128. doi: 10.1007/s42995-019-00004-3
Liu, J., Zheng, Y., Lin, H., Wang, X., Li, M., Liu, Y., et al. (2019b). Proliferation of hydrocarbon-degrading microbes at the bottom of the Mariana Trench. Microbiome 7:47. doi: 10.1186/s40168-019-0652-3
Liu, R., Wang, L., Wei, Y., and Fang, J. (2018). The hadal biosphere: recent insights and new directions. Deep Sea Res. II. 155, 11–18. doi: 10.1016/j.dsr2.2017.04.015
Mirdita, M., von den Driesch, L., Galiez, C., Martin, M. J., Söding, J., and Steinegger, M. (2017). Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 45, D170–D176. doi: 10.1093/nar/gkw1081
Not, F., Gausling, R., Azam, F., Heidelberg, J. F., and Worden, A. Z. (2007). Vertical distribution of picoeukaryotic diversity in the Sargasso Sea. Environ. Microbiol. 9, 1233–1252. doi: 10.1111/j.1462-2920.2007.01247.x
Nunoura, T., Takaki, Y., Hirai, M., Shimamura, S., Makabe, A., Koide, O., et al. (2015). Hadal biosphere: insight into the microbial ecosystem in the deepest ocean on Earth. Proc. Natl. Acad. Sci. U.S.A. 112, E1230–E1236. doi: 10.1073/pnas.1421816112
Obiol, A., Giner, C. R., Sánchez, P., Duarte, C. M., Acinas, S. G., and Massana, R. (2020). A metagenomic assessment of microbial eukaryotic diversity in the global ocean. Mol. Ecol. Resour. 20, 718–731. doi: 10.1111/1755-0998.13147
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P., O’hara, R., et al. (2016). Vegan: Community Ecology Package. Version 2.4-1. Available online at: http://CRAN.Rproject.org/package=vegan (accessed December 21, 2016).
Pasulka, A., Hu, S. K., Countway, P. D., Coyne, K. J., Cary, S. C., Heidelberg, K. B., et al. (2019). SSU-rRNA gene sequencing survey of benthic microbial eukaryotes from Guaymas Basin hydrothermal vent. J. Eukaryot. Microbiol. 66, 637–653. doi: 10.1111/jeu.12711
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. doi: 10.1038/nmeth.4197
Pernice, M. C., Forn, I., Gomes, A., Lara, E., Alonso-Sáez, L., Arrieta, J. M., et al. (2015a). Global abundance of planktonic heterotrophic protists in the deep ocean. ISME J. 9, 782–792. doi: 10.1038/ismej.2014.168
Pernice, M. C., Giner, C. R., Logares, R., Perera-Bel, J., Acinas, S. G., Duarte, C. M., et al. (2015b). Large variability of bathypelagic microbial eukaryotic communities across the world’s oceans. ISME J. 10, 945–958. doi: 10.1038/ismej.2015.170
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Settembre, C., Fraldi, A., Medina, D. L., and Ballabio, A. (2013). Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 14, 283–296. doi: 10.1038/nrm3565
Sherr, E. B., and Sherr, B. F. (1994). Bacterivory and herbivory: key roles of phagotrophic protists in pelagic food webs. Microb. Ecol. 28, 223–235. doi: 10.1007/BF00166812
Sherr, E. B., and Sherr, B. F. (2002). Significance of predation by protists in aquatic microbial food webs. Antonie van Leeuwenhoek 81, 293–308. doi: 10.1023/A:1020591307260
Steele, J. A., Countway, P. D., Xia, L., Vigil, P. D., Beman, J. M., Kim, D. Y., et al. (2011). Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME J. 5:1414. doi: 10.1038/ismej.2011.24I
Steinegger, M., Mirdita, M., and Söding, J. (2019). Protein-level assembly increases protein sequence recovery from metagenomic samples manyfold. Nat. Methods 16, 603–606. doi: 10.1101/386110
Stoeck, T., Hayward, B., Taylor, G. T., Varela, R., and Epstein, S. S. (2006). A multiple PCR-primer approach to access the microeukaryotic diversity in environmental samples. Protist 157, 31–43. doi: 10.1016/j.protis.2005.10.004
Stoecker, D. K., Hansen, P. J., Caron, D. A., and Mitra, A. (2017). Mixotrophy in the marine plankton. Ann. Rev. Mar. Sci. 9, 311–335. doi: 10.1146/annurev-marine-010816-060617
Taira, K., Yanagimoto, D., and Kitagawa, S. (2005). Deep CTD casts in the challenger deep. Mariana Trench. J. Oceanogr. 61, 447–454. doi: 10.1007/s10872-005-0053-z
Tian, J., Fan, L., Liu, H., Liu, J., Li, Y., Qin, Q., et al. (2018). A nearly uniform distributional pattern of heterotrophic bacteria in the Mariana Trench interior. Deep Sea Res. I. 142, 116–126. doi: 10.1016/j.dsr.2018.10.002
Turner, J. T. (2015). Zooplankton fecal pellets, marine snow, phytodetritus and the ocean’s biological pump. Prog. Oceanogr. 130, 205–248. doi: 10.1016/j.pocean.2014.08.005
Wang, Z., Liu, Z., Wang, Y., Bi, W., Liu, L., Wang, H., et al. (2019). Fungal community analysis in seawater of the Mariana Trench as estimated by Illumina HiSeq. RSC Adv. 9, 6956–6964. doi: 10.1039/c8ra10142f
Ward, B. A., and Follows, M. J. (2016). Marine mixotrophy increases trophic transfer efficiency, mean organism size, and vertical carbon flux. Proc. Natl. Acad. Sci. U.S.A. 113, 2958–2963. doi: 10.1073/pnas.1517118113
West, P. T., Probst, A. J., Grigoriev, I. V., Thomas, B. C., and Banfield, J. F. (2018). Genome-reconstruction for eukaryotes from complex natural microbial communities. Genome Res. 28, 569–580. doi: 10.1101/gr.228429.117
Worden, A. Z., Follows, M. J., Giovannoni, S. J., Wilken, S., Zimmerman, A. E., and Keeling, P. J. (2015). Rethinking the marine carbon cycle: factoring in the multifarious lifestyles of microbes. Science 347:1257594. doi: 10.1126/science.1257594
Xu, D., Li, R., Hu, C., Sun, P., Jiao, N., and Warren, A. (2017). Microbial eukaryote diversity and activity in the water column of the South China Sea based on DNA and RNA high throughput sequencing. Front. Microbiol. 8:1121. doi: 10.3389/fmicb.2017.01121
Xu, Z., Wang, M., Wu, W., Li, Y., Liu, Q., Han, Y., et al. (2018). Vertical distribution of microbial eukaryotes from surface to the hadal zone of the Mariana Trench. Front. Microbiol. 9:2023. doi: 10.3389/fmicb.2018.02023
Zhang, H., Yohe, T., Huang, L., Entwistle, S., Wu, P., Yang, Z., et al. (2018). dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–W101. doi: 10.1093/nar/gky418
Zheng, Y. F., Wang, J. Y., Zhou, S., Zhang, Y. H., Liu, J., Xue, C. X., et al. (2020). Bacteria are important dimethylsulfoniopropionate producers in marine aphotic and high-pressure environments. Nat. commun. 11:4658. doi: 10.1038/s41467-020-18434-4
Zhong, H., Lehtovirta-Morley, L., Liu, J., Zheng, Y., Lin, H., Song, D., et al. (2020). Novel insights into the Thaumarchaeota in the deepest oceans: their metabolism and potential adaptation mechanisms. Microbiome 8:78. doi: 10.1186/s40168-020-00849-2
Živaljić, S., Schoenle, A., Nitsche, F., Hohlfeld, M., Piechocki, J., Reif, F., et al. (2018). Survival of marine heterotrophic flagellates isolated from the surface and the deep sea at high hydrostatic pressure: literature review and own experiments. Deep Sea Res. II. 148, 251–259. doi: 10.1016/j.dsr2.2017.04.022
Keywords: Mariana Trench, microbial eukaryotes, metagenomics, metabolic potentials, diversity
Citation: Zhu X-Y, Liu J, Xue C-X, Tian J and Zhang X-H (2021) Shift and Metabolic Potentials of Microbial Eukaryotic Communities Across the Full Depths of the Mariana Trench. Front. Microbiol. 11:603692. doi: 10.3389/fmicb.2020.603692
Received: 07 September 2020; Accepted: 15 December 2020;
Published: 18 January 2021.
Edited by:
Fabiano Thompson, Alberto Luiz Coimbra Institute for Graduate Studies and Engineering Research, Federal University of Rio de Janeiro, BrazilReviewed by:
Yong Wang, Institute of Deep-Sea Science and Engineering (CAS), ChinaRulong Liu, Shanghai Ocean University, China
Copyright © 2021 Zhu, Liu, Xue, Tian and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-Hua Zhang, eGh6aGFuZ0BvdWMuZWR1LmNu
†These authors have contributed equally to this work