
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 08 January 2021
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.598291
 Congjuan Xu1†Huimin Liu2†
Congjuan Xu1†Huimin Liu2† Xiaolei Pan1Zhenzhen Ma1Dan Wang1Xinxin Zhang1Guangbo Zhu2
Xiaolei Pan1Zhenzhen Ma1Dan Wang1Xinxin Zhang1Guangbo Zhu2 Fang Bai1Zhihui Cheng1
Fang Bai1Zhihui Cheng1 Weihui Wu1
Weihui Wu1 Yongxin Jin1*
Yongxin Jin1*Treatment of infections by Pseudomonas aeruginosa is difficult due to its high intrinsic and acquired antibiotic resistance. Upon colonization in the human hosts, P. aeruginosa accumulates genetic mutations that confer the bacterium antibiotic resistance and ability to better live in the host environment. Characterizing the evolutionary traits would provide important insights into the development of effective combinatory antibiotic therapies to cure P. aeruginosa infections. In this work, we performed a detailed analysis of the molecular mechanisms by which a clinical isolate (CSP18) yields a ciprofloxacin-resistant derivative (CRP42). Genomic DNA re-sequencing and RNAseq were carried out to compare the genomic mutational signature and transcriptional profiles between the two isolates. The results indicated that D87G mutation in GyrA, together with MexEF-OprN hyper-expression caused by F7S mutation in MexS, was responsible for the increased resistance to ciprofloxacin in the isolate CRP42. Further simulation of CRP42 by gene editing in CSP18 demonstrated that D87G mutation in GyrA rendered CSP18 a fourfold increase in minimum inhibitory concentration against ciprofloxacin, while F7S mutation in MexS conferred an additional eightfold increase. Our experimental results demonstrate for the first time that the clinically relevant F7S point mutation in MexS results in hyper-expression of the mexEF-oprN and thus confers P. aeruginosa resistance to ciprofloxacin.
Pseudomonas aeruginosa is an opportunistic human pathogen and one of the leading causes of nosocomial infections all over the world (Vincent et al., 1995). Infections by P. aeruginosa are often challenging to cure due to its intrinsic and acquired resistance to a wide variety of antibiotics, leaving a limited number of effective antimicrobial agents. Ciprofloxacin is one of the most important antibiotics used for the treatment of P. aeruginosa infections (Andriole, 2005), including children and adults with cystic fibrosis (Doring et al., 2000). However, ciprofloxacin resistance of clinical P. aeruginosa isolates has been increasingly reported worldwide (Pitt et al., 2003; Landman et al., 2007). For example, 30% of clinical P. aeruginosa isolated from cystic fibrosis patients were ciprofloxacin resistant in a study from the United Kingdom (Pitt et al., 2003), while in a study from New York, it was 40% (Landman et al., 2007).
The mechanisms of ciprofloxacin resistance in P. aeruginosa are usually multifactorial, which include the following: (i) mutation of ciprofloxacin target-encoding genes gyrAB (encoding DNA gyrase) or/and parCE (encoding topoisomerase; Lee et al., 2005; Feng et al., 2019); (ii) overexpression of efflux pump encoding genes to increase the expulsion of ciprofloxacin from P. aeruginosa cells, including mexEF-oprN, mexAB-oprM, mexXY, and mexCD-oprJ (Kohler et al., 1997; Masuda et al., 2000; Rehman et al., 2019); and (iii) acquisition of ciprofloxacin-resistant genes through horizontal gene transfer (Chavez-Jacobo et al., 2018; Liu et al., 2018). Although these and other studies have associated the mechanisms of ciprofloxacin resistance among clinical P. aeruginosa strains, there is little information about the molecular details resulting in the evolutionary dynamics of clinical isolates of P. aeruginosa from ciprofloxacin susceptible to resistant, as well as the relative quantitative contribution of each of the resistant mechanisms mentioned above.
In this work, two clinical strains of P. aeruginosa were obtained from sputum samples of the same patient with ulcerative colitis before and after treatment first with cefoxitin for 7 days and then imipenem-cilastatin sodium and ciprofloxacin for another 8 days. The first strain was isolated soon after the patient was admitted to the hospital, while the second one was obtained 15 days after the antibiotics treatment. The first isolate, CSP18, was ciprofloxacin susceptible, whereas the latter one, CRP42, was ciprofloxacin resistant. Therefore, our aim was to characterize the molecular mechanisms for the ciprofloxacin resistance being developed in the clinical setting. The results presented in this study demonstrated that a D87G mutation in GyrA combined with hyper-expression of mexEF-oprN caused by an F7S mutation in MexS are the contributory factors for the conversion. Furthermore, we present experimental evidence that D87G mutation of GyrA contributes a fourfold increase in minimum inhibitory concentration (MIC) to ciprofloxacin, while MexEF-OprN hyper-expression caused by the F7S mutation in MexS contributes a further eightfold increase of MIC to ciprofloxacin in the CRP42 strain. These findings provide novel insights into the mexEF-oprN overexpression and ciprofloxacin resistance in P. aeruginosa.
Bacterial strains and plasmids used in this work are shown in Supplementary Table 1. Ciprofloxacin-susceptible (CSP18) and -resistant (CRP42) P. aeruginosa strains characterized here were isolated from sputum samples of a patient with ulcerative colitis before and after treatment with antibiotics for 15 days at the Nankai University Affiliated Hospital, Tianjin, China. The 16S rDNA was PCR amplified with primers (Supplementary Table 2) and sequenced to identify the species of the two isolates (Spilker et al., 2004). RAPD (Random amplified polymorphic DNA) typing was performed with primer 272 following a previous description (Mahenthiralingam et al., 1996). Multilocus sequence typing (MLST) was carried out as described previously with minor modification to confirm the allelic profiles of these two isolates (Curran et al., 2004). Briefly, chromosomal DNA, purified from overnight cultured bacteria with DNA purification kit (Tiangen Biotec, Beijing, China), was used as PCR template. The internal fragments of aroE, acsA, mutL, guaA, ppsA, nuoD, and trpE genes were PCR-amplified, sequenced using primers (Supplementary Table 2) as described previously and then the sequences were submitted to the P. aeruginosa MLST database1 to obtain the allelic numbers. A sequence type (ST) was assigned to each P. aeruginosa isolate by combination of the seven allelic numbers. MIC of antibiotics was examined by the twofold serial dilution method described previously except for the bacteria being grown in LB broth (Richardot et al., 2016), and susceptibility was interpreted based on the Clinical and Laboratory Standards Institute guidelines (CLSI 2011–2018).
For expression of gyrA, a 3338-bp gyrA-containing fragment with its putative promoter and SD (Shine-Dalgarno) sequence was amplified with CSP18 and CRP42 chromosomal DNA as templates (primers listed in Supplementary Table 2). The PCR products were treated with SacI and HindIII and then cloned into vector pUC18T-mini-Tn7T, leading to pUC18T-gyrACSP18 and pUC18T-gyrACRP42, respectively.
To delete the gyrA gene, a 924-bp fragment immediately upstream of the gyrA gene and an 836-bp fragment downstream of the gyrA were amplified (primers listed in Supplementary Table 2). The two fragments were digested with EcoRI–KpnI and KpnI–BamHI, respectively, and then ligated into pEX18Tc that was treated with EcoRI–BamHI, leading to pEX18-gyrA. To make the mexS (MexS-F7S) point mutation construct, F7S point mutated sites were included in the reverse primer of upstream and forward primer of downstream of mexS (primers shown in Supplementary Table 2), and the upstream and downstream fragments were digested with EcoRI–BglII and BglII–HindIII, and then ligated into pEX18Tc vector, leading to pEX18-mexS. Gene editing in P. aeruginosa was performed by conjugal transfer of these constructs and then selecting for single and double crossovers, as described previously (Schweizer, 1992).
Cultured P. aeruginosa strains were subcultured to an optical density of 1.0 (wavelength of 600 nm). RNA was purified with an RNAprep Pure Cell/Bacteria Kit (Tiangen Biotec, Beijing, China) and then reverse transcribed to cDNA with PrimeScript Reverse Transcriptase and random primers (Takara). The cDNA was added into the mixture of indicated qRT-PCR primers (shown in Supplementary Table 2) and SYBR premix Ex Taq II (Takara) and then quantitatively PCR amplified with a CFX Connect Real-time system (Bio-Rad, United States). A 30S ribosomal protein gene rpsL was utilized as an internal control.
The RNAseq analysis was carried out by GENEWIZ (Suzhou, China) as previously described (Xu et al., 2020). Briefly, total RNA was isolated from P. aeruginosa strains as described above, qualified and quantified by Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, United States), NanoDrop, and electrophoresis on 1% (wt/vol) agarose gel. After removing the rRNA by Ribo-Zero rRNA Removal Kit (Illumina for bacteria), the mRNA was fragmented and reverse transcribed. Then, the purified double-strand cDNA was subjected to end repair and ligation with adaptors. Following 11 cycles of amplification with PCR, the products were cleaned, validated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, United States), and further quantified with a Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, United States). The generating libraries were sequenced on an Illumina HiSeq 2500 platform with a 2 × 150 paired-end (PE) configuration.
Sequence reads were aligned to PAO1 reference genome (NC_002516.2) using software Bowtie2 (v2.1.0). RNA expressional levels were analyzed by HTSeq (v0.6.1p1). Genes with differential expression were identified using the DESeq Bioconductor package, with the fold change larger than 2 and P value no more than 0.05 as cutoff values.
Genomic DNA isolation and re-sequencing were carried out as described previously (Xu et al., 2020). Briefly, genomic DNA of P. aeruginosa strains was purified with DNA purification kit (Tiangen Biotec, Beijing, China). Two hundred nanograms of genomic DNA was used to generate fragments smaller than 500 bp with sonication (Covaris S220). After end treatment and adaptor ligation, the DNA fragments of about 470 bp were purified and amplified with PCR for six cycles. The PCR products were then cleaned up using beads, validated, and quantified using Qubit3.0 Fluorometer (Invitrogen, Carlsbad, United States). Sequencing was performed on an Illumina Hiseq instrument with a 2 × 150 PE configuration following the manufacturer’s instructions (Illumina, San Diego, United States). The sequences were mapped onto the PAO1 reference genome (NC_002516.2) with software BW2 (version 0.7.12). InDel mutation and single nucleotide variation (SNV) were analyzed via the Unified Genotyper module from GATK and the software Samtools (version 1.1).
The full-length mexS gene with its 85-bp upstream and 87-bp downstream region was PCR amplified using primers displayed in Supplementary Table 2, cloned into pUCP20. Similarly, the mexT gene with its 114-bp upstream and 77-bp downstream region was PCR amplified and cloned into pUCP20. After sequencing, the mexS or mexT gene sequence from CSP18 or CRP42 was aligned with the mexS or mexT gene of the PAO1 and PA14 reference strains2. For gyrA, the gyrA gene with its 467-bp upstream and 99-bp downstream region was PCR amplified, cloned into pUC18T-mini-Tn7T, and analyzed by sequencing. The gryA gene sequence from CSP18 or CRP42 was aligned with the gryA gene of the PAO1 reference strain (see text footnote 2).
The pyocyanin production of P. aeruginosa was determined as previously described with minor modification (Tan et al., 2016). Briefly, overnight bacterial culture was inoculated with 50-fold dilution and grown for 24 h. One-milliliter supernatant of each bacterial culture was extracted into 0.5 ml of chloroform, 0.4 ml of which was then re-extracted into 0.3 ml of HCl (0.2 N), and absorbance at 520 nm was then determined with Varioskan Flash (Thermo Scientific, Netherlands).
Rhamnolipid production was observed by growing bacteria on an M8-based agar plate as previously described (Chen et al., 2019). Bacteria were cultured in LB medium overnight and 1 μl of the culture was inoculated onto the plate and grown for 24 h at 37°C. After that, the plate was further kept at room temperature for more than 72 h until a blue halo appeared surrounding the colony. The relative production level of rhamnolipids was examined by measuring the diameter of each halo divided by the diameter of its colony.
Statistical analyses were performed with GraphPad software. The real-time qPCR results were analyzed by Student’s t test (two-tailed).
We have a waiver from the medical ethics committee of Tianjin Union Medical Center, exempting this work from the requirement for ethics approval and written informed consent as the clinical strains used in the study come from the routine procedures of the clinical laboratory rather than the clinical trials.
The datasets generated for the present work can be found in NCBI, under accession PRJNA638759.
Sputum samples from a patient with ulcerative colitis were collected before and after treatment first with cefoxitin for 7 days and then imipenem-cilastatin sodium and ciprofloxacin for another 8 days. Two isolates were obtained, one before the antibiotics treatment that was sensitive to ciprofloxacin (CSP18), and the other one after the antibiotics treatment that displayed resistance against the ciprofloxacin (CRP42; Table 1). PCR amplification and sequencing analysis of the 16S rDNAs demonstrated that they were both P. aeruginosa (Supplementary Figure 1A). The MLST analysis for both strains revealed an allelic profile for aroE, acsA, mutL, guaA, ppsA, nuoD and trpE as 5, 17, 98, 12, 14, 4 and 10, respectively, corresponding to the same ST, ST611. A RAPD typing was further carried out on the CSP18 and CRP42 strains and shown in Supplementary Figure 1B. MLST analysis and RAPD typing results indicated that they belonged to the same clone lineage (Diene et al., 2013; Wang et al., 2017; Fraile-Ribot et al., 2018).
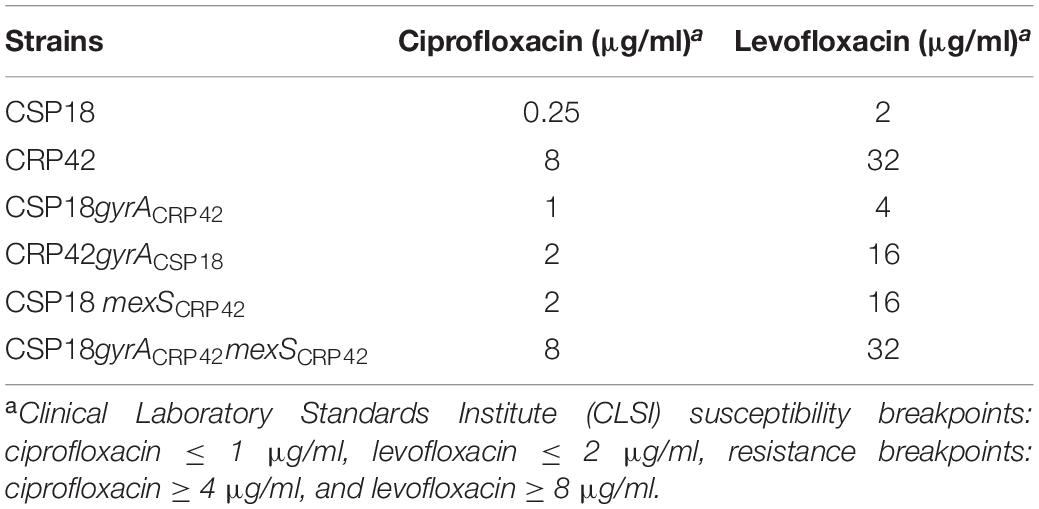
Table 1. MICs (μg/ml) of indicated P. aeruginosa strains.
Ciprofloxacin is commonly used to treat serious P. aeruginosa infections, and ciprofloxacin susceptibility of CSP18 and CRP42 was found different according to the preliminary analysis with VITEK automatic microbe analysis instrument (data not shown). Therefore, we further tested their susceptibility to ciprofloxacin by the twofold serial dilution method (Richardot et al., 2016). As shown in Table 1, initial isolate (CSP18) was found to be susceptible to ciprofloxacin, while the later isolate (CRP42) was resistant to the antibiotic, with a 32-fold increase in MIC of ciprofloxacin. Levofloxacin, another fluoroquinolone, was also examined. Similar to ciprofloxacin, CSP18 was susceptible to levofloxacin, while CRP42 showed resistance to levofloxacin, with a 16-fold increase in MIC of levofloxacin (Table 1). Besides, CRP42 displayed a reduced susceptibility to chloramphenicol, nalidixic acid, norfloxacin, and imipenem, and an increased susceptibility to gentamicin and amikacin, as well as a same susceptibility to tetracycline (Table 2).

Table 2. MICs (μg/ml) of CSP18 and CRP42 strains.
To elucidate the mechanism of reduced susceptibility to ciprofloxacin in isolate CRP42, genome re-sequencing was carried out to identify mutations in the genome of CRP42 relative to that of CSP18, using PAO1 as reference genome (see text footnote 2). Between strains CSP18 and CRP42, 17 genes had frameshifts (insertion/deletion), while 35 genes had non-synonymous SNV (Supplementary Table 3). Among them, the sequence of gyrA, encoding DNA gyrase subunit A, which has been reported to be involved in ciprofloxacin resistance (Higgins et al., 2003), was identical to that of PAO1 in CSP18, while in strain CRP42, an “A” was substituted by “G” at the 260th position, resulting in a D87G substitution in GyrA. To confirm the mutation, gyrA genes were amplified and cloned from the genomic DNA of strains CSP18 and CRP42. Sequencing analysis revealed that gyrA in CSP18 was the same as PAO1, while gyrA from CRP42 displayed a D87G substitution, which confirmed the genomic re-sequencing results. To test if the D87G substitution in GyrA contributed to the decreased susceptibility of the CRP42 to ciprofloxacin, the gyrACRP42 gene with its native promoter was introduced into the genome of CSP18 via pUC18T-mini-Tn7T plasmid and then the native gyrA was knocked out in CSP18 by chromosomal recombination. As shown in Table 1, replacement of the gyrACSP18 with gyrACRP42 resulted in a fourfold increase in MIC against ciprofloxacin in CSP18. Similarly, replacement of the gyrACRP42 with gyrACSP18 decreased the MIC of ciprofloxacin in CRP42 by fourfold. These results indicated that the D87G substitution in GyrA contributed to the reduced susceptibility to ciprofloxacin in strain CRP42 and also suggested the presence of other ciprofloxacin resistance mechanisms in CRP42.
To look for the other possible ciprofloxacin resistance mechanisms, we further compared the global gene expression profiles between strain CSP18 and CRP42. Expression levels of 48 genes were altered between the two strains (Supplementary Table 4). Among them, MexEF-OprN efflux pump encoding genes mexE, mexF, and oprN displayed 239-, 223-, and 169-fold higher mRNA levels, respectively, in CRP42 than those in CSP18 (Supplementary Table 4). To confirm the transcriptional up-regulation, the mRNA levels of mexEF-oprN operon were further examined and compared between CSP18 and CRP42 by real-time qPCR. As displayed in Figure 1, consistent with the RNAseq results, the relative mRNA level of mexE showed a 370-fold increase in the strain CRP42 compared to that in CSP18. To further elucidate the molecular mechanism of the reduced susceptibility to ciprofloxacin in CRP42, as well as the increased transcriptional levels of the mexEF-oprN operon, we further examined the genomic re-sequencing results. Among them, a T20C substitution in the mexS gene resulted in an F7S substitution in the MexS, a putative oxidoreductase of the CRP42 strain in comparison to that of CSP18 (Supplementary Table 3). This was further verified by sequencing of the PCR amplicons. To assess if the F7S substitution in MexS contributed to the increased mexEF-oprN expression and the decreased susceptibility to ciprofloxacin in CRP42, the chromosomal mexS gene was replaced by mexSCRP42 in the CSP18 strain. The mRNA levels of mexE were compared by real-time qPCR. As the results shown in Figure 1, the replacement by mexSCRP42 led to a 439-fold increase in the relative mRNA level of mexE in the CSP18 strain, while no significant change of mexE mRNA levels was observed with GyrA D87G substitution (Supplementary Figure 2F). To further confirm the increased expression of mexE gene, mexE-lacZ reporter construct was further introduced into the CSP18mexSCRP42 strain. Consistent with the real-time qPCR results, the mexS replacement rendered a significant increase in beta-galactosidase activity compared to that of CSP18 strain (Supplementary Figure 2A). Furthermore, as shown in Table 1, replacement of chromosomal MexS with F7S-substituted MexS conferred CSP18 an eightfold increased MIC against ciprofloxacin. These data demonstrated that the F7S substitution in MexS is the cause of the dramatic increase in the expression of mexEF-oprN seen in the CRP42 strain. Clearly, the F7S substitution disrupted the repressor function of the MexS. This is a new finding, since F7S point mutation in MexS has not been described to affect the expression of MexEF-OprN up to now.
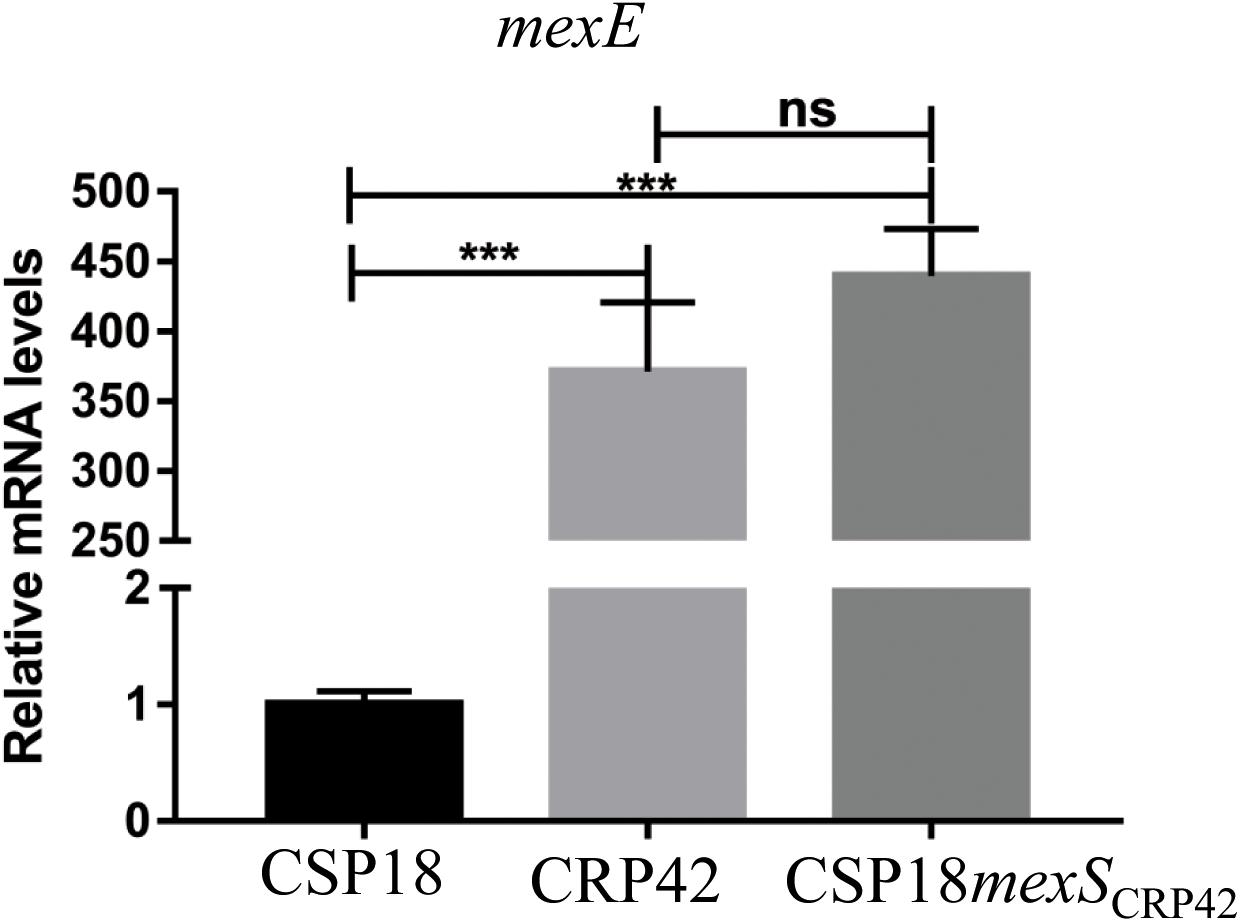
Figure 1. Transcriptional levels of mexE gene in indicated strains. Total RNA was extracted from indicated P. aeruginosa strains at OD600 of 1.0, and the relative mRNA levels of mexE were determined by real-time qPCR with rpsL as an internal control. ns, not significant, ***P < 0.001, by Student’s t test.
It has been demonstrated that mexT, located downstream of the mexS, encodes a LysR-family transcriptional activator for the mexEF-oprN (Kohler et al., 1997). We addressed if the MexT is involved in the up-regulation of mexEF-oprN and thus the reduced susceptibility to ciprofloxacin in CRP42. The mexT genes were PCR amplified from genomes of CSP18 and CRP42 and subjected to sequencing analysis. Consistent with the genomic re-sequencing results, mexT in strain CSP18 shares an identical sequence with that of CRP42, with an 8-bp deletion (CGGCCAGC) spanning from nucleotide 225 to 232 (104 to 111) in comparison to the reference strain PAO1 (see text footnote 2; PAO1 lab strain; Jin et al., 2011), while in comparison to PA14, 12 SNVs were observed in nucleotide sequence of mexT, resulting in no amino acid change of MexT in both CSP18 and CRP42 strains. Furthermore, no change at the transcriptional level of the mexT was observed between CSP18 and CRP42 (Supplementary Figure 2B), which is consistent with previous reports that the impact of mexS mutation on mexEF-oprN expression was not related to changes in MexT levels (e.g., Sobel et al., 2005; Richardot et al., 2016). These results suggested that MexT was the cause of neither the elevated mexEF-oprN transcription nor the increased resistance to ciprofloxacin in CRP42.
Besides MexEF-OprN, MexAB-OprM, MexCD-OprJ and MexXY are involved in ciprofloxacin resistance (Kohler et al., 1997; Masuda et al., 2000; Morita et al., 2012; Rehman et al., 2019). While as shown in Supplementary Figure 2, expression levels of mexAB-oprM, mexCD-oprJ and mexXY are the same between CSP18 and CRP42, and no mutation was found in these genes (data not shown). Thus, they are not responsible for the reduced susceptibility to ciprofloxacin in CRP42.
We further assessed if the D87G-substituted GyrA, together with the F7S-substituted MexS, was responsible for the reduced susceptibility to ciprofloxacin in strain CRP42. In the CSP18gyrACRP42 background, the mexSCSP18 gene was further replaced by mexSCRP42. As shown in Table 1, replacement of the mexSCSP18 with mexSCRP42 conferred CSP18gyrACRP42 strain (CSP18gyrACRP42mexSCRP42) an identical MIC against ciprofloxacin as the CRP42 strain. Replacement of gyrACSP18 with gyrACRP42 in CSP18 (CSP18gyrACRP42) resulted in a fourfold increase in MIC against ciprofloxacin (from 0.25 to 1.0 μg/ml), and the additional replacement of mexSCSP18 with mexSCRP42 (CSP18gyrACRP42mexSCRP42) conferred a further eightfold increase in the MIC (from 1 to 8 μg/ml; Table 1).
Our RNAseq results also revealed decreased transcriptional levels of phzM, phzA1-phzB1, phzG1, and phzS, and rhlA and rhlB (Supplementary Table 4), which are involved in pyocyanin and rhamnolipids production, respectively, (Mavrodi et al., 2001; Zhu and Rock, 2008). Accordingly, we examined and compared the pyocyanin and rhamnolipids production between CSP18 and CRP42 strains. As the results shown in Supplementary Figure 3, the CRP42 strain produced strongly decreased pyocyanin and rhamnolipids than the CSP18 strain. Furthermore, replacement of mexS by mexSCRP42 decreased the pyocyanin and rhamnolipids production of CSP18 almost to the levels of CRP42 (Supplementary Figure 3), while no obvious change of pyocyanin or rhamnolipids production was observed with gyrA substitution (Supplementary Figure 3).
Pseudomonas aeruginosa PAO1 genome encodes 12 RND-type efflux pumps (Stover et al., 2000). Among them, MexEF-OprN can exclude ciprofloxacin, chloramphenicol and trimethoprim from bacterial cell (Kohler et al., 1997). Its expression is controlled by multiple factors. mexT, located immediately upstream of and transcribed in the same direction as the mexEF-oprN, encodes a LysR family transcriptional regulator, capable of activating mexEF-oprN operon expression (Kohler et al., 1997). In the strain CSP18, mexT shares an identical sequence to that in CRP42, which has an 8-bp deletion (CGGCCAGC) at the 225th position in comparison to that of reference strain PAO1 (see text footnote 2). According to previous reports, in the absence of this 8-bp fragment, MexT is functional (Maseda et al., 2000; Richardot et al., 2016). Therefore, MexT is functional and responsible for the increased expression of mexEF-oprN due to the inactivation of MexS in CRP42.
mexS, located upstream of and activated by MexT, encodes a putative oxidoreductase/dehydrogenase homology (see text footnote 2). First of all, MexS was found to repress mexEF-orN expression independent of the changes in MexT levels in a clinical isolate of P. aeruginosa (Sobel et al., 2005). Consistent with that study, F7S substitution in MexS caused a hyper-expression of mexEF-oprN in CRP42, with no transcriptional level change in mexT. However, the effect of MexS on mexEF-oprN is complex. In our previous study with a lab strain PAO1, MexS positively regulated the expression of mexEF-oprN (Jin et al., 2011). A more recent study demonstrated that there are two separate pathways regulating the mexEF-oprN expression, a MexS-mediated and a MexS-bypassed pathway (Uwate et al., 2013). The complicated regulation of mexEF-oprN mediated by MexS may reflect the differences observed in the different P. aeruginosa strains. Of note, single point mutation in MexS (A155V/L263Q/S60P/A166P/C245G/F185L/S60F/D44E/F253L), resulting in partially or completely deficient MexS activity as well as mexEF-oprN overexpression, has recently been reported in clinical P. aeruginosa (Morita et al., 2015; Richardot et al., 2016). Our current study demonstrated that a novel single point mutation, resulting in F7S substitution in MexS, a putative oxidoreductase, caused a significant up-regulation of mexEF-oprN in both clinical P. aeruginosa strain CRP42 and CSP18mexSCRP42. In fact, the F7S substitution was predicted to be deleterious to the MexS’s function using the SIFT algorithm (data not shown)3 (Kumar et al., 2009). Consistent with previous reports that mutations in MexS decreased production of outer membrane protein OprD, concomitant with enhanced resistance to imipenem in P. aeruginosa (Sobel et al., 2005), CRP42 strain displayed a 3.6-fold decreased mRNA level of oprD (data not shown) and a reduced susceptibility to imipenem compared to CSP18 (Table 2).
Expression of mexEF-oprN is also regulated by MvaT, a global regulator of P. aeruginosa virulence genes. MvaT negatively regulates mexEF-oprN expression independent of mexT and mexS (Westfall et al., 2006). Besides, CmrA, an AraC-like transcriptional regulator, negatively controls mexEF-oprN expression through mexS and mexT (Juarez et al., 2017). MxtR, a sensor kinase, has recently been reported to repress mexEF-oprN via MexT in P. aeruginosa (Zaoui et al., 2012). In the present study, none of the MvaT, CmrA or MxtR encoding genes displayed different nucleotide sequences between CSP18 and CRP42 (data not shown), thus unlikely the cause of increased mexEF-oprN expression in CRP42.
It has been reported that MexEF-OprN can also export signal molecules of quorum sensing, and hyper-production of MexEF-OprN reduced production of several quorum sensing-dependent extracellular virulence factors, such as pyocyanin and rhamnolipids (Kohler et al., 2001). Consistent with the previous study, transcriptional levels of genes related to phenazine biosynthesis displayed 6.8- to 30-fold decreases in CRP42 (Supplementary Table 4), and production of pyocyanin in CRP42 was significantly decreased than that in CSP18 (Supplementary Figure 3). Similarly, the CRP42 strain displayed a 10-fold decreased transcriptional level in rhlA and rhlB, two rhamnolipids-related genes (Supplementary Table 4), and a strongly decreased production of rhamnolipids. Replacement by mexSCRP42 conferred CSP18 strain a CRP42-like expression level of mexEF-oprN (Figure 1), as well as production of pyocyanin and rhamnolipids (Supplementary Figure 3).
Previous studies have shown that T83I substitution in GyrA is the most common alteration associated with ciprofloxacin resistance in P. aeruginosa clinical isolates (Higgins et al., 2003; Lee et al., 2005; Pasca et al., 2012; Bruchmann et al., 2013). Substitutions of aspartate at position 87 with asparagine, tyrosine, or glycine residues have also been reported among ciprofloxacin-resistant clinical P. aeruginosa isolates (Higgins et al., 2003; Lee et al., 2005; Pasca et al., 2012; Bruchmann et al., 2013). Although the possible contribution of the D87G substitution in GyrA to ciprofloxacin resistance had been suggested, the quantitative contribution of the D87G substitution in ciprofloxacin resistance has not been examined for P. aeruginosa. Our study demonstrated that D87G mutation in GyrA renders a fourfold increase in MIC of ciprofloxacin.
The previous study used the well-known RND-type multidrug efflux pump inhibitor Phe-Arg-β-naphthylamide (PAβN) to study relative contribution of efflux pump up-regulation and target gene mutations to ciprofloxacin resistance in clinical P. aeruginosa and found that it was complex (Dunham et al., 2010). In the present study, we simulated the point mutation of genes gyrA and mexS in the ciprofloxacin-susceptible strain CSP18 by gene editing to its ciprofloxacin-resistant derivative and demonstrated the detailed quantitative contribution of the D87G point mutation of target gene gyrA and MexEF-OprN hyper-expression caused by F7S point mutation in MexS.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
YJ conceived and designed the experiments. CX, HL, XP, ZM, DW, XZ, and GZ performed the experiments. YJ, FB, ZC, and WW analyzed the data. YJ wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Science Foundation of China (31600110, 31870130, 31670130, 81670766, and 31970680), the Science and Technology Committee of Tianjin (17JCQNJC09200 and 19JCYBJC24700), the National Key Research and Development Project of China (2017YFE0125600), and the Science and Technology Program of Sichuan Province (2018JZ0069).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.598291/full#supplementary-material
Supplementary Figure 1 | PCR results of indicated strains. (A) 16S rDNA gene amplification; (B) RAPD typing of indicated strains.
Supplementary Figure 2 | Transcriptional levels of indicated genes in indicated strains. (A) β-galactosidase assay was conducted to determine the transcriptional activity of mexE gene promoter fused to a lacZ gene in indicated strains. (B–E) Relative mRNA levels of mexT (B), mexB (C), mexC (D), mexY (E), and mexE (F) in indicated strains; Total RNA was extracted from indicated P. aeruginosa strains at OD600 of 1.0, and the relative mRNA levels of indicated genes were determined by real-time qPCR with rpsL as an internal control. ns, not significant, ∗∗∗P < 0.001, by student’s t test.
Supplementary Figure 3 | Pyocyanin and rhamnolipids production by indicated strains. Overnight culture of indicated strains (A) were diluted 50-fold and grown for 24 h. (B) 1 mL supernatant of each bacterial culture was extracted into 0.5 mL of chloroform, then 0.4 mL of which was re-extracted into 0.3 mL of HCl (0.2N), and subjected to measurement at OD520. ns, not significant, ∗∗P < 0.01 by Student’s t test. (C) Rhamnolipids production were determined on the plate. 1 microliter culture of the indicated P. aeruginosa strain was inoculated onto the plate, grown at 37°C for 24 h and then at room temperature for another 72 h. The presence of a halo around the colonies suggests the production of rhamnolipids. The diameter ratio means the diameter of each halo divided by the diameter of its colony. The data shown represent means ± standard errors.
Supplementary Table 1 | Strains and plasmids used in this work.
Supplementary Table 2 | Primers used in the work.
Supplementary Table 3 | SNVs identified by genomic re-sequencing with elimination of SNVs with synonymous mutation and the same mutations between CSP18 and CRP42.
Supplementary Table 4 | Transcriptome analysis: differentially expressed genes by RNAseq.
Andriole, V. T. (2005). The quinolones: past, present, and future. Clin. Infect. Dis. 41(Suppl. 2), S113–S119. doi: 10.1086/428051
Bruchmann, S., Dotsch, A., Nouri, B., Chaberny, I. F., and Haussler, S. (2013). Quantitative contributions of target alteration and decreased drug accumulation to Pseudomonas aeruginosa fluoroquinolone resistance. Antimicrob. Agents Chemother. 57, 1361–1368. doi: 10.1128/aac.01581-1512
Chavez-Jacobo, V. M., Hernandez-Ramirez, K. C., Romo-Rodriguez, P., Perez-Gallardo, R. V., Campos-Garcia, J., Gutierrez-Corona, J. F., et al. (2018). CrpP is a novel ciprofloxacin-modifying enzyme encoded by the Pseudomonas aeruginosa pUM505 plasmid. Antimicrob. Agents Chemother. 62:e002629-17. doi: 10.1128/aac.02629-2617
Chen, R., Wei, X., Li, Z., Weng, Y., Xia, Y., Ren, W., et al. (2019). Identification of a small RNA that directly controls the translation of the quorum sensing signal synthase gene rhlI in Pseudomonas aeruginosa. Environ. Microbiol. 21, 2933–2947. doi: 10.1111/1462-2920.14686
Curran, B., Jonas, D., Grundmann, H., Pitt, T., and Dowson, C. G. (2004). Development of a multilocus sequence typing scheme for the opportunistic pathogen Pseudomonas aeruginosa. J. Clin. Microbiol. 42, 5644–5649. doi: 10.1128/jcm.42.12.5644-5649.2004
Diene, S. M., L’Homme, T., Bellulo, S., Stremler, N., Dubus, J. C., Mely, L., et al. (2013). ISPa46, a novel insertion sequence in the oprD porin gene of an imipenem-resistant Pseudomonas aeruginosa isolate from a cystic fibrosis patient in Marseille, France. Int. J. Antimicrob. Agents 42, 268–271. doi: 10.1016/j.ijantimicag.2013.06.001
Doring, G., Conway, S. P., Heijerman, H. G., Hodson, M. E., Hoiby, N., Smyth, A., et al. (2000). Antibiotic therapy against Pseudomonas aeruginosa in cystic fibrosis: a European consensus. Eur. Respir. J. 16, 749–767. doi: 10.1034/j.1399-3003.2000.16d30.x
Dunham, S. A., McPherson, C. J., and Miller, A. A. (2010). The relative contribution of efflux and target gene mutations to fluoroquinolone resistance in recent clinical isolates of Pseudomonas aeruginosa. Eur. J. Clin. Microbiol. Infect. Dis. 29, 279–288. doi: 10.1007/s10096-009-0852-z
Feng, X., Zhang, Z., Li, X., Song, Y., Kang, J., Yin, D., et al. (2019). Mutations in gyrB play an important role in ciprofloxacin-resistant Pseudomonas aeruginosa. Infect. Drug Resist. 12, 261–272. doi: 10.2147/idr.s182272
Fraile-Ribot, P. A., Cabot, G., Mulet, X., Periañez, L., Martín-Pena, M. L., Juan, C., et al. (2018). Mechanisms leading to in vivo ceftolozane/tazobactam resistance development during the treatment of infections caused by MDR Pseudomonas aeruginosa. J. Antimicrob. Chemother. 73, 658–663. doi: 10.1093/jac/dkx424
Higgins, P. G., Fluit, A. C., Milatovic, D., Verhoef, J., and Schmitz, F. J. (2003). Mutations in GyrA, ParC, MexR and NfxB in clinical isolates of Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 21, 409–413. doi: 10.1016/s0924-8579(03)00009-8
Jin, Y., Yang, H., Qiao, M., and Jin, S. (2011). MexT regulates the type III secretion system through MexS and PtrC in Pseudomonas aeruginosa. J. Bacteriol. 193, 399–410. doi: 10.1128/jb.01079-1010
Juarez, P., Jeannot, K., Plesiat, P., and Llanes, C. (2017). Toxic electrophiles induce expression of the multidrug efflux pump MexEF-OprN in Pseudomonas aeruginosa through a novel transcriptional regulator, CmrA. Antimicrob. Agents Chemother. 61:e00585-17. doi: 10.1128/aac.00585-17
Kohler, T., Michea-Hamzehpour, M., Henze, U., Gotoh, N., Curty, L. K., and Pechere, J. C. (1997). Characterization of MexE-MexF-OprN, a positively regulated multidrug efflux system of Pseudomonas aeruginosa. Mol. Microbiol. 23, 345–354. doi: 10.1046/j.1365-2958.1997.2281594.x
Kohler, T., van Delden, C., Curty, L. K., Hamzehpour, M. M., and Pechere, J. C. (2001). Overexpression of the MexEF-OprN multidrug efflux system affects cell-to-cell signaling in Pseudomonas aeruginosa. J. Bacteriol. 183, 5213–5222. doi: 10.1128/jb.183.18.5213-5222.2001
Kumar, P., Henikoff, S., and Ng, P. C. (2009). Prdicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081. doi: 10.1038/nprot.2009.86
Landman, D., Bratu, S., Kochar, S., Panwar, M., Trehan, M., Doymaz, M., et al. (2007). Evolution of antimicrobial resistance among Pseudomonas aeruginosa, Acinetobacter baumannii and Klebsiella pneumoniae in Brooklyn, NY. J. Antimicrob. Chemother. 60, 78–82. doi: 10.1093/jac/dkm129
Lee, J. K., Lee, Y. S., Park, Y. K., and Kim, B. S. (2005). Alterations in the GyrA and GyrB subunits of topoisomerase II and the ParC and ParE subunits of topoisomerase IV in ciprofloxacin-resistant clinical isolates of Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 25, 290–295. doi: 10.1016/j.ijantimicag.2004.11.012
Liu, J., Yang, L., Chen, D., Peters, B. M., Li, L., Li, B., et al. (2018). Complete sequence of pBM413, a novel multidrug resistance megaplasmid carrying qnrVC6 and blaIMP-45 from pseudomonas aeruginosa. Int. J. Antimicrob. Agents 51, 145–150. doi: 10.1016/j.ijantimicag.2017.09.008
Mahenthiralingam, E., Campbell, M. E., Foster, J., Lam, J. S., and Speert, D. P. (1996). Random amplified polymorphic DNA typing of Pseudomonas aeruginosa isolates recovered from patients with cystic fibrosis. J. Clin. Microbiol. 34, 1129–1135.
Maseda, H., Saito, K., Nakajima, A., and Nakae, T. (2000). Variation of the mexT gene, a regulator of the MexEF-oprN efflux pump expression in wild-type strains of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 192, 107–112. doi: 10.1111/j.1574-6968.2000.tb09367.x
Masuda, N., Sakagawa, E., Ohya, S., Gotoh, N., Tsujimoto, H., and Nishino, T. (2000). Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-oprM efflux pumps in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 44, 3322–3327. doi: 10.1128/aac.44.12.3322-3327.2000
Mavrodi, D. V., Bonsall, R. F., Delaney, S. M., Soule, M. J., Phillips, G., and Thomashow, L. S. (2001). Functional analysis of genes for biosynthesis of pyocyanin and phenazine-1-carboxamide from Pseudomonas aeruginosa PAO1. J. Bacteriol. 183, 6454–6465. doi: 10.1128/jb.183.21.6454-6465.2001
Morita, Y., Tomida, J., and Kawamura, Y. (2012). Primary mechanisms mediating aminoglycoside resistance in the multidrug-resistant Pseudomonas aeruginosa clinical isolate PA7. Microbiology 158(Pt 4), 1071–1083. doi: 10.1099/mic.0.054320-54320
Morita, Y., Tomida, J., and Kawamura, Y. (2015). Efflux-mediated fluoroquinolone resistance in the multidrug-resistant Pseudomonas aeruginosa clinical isolate PA7: identification of a novel MexS variant involved in upregulation of the mexEF-oprN multidrug efflux operon. Front. Microbiol. 6:8. doi: 10.3389/fmicb.2015.00008
Pasca, M. R., Dalla Valle, C., De Jesus Lopes, O., Ribeiro, A. L., Buroni, S., Papaleo, M. C., et al. (2012). Evaluation of fluoroquinolone resistance mechanisms in Pseudomonas aeruginosa multidrug resistance clinical isolates. Microb. Drug Resist. 18, 23–32. doi: 10.1089/mdr.2011.0019
Pitt, T. L., Sparrow, M., Warner, M., and Stefanidou, M. (2003). Survey of resistance of Pseudomonas aeruginosa from UK patients with cystic fibrosis to six commonly prescribed antimicrobial agents. Thorax 58, 794–796. doi: 10.1136/thorax.58.9.794
Rehman, A., Patrick, W. M., and Lamont, I. L. (2019). Mechanisms of ciprofloxacin resistance in Pseudomonas aeruginosa: new approaches to an old problem. J. Med. Microbiol. 68, 1–10. doi: 10.1099/jmm.0.000873
Richardot, C., Juarez, P., Jeannot, K., Patry, I., Plesiat, P., and Llanes, C. (2016). Amino acid substitutions account for most MexS alterations in clinical nfxC mutants of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 60, 2302–2310. doi: 10.1128/aac.02622-2615
Schweizer, H. P. (1992). Allelic exchange in Pseudomonas aeruginosa using novel ColE1-type vectors and a family of cassettes containing a portable oriT and the counter-selectable Bacillus subtilis sacB marker. Mol. Microbiol. 6, 1195–1204. doi: 10.1111/j.1365-2958.1992.tb01558.x
Sobel, M. L., Neshat, S., and Poole, K. (2005). Mutations in PA2491 (mexS) promote MexT-dependent mexEF-oprN expression and multidrug resistance in a clinical strain of Pseudomonas aeruginosa. J. Bacteriol. 187, 1246–1253. doi: 10.1128/jb.187.4.1246-1253.2005
Spilker, T., Coenye, T., Vandamme, P., and LiPuma, J. J. (2004). PCR-based assay for differentiation of Pseudomonas aeruginosa from other Pseudomonas species recovered from cystic fibrosis patients. J. Clin. Microbiol. 42, 2074–2079.
Stover, C. K., Pham, X. Q., Erwin, A. L., Mizoguchi, S. D., Warrener, P., Hickey, M. J., et al. (2000). Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406, 959–964. doi: 10.1038/35023079
Tan, H., Zhang, L., Zhao, Q., Chen, R., Liu, C., Weng, Y., et al. (2016). DeaD contributes to Pseudomonas aeruginosa virulence in a mouse acute pneumonia model. FEMS Microbiol. Lett. 363:fnw227. doi: 10.1093/femsle/fnw227
Uwate, M., Ichise, Y. K., Shirai, A., Omasa, T., Nakae, T., and Maseda, H. (2013). Two routes of MexS-MexT-mediated regulation of MexEF-OprN and MexAB-OprM efflux pump expression in Pseudomonas aeruginosa. Microbiol. Immunol. 57, 263–272. doi: 10.1111/1348-0421.12032
Vincent, J. L., Bihari, D. J., Suter, P. M., Bruining, H. A., White, J., Nicolas-Chanoin, M. H., et al. (1995). The prevalence of nosocomial infection in intensive care units in Europe. Results of the european prevalence of infection in intensive care (EPIC) Study. EPIC international advisory committee. JAMA 274, 639–644.
Wang, K., Chen, Y. Q., Salido, M. M., Kohli, G. S., Kong, J. L., Liang, H. J., et al. (2017). The rapid in vivo evolution of Pseudomonas aeruginosa in ventilator-associated pneumonia patients leads to attenuated virulence. Open Biol. 7:170029. doi: 10.1098/rsob.170029
Westfall, L. W., Carty, N. L., Layland, N., Kuan, P., Colmer-Hamood, J. A., and Hamood, A. N. (2006). mvaT mutation modifies the expression of the Pseudomonas aeruginosa multidrug efflux operon mexEF-oprN. FEMS Microbiol. Lett. 255, 247–254. doi: 10.1111/j.1574-6968.2005.00075.x
Xu, C., Wang, D., Zhang, X., Liu, H., Zhu, G., Wang, T., et al. (2020). Mechanisms for rapid evolution of carbapenem resistance in a clinical isolate of Pseudomonas aeruginosa. Front. Microbiol. 11:1390. doi: 10.3389/fmicb.2020.01390
Zaoui, C., Overhage, J., Lons, D., Zimmermann, A., Musken, M., Bielecki, P., et al. (2012). An orphan sensor kinase controls quinolone signal production via MexT in Pseudomonas aeruginosa. Mol. Microbiol. 83, 536–547. doi: 10.1111/j.1365-2958.2011.07947.x
Keywords: Pseudomonas aeruginosa, ciprofloxacin resistance, mexS, gyrA, MexEF-OprN
Citation: Xu C, Liu H, Pan X, Ma Z, Wang D, Zhang X, Zhu G, Bai F, Cheng Z, Wu W and Jin Y (2021) Mechanisms for Development of Ciprofloxacin Resistance in a Clinical Isolate of Pseudomonas aeruginosa. Front. Microbiol. 11:598291. doi: 10.3389/fmicb.2020.598291
Received: 24 August 2020; Accepted: 24 November 2020;
Published: 08 January 2021.
Edited by:
Yuji Morita, Meiji Pharmaceutical University, JapanReviewed by:
Catherine Llanes, University of Franche-Comté, FranceCopyright © 2021 Xu, Liu, Pan, Ma, Wang, Zhang, Zhu, Bai, Cheng, Wu and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongxin Jin, eXhqaW5AbmFua2FpLmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.