 Jéssyca Freitas-Silva1†
Jéssyca Freitas-Silva1† Bruno Francesco Rodrigues de Oliveira1,2†
Bruno Francesco Rodrigues de Oliveira1,2† Felipe de Mello Vigoder3
Felipe de Mello Vigoder3 Guilherme Muricy4
Guilherme Muricy4 Alan D. W. Dobson2,5*
Alan D. W. Dobson2,5* Marinella Silva Laport1*
Marinella Silva Laport1*- 1Institute of Microbiology Paulo de Góes, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
- 2School of Microbiology, University College Cork, Cork, Ireland
- 3Department of Genetics, Institute of Biology, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
- 4Department of Invertebrates, National Museum, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
- 5Environmental Research Institute, University College Cork, Cork, Ireland
Bacillus pumilus 64-1, a bacterial strain isolated from the marine sponge Plakina cyanorosea, which exhibits antimicrobial activity against both pathogenic and drug-resistant Gram-positive and Gram-negative bacteria. This study aimed to conduct an in-depth genomic analysis of this bioactive sponge-derived strain. The nearly complete genome of strain 64-1 consists of 3.6 Mbp (41.5% GC), which includes 3,705 coding sequences (CDS). An open pangenome was observed when limiting to the type strains of the B. pumilus group and aquatic-derived B. pumilus representatives. The genome appears to encode for at least 12 potential biosynthetic gene clusters (BGCs), including both types I and III polyketide synthases (PKS), non-ribosomal peptide synthetases (NRPS), and one NRPS-T1PKS hybrid, among others. In particular, bacilysin and other bacteriocin-coding genes were found and may be associated with the detected antimicrobial activity. Strain 64-1 also appears to possess a broad repertoire of genes encoding for plant cell wall-degrading carbohydrate-active enzymes (CAZymes). A myriad of genes which may be involved in various process required by the strain in its marine habitat, such as those encoding for osmoprotectory transport systems and the biosynthesis of compatible solutes were also present. Several heavy metal tolerance genes are also present, together with various mobile elements including a region encoding for a type III-B Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) region, four prophage segments and transposase elements. This is the first report on the genomic characterization of a cultivable bacterial member of the Plakina cyanorosea holobiont.
Introduction
The symbiotic relationship between marine invertebrates and microorganisms is among the oldest, most faithfully maintained, and functionally sophisticated within the realm of animal microbiomes (Petersen and Osvatic, 2018). The increased availability of -omics data continues to provide valuable information on the microbial ecology of bacterial sponge communities and their complex microbial-host interactions (Thomas et al., 2016; Moitinho-Silva et al., 2017). Nevertheless, it is also still important to isolate and genetically characterize sponge-associated bacteria to gain further insights into their role within the overall functioning of the sponge holobiont (Laport, 2018; Pita et al., 2018) as well as their potential to produce bioactive compounds with pharmaceutical applications, together with enzymes and biosurfactants with industrial utility (Santos-Gandelman et al., 2014).
From a biotechnological perspective, sponge-derived cultivable members of the phyla Proteobacteria, Actinobacteria and Firmicutes, such as Pseudovibrio (O’Halloran et al., 2011; Buijs et al., 2019), Streptomyces (Jackson et al., 2018; Yang et al., 2019) and Bacillus (Phelan et al., 2014), have been the main microbial sources targeted for the isolation and characterization of bioactive compounds. Indeed, Bacillus species are well-known producers of secondary metabolites, namely non-ribosomal peptides, lanthipeptides and, in particular; a wide variety of bacteriocins (Ortiz and Sansinenea, 2019). In addition, a diverse range of antibacterial and antifungal substances, together with enzymes and inhibitors, biosurfactants and compounds with bioremediation, bioleaching or cytotoxic potentials have been isolated from sponge-derived Bacillus (Santos-Gandelman et al., 2014; Indraningrat et al., 2016; Laport, 2018; Ortiz and Sansinenea, 2019).
Following a screening regime of bacteria isolated from the recently described marine sponge Plakina cyanorosea (Muricy et al., 2019), a Bacillus pumilus strain, identified as the number 64-1, displayed antimicrobial activity against multidrug-resistant (MDR) and medically relevant bacterial strains (Freitas-Silva et al., 2020). This motivated us to move forward with the genome sequencing of this bacterial strain to better access its potential to produce bioactive microbial metabolites. In this study, we explored the B. pumilus 64-1 genome isolated from P. cyanorosea to identify biosynthetic gene clusters (BGCs) that may be associated with its strong antimicrobial potential. Through an ecological perspective, we also explored the relevant genes potentially involved with survival in the marine environment. In addition, other biotechnological potentialities were also uncovered in this sponge-derived Bacillus strain.
Materials and Methods
Bacterial Strain
Bacillus pumilus 64-1 was isolated from the marine sponge P. cyanorosea as previously described (Freitas-Silva et al., 2020). Briefly, the sponge sample was collected manually, washed with artificial sea water [NaCl 2.34% (w/v), MgSO4.7H2O 0.49% (w/v), MgCl2.6H2O 0.4% (w/v), CaCl2.H2O 0.15% (w/v), KCl 0.075% (w/v), NaHCO3 0.017% (w/v)] and immediately transferred into brain heart infusion (BHI). The sample was processed under aseptic conditions by initial maceration with glass beads. Serial dilutions (10–3 to 10–5) were prepared from sponge macerate and 100 μL aliquots of each dilution were plated in duplicate onto the BHI agar. The plates were incubated for up to seven days at 25°C and the colonies were isolated, purified, and cryopreserved at −20°C for further analysis.
DNA Extraction
Genomic DNA was extracted from a pure culture of B. pumilus 64-1 following growth for 24 h on BHI medium using a modified guanidine method (Pitcher et al., 1989). These adaptations include: before the addition of lysozyme, the cells were centrifuged at 8,000 × g for 15 min and washed in Tris–EDTA (TE) buffer 1X (pH 8.0); thereafter, the pellet was submitted to new centrifugation at 8,000 × g for 5 min; and detergent polyoxyethylene lauryl ether (BRIJ) solution and chloroform-isoamyl alcohol were used instead of sarkosyl and two-propanol from the original method. The quality and purity of the DNA were checked on 0.8% (w/v) agarose gel electrophoresis, while the quantity was determined using NanoVueTM Plus Spectrophotometer (GE Healthcare, United States). The extraction of plasmid DNA was performed with the Wizard® Plus SV Minipreps DNA Purification System Protocol kit with lysozyme (20 mg/ml), in accordance with the manufacturer’s instructions. Klebsiella pneumoniae Kp13 plasmids (Ramos et al., 2014) were used as a marker of the plasmid’s molecular size. Plasmid DNA profiles were observed on 0.8% agarose gel electrophoresis with a UV transilluminator.
Genome Sequencing and Assembly
A workflow detailing the bioinformatic tools adopted for the assembly and annotation of the B. pumilus 64-1 genome is presented in the Supplementary Figure 1. The fragmentation of genomic DNA and preparation of the sequencing library were achieved using a Nextera XT Sample Preparation, Nextera XT Index kits (Illumina, United States). Following purification with Agencourt AMPure XP beads (Beckman Coulter, United States) and library quantification by qPCR (KAPA-KK4824 - Illumina/Universal), whole-genome sequencing was performed on the Illumina NextSeq 500 System, with the NextSeq 500 MID Output kit (300 cycles). The quality of the generated raw paired-end reads was checked by the A5-miseq pipeline tool (Coil et al., 2015) and monitored with the FastQC program (Andrews, 2010). After trimming and the removal of adaptors/barcode sequences, de novo genome assembly was achieved using SPAdes 3.13.0 (Bankevich et al., 2012) and evaluated with the QUAST 5.0.2 tool (Gurevich et al., 2013). Contigs with length equal or inferior to 400 bp were removed from the scaffolds output archive. Completeness and contamination were then determined with the CheckM v1.0.18 software (Parks et al., 2015).
Gene Prediction and Functional Annotation
The overall structural annotation was performed using the myRAST application (Overbeek et al., 2013) using reference genomes that are phylogenetically close to the genome to be annotated and the DDBJ Fast Annotation and Submission Tool (DFAST) that allows the detection of coding sequences (CDS), ribosomal (rRNA) and transporter (tRNA) RNA genes (Tanizawa et al., 2018). The translated protein sequences were submitted for functional categorization as clusters of orthologous groups (COG) within the eggNOG database (Huerta-Cepas et al., 2019). The eggNOG functional annotation was further used for validation of the explored ecological- and biotechnologically relevant genes. SignalP-4.1 (Nielsen, 2017) and TMHMM v.2.0 (Krogh et al., 2001) were chosen for the identification of signal peptides and transmembrane helices in the predicted proteins. PSORTb 3.0.2 was applied to infer their subcellular localization (Yu et al., 2010). The presence of putative plasmid sequences was carried on with PlasmidFinder 2.0 (Carattoli et al., 2014).
The identification, annotation and analysis of BGCs were performed using the antibiotics and Secondary Metabolite Analysis SHell (antiSMASH) version 5.0.0 beta (Blin et al., 2019). Moreover, the BAGEL4 webserver was used for automated identification of genes encoding for bacteriocins and (non-) bactericidal post-translationally modified peptides, with default parameters (van Heel et al., 2018).
Carbohydrate-active enzymes (CAZymes) were identified and annotated with HMMER, DIAMOND and Hotpep algorithms from the dbCAN2 metaserver (Zhang et al., 2018). HMMER webserver 2.40 based searches were performed using the phmmer parameter (Potter et al., 2018) against the MEROPS complete sequence library (Rawlings et al., 2018) to track peptidase and their inhibitors.
Groups of CDS with potential roles with survival in marine habitats, such as osmoregulatory transport systems and biosynthetic enzymes for compatible solutes (Jackson et al., 2018), were identified in the eggNOG functional annotation files. The detected transporter systems were then additionally interrogated using the Transporter Classification Database (TCDB) (Saier et al., 2016) and the Archaeal and Bacterial ABC Systems Database (ABCdb) (Fichant et al., 2006).
Acquired antimicrobial resistance genes (ARG) and/or chromosomal mutations were scanned automatically with the Resistance Gene Identifier tool (RGI 5.1.0) from the Comprehensive Antibiotic Resistance Database (CARD 3.0.7) (Alcock et al., 2020), narrowing the criteria for “perfect and strict hits only” and the sequence quality for “high quality/coverage”. Putative genes for modification or detoxification enzymes acting specifically on antimicrobials from different classes and dedicated to metal tolerance were manually identified from the eggNOG annotation results.
The identification of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) and cas genes was performed with the CRISPRCasFinder program (Couvin et al., 2018). BLASTn searches (Altschul et al., 1990) were performed with the identified direct repeats (DR) regions. The Artemis version 16.0.0 (Carver et al., 2008) was then used for the annotation and visualization of the genomic organization of the detected CRISPR regions and, finally, the RNAfold web server program was applied to reconstruct the putative secondary structures of the DR sequences (Lorenz et al., 2011). IslandViewer 4 (Bertelli et al., 2017) was employed for the integrated prediction of genomic islands, with the complete chromosomes of B. pumilus NCTCC 10337 (=ATCC 7061T) (NZ_LT906438.1) and B. pumilus PDSLzg-1 (NZ_CP016784.1) used as references to reorder the contigs from the B. pumilus 64-1 draft genome. The identification of prophage elements was performed with the PHASTER webserver (Arndt et al., 2016). Insertion sequences (ISs) were initially detected using the BLASTn searches against the ISfinder database (Siguier et al., 2006). For the additional detection of potential mobilome elements, manual curation was also performed for the localization of the keywords “phage,” “transponsase,” “integrase,” and “insertion” through the eggNOG annotation files.
Phylogenetic Analyses
The complete 16S rRNA sequence (1,548 bp) of the B. pumilus 64-1 strain was recovered from its draft genome (section “Gene Prediction and Functional Annotation”). For further phylogenetic analyses, we prioritized the 16S rRNA sequences of closely related species/representatives with identity scores ≥98.7% (Rosselló-Móra and Amann, 2015) and from valid type strains from the B. pumilus clade: B. pumilus ATCC 7061T (NR_043242.1), Bacillus safensis NBRC100820 (AB681259.1), Bacillus zhangzhouensis MCCC 1A05787 (JX680098.1), Bacillus altitudinis 41KF2bT (ASJC01000029.1), Bacillus australimaris MCCC 1A05787 (JX680098.1), Bacillus xiamenensis MCCC 1A00008 (JX680066.1). The 16S rRNA gene sequences from aquatic-derived B. pumilus strains with deposited genome sequences (Table 1) were also retrieved from the National Center for Biotechnology Information (NCBI) GenBank database and incorporated for this analysis: B. pumilus 145 (CP027116.1), B. pumilus 150a (CP027034.1), B. pumilus PDSLzg-1 (CP016784.1), B. pumilus PE09-72 (NZ MIJA01000046.1), B. pumilus RI06-95 (NZ_LFGZ01000009.1), B. pumilus Ha06YP001 (NZ PTXV01000013.1) and B. pumilus SF214 (FJ977607.1).
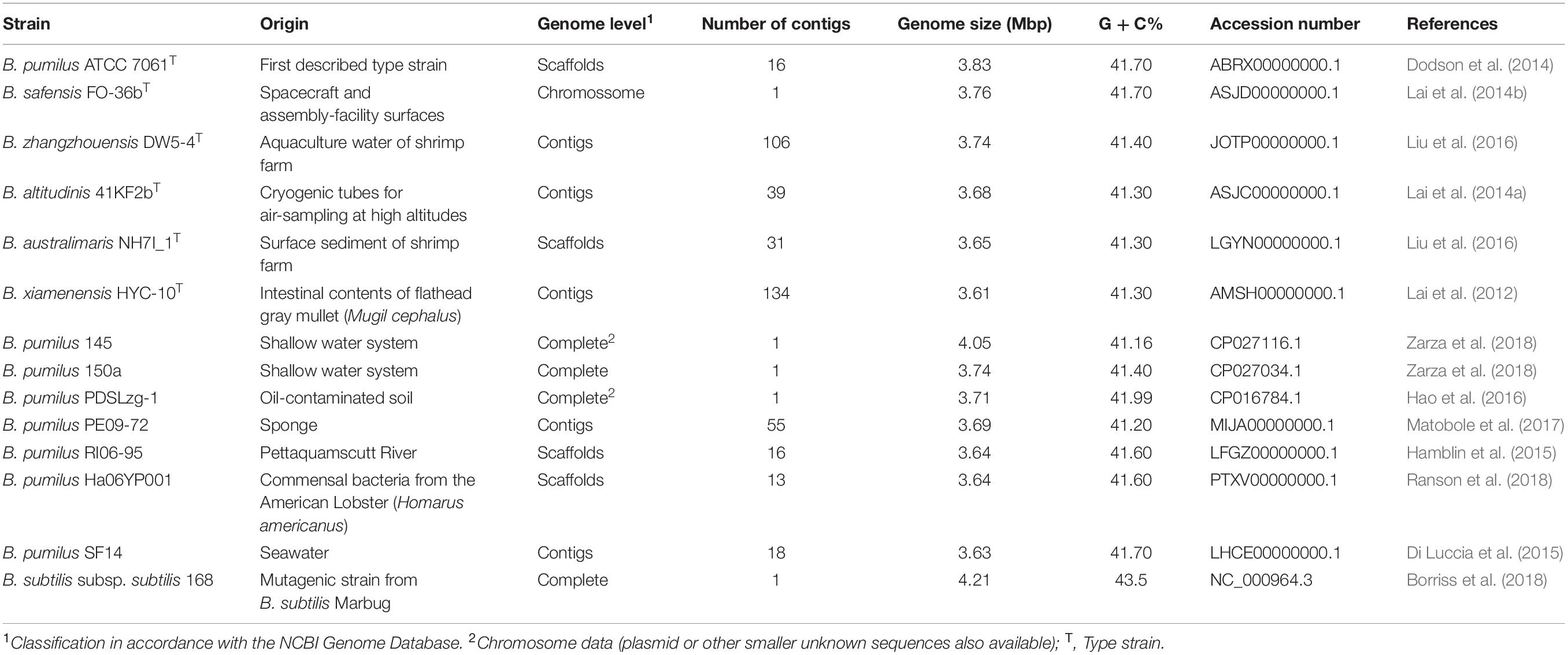
Table 1. List of Bacillus genomes selected for phylogenetic analyses and the overall genome-related index (OGRIs) calculation.
Among seven different housekeeping genes, Liu et al. (2013) highlighted the gyrase B-coding gene as a valuable alternative for the assessment of the phylogenetic diversity of bacteria in the B. pumilus clade, when analyzing marine and terrestrial-derived strains. In this sense, it is important to use at least another reliable marker gene for accurate phylogenetic inference, particularly for the B. pumilus clade, whose taxonomy has been extensively restructured within the last few years (Branquinho et al., 2015; Liu et al., 2015, 2016). Owing to this confirmed higher resolution of the gyrB housekeeping gene in distinguishing marine strains from the B. pumilus group (Liu et al., 2013), we performed the phylogenetic analyses with the gyrB sequences from the members of this clade and the marine-derived B. pumilus strains: B. pumilus ATCC 7061T (ABRX01000004.1), B. safensis F0-036b (AY167867.1), B. zhangzhouensis DW5-4T (JOTP01000022.1), B. altitudinis 41KF2bT (ASJC01000015.1), B. australimaris NH7I 1 (LGYN01000023.1), B. xiamenensis HYC-10T (AMSH01000062.1), B. pumilus 145 (CP027116.1), B. pumilus 150a (CP027034.1), B. pumilus PDSLzg-1 (CP016784.1), B. pumilus PE09-72 (NZ MIJA01000032.1), B. pumilus RI06-95 (NZ_LFGZ01000003.1), B. pumilus Ha06YP001 (NZ PTXV01000002.1), and B. pumilus SF214 (LHCE01000018.1).
These sequences were all aligned with the ClustalW tool within the interface of the MEGAX software (Kumar et al., 2018). The neighbor-joining (NJ) method (Saitou and Nei, 1987) was selected for the 16S rRNA coding sequence and the maximum likelihood (ML) method (Felsenstein, 1981) for the gyrB marker. Following the best-fit substitution model, NJ and ML trees were constructed using Tamura-Nei (Tamura and Nei, 1993) and Kimura-2 (Kimura, 1980) models, respectively. Tree topologies were guaranteed by the bootstrap test with 1000 (NJ) and 100 replicates (ML). Four 16S rRNA gene sequences from different microbial strains were used as outgroups: Bacillus subtilis subsp. subtilis 168 (NC 000964.3) as a genus relative, Clostridium oceanicum DSM 1290T (FR749925.1) as a Firmicutes counterpart, Ruegeria pomeroyi DSS-3 (NR_028727.1) as a Gram-negative representative and Thermococcus litoralis JCM 8560 (AB603515.1) as the ultimate outgroup. The gyrB gene sequences from B. subtilis subsp. subtilis 168 (NC_000964.3) and Haloferax mediterranei ATCC 33500 (AOLO01000002.1) were selected as outgroups. Finally, the trees were depicted with FigTree1.
Genome-Based Taxonomy
The overall genome-related index (OGRIs) (Chun and Rainey, 2014) was calculated to evaluate potential relatedness of the sponge-associated B. pumilus 64-1 strain with those from the B. pumilus group. Seven genomes of aquatic-derived B. pumilus isolates deposited in the GenBank database2 were incorporated into this analysis. The complete genome sequence of the B. subtilis 168 strain consisted of the external group. A summary of basic genomic aspects of the elected Bacillus type strains can be found in Table 1.
Average nucleotide identity (ANI) values were determined by the ANI calculator, part of the enveomics toolbox (Rodriguez-R and Konstantinidis, 2016) and the JSpeciesWS (Richter et al., 2016). The identity thresholds of ≥95% for ANIb (estimated by the BLAST algorithm) and ≥96% for ANIm (calculated by the MUMmer algorithm) were adopted for species delineation (Rosselló-Móra and Amann, 2015). A Pearson correlation matrix of the ANIm values (JSpeciesWS) was built and finally plotted hierarchically with the R corrplot package (Wei and Simko, 2017).
JSpeciesWS was also employed for estimation of correlation indexes of tetra-nucleotide signatures, in which only regression values above 0.999 were taken in consideration to group strains in the same species (Rosselló-Móra and Amann, 2015). The Genome-to-Genome Distance Calculator (GGDC 2.1) was applied to the assessment of the digital DNA-DNA hybridization (dDDH), assuming a 70% cutoff (Meier-Kolthoff et al., 2013) and a maximum degree of 1.0% for variation in the genomic G + C content within the same species (Meier-Kolthoff et al., 2014). The Microbial Genome Atlas (MiGA) webserver (Rodriguez-R et al., 2018) was also employed for taxonomic classification of the B. pumilus 64-1 genome against the updated databases of reference genome sequences (NCBI Prok) and type strains genomes (TypeMat). The BLAST Ring Image Generation (BRIG) program was applied to depict comparisons between the B. pumilus 64-1 genome with those strains for which the highest OGRIs was obtained (Alikhan et al., 2011).
Core and Pangenome Analyses
The pangenome of this group of strains (B. pumilus 64-1, aquatic-derived B. pumilus, and B. pumilus ATCC 7061T) was estimated by a CDS multiclustering approach from the GET_HOMOLOGUES package (Contreras-Moreira and Vinuesa, 2013), using a 75% of pairwise alignment coverage and e-value set at 10−5. The core genome was calculated with all three clustering algorithms: bidirectional best hit (BDBH), COGtriangles and OrthoMCL, the pangenome with the COGtriangles and OrthoMCL algorithms. The Venn diagrams for the clustered orthologous genes were plotted with the InteractiVenn tool (Heberle et al., 2015). The pangenome compartments, “core,” “soft core,” “cloud,” and “shell,” were obtained with the GET_HOMOLOGUES script “parse pangenome matrix.pl.” Briefly, the “core” comprises the genes common to all the analyzed genomes; the “soft-core,” all the genes found in 95% of the genomes, the “cloud” component only the genes present in a minority of genomes and the “shell,” the rest of the genes found in several genomes (Contreras-Moreira and Vinuesa, 2013). The theoretical sizes of the core- and pan-genomes were estimated with OrthoMCL derived gene families, applying both the Tetellin and Willenbrock exponential models. To exclude any bias due to the differences between the original programs used to annotate these B. pumilus genomes, they were priory reannotated with the DFAST software, the same employed for the structural annotation of the B. pumilus 64-1 strain.
Nucleotide Sequence Accession Number
The draft genome sequence of B. pumilus 64-1 strain was submitted to the NCBI GenBank database under the accession number NZ_VSRW00000000.1 and the version described in this paper is the first version.
Results and Discussion
Genome Properties
The B. pumilus genome was assembled into 16 contigs with an N50 of 92,464 bp and coverage of 190×. The final assembly had a very high level of completeness (99.59%) with no contamination following evaluation with the CheckM tool. The genome has approximately 3.66 Mbp with a 41.5% GC content. A total of 3,705 CDS, five rRNA and 45 tRNA-coding genes were assigned, with an estimated coding ratio of 88.3% (Supplementary Table 1). These results were comparable with data derived from other B. pumilus genomes deposited in the NCBI GenBank database, for which the average values comprise a genome size of 3.83 Mbp, with a GC content of 41.3% and around 3,784 protein-coding genes. Plasmid sequences were not found using the PlasmidFinder 2.0 tool. Plasmid DNA extraction further confirmed the absence of plasmids in the B. pumilus 64-1 strain (data not shown).
From the 3,589 CDS scanned in the eggNOG database, a total of 94.12% could be assigned to one of the functional COG classes. Even though the majority were classified to the Function unknown (“S”) category (Table 2). A considerable number of genes within this category had at least a broad function attributed to them, such as cellular localization or even an enzyme class. Excluding the Function unknown (“S”) class, the other common categories were Transcription (“K”), Amino acid transport and metabolism (“E”) and Cell wall/membrane/envelope biogenesis (“M”).
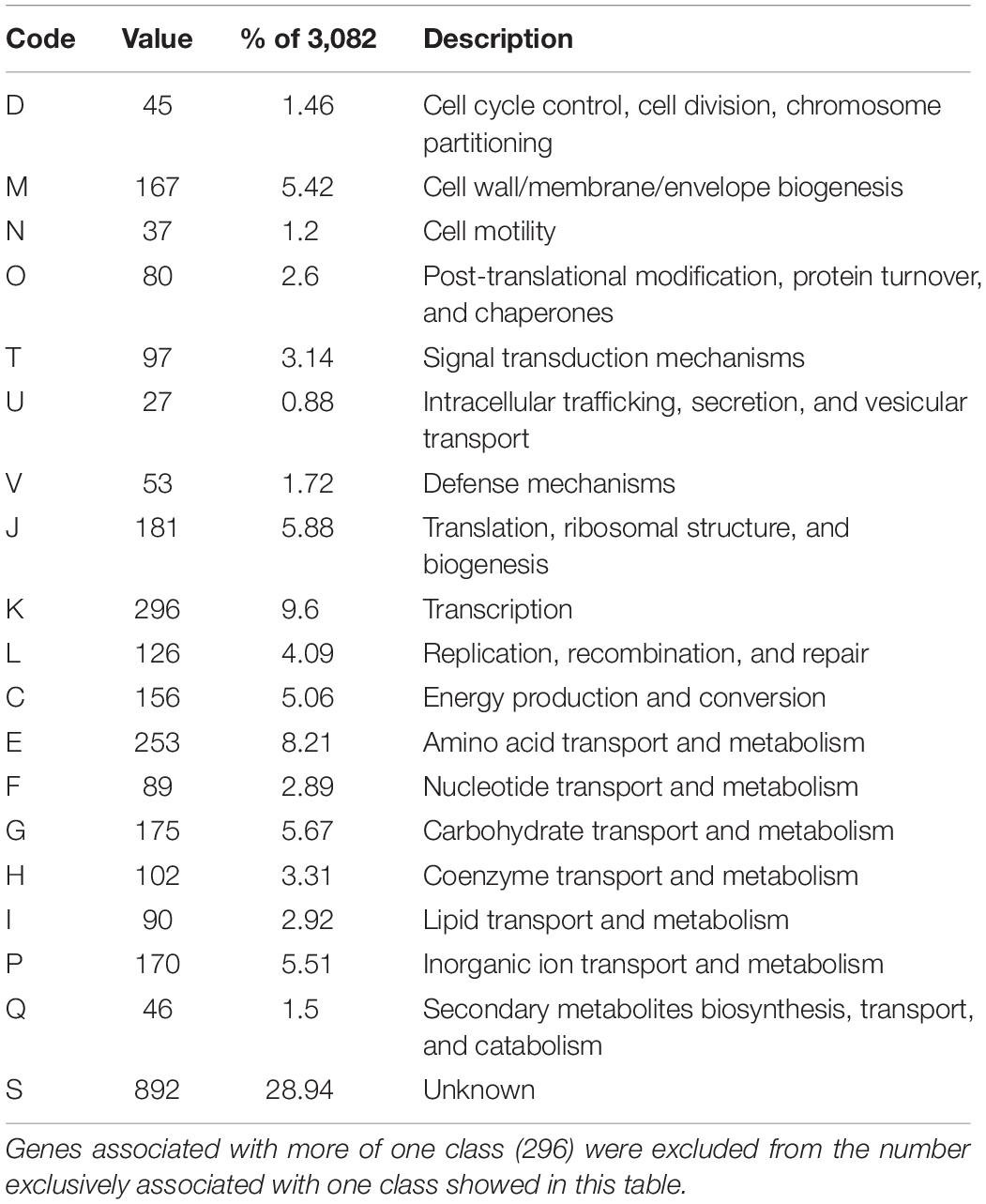
Table 2. Functional annotation of orthological groups.
Phylogenetic and Genome-Based Taxonomy Analyses
The 16S rRNA phylogeny reconstruction resulted in the confirmation of a closer association of the B. pumilus 64-1 strain with B. pumilus ATCC 7061T, while the aquatic-derived B. pumilus were present onanother major tree branch (Figure 1, Panel Aa). The analysis was replicated using different evolutionary models and the same clade arrangement was verified for the NJ trees. The outcome was somewhat different using the gyrB marker gene: B. pumilus 64-1 clustered together with the B. pumilus 150a and B. pumilus PE09-72 (Figure 1, Panel Ab). All B. pumilus strains are also distinctively grouped when compared with the other species of the B. pumilus clade in the gyrB tree (Figure 1, Panel Ab).
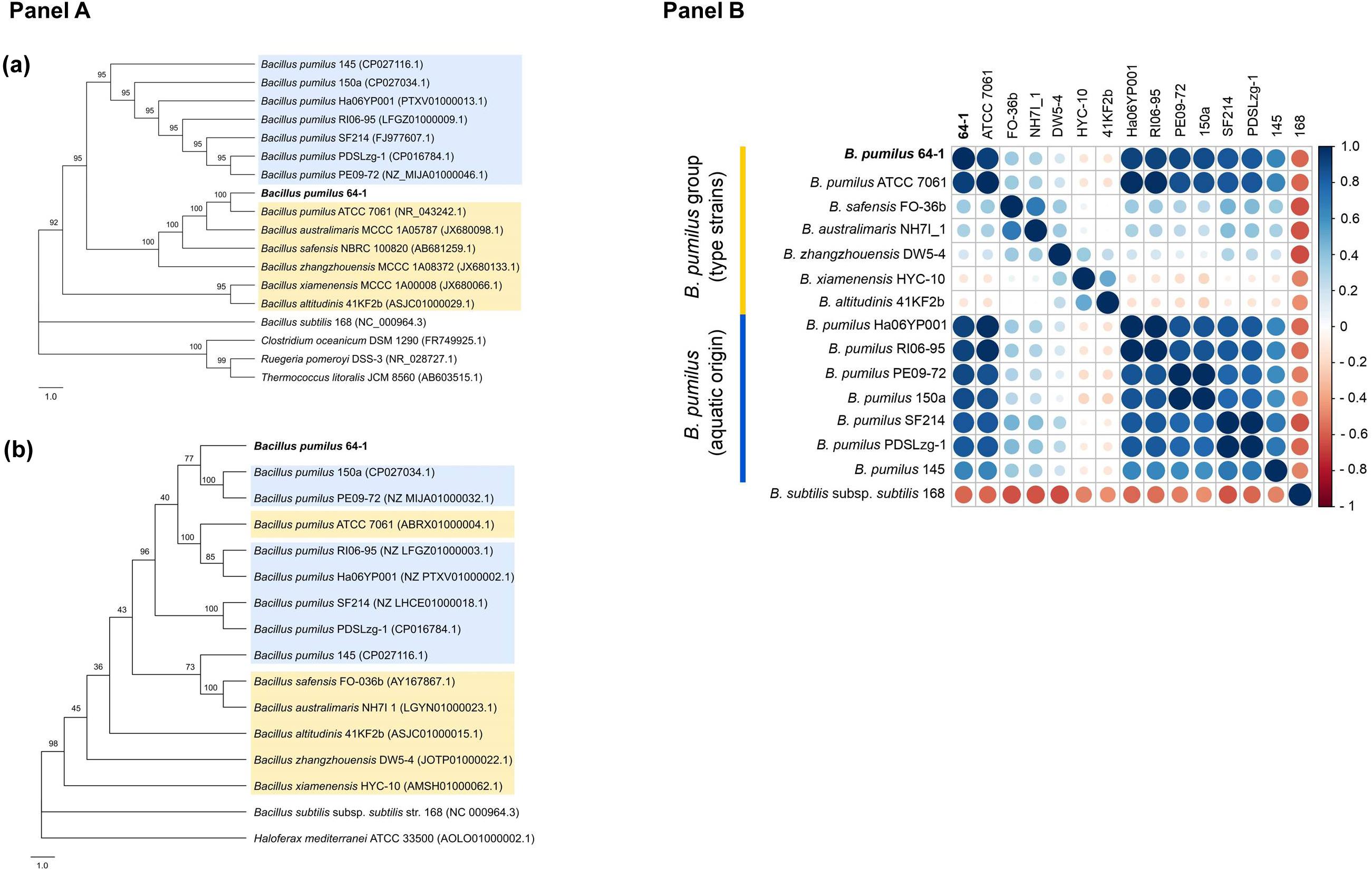
Figure 1. (Panel A): Phylogenetic trees with the B. pumilus 64-1 with selected representatives (B. pumilus clade shaded in yellow and aquatic-derived B. pumilus shaded in blue) based on (a) its 16S rRNA and (b) gyrB gene sequences, using the NJ and ML methods, respectively. Bootstrap values are shown at branch nodes. Bar, 1.0 nucleotide substitution (Kunits) rates. For the 16S rRNA NJ tree, B. subtilis subsp. subtilis 168 (NC 000964.3), C. oceanicum DSM 1290T (FR749925.1), R. pomeroyi DSS-3 (NR 028727.1) and T. litoralis JCM 8560 (AB603515.1) were chosen as outgroups. For the gyrB ML tree, B. subtilis subsp. subtilis 168 (NC 000964.3) and H. mediterranei ATCC 33500 (AOLO01000002.1) were set as outgroups. Trees were generated by MEGAX software and depicted with FigTree. (Panel B): Correlation plot based on ANIm values (%) obtained by the JSpeciesWS. The R program was used to the construction of a Pearson correlation matrix using the ANIm values derived from the JSpeciesWS and, posteriorly, for it’s plotting with the corrplot package. The size of the circles is proportional to the ANIm identity value (%). On the left, the key color (red to blue) refers to the increasing level of ANI identity between the pair of isolates.
To provide a clearer picture of the taxonomic status of B. pumilus 64-1 and its closely selected relatives and, in particular, a possible genomic identity of the hitherto described aquatic-derived B. pumilus, several genomes alignment-dependent (ANIm, ANIb, and dDDH) and alignment-independent (tetranucleotide composition and GC content) methods were employed. We also used the an OGRI-based genome classifier (MiGA) to ascertain the species affiliation of strain 64-1 and its relatedness with other B. pumilus and Bacillus spp.
In agreement with the molecular identification, the isolate 64-1 was confirmed to belong to the B. pumilus species. Sharing ANIm and ANIb values above the accepted ≥95–95 – 96% threshold for species delineation (Rosselló-Móra and Amann, 2015), the P. cyanorosea-isolated strain has as the closest genome relatives the aquatic-derived B. pumilus RI06-95 and B. pumilus Ha06YP001, the reference strain B. pumilus ATCC 7061T, followed by two other aquatic strains: B. pumilus 150a and B. pumilus PE09-72 (Figure 1, Panel B). Accordingly, the tetranucleotide regression values and the dDDH percentages were also higher for B. pumilus RI06-95 and B. pumilus Ha06YP001. The other species from the B. pumilus clade had the lowest calculated metrics (Supplementary Table 2).
Apart from the type strain B. pumilus ATCC 7061T, the genomic metrics point out that the B. pumilus 64-1 isolate clustered together with aquatic-derived B. pumilus strains (Supplementary Table 2), none of which had been recovered from sponge samples. The first one, B. pumilus RI06-95, is from a freshwater environment and with relevant probiotic potential in shellfish aquaculture systems (Hamblin et al., 2015), while the second closest relative is B. pumilus Ha06YP001, a marine bacterium isolated from an American lobster (Ranson et al., 2018). The B. pumilus 150a, isolated from a shallow-water system (Zarza et al., 2018), and the B. pumilus PE09-72, a symbiont from a marine demosponge (Matobole et al., 2017), clustered together with the P. cyanorosea-derived strain in the gyrB phylogenetic inference. In summary, genome-based analyses have corroborated the taxonomy of strain 64-1 as belonging to the B. pumilus species and guided us to select these aquatic-derived B. pumilus isolates, together with the B. pumilus ATCC 7061T, for further short-scale pangenomic assessment.
Core and Pangenome Analyses
The orthologous clustering of 33,938 CDS from the nine genomes resulted in 5,592 consensus gene clusters, the calculated pangenome (Supplementary Figure 2A). Of these, 2,984 clusters (53.36%) were found in all the studied bacilli, constituting the core genome (Supplementary Figure 2B). The computation of the flexible genome corroborated this result, with the “cloud” compartment summing up the genes found on two or fewer genomes, the “shell” as the complement of genes present on seven genomes; and the “soft core” refereeing to those encountered on eight genomes (Supplementary Figure 3).
While the relatively high proportion of the core genome might be explained by the analyses being conducted with a restricted number of strains from the same species, subsequent analyses indicate that the pangenome of these B. pumilus strains is open (Supplementary Figures 2C,D). Recent pangenome analyses for the other major Bacillus clades – B. subtilis (Wu et al., 2020), B. cereus (Bazinet, 2017), and B. amyloliquefaciens (Chun et al., 2018) – also indicate the presence of open pangenomes with a smaller set of the core genetic repertoire due to the increased number of genomes analyzed, endorsing the huge and widely known metabolic diversity in these ubiquitary Bacillus groups.
Analyzing the B. pumilus specific genes, none of them were associated with survival in the marine and sponge niches. In fact, most of these strain specific genes were either annotated as hypothetical proteins, assigned to the Function unknown (“S”) class or with no function attributed during the eggNOG functional characterization; or consisted of a prophage or transposable-related gene (data not shown). Therefore, with the exception of particular mobilome components, the further addressed potential ecological and biotechnological relevant genes seem to not be part of the accessory component of the B. pumilus 64-1 genome.
Prediction of Secondary Metabolite Gene Clusters
Twelve BGCs were identified in the B. pumilus 64-1 strain by the antiSMASH software. These included clusters potentially encoding for five non-ribosomal peptide synthetases (NRPS), one type III polyketide synthase (PKS), one hybrid NRPS-T1PKS; one bacteriocin, two betalactones and two terpenes. One BGC had high homology with the bacilysin gene clusters in B. velezensis FZB42 (BGC0001184.1) and Bacillus sp. CS93 (BGC0000888.1), displaying 85 and 80% similarity, respectively, (Table 3 and Supplementary Figure 4A). Bacilysin is a non-ribosomally synthesized dipeptide composed of an N-terminal L-alanine residue and a C-terminal L-anticapsin residue. Released upon the action of peptidases, it acts as a competitive inhibitor of glucosamine-6-phosphate synthetase, affecting the supply of a key monomeric precursor of bacterial and fungal cell walls, which leads to cell lysis and explains its activity against a wide range of microbial strains (Wang et al., 2018).
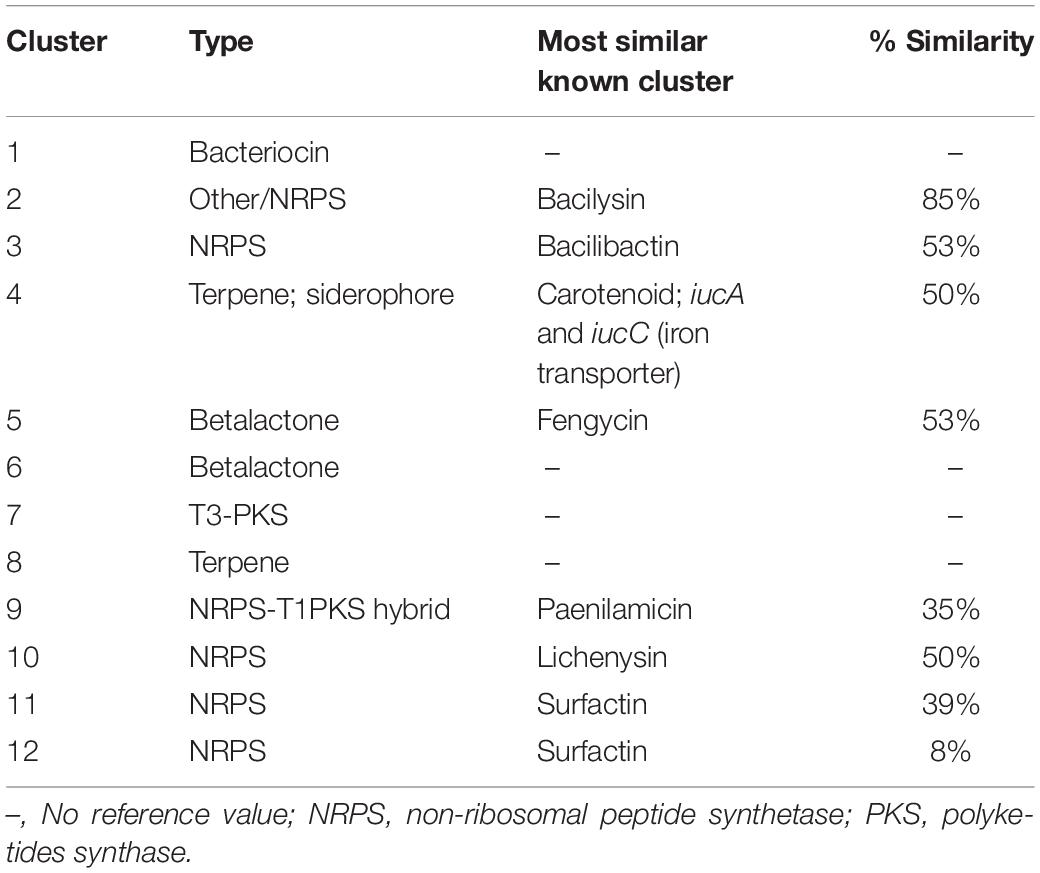
Table 3. BGCs clusters identified in the B. pumilus 64-1 genome by the antiSMASH platform.
Bacilysin is encoded by the bacABCDE operon and its synthesis involves at least six different enzymatic reactions starting with a prephenate intermediate in all Bacillus species in which this BGC has been detected (Steinborn et al., 2005). Each enzyme is encoded for by its respective gene within the core cluster, apart from the bacE gene, which has been categorized as an L-amino acid ligase participating in the export of bacilysin. Additionally, the bacF gene encodes the transaminase BacF which is involved in the penultimate step in the formation of the dipeptide, and the bacG gene, encoding the NADPH-dependent oxidoreductase BacG, which provides the epoxide substrate for the BacF enzyme (Özcengiz and Öğülür, 2015). Apart from the bacE gene, all the bacABCDE operon genes were present as a cluster in the B. pumilus 64-1 genome (Supplementary Figure 4A). As previously mentioned, the cluster displayed a high level of identity with the bacilysin gene clusters in B. velezensis FZB42 and Bacillus sp. CS93. The only differences between the bacilysin gene cluster in B. pumilus 64-1 are the presence of the bacE gene in the B. velezensis FZB42 bac operon and the absence of the bacA gene in the Bacillus sp. CS93 bac operon. There is the possibility that the bacE gene may be located in another region of the B. pumilus 64-1 genome, typically mediated by bacE in other Bacillus strains such as B. velezensis, or that the role in bacilysin transport is being undertaken by the product of another as yet unidentified gene. A B. zhangzhouensis PE09-72, isolated from the Demospongiae Isodictya compressa collected in Algoa Bay, harbored at least eight secondary metabolic pathways included three NRPS, of which one PKS/NRPS hybrid, one terpene and two bacteriocins namely, bacilysin and lichenysin. Interestingly similar to the B. pumilus 64-1 strain, the bacE gene was also not present within the bacABCDE operon in the B. zhangzhouensis PE09-72 genome (Matobole et al., 2017).
Following analysis using the BAGEL 4 software, two contigs encoding genes potentially related to the biosynthesis of amylocyclicin, sactipeptides and the bacteriocin UviB precursor were identified in our strain (Supplementary Figure 4B). Analyzing the plant-associated B. amyloliquefaciens FZB42, Scholz et al. (2014) elucidated a cluster of six genes responsible for the metabolism and export of amylocyclicin, which is a ribosomally synthesized class II bacteriocin with strong inhibitory activity against several Gram-positive bacteria. The UviB protein was first described in a Clostridium perfringens plasmid pIP404, encoded by a transcriptional unit, uviABC, whose function was related to assisting in the secretion of the UV-inducible bacteriocin BCN5 (Gamier and Cole, 1988). However, the isolated presence of a single UviB-like protein may not correspond to a functionally operative uviABC operon. But is important to emphasize that the uviB gene identified by the BAGEL4 software encodes a membrane-disrupting holin, previously described in a B. pumilus strain and which shares high similarity to the bacteriocin UviB of C. perfringens (Aunpad and Panbangred, 2012). The sporulation killing factor (SKF) is one of the three types of sactipeptides already identified in Bacillus spp., including in B. pumilus, such as identified in the strain 64-1 by BAGEL4 in the present study. In high concentrations, this peptide has been proved to inhibit the growth of other bacteria (Zhao and Kuipers, 2016).
Together with metabolomics data, “genome scanning” applied to aquatic or marine-derived B. pumilus has proven useful in unveiling a number of BGCs that encode for antimicrobial metabolites, such as amicoumacin (Hamblin et al., 2015), pumilacidin (Saggese et al., 2018), amides (Zhou et al., 2018), and bacteriocins (Zarza et al., 2018). Our results reinforce the potential that B. pumilus 64-1 strain may possess the capacity to produce a range of different bioactive peptides, which may find utility in the treatment of MDR microbial infections (Freitas-Silva et al., 2020), particularly given that sponge-associated strains of B. pumilus, including B. pumilus 64-1, have previously been reported to have activity against MDR strains of Escherichia coli, Citrobacter freundii, K. pneumoniae, Neisseria gonorrhoeae, Staphylococcus spp., Streptococcus spp., and vancomycin-resistant Enterococcus (VRE) (Santos et al., 2010; Freitas-Silva et al., 2020).
Screening for Genes Encoding Carbohydrate-Active Enzymes (CAZymes) and the Proteolytic Repertoire
In total, 77 genes potentially encoding CAZymes – mostly from the glycoside hydrolase (GH) and glycosyltransferase (GT), followed by carbohydrate esterase (CE) and polysaccharide lyase (PL) classes – were identified in the B. pumilus 64-1 genome (Supplementary Figure 5A). Apart from the presence of GHs potentially involved in starch (GH126 and GH13) and cellulose breakdown (GH9 and GH48), it is noteworthy that complete sets of genes were present for the degradation of pectin (GH28, GH105, PL1, PL6, and CE8), chitin (GH18, CE4, CE6, and CE14) and xylan (GH30, GH43, GH51, GH10, GH11, CE1, and CE7) (Supplementary Table 3). Interestingly, we have found potential xylanase-coding genes (GH10 and GH11 families), some of them co-located with the cellulolytic GH genes (Supplementary Figure 5B), indicating the likelihood of their potential coordinated co-expression for the breakdown of plant-derived polysaccharides. Novel xylanases have been reported from some marine-derived Bacillus spp. for a range of applications including the enzymatic hydrolysis of seaweed biomass (Parab et al., 2017) and the production of xylobiose from agricultural residues (Khandeparker et al., 2017). Given the broad applicability of these enzymes, further heterologous expression of the GH10 and GH11 (endo-1,4-β-xylanases) genes and their subsequent biochemical characterization from this sponge-derived B. pumilus 64-1 strain may prove fruitful.
Other genes possibly linked to xylan metabolism were also identified including a sialic acid-specific acetylesterase from the carboxyethylesterase family 6 (axeA), which appears to contain specific domains for the esterification of this acidic sugar. Sialidases and sialic-acid modifying enzymes have been characterized as important effectors for evasion of the human host immune response in bacterial pathogens (Severi et al., 2007) or in assisting members of the human microbiome in establishing on the intestinal mucosa (Vimr, 2013). Sialylated molecules are nevertheless likely to be present in the sponge mesohyl (Müller et al., 1976), and genomes of other sponge microbial symbionts harbored putative sialidases, with the suggestion of this being linked to their capacity to degrade these sialylated molecules in the sponge extracellular matrix (Kamke et al., 2013; Bayer et al., 2018; Podell et al., 2019).
In addition, approximately 6.5% of the CDS were affiliated to various peptidase families and/or to peptidase inhibitors, with the majority being associated with some essential metabolic process in the B. pumilus species. In particular, we identified an intracellular collagenase from the ample peptidase U32 family (MER0123646). The majority of U32 collagenolytic proteases are known to function as virulence factors in human pathogenic bacteria (Zhang et al., 2015). Furthermore, the collagenase annotated from B. pumilus 64-1 shares 35.4% similarity with a spongin-degrading collagenase from the Pseudoalteromonas agarovirans NW4327, a primary pathogen of the Great Barrier Reef sponge Rhopaloeides odorabile (Mukherjee et al., 2009). Further work will be required to determine whether B. pumilus 64-1 does possess in vitro collagenolytic activity to subsequently assign a role to this enzyme in the association of B. pumilus 64-1 with the P. cyanorosea host.
Genomic Features Potentially Associated With Survival in the Marine Environment
For bacteria to survive in marine environments at salinity levels of approximately 3.5%, they must be able to overcome stresses due to both high Na+ concentrations as well as high osmotic pressure. They typically respond to variations in external osmotic pressure by relying upon diverse and integrated membrane transport systems and the biosynthesis and accumulation of certain compatible solutes (Yaakop et al., 2016). In this context, the B. pumilus 64-1 genome has a number of genes potentially associated with marine survival directly related to osmoregulation and/or homeostasis of intracellular salinity level (data not shown). These genes include those encoding for: (i) mechanosensitive channels from the MscS (yhdY, ykuT, and yfkC) and MscL families (mscL); (ii) an antiporter complex involved in resistance to increased cation concentration and alkali conditions (phaA, mrpB, phaC, mrpD, mrpE, phaF, and phaG); (iii) components of the potassium importer KtrA (ktrA and ktrB); and (iv) CrcB fluoride exporters (crcB). Control of the biosynthesis of cellular L-proline and glycine betaine pools appears to be important in the B. pumilus 64-1 strain given the presence of genes for: (i) the OpuA, OpuB, and OpuC glycine betaine transporters (opuAC and opuAB); (ii) the osmoprotectant ABC-type proline betaine transport system (proV, opuCD, opuCC, opuCB, opuCA, proWX, and proV); (iii) the glycerol uptake facilitator protein (glpF); (iv) and enzymes for glycine betaine (gbsA and gbsB) and proline metabolism (proA, proB, proC, and ycgM) (data not shown).
Mechanosensitive channels are recognized as having a role in protecting bacterial cells from the effects of sudden hypotonic shocks (Booth, 2014). The B. pumilus 64-1 genome appears to possess at least three different copies of genes potentially encoding for members of the MscS family in addition to one set of genes encoding an MscL channel. Both sets of these families have previously been reported to play important roles in a protective effect against osmotic shock (Booth and Blount, 2012). These mechanosensitive channels, together with Ktr components, the Opu glycine betaine uptake systems and the fine cytoplasmic control of glycine betaine and proline levels are fundamental to the “salt out” tactics employed by B. subtilis (Hoffmann and Bremer, 2017) and in B. licheniformis (Schroeter et al., 2013). A similar set of protective actions has not to date been comprehensively described for any member of the B. pumilus group. To our knowledge, the only report where a terrestrial and marine Bacillus strains were compared in terms of survival under hyperosmolarity was 20 years ago: the genome of an alkalophilic Bacillus halodurans strain was enriched in ABC transport systems when compared to a soil-born B. subtilis (Takami et al., 2000).
Identification of Antimicrobial Resistance and Metal Tolerance Genes
The cat-86 gene, encoding a chloramphenicol acetyltransferase (CAT), was identified along with an extended-spectrum BPU (from B. pumilus) β-lactamase, an identity level superior to 90% as expressed by the RGI tool. However, the functional annotation reveals the theoretical capacity of the B. pumilus 64-1 strain to be resistant to representatives of other antimicrobial classes, including chloramphenicol, aminoglycosides, fosfomycin, tetraciclins, cyclic peptides, glycopeptides and betalactams (Supplementary Table 4). No resistance phenotype was however observed after disk diffusion susceptibility tests with the strain 64-1 (data not shown); although, this may mean that the genotype was not expressed under the tested conditions. Future application of more discriminatory antimicrobial susceptibility tests, such as microdilution-based methods using increasing concentrations of antimicrobials from each representative class, would confirm this resistance profile.
Genes for transcriptional regulators involved with antibiotic resistance, such as TetR and MarR, and for the response regulator OmpR, were also identified in the B. pumilus 64-1 genome (Supplementary Table 4), showing that the B. pumilus 64-1 strain may be able to control the expression of these genes under the conditions imposed by these antagonist compounds. Moreover, the functional annotation of multidrug resistance proteins (ykkC, ykkD, ebrA, ebrB, mdtI, and sugE) and multidrug efflux pumps (yhbJ and norM), including those from the multidrug and toxic compound extrusion (MATE) family (yisQ and yoeA) and the RND (resistance-nodulation-cell division) drug exporters (ydgH, mdtC, and swrC), suggest a wider intrinsic resistance, as verified for the B. cereus group, either for their chloramphenicol and tetracycline genetic repertoire (Glenwright et al., 2017) or by the conservation of these small molecules efflux pumps (Hassan et al., 2017). This apparent intrinsic resistance genotype of the strain 64-1 may not only form part of defense strategies against the ever-changing environmental conditions but also for physiological processes, considering that some of the substrates for these multidrug transporters are cations, such as Na+ and H+ (Du et al., 2018).
The B. pumilus 64-1 also appears to possess an extensive genetic reservoir for potential tolerance to several heavy metals, comprising arsenic, mercury, tellurium, cobalt, cooper and chromate (Supplementary Table 4). Likely arsenic resistance is particularly evident, with the presence of an ars operon, including all the genetic elements required for cellular detoxification of the metal. The presence of these genes in B. pumilus 64-1 is perhaps not surprising given that arsenic tolerance has previously been reported in B. pumilus strains isolated from various environments (Raja and Omine, 2012; Khowal et al., 2017; Nithya et al., 2017; Titah et al., 2018; Dolphen and Thiravetyan, 2019). Additionally, marine sponge-associated Streptomyces strains have also previously been reported to be enriched in arsenic transport genes (Jackson et al., 2018) and the sponge-associated Entotheonella spp. are known to sequester arsenic in intracellular vesicles whilst residing in their sponge host, Theonella swinhoei (Keren et al., 2017). Future work on the potential degradation of arsenic by the B. pumilus 64-1 strain might clarify its applicability for bioremediation or biosorption processes in marine environments.
CRISPR-Cas System and Mobilome Elements
CRISPR–Cas systems are known to be part of the molecular armamentarium employed by sponge microbial symbionts against viral attack. Ultimately, these sequences can influence the evolution of their genomes (Horn et al., 2016; Alex and Antunes, 2018; Burgsdorf et al., 2019). A functional class 1 CRISPR was detected on the B. pumilus 64-1 genome, being classified as a type III-B CRISPR-Cas10 (Couvin et al., 2018). This region harbors the type III-signature protein, the ssDNase Cas10, and the five spacers, coding for the Cmr1, Cmr3, Cmr4, Cmr5, and Cmr6 proteins, with conservation DNA identities of 86.14% for the DR and 7.14% for the spacers. The Cmr5 protein was confirmed during the annotation step (Supplementary Figures 6A,B). One spacer showed similarity with spacers from the genome of B. safensis KCTC 12796BP, a strain which was also isolated from a marine sponge in the Jeju Island, South Korea (Hanh et al., 2018). Type III-B systems shows in vivo specificity toward the mRNA of invasive virus and foreign DNA by an RNA-targeted mode (Tamulaitis et al., 2017). Therefore, there is the likelihood that B. pumilus 64-1 may use this CRISPR as an additional defense system against phages and/or other mobile elements.
No entire genomic island (GI) was identified in the B. pumilus 64-1 genome. Four incomplete regions for temperate prophages were detected by the PHASTER server; one assigned to the family Myoviridae, Brevibacillus phage Jimmer1, and the other three to the family Siphoviridae: Bacillus phage phi105, Bacillus phage Wbeta, Bacillus phage G (data not shown). Using the ISfinder database, BLASTx searches produced significant alignments with IS1182-like elements, all originally characterized in Bacillaceae, including the ISBpu1, specific for the B. pumilus species (data not shown). All these IS harbor one single ORF for a transposase (Siguier et al., 2015) and were previously shown to mediate the transfer of antimicrobial resistance and toxin genes in clinically important Firmicutes pathogens (Mikalsen et al., 2015; Smith et al., 2015; Furi et al., 2016). A manual inspection of the eggNOG annotation files allowed the identification of 76 prophage genes (Supplementary Table 5), some of them matching exactly to the segments confirmed after scanning by PHASTER program. Genes for an IS66 transposase and three putative transposases were also observed during the curation of the functional annotation (Supplementary Table 5).
Bacillus pumilus was found to have a percentage of horizontally transferred genes (%HGT) of around 2.16% which is similar to the 2.52% for the type-strain B. pumilus ATCC 7061T (IMG Genome ID: 642791616), and higher than all the twenty B. pumilus genomes with calculated %HGT upon comparison with the [Integrated Microbial Genomes and Microbiomes (IMG/M) Database v. 5.0] (Chen et al., 2019). HGT has been credited to be the additional important component in the cross-regulation of marine animal-bacterial interactions (Degnan, 2014) and may even engage bacteria in symbioses with some eukaryotic hosts (Tong et al., 2020). A higher mobilome content may thus reflect the ability of the B. pumilus 64-1 strain to continuously acquire and exchange new and advantageous metabolic functions from the other members constituting the sponge-associated and the environmental microbiomes and, even, the invertebrate host.
Conclusion
Owing to its previously reported antimicrobial activity, the genome of the sponge-derived B. pumilus 64-1 strain was sequenced and subsequently analyzed employing a variety of different bioinformatic tools. The isolate was confirmed to have the type strain of the B. pumilus clade (B. pumilus ATCC 7061T) and other aquatic-derived B. pumilus strains (B. pumilus RI06-95 and B. pumilus Ha06YP001) as its closest relatives, following phylogenetic and genome-based taxonomic analyses. In addition, together with these related strains, it appears to have an open pangenome, with a dominant core component and specific accessory genes distributed throughout these strains.
A number of BGCs were identified one of which potentially encodes for the antimicrobial peptide bacilysin. This gene cluster exhibited high similarity (85%) to the corresponding bac operon and may be responsible for the bioactivity previously reported for this isolate. Further genome mining revealed a variety of metabolic traits with interesting ecophysiological and biotechnological significance. The enhanced survival of the strain in the marine habitat is likely to be due to a high number of genes which encode for transporters and osmolytes. Sequences encoding for lignocellulose-degrading CAZymes depicts the potential applicability of this strain for the bioconversion of plant-derived feedstock in biofuels. Future confirmation of complete arsenic metabolism may also shed more light on the prospective use of the strain 64-1 for bioremediation purposes, once all genes for detoxification of this heavy metal were present in its genome.
The first report on the genomic characterization of a microbial isolate from a Homoscleromorpha sponge was for the Bacillus plakortidis P203 (Wang et al., 2016), a strain isolated from the marine sponge Plakortis simplex (Borchert et al., 2007). This current work is the second to present the genome of a bacterial isolate from another homoscleromorph sponge and the first for the recently described P. cyanorosea species (Muricy et al., 2019), and for the Plakina genus. Increased information on cultured isolates from different sponge species will shed further light on marine sponge microbiomes (Laport, 2018), considering the growing knowledge on the biology of microbe-sponge interactions (Pita et al., 2018); particularly in often-neglected bacterial taxa studied in this holobiont, namely the Firmicutes. As suggested future research, a comparative genomics between B. pumilus isolated from sponges, isolated from the aquatic environment, and isolated from the terrestrial environment may prove fruitful. This will allow a further analysis for the enrichment of sponge-host dependent lifestyle genes and marine adaptation in these sponge-derived Bacillus. In addition, the bioactive substances produced by B. pumilus 64-1 are currently being isolated, purified for further evaluantion of its molecular mechanism of antimicrobial action. All CAZymes activities can also be explored, followed by heterologous expression assays, mainly the ones with degrading abilities in plant biomass.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
JF-S and ML conceived the study and designed the experiments. JF-S, GM, and ML performed the experiments. JF-S, BO, and FV analyzed the data. BO, JF-S, AD, and ML wrote the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Funding
JF-S and BO acknowledges the doctoral scholarships granted by the Brazilian National Council for Scientific and Technological Development (CNPq) (grant numbers: 140046/2020-8 and 140840/2018-4). BO was recipient of a Sandwich doctoral scholarship by the Coordination for the Improvement of Higher Education Personnel (CAPES) (grant number: 88887.341847/2019-00). This work was supported by the Rio de Janeiro State Foundation Support Research (FAPERJ) (grant numbers: E-26/203.320/2017 and E-26/202.898/2018), and CNPq (grant numbers: 304477/2015-0 and 170521/2017-6). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge Dr. Ciro Rossi for the help with the pictures of CRISPR sequences.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.592735/full#supplementary-material
Supplementary Figure 1 | Workflow of the bioinformatic tools used during the assembly (colored in red) and the annotation (colored in blue) of the B. pumilus 64-1 genome. Details about the identification of genes potentially associated, resistome and mobilome elements from the eggNOG functional annotation files are depicted in gray, and black, respectively. ARG, antimicrobial resistance genes; BGCs, biosynthetic gene clusters; CAZymes, carbohydrate-active enzymes; IS, insertion sequences.
Supplementary Figure 2 | Estimation of the core and pangenome sizes: Venn diagrams depicting the (A) pangenome and (B) core genome differentially calculated for the B. pumilus 64-1 and the genomic-related B. pumilus strains by the GET_HOMOLOGUES software; (C) theoretical pangenome size and (D) theoretical core genome size applying the Tettellin (blue) and Willenbrock (red) fits by the OrtoMCL algorithm.
Supplementary Figure 3 | Barplot with the frequencies of orthologs clusters assigned to core, soft core, shell and cloud categories according to the orthoMCL algorithm by the GET_HOMOLOGUES software.
Supplementary Figure 4 | Genomic architecture of the: (A) bacilysin gene cluster and its closest BGCs matches (MiBIG accession numbers parenthetical) according to the antiSMASH tool; (B) bioactive peptides-encoding genes within the areas of interest (AOIs) detected by the BAGEL4 software.
Supplementary Figure 5 | Relevant outcomes about the CAZyme repertoire of the B. pumilus 64-1 genome: (A) Distribution of CAZymes classes by the dbCAN 2 metaserver. GHs, glycoside hydrolases; GTs, glicosyltransferases; PLs, polysacharides lyases; CEs, carbohydrate esterases. (B) Concatenated organization of four putative extracellular cellulases (GH9, GH48, and GH5) and xylanase (GH10) genes.
Supplementary Figure 6 | Type III-B CRISPR-Cas10 system identified in the B. pumilus 64-1 genome: (A) genomic organization adapted by the CRISPRCasFinder program and Artemis. Highlighted in brown the spacer sequence showing similarity with the B. safensis KCTC 12796BP genome; (B) RNA structure (colored by base-pairing probability) predicted by RNAFolder web server.
Supplementary Table 1 | Genome properties of B. pumilus 64-1.
Supplementary Table 2 | The overall genome-related index (OGRIs) values from different in silico bioinformatics tools among B. pumilus 64-1 versus members from the B. pumilus group and aquatic-derived B. pumilus strains.
Supplementary Table 3 | List of relevant polyssacharide-degrading CAZymes identified in the B. pumilus 64-1 genome.
Supplementary Table 4 | List of genes related to antimicrobial resistance and metal tolerance/detoxification annotated in the B. pumilus 64-1 draft genome.
Supplementary Table 5 | List of mobilome-related elements in the B. pumilus 64-1 draft genome detected by the eggNOG-mapper v2.
Footnotes
References
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525. doi: 10.1093/nar/gkz935
Alex, A., and Antunes, A. (2018). Genus-wide comparison of Pseudovibrio bacterial genomes reveal diverse adaptations to different marine invertebrate hosts. PLoS One 13:e0194368. doi: 10.1371/journal.pone.0194368
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Andrews, S. (2010). FASTQC. A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed July 20, 2019).
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Aunpad, R., and Panbangred, W. (2012). Evidence for two putative holin-like peptides encoding genes of Bacillus pumilus strain WAPB4. Curr. Microbiol. 64, 343–348. doi: 10.1007/s00284-011-0074-3
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bayer, K., Jahn, M. T., Slaby, B. M., Moitinho-Silva, L., and Hentschel, U. (2018). Marine sponges as Chloroflexi hot spots: genomic insights and high-resolution visualization of an abundant and diverse symbiotic clade. mSystems 3:e00150-18. doi: 10.1128/mSystems.00150-18
Bazinet, A. (2017). Pan-genome and phylogeny of Bacillus cereus sensu lato. BMC Evol. Biol. 17:176. doi: 10.1186/s12862-017-1020-1
Bertelli, C., Laird, M. R., Williams, K. P., Simon Fraser University Research Computing Group, Lau, B. Y., Hoad, G., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Blin, K., Shaw, S., Steinke, K., Villebro, R., Ziemert, N., Lee, S. Y., et al. (2019). antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–W87. doi: 10.1093/nar/gkz310
Booth, I. R. (2014). Bacterial mechanosensitive channels: progress towards an understanding of their roles in cell physiology. Curr. Opin. Microbiol. 18, 16–22. doi: 10.1016/j.mib.2014.01.005
Booth, I. R., and Blount, P. (2012). The MscS and MscL families of mechanosensitive channels act as microbial emergency release valves. J. Bacteriol. 194, 4802–4809. doi: 10.1128/jb.00576-12
Borchert, M. S., Nielsen, P., Graeber, I., Kaesler, I., Szewzyk, U., Pape, T., et al. (2007). Bacillus plakortidis sp. nov. and Bacillus murimartini sp. nov., novel alkalitolerant members of rRNA group 6. Int. J. Syst. Evol. Microbiol. 57, 2888–2893. doi: 10.1099/ijs.0.65177-0
Borriss, R., Danchin, A., Harwood, C. R., Medigue, C., Rocha, E. P. C., Sekowska, A., et al. (2018). Bacillus subtilis, the model Gram-positive bacterium: 20 years of annotation refinement. Microb. Biotechnol. 11, 3–17. doi: 10.1111/1751-7915.13043
Branquinho, R., Klein, G., Kämpfer, P., and Peixe, L. V. (2015). The status of the species Bacillus aerophilus and Bacillus stratosphericus, request for an opinion. Int. J. Syst. Evol. Microbiol. 65:1101. doi: 10.1099/ijs.0.000004
Buijs, Y., Bech, P. K., Vazquez-Albacete, D., Bentzon-Tilia, M., Sonnenschein, E. C., Gram, L., et al. (2019). Marine Proteobacteria as a source of natural products: advances in molecular tools and strategies. Nat. Prod. Rep. 36, 1333–1350. doi: 10.1039/c9np00020h
Burgsdorf, I., Handley, K. M., Bar-Shalom, R., Erwin, P. M., and Steindler, L. (2019). Life at home and on the roam: genomic adaptions reflect the dual lifestyle of an intracellular, facultative symbiont. mSystems 4:e00057-19. doi: 10.1128/mSystems.00057-19
Carattoli, A., Zankari, E., García-Fernández, A., Voldby, L. M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903.
Carver, T., Berriman, M., Tivey, A., Patel, C., Böhme, U., Barrell, B. G., et al. (2008). Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 24, 2672–2676. doi: 10.1093/bioinformatics/btn529
Chen, I. A., Chu, K., Palaniappan, K., Pillay, M., Ratner, A., Huang, J., et al. (2019). IMG/M v.5.0: an integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res. 47, D666–D677. doi: 10.1093/nar/gky901
Chun, B. H., Kim, K. H., Jeong, S. E., and Jeon, C. O. (2018). Genomic and metabolic features of the Bacillus amyloliquefaciens group - B. amyloliquefaciens, B. velezensis, and B. siamensis - revealed by pan-genome analysis. Food Microbiol. 77, 146–157. doi: 10.1016/j.fm.2018.09.001
Chun, J., and Rainey, F. A. (2014). Integrating genomics into the taxonomy and systematics of the Bacteria and Archaea. Int. J. Syst. Evol. Microbiol. 64, 316–324. doi: 10.1099/ijs.0.054171-0
Coil, D., Jospin, G., and Darling, A. E. (2015). A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 31, 587–589. doi: 10.1093/bioinformatics/btu661
Contreras-Moreira, B., and Vinuesa, P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701. doi: 10.1128/AEM.02411-13
Couvin, D., Bernheim, A., Toffano-Nioche, C., Touchon, M., Michalik, J., Néron, B., et al. (2018). CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 46, W246–W251. doi: 10.1093/nar/gky425
Degnan, S. M. (2014). Think laterally: horizontal gene transfer from symbiotic microbes may extend the phenotype of marine sessile hosts. Front. Microbiol. 19:638. doi: 10.3389/fmicb.2014.00638
Di Luccia, B., Riccio, A., Vanacore, A., Baccigalupi, L., Molinaro, A., and Ricca, E. (2015). Matrix production, pigment synthesis, and sporulation in a marine isolated strain of Bacillus pumilus. Mar. Drugs 13, 6472–6488. doi: 10.3390/md13106472
Dodson, R. J., Munk, A. C., Tapia, R., Green, L., Rogers, Y., Detter, J. C., et al. (2014). Genome Sequence of Bacillus pumilis ATCC 7061T. NCBI GenBank Database Genomes. Available online at: https://www.ncbi.nlm.nih.gov/nuccore/ABRX00000000 (accessed August 29, 2019).
Dolphen, R., and Thiravetyan, P. (2019). Reducing arsenic in rice grains by leonardite and arsenic-resistant endophytic bacteria. Chemosphere 223, 448–454. doi: 10.1016/j.chemosphere.2019.02.054
Du, D., Wang-Kan, X., Neuberger, A., van Veen, H. W., Pos, K. M., Piddock, L. J. V., et al. (2018). Multidrug efflux pumps: structure, function and regulation. Nat. Rev. Microbiol. 16, 523–539. doi: 10.1038/s41579-018-0048-6
Felsenstein, J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17, 368–376. doi: 10.1007/BF01734359
Fichant, G., Basse, M. J., and Quentin, Y. (2006). ABCdb: an online resource for ABC transporter repertories from sequenced archaeal and bacterial genomes. FEMS Microbiol. Lett. 256, 333–339. doi: 10.1111/j.1574-6968.2006.00139.x
Freitas-Silva, J., Oliveira, T. S., Muricy, G., and Laport, M. S. (2020). Bacillus strains associated to Homoscleromorpha sponges are highly active against multidrug resistant bacteria. Curr. Microbiol. 77, 807–815. doi: 10.1007/s00284-019-01870-x
Furi, L., Haigh, R., Al Jabri, Z. J., Morrissey, I., Ou, H. Y., León-Sampedro, R., et al. (2016). Dissemination of novel antimicrobial resistance mechanisms through the insertion sequence mediated spread of metabolic genes. Front. Microbiol. 7:1008. doi: 10.3389/fmicb.2016.01008
Gamier, T., and Cole, S. T. (1988). Studies of UV-inducible promoters from Clostridium perfringens in vivo and in vitro. Mol. Microbiol. 2, 607–614. doi: 10.1111/j.1365-2958.1988.tb00069.x
Glenwright, H., Pohl, S., Navarro, F., Miro, E., Jiménez, G., Blanch, A. R., et al. (2017). The identification of intrinsic chloramphenicol and tetracycline resistance genes in members of the Bacillus cereus group (sensu lato). Front. Microbiol. 7:2122.
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hamblin, M., Spinard, E., Gomez-Chiarri, M., Nelson, D. R., and Rowley, D. C. (2015). Draft genome sequence of the shellfish larval probiotic Bacillus pumilus RI06-95. Genome Announc. 3:e00858-15. doi: 10.1128/genomeA.00858-15
Hanh, N. P. K., Kim, S. H., Kim, G. J., Choi, H., and Nam, D. H. (2018). The complete genome sequence of a marine sponge-associated bacteria, Bacillus safensis KCTC 12796BP, which produces the anti-allergic compounds. Korean J. Microbiol. 54, 448–452. doi: 10.7845/kjm.2018.8069
Hao, K., Li, H., Li, F., and Guo, P. (2016). Complete genome sequence of Bacillus pumilus PDSLzg-1, a hydrocarbon-degrading bacterium isolated from oil-contaminated soil in China. Genome Announc. 4:e01079-16. doi: 10.1128/genomeA.01079-16
Hassan, K. A., Fagerlund, A., Elbourne, L. D. H., Vörös, A., Kroeger, J. K., Simm, R., et al. (2017). The putative drug efflux systems of the Bacillus cereus group. PLoS One 12:e0176188.
Heberle, H., Meirelles, G. V., da Silva, F. R., Telles, G. P., and Minghim, R. (2015). InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics 16:169.
Hoffmann, T., and Bremer, E. (2017). Guardians in a stressful world: the Opu family of compatible solute transporters from Bacillus subtilis. Biol. Chem. 398, 193–214. doi: 10.1515/hsz-2016-0265
Horn, H., Slaby, B. M., Jahn, M. T., Bayer, K., Moitinho-Silva, L., Förster, F., et al. (2016). An enrichment of CRISPR and other defense-related features in marine sponge-associated microbial metagenomes. Front. Microbiol. 8:1751. doi: 10.3389/fmicb.2016.01751
Huerta-Cepas, J., Szklarczyk, D., Heller, D., Hernández-Plaza, A., Forslund, S. K., Cook, H., et al. (2019). eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314. doi: 10.1093/nar/gky1085
Indraningrat, A. A., Smidt, H., and Sipkema, D. (2016). Bioprospecting sponge-associated microbes for antimicrobial compounds. Mar. Drugs 14:E87. doi: 10.3390/md14050087
Jackson, S. A., Crossman, L., Almeida, E. L., Margassery, L. M., Kennedy, J., and Dobson, A. D. W. (2018). Diverse and abundant secondary metabolism biosynthetic gene clusters in the genomes of marine sponge-derived Streptomyces spp. isolates. Mar. Drugs 16:67. doi: 10.3390/md16020067
Kamke, J., Sczyrba, A., Ivanova, N., Schwientek, P., Rinke, C., Mavromatis, K., et al. (2013). Single-cell genomics reveals complex carbohydrate degradation patterns in poribacterial symbionts of marine sponges. ISME J. 7, 2287–2300. doi: 10.1038/ismej.2013.111
Keren, R., Mayzel, B., Lavy, A., Polishchuk, I., Levy, D., Fakra, S. C., et al. (2017). Sponge-associated bacteria mineralize arsenic and barium on intracellular vesicles. Nature Commun. 8:14393. doi: 10.1038/ncomms14393
Khandeparker, R., Parab, P., and Amberkar, U. (2017). Recombinant xylanase from Bacillus tequilensis BT21: biochemical characterisation and its application in the production of xylobiose from agricultural residues. Food Technol. Biotechnol. 55, 164–172. doi: 10.17113/ftb.55.02.17.4896
Khowal, S., Siddiqui, M. Z., Ali, S., Khan, M. T., Khan, M. A., Naqvi, S. H., et al. (2017). A report on extensive lateral genetic reciprocation between arsenic resistant Bacillus subtilis and Bacillus pumilus strains analyzed using RAPD-PCR. Mol. Phylogenet. Evol. 107, 443–454. doi: 10.1016/j.ympev.2016.12.010
Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. doi: 10.1007/BF01731581
Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msy096
Lai, Q., Liu, Y., and Shao, Z. (2012). Genome sequence of Bacillus sp. strain HYC-10, isolated from intestinal tract contents from a marine fish Mugil cephalus. J. Bacteriol. 194:6991. doi: 10.1128/JB.01920-12
Lai, Q., Liu, Y., and Shao, Z. (2014a). Genome Sequence of Bacillus altitudinis 41KF2b. NCBI GenBank Database Genomes. Available online at: https://www.ncbi.nlm.nih.gov/nuccore/ASJC00000000 (accessed August 29, 2019).
Lai, Q., Liu, Y., and Shao, Z. (2014b). Genome Sequence of Bacillus safensis FO-36b. NCBI GenBank Database Genomes. Available online at: https://www.ncbi.nlm.nih.gov/nuccore/ASJD00000000 (accessed August 29, 2019).
Laport, M. S. (2018). Isolating bacteria from sponges: why and how? Curr. Pharm. Biotechnol. 18, 1224–1236. doi: 10.2174/1389201019666180329111327
Liu, Y., Lai, Q., Dong, C., Sun, F., Wang, L., Li, G., et al. (2013). Phylogenetic diversity of the Bacillus pumilus group and the marine ecotype revealed by multilocus sequence analysis. PLoS One 8:e80097. doi: 10.1371/journal.pone.0080097
Liu, Y., Lai, Q., Du, J., and Shao, Z. (2016). Bacillus zhangzhouensis sp. nov. and Bacillus australimaris sp. nov. Int. J. Syst. Evol. Microbiol. 66, 1193–1199. doi: 10.1099/ijsem.0.000856
Liu, Y., Ramesh Kumar, N., Lai, Q., Du, J., Dobritsa, A. P., Samadpour, M., et al. (2015). Identification of strains Bacillus aerophilus MTCC 7304T as Bacillus altitudinis and Bacillus stratosphericus MTCC 7305T as a Proteus sp. and the status of the species Bacillus aerius Shivaji et al. (2006). Request for an Opinion. Int. J. Syst. Evol. Microbiol. 65, 3228–3231. doi: 10.1099/ijsem.0.000408
Lorenz, R., Bernhart, S. H., Höner, Zu Siederdissen, C., Tafer, H., Flamm, C., et al. (2011). ViennaRNA Package 2.0. Algorithms Mol. Biol. 6:26. doi: 10.1186/1748-7188-6-26
Matobole, R. M., van Zyl, L., Parker-Nance, S., Davies-Coleman, M. T., and Trindade, M. (2017). Antibacterial activities of bacteria isolated from the marine sponges Isodictya compressa and Higginsia bidentifera collected from Algoa Bay. South Africa. Mar. Drugs 15:47. doi: 10.3390/md15020047
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Goker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 14:60. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Göker, M. (2014). Taxonomic use of DNA G+C content and DNA–DNA hybridization in the genomic age. BMC Bioinformatics 64:352–356. doi: 10.1099/ijs.0.056994-0
Mikalsen, T., Pedersen, T., Willems, R., Coque, T. M., Werner, G., Sadowy, E., et al. (2015). Investigating the mobilome in clinically important lineages of Enterococcus faecium and Enterococcus faecalis. BMC Genomics 16:282. doi: 10.1186/s12864-015-1407-6
Moitinho-Silva, L., Díez-Vives, C., Batani, G., Esteves, A. I., Jahn, M. T., and Thomas, T. (2017). Integrated metabolism in sponge-microbe symbiosis revealed by genome-centered metatranscriptomics. ISME J. 11, 1651–1666. doi: 10.1038/ismej.2017.25
Mukherjee, J., Webster, N., and Llewellyn, L. E. (2009). Purification and characterization of a collagenolytic enzyme from a pathogen of the Great Barrier Reef Sponge, Rhopaloeides odorabile. PLoS One 4:e7177. doi: 10.1371/journal.pone.0007177
Müller, W. E., Müller, I., Zahn, R. K., and Kurelec, B. (1976). Species-specific aggregation factor in sponges. VI. Aggregation receptor from the cell surface. J. Cell. Sci. 21, 227–241.
Muricy, G., Domingos, C., Lage, A., Lanna, E., Hardoim, C., Laport, M., et al. (2019). Integrative taxonomy widens our knowledge of the diversity, distribution and biology of the genus Plakina Homosclerophorida: Plakinidae. Invertebr. Syst. 33, 367–401. doi: 10.1071/IS18027
Nielsen, H. (2017). Predicting secretory proteins with SignalP. Methods Mol. Biol. 1611, 59–73. doi: 10.1007/978-1-4939-7015-5_6
Nithya, C., Gnanalakshmi, B., and Pandian, S. K. (2017). Assessment and characterization of heavy metal resistance in Palk Bay sediment bacteria. Mar. Environ. Res. 71, 283–294. doi: 10.1016/j.marenvres.2011.02.003
O’Halloran, J. A., Barbosa, T. M., Morrissey, J. P., Kennedy, J., O’Gara, F., and Dobson, A. D. (2011). Diversity and antimicrobial activity of Pseudovibrio spp. from Irish marine sponges. J. Appl. Microbiol. 110, 1495–1508. doi: 10.1111/j.1365-2672.2011.05008.x
Ortiz, A., and Sansinenea, E. (2019). Chemical compounds produced by Bacillus sp. factories and their role in nature. Mini-Rev. Med. Chem. 19, 373–380. doi: 10.2174/1389557518666180829113612
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2013). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Özcengiz, G., and Öğülür, İ (2015). Biochemistry, genetics and regulation of bacilysin biosynthesis and its significance more than an antibiotic. N. Biotechnol 32, 612–619. doi: 10.1016/j.nbt.2015.01.006
Parab, P., Khandeparker, R., Amberkar, U., and Khodse, V. (2017). Enzymatic saccharification of seaweeds into fermentable sugars by xylanase from marine Bacillus sp. strain BT21. 3 Biotech. 7:296. doi: 10.1007/s13205-017-0921-4
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Petersen, J. M., and Osvatic, J. (2018). Microbiomes In Natura: importance of invertebrates in understanding the natural variety of animal-microbe interactions. mSystems 3:e00179-17.
Phelan, R. W., Barret, M., Cotter, P. D., O’Connor, P. M., Chen, R., Morrissey, J. P., et al. (2014). Subtilomycin: a new lantibiotic from Bacillus subtilis strain MMA7 isolated from the marine sponge Haliclona simulans. Mar. Drugs 11, 1878–1898. doi: 10.3390/md11061878
Pita, L., Rix, L., Slaby, B. M., Franke, A., and Hentschel, U. (2018). The sponge holobiont in a changing ocean: from microbes to ecosystems. Microbiome 6:46. doi: 10.1186/s40168-018-0428-1
Pitcher, D. G., Saunders, N. A., and Owen, R. J. (1989). Rapid extraction of bacterial genomic DNA with guanidium thiocyanate. Lett. Appl. Microbiol. 8, 151–156. doi: 10.1111/j.1472-765X.1989.tb00262.x
Podell, S., Blanton, J. M., Neu, A., Agarwal, V., Biggs, J. S., Moore, B. S., et al. (2019). Pangenomic comparison of globally distributed Poribacteria associated with sponge hosts and marine particles. ISME J. 13, 468–481. doi: 10.1038/s41396-018-0292-9
Potter, S. C., Luciani, A., Eddy, S. R., Park, Y., Lopez, R., and Finn, R. D. (2018). HMMER web server: 2018 update. Nucleic Acids Res. 46, W200–W204.
Raja, C. E., and Omine, K. (2012). Characterization of boron resistant and accumulating bacteria Lysinibacillus fusiformis M1, Bacillus cereus M2, Bacillus cereus M3, Bacillus pumilus M4 isolated from former mining site, Hokkaido, Japan. J. Environ. Sci. Health. A. Tox. Hazard. Subst. Environ. Eng. 47, 1341–1349. doi: 10.1080/10934529.2012.672299
Ramos, P. I. P., Picão, R. C., Almeida, L. G. P., Lima, N. C. B., Girardello, R., Vivan, A. C. P., et al. (2014). Comparative analysis of the complete genome of KPC-2 producing Klebsiella pneumoniae Kp13 reveals remarkable genome plasticity and a wide repertoire of virulence and resistance mechanisms. BMC Genomics 15:54. doi: 10.1186/1471-2164-15-54
Ranson, H. J., LaPorte, J., Spinard, E., Gomez-Chiarri, M., Nelson, D., and Rowley, D. C. (2018). Draft Genome of the Marine Bacterium Bacillus pumilus strains Ha06YP001. NCBI GenBank Database Genomes. Available online at: https://www.ncbi.nlm.nih.gov/nuccore/PTXV00000000 (accessed August 29, 2019).
Rawlings, N. D., Barrett, A. J., Thomas, P. D., Huang, X., Bateman, A., and Finn, R. D. (2018). The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acid Res. 46, D624–D632. doi: 10.1093/nar/gkx1134
Richter, M., Rosselló-Móra, R., Oliver Glöckner, F., and Peplies, J. (2016). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Rodriguez-R, L. M., Gunturu, S., Harvey, W. T., Rosselló-Mora, R., Tiedje, J. M., Cole, J. R., et al. (2018). The Microbial Genomes Atlas (MiGA) webserver: taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 4, W282–W288. doi: 10.1093/nar/gky467
Rodriguez-R, L. M., and Konstantinidis, K. T. (2016). The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ. Prepr. 4:e1900v1. doi: 10.7287/peerj.preprints.1900v1
Rosselló-Móra, R., and Amann, R. (2015). Past and future species definitions for Bacteria and Archaea. Syst. Appl. Microbiol. 38, 209–216. doi: 10.1016/j.syapm.2015.02.001
Saggese, A., Culurciello, R., Casillo, A., Corsaro, M. M., Ricca, E., and Baccigalupi, L. (2018). A marine isolate of Bacillus pumilus secretes a pumilacidin active against Staphylococcus aureus. Mar. Drugs 16:180. doi: 10.3390/md16060180
Saier, M. H., Reddy, V. S., Tsu, B. V., Ahmed, M. S., Li, C., and Moreno-Hagelsieb, G. (2016). The Transporter Classification Database (TCDB): recent advances. Nucleic Acids Res. 44, D372–D379. doi: 10.1093/nar/gkv1103
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Santos, O. C. S., Pontes, P. V. M. L., Santos, J. F. M., Muricy, G., Giambiagi-de-Marval, M., and Laport, M. S. (2010). Isolation, characterization and phylogeny of sponge-associated bacteria with antimicrobial activities from Brazil. Res. Microbiol. 161, 604–612. doi: 10.1016/j.resmic.2010.05.013
Santos-Gandelman, J. F., Giambiagi-deMarval, M., Oelemann, W. M., and Laport, M. S. (2014). Biotechnological potential of sponge-associated bacteria. Curr. Pharm. Biotechnol. 15, 143–155. doi: 10.2174/1389201015666140711115033
Scholz, R., Vater, J., Budiharjo, A., Wang, Z., He, Y., Dietel, K., et al. (2014). Amylocyclicin, a novel circular bacteriocin produced by Bacillus amyloliquefaciens FZB42. J. Bacteriol. 196, 1842–1852. doi: 10.1128/JB.01474-14
Schroeter, R., Hoffmann, T., Voigt, B., Meyer, H., Bleisteiner, M., Muntel, J., et al. (2013). Stress responses of the industrial workhorse Bacillus licheniformis to osmotic challenges. PLoS One 8:e80956. doi: 10.1371/journal.pone.0080956
Severi, E., Hood, D. W., and Thomas, G. H. (2007). Sialic acid utilization by bacterial pathogens. Microbiology 153, 2817–2822. doi: 10.1099/mic.0.2007/009480-0
Siguier, P., Gourbeyre, E., Varani, A., Ton-Hoang, B., and Chandler, M. (2015). Everyman’s guide to bacterial insertion sequences. Microbiol. Spectr. 32:MDNA3-0030-2014. doi: 10.1128/microbiolspec.MDNA3-0030-2014
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006). ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36. doi: 10.1093/nar/gkj014
Smith, T. J., Hill, K. K., Xie, G., Foley, B. T., Williamson, C. H. D., Foster, J. T., et al. (2015). Genomic sequences of six botulinum neurotoxin-producing strains representing three clostridial species illustrate the mobility and diversity of botulinum neurotoxin genes. Infect. Genet. Evol. 30, 102–113. doi: 10.1016/j.meegid.2014.12.002
Steinborn, G., Hajirezaei, M. R., and Hofemeister, J. (2005). bac genes for recombinant bacilysin and anticapsin production in Bacillus host strains. Arch. Microbiol. 183, 71–79. doi: 10.1007/s00203-004-0743-8
Takami, H., Nakasone, K., Takaki, Y., Maeno, G., Sasaki, R., Masui, N., et al. (2000). Complete genome sequence of the alkaliphilic bacterium Bacillus halodurans and genomic sequence comparison with Bacillus subtilis. Nucleic Acids Res. 28, 4317–4331. doi: 10.1093/nar/28.21.4317
Tamulaitis, G., Venclovas, Č, and Siksnys, V. (2017). Type III CRISPR-Cas immunity: major differences brushed aside. Trends Microbiol. 25, 49–61. doi: 10.1016/j.tim.2016.09.012
Tamura, K., and Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526. doi: 10.1093/oxfordjournals.molbev.a040023
Tanizawa, Y., Fujisawa, T., and Nakamura, Y. (2018). DFAST: a flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics 34, 1037–1039. doi: 10.1093/bioinformatics/btx713
Thomas, T., Moitinho-Silva, L., Lurgi, M., Björk, J. R., Easson, C., Astudillo-García, C., et al. (2016). Diversity, structure and convergent evolution of the global sponge microbiome. Nat. Commun. 16:11870. doi: 10.1038/ncomms11870
Titah, H. S., Abdullah, S., Idris, M., Anuar, N., Basri, H., Mukhlisin, M., et al. (2018). Arsenic resistance and biosorption by isolated rhizobacteria from the roots of Ludwigia octovalvis. Int. J. Microbiol. 2018:3101498. doi: 10.1155/2018/3101498
Tong, W., Li, X., Wang, E., Cao, Y., Chen, W., Tao, S., et al. (2020). Genomic insight into the origins and evolution of symbiosis genes in Phaseolus vulgaris microsymbionts. BMC Genomics 21:186. doi: 10.1186/s12864-020-6578-0
van Heel, A. J., de Jong, A., Song, C., Viel, J. H., Kok, J., and Kuipers, O. P. (2018). BAGEL4: a user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 46, W278–W281. doi: 10.1093/nar/gky383
Vimr, E. R. (2013). Unified theory of bacterial sialometabolism: how and why bacteria metabolize host sialic acids. Int. Sch. Res. Notices 2013:816713. doi: 10.1155/2013/816713
Wang, J. P., Liu, B., Liu, G. H., Ge, C. B., Xiao, R. F., Zheng, X. F., et al. (2016). Draft genome sequence of Bacillus plakortidis P203T (DSM 19153), an alkali- and salt-tolerant marine bacterium. Genome Announc. 4:e01690-15. doi: 10.1128/genomeA.01690-15
Wang, T., Liu, X. H., Wu, M. B., and Ge, S. (2018). Molecular insights into the antifungal mechanism of bacilysin. J. Mol. Model. 24:118. doi: 10.1007/s00894-018-3645-4
Wei, T., and Simko, V. (2017). R package “corrplot”: Visualization of a Correlation Matrix Version 0.84. Available online at: https://github.com/taiyun/corrplot (accessed August 30, 2019).
Wu, H., Wang, D., and Gao, F. (2020). Toward a high-quality pan-genome landscape of Bacillus subtilis by removal of confounding strains. Brief Bioinform bbaa013. doi: 10.1093/bib/bbaa013
Yaakop, A. S., Chan, K. G., Ee, R., Lim, Y. L., Lee, S. K., Manan, F. A., et al. (2016). Characterization of the mechanism of prolonged adaptation to osmotic stress of Jeotgalibacillus malaysiensis via genome and transcriptome sequencing analyses. Sci. Rep. 19:33660. doi: 10.1038/srep33660
Yang, Z., He, J., Wei, X., Ju, J., and Ma, J. (2019). Exploration and genome mining of natural products from marine Streptomyces. Appl. Microbiol. Biotechnol. 104, 67–76. doi: 10.1007/s00253-019-10227-0
Yu, N. Y., Wagner, J. R., Laird, M. R., Melli, G., Rey, S., Lo, R., et al. (2010). PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615. doi: 10.1093/bioinformatics/btq249
Zarza, E., Alcaraz, L. D., Aguilar-Salinas, B., Islas, A., and Olmedo-Álvarez, G. (2018). Complete genome sequences of two Bacillus pumilus strains from Cuatrociénegas, Coahuila, Mexico. Genome Announc. 6:e00364-18. doi: 10.1128/genomeA.00364-18
Zhang, H., Yohe, T., Huang, L., Entwistle, S., Wu, P., Yang, Z., et al. (2018). dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–W101. doi: 10.1093/nar/gky418
Zhang, Y. Z., Ran, L. Y., Li, C. Y., and Chen, X. L. (2015). Diversity, structures, and collagen-degrading mechanisms of bacterial collagenolytic proteases. Appl. Environ. Microbiol. 81, 6098–6107. doi: 10.1128/AEM.00883-15
Zhao, X., and Kuipers, O. P. (2016). Identification and classification of known and putative antimicrobial compounds produced by a wide variety of Bacillales species. BMC Genomics 17:882. doi: 10.1186/s12864-016-3224-y
Keywords: antimicrobials, Bacillus pumilus, genome mining, Plakina cyanorosea, sponge-associated bacteria
Citation: Freitas-Silva J, de Oliveira BFR, Vigoder FM, Muricy G, Dobson ADW and Laport MS (2021) Peeling the Layers Away: The Genomic Characterization of Bacillus pumilus 64-1, an Isolate With Antimicrobial Activity From the Marine Sponge Plakina cyanorosea (Porifera, Homoscleromorpha). Front. Microbiol. 11:592735. doi: 10.3389/fmicb.2020.592735
Received: 07 August 2020; Accepted: 09 December 2020;
Published: 08 January 2021.
Edited by:
Vasco Ariston De Carvalho Azevedo, Federal University of Minas Gerais, BrazilReviewed by:
Jinshui Zheng, Huazhong Agricultural University, ChinaGabriela Olmedo-Alvarez, Unidad Irapuato (CINVESTAV), Mexico
Copyright © 2021 Freitas-Silva, de Oliveira, Vigoder, Muricy, Dobson and Laport. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marinella Silva Laport, bWFyaW5lbGxhQG1pY3JvLnVmcmouYnI=; Alan D. W. Dobson, YS5kb2Jzb25AdWNjLmll
†These authors have contributed equally to this work