
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol., 16 November 2020
Sec. Microbiotechnology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.590049
This article is part of the Research TopicNew Insights into the Biodegradation of Organic Contaminants in Subsurface Ecosystems: Approaches and Achievements of the Multiomics EraView all 9 articles
 Krisztián Laczi1
Krisztián Laczi1 Ágnes Erdeiné Kis1,2
Ágnes Erdeiné Kis1,2 Árpád Szilágyi1Naila Bounedjoum1,3
Árpád Szilágyi1Naila Bounedjoum1,3 Attila Bodor1,2,3György Erik Vincze1Tamás Kovács4
Attila Bodor1,2,3György Erik Vincze1Tamás Kovács4 Gábor Rákhely1,2,3*†
Gábor Rákhely1,2,3*† Katalin Perei1,3†
Katalin Perei1,3†The accumulation of petroleum hydrocarbons in the environment substantially endangers terrestrial and aquatic ecosystems. Many microbial strains have been recognized to utilize aliphatic and aromatic hydrocarbons under aerobic conditions. Nevertheless, most of these pollutants are transferred by natural processes, including rain, into the underground anaerobic zones where their degradation is much more problematic. In oxic zones, anaerobic microenvironments can be formed as a consequence of the intensive respiratory activities of (facultative) aerobic microbes. Even though aerobic bioremediation has been well-characterized over the past few decades, ample research is yet to be done in the field of anaerobic hydrocarbon biodegradation. With the emergence of high-throughput techniques, known as omics (e.g., genomics and metagenomics), the individual biodegraders, hydrocarbon-degrading microbial communities and metabolic pathways, interactions can be described at a contaminated site. Omics approaches provide the opportunity to examine single microorganisms or microbial communities at the system level and elucidate the metabolic networks, interspecies interactions during hydrocarbon mineralization. Metatranscriptomics and metaproteomics, for example, can shed light on the active genes and proteins and functional importance of the less abundant species. Moreover, novel unculturable hydrocarbon-degrading strains and enzymes can be discovered and fit into the metabolic networks of the community. Our objective is to review the anaerobic hydrocarbon biodegradation processes, the most important hydrocarbon degraders and their diverse metabolic pathways, including the use of various terminal electron acceptors and various electron transfer processes. The review primarily focuses on the achievements obtained by the current high-throughput (multi-omics) techniques which opened new perspectives in understanding the processes at the system level including the metabolic routes of individual strains, metabolic/electric interaction of the members of microbial communities. Based on the multi-omics techniques, novel metabolic blocks can be designed and used for the construction of microbial strains/consortia for efficient removal of hydrocarbons in anaerobic zones.
Various hydrocarbon (HC) compounds, derived from crude oil (n-alkanes; other aliphatics; mono-, di- and polyaromatic compounds; heterocyclic aromatics), are the most abundant hazardous organic wastes which are mostly released during the extraction processes, drilling of wells, transportation and unsuitable storage of oil, even in the immediate vicinity of filling stations (Lim et al., 2016; Bacosa et al., 2018; Xu et al., 2018). Usually, these pollutants are mixtures of various compounds including linear, branched and cyclic alkanes, monoaromatic and polyaromatic molecules. In the case of land contamination, oil can migrate into the soil and adsorb to its particles, resulting in reduced soil quality. Hydrocarbons, reaching the groundwater, can spread, causing immense contamination (Xu et al., 2018). The effect of spilled oil on the marine environment is also immensely remarkable (Overholt et al., 2016). Some of the oil sinks to the bottom of the sea, endangering deep-water organisms (Lim et al., 2016). Due to wind-induced waves, oil breaks down into small drops, forming an emulsion in the waterbody, whereas oil spills on the water surface block the oxygen transfer/flow to the pelagic organisms of the sea. The challenge of oil removal from the environment ensued from the diverse composition and hydrophobic properties of the contaminant and weathering effects, as well (Jiang et al., 2016; Lim et al., 2016; Ławniczak et al., 2020).
In an ecosystem, microbes have well-known roles in the conversion of hydrocarbons. Their extensive metabolic activity makes the inexpensive and efficient cleanup of contaminated sites possible. The organic pollutant is commonly utilized as an energy and nutrient source in metabolic processes. Bioconversion of contaminants can take place in either aerobic or anaerobic environments (Meckenstock et al., 2015; Bacosa et al., 2018).
By the end of the 20th century, in many remediation technologies, pure microbial cultures or simple consortia isolated by classical isolation methods have been applied for removal of contaminants, such as hydrocarbons (Löffler and Edwards, 2006). However, it has been recognized that most of the bacteria living in our environment can not be studied by the classical cultivation methods (Amann et al., 1995; Oberhardt et al., 2015). It was frequently observed that a process operating optimally under laboratory conditions did not work in the field and vice versa. The background of these facts was examined, and among others, the effect of environmental factors, horizontal gene transfer, metabolites or other unknown components, derived from the partner microbes, were suggested to be responsible for these phenomena (National Research Council, 1993). In the absence of modern, new generation molecular biological methods, it was tough to resolve the contradictory results obtained in laboratory conditions and on-site experiments.
It is well-known that many properties of conventionally isolated microorganisms in their original environment might be overlooked, which may be owing to the targeted isolation methods. On the other hand, an isolated microbe as monoculture might be inefficient degrader in the lab since its essential synergistic partners were removed during the isolation process (François et al., 2016). Therefore, approaches capable of investigating the processes at the system level have been necessary.
Advanced molecular technologies allow us to disclose metabolic functions, routes, interactions and networks; therefore, their application in biodegradation might revolutionize the bioremediation technologies (Lovley, 2003).
Aerobic biodegradation of aliphatic and aromatic compounds (mono- and polycyclic) has been prominent and documented in several studies (Brzeszcz and Kaszycki, 2018; Kuang et al., 2018; Révész et al., 2018; Xu et al., 2018; Mejeha et al., 2019; Steliga et al., 2020). The microorganisms use molecular oxygen as a terminal electron acceptor under aerobic conditions. Molecular oxygen functions as a trigger component in the activation mechanisms. Oxygenases (monooxygenases and dioxygenases), which transfer one or two oxygen atoms onto the substrate, have a central role in the oxic bioconversion of hydrocarbons (Karigar and Rao, 2011; Hua and Wang, 2014; Bacosa et al., 2018).
It is common knowledge that several microorganisms can utilize aromatic and aliphatic hydrocarbons for their growth under anoxic conditions. This topic was reviewed several times in the last decade (Carmona et al., 2009; Fuchs et al., 2011; Rabus et al., 2016). The identification and characterization of microorganisms capable of anaerobic hydrocarbon mineralization have been a major challenge for researchers because most of these microorganisms exist in a consortium. They are usually not culturable by the classical methods; it is true, especially for the methanogenic members of the hydrocarbon-degrading communities (Kleinsteuber et al., 2012; Laso-Pérez et al., 2019). Even so, a number of research groups have recently published their findings regarding HC-degrading microorganisms in sediments or subsurfaces under limited oxygen conditions (Yang et al., 2016; Bacosa et al., 2018; Espínola et al., 2018; Roy et al., 2018; Sperfeld et al., 2018; Liu et al., 2019; Marić et al., 2019; Miller et al., 2019; Pilloni et al., 2019; Song et al., 2019; Sun and Kostka, 2019; Li et al., 2020; Révész et al., 2020). Several recent review articles have summarized the microorganisms and metabolic pathways involved in the anaerobic hydrocarbon biodegradation. Various microbes, metabolic pathways linked to different redox compounds might coexist in the environment (Boll et al., 2014). Based on an interdisciplinary collaboration, pure and enriched cultures of bacteria and their key enzymatic reactions involved in anaerobic hydrocarbon degradation were reviewed (Rabus et al., 2016). This synopsis review (referring 14 other reviews on the comprehensive work of a large consortium about anaerobic hydrocarbon degradation) primarily focused on the characterization of toluene-activating benzylsuccinate synthase, phylogenetic classifying of alkyl-/arylalkylsuccinate synthases, stereochemical and co-metabolic insights into n-alkane-activating (methylalkyl)succinate synthases and Mo-cofactor containing dehydrogenases. A review by T. Lueders focusing on BTEX biodegradation summarizes the microorganisms and microbial interactions involved in the process (Lueders, 2017). Nevertheless, to develop effective bioremediation technologies using such microorganisms, there is a need for a better insight into the molecular mechanisms/events on both cell and community level. Exploring the composition and critical players of the microbial community is essential for their applications in the bioremediation technologies (Kleinsteuber et al., 2012; Bacosa et al., 2018). Considering the environmental parameters and available microorganisms (native or non-native), Ławniczak and colleagues reviewed the current hydrocarbon bioremediation strategies (Ławniczak et al., 2020).
With the help of genomics, transcriptomics and proteomics, a deeper understanding of hydrocarbon biodegradation in pure cultures can be achieved. Additionally, metabolomics can elucidate the metabolites present during hydrocarbon biodegradation in pure cultures or consortia. However, culture-dependent methods have severe limitations when it comes to the investigation of microbial communities. Most of the microorganisms are difficult or even impossible to culture under laboratory conditions. Thus, their functions cannot be fully resolved by classical, standard approaches. The constant development of high throughput data generation and assessment has opened new frontiers in the research of hydrocarbon biodegradation. For examination of entire hydrocarbon-degrading communities, “meta-omics” approaches, including metagenomics, metatranscriptomics, metaproteomics, have been developed.
In the next chapters, the current knowledge about anaerobic hydrocarbon-degrading microorganisms, metabolic pathways, microbial interactions in the multi-omics era is overviewed.
The classical culture-based microbiological, molecular biological and biochemical methods have several limitations, including the problems regarding the unculturable microbes or studying a single microbe or a community at the system level. Powerful techniques of the multi-omics era overcome the problems of conventional biochemical and microbiological methods to provide a system-wide picture of the capabilities, molecular mechanisms and interactions of the cells and consortia during hydrocarbon mineralization. Most of the pollutants are composed of various hydrocarbons; their bioconversions require various metabolic pathways usually present in distinct microbes. Therefore, complex contaminations can only be eliminated by the concerted action of diverse microbial communities. Understanding such complicated systems needs high throughput approaches enabling to study the processes at the system level. In this chapter, we present a brief overview of the widespread branches of the omics. Table 1 summarizes recent studies on anaerobic hydrocarbon biodegradation utilizing multi-omics techniques.
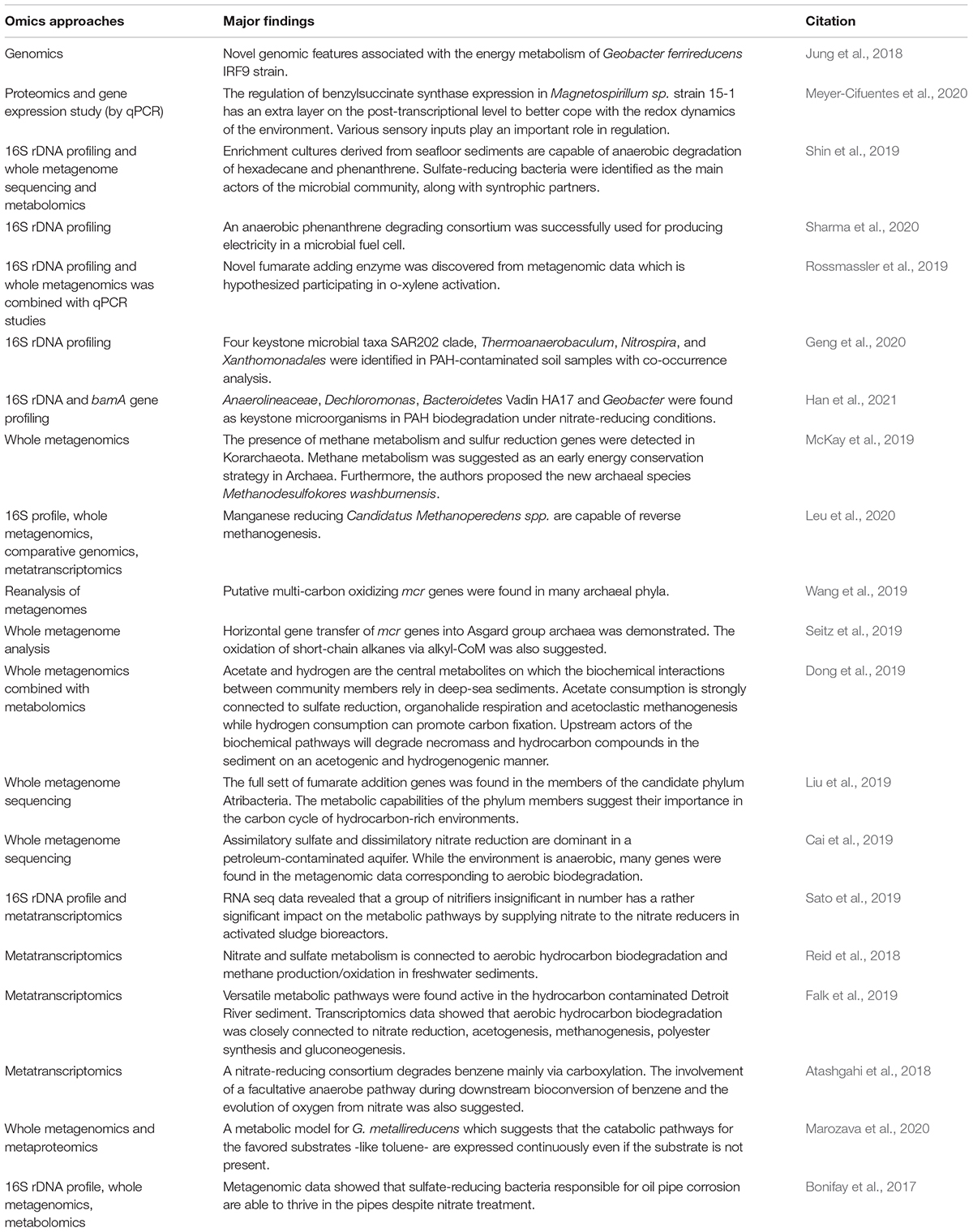
Table 1. Recent studies using omics approaches to characterize anaerobic hydrocarbon biodegrading microbial communities.
After isolating a hydrocarbon-degrading microorganism, its metabolic potential is investigated via whole-genome sequencing (WGS). The assembled genomes provide a blueprint of the organism and all the biochemical pathways it possesses. Moreover, knowledge of the full genome sequence makes taxonomic identification easier and the databases built from the genomes serve as templates in the metagenomic databases, analysis. Many genomes of anaerobic hydrocarbon biodegraders have been published (Rabus et al., 2005; Mattes et al., 2008; Aklujkar et al., 2009; Selesi et al., 2010; Jiang et al., 2012; Martín-Moldes et al., 2015; Yin et al., 2017). Comparative analysis of the genome of the iron-reducing bacterium Geosporobacter ferrireducens IRF9 identified multiple anaerobic hydrocarbon activating genes (alkylsuccinate synthase) harbored by the strain (Jung et al., 2018).
The genome of an organism harbors vast numbers of enzymes for many metabolic pathways, but specific the DNA sequence itself does not reveal their activity under certain conditions or in the presence of specific carbon sources. Transcriptomic and proteomic analysis shed light on the active pathways during hydrocarbon biodegradation. Numerous studies have been published on whole-cell transcriptomic responses of pure aerobic cultures to the presence of hydrocarbons and other xenobiotics (Sabirova et al., 2006, 2011; Laczi et al., 2015; Hegedüs et al., 2018). An early study of the marine sulfate-reducing strain NaphS2 revealed the key enzymes of anaerobic naphthalene biodegradation with the combined power of draft genome sequencing, RNA microarray and proteomics (Di Donato et al., 2010). In a proteomics study, Ralf Rabus and colleagues elucidated the catabolic network of Aromatoleum aromaticum EbN1 during aromatics biodegradation applying more than 50 growth conditions. They identified 20 new proteins participating in the peripheral pathway of anaerobic aromatics mineralization and tested the effect of environmental conditions such as carbon limitation or solvent toxicity on the proteome of A. aromaticum EbN1 (Rabus et al., 2014). Microarray analysis and qPCR experiments shed light on the transcriptional regulation of gene products involved in alkylsuccinate metabolism in Desulfatibacillum alkenivorans AK-01 (Herath et al., 2016). Gene expression experiments combined with proteomics recently provided evidence on the post-translational regulation of benzylsuccinate synthase in Magnetospirillum sp. 15-1 (Meyer-Cifuentes et al., 2020). However, combined studies of transcriptomics and proteomics are still rare. Transcriptomic data, as the final output of gene expression, must be handled carefully, since mRNA levels and protein levels are not always proportional. Buccitelli and Selbach argue in their recent review that integrated transcriptomics and proteomics should become a common integrated approach and quantifying protein and mRNA levels (which are the result of synthesis and degradation) would provide a complete picture on gene expression dynamics (Buccitelli and Selbach, 2020).
Although studying individual isolates is an important way to reveal molecular mechanisms behind anaerobic hydrocarbon bioconversion, usually the microorganisms do not act alone in the environment. Therefore, the anaerobic biodegradation of hydrocarbons is mainly studied in microbial communities. We discuss further examples and techniques in the following chapters.
A cost-effective technique to elucidate the composition and functional capabilities of a hydrocarbon-degrading microbial community is metagenomics. There are two major approaches (1) amplicon sequencing (or targeted metagenome sequencing) and (2) whole metagenome sequencing (mWGS) or shotgun metagenome sequencing. In the case of targeted metagenome sequencing, specific genes (targets) are amplified by PCR then sequenced on second- or third-generation sequencing platforms. The most common target is the V3–V4 hypervariable region of the 16S rRNA gene, but other regions, including V1–V2, V6–V8 or even the whole sequences (Johnson et al., 2019), are also used. The choice of the region can have a significant effect on taxonomic resolution (Graspeuntner et al., 2018; Bukin et al., 2019; Johnson et al., 2019). With a 16S rDNA gene profile, we can get a picture of the taxonomic composition of hydrocarbon-degrading bacterial communities. Besides the taxonomic classification, the richness and diversity of the samples can also be estimated (Potts et al., 2019). Co-occurrence of microorganisms in multiple hydrocarbon-contaminated samples suggests their association and microbial networks can be built from co-occurrence studies (Geng et al., 2020; Tikariha and Purohit, 2020; Han et al., 2021).
Genes of selected function are also utilized in targeted metagenomics, which narrows the analysis to particular group of microbes and/or a function in the community. Such genes can be, for example, mcrA for methanogenic archaea (Denonfoux et al., 2013), the catalytic domain of alkyl- and benzylsuccinate synthases (assA/masD, bssA) and the 6-oxocyclohex-1-ene-1-carbonyl-coenzyme A hydrolases (bamA) of anaerobic hydrocarbon degraders (Kuntze et al., 2008; Stagars et al., 2016; Sperfeld et al., 2018). Using functional genes in amplicon sequencing, one can predict the hydrocarbon-degrading potential of a microbial community.
For evaluation of 16S and other target gene profiles, there are many bioinformatical tools. Table 2 reports the most frequently used programs such as BLAST or MALT combined with Megan (Huson et al., 2007, 2016). Qiime 2, for example, is a recently developed pipeline (Hall and Beiko, 2018; Bolyen et al., 2019). Because it is robust, Qiime 2, and its predecessor Qiime, are widely used in 16S profile analysis of hydrocarbon-degrading microbiomes (Mejeha et al., 2019; Shin et al., 2019; BenIsrael et al., 2020; Sharma et al., 2020) and other microbial communities.
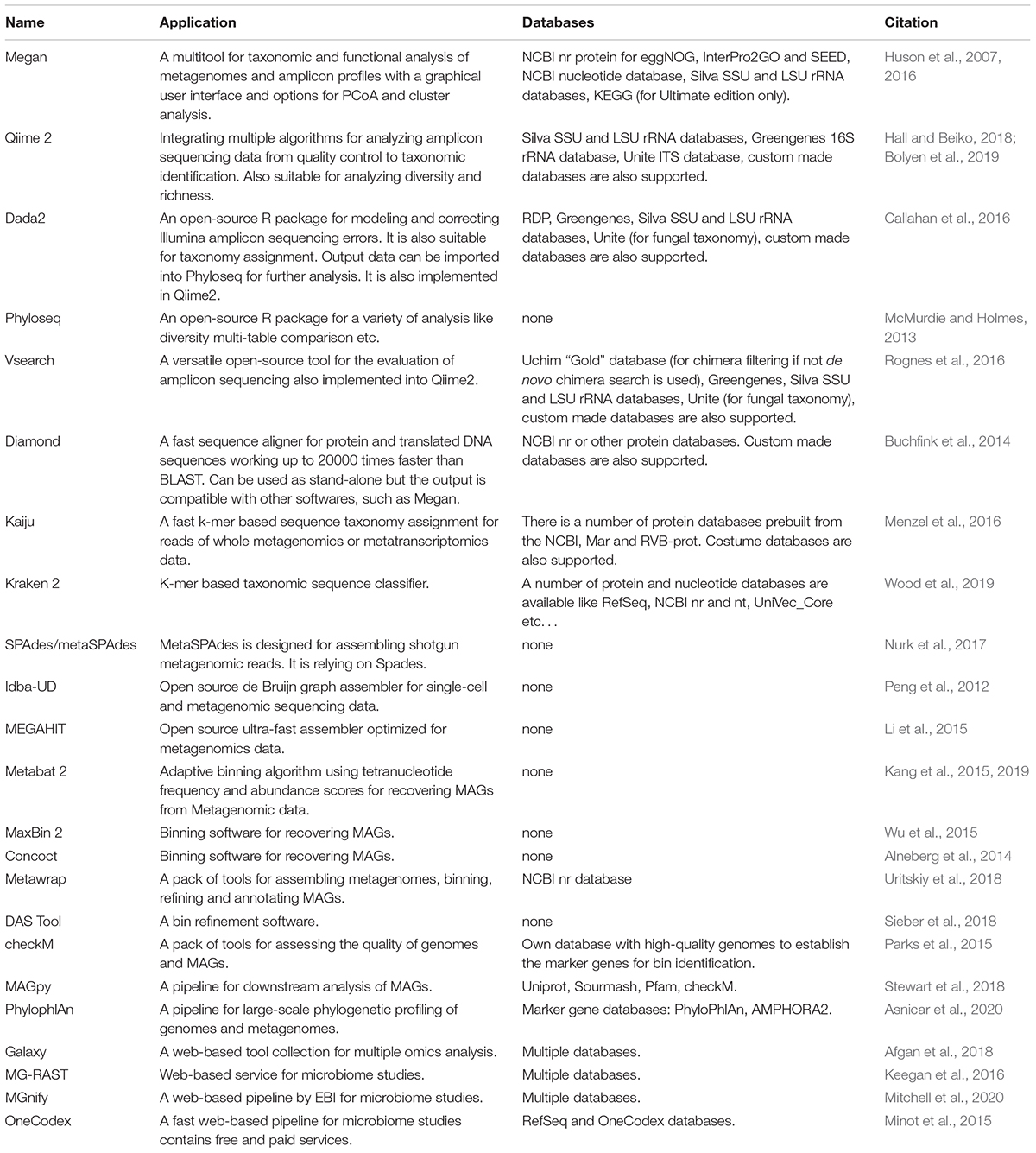
Table 2. Bioinformatic tools used in the multi-omics analysis.
If we want to inspect the overall metabolic potential of a hydrocarbon-degrading community, we need to apply the mWGS method. Shotgun sequencing data can be processed in two ways (1) read-based analysis and (2) assembly based analysis.
In the first case, the reads are utilized to obtain taxonomic and functional information. Reads are aligned with the NCBI RefSeq or the NCBI nr protein database. Diversity also can be calculated from the read count. In the second case, the reads are assembled into contigs with an appropriate assembler (Ayling et al., 2019), and individual genomes are recovered with binning methods (Sangwan et al., 2016; Sieber et al., 2018; Uritskiy et al., 2018). The quality of the bins is characterized by completeness and contamination (Parks et al., 2015).
Metagenomic approaches are the most frequently used techniques among the “omics” for investigation of - hydrocarbon-degrading - microbial communities. In the first half of the last decade, many microbiomes were studied in various anaerobic hydrocarbon-contaminated niches, including crude oil (An et al., 2013b; Hu et al., 2016; Nie et al., 2016) and coal reservoirs (An et al., 2013b; Lawson et al., 2015), hydrothermal vents (He et al., 2013, 2015; Oulas et al., 2016), natural oil seeps (Håvelsrud et al., 2011; Hawley et al., 2013, 2014), hydrocarbon-contaminated terrestrial (Abbai and Pillay, 2013) and marine environments (Kimes et al., 2014). Metagenomic data can provide valuable information on the metabolic capabilities of known and newly discovered, culturable and non-culturable microorganisms as well. Some examples are discussed here from recent years. Metagenome-based evidence on the utilization of hydrocarbons via fumarate addition in Smithella sp. and members of the Anaerolineaceae family has been published (Tan et al., 2014; Rossmassler et al., 2019). There is a vast number of uncultured bacterial species in hydrocarbon-associated deep-sea sediments. The members of two candidate phyla, Ca. Atribacteria and Ca. Bathyarchaeota, were found predominantly in the hydrocarbon-containing sediments of the Gulf of Mexico. 13 metagenome-assembled genomes (MAGs) (Chloroflexi, Aminicenantes, Aerophobetes, Actinobacteria, Ca. Bathyarchaeota, Thorarchaeota, and Lokiarchaeota) harbored glycyl-radical enzymes catalyzing the fumarate addition pathway (Dong et al., 2019). In another recent study, MAGs and SAGs (single-cell amplified genomes) were reanalyzed and linked anaerobic hydrocarbon biodegradation by fumarate addition to Ca. Atribacteria. The authors identified four novel Atribacterial lineages and analyzed their metabolic capabilities (Liu et al., 2019). In a petroleum-contaminated aquifer, nitrate and sulfate reducers dominated the community. Although the aquifer was anaerobic, multiple aerobic biodegradation genes were found in the samples, but genes of enzymes involved in methanogenesis were lacking (Cai et al., 2019). In certain environments having fluctuating levels of dissolved oxygen, both anaerobic and aerobic metabolic pathways could be detected in the metagenomes. For example, the co-occurrence of aerobic and anaerobic aromatics degraders in oil-impacted mangrove sediments was recently reported (Sousa et al., 2020).
Shotgun metagenome sequencing provides an overwhelming amount of information on the composition and potential metabolic pathways existing in a microbiome. On the other hand, it fails to fully resolve the active members, components and pathways of the community and distinguish them from the inactive ones. The taxonomic profile will elucidate the most active community members, while assembly of transcriptomic data can provide information on the active pathways at the cell or community level (Shakya et al., 2019). In a recent study, Sato and colleagues showed that, contrary to their low abundance, nitrifiers play an important role in heavy oil biodegradation by providing nitrate to denitrifying hydrocarbon degraders in activated sludge reactors (Sato et al., 2019). Another study showed both nitrogen and sulfur cycles are active alongside methanogenesis and methane oxidation in hydrocarbon-containing freshwater sediments (Reid et al., 2018). Falk and colleagues reported that nitrate reduction and methanogenesis are the most abundant pathways in the hydrocarbon-contaminated Detroit River (Falk et al., 2019). Another metatranscriptomic study suggests the co-occurrence of aerobic and anaerobic benzene degradation pathways, which are enabled by nitrate/nitrite reduction driven oxygen evolution catalyzed by a nitric oxide dismutase (NOD) (Atashgahi et al., 2018).
The metaproteome is the expressed protein complement of a microbiome (Rodríguez-Valera, 2004; Wilmes and Bond, 2004; Pieper et al., 2013). Since proteins provide biological functions for an organism, metaproteomics shed light on the metabolic activity and dynamics of a microbial community at the protein level (Pieper et al., 2013). Besides, protein-based stable isotope probing (protein-SIP) of a metaproteome yields information on protein activity and turnover (Jehmlich et al., 2016), while stable isotope fingerprinting (SIF) will elucidate carbon sources and assimilation pathways used by the community members (Kleiner et al., 2018). Moreover, a method based on protein abundance has been recently developed for accurate assessment of the microbial community structure and species biomass contributions (Kleiner et al., 2017). With the development of tandem LC-MS/MS methods, new opportunities have opened for fast and accurate proteomic analysis. In the last decade, 2D gel electrophoresis has been replaced by more rapid online liquid chromatography separation. A recently published paper evaluates multiple online and offline LC-MS/MS pipelines and provides a decision chart for the most appropriate choice of method for any budget and purpose (Hinzke et al., 2019). Although metaproteomics is a powerful method to study the biochemical functions of microbial communities, it is in its infancy and has some limitations (Abiraami et al., 2020). Therefore, only a few studies used metaproteomics to examine anaerobic hydrocarbon biodegradation.
Benndorf and colleagues investigated the efficiency of metaproteomics on anaerobic benzene degrading communities (Benndorf et al., 2009). They concluded that metaproteomics is a useful method to examine unculturable microbes; however, the protein extraction methods should be adapted to the slow-growing anaerobic microbial communities. Bargiela and colleagues combined metaproteomics and metabolomics in a study of three chronically polluted sites in the Mediterranean sea. Their results revealed the prevalence of C1-compound metabolism. Methane oxidation was detected on the oxygen-rich site while methanol catabolism was observed on all polluted sites (Bargiela et al., 2015). In a recent study, Marozava and colleagues used metagenomics and metaproteomics to establish a specific expression profile of catabolic pathways for aromatics biodegradation by Geobacter metallireducens during sessile growth (Marozava et al., 2020).
The collection of all biochemical molecules, substrates, products, intermediates existing in a living organism is called the metabolome. Similarly to the transcriptome and proteome, it is dynamic in time; moreover the metabolome might be considered as the most sensitive, most dynamic system in the cells. Metabolome, the result of cell metabolism, serves as the “ultimate proof” for biochemical reactions taking place in living systems (Liu and Locasale, 2017).
Metabolome research can be divided into three parts: non-diffusible, diffusible and epi-metabolome. The non-diffusible metabolome will never leave the cell, while the diffusible metabolome can escape the cell occasionally depending on the metabolic rate of the cell. From the three parts, the epi-metabolome has the lowest conversion rate so it can diffuse out, or be actively transported into the environment then captured and further metabolized by other members of the community. The epi-metabolome is the only part of the metabolome which moves freely at the contaminated site. Molecules belonging to the epi-metabolome are metabolized by the microbial network (De Lorenzo, 2008). The redox electron carriers of the epi-metabolome have outstanding importance in the case of exo-electrogenic microbes (see below).
In anaerobic hydrocarbon biodegradation studies, metabolomics is usually used to search for signature molecules of hydrocarbon degradation pathways. The hydrocarbon substituted succinate derivatives are excellent candidates for signature molecules since they are unique to anaerobic hydrocarbon biodegradation (Bian et al., 2014, 2015; Chen et al., 2020). Metabolomic studies are often combined with metagenomic approaches. In a recent study, Dong and colleagues investigated oil seepage in deep-sea sediments and concluded that acetate and hydrogen are central intermediates, supporting metabolic interactions between community members (Dong et al., 2019). Oil companies often use nitrate as an additive to fight off sulfate-reducing bacteria, thus preventing crude oil souring and corrosion of transport and storage vessels. Bonifay and colleagues investigated the corrosion of oil production pipelines with the help of metagenomics combined with metabolomics. They found that the addition of nitrate is not enough to prevent oil pipeline corrosion because sulfate reducers can survive in biofilms, thus evading the effect of nitrate and facilitating oil souring and pipe corrosion (Bonifay et al., 2017).
Microbial biodegradation of hydrocarbons starts with an activation reaction, both aerobically and anaerobically. In aerobic pathways, mono- and dioxygenases catalyze this reaction by utilizing molecular oxygen to synthesize the corresponding alcohol from the hydrocarbon. On the other hand, anaerobic activation of hydrocarbons can happen through several distinct reactions: fumarate addition, oxygen-independent hydroxylation, direct carboxylation, hydration and reverse methanogenesis.
Although the primary topic of this review is the anaerobic biodegradation of hydrocarbons, the importance of hydrocarbon utilization under aerobic conditions cannot be ignored. Thus, in the following paragraphs, a brief summary of aerobic hydrocarbon oxidation pathways and enzymes is given.
The initial reaction of aerobic alkane biodegradation is catalyzed by various monooxygenases (also known as alkane hydroxylases) depending on the chain lengths of the alkanes (Van Beilen and Funhoff, 2007; Wang and Shao, 2013). These include methane monooxygenases [either membrane-bound (pMMO) or soluble (sMMO)], propane and butane monooxygenases (Van Beilen and Funhoff, 2007), the CYP153 family of Cytochrome P450 enzymes (Funhoff et al., 2006) and alkane-1-monooxygenases (AlkB).
The biodegradation of aromatics can be divided into two subsequent pathways (Fuchs et al., 2011). In the peripheral pathway, the aromatic ring is converted into catechol or protocatechuate by ring-hydroxylating dioxygenases or monooxygenases. In the central pathway, the ring is opened by ring-cleaving dioxygenases in ortho meta or para positions. Then the catechol/protocatechuate is transformed into acetaldehyde/pyruvate or into β-ketoadipate, which is further converted into succinate and Ac-CoA.
The most common and best-described anaerobic alkane activation reaction is fumarate addition (Figure 1A). Fumarate is fused with the alkane chain at the second carbon atom, forming a (1-methylalkyl)-succinate. The product is attached to an acetyl-coenzyme A and converted into (2-methylalkyl)-malonyl-CoA through a carbon skeleton rearrangement. Later 4-methylalkyl-CoA is formed and utilized in β-oxidation. In the first two cycles of β-oxidation, acetate and propionate are cleaved off the molecule. The propionate is recycled in the methylmalonyl-CoA pathway to form fumarate, which can be reutilized in (1-methylalkyl)-succinate synthesis (Davidova et al., 2005). The key enzyme of the fumarate addition reaction is identified as a glycyl radical enzyme named as alkyl succinate synthase or (methylalkyl)-succinase (Ass/Mas). The enzyme has three different subunits (AssABC). Mas genes were first described in the denitrifying Aromatoleum sp. strain HxN1 (Grundmann et al., 2008). Two genes (assA1 and assA2), coding for the catalytic subunit of Ass, were found in various loci in the genome of D. alkenivorans AK-01 (Callaghan et al., 2012). Metagenomic analysis revealed that Smithella sp., and Anaerolinea sp. also harbor ass genes (Tan et al., 2014; Rossmassler et al., 2019). In a recent study, ass and bss genes were detected from MAGs of the Asgard archaea (Farag et al., 2020).
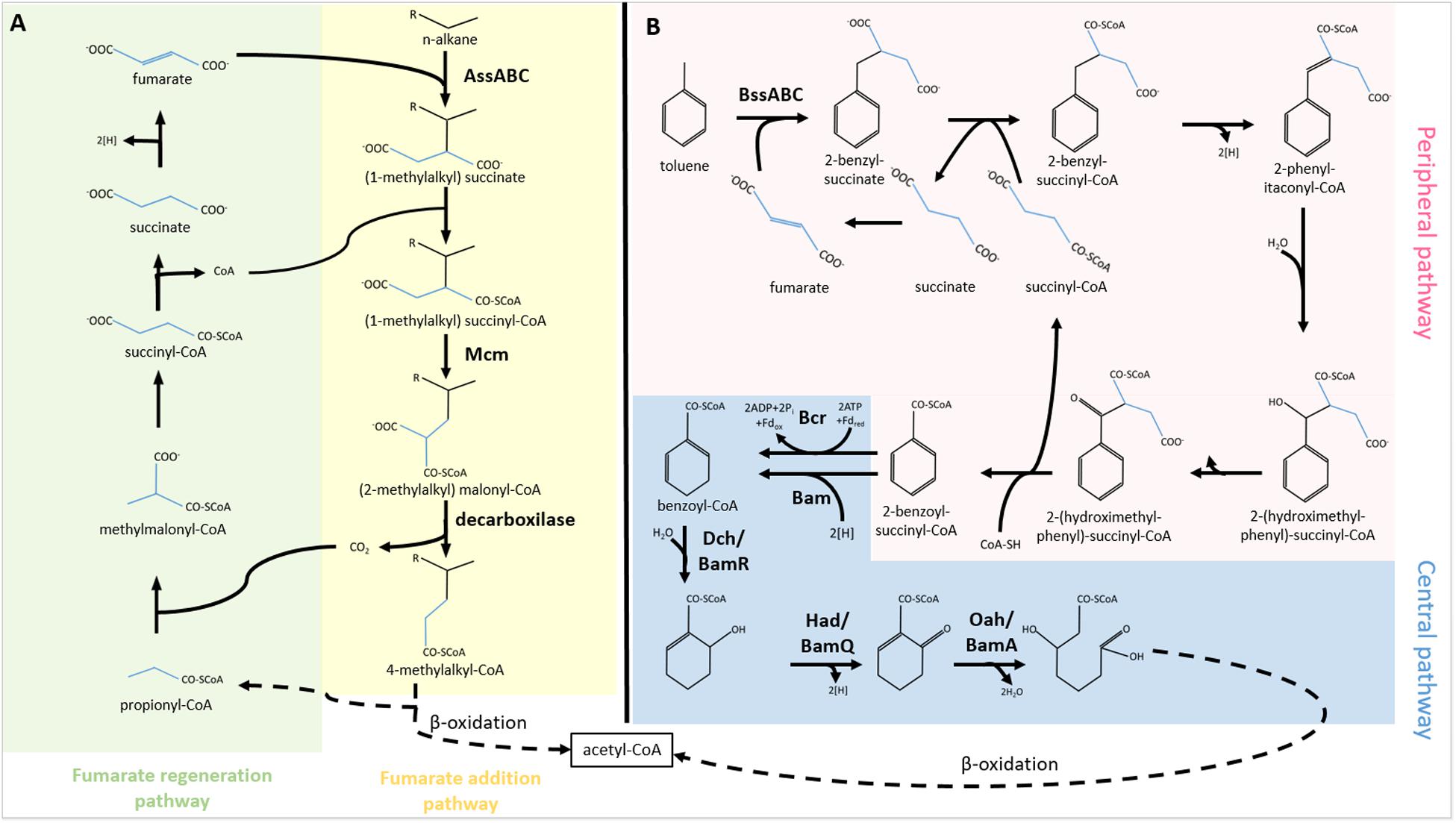
Figure 1. Activation of (A) alkanes and (B) aromatics through fumarate addition and their biodegradation under anaerobic conditions. AssABC, alkylsuccinate synthase; Mcm, methylmalonyl-CoA mutase; BssABC, benzyl-succinate synthase; Bcr/Bam, benzoyl-CoA reductase; Dch/BamR, dienoyl-CoA hydratase; Had/BamQ, hydroxyacyl-CoA dehydrogenase; Oah/BamA, oxoacyl-CoA hydrolase; Fdox, oxidized ferredoxin; Fdred, reduced ferredoxin.
Methanotrophic archaea can oxidize methane under anoxic conditions via reverse methanogenesis. The mechanism was predicted by Hallam and colleagues based on genomic data (Hallam et al., 2004). They found the gene of methyl-CoM reductase (Mcr) in the genome of methanotrophic archaea, which catalyze the final reaction of methanogenesis. The enzyme reduces methyl-CoM, resulting in methane and a heterodisulphide composed of coenzyme M and coenzyme B. Under suitable conditions, the process can be reversed by Mcr to form methyl-CoM. This hypothesis was proven by Scheller and colleagues (Scheller et al., 2010). Metagenomic analysis of a hot spring shed light on the coupling of methane and dissimilatory sulfur metabolism in Korarchaeota (McKay et al., 2019). A combined metagenomic and metatranscriptomic study showed that Candidatus Methanoperedens spp. is capable of reverse methanogenesis during manganese reduction (Leu et al., 2020).
Additionally, an alternative pathway for butane activation by Mcr proteins in thermophilic archaea has been proposed from a metagenomic analysis (Laso-Pérez et al., 2016), indicating possible activation pathways of non-methane alkanes. Wang and colleagues reanalyzed 64 metagenomes and found putative multi-carbon oxidizing mcr genes among many archaeal phyla (Wang et al., 2019). Horizontal gene transfer of mcr genes into the Asgard archaea was justified by metagenome analysis. The authors also suggested the oxidation of short-chain alkanes via alkyl-CoM in Helarchaeota (Seitz et al., 2019).
Two alternative activation pathways of alkanes were proposed in Desulfococcus oleovorans. In the first scenario, the carbon chain is carboxylated at the third carbon atom, forming a 1-ethyl alkanoate, which is further converted into acetate in β-oxidation (Meyer et al., 2003). In the other scenario, a subterminal hydroxylation occurs at C3, which is transformed into a branched fatty acid through a ketone intermediate. Genomic data support the latter since an ethylbenzene dehydrogenase (EBDH)-like enzyme is encoded in the genome of D. oleovorans (Rabus et al., 2016).
Similarly to aerobic biodegradation, utilization of aromatic compounds under anoxic conditions can be split into peripheral and central pathways (Fuchs et al., 2011; Figure 1B). In the peripheral pathways, the activation of the aromatic compound takes place through distinct enzymatic reactions. Under anoxic conditions, the aromatic molecules are converted into the central metabolite, benzoyl-CoA or its derivatives through several steps. First, fumarate is added to the aromatic compound by the glycyl radical enzyme, benzylsuccinate synthase (Bss). The enzyme occurred in several denitrifying bacteria, e.g., Azoarcus sp. (Achong et al., 2001) recently renamed to Aromatoleum sp. (Rabus et al., 2019) and Thauera aromatica (Leuthner and Heider, 2000). The enzyme is encoded by the bssABC genes forming an operon (Leuthner et al., 1998; Hermuth et al., 2002). Bss protein is one of the few enzymes involved in anaerobic hydrocarbon activation of which 3D structure has been determined. Based on X-ray diffraction study, the structure of Bss is a heterohexamer (αβγ)2 (Funk et al., 2014). The small subunits (β and γ) comprise 4Fe-4S clusters coordinated by four cysteines, whilst the large subunit (α) harbors the catalytic site (Funk et al., 2014). Another gene codes for a glycyl radical enzyme-activating protein, BssD (Leuthner et al., 1998; Hermuth et al., 2002). This S-adenosyl-methionine-dependent protein plays a role in the Bss precursor activation (Hermuth et al., 2002). The benzylsuccinate formed in the reaction is further converted into the central metabolite, benzoyl-CoA, by the enzymes encoded in the bbs operon (Leuthner and Heider, 2000).
Furthermore, the activation of polyaromatic hydrocarbons via fumarate addition has been suggested (Annweiler et al., 2000). Another glycyl radical enzyme, naphtyl-2-methylsuccinate synthase (Nms), was identified in a sulfate-reducing enrichment culture (Selesi et al., 2010). Nms is closely related to Bss; however, they are clustered into two separate groups on the phylogenetic tree (Von Netzer et al., 2013; Heider et al., 2016).
On the other hand, the activation of aromatics can be performed through oxygen-independent hydroxylation. The mechanism was studied on ethyl- and propylbenzene. A molybdenum/iron-sulfur/heme-enzyme, ethylbenzene dehydrogenase (EBDH), catalyses the reaction in the periplasm of A. aromaticum EbN1 (Kniemeyer and Heider, 2001). The enzyme consists of three subunits, two of which (αβ) have five iron-sulfur clusters together. The molybdenum-BisMGD cofactor can be found in the α subunit, along with one of the five iron-sulfur clusters (FS0). The γ subunit harbors the heme b cofactor (Kloer et al., 2006). The enzyme can convert a broad range of substrates (Szaleniec et al., 2007; Knack et al., 2012) and has a higher binding affinity toward hydrophobic compounds; nevertheless, substrates with electron-donating substituents in the para position can elevate the conversion rate (Knack et al., 2012). EBDH and similar enzymes, as well as their roles in oxygen-independent hydroxylation, have been reviewed (Heider et al., 2016).
Direct carboxylation of benzene has also been reported in a few studies (Musat and Widdel, 2007; Kunapuli et al., 2008; Abu Laban et al., 2009). However, benzoic acid, which was detected as an intermediate, is also the central metabolite in the anaerobic biodegradation of aromatic compounds (Fuchs et al., 2011); therefore, other activation mechanisms could not be excluded. The proteomic study of an iron-reducing enrichment culture revealed the benzene-dependent induction of carboxylase-like proteins (Abu Laban et al., 2010). Similarly, in a metatranscriptomic study of a nitrate-reducing enrichment, the authors found benzene-induced expression of carboxylases (Luo et al., 2014). These findings support the hypothesis of direct carboxylation as an alternative activation reaction, although no direct evidence for the existence of this mechanism was reported.
There are two different routes for the central pathway for anaerobic biodegradation of aromatics (Figure 1B). One is ATP dependent, whilst the other is ATP independent. The ATP-dependent pathway begins with a reduction reaction catalyzed by a class I benzoyl-CoA reductase (Bcr) (Boll and Fuchs, 1995). The four subunits of the heterotetrameric enzyme are encoded by the bcrABCD genes, which are organized into an operon in Thauera aromatica. The enzyme cleaves ATP into ADP + Pi during the reduction process and accepts ferredoxin as an electron donor. The presence of an ATP-dependent benzoyl-CoA reductase in the hyperthermophilic archaeon Ferroglobus placidus was recently demonstrated (Schmid et al., 2016). Apart from the central dearomatisation reaction, the role of Bcr in dehalogenation of chlorinated, fluorinated and brominated aromatics was shown (Tiedt et al., 2016).
The ATP-independent pathway is catalyzed by a class II benzoyl-CoA reductase (Bam). The function of the Bam protein was first suggested by Laempe and colleagues (Laempe et al., 1999). This enzyme class is found in strictly anaerobic bacteria, including the iron reducer G. metallireducens (Wischgoll et al., 2005). The enzyme is composed of eight subunits which are encoded by bamBCDEFGHI genes in one operon with bamA which is a hydrolase and catalyses the final ring cleavage in the ATP-independent pathway (Löffler et al., 2011). A differential membrane proteome analysis of G. metallireducens revealed the association of Bam protein complex to the membrane (Heintz et al., 2009). The Bam enzyme was identified distinctly in iron-reducing (Wischgoll et al., 2005) and sulfate-reducing bacteria (Kim et al., 2014; Dong et al., 2016). The structure and function of Bam have been reviewed (Boll et al., 2016).
The compositions of microbial communities present in hydrocarbon-contaminated sites are highly dependent on the composition of the pollutants, physico-chemical properties of the environment and electron acceptors available. Electron acceptors, including nitrate, iron and sulfate, can be depleted relatively rapidly. Under these conditions, a syntrophy between hydrocarboclastic bacteria, methanogenic archaea and other microbial partners is established. Nevertheless, syntrophy is a crucial mechanism of anaerobic hydrocarbon biodegradation not just under metanogenic conditions but in the presence of sulfate, ferric ion or even nitrate (Kleinsteuber et al., 2012). Even though syntrophic communities are common under anaerobic hydrocarbon-degrading conditions, there is limited information as regards the interactions between community members (Gieg et al., 2014). With the emergence of new high-throughput sequencing techniques in the last decade, new perspectives have opened for the study of microbial communities. Metagenomic analysis of samples derived from hydrocarbon-contaminated environments highlighted the role of the δ-proteobacterial class Syntrophaceae in hydrocarbon biodegradation under methanogenic conditions (Siddique et al., 2011; An et al., 2013a; Embree et al., 2014; Wawrik et al., 2016). The role of Peptococcaceae in methanogenesis coupled to n-alkane biodegradation has also been endorsed (Abu Laban et al., 2015; Tan et al., 2015; Mohamad Shahimin et al., 2016; Mohamad Shahimin and Siddique, 2017a,b). Also, oil degraders belonging to other bacterial groups, for instance, members of Chloroflexi, like Anaerolinea, have been identified. These bacteria are thought to be scavenging dead cells and metabolites that are derived from hydrocarbon biodegradation (Gieg et al., 2014). The involvement of Anaerolinea in the primary degradation of hydrocarbons has been proposed (Liang et al., 2015, 2016; Mohamad Shahimin and Siddique, 2017a,b). Both acetoclastic and hydrogenotrophic methanogenic archaea are found in hydrocarbon-contaminated soil and sediments. Nonetheless, their ratio depends on the composition of the entire community (Siddique et al., 2011; Wawrik et al., 2016; Mohamad Shahimin and Siddique, 2017a).
The energy transfer between hydrocarbon degraders and methanogenic archaea is complex and not well-understood. According to the model (Embree et al., 2014; Gieg et al., 2014; Wawrik et al., 2016; Figure 2), the hydrocarbon degrader may provide electrons to methanogens through H2/formate. Hydrogen, along with carbon dioxide, is utilized by the hydrogenotrophic methanogenic archaea for methane evolution. Acetoclastic methanogens utilize acetate derived from hydrocarbon biodegradation. Additionally, other microorganisms, such as Desulfuromonas sp. (Wawrik et al., 2016), can also utilize acetate for hydrogen evolution, supplying the hydrogenotrophic methanogens. Hydrogenase and dehydrogenase enzymes are important participants in syntrophic communities (Wawrik et al., 2016), so a statistical method was developed to recognize communities with syntrophic potential from metagenomic data (Oberding and Gieg, 2016).
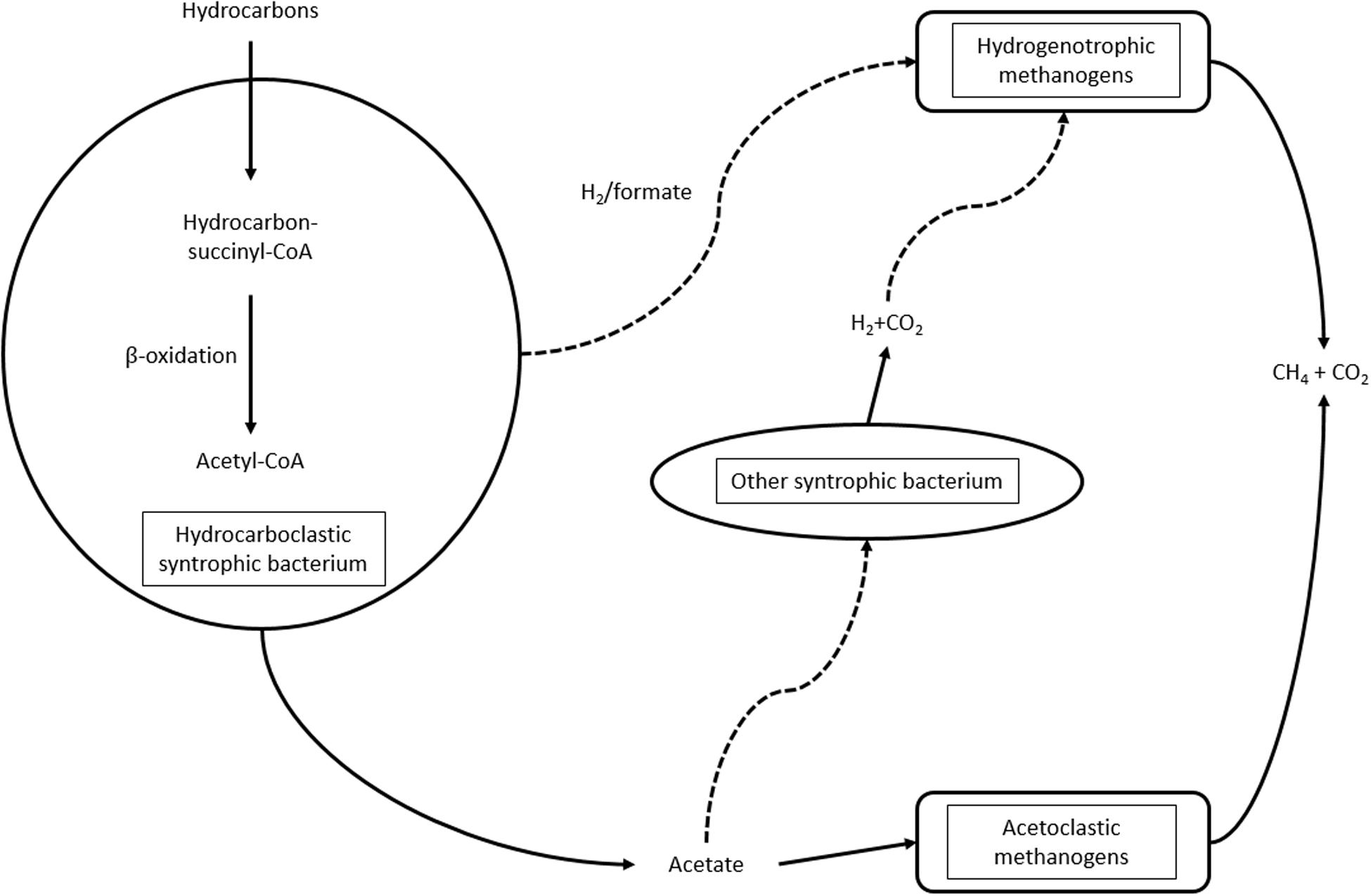
Figure 2. Syntrophic interactions during hydrocarbon biodegradation under methanogenic conditions.
In the case of contaminants spread over a large area, the application of phyto-micro- bioremediation can be a cost and environmentally sound technology. Phytoremediation is a well-known technology in which microorganisms complement the role of plants. The interactions between the microbes and plants can be fruitful cooperation not only in the aerobic zone but the deeper level of soils and waters. Roots of plants provide nutrients and can improve the soil consistency that is especially beneficial in the deeper soil region. There are only a few species of plants with long roots able to achieve the 7–8 m depth (poplar, willow). Spartina species used in the remediation of oil-contaminated wetland enhanced the aerobic biodegradation by transferring oxygen to their roots (Lin and Mendelssohn, 1998).
Bioaugmentation of contaminated sites sometimes leads to unexpected modification in the rhizosphere. Recently published results justify that horizontal gene transfer occurred during rhizoremediation of organic compounds (Fernández et al., 2012; Segura and Ramos, 2013). In these cases, the microbe(s) used for biodegradation can not survive in the target environment, but the genes, metabolic routes of biodegradation are transferred into the local microbes; thus the bioconversion can take place. On the other hand, phytoremediation alone cannot be purely assigned to the metabolic activity of the plants; several studies linked the success of phytoremediation to the microbial activities of the rhizomicrobiome, but more likely it should be studied at metaorganism (host + microbiome) level (Thijs et al., 2016). Since the rhizosphere microbiome is sensitive to both pollutants and treatments, a competitive plant-growth-promoting and catabolic microbiome should be selected for efficient remediation and plant growth (Thijs et al., 2016). However, this makes the picture most complex since host-microbe and microbe-microbe interactions must be considered.
Methanogenic archaea live in syntrophy with bacteria that can break down complex organic compounds, such as plant parts or diesel oil, into simple molecules including acetate. Interspecies electron transfer (IET) is a critical element of syntrophic microbial communities. IET is a mechanism wherein the syntrophic partners share electrons through mediators (Mediated Interspecies Electron Transfer or MIET) or directly (Direct Interspecies Electron Transfer or DIET). Hydrogen functions as the dominant mediator in such communities, even though formate is energetically more favorable (Storck et al., 2016). Apart from this, quinone-mediated interspecies electron transfer (QUIET) was recently proposed for G. metallireducens/G. sulfurreducens co-cultures (Smith et al., 2015). Quinone moieties are abundant in many soil types as components of humic substances (Gralnick and Newman, 2007). In the case of QUIET, these quinones are utilized by the microbes as electron shuttles (Smith et al., 2015). In another study, the transition of electrons from the donor species to the acceptor species via conductive minerals has been reported (Kato et al., 2012). Recent studies disclosed that the DIET process might take place in co-cultures of various microorganisms in which Geobacter species play the leading role as primary degraders and electron donors for methanogens or other syntrophic partners (Rotaru et al., 2014a,b; Wang et al., 2016). Based on this knowledge, Geobacter strains appear to be potential vital partners in anaerobic bioremediation processes. Additionally, a recent study validated that DIET also works with cytochrome type C with no conductive pili between pili-deficient Geobacter species (Liu et al., 2018). The findings propose that DIET is more common and widespread among syntrophic communities than first presumed.
In exceptional cases, such as in seasonally anoxic sediments, the electron donor and the electron acceptor are spatially separated. Members of the Desulfobulbaceae family, however, can bridge the distance between the electron donor and acceptor with long-distance electron transfer (LDET) (Pfeffer et al., 2012; Bjerg et al., 2018). These so-called cable bacteria live in coastal marine or freshwater sediments. They form several centimeter-long filamentous multicellular structures with thousands of cells in them. The electron donor for the reaction is sulfide, which is oxidized by the cable bacteria several centimeters deep in the sediment. Electrons are then transferred via the suboxic zone, where both sulfide and oxygen are lacking. The terminal electron acceptor for cable bacteria is oxygen; however, they are also capable of nitrate respiration (Marzocchi et al., 2014; Seitaj et al., 2015). They have a thick-ridged envelope along the filament. It was suggested that the envelope plays a dual role as a brace and an electron conductor (Cornelissen et al., 2018). Although the exact mechanism of LDET is not well-understood, recent studies have indicated that cytochrome type C plays an essential role in it (Bjerg et al., 2018). Interactions between cable bacteria and other microorganisms have yet to be investigated; nonetheless, several models have been suggested. These models include MIET and DIET, where bacterial partners pass their electrons to cable bacteria (Meysman, 2018). By participating in the sulfur cycle, cable bacteria can facilitate the biodegradation of hydrocarbons and other pollutants by sulfate-reducing microorganisms. Despite the potential of cable bacteria in hydrocarbon removal, only a few studies were performed in this field (Müller et al., 2016; Matturro et al., 2017). This process can be enhanced by applying a bioelectrochemical snorkel, which provides an artificial electron channel toward the oxic zone (Matturro et al., 2017).
An indirect approach for electron transfer is based on bioelectrochemical (BES) systems, such as microbial fuel cells (MFC), microbial electrolysis cells (MEC) and their sediment variants (sMFC and sMEC) (Pisciotta and Dolceamore, 2016). In MFCs, microbes oxidize the organic pollutants and donate electrons to the anode generating current through an external circuit (Morris et al., 2009). In contrast, MEC requires an input of electrical energy to promote reactions otherwise not taking place. MFC might play a dual role; it can serve as an electron acceptor at the anode and produce energy as electric current. The BES systems usually require exoelectrogens microbes, such as G. metallireducens or Shewanella oneidensis, capable of extracellular electron transfer (Pisciotta and Dolceamore, 2016). Nevertheless, the anodic microbial communities used to be adapted to the pollutants and environmental conditions, including the externally added surfactants (Li et al., 2018).
Various omics approaches can be used for studying the composition of the microbial communities, metabolic interactions and networks formed during interspecies electron transfer. For instance, a transcriptomic study showed the importance of pili, flagella and a cytochrome type-c protein during DIET in G. metallireducens/G. sulfurreducens co-cultures. The authors also observed different gene expression patterns between DIET and hydrogen- coupled MIET (Shrestha et al., 2013). A metatranscriptomic analysis further proved the DIET between Geobacter and Methanothrix spp (Holmes et al., 2017). A recent review summarizes how DIET can be detected with meta-omics techniques and also implies the importance of combining omics with other methods to evaluate the metabolic network (Van Steendam et al., 2019). With the combination of genomics and metagenomics with substrate turnover measurements, a new model for energy conservation in cable bacteria was proposed (Müller et al., 2020).
Systems biology integrates the multi-omics data to analyze and build models of complex biological systems, such as microbial consortia; thus, their metabolic networks can be examined as a whole. The use of systems biology approaches in bioremediation is indispensable (Dangi et al., 2019; Jaiswal et al., 2019). Nonetheless, considering only the biological aspects of bioremediation is not enough for a successful treatment. Besides modeling cellular and community functions, geochemical and ecological models of the site should be built in an environmental systems biology approach (Hazen, 2019). The knowledge on metabolic networks allows us to fine-tune the processes in the network. Employing metabolic engineering, we can add delete or duplicate genes or entire gene clusters, alter the regulation of pathways or stimulate pathways with precursors, thus tailor a better, more efficient system (Dangi et al., 2019). Metabolic engineering is a relatively young concept; therefore, it is yet rarely applied in anaerobic hydrocarbon biodegradation. In a recent study, a broad-host-range benzoyl-CoA expression cassette was developed for benzoate biodegradation, which might work in the original host or can also be naturally transferred into the indigenous microbes of the contaminated site (Zamarro et al., 2017). Stimulating metabolic pathways is the other aspect of metabolic engineering. The stimulating effect of activated carbon on anaerobic naphthalene biodegradation was recently demonstrated. Besides, the authors hypothesized that activated carbon also stimulates the DIET (Bonaglia et al., 2020). Stimulating DIET by electroconductive materials may be the driving force of anaerobic biodegradation processes (Aulenta et al., 2020).
Given that hydrocarbons are increasingly spreading into anoxic environments, the necessity of a deeper understanding of the anaerobic degradation pathways is steadily growing.
Investigation of the anaerobic biodegradation of petroleum hydrocarbons has disclosed surprisingly diverse and divergent metabolisms in comparison with aerobic pathways and revealed novel biochemical microbial profiles (Heider et al., 1998). Recent advances widened the scope of anaerobic bioremediation technologies. The exploitation of such degradative capacities may aid in various cases as follows:
(a) Decontamination of anoxic environments, anaerobic treatments of aquifers, oceanic water column, wastewater and sewage sludge. Anaerobic biomineralisation of aliphatic and aromatic compounds is a promising alternative to oxic biodegradation strategies (Holliger and Zehnder, 1996). Because degradation processes under anaerobic conditions are acknowledged to be less efficacious and slower than under aerobic conditions, this is only appropriate at contaminated sites with low air contact or which are barely aerated (Heider et al., 1998). The processes can be accelerated by adding external electron acceptor molecules such as nitrate, sulfate, etc., but these might be quite costly solutions and have severe environmental impacts.
(b) Bioenergy recovery. G. metallireducens GS-15 is a toluene-degrading strain under iron-reducing conditions that was proven to be also an “electricigen.” In other words, this strain may be utilized for low-power generation (Lovley, 2006; Foght, 2008). Syntrophic microbial communities can convert contaminants into methane. This process can also be a promising tool in microbial energy recovery, but unfortunately, it is a relatively slow process (Suflita et al., 2004; Gieg et al., 2008; Gray et al., 2010; Xia et al., 2016). Another promising approach to recover energy derived from the oxidation of organic pollutants applies BES-coupled, especially MFC-coupled bioremediation technologies (Morris et al., 2009; Pisciotta and Dolceamore, 2016; Li et al., 2018). MFCs have dual advantages: (i) the anode can serve as an electron acceptor and thus accelerates the bioremediation processes; (ii) it is integrated into an electric circuit to generate electric current. A severe technical limitation of this approach is to construct an electric network covering large contaminated areas.
(c) The remarkable diversity of biological catalysts involved in the anaerobic metabolism of hydrocarbons offers a broader range of applications by overcoming several limiting abiotic and biotic factors. The use of isolated enzymes mitigates microbial competitiveness, limiting nutrients, etc. However, the biocatalyst approach is still costly (low yield of enzyme production), and the enzyme integrity needs to be improved (Setti et al., 1997; Sutherland et al., 2004; Alcalde et al., 2006; Peixoto et al., 2011).
Many new insights are needed to provide improvements to anaerobic biodegradation treatments. Physicochemical characterization of the polluted sites is a critical step for the application of the most efficacious approach. However, characterization of the natural local microflora is at least as necessary. Employing multi-omics techniques, the composition of the microbial community and the metabolic interactions of the community members, as well as other organisms, such as plants, can be discovered. So, cost-effective strategies can be planned for the remediation of hydrocarbon-contaminated sites. The recovery and the characterization of potential hydrocarbon degraders (in pure and mixed cultures) are important to deepen the understanding of their metabolic pathways, for the enhancement of the bioremediation efficiency and identification of interesting functional genes. In many cases, the microbes actively participating in biodegradation can not be isolated and cultured; however, the omics techniques enable us to characterize the metabolic activities of environmental samples which can be further used as inoculum in other remediation projects.
Furthermore, omics techniques allow us to study the biodegradation processes at the system level, metabolic blocks, “metabolic bricks” can be generated and used in metabolic engineering processes for the construction of targeted or multipurpose strains/microbial consortia.
The horizontal gene transfer events taking place in the environment might spread these metabolic bricks to the indigenous microbes and thus increasing the lifetime of a newly introduced metabolic route in the environment.
The multi-omics era provides a multitude of opportunities for the examination of individual microbes, microbial communities, their metabolism and interactions. Genomics and metagenomics characterize the metabolic potential of the microorganisms; we can identify new microbes in a community and elucidate new pathways and genes which are responsible for hydrocarbon biodegradation. Transcriptomics and metatranscriptomics can further deepen our knowledge by highlighting active pathways in a genome or metagenome under given circumstances. It can even elucidate the role of less abundant members of a microbial community, and it highlights the importance of metabolic networks between community members. Similar results are provided by proteomic and metaproteomic studies, while metabolomics will confirm the chemical reactions occurring during hydrocarbon biodegradation and anaerobic respiration. The omics approaches, alone or in combination, are powerful tools to characterize microbial communities of hydrocarbon-contaminated sites and see the whole network as one big picture. Characterization of the microbiome at polluted sites is a crucial step for the application of the most efficacious remediation approaches. Based on the data provided by omics approaches, cost-effective strategies can be planned for the remediation of hydrocarbon-contaminated sites. The recovery and characterization of potential hydrocarbon-degraders (in pure and mixed cultures) are essential to broaden our understanding of their metabolic pathways, in order to enhance bioremediation efficiency and to identify interesting functional genes. Moreover, in combination with genetic/genomic/metabolic engineering, the stability, the catalytic potential and the enzyme production yield may be increased to promote successful bioremediation strategies. Most of the contaminants are mixtures of various hazardous materials; therefore, complex metabolic activities are required for the complete elimination of these pollutants. The revolution of the new high throughput technologies enables to study the molecular events at the system level and to design synthetic microbiome for targeted challenges, to combine remediation to energy recovery and to redirect the processes from simple elimination of pollutants to production of added-value materials (De Vrieze et al., 2018).
KL wrote the dominant part of the draft manuscript including the chapters on omics technologies. ÁEK and ÁS contributed to the section of “Introduction,” NB and GV were involved in the section of “Electron transfer between bacterial cells during hydrocarbon biodegradation.” TK wrote the draft version of the section “Applications and prospects,” AB and KP contributed to the part of “Metabolic aspects of hydrocarbon biodegradation, KP provided input to the sections “Introduction” and “Phytomicroremediation: plant-microbe, microbe-microbe interactions,” GR contributed to the part of “Syntrophy in hydrocarbon-degrading microbial communities,” “Phytomicroremediation: plant-microbe, microbe-microbe interactions and Conclusion.” KL, GR, and KP conceptualized the manuscript. GR and KP provided critical feedback and made the final improvement of the manuscript. All authors contributed to the figures, tables, and literature search and contributed to the article and approved the submitted version.
The project was supported by the European Union and co-financed by the European Social Fund (grant agreement no. EFOP-3.6.2-16-2017-00010) in the frame of the Széchényi Program of the Hungarian State.
TK was employed by the company Enviroinvest Zrt.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbai, N. S., and Pillay, B. (2013). Analysis of hydrocarbon-contaminated groundwater metagenomes as revealed by high-throughput sequencing. Mol. Biotechnol. 54, 900–912. doi: 10.1007/s12033-012-9639-z
Abiraami, T. V., Singh, S., and Nain, L. (2020). Soil metaproteomics as a tool for monitoring functional microbial communities: promises and challenges. Rev. Environ. Sci. Biotechnol. 19, 73–102. doi: 10.1007/s11157-019-09519-8
Abu Laban, N., Dao, A., Semple, K., and Foght, J. (2015). Biodegradation of C7 and C8 iso -alkanes under methanogenic conditions. Environ. Microbiol. 17, 4898–4915. doi: 10.1111/1462-2920.12643
Abu Laban, N., Selesi, D., Jobelius, C., and Meckenstock, R. U. (2009). Anaerobic benzene degradation by Gram-positive sulfate-reducing bacteria. FEMS Microbiol. Ecol. 68, 300–311. doi: 10.1111/j.1574-6941.2009.00672.x
Abu Laban, N., Selesi, D., Rattei, T., Tischler, P., and Meckenstock, R. U. (2010). Identification of enzymes involved in anaerobic benzene degradation by a strictly anaerobic iron-reducing enrichment culture. Environ. Microbiol. 12:2248. doi: 10.1111/j.1462-2920.2010.02248.x
Achong, G. R., Rodriguez, A. M., and Spormann, A. M. (2001). Benzylsuccinate synthase of Azoarcus sp. strain T: cloning, sequencing, transcriptional organization, and its role in anaerobic toluene and m-xylene mineralization. J. Bacteriol. 183, 6763–6770. doi: 10.1128/JB.183.23.6763-6770.2001
Afgan, E., Baker, D., Batut, B., van den Beek, M., Bouvier, D., Čech, M., et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544. doi: 10.1093/nar/gky379
Aklujkar, M., Krushkal, J., Dibartolo, G., Lapidus, A., Land, M. L., and Lovley, D. R. (2009). The genome sequence of Geobacter metallireducens: Features of metabolism, physiology and regulation common and dissimilar to Geobacter sulfurreducens. BMC Microbiol. 9:1–22. doi: 10.1186/1471-2180-9-109
Alcalde, M., Ferrer, M., Plou, F. J., and Ballesteros, A. (2006). Environmental biocatalysis: from remediation with enzymes to novel green processes. Trends Biotechnol. 24, 281–287. doi: 10.1016/j.tibtech.2006.04.002
Alneberg, J., Bjarnason, B. S., De Bruijn, I., Schirmer, M., Quick, J., Ijaz, U. Z., et al. (2014). Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146. doi: 10.1038/nmeth.3103
Amann, R. I., Ludwig, W., and Schleifer, K. H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59, 143–169.
An, D., Brown, D., Chatterjee, I., Dong, X., Ramos-Padron, E., Wilson, S., et al. (2013a). Microbial community and potential functional gene diversity involved in anaerobic hydrocarbon degradation and methanogenesis in an oil sands tailings pond 1. Genome 56, 612–618. doi: 10.1139/gen-2013-0083
An, D., Caffrey, S. M., Soh, J., Agrawal, A., Brown, D., Budwill, K., et al. (2013b). Metagenomics of hydrocarbon resource environments indicates aerobic taxa and genes to be unexpectedly common. Environ. Sci. Technol. 47, 10708–10717. doi: 10.1021/es4020184
Annweiler, E., Materna, A., Safinowski, M., Kappler, A., Richnow, H. H., Michaelis, W., et al. (2000). Anaerobic degradation of 2-methylnaphthalene by a sulfate-reducing enrichment culture. Appl. Environ. Microbiol. 66, 5329–5333.
Asnicar, F., Thomas, A. M., Beghini, F., Mengoni, C., Manara, S., Manghi, P., et al. (2020). Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat. Commun. 11, 1–10. doi: 10.1038/s41467-020-16366-7
Atashgahi, S., Hornung, B., Van Der Waals, M. J., Da Rocha, U. N., Hugenholtz, F., Nijsse, B., et al. (2018). A benzene-degrading nitrate-reducing microbial consortium displays aerobic and anaerobic benzene degradation pathways. Sci. Rep. 8, 1–12. doi: 10.1038/s41598-018-22617-x
Aulenta, F., Tucci, M., Cruz, V. C., Dolfing, J., Head, I. M., and Rotaru, A. (2020). An underappreciated DIET for anaerobic petroleum hydrocarbon-degrading microbial communities. Microb. Biotechnol. 20, 1751–7915.13654. doi: 10.1111/1751-7915.13654
Ayling, M., Clark, M. D., and Leggett, R. M. (2019). New approaches for metagenome assembly with short reads. Brief. Bioinform. 21, 584–594. doi: 10.1093/bib/bbz020
Bacosa, H. P., Erdner, D. L., Rosenheim, B. E., Shetty, P., Seitz, K. W., Baker, B. J., et al. (2018). Hydrocarbon degradation and response of seafloor sediment bacterial community in the northern Gulf of Mexico to light Louisiana sweet crude oil. ISME J. 12, 2532–2543. doi: 10.1038/s41396-018-0190-1
Bargiela, R., Herbst, F.-A., Martínez-Martínez, M., Seifert, J., Rojo, D., Cappello, S., et al. (2015). Metaproteomics and metabolomics analyses of chronically petroleum-polluted sites reveal the importance of general anaerobic processes uncoupled with degradation. Proteomics 15, 3508–3520. doi: 10.1002/pmic.201400614
BenIsrael, M., Wanner, P., Fernandes, J., Burken, J. G., Aravena, R., Parker, B. L., et al. (2020). Quantification of toluene phytoextraction rates and microbial biodegradation functional profiles at a fractured bedrock phytoremediation site. Sci. Total Environ. 707:135890. doi: 10.1016/j.scitotenv.2019.135890
Benndorf, D., Vogt, C., Jehmlich, N., Schmidt, Y., Thomas, H., Woffendin, G., et al. (2009). Improving protein extraction and separation methods for investigating the metaproteome of anaerobic benzene communities within sediments. Biodegradation 20, 737–750. doi: 10.1007/s10532-009-9261-3
Bian, X. Y., Mbadinga, S. M., Liu, Y. F., Yang, S. Z., Liu, J. F., Ye, R. Q., et al. (2015). Insights into the anaerobic biodegradation pathway of n-Alkanes in oil reservoirs by detection of signature metabolites. Sci. Rep. 5, 1–12. doi: 10.1038/srep09801
Bian, X.-Y., Mbadinga, S. M., Yang, S.-Z., Gu, J.-D., Ye, R.-Q., and Mu, B.-Z. (2014). Synthesis of anaerobic degradation biomarkers Alkyl-, Aryl- and cycloalkylsuccinic acids and their mass spectral characteristics. Eur. J. Mass Spectrom. 20, 287–297. doi: 10.1255/ejms.1280
Bjerg, J. T., Boschker, H. T. S., Larsen, S., Berry, D., Schmid, M., Millo, D., et al. (2018). Long-distance electron transport in individual, living cable bacteria. Proc. Natl. Acad. Sci. U.S.A. 115, 5786–5791. doi: 10.1073/pnas.1800367115
Boll, M., and Fuchs, G. (1995). Benzoyl-coenzyme A reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism. ATP dependence of the reaction, purification and some properties of the enzyme from Thauera aromatica strain K172. Eur. J. Biochem. 234, 921–933.
Boll, M., Einsle, O., Ermler, U., Kroneck, P. M. H., and Ullmann, G. M. (2016). Structure and function of the unusual tungsten enzymes acetylene hydratase and class II benzoyl-coenzyme A reductase. J. Mol. Microbiol. Biotechnol. 26, 119–137. doi: 10.1159/000440805
Boll, M., Löffler, C., Morris, B. E. L., and Kung, J. W. (2014). Anaerobic degradation of homocyclic aromatic compounds via arylcarboxyl-coenzyme A esters: organisms, strategies and key enzymes. Environ. Microbiol. 16, 612–627. doi: 10.1111/1462-2920.12328
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Bonaglia, S., Broman, E., Brindefalk, B., Hedlund, E., Hjorth, T., Rolff, C., et al. (2020). Activated carbon stimulates microbial diversity and PAH biodegradation under anaerobic conditions in oil-polluted sediments. Chemosphere 248:126023. doi: 10.1016/j.chemosphere.2020.126023
Bonifay, V., Wawrik, B., Sunner, J., Snodgrass, E. C., Aydin, E., Duncan, K. E., et al. (2017). Metabolomic and metagenomic analysis of two crude oil production pipelines experiencing differential rates of corrosion. Front. Microbiol. 8:99. doi: 10.3389/fmicb.2017.00099
Brzeszcz, J., and Kaszycki, P. (2018). Aerobic bacteria degrading both n-alkanes and aromatic hydrocarbons: an undervalued strategy for metabolic diversity and flexibility. Biodegradation 29, 359–407. doi: 10.1007/s10532-018-9837-x
Buccitelli, C., and Selbach, M. (2020). mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 21, 630–644. doi: 10.1038/s41576-020-0258-4
Buchfink, B., Xie, C., and Huson, D. H. (2014). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Bukin, Y. S., Galachyants, Y. P., Morozov, I. V., Bukin, S. V., Zakharenko, A. S., and Zemskaya, T. I. (2019). The effect of 16s rRNA region choice on bacterial community metabarcoding results. Sci. Data 6, 1–14. doi: 10.1038/sdata.2019.7
Cai, P., Ning, Z., Zhang, N., Zhang, M., Guo, C., Niu, M., et al. (2019). Insights into biodegradation related metabolism in an abnormally low dissolved inorganic carbon (DIC) petroleum-contaminated aquifer by metagenomics analysis. Microorganisms 7:412. doi: 10.3390/microorganisms7100412
Callaghan, A. V., Morris, B. E. L., Pereira, I. A. C., McInerney, M. J., Austin, R. N., Groves, J. T., et al. (2012). The genome sequence of desulfatibacillum alkenivorans AK-01: a blueprint for anaerobic alkane oxidation. Environ. Microbiol. 14, 101–113. doi: 10.1111/j.1462-2920.2011.02516.x
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Carmona, M., Zamarro, M. T., Blázquez, B., Durante-Rodríguez, G., Juárez, J. F., Valderrama, J. A., et al. (2009). Anaerobic catabolism of aromatic compounds: a genetic and genomic view. Microbiol. Mol. Biol. Rev. 73, 71–133. doi: 10.1128/mmbr.00021-08
Chen, J., Zhou, L., Liu, Y. F., Hou, Z. W., Li, W., Mbadinga, S. M., et al. (2020). Synthesis and mass spectra of rearrangement bio-signature metabolites of anaerobic alkane degradation via fumarate addition. Anal. Biochem. 600, 113746. doi: 10.1016/j.ab.2020.113746
Cornelissen, R., Bøggild, A., Thiruvallur Eachambadi, R., Koning, R. I., Kremer, A., Hidalgo-Martinez, S., et al. (2018). The cell envelope structure of cable bacteria. Front. Microbiol. 9:3044. doi: 10.3389/fmicb.2018.03044
Dangi, A. K., Sharma, B., Hill, R. T., and Shukla, P. (2019). Bioremediation through microbes: systems biology and metabolic engineering approach. Crit. Rev. Biotechnol. 39, 79–98. doi: 10.1080/07388551.2018.1500997
Davidova, I. A., Gieg, L. M., Nanny, M., Kropp, K. G., and Suflita, J. M. (2005). Stable isotopic studies of n-alkane metabolism by a sulfate-reducing bacterial enrichment culture. Appl. Environ. Microbiol. 71, 8174–8182. doi: 10.1128/AEM.71.12.8174-8182.2005
De Lorenzo, V. (2008). Systems biology approaches to bioremediation. Curr. Opin. Biotechnol. 19, 579–589. doi: 10.1016/j.copbio.2008.10.004
De Vrieze, J., Boon, N., and Verstraete, W. (2018). Taking the technical microbiome into the next decade. Environ. Microbiol. 20, 1991–2000. doi: 10.1111/1462-2920.14269
Denonfoux, J., Parisot, N., Dugat-Bony, E., Biderre-Petit, C., Boucher, D., Morgavi, D. P., et al. (2013). Gene capture coupled to high-throughput sequencing as a strategy for targeted metagenome exploration. DNA Res. 20, 185–196. doi: 10.1093/dnares/dst001
Di Donato, R. J., Young, N. D., Butler, J. E., Chin, K.-J., Hixson, K. K., Mouser, P., et al. (2010). Genome sequence of the deltaproteobacterial strain NaphS2 and analysis of differential gene expression during anaerobic growth on naphthalene. PLoS One 5:e14072. doi: 10.1371/journal.pone.0014072
Dong, X., Dröge, J., von Toerne, C., Marozava, S., McHardy, A. C., and Meckenstock, R. U. (2016). Reconstructing metabolic pathways of a member of the genus Pelotomaculum suggesting its potential to oxidize benzene to carbon dioxide with direct reduction of sulfate. FEMS Microbiol. Ecol. 62:fiw254. doi: 10.1093/femsec/fiw254
Dong, X., Greening, C., Rattray, J. E., Chakraborty, A., Chuvochina, M., Mayumi, D., et al. (2019). Metabolic potential of uncultured bacteria and archaea associated with petroleum seepage in deep-sea sediments. Nat. Commun. 10, 1–12. doi: 10.1038/s41467-019-09747-0
Embree, M., Nagarajan, H., Movahedi, N., Chitsaz, H., and Zengler, K. (2014). Single-cell genome and metatranscriptome sequencing reveal metabolic interactions of an alkane-degrading methanogenic community. ISME J. 8, 757–767. doi: 10.1038/ismej.2013.187
Espínola, F., Dionisi, H. M., Borglin, S., Brislawn, C. J., Jansson, J. K., Mac Cormack, W. P., et al. (2018). Metagenomic analysis of subtidal sediments from polar and subpolar coastal environments highlights the relevance of anaerobic hydrocarbon degradation processes. Microb. Ecol. 75, 123–139. doi: 10.1007/s00248-017-1028-5
Falk, N., Reid, T., Skoyles, A., Grgicak-Mannion, A., Drouillard, K., and Weisener, C. G. (2019). Microbial metatranscriptomic investigations across contaminant gradients of the Detroit River. Sci. Total Environ. 690, 121–131. doi: 10.1016/j.scitotenv.2019.06.451
Farag, I. F., Biddle, J. F., Zhao, R., Martino, A. J., House, C. H., and León-Zayas, R. I. (2020). Metabolic potentials of archaeal lineages resolved from metagenomes of deep Costa Rica sediments. ISME J. 14, 1345–1358. doi: 10.1038/s41396-020-0615-5
Fernández, M., Niqui-Arroyo, J. L., Conde, S., Ramos, J. L., and Duque, E. (2012). Enhanced tolerance to naphthalene and enhanced rhizoremediation performance for Pseudomonas putida KT2440 via the NAH7 catabolic plasmid. Appl. Environ. Microbiol. 78, 5104–5110. doi: 10.1128/AEM.00619-12
Foght, J. (2008). Anaerobic biodegradation of aromatic hydrocarbons: pathways and prospects. J. Mol. Microbiol. Biotechnol. 15, 93–120. doi: 10.1159/000121324
François, B., Armand, M., Marie-José, D., and Thouand, G. (2016). From laboratory to environmental conditions: a new approach for chemical’s biodegradability assessment. Environ. Sci. Pollut. Res. 23, 18684–18693. doi: 10.1007/s11356-016-7062-x
Fuchs, G., Boll, M., and Heider, J. (2011). Microbial degradation of aromatic compounds – from one strategy to four. Nat. Rev. Microbiol. 9, 803–816. doi: 10.1038/nrmicro2652
Funhoff, E. G., Bauer, U., García-Rubio, I., Witholt, B., and van Beilen, J. B. (2006). CYP153A6, a soluble P450 oxygenase catalyzing terminal-alkane hydroxylation. J. Bacteriol. 188, 5220–5227. doi: 10.1128/JB.00286-06
Funk, M. A., Judd, E. T., Marsh, E. N. G., Elliott, S. J., and Drennan, C. L. (2014). Structures of benzylsuccinate synthase elucidate roles of accessory subunits in glycyl radical enzyme activation and activity. Proc. Natl. Acad. Sci. 111, 10161–10166. doi: 10.1073/pnas.1405983111
Geng, S., Cao, W., Yuan, J., Wang, Y., Guo, Y., Ding, A., et al. (2020). Microbial diversity and co-occurrence patterns in deep soils contaminated by polycyclic aromatic hydrocarbons (PAHs). Ecotoxicol. Environ. Saf. 203:110931. doi: 10.1016/j.ecoenv.2020.110931
Gieg, L. M., Duncan, K. E., and Suflita, J. M. (2008). Bioenergy production via microbial conversion of residual oil to natural gas. Appl. Environ. Microbiol. 74, 3022–3029. doi: 10.1128/AEM.00119-08
Gieg, L. M., Fowler, S. J., and Berdugo-Clavijo, C. (2014). Syntrophic biodegradation of hydrocarbon contaminants. Curr. Opin. Biotechnol. 27:1016. doi: 10.1016/j.copbio.2013.09.002
Gralnick, J. A., and Newman, D. K. (2007). Extracellular respiration. Mol. Microbiol. 65, 1–11. doi: 10.1111/j.1365-2958.2007.05778.x
Graspeuntner, S., Loeper, N., Künzel, S., Baines, J. F., and Rupp, J. (2018). Selection of validated hypervariable regions is crucial in 16S-based microbiota studies of the female genital tract. Sci. Rep. 8, 1–7. doi: 10.1038/s41598-018-27757-8
Gray, N. D., Sherry, A., Hubert, C., Dolfing, J., and Head, I. M. (2010). Methanogenic degradation of petroleum hydrocarbons in subsurface environments: remediation, heavy oil formation, and energy recovery. Adv. Appl. Microbiol. 72, 137–161. doi: 10.1016/S0065-2164(10)72005-0
Grundmann, O., Behrends, A., Rabus, R., Amann, J., Halder, T., Heider, J., et al. (2008). Genes encoding the candidate enzyme for anaerobic activation of n-alkanes in the denitrifying bacterium, strain HxN1. Environ. Microbiol. 10, 376–385. doi: 10.1111/j.1462-2920.2007.01458.x
Hall, M., and Beiko, R. G. (2018). 16S rRNA Gene Analysis with QIIME2: Methods in Molecular Biology. Totowa, NJ: Humana Press Inc, 113–129. doi: 10.1007/978-1-4939-8728-3_8
Hallam, S. J., Putnam, N., Preston, C. M., Detter, J. C., Rokhsar, D., Richardson, P. M., et al. (2004). Reverse methanogenesis: testing the hypothesis with environmental genomics. Science 305, 1457–1462. doi: 10.1126/science.1100025
Han, X., Wang, F., Zhang, D., Feng, T., and Zhang, L. (2021). Nitrate-assisted biodegradation of polycyclic aromatic hydrocarbons (PAHs) in the water-level-fluctuation zone of the three Gorges Reservoir, China: Insights from in situ microbial interaction analyses and a microcosmic experiment. Environ. Pollut. 268:115693. doi: 10.1016/j.envpol.2020.115693
Håvelsrud, O. E., Haverkamp, T. H. A., Kristensen, T., Jakobsen, K. S., and Rike, A. G. (2011). A metagenomic study of methanotrophic microorganisms in Coal Oil Point seep sediments. BMC Microbiol. 11:221. doi: 10.1186/1471-2180-11-221
Hawley, E. R., Malfatti, S. A., Pagani, I., Huntemann, M., Chen, A., Foster, B., et al. (2014). Metagenomes from two microbial consortia associated with Santa Barbara seep oil. Mar. Genomics 18, 97–99. doi: 10.1016/j.margen.2014.06.003
Hawley, E. R., Piao, H., Scott, N. M., Malfatti, S., Pagani, I., Huntemann, M., et al. (2013). Metagenomic analysis of microbial consortium from natural crude oil that seeps into the marine ecosystem offshore Southern California. Stand. Genomic Sci. 9:1259. doi: 10.4056/sigs.5029016
Hazen, T. C. (2019). Environmental Systems Biology Approach to Bioremediation. Cham: Springer, 103–127. doi: 10.1007/978-3-030-10777-2_4
He, Y., Feng, X., Fang, J., Zhang, Y., and Xiao, X. (2015). Metagenome and metatranscriptome revealed a highly active and intensive sulfur cycle in an oil-immersed hydrothermal chimney in Guaymas Basin. Front. Microbiol. 6:1236. doi: 10.3389/fmicb.2015.01236
He, Y., Xiao, X., and Wang, F. (2013). Metagenome reveals potential microbial degradation of hydrocarbon coupled with sulfate reduction in an oil-immersed chimney from Guaymas Basin. Front. Microbiol. 4:148. doi: 10.3389/fmicb.2013.00148
Hegedüs, B., Kós, P. B., Bende, G., Bounedjoum, N., Maróti, G., Laczi, K., et al. (2018). Starvation- and xenobiotic-related transcriptomic responses of the sulfanilic acid-degrading bacterium, Novosphingobium resinovorum SA1. Appl. Microbiol. Biotechnol. 102, 305–318. doi: 10.1007/s00253-017-8553-5
Heider, J., Spormann, A. M., Beller, H. R., and Widdel, F. (1998). Anaerobic bacterial metabolism of hydrocarbons. FEMS Microbiol. Rev. 22, 459–473. doi: 10.1111/j.1574-6976.1998.tb00381.x
Heider, J., Szaleniec, M., Martins, B. M., Seyhan, D., Buckel, W., and Golding, B. T. (2016). Structure and function of benzylsuccinate synthase and related fumarate-adding glycyl radical enzymes. J. Mol. Microbiol. Biotechnol. 26, 29–44. doi: 10.1159/000441656
Heintz, D., Gallien, S., Wischgoll, S., Ullmann, A. K., Schaeffer, C., Kretzschmar, A. K., et al. (2009). Differential membrane proteome analysis reveals novel proteins involved in the degradation of aromatic compounds in Geobacter metallireducens. Mol. Cell. Proteomics 8, 2159–2169. doi: 10.1074/mcp.M900061-MCP200
Herath, A., Wawrik, B., Qin, Y., Zhou, J., and Callaghan, A. V. (2016). Transcriptional response of Desulfatibacillum alkenivorans AK-01 to growth on alkanes: insights from RT-qPCR and microarray analyses. FEMS Microbiol. Ecol. 92:fiw062. doi: 10.1093/femsec/fiw062
Hermuth, K., Leuthner, B., and Heider, J. (2002). Operon structure and expression of the genes for benzylsuccinate synthase in Thauera aromatica strain K172. Arch. Microbiol. 177, 132–138. doi: 10.1007/s00203-001-0375-1
Hinzke, T., Kouris, A., Hughes, R. A., Strous, M., and Kleiner, M. (2019). More is not always better: evaluation of 1D and 2D-LC-MS/MS methods for metaproteomics. Front. Microbiol. 10:238. doi: 10.3389/fmicb.2019.00238
Holliger, C., and Zehnder, A. J. (1996). Anaerobic biodegradation of hydrocarbons. Curr. Opin. Biotechnol. 7, 326–330. doi: 10.1016/S0958-1669(96)80039-5
Holmes, D. E., Shrestha, P. M., Walker, D. J. F., Dang, Y., Nevin, K. P., Woodard, T. L., et al. (2017). Metatranscriptomic evidence for direct interspecies electron transfer between Geobacter and Methanothrix species in methanogenic rice paddy soils. Appl. Environ. Microbiol. 83, 223–240. doi: 10.1128/AEM.00223-17
Hu, P., Tom, L., Singh, A., Thomas, B. C., Baker, B. J., Piceno, Y. M., et al. (2016). Genome-resolved metagenomic analysis reveals roles for candidate phyla and other microbial community members in biogeochemical transformations in oil reservoirs. MBio 7:e01669-15. doi: 10.1128/mBio.01669-15
Hua, F., and Wang, H. Q. (2014). Uptake and trans-membrane transport of petroleum hydrocarbons by microorganisms. Biotechnol. Biotechnol. Equip. 28, 165–175. doi: 10.1080/13102818.2014.906136
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Huson, D. H., Beier, S., Flade, I., Górska, A., El-Hadidi, M., Mitra, S., et al. (2016). MEGAN community edition–interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 12:4957. doi: 10.1371/journal.pcbi.1004957
Jaiswal, S., Singh, D. K., and Shukla, P. (2019). Gene editing and systems biology tools for pesticide bioremediation: a review. Front. Microbiol. 10:87. doi: 10.3389/fmicb.2019.00087
Jehmlich, N., Vogt, C., Lünsmann, V., Richnow, H. H., and von Bergen, M. (2016). Protein-SIP in environmental studies. Curr. Opin. Biotechnol. 41, 26–33. doi: 10.1016/j.copbio.2016.04.010
Jiang, K., Sanseverino, J., Chauhan, A., Lucas, S., Copeland, A., Lapidus, A., et al. (2012). Complete genome sequence of Thauera aminoaromatica strain MZ1T. Stand. Genomic Sci. 6, 325–335. doi: 10.4056/sigs.2696029
Jiang, Y., Brassington, K. J., Prpich, G., Paton, G. I., Semple, K. T., Pollard, S. J. T., et al. (2016). Insights into the biodegradation of weathered hydrocarbons in contaminated soils by bioaugmentation and nutrient stimulation. Chemosphere 161, 300–307. doi: 10.1016/j.chemosphere.2016.07.032
Johnson, J. S., Spakowicz, D. J., Hong, B. Y., Petersen, L. M., Demkowicz, P., Chen, L., et al. (2019). Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 10, 1–11. doi: 10.1038/s41467-019-13036-1
Jung, M. Y., Kim, S. J., Kim, J. G., Hong, H., Gwak, J. H., Park, S. J., et al. (2018). Comparative genomic analysis of Geosporobacter ferrireducens and its versatility of anaerobic energy metabolism. J. Microbiol. 56, 365–371. doi: 10.1007/s12275-018-7451-6
Kang, D. D., Froula, J., Egan, R., and Wang, Z. (2015). MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015:e1165. doi: 10.7717/peerj.1165
Kang, D., Li, F., Kirton, E. S., Thomas, A., Egan, R. S., An, H., et al. (2019). MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7:e7359. doi: 10.7717/peerj.7359
Karigar, C. S., and Rao, S. S. (2011). Role of microbial enzymes in the bioremediation of pollutants: a review. Enzyme Res. 2011:805187. doi: 10.4061/2011/805187
Kato, S., Hashimoto, K., and Watanabe, K. (2012). Microbial interspecies electron transfer via electric currents through conductive minerals. Proc. Natl. Acad. Sci. U.S.A. 109, 10042–10046. doi: 10.1073/pnas.1117592109
Keegan, K. P., Glass, E. M., and Meyer, F. (2016). MG-RAST, A Metagenomics Service for Analysis of Microbial Community Structure and Function: Methods in Molecular Biology. Totowa, NJ: Humana Press Inc, 207–233. doi: 10.1007/978-1-4939-3369-3_13
Kim, S.-J., Park, S.-J., Jung, M.-Y., Kim, J.-G., Min, U.-G., Hong, H.-J., et al. (2014). Draft genome sequence of an aromatic compound-degrading bacterium, Desulfobacula sp. TS, belonging to the Deltaproteobacteria. FEMS Microbiol. Lett. 360, 9–12. doi: 10.1111/1574-6968.12591
Kimes, N. E., Callaghan, A. V., Suflita, J. M., and Morris, P. J. (2014). Microbial transformation of the deepwater horizon oil spill-past, present, and future perspectives. Front. Microbiol. 5:603. doi: 10.3389/fmicb.2014.00603
Kleiner, M., Dong, X., Hinzke, T., Wippler, J., Thorson, E., Mayer, B., et al. (2018). Metaproteomics method to determine carbon sources and assimilation pathways of species in microbial communities. Proc. Natl. Acad. Sci. U.S.A. 115, E5576–E5584. doi: 10.1073/pnas.1722325115
Kleiner, M., Thorson, E., Sharp, C. E., Dong, X., Liu, D., Li, C., et al. (2017). Assessing species biomass contributions in microbial communities via metaproteomics. Nat. Commun. 8, 1–14. doi: 10.1038/s41467-017-01544-x
Kleinsteuber, S., Schleinitz, K. M., and Vogt, C. (2012). Key players and team play: Anaerobic microbial communities in hydrocarbon-contaminated aquifers. Appl. Microbiol. Biotechnol. 94, 851–873. doi: 10.1007/s00253-012-4025-0
Kloer, D. P., Hagel, C., Heider, J., and Schulz, G. E. (2006). Crystal structure of ethylbenzene dehydrogenase from aromatoleum aromaticum. Structure 14, 1377–1388. doi: 10.1016/j.str.2006.07.001
Knack, D., Hagel, C., Szaleniec, M., Dudzik, A., Salwinski, A., and Heider, J. (2012). Substrate and inhibitor spectra of ethylbenzene dehydrogenase: perspectives on application potential and catalytic mechanism. Appl. Environ. Microbiol. 78, 6475–6482. doi: 10.1128/AEM.01551-12
Kniemeyer, O., and Heider, J. (2001). Ethylbenzene dehydrogenase, a novel hydrocarbon-oxidizing molybdenum/iron-sulfur/heme enzyme. J. Biol. Chem. 276, 21381–21386. doi: 10.1074/jbc.M101679200
Kuang, S., Su, Y., Wang, H., Yu, W., Lang, Q., and Matangi, R. (2018). Soil microbial community structure and diversity around the aging oil sludge in yellow river delta as determined by high-throughput sequencing. Archaea 2018:7861805. doi: 10.1155/2018/7861805
Kunapuli, U., Griebler, C., Beller, H. R., and Meckenstock, R. U. (2008). Identification of intermediates formed during anaerobic benzene degradation by an iron-reducing enrichment culture. Environ. Microbiol. 10, 1703–1712. doi: 10.1111/j.1462-2920.2008.01588.x
Kuntze, K., Shinoda, Y., Moutakki, H., McInerney, M. J., Vogt, C., Richnow, H. H., et al. (2008). 6-Oxocyclohex-1-ene-1-carbonyl-coenzyme A hydrolases from obligately anaerobic bacteria: characterization and identification of its gene as a functional marker for aromatic compounds degrading anaerobes. Environ. Microbiol. 10, 1547–1556. doi: 10.1111/j.1462-2920.2008.01570.x
Laczi, K., Kis, Á, Horváth, B., Maróti, G., Hegedüs, B., Perei, K., et al. (2015). Metabolic responses of Rhodococcus erythropolis PR4 grown on diesel oil and various hydrocarbons. Appl. Microbiol. Biotechnol. 99, 9745–9759. doi: 10.1007/s00253-015-6936-z
Laempe, D., Jahn, M., and Fuchs, G. (1999). 6-Hydroxycyclohex-1-ene-1-carbonyl-CoA dehydrogenase and 6-oxocyclohex- 1-ene-1-carbonyl-CoA hydrolase, enzymes of the benzoyl-CoA pathway of anaerobic aromatic metabolism in the denitrifying bacterium Thauera aromatica. Eur. J. Biochem. 263, 420–429. doi: 10.1046/j.1432-1327.1999.00504.x
Laso-Pérez, R., Hahn, C., Van Vliet, D. M., Tegetmeyer, H. E., Schubotz, F., Smit, N. T., et al. (2019). Anaerobic degradation of non-methane alkanes by “candidatus methanoliparia” in hydrocarbon seeps of the gulf of Mexico. MBio 10, 1814–1819. doi: 10.1128/mBio.01814-19
Laso-Pérez, R., Wegener, G., Knittel, K., Widdel, F., Harding, K. J., Krukenberg, V., et al. (2016). Thermophilic archaea activate butane via alkyl-coenzyme M formation. Nature 539, 396–401. doi: 10.1038/nature20152
Ławniczak, Ł, Woźniak-Karczewska, M., Loibner, A. P., Heipieper, H. J., and Chrzanowski, Ł (2020). Microbial degradation of hydrocarbons—basic principles for bioremediation: a review. Molecules 25:856. doi: 10.3390/molecules25040856
Lawson, C. E., Strachan, C. R., Williams, D. D., Koziel, S., Hallam, S. J., and Budwill, K. (2015). Patterns of endemism and habitat selection in coalbed microbial communities. Appl. Environ. Microbiol. 81, 7924–7937. doi: 10.1128/AEM.01737-15
Leu, A. O., Cai, C., McIlroy, S. J., Southam, G., Orphan, V. J., Yuan, Z., et al. (2020). Anaerobic methane oxidation coupled to manganese reduction by members of the Methanoperedenaceae. ISME J. 14, 1030–1041. doi: 10.1038/s41396-020-0590-x
Leuthner, B., and Heider, J. (2000). Anaerobic toluene catabolism of Thauera aromatica: the bbs operon codes for enzymes of β oxidation of the intermediate benzylsuccinate. J. Bacteriol. 182, 272–277. doi: 10.1128/JB.182.2.272-277.2000
Leuthner, B., Leutwein, C., Schulz, H., Horth, P., Haehnel, W., Schiltz, E., et al. (1998). Biochemical and genetic characterization of benzylsuccinate synthase from Thauera aromatica: a new glycyl radical enzyme catalysing the first step in anaerobic toluene metabolism. Mol. Microbiol. 28, 615–628. doi: 10.1046/j.1365-2958.1998.00826.x
Li, D., Liu, C.-M., Luo, R., Sadakane, K., and Lam, T.-W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, X., Zhang, X., Li, L., Lin, C., Dong, W., Shen, W., et al. (2020). Anaerobic biodegradation of pyrene by Klebsiella sp. LZ6 and its proposed metabolic pathway. Environ. Technol. (U. K.) 41, 2130–2139. doi: 10.1080/09593330.2018.1556348
Li, X., Zhao, Q., Wang, X., Li, Y., and Zhou, Q. (2018). Surfactants selectively reallocated the bacterial distribution in soil bioelectrochemical remediation of petroleum hydrocarbons. J. Hazard. Mater. 344, 23–32. doi: 10.1016/j.jhazmat.2017.09.050
Liang, B., Wang, L.-Y., Mbadinga, S. M., Liu, J.-F., Yang, S.-Z., Gu, J.-D., et al. (2015). Anaerolineaceae and Methanosaeta turned to be the dominant microorganisms in alkanes-dependent methanogenic culture after long-term of incubation. AMB Express 5:117. doi: 10.1186/s13568-015-0117-4
Liang, B., Wang, L.-Y., Zhou, Z., Mbadinga, S. M., Zhou, L., Liu, J.-F., et al. (2016). High frequency of thermodesulfovibrio spp. and anaerolineaceae in association with methanoculleus spp. in a long-term incubation of n-alkanes-degrading methanogenic enrichment culture. Front. Microbiol. 7:1431. doi: 10.3389/fmicb.2016.01431
Lim, M. W., Lau, E., Von, and Poh, P. E. (2016). A comprehensive guide of remediation technologies for oil contaminated soil — Present works and future directions. Mar. Pollut. Bull. 109, 14–45. doi: 10.1016/j.marpolbul.2016.04.023
Lin, Q., and Mendelssohn, I. A. (1998). The combined effects of phytoremediation and biostimulation in enhancing habitat restoration and oil degradation of petroleum contaminated wetlands. Ecol. Eng. 10, 263–274. doi: 10.1016/S0925-8574(98)00015-9
Liu, X., and Locasale, J. W. (2017). Metabolomics: a primer. Trends Biochem. Sci. 42, 274–284. doi: 10.1016/j.tibs.2017.01.004
Liu, X., Zhuo, S., Rensing, C., and Zhou, S. (2018). Syntrophic growth with direct interspecies electron transfer between pili-free Geobacter species. ISME J. 12, 2142–2151. doi: 10.1038/s41396-018-0193-y
Liu, Y. F., Qi, Z. Z., Shou, L., Bin, Liu, J. F., Yang, S. Z., et al. (2019). Anaerobic hydrocarbon degradation in candidate phylum ‘Atribacteria’ (JS1) inferred from genomics. ISME J. 13, 2377–2390. doi: 10.1038/s41396-019-0448-2
Löffler, C., Kuntze, K., Vazquez, J. R., Rugor, A., Kung, J. W., Böttcher, A., et al. (2011). Occurrence, genes and expression of the W/Se-containing class II benzoyl-coenzyme A reductases in anaerobic bacteria. Environ. Microbiol. 13, 696–709. doi: 10.1111/j.1462-2920.2010.02374.x
Löffler, F. E., and Edwards, E. A. (2006). Harnessing microbial activities for environmental cleanup. Curr. Opin. Biotechnol. 17, 274–284. doi: 10.1016/j.copbio.2006.05.001
Lovley, D. R. (2003). Cleaning up with genomics: applying molecular biology to bioremediation. Nat. Rev. Microbiol. 1, 35–44. doi: 10.1038/nrmicro731
Lovley, D. R. (2006). Bug juice: harvesting electricity with microorganisms. Nat. Rev. Microbiol. 4, 497–508. doi: 10.1038/nrmicro1442
Lueders, T. (2017). The ecology of anaerobic degraders of BTEX hydrocarbons in aquifers. FEMS Microbiol. Ecol. 93:Fiw220. doi: 10.1093/femsec/fiw220
Luo, F., Gitiafroz, R., Devine, C. E., Gong, Y., Hug, L. A., Raskin, L., et al. (2014). Metatranscriptome of an anaerobic benzene-degrading, nitrate-reducing enrichment culture reveals involvement of carboxylation in benzene ring activation. Appl. Environ. Microbiol. 80, 4095–4107. doi: 10.1128/AEM.00717-14
Marić, N., Štrbaèki, J., Kurilić, S., Beškoski, V. P., Nikić, Z., Ignjatović, S., et al. (2019). Hydrochemistry of groundwater contaminated by petroleum hydrocarbons: the impact of biodegradation (Vitanovac, Serbia). Environ. Geochem. Health 19, 1–15. doi: 10.1007/s10653-019-00462-9
Marozava, S., Merl-Pham, J., Müller, H., and Meckenstock, R. U. (2020). Adaptation of carbon source utilization patterns of geobacter metallireducens during sessile growth. Front. Microbiol. 11:1271. doi: 10.3389/fmicb.2020.01271
Martín-Moldes, Z., Zamarro, M. T., del Cerro, C., Valencia, A., Gómez, M. J., Arcas, A., et al. (2015). Whole-genome analysis of Azoarcus sp. strain CIB provides genetic insights to its different lifestyles and predicts novel metabolic features. Syst. Appl. Microbiol. 38, 462–471. doi: 10.1016/j.syapm.2015.07.002
Marzocchi, U., Trojan, D., Larsen, S., Louise Meyer, R., Peter Revsbech, N., Schramm, A., et al. (2014). Electric coupling between distant nitrate reduction and sulfide oxidation in marine sediment. ISME J. 8, 1682–1690. doi: 10.1038/ismej.2014.19
Mattes, T. E., Alexander, A. K., Richardson, P. M., Munk, A. C., Han, C. S., Stothard, P., et al. (2008). The genome of Polaromonas sp. strain JS666: Insights into the evolution of a hydrocarbon- and xenobiotic-degrading bacterium, and features of relevance to biotechnology. Appl. Environ. Microbiol. 74, 6405–6416. doi: 10.1128/AEM.00197-08
Matturro, B., Cruz Viggi, C., Aulenta, F., and Rossetti, S. (2017). Cable bacteria and the bioelectrochemical snorkel: the natural and engineered facets playing a role in hydrocarbons degradation in marine sediments. Front. Microbiol. 8:952. doi: 10.3389/fmicb.2017.00952
McKay, L. J., Dlakić, M., Fields, M. W., Delmont, T. O., Eren, A. M., Jay, Z. J., et al. (2019). Co-occurring genomic capacity for anaerobic methane and dissimilatory sulfur metabolisms discovered in the Korarchaeota. Nat. Microbiol. 4, 614–622. doi: 10.1038/s41564-019-0362-4
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Meckenstock, R. U., Elsner, M., Griebler, C., Lueders, T., Stumpp, C., Aamand, J., et al. (2015). Biodegradation: updating the concepts of control for microbial cleanup in contaminated aquifers. Environ. Sci. Technol. 49, 7073–7081. doi: 10.1021/acs.est.5b00715
Mejeha, O. K., Head, I. M., Sherry, A., McCann, C. M., Leary, P., Jones, D. M., et al. (2019). Beyond N and P: the impact of Ni on crude oil biodegradation. Chemosphere 237:124545. doi: 10.1016/j.chemosphere.2019.124545
Menzel, P., Ng, K. L., and Krogh, A. (2016). Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 7, 1–9. doi: 10.1038/ncomms11257
Meyer, F., Goesmann, A., McHardy, A. C., Bartels, D., Bekel, T., Clausen, J., et al. (2003). GenDB–an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 31, 2187–2195. doi: 10.1093/nar/gkg312
Meyer-Cifuentes, I., Gruhl, S., Haange, S.-B., Lünsmann, V., Jehmlich, N., von Bergen, M., et al. (2020). Benzylsuccinate synthase is post-transcriptionally regulated in the toluene-degrading denitrifier Magnetospirillum sp. Strain 15-1. Microorganisms 8:681. doi: 10.3390/microorganisms8050681
Meysman, F. J. R. (2018). Cable bacteria take a new breath using long-distance electricity. Trends Microbiol. 26, 411–422. doi: 10.1016/j.tim.2017.10.011
Miller, J. I., Techtmann, S., Fortney, J., Mahmoudi, N., Joyner, D., Liu, J., et al. (2019). Oil hydrocarbon degradation by caspian sea microbial communities. Front. Microbiol. 10:995. doi: 10.3389/fmicb.2019.00995
Minot, S. S., Krumm, N., and Greenfield, N. B. (2015). One codex: a sensitive and accurate data platform for genomic microbial identification. bioRxiv [Preprint]. doi: 10.1101/027607
Mitchell, A. L., Almeida, A., Beracochea, M., Boland, M., Burgin, J., Cochrane, G., et al. (2020). MGnify: the microbiome analysis resource in 2020. Nucleic Acids Res. 48, D570–D578. doi: 10.1093/nar/gkz1035
Mohamad Shahimin, M. F., and Siddique, T. (2017a). Methanogenic biodegradation of paraffinic solvent hydrocarbons in two different oil sands tailings. Sci. Total Environ. 583, 115–122. doi: 10.1016/j.scitotenv.2017.01.038
Mohamad Shahimin, M. F., and Siddique, T. (2017b). Sequential biodegradation of complex naphtha hydrocarbons under methanogenic conditions in two different oil sands tailings. Environ. Pollut. 221, 398–406. doi: 10.1016/j.envpol.2016.12.002
Mohamad Shahimin, M. F., Foght, J. M., and Siddique, T. (2016). Preferential methanogenic biodegradation of short-chain n -alkanes by microbial communities from two different oil sands tailings ponds. Sci. Total Environ. 553, 250–257. doi: 10.1016/j.scitotenv.2016.02.061
Morris, J. M., Jin, S., Crimi, B., and Pruden, A. (2009). Microbial fuel cell in enhancing anaerobic biodegradation of diesel. Chem. Eng. J. 146, 161–167. doi: 10.1016/j.cej.2008.05.028
Müller, H., Bosch, J., Griebler, C., Damgaard, L. R., Nielsen, L. P., Lueders, T., et al. (2016). Long-distance electron transfer by cable bacteria in aquifer sediments. ISME J. 10, 2010–2019. doi: 10.1038/ismej.2015.250
Müller, H., Marozava, S., Probst, A. J., and Meckenstock, R. U. (2020). Groundwater cable bacteria conserve energy by sulfur disproportionation. ISME J. 14, 623–634. doi: 10.1038/s41396-019-0554-1
Musat, F., and Widdel, F. (2007). Anaerobic degradation of benzene by a marine sulfate-reducing enrichment culture, and cell hybridization of the dominant phylotype. Environ. Microbiol. 14, 101–113. doi: 10.1111/j.1462-2920.2007.01425.x
National Research Council (1993). In Situ Bioremediation: When Does it Work?. Washington, DC: The National Academies Press, doi: 10.17226/2131
Nie, Y., Zhao, J.-Y., Tang, Y.-Q., Guo, P., Yang, Y., Wu, X.-L., et al. (2016). Species Divergence vs. functional convergence characterizes crude oil microbial community assembly. Front. Microbiol. 7:1254. doi: 10.3389/fmicb.2016.01254
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). MetaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Oberding, L., and Gieg, L. M. (2016). Metagenomic analyses reveal that energy transfer gene abundances can predict the syntrophic potential of environmental microbial communities. Microorganisms 4:10005. doi: 10.3390/microorganisms4010005
Oberhardt, M. A., Zarecki, R., Gronow, S., Lang, E., Klenk, H. P., Gophna, U., et al. (2015). Harnessing the landscape of microbial culture media to predict new organism-media pairings. Nat. Commun. 6, 1–14. doi: 10.1038/ncomms9493
Oulas, A., Polymenakou, P. N., Seshadri, R., Tripp, H. J., Mandalakis, M., Paez-Espino, A. D., et al. (2016). Metagenomic investigation of the geologically unique hellenic volcanic arc reveals a distinctive ecosystem with unexpected physiology. Environ. Microbiol. 18, 1122–1136. doi: 10.1111/1462-2920.13095
Overholt, W. A., Marks, K. P., Romero, I. C., Hollander, D. J., Snell, T. W., and Kostka, J. E. (2016). Hydrocarbon-degrading bacteria exhibit a species-specific response to dispersed oil while moderating ecotoxicity. Appl. Environ. Microbiol. 82, 518–527. doi: 10.1128/AEM.02379-15
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Peixoto, R. S., Vermelho, A. B., and Rosado, A. S. (2011). Petroleum-degrading enzymes: bioremediation and new prospects. Enzyme Res. 2011, 1–7. doi: 10.4061/2011/475193
Peng, Y., Leung, H. C. M., Yiu, S. M., and Chin, F. Y. L. (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428. doi: 10.1093/bioinformatics/bts174
Pfeffer, C., Larsen, S., Song, J., Dong, M., Besenbacher, F., Meyer, R. L., et al. (2012). Filamentous bacteria transport electrons over centimetre distances. Nature 491, 218–221. doi: 10.1038/nature11586
Pieper, R., Huang, S.-T., and Suh, M.-J. (2013). Proteomics and Metaproteomics: Encyclopedia of Metagenomics. New York, NY: Springer, 1–11. doi: 10.1007/978-1-4614-6418-1_690-9
Pilloni, G., Bayer, A., Ruth-Anneser, B., Fillinger, L., Engel, M., Griebler, C., et al. (2019). Dynamics of hydrology and anaerobic hydrocarbon degrader communities in a tar-oil contaminated aquifer. Microorganisms 7:46. doi: 10.3390/microorganisms7020046
Pisciotta, J. M., and Dolceamore, J. J. Jr. (2016). Bioelectrochemical and conventional bioremediation of environmental pollutants. J. Microb. Biochem. Technol. 8:306. doi: 10.4172/1948-5948.1000306
Potts, L. D., Perez Calderon, L. J., Gubry-Rangin, C., Witte, U., and Anderson, J. A. (2019). Characterisation of microbial communities of drill cuttings piles from offshore oil and gas installations. Mar. Pollut. Bull. 142, 169–177. doi: 10.1016/j.marpolbul.2019.03.014
Rabus, R., Boll, M., Heider, J., Meckenstock, R. U., Buckel, W., Einsle, O., et al. (2016). Anaerobic microbial degradation of hydrocarbons: from enzymatic reactions to the environment. J. Mol. Microbiol. Biotechnol. 26, 5–28. doi: 10.1159/000443997
Rabus, R., Kube, M., Heider, J., Beck, A., Heitmann, K., Widdel, F., et al. (2005). The genome sequence of an anaerobic aromatic-degrading denitrifying bacterium, strain EbN1. Arch. Microbiol. 183, 27–36. doi: 10.1007/s00203-004-0742-9
Rabus, R., Trautwein, K., and Wöhlbrand, L. (2014). Towards habitat-oriented systems biology of “aromatoleum aromaticum” EbN1: chemical sensing, catabolic network modulation and growth control in anaerobic aromatic compound degradation. Appl. Microbiol. Biotechnol. 98, 3371–3388. doi: 10.1007/s00253-013-5466-9
Rabus, R., Wöhlbrand, L., Thies, D., Meyer, M., Reinhold-Hurek, B., and Kampfer, P. (2019). Aromatoleum gen. Nov., a novel genus accommodating the phylogenetic lineage including azoarcus evansii and related species, and proposal of aromatoleum aromaticum sp. nov., aromatoleum petrolei sp. nov., aromatoleum bremense sp. nov., aromatoleum toluolicum sp. nov. and aromatoleum diolicum sp. nov. Int. J. Syst. Evol. Microbiol. 69, 982–997. doi: 10.1099/ijsem.0.003244
Reid, T., Chaganti, S. R., Droppo, I. G., and Weisener, C. G. (2018). Novel insights into freshwater hydrocarbon-rich sediments using metatranscriptomics: opening the black box. Water Res. 136, 1–11. doi: 10.1016/j.watres.2018.02.039
Révész, F., Figueroa-Gonzalez, P. A., Probst, A. J., Kriszt, B., Banerjee, S., Szoboszlay, S., et al. (2020). Microaerobic conditions caused the overwhelming dominance of Acinetobacter spp. and the marginalization of Rhodococcus spp. in diesel fuel/crude oil mixture-amended enrichment cultures. Arch. Microbiol. 202, 329–342. doi: 10.1007/s00203-019-01749-2
Révész, F., Tóth, E. M., Kriszt, B., Bóka, K., Benedek, T., Sárkány, O., et al. (2018). Sphingobium aquiterrae sp. Nov., a toluene, meta- and para-xylene-degrading bacterium isolated from petroleum hydrocarbon-contaminated groundwater. Int. J. Syst. Evol. Microbiol. 68, 2807–2812. doi: 10.1099/ijsem.0.002898
Rodríguez-Valera, F. (2004). Environmental genomics, the big picture? FEMS Microbiol. Lett. 231, 153–158. doi: 10.1016/S0378-1097(04)00006-0
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). Vsearch: a versatile open source tool for metagenomics. PeerJ 2016:e2584. doi: 10.7717/peerj.2584
Rossmassler, K., Snow, C. D., Taggart, D., Brown, C., and De Long, S. K. (2019). Advancing biomarkers for anaerobic o-xylene biodegradation via metagenomic analysis of a methanogenic consortium. Appl. Microbiol. Biotechnol. 103, 4177–4192. doi: 10.1007/s00253-019-09762-7
Rotaru, A.-E., Shrestha, P. M., Liu, F., Markovaite, B., Chen, S., Nevin, K. P., et al. (2014a). Direct interspecies electron transfer between Geobacter metallireducens and Methanosarcina barkeri. Appl. Environ. Microbiol. 80, 4599–4605. doi: 10.1128/AEM.00895-14
Rotaru, A.-E., Shrestha, P. M., Liu, F., Shrestha, M., Shrestha, D., Embree, M., et al. (2014b). A new model for electron flow during anaerobic digestion: direct interspecies electron transfer to Methanosaeta for the reduction of carbon dioxide to methane. Energy Environ. Sci. 7, 408–415. doi: 10.1039/C3EE42189A
Roy, A., Sar, P., Sarkar, J., Dutta, A., Sarkar, P., Gupta, A., et al. (2018). Petroleum hydrocarbon rich oil refinery sludge of North-East India harbours anaerobic, fermentative, sulfate-reducing, syntrophic and methanogenic microbial populations. BMC Microbiol. 18:151. doi: 10.1186/s12866-018-1275-8
Sabirova, J. S., Becker, A., Lünsdorf, H., Nicaud, J. M., Timmis, K. N., and Golyshin, P. N. (2011). Transcriptional profiling of the marine oil-degrading bacterium Alcanivorax borkumensis during growth on n-alkanes. FEMS Microbiol. Lett. 319, 160–168. doi: 10.1111/j.1574-6968.2011.02279.x
Sabirova, J. S., Ferrer, M., Regenhardt, D., Timmis, K. N., and Golyshin, P. N. (2006). Proteomic insights into metabolic adaptations in Alcanivorax borkumensis induced by alkane utilization. J. Bacteriol. 188, 3763–3773. doi: 10.1128/JB.00072-06
Sangwan, N., Xia, F., and Gilbert, J. A. (2016). Recovering complete and draft population genomes from metagenome datasets. Microbiome 4, 1–11. doi: 10.1186/s40168-016-0154-5
Sato, Y., Hori, T., Koike, H., Navarro, R. R., Ogata, A., and Habe, H. (2019). Transcriptome analysis of activated sludge microbiomes reveals an unexpected role of minority nitrifiers in carbon metabolism. Commun. Biol. 2, 1–8. doi: 10.1038/s42003-019-0418-2
Scheller, S., Goenrich, M., Boecher, R., Thauer, R. K., and Jaun, B. (2010). The key nickel enzyme of methanogenesis catalyses the anaerobic oxidation of methane. Nature 465, 606–608. doi: 10.1038/nature09015
Schmid, G., Auerbach, H., Pierik, A. J., Schünemann, V., and Boll, M. (2016). ATP-dependent electron activation module of benzoyl-coenzyme a reductase from the hyperthermophilic archaeon Ferroglobus placidus. Biochemistry 55, 5578–5586. doi: 10.1021/acs.biochem.6b00729
Segura, A., and Ramos, J. L. (2013). Plant-bacteria interactions in the removal of pollutants. Curr. Opin. Biotechnol. 24, 467–473. doi: 10.1016/j.copbio.2012.09.011
Seitaj, D., Schauer, R., Sulu-Gambari, F., Hidalgo-Martinez, S., Malkin, S. Y., Burdorf, L. D. W., et al. (2015). Cable bacteria generate a firewall against euxinia in seasonally hypoxic basins. Proc. Natl. Acad. Sci. U.S.A. 112:13278. doi: 10.1073/PNAS.1510152112
Seitz, K. W., Dombrowski, N., Eme, L., Spang, A., Lombard, J., Sieber, J. R., et al. (2019). Asgard archaea capable of anaerobic hydrocarbon cycling. Nat. Commun. 10, 1–11. doi: 10.1038/s41467-019-09364-x
Selesi, D., Jehmlich, N., Von Bergen, M., Schmidt, F., Rattei, T., Tischler, P., et al. (2010). Combined genomic and proteomic approaches identify gene clusters involved in anaerobic 2-methylnaphthalene degradation in the sulfate-reducing enrichment culture N47. J. Bacteriol. 192, 295–306. doi: 10.1128/JB.00874-09
Setti, L., Lanzarini, G., and Pifferi, P. G. (1997). Whole cell biocatalysis for an oil desulfurization process. Fuel Process. Technol. 52, 145–153. doi: 10.1016/S0378-3820(97)00023-4
Shakya, M., Lo, C. C., and Chain, P. S. G. (2019). Advances and challenges in metatranscriptomic analysis. Front. Genet. 10:904. doi: 10.3389/fgene.2019.00904
Sharma, M., Nandy, A., Taylor, N., Venkatesan, S. V., Ozhukil Kollath, V., Karan, K., et al. (2020). Bioelectrochemical remediation of phenanthrene in a microbial fuel cell using an anaerobic consortium enriched from a hydrocarbon-contaminated site. J. Hazard. Mater. 389:121845. doi: 10.1016/j.jhazmat.2019.121845
Shin, B., Kim, M., Zengler, K., Chin, K. J., Overholt, W. A., Gieg, L. M., et al. (2019). Anaerobic degradation of hexadecane and phenanthrene coupled to sulfate reduction by enriched consortia from northern Gulf of Mexico seafloor sediment. Sci. Rep. 9, 1–13. doi: 10.1038/s41598-018-36567-x
Shrestha, P. M., Rotaru, A. E., Summers, Z. M., Shrestha, M., Liu, F., and Lovley, D. R. (2013). Transcriptomic and genetic analysis of direct interspecies electron transfer. Appl. Environ. Microbiol. 79, 2397–2404. doi: 10.1128/AEM.03837-12
Siddique, T., Penner, T., Semple, K., and Foght, J. M. (2011). Anaerobic biodegradation of longer-chain n -Alkanes coupled to methane production in oil sands tailings. Environ. Sci. Technol. 45, 5892–5899. doi: 10.1021/es200649t
Sieber, C. M. K., Probst, A. J., Sharrar, A., Thomas, B. C., Hess, M., Tringe, S. G., et al. (2018). Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 3, 836–843. doi: 10.1038/s41564-018-0171-1
Smith, J. A., Nevin, K. P., and Lovley, D. R. (2015). Syntrophic growth via quinone-mediated interspecies electron transfer. Front. Microbiol. 6:121. doi: 10.3389/fmicb.2015.00121
Song, B., Tang, J., Zhen, M., and Liu, X. (2019). Effect of rhamnolipids on enhanced anaerobic degradation of petroleum hydrocarbons in nitrate and sulfate sediments. Sci. Total Environ. 678, 438–447. doi: 10.1016/j.scitotenv.2019.04.383
Sousa, S. T. P., de, Cabral, L., Lacerda-Júnior, G. V., Noronha, M. F., Ottoni, J. R., et al. (2020). Exploring the genetic potential of a fosmid metagenomic library from an oil-impacted mangrove sediment for metabolism of aromatic compounds. Ecotoxicol. Environ. Saf. 189:109974. doi: 10.1016/j.ecoenv.2019.109974
Sperfeld, M., Rauschenbach, C., Diekert, G., and Studenik, S. (2018). Microbial community of a gasworks aquifer and identification of nitrate-reducing Azoarcus and Georgfuchsia as key players in BTEX degradation. Water Res. 132, 146–157. doi: 10.1016/j.watres.2017.12.040
Stagars, M. H., Emil Ruff, S., Amann, R., and Knittel, K. (2016). High diversity of anaerobic alkane-degrading microbial communities in marine seep sediments based on (1-methylalkyl)succinate synthase genes. Front. Microbiol. 6:1511. doi: 10.3389/fmicb.2015.01511
Steliga, T., Wojtowicz, K., Kapusta, P., and Brzeszcz, J. (2020). Assessment of biodegradation efficiency of polychlorinated biphenyls (PCBs) and Petroleum Hydrocarbons (TPH) in Soil Using Three Individual Bacterial Strains and Their Mixed Culture. Molecules 25:709. doi: 10.3390/molecules25030709
Stewart, R. D., Auffret, M. D., Snelling, T. J., Roehe, R., and Watson, M. (2018). MAGpy: a reproducible pipeline for the downstream analysis of metagenome-assembled genomes (MAGs). Bioinformatics 35, 2150–2152. doi: 10.1093/bioinformatics/bty905
Storck, T., Virdis, B., and Batstone, D. J. (2016). Modelling extracellular limitations for mediated versus direct interspecies electron transfer. ISME J. 10, 621–631. doi: 10.1038/ismej.2015.139
Suflita, J. M., Davidova, I. A., Gieg, L. M., Nanny, M., and Prince, R. C. (2004). Chapter 10 Anaerobic hydrocarbon biodegradation and the prospects for microbial enhanced energy production. Stud. Surf. Sci. Catal. 151, 283–305. doi: 10.1016/S0167-2991(04)80151-4
Sun, X., and Kostka, J. E. (2019). Hydrocarbon-degrading microbial communities are site specific, and their activity is limited by synergies in temperature and nutrient availability in surface ocean waters. Appl. Environ. Microbiol. 85:e00443-19. doi: 10.1128/AEM.00443-19
Sutherland, T., Horne, I., Weir, K., Coppin, C., Williams, M., Selleck, M., et al. (2004). Enzymatic bioremediation: from enzyme discivery to applications. Clin. Exp. Pharmacol. Physiol. 31, 817–821. doi: 10.1111/j.1440-1681.2004.04088.x
Szaleniec, M., Hagel, C., Menke, M., Nowak, P., Malgorzata Witko, A., and Heider, J. (2007). Kinetics and mechanism of oxygen-independent hydrocarbon hydroxylation by ethylbenzene dehydrogenase. Biochemistry 46, 7637–7646. doi: 10.1021/BI700633C
Tan, B., Jane Fowler, S., Laban, N. A., Dong, X., Sensen, C. W., Foght, J., et al. (2015). Comparative analysis of metagenomes from three methanogenic hydrocarbon-degrading enrichment cultures with 41 environmental samples. ISME J. 9, 2028–2045. doi: 10.1038/ismej.2015.22
Tan, B., Nesbø, C., and Foght, J. (2014). Re-analysis of omics data indicates Smithella may degrade alkanes by addition to fumarate under methanogenic conditions. ISME J. 887, 2353–2356. doi: 10.1038/ismej.2014.87
Thijs, S., Sillen, W., Rineau, F., Weyens, N., and Vangronsveld, J. (2016). Towards an enhanced understanding of plant-microbiome interactions to improve phytoremediation: Engineering the metaorganism. Front. Microbiol. 7:341. doi: 10.3389/fmicb.2016.00341
Tiedt, O., Mergelsberg, M., Boll, K., Müller, M., Adrian, L., Jehmlich, N., et al. (2016). ATP-dependent C-F bond cleavage allows the complete degradation of 4-fluoroaromatics without oxygen. MBio 7:e00990-16. doi: 10.1128/mBio.00990-16
Tikariha, H., and Purohit, H. J. (2020). Unfolding microbial community intelligence in aerobic and anaerobic biodegradation processes using metagenomics. Arch. Microbiol. 202, 1269–1274. doi: 10.1007/s00203-020-01839-6
Uritskiy, G. V., Diruggiero, J., and Taylor, J. (2018). MetaWRAP – A flexible pipeline for genome-resolved metagenomic data analysis 08 Information and Computing Sciences 0803 Computer Software 08 Information and Computing Sciences 0806 Information Systems. Microbiome 6:158. doi: 10.1186/s40168-018-0541-1
Van Beilen, J. B., and Funhoff, E. G. (2007). Alkane hydroxylases involved in microbial alkane degradation. Appl. Microbiol. Biotechnol. 74, 13–21. doi: 10.1007/s00253-006-0748-0
Van Steendam, C., Smets, I., Skerlos, S., and Raskin, L. (2019). Improving anaerobic digestion via direct interspecies electron transfer requires development of suitable characterization methods. Curr. Opin. Biotechnol. 57, 183–190. doi: 10.1016/j.copbio.2019.03.018
Von Netzer, F., Pilloni, G., Kleindienst, S., Krüger, M., Knittel, K., Gründger, F., et al. (2013). Enhanced gene detection assays for fumarate-adding enzymes allow uncovering of anaerobic hydrocarbon degraders in terrestrial and marine systems. Appl. Environ. Microbiol. 79, 543–552. doi: 10.1128/AEM.02362-12
Wang, L.-Y., Nevin, K. P., Woodard, T. L., Mu, B.-Z., and Lovley, D. R. (2016). Expanding the diet for DIET: electron donors supporting direct interspecies electron transfer (DIET) in defined Co-cultures. Front. Microbiol. 7:236. doi: 10.3389/fmicb.2016.00236
Wang, W., and Shao, Z. (2013). Enzymes and genes involved in aerobic alkane degradation. Front. Microbiol. 4:116. doi: 10.3389/fmicb.2013.00116
Wang, Y., Wegener, G., Hou, J., Wang, F., and Xiao, X. (2019). Expanding anaerobic alkane metabolism in the domain of Archaea. Nat. Microbiol. 4, 595–602. doi: 10.1038/s41564-019-0364-2
Wawrik, B., Marks, C. R., Davidova, I. A., McInerney, M. J., Pruitt, S., Duncan, K. E., et al. (2016). Methanogenic paraffin degradation proceeds via alkane addition to fumarate by ‘Smithella’ spp. mediated by a syntrophic coupling with hydrogenotrophic methanogens. Environ. Microbiol. 18, 2604–2619. doi: 10.1111/1462-2920.13374
Wilmes, P., and Bond, P. L. (2004). The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms. Environ. Microbiol. 6, 911–920. doi: 10.1111/j.1462-2920.2004.00687.x
Wischgoll, S., Heintz, D., Peters, F., Erxleben, A., Sarnighausen, E., Reski, R., et al. (2005). Gene clusters involved in anaerobic benzoate degradation of Geobacter metallireducens. Mol. Microbiol. 58, 1238–1252. doi: 10.1111/j.1365-2958.2005.04909.x
Wood, D. E., Lu, J., and Langmead, B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20:257. doi: 10.1186/s13059-019-1891-0
Wu, Y.-W., Simmons, B. A., and Singer, S. W. (2015). MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607. doi: 10.1093/bioinformatics/btv638
Xia, W., Shen, W., Yu, L., Zheng, C., Yu, W., and Tang, Y. (2016). Conversion of petroleum to methane by the indigenous methanogenic consortia for oil recovery in heavy oil reservoir. Appl. Energy 171, 646–655. doi: 10.1016/J.APENERGY.2016.03.059
Xu, X., Liu, W., Tian, S., Wang, W., Qi, Q., Jiang, P., et al. (2018). Petroleum hydrocarbon-degrading bacteria for the remediation of oil pollution under aerobic conditions: a perspective analysis. Front. Microbiol. 9:2885. doi: 10.3389/fmicb.2018.02885
Yang, S. C., Song, Y., Wang, D., Wei, W. X., Yang, Y., Men, B., et al. (2016). Application of nitrate to enhance biodegradation of gasoline components in soil by indigenous microorganisms under anoxic condition. Environ. Technol. (U. K.) 37, 1045–1053. doi: 10.1080/09593330.2015.1098731
Yin, Y., Wang, Y., Tang, W., and Song, L. (2017). Thauera phenolivorans sp. nov., a phenol degrading bacterium isolated from activated sludge. Antonie Van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 110, 1681–1690. doi: 10.1007/s10482-017-0918-3
Keywords: anoxic biodegradation, hydrocarbon-degrading microorganisms and microbial communities, catabolic pathways, high throughput technologies, functional genomics/metagenomics
Citation: Laczi K, Erdeiné Kis Á, Szilágyi Á, Bounedjoum N, Bodor A, Vincze GE, Kovács T, Rákhely G and Perei K (2020) New Frontiers of Anaerobic Hydrocarbon Biodegradation in the Multi-Omics Era. Front. Microbiol. 11:590049. doi: 10.3389/fmicb.2020.590049
Received: 31 July 2020; Accepted: 26 October 2020;
Published: 16 November 2020.
Edited by:
András Táncsics, Szent István University, HungaryReviewed by:
Naresh Singhal, The University of Auckland, New ZealandCopyright © 2020 Laczi, Erdeiné Kis, Szilágyi, Bounedjoum, Bodor, Vincze, Kovács, Rákhely and Perei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gábor Rákhely, cmFraGVseS5nYWJvckBiaW8udS1zemVnZWQuaHU=; cmFraGVseS5nYWJvckBicmMuaHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.