 Sharon Ruiz-Lopez
Sharon Ruiz-Lopez Lynn Foster
Lynn Foster Chris Boothman
Chris Boothman Nick Cole2
Nick Cole2 Katherine Morris
Katherine Morris Jonathan R. Lloyd
Jonathan R. Lloyd- 1Department of Earth and Environmental Sciences, University of Manchester (UoM), Manchester, United Kingdom
- 2Sellafield Ltd., Warrington, United Kingdom
The use of nuclear power has been a significant part of the United Kingdom’s energy portfolio with the Sellafield site being used for power production and more recently reprocessing and decommissioning of spent nuclear fuel activities. Before being reprocessed, spent nuclear fuel is stored in water ponds with significant levels of background radioactivity and in high alkalinity (to minimize fuel corrosion). Despite these challenging conditions, the presence of microbial communities has been detected. To gain further insight into the microbial communities present in extreme environments, an indoor, hyper-alkaline, oligotrophic, and radioactive spent fuel storage pond (INP) located on the Sellafield site was analyzed. Water samples were collected from sample points within the INP complex, and also the purge water feeding tank (FT) that supplies water to the pond, and were screened for the presence of the 16S and 18S rRNA genes to inform sequencing requirements over a period of 30 months. Only 16S rRNA genes were successfully amplified for sequencing, suggesting that the microbial communities in the INP were dominated by prokaryotes. Quantitative Polymerase Chain Reaction (qPCR) analysis targeting 16S rRNA genes suggested that bacterial cells in the order of 104–106 mL–1 were present in the samples, with loadings rising with time. Next generation Illumina MiSeq sequencing was performed to identify the dominant microorganisms at eight sampling times. The 16S rRNA gene sequence analysis suggested that 70% and 91% from of the OTUs samples, from the FT and INP respectively, belonged to the phylum Proteobacteria, mainly from the alpha and beta subclasses. The remaining OTUs were assigned primarily to the phyla Acidobacteria, Bacteroidetes, and, Cyanobacteria. Overall the most abundant genera identified were Hydrogenophaga, Curvibacter, Porphyrobacter, Rhodoferax, Polaromonas, Sediminibacterium, Roseococcus, and Sphingomonas. The presence of organisms most closely related to Hydrogenophaga species in the INP areas, suggests the metabolism of hydrogen as an energy source, most likely linked to hydrolysis of water caused by the stored fuel. Isolation of axenic cultures using a range of minimal and rich media was also attempted, but only relatively minor components (from the phylum Bacteroidetes) of the pond water communities were obtained, emphasizing the importance of DNA-based, not culture-dependent techniques, for assessing the microbiome of nuclear facilities.
Introduction
Nuclear power supplies about 11% of the world’s electricity (WNA, 2016), and with increasing global energy demands this seems unlikely to decline. Although considered a “low carbon” generating energy source, radioactive waste is produced, including spent fuels that need storage prior to reprocessing and final disposal (Deutch et al., 2009). In the United Kingdom, this task is performed at Sellafield, one of the largest and most complex nuclear sites in Europe. With over 1,400 discrete operations, handling 240 nuclear materials, it is located in Cumbria on the North West coast of England and has been operated by the Nuclear Decommissioning Authority (NDA) since 2005 (Baldwin, 2003; WNA, 2018a). Calder Hall, located on the site, was the world’s first commercial nuclear power station, and here, energy was generated from 1956 to 2003. The Sellafield site also contains a range of storage ponds built during the 1950s which were intended to support the production of weapon-grade plutonium, and more recently fuels from the United Kingdom’s fleet of nuclear power stations (Reddy et al., 2012; WNA, 2018b). The legacy of activities have left a complex range of nuclear operations at Sellafield, including the decommissioning of redundant facilities associated with the site’s early defense work, and spent fuel management including Magnox and Oxide fuel reprocessing (Gov UK, 2018).
Prior to reprocessing, all irradiated fuel delivered to Sellafield is stored for a period of at least 100 days in water-filled reinforced concrete ponds that allow the decay of short-lived radioisotopes. During storage, the degree of corrosion experienced by the fuel is monitored to determine storage life and optimize water chemistry (Shaw, 1990). The temperature within the ponds is controlled by refrigerant chillers to further limit fuel corrosion, while the levels of both radioactive and non-radioactive ions in the pond waters are controlled by purging cycles of demineralised water adjusted to pH 11.1–11.6 with the addition of sodium hydroxide (Howden, 1987). The main pre-reprocessing storage pond at the Sellafield site is the indoor alkaline storage pond (INP), a concrete wall pond filled with demineralised water, responsible for receiving, storing and mechanically processing spent nuclear fuel (SNF). The SNF, defined as nuclear reactor fuel that has been used to the extent that it can no longer effectively sustain a chain reaction, is received and handled in Sellafield from Magnox and Advanced Gas-cooled Reactor (AGR) stations from across the United Kingdom (Sellafield Ltd, 2015).
Although Sellafield’s nuclear facilities, including the INP, are considered to be oligotrophic with high background levels of radiation, microorganisms have been shown to colonize these inhospitable environments (MeGraw et al., 2018). The presence of diverse microbial communities may impact on site operations and fuel stability. Microorganisms can also play a significant role in the transformations of radionuclides in the environment by altering their chemical speciation, solubility and sorption properties, ultimately impacting their environmental mobility and bioavailability (Lloyd and Renshaw, 2005; Francis, 2012; Newsome et al., 2014a; MeGraw et al., 2018). For example, the interactions between microbial populations and soluble radionuclides in groundwater can lead to precipitation reactions [e.g., via U (VI) or Tc (VII) bioreduction] and subsequent bioremediation (Newsome et al., 2014b). Of particular note within these pond environments is the fate of 90Sr and 137Cs, which dominate the radionuclide inventory in the water columns in storage ponds at Sellafield (Lang et al., 2019). Previous studies showed that seasonal blooms dominated by the alga Haematococcus, have adapted to survive in a circumneutral pH outdoor spent fuel storage pond at Sellafield, and are able to accumulate high levels of these radionuclides (MeGraw et al., 2018).
The accumulation of radionuclides by microbial cells can be driven by a range of processes including biosorption, biomineralization and bioprecipitation (Gadd, 2009), although these are poorly defined in nuclear storage ponds. Biosorption is species-specific and is affected by the chemistry and the pH of the solution, the physiological state of the cells, the cell wall architecture, and the presence of extracellular polymeric substances (EPS) (Comte et al., 2008; Merroun and Selenska-Pobell, 2008). The EPS is especially important, being mainly composed of polysaccharides, proteins, humic substances, uronic acids, nucleic acids, and lipids (Wingender et al., 1999), and containing ionisable functional groups that represent potential binding sites for the sequestration of metal ions (Brown and Lester, 1982; Lawson et al., 1984). Biosorption of divalent cations such as Sr2+ is well known (White and Gadd, 1990; Gadd, 2009; Liu et al., 2014), and would be favored in high pH pond systems (Ghorbanzadeh and Mohammad, 2009). Monovalent cations such as Cs+ would sorb less strongly than divalent cations (Andrès et al., 2001), although can bioaccumulate in biomass being transported into microbial cells, such as Rhodococcus, via potassium transport systems (Tomioka et al., 1992; Avery, 1995a,b). Recent work on a legacy high pH outdoor storage system at Sellafield, identified a Pseudanabaena species as the dominant photosynthetic microorganism (Foster et al., 2020a), and lab-based experiments on a culture dominated by a close relative showed increased polysaccharide production following irradiation treatments (Foster et al., 2020b). The polysaccharide production can promote the EPS formation which eventually can impact on 90Sr sorption-desorption behavior at alkaline environmental conditions under pond water conditions (Ashworth et al., 2018; MeGraw et al., 2018; Foster et al., 2020b).
Biomineralization reactions can also be linked to radionuclide fate (reviewed by Lloyd and Macaskie, 2000), due to local redox changes (e.g., bioreduction of actinides or key fission products) (Lloyd, 2003), localized alkalinisation at the cell surface (Van Roy et al., 1997) or the accumulation of microbially generated ligands (e.g., phosphate, sulfide, oxalate or carbonate) (White and Gadd, 1990; Macaskie et al., 1992; White et al., 1998; Boswell et al., 2001; Lloyd and Macaskie, 2002). For the latter, induced or mediated carbonate mineralization (MICP) (Braissant et al., 2002), can affect the mobility and sequestration of radionuclides in the near-surface environment (Ferris et al., 1994; Reeder et al., 2001), and has been studied widely due to its importance in the remediation on contaminated Sr systems (Mortensen et al., 2011). Additionally, a variety of microorganisms are able to drive MICP via urea hydrolysis (Fujita et al., 2004; Achal et al., 2012; Bhaduri et al., 2016) or via photosynthetic processes (Ferris et al., 1994; Dittrich et al., 2003; Lee et al., 2014; Zhu and Dittrich, 2016).
Finally microorganisms can affect the physical chemistry of the water-fuel interactions, leading to microbial-influenced corrosion (MIC) and hence fuel material degradation and radionuclide release (Shaw, 1990; Springell et al., 2014; Rajala et al., 2017). In open storage systems, the proliferation of microorganisms (together with the accumulation of radioactive sludge as a result of corrosion in spent fuel ponds) can also adversely impact on pond visibility, increasing the costs of fuel storage, hampering decommissioning operations and also increasing the exposure time to personnel (Wolfram et al., 1996; Jackson et al., 2014).
Recent publications have shown the presence of wide diversity of microorganisms living in SNF ponds, mainly bacteria, photosynthetic cyanobacteria and eukaryotic algae (Chicote et al., 2004, 2005; Sarró et al., 2005; Sellafield Ltd, 2011; Tišáková et al., 2013; Karley et al., 2018; MeGraw et al., 2018; Pipíška et al., 2018; Silva et al., 2018). The observed adaptation mechanisms include biofilm formation (Santo Domingo et al., 1998; Sarró et al., 2005; Bruhn et al., 2009), interactions with radionuclides via biosorption (Tomioka et al., 1992; Adam and Garnier-Laplace, 2003; Ghorbanzadeh and Mohammad, 2009; Dekker et al., 2014) and bioprecipitation (Ferris et al., 1994; Achal et al., 2012; Bhaduri et al., 2016; Zhu and Dittrich, 2016; Bagwell et al., 2018). To date, most published work on the Sellafield site has been on legacy outdoor pond systems (MeGraw et al., 2018; Foster et al., 2020a,b) which are open to external energy sources (including daylight, supporting photosynthetic primary colonizers). Indoor pond systems on the Sellafield site, with lower light intensities, and reduced inputs from atmospheric deposition, have not been studied in such detail.
The aim of this study was to characterize microbial communities of an indoor alkaline spent fuel storage pond (INP) on the Sellafield site, to help understand the microbial ecology of this facility, and the potential forms of metabolism that could underpin colonization. An additional goal was to provide baseline microbial community data, so that the impact of receiving new fuels and stored waste material during upcoming and extensive site-wide decommissioning activities across the Sellafield site can be assessed. The findings of this 30-month survey are discussed in relation to microbial survival to extreme environments (including potential energy sources) and how the extant microbiomes may potentially impact pond management. Microbial communities in the feeding tank supplying the pond system were identified and compared to those in the main and subponds containing spent fuel, to determine which organisms were uniquely adapted to the extreme pond chemistry (e.g., high pH) and high background radiation levels. Throughout the sampling campaign, the presence of hydrogen-oxidizing bacteria (affiliated with the genus Hydrogenophaga) in the INP, was consistent with the existence of hydrogen-oxidizing ecosystem, potentially linked to radiolysis in the fuel storage pond.
Materials and Methods
Indoor Nuclear Fuel Storage Pond
The INP is an indoor pond complex divided into three main ponds and three subponds linked by a transfer channel that enables water flow (Figure 1). In order to control the pond-water activity and quality, there is a continuous “once through” purge flow; pond-water from the main ponds flows into the transfer channel and enters the recirculation pump chamber where it is continuously pumped round a closed circulation loop and through a heat exchanger system, which cools the pond-water before it is recycled into the main ponds. Through the control feed, purge and re-circulation flow rates, the water depth is maintained at 7 ± 0.05 m. The purge flow can be either from a donor plant or from other hydraulically linked ponds within the Sellafield complex. The temperature and pH are controlled at 15°C and 11.6, respectively. Analyzed samples were taken from three designated main areas: main ponds (MP2 and MP3), subponds (SP1 and SP2), and from the Feeding tank (FT) of the donor plant, where the demineralised water used to feed the INP is stored.
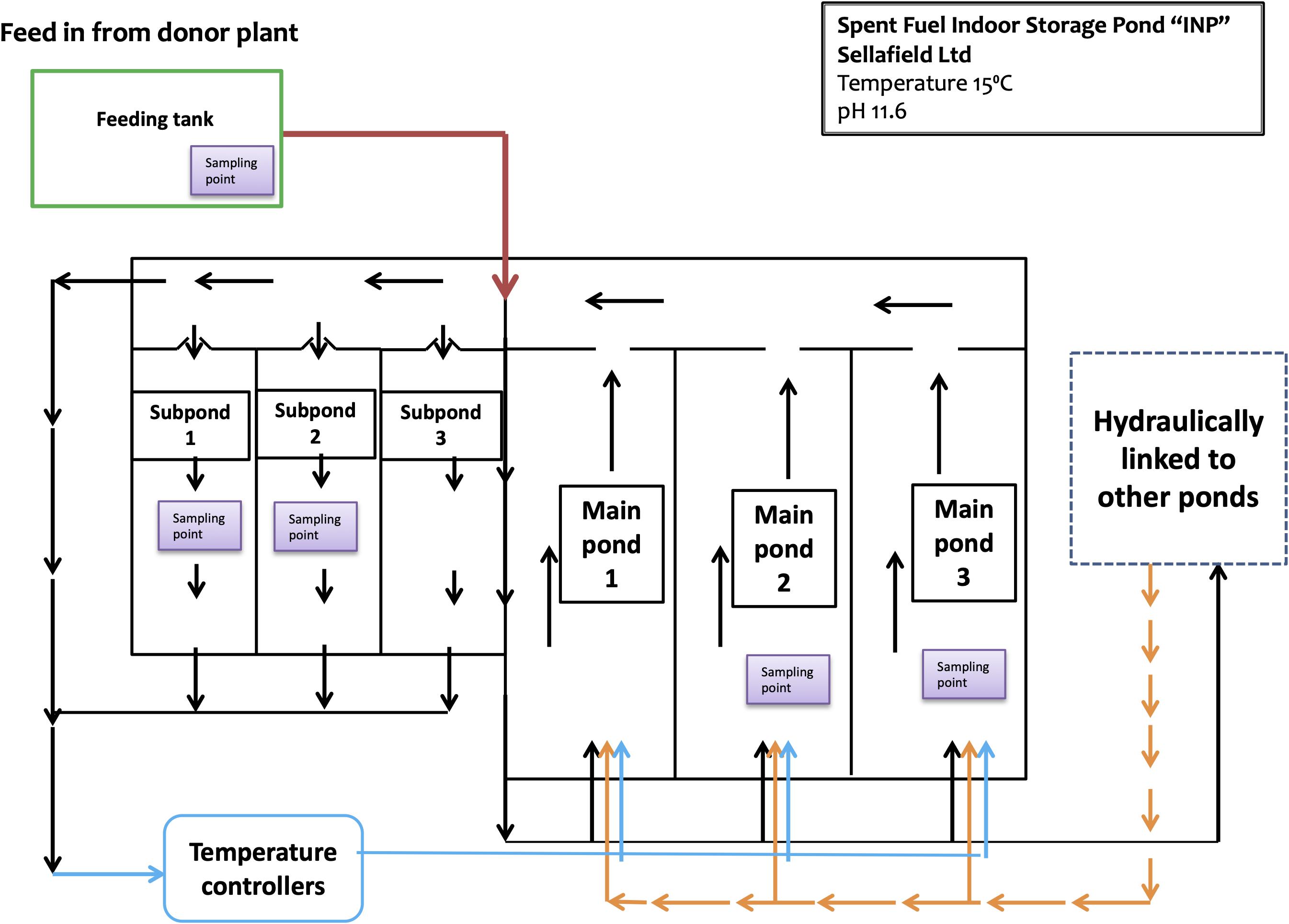
Figure 1. Diagram of the spent fuel indoor storage pond “INP.” It consists of 3 main ponds and 3 subponds linked by a transfer channel which enables water flow. The sampling points are located at the main ponds 2 and 3; subponds 1 and 2; and the head feeding tank (at the top of the pond).
Samples
Thirty samples were taken from three designated areas resulting on five sampling points (Table 1) for a period of 30 months (October 2016–April 2019).
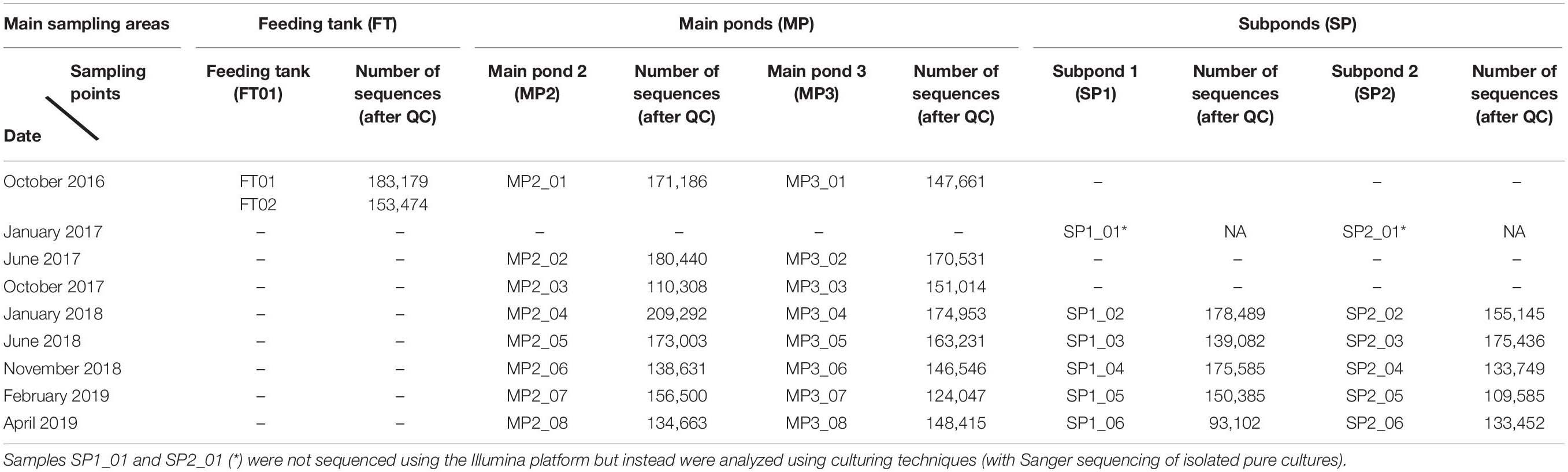
Table 1. Distribution of samples taken for a period of 30 months from different areas within the SNF pond, and analyzed using high-throughput (Illumina) DNA microbial profiling.
Water samples from the FT were considered non-active and were shipped directly to the University of Manchester in October 2016 and stored in the dark at 10°C. Water samples from the MP 2 and 3 and SP 1 and 2 were radioactive, hence appropriate handling procedures were required. The protocols for these samples were developed and applied under Command and Control regimes by Sellafield Ltd. and NNL, with samples transferred directly from the pond to the NNL Central Laboratory (National Nuclear Laboratory, Cumbria, United Kingdom), where DNA was extracted and the samples were checked for radioactivity in line with the Environmental Permits and Nuclear Site licenses held by Sellafield Ltd. Extracted DNA samples free from significant radionuclide contamination were shipped to the University of Manchester and stored in the freezer (−20°C) until use.
In addition to microbial profiling via DNA analyses, a complementary “cultivation-dependent” approach was also adopted to help further characterize the pond microbial community composition. Two low-volume samples (approximately 5 mL) from subponds 1 and 2 (Figure 1) were analyzed by traditional culturing techniques. The subponds are more radioactive than the main ponds, but the temperature and pH values are maintained at the same values as the main ponds, 21°C and 11.6, respectively. The typical pond water activities are 1,000 Bq/ml β (Table 2).
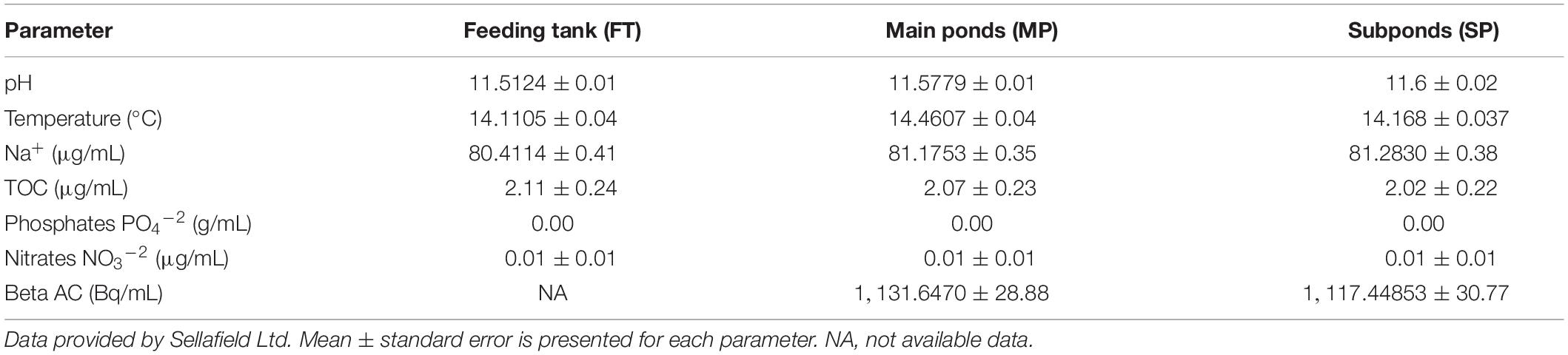
Table 2. Parameters measured in the indoor alkaline spent fuel storage pond (INP).
Cultivation-Independent DNA Analyses of Microbial Communities
DNA Extraction and PCR Amplifications
The MoBio PowerWater DNA isolation kit (MoBio Laboratories, Inc., Carlsbad, CA, United States), was used to extract DNA from water samples of approximately 1 L. The DNA was eluted to a final volume of 100 μL, and stored at 4°C until they were transported to UoM, where it was kept at −20°C to await further analyses.
Polymerase Chain Reaction (PCR) amplification was performed from the extracted DNA using a Techne Thermocycler (Cole-Parmer, Staffordshire, United Kingdom). Primers used for detection of bacterial 16S rRNA gene amplification were the broad-specificity 8F forward primer and the reverse primer 1492R (Eden et al., 1991). The primers used to detect eukaryotic organisms, targeting the 18S rRNA gene, were Euk F forward primer and the reverse primer Euk R (DeLong, 1992) whilst the archaeal primers that targeted the 16S rRNA gene, were forward primer 21F and reverse primer 958R (DeLong, 1992). The PCR reaction mixtures contained; 5 μL 10× PCR buffer, 4 μL 10 mM dNTP solution (2.5 mM each nucleotide), 1 μL of 25 μM forward primer, 1 μL of 25 μM reverse primer, and 0.3 μL Ex Takara Taq DNA Polymerase. The final volume was made up to 50 μL with PCR grade water, which included the addition of 2 μL of sample. The thermal cycling protocol used was as follows for the bacterial 8F and 1492R primers; initial denaturation at 94°C for 4 min, melting at 94°C for 30 s, annealing at 55°C for 30 s, extension at 72°C for 1 min (35 cycles with a final extension at 72°C for 5 min) (Eden et al., 1991). For the eukaryotic 18S rRNA gene amplification, the temperature cycle was; initial denaturation at 94°C for 2 min, melting at 94°C for 30 s, annealing at 55°C for 1.5 min, extension at 72°C for 1.5 min for a total of 30 cycles, and final extension at 72°C for 5 min (DeLong, 1992). For archaeal 16S rRNA genes the thermal cycle protocol consisted of an initial denaturation step at 94°C for 4 min, melting at 94°C for 45 s, annealing at 55°C for 30 s, extension at 72°C for 1 min (for a total of 30 cycles) and a final extension step at 72°C for 5 min (DeLong, 1992).
The purity of the amplified PCR products were checked by electrophoresis using a 1% (w/v) agarose gel in 1X TAE buffer (Tris-acetic acid-EDTA). DNA was stained with SYBER safe DNA gel stain (Thermofisher), and then viewed under short-wave UV light using a BioRad Geldoc 2000 system (BioRad, Hemel Hempstead, Herts, United Kingdom).
Quantitative Polymerase Chain Reaction
Quantitative Polymerase Chain Reaction (qPCR) of the prokaryotic 16S rRNA gene was performed by using Brilliant II Sybr Green qPCR Master Mix and the MX3000P qPCR System (Agilent Genomics, Headquarters, Santa Clara, CA, United States). The qPCR master mix contained 0.4 μL 8F forward primer (25 μM), and 0.4 μL 519R reverse primer (25 μM) (Turner et al., 1999), 0.4 μL of one in five diluted Rox reference dye, 12.5 μL of 2x qPCR Sybr green master mix, and Roche PCR Grade water to make up a final volume of 23 μL. Finally, 2 μL of sample was added. A standard curve from known serial dilutions of template DNA was constructed by plotting the CT (cycle threshold) values to verify the presence of a single gene-specific peak and the absence of primer dimer. The cycling conditions consisted of one cycle of denaturation at 94°C for 10 min, followed by 35 three-segment cycles of amplification (94°C for 30 s, 50°C for 30 s and 72°C for 45 s). Fluorescence was automatically measured during the PCR amplification, and one three-segment cycle of product melting (94°C for 10 min, 50°C for 30 s and 94°C for 30 s). Gene quantification was achieved by determining the threshold cycle (CT) of the unknown samples compared to the standard curve. The baseline adjustment method for the Mx3000 (Agilent) software was used to determine the Ct in each reaction. All samples were amplified in triplicate, and the mean was used for further analysis. In order to quantify the concentration of target genes, the absolute quantification by the standard-curve (SC) method was used (Brankatschk et al., 2012). To determine the abundance of cells mL–1 of sample, the total number of 16S rRNA genes determined by qPCR was adjusted to the approximated number of 16S rRNA gene copy numbers reported for members of the Proteobacteria; specifically for classes alpha and beta the average number of copies is reported to be 4 (Větrovský and Baldrian, 2013). A paired-samples (one-tailed) t-test (Sullivan, 2017) was conducted to compare the number of DNA copies over time in the MP and SP areas). Analysis was carried out on MP samples that were collected between 2016 and 2019, whilst the SP samples were collected between 2018 and 2019.
DNA Sequencing
Sequencing of 16S rRNA gene PCR amplicons was conducted using the Illumina MiSeq platform (Illumina, San Diego, CA, United States) targeting the V4 hyper variable region (forward primer, 515F, 5′-GTGYCAGCMGCCGCGGTAA-3′; reverse primer, 806R, 5′-GGACTACHVGGGTWTCTAAT-3′) for 2 × 250-bp paired-end sequencing (Illumina, San Diego, CA, United States) (Caporaso et al., 2011, 2012). PCR amplification was performed using the Roche FastStart High Fidelity PCR System (Roche Diagnostics Ltd., Burgess Hill, United Kingdom) in 50 μL reactions under the following conditions; initial denaturation at 95°C for 2 min, followed by 36 cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 1 min, and a final extension step of 5 min at 72°C. The PCR products were purified and normalized to ∼20 ng each using the SequalPrep Normalization Kit (Fisher Scientific, Loughborough, United Kingdom). The PCR amplicons from all samples were pooled in equimolar ratios. The run was performed using a 4 pM sample library spiked with 4 pM PhiX to a final concentration of 10% following the method of Schloss and Kozich (Kozich et al., 2013).
Raw sequences were divided into samples by barcodes index I5 and I7 (up to one mismatch was permitted) using a sequencing pipeline. Quality control and trimming (Q score of 20, and a minimum length of 250 base pairs) was performed using Cutadapt (Martin, 2011), FastQC (Bioinformatics, 2018), and Sickle (Joshi and Fass, 2011). MiSeq error correction was performed using SPADes (Nurk et al., 2013). Forward and reverse reads were incorporated into full-length sequences with Pandaseq (Masella et al., 2012). Chimeras were removed using ChimeraSlayer (Edgar et al., 2011), and OTU’s were generated with UPARSE (Edgar, 2013). OTUs were classified by Usearch (Edgar, 2010) at the 97% similarity level, and singletons were removed. Rarefaction analysis was conducted using the original detected OTUs in Qiime (Caporaso et al., 2010). The taxonomic assignment was performed by the RDP classifier (Wang et al., 2007). OTU sequences were submitted to the NCBI GenBank repository under the Bioproject number PRJNA660452, detailed accession numbers are indicated in Supplementary Table 2.
Culturing and Identification of the Pond Microorganisms
A complementary culture-dependent approach was used to help characterize the microorganisms present. To facilitate this, a series of 10-fold dilutions of water samples from the subponds 1 and 2 were plated onto fresh solid media. A range of complex or semi-defined solid media (at 10, 50, and 100% concentrations) were used (Supplementary Table 3) including Luria Bertani (LB) (Sezonov et al., 2007), Nutrient Agar (NA) (Misal et al., 2013), and Minimum medium DL (Lovley et al., 1984) at a range of pH values (7, 10, and 11). The marine medium of Zobell was also selected for isolation of alpha and gammaproteobacteria (Brettar et al., 2004) that had been detected in the pond using cultivation-independent DNA sequencing. Finally the fully defined minimal medium M9 (Neidhart et al., 1974) was also used at a range of concentrations and pH. The M9 medium contained no added carbon, selecting for autotrophic oligotrophs.
The isolated colonies were then resuspended in 10 mL of fresh liquid media and grown aerobically for 48 h. Cells were harvested by centrifuging at 3,500 g for 10 min, and supernatant was removed leaving the cell pellet and 100 μL of culture medium. DNA was extracted separately from the cell pellets using the Power Biofilm DNA Isolation Kit (MoBio Laboratories, Inc., Carlsbad, CA, United States). The DNA was eluted to a final volume of 100 μL, and stored at 4°C until use.
The 16S rRNA gene sequences of the isolates were determined by the chain termination sequencing method to facilitate phylogenetic analyses of the pure cultures (Slatko et al., 1999). PCR amplification was performed from the extracted DNA using a Techne Thermocycler (Cole-Parmer, Staffordshire, United Kingdom). Two PCR mixtures were prepared (one for each primer) and contained 3.5 μL 5X PCR buffer, 0.15 μL of 25 μM primer, and 1 μL Terminator BigDye (Thermo Fisher Scientific, Waltham, MA, United States), 1 μL of DNA was added to each tube, and was made to a final volume of 15 μL with PCR grade water. The thermal cycling protocol used was adapted for the primers as follows; initial denaturation at 96°C for 6 min, melting at 94°C for 40 s, annealing at 55°C for 15 s, extension at 60°C for 3 min; 30 cycles, and a final extension at 60°C for 5 min (Lorenz, 2012). The resulting PCR products were purified using the GlycoBlue coprecipitant protocol AM9516 (Thermo Fisher Scientific, Waltham, MA, United States), and the resulting pellets were then sequenced. An ABI Prism BigDye Terminator Cycle Sequencing Kit was used in combination with an ABI Prism 3730XL Capillary DNA Analyzer (Applied Biosystems, Warrington, United Kingdom). The primers 8F and 1592R were used for initial amplification and sequencing: 8F 5′-AGA GTT TGATCC TGG CTC AG-3′, and 1492R 5′-TAC GGY TAC CTT GTTACG ACT T-3′ (Lane et al., 1986). Sequences (typically 950 base pairs in length) were aligned with Muscle (Edgar, 2004) via the MEGA software (Kumar et al., 2016) version X (Kumar et al., 2018), then sequences were compared against the GenBank NCBI database using the BLAST program packages and matched to the most similar known 16S rRNA gene sequences (affiliations are detailed on Supplementary Figure 1).
Results
The aim of this study was to characterize the microbial populations living under the inhospitable high pH, oligotrophic and high background radiation conditions within an INP at the Sellafield complex. Duplicate samples of the purge waters of the FT were collected in October 2016. This non-radioactive purge water feeds into the INP, and therefore the analysis set out to determine the microbial community present in these samples that could seed the INP. We received an initial sample from the SP (in duplicate) in which the sole purpose was try to get the microorganisms present in the pond into culture. We were then provided with further samples (January 2018 onward) from this region of the INP facility to carry out next generation sequencing on. Since culture dependent techniques do not reveal the whole microbial community, it was important to use DNA sequencing techniques to better understand what microorganisms inhabit the SP.
Cell Density
In order to get an estimate of the overall cell density of the microorganisms that inhabit this facility, qPCR was carried out. Only the 16S rRNA gene could be detected in the extracted DNA samples, PCR using the eukaryotic 18S rRNA and the archaeal specific 16S rRNA primers did not result in any detectable amplification. Copy numbers were lower in the FT (μ = 8.1 × 104, SE = 3.9 × 104 DNA copies) and SP (μ = 2.5 × 105, SE = 8.8 × 104 DNA copies), while MP values ranged from 2.5 × 105 to 1.5 × 106 DNA copies (Figure 2), peaking in MP2_08 and MP3_08 (1.4 × 106 and 1.56 × 106 DNA copies, respectively; April 2019) and in MP3_06 (1.1 × 106 DNA copies, November 2018). The results showed that the number of DNA copies in the MP from the period 2016–2017 (μ = 2.9 × 105, SD = 1.1 × 105) to the period 2018–2019 (μ = 1.4 × 105, SD = 5.5 × 105) increased significantly [t(16) = 2.09, p < 0.05]. Likewise, the results on the SP showed that the number of DNA copies from 2018 (μ = 7.8 × 104, SD = 5.7 × 104) to 2019 (μ = 7.8 × 105, SD = 2.7 × 105) increased significantly [t(10) = 2.71, p < 0.05] (detailed statistical data is provided in Supplementary Table 1).
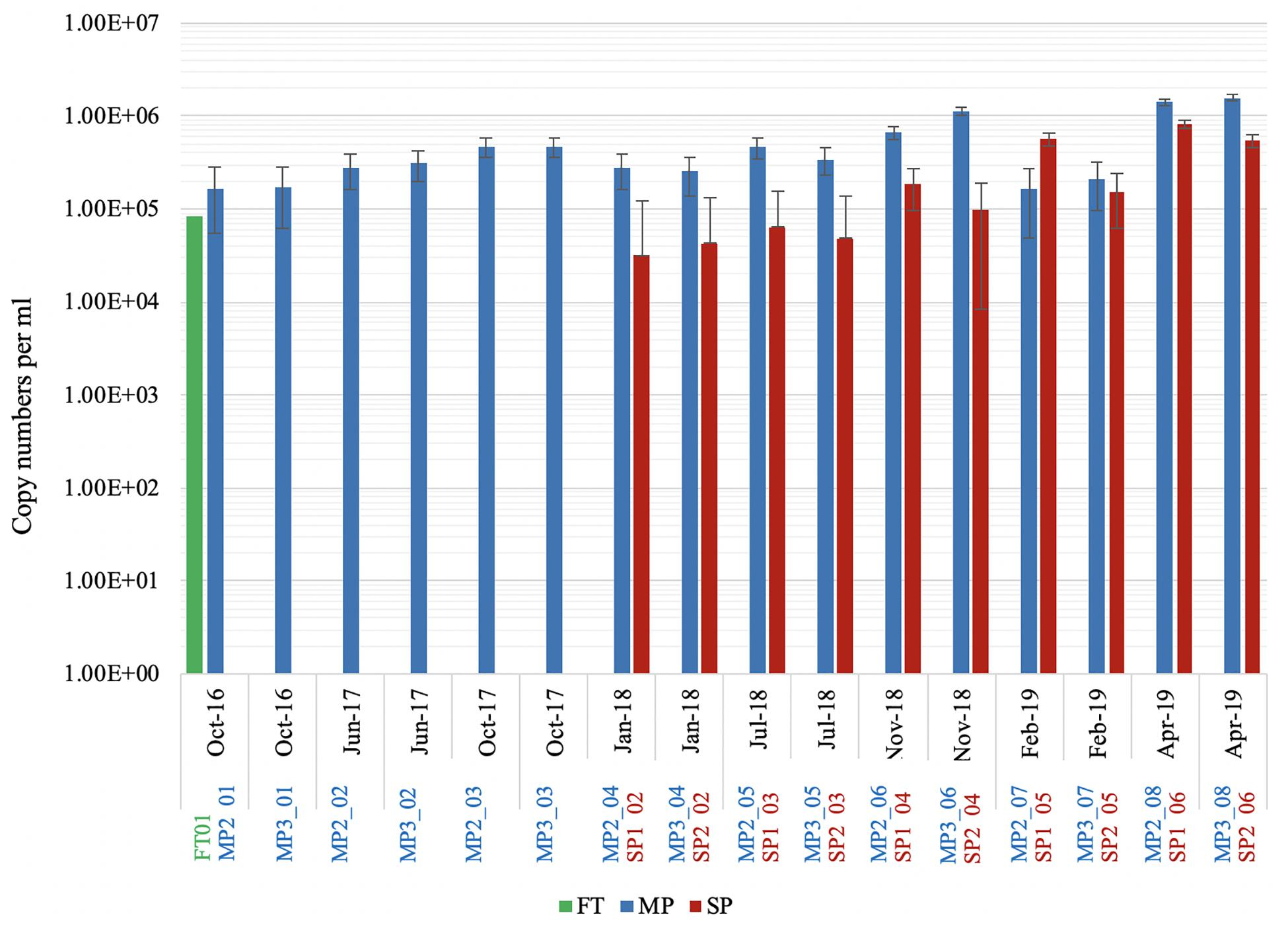
Figure 2. QPCR results show the average number of copies per mL per sampling site and time (FT: feeding tank, MP: main ponds and SP: subponds). Copy numbers are expressed on log scale and error bars represent standard error. Missing bars represent lack of samples for certain sampling dates (FT and SP). A standard curve for QPCR reaction was calculated at concentration ranging from 7.53 × 10−4 to 7.530 × 103 ng mL−1 (2.8 × 103−2.8×1012 DNA copies) to estimate the concentration of DNA in the samples.
Identification of Microorganisms by Next Generation DNA Sequencing
Over a 30-month sampling campaign a total of 30 samples (Table 1) from three sampling areas (FT, MP, and SP) were analyzed by 16S rRNA gene sequencing on the Illumina MiSeq. The initial sampling (by duplicate) was taken from the feeding head tank (FT) supplying the pond complex with demineralised water adjusted to pH 11.6 in October 2016, to help identify organisms present in the background waters, and hence (by comparison) help identify the organisms that were exclusively present in the INP main and subponds (Figure 3).
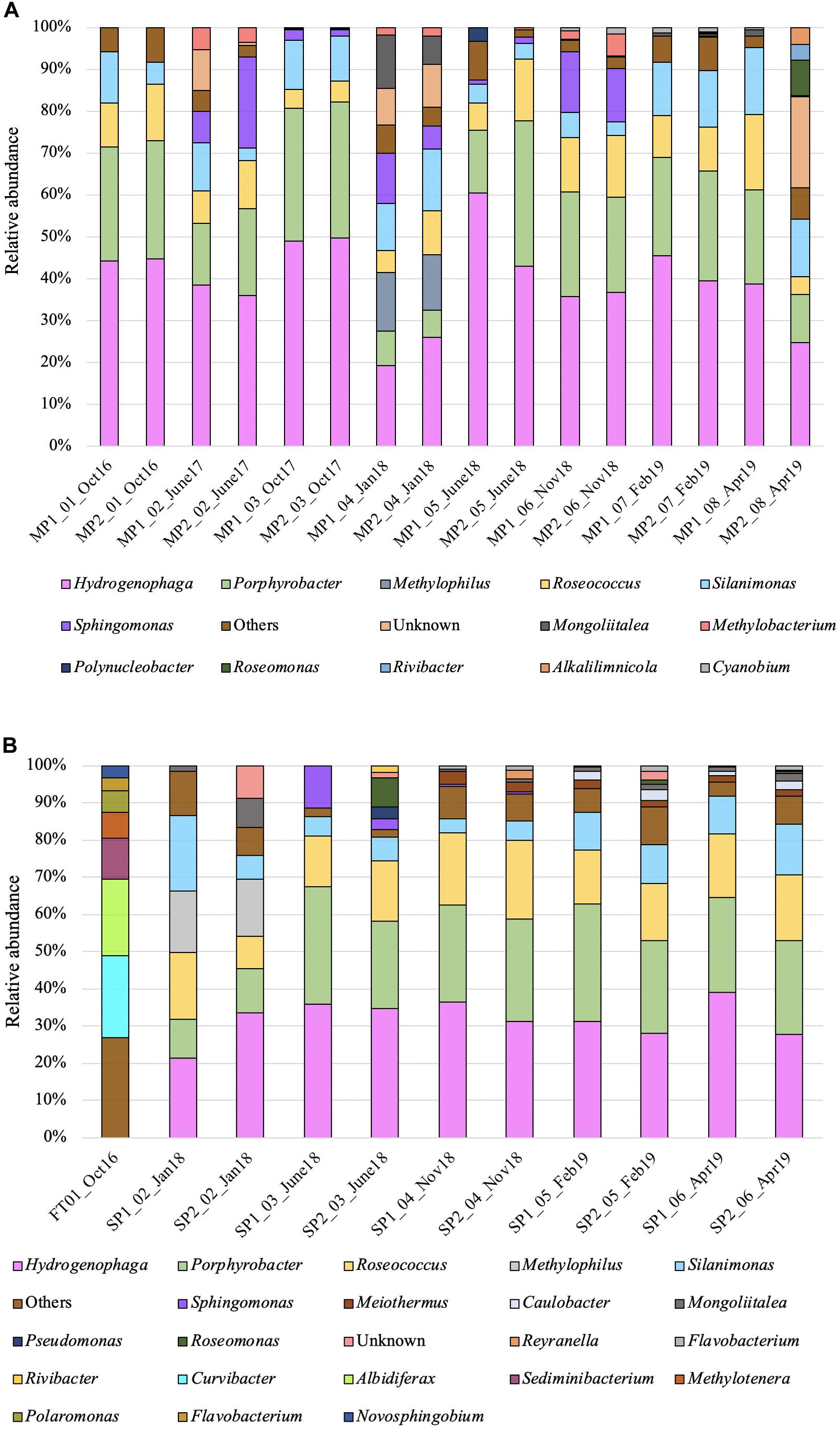
Figure 3. Phylogenetic affiliations (closest known genera) of microorganisms detected in the indoor nuclear pond (INP) at Sellafield: (A) microbial community in the main pond (MP); (B) microbial community in the feeding tank (FT) and subponds (SP), based on Illumina sequencing of the prokaryote 16S rRNA gene. Only genera that contained more than 1.5% of the total number of sequences are shown.
DNA extracted from the pond samples were assessed using PCR with 3 primer sets to screen for the presence of the prokaryotic and archaeal 16S rRNA genes and the eukaryotic 18S rRNA gene. However, only prokaryotic 16S rRNA gene amplification products were detected, and it was therefore concluded that eukaryotic and archaeal microorganisms were absent, or below the limit of detection. The 16S rRNA gene was targeted for sequencing using the Illumina MiSeq next generation sequencing platform, and analyzed using a bespoke bioinformatics platform which included comparison to prokaryotic gene sequences deposited in the NCBI databases.
Samples from the main ponds (MP) were dominated [mean ± standard deviation (SD)] by Proteobacteria (92.68 ± 6.26%) and Bacteroidetes (5.25 ± 6.93%). Organisms affiliated with the phylum Cyanobacteria were not detected in the initial samples, but were detected at subsequent times (from October 2017 to April 2019), although at a relative abundance of less than 3%. Samples from the subponds (SP) were also dominated (mean ± SD) by Proteobacteria (90.33 ± 4.01%) and Bacteroidetes (3.67 ± 2.19%), while the relative abundance of Cyanobacteria was again low (0.88 ± 0.52%). In addition, other phyla were detected at higher levels at specific dates in the subponds including organisms affiliated with the Actinobacteria (6.96 ± 2.13%, January 2018), Armatimonadetes (5.54 ± 1.78%, June 2018) and Deinococcus-Thermus groups (3.5 ± 0.95% from November 2018). Samples from the feeding tank (FT) were also dominated (mean ± SD) by Proteobacteria (73.3 ± 3.36%), Bacteroidetes (16.62 ± 3.6%) and Actinobacteria (2.58 ± 1.23%) (Supplementary Figure 2).
At the genus level the FT was dominated (mean ± SD) by close relatives of Curvibacter (21.4 ± 1.33%, Betaproteobacteria, 1 OTU), Rhodoferax (19.97 ± 0.51%, Betaproteobacteria, 1 OTU), Sediminibacterium (10.99 ± 0.85 %, Bacteroidetes, 2 OTUs), Polaromonas (5.51 ± 5.74%, Betaproteobacteria, 2 OTUs), Methylotenera (6.74 ± 6.29%, Betaproteobacteria, 2 OTUs), Novosphingobium (3.26 ± 1.87%, Alphaproteobacteria, 2 OTUs), Flavobacterium (3.40 ± 3.33%, Bacteroidetes, 2 OTUs), Unidibacterium (3.11 ± 0.52%, Betaproteobacteria, 2 OTUs). Approximately 26.25 ± 1.33% of the total OTUs (26) represented unidentified organisms.
Microbial distributions were consistent at all sampling times within the main ponds (MP) (Figure 3A). Overall at the genus level the microbial diversity was dominated (mean ± SD) by 1 OTU belonging to genus Hydrogenophaga (Betaproteobacteria), representing up to 38.76 ± 10.03% of the total population. The remaining community consisted of Porphyrobacter (21.57 ± 8.44%, Alphaproteobacteria, 1 OTU), Roseococcus (9.82 ± 4.15%, Alphaproteobacteria, 3 OTUs), Silanimonas (9.49 ± 4.50%, Gammaproteobacteria, 1 OTU), Sphingomonas (7.84 ± 6.82%, Alphaproteobacteria, 2 OTUs), and Synechococcus (0.91 ± 0.49%, Cyanophyceae, 3 OTUs). The exception was one set of samples taken on January 2018 (MP2_04 and MP3_04), where broad microbial diversity was recorded and the abundance of Hydrogenophaga, and Porphyrobacter dropped to 22.52 ± 4.73% and 7.40 ± 1.19%, respectively. Additionally, representatives of the genera Methylophilus (13.61 ± 0.69%, Betaproteobacteria, 1 OTU) and Mongoliitalea (9.80 ± 4.37%, Bacteroidetes, 2 OTUs) were exclusively identified at this sampling time. Unidentified (uncultured) sequences, although detected at all sampling times, represented more than 2% of the total community in samples MP2_02 (June 2017, 8.84 ± 6.77%, 24 OTUs), MP2_04 and MP3_04 (January 2018, 8.67 ± 1.08% and 10.19 ± 6.77%, 38 OTUs) and MP3_08 (April 2019, 21.48 ± 6.77%, 47 OTUs).
The microbial profiles of the subponds (SP) were similar to the main ponds (MP), and were dominated (mean ± SD) by representatives of the genera Hydrogenophaga (30.55 ± 5.73%, Betaproteobacteria, 1 OTU), Porphyrobacter (23.06 ± 7.52%, Alphaproteobacteria, 1 OTU), Roseococcus (15.44 ± 3.45%, Alphaproteobacteria, 2 OTUs), Silanimonas (8.73 ± 4.34%, Gammaproteobacteria, 2 OTUs), and Sphingomonas (2.4 ± 4.21%, Alphaproteobacteria, 3 OTUs). Samples SP1_02 and SP2_02 (January, 2018) showed a few differences with close relatives affiliated to genus Methylophilus (13.61 ± 0.69%, Alphaproteobacteria, 1 OTU) detected in these samples only (Figure 3B).
When viewed at the phylum level all three sampling areas (5 sampling points in total) showed similar profiles, as they were dominated by Proteobacteria, however, when looking at the affiliations at the genus level the microbial communities at each sampling point could be seen to differ substantially. Data would seem to suggest that the microbial community compositions in the MP, SP and FT samples represent distinct ecosystems, most likely linked to the impacts of the spent fuel in the INP environment.
Cultivation-Dependent Analysis for Determining Microbial Diversity in the INP
After 7 days of incubation, growth was detected exclusively in the undiluted samples (100) from plates containing non-defined complex media (DL, NA, and Zobell media; Supplementary Table 3). CFU mL–1 were between 700 and 1,000 mL–1 for each medium and eleven distinct colony morphologies were noted. Representative single colonies were isolated and identified by sequencing using the dideoxynucleotide technique. The presence of colonies was not detected on the fully defined media (minimal media M9).
Overall, representatives of four different genera were identified by 16S rRNA gene sequencing. Representatives most closely related to species of the genus Algoriphagus (isolates S01, 91.5% similarity; S05, 91% similarity; S06, 91.5% similarity; and S07, 89.5% similarity) were isolated on DL and NA agars, and produced light pink-colored, rod-shaped and raised colonies (1–2 mm diameter). Organisms most closely related to members of Echinicola genus (isolates S02, 91% similarity; S08, 88% similarity; and S09, 93.5% similarity) were obtained on the DL and NA agar plates, and produced red-colored colonies, that were rod-shaped with raised elevation (2–3 mm diameter). Strains S03, S10, and S11 were isolated from DL, NA and Zobell plates; were rod-shaped, translucent and had raised colonies (2–3 mm diameter) and were affiliated to an unclassified genus from the family Cyclobacteriaceae (S03, 93.5% similarity; S10, 85% similarity and S11, 91% similarity). Finally, a close relative to genus Bacteroides (strain S04, 91.5% similarity) was isolated from the DL plates and produced short round-shaped, bright-orange raised colonies (1–2 mm diameter). All eleven isolated strains belonged to the phylum Bacteroidetes (specific details on similarity and media are shown on Supplementary Table 4).
Members belonging to genus Echinicola (phylum Bacteroidetes) were previously detected in the MP and SP samples by DNA-based techniques; however, they did not represent a major component of the community. More precisely, members of the genus Echinicola were detected in samples MP2_03 and MP3_03 (October 2017) at a relative abundance of 0.28 and 0.39%, respectively (Supplementary Table 5).
Discussion
The present research was focused on characterizing the microbial community of a Sellafield INP complex containing main ponds (MP), subponds (SP) and a feeding head tank (FT) over a period of 30 months. The results showed that bacteria affiliated with a range of phylogenetic groups are able to survive and colonize the different areas across the INP complex.
Microbial diversity within the FT, an oligotrophic and hyper-alkaline environment, was dominated by members belonging to the Proteobacteria and Bacteroidetes. Previous studies showed that oligotrophic conditions do not prevent microbial colonization and allow microbial communities to display diverse adaptation mechanisms (Kawai et al., 2002; Kulakov et al., 2002; Chen et al., 2004). Specifically, organisms associated to Proteobacteria and Bacteroidetes have been identified previously in similar oligotrophic environments, including industrial ultrapure water (Galès et al., 2004; Bohus et al., 2011; Proctor et al., 2015). Microbial colonization in such environments has been linked to low levels of residual organic matter in the system, originating from dead microbial cells that and to biofilm formation on the walls, linked to planktonic cells delivered by water recirculation in the pond areas (Bohus et al., 2011). Organisms detected in the FT area are reported to support diverse forms of heterotrophic metabolism, which could occur within the FT. For example, members of the genera Rhodoferax (Finneran et al., 2003; Risso et al., 2009), Curvibacter and Sediminibacterium (Qu and Yuan, 2008; Ding and Yokota, 2010; Kang et al., 2013, 2014; Ma et al., 2016) are able to oxidize a range of complex organic compounds, while Methylotenera can utilise reduced one-carbon compounds (methylotrophy) such as methanol as energy sources (Kalyuzhnaya et al., 2006, 2011). However, the source of carbon and the source of energy microorganisms use in the FT remains to be investigated.
Although the INP has a continuous pond purge, the main ponds (MP) and subponds (SP) contained stable microbial populations with similar community profiles, which contrasted with the distinct microbiome of the FT. Key organisms detected in MP and SP samples included species of Hydrogenophaga, Silanimonas, Porphyrobacter, and Roseococcus.
In addition to the oligotrophic and hyper-alkaline characteristics of the MP and SP areas, spent nuclear fuel results in high background radioactivity, which further challenges the microbial community in the pond. Despite these adverse conditions, microbial colonization of similar spent fuel storage systems has been documented (Santo Domingo et al., 1998; Galès et al., 2004; Bruhn et al., 2009), and dominated by organisms associated to the phyla Proteobacteria (Chicote et al., 2004; Bagwell et al., 2018; MeGraw et al., 2018; Silva et al., 2018), Firmicutes (Sarró et al., 2005), Actinobacteria (Sarró et al., 2005), Cyanobacteria (MeGraw et al., 2018; Silva et al., 2018; Foster et al., 2020a), and Deinococcus-Thermus (Masurat et al., 2005). Whilst it was not possible to identify any eukaryotic organisms in the INP, other studies have identified fresh water microalgae (Rivasseau et al., 2016; MeGraw et al., 2018) and Fungi (Chicote et al., 2004; Silva et al., 2018) in both indoor and outdoor facilities. Although the energy sources supporting microbial growth in these systems remains largely uncharacterized, it is possible that radiolysis could play a direct role in supporting microbial growth. The presence of alpha, beta and gamma radiation from the spent fuel can promote the radiolysis of water, driving the formation of short-lived, highly oxidizing free radical species, such as -OH and H2O2 (Shoesmith, 2000; Jonsson et al., 2007) and also the production of H2 (Brodie et al., 2006; Libert et al., 2011) that could be utilized by hydrogen-oxidizing bacteria (Knallgas bacteria) (Yu, 2018). The most abundant organism in the MP and SP areas in this study were affiliated with the genus Hydrogenophaga (35.61 ± 9.42%), which comprise aerobic, chemoorganotrophic organisms that use hydrogen as an energy source (Willems et al., 1989; Kampfer et al., 2005; Yoon et al., 2008). Members of genus Hydrogenophaga are present in a variety of natural and engineered (e.g., waste water) environments (Lambo and Patel, 2006; Fahy et al., 2008; Yoon et al., 2008; Schwartz et al., 2013), including hyper alkaline sites such as Allas Springs, Cyprus where the pH was 11.9, similar to the alkaline conditions to the INP waters (pH 11.6) (Rizoulis et al., 2016) and serpentinizing springs (pH 11.6, The Cedars, Los Angeles, CA, United States) (Suzuki et al., 2014). The presence of Hydrogenophaga as a key microbial component during all the sampling times suggests that the metabolism of H2 may be occurring within the pond, which is of particular interest since oxidation of hydrogen could also be linked to the reduction of a range of electron acceptors, including radionuclides (Lloyd, 2003).
Hydrogen metabolism has not been reported for the remaining microbial community identified in the INP. Porphyrobacter, an aerobic anoxygenic phototrophic bacteria (AAP), has the ability to harvest energy photosynthetically (Hanada et al., 1997; Yoon et al., 2004; Liu et al., 2017); however, given the limited light availability in the pond, it is unlikely to be photosynthetically active in the INP. Members of this genus have been shown to be well-adapted to life in environments with light restrictions using light energy via Bacteriochlorophyll α synthesized in the dark (Fuerst et al., 1993; Yoon et al., 2008; Liu et al., 2017). Members of the Roseococcus genus, are obligate aerobes and chemoorganotrophic, they contain Bacteriochlorophyll α and carotenoid pigments (Boldareva et al., 2009; Yurkov, 2015), and are also able to grow in the dark (Yurkov et al., 1994). Sphingomonas species are metabolically versatile and can use a wide range of compounds as energy sources (Lee et al., 2001; Feng et al., 2014; Singh et al., 2015) such as polycyclic aromatic hydrocarbons (Leys et al., 2004); and contains ubiquinone Q-10, a molecule involved in respiratory functions (Niharika et al., 2012) where hydrogen, a potentially abundant energy source in the MP and SP areas, is required. Roseomonas species also contain ubiquinone Q-10 (Kim et al., 2009; Wang et al., 2016), and have the ability to grow on biofilms to protect themselves from adverse conditions (Diesendorf et al., 2017), such as those present in this radioactive facility. Microorganisms associated with the oxygenic and phototrophic phylum Cyanbacteria (Peschek, 1999), were much less abundant (identified as genera Synecochoccus and Cyanobium), which is likely to be a result of the low levels of light in the INP. The metabolic pathways utilized in the pond to facilitate their growth are not known yet and further work is required to better understand this.
Finally, agar-based cultivation approaches were tested alongside DNA-based approaches in this study, and resulted in the isolation of bacteria from the family Cyclobacteriacea, but proved unsuccessful for targeting organisms that were numerically dominant within the INP complex. Whilst the isolated organisms do not represent the major components of the pond microbial communities identified by NGS techniques, these new findings showed that organisms affiliated with the genera Algoriphagus and Echinicola were able to tolerate alkaline conditions (and given the source of inocula, presumably high levels of radioactivity and oligotrophic nutrient conditions), in stark contrast to the neutral pH environments they are normally associated with (Tiago et al., 2004; Yoon et al., 2004; Alegado et al., 2013; Kang et al., 2013; Misal et al., 2013; Glaring et al., 2015).
Overall this study reinforces the view that cultivation-independent molecular ecology techniques are crucial first steps in understanding the microbial dynamics in oligotrophic SNPs, offering the benefits of high-throughput sequencing of DNA that has been purified away from contaminating radionuclides present in the pond waters. This opens up the way for more detailed metagenomic analyses which are ongoing in our laboratories, alongside more targeted research on the impact of extant microbiomes within spent nuclear fuel storage ponds on the speciation and fate of key radionuclides present within the pond systems, and also the integrity of stored fuel materials.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
SR-L developed the concept, analyzed and interpreted data, and wrote the manuscript. CB performed the DNA sequencing runs. LF performed the DNA extractions at NNL labs. KM contributed to concept development. NC data curation and provision of samples from the facility and reviewed the manuscript. JL developed the concept and extensively reviewed the manuscript. All the authors read and approved the final manuscript.
Funding
This work was funded from a Ph.D. program funded by the National Mexican Council of Science and Technology (CONACyT). This work was also supported by funding from Sellafield Limited and the Royal Society to JL. LF was supported by an EPSRC CASE Ph.D. and IAA funding.
Conflict of Interest
Additional funding was provided by Sellafield. The funder played no role in the study design and analysis, decision to publish, or preparation of the manuscript. Sellafield Ltd., did provide pond samples and detailed pond data was collected as part of routine operations.
NC was employed by Sellafield Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author would like to thank to the National Council of Science and Technology (CONACyT) for providing the funding. In addition to the staff at NNL central laboratory and Sellafield Ltd., for their assistance with handling and transferring the samples.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.587556/full#supplementary-material
References
Achal, V., Pan, X., and Zhang, D. (2012). Bioremediation of strontium (Sr) contaminated aquifer quartz sand based on carbonate precipitation induced by Sr resistant Halomonas sp. Chemosphere 89, 764–768. doi: 10.1016/j.chemosphere.2012.06.064
Adam, C., and Garnier-Laplace, J. (2003). Bioaccumulation of silver-110m, cobalt-60, cesium-137, and manganese-54 by the freshwater algae Scenedesmus obliquus and Cyclotella meneghiana and by suspended matter collected during a summer bloom event. Limnol. Oceanogr. 48, 2303–2313.
Alegado, R. A., Grabenstatter, J. D., Zuzow, R., Morris, A., Huang, S. Y., Summons, R. E., et al. (2013). Algoriphagus machipongonensis sp. nov., co-isolated with a colonial choanoflagellate. Int. J. Syst. Evol. Microbiol. 63, 163–168.
Andrès, Y., Redercher, S., Gerente, C., and Thouand, G. (2001). Contribution of biosorption to the behavior of radionuclides in the environment. J. Radioanal. Nucl. Chem. 247, 89–93. doi: 10.1023/A:1006763030854
Ashworth, H., Abrahamsen-Mills, L., Bryan, N., Foster, L., Lloyd, J. R., Kellet, S., et al. (2018). Effect of humic acid and bacterial exudates on sorption-desorption interactions of 90Sr with brucite. Environ. Sci. Process. Impacts 20, 956–964. doi: 10.1039/c8em00073e
Avery, S. V. (1995a). Caesium accumulation by microorganisms: uptake mechanisms, cation competition, compartmentalization and toxicity. J. Ind. Microbiol. 14, 76–84. doi: 10.1007/BF01569888
Avery, S. V. (1995b). Microbial interactions with caesium—implications for biotechnology. J. Chem. Technol. Biotechnol. 62, 3–16. doi: 10.1002/jctb.280620102
Bagwell, C. E., Noble, P. A., Milliken, C. E., Li, D., and Kaplan, D. I. (2018). Amplicon sequencing reveals microbiological signatures in spent nuclear fuel storage basins. Front. Microbiol. 9:377. doi: 10.3389/fmicb.2018.00377
Baldwin, N. D. (2003). “Remediating Sellafield, a new focus for the site,” in Proceedings of the Waste Management Conference, Tucson, AZ.
Bhaduri, S., Debnath, N., Mitra, S., Liu, Y., and Kumar, A. (2016). Microbiologically induced calcite precipitation mediated by Sporosarcina pasteurii. J. Vis. Exp. 2016, 1–7. doi: 10.3791/53253
Bioinformatics, B. (2018). Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data. Available online at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed December 18, 2019).
Bohus, V., Kéki, Z., Márialigeti, K., Baranyi, K., Patek, G., Schunk, J., et al. (2011). Bacterial communities in an ultrapure water containing storage tank of a power plant. Acta Microbiol. Immunol. Hung. 58, 371–382. doi: 10.1556/AMicr.58.2011.4.12
Boldareva, E. N., Tourova, T. P., Kolganova, T. V., Moskalenko, A. A., Makhneva, Z. K., and Gorlenko, V. M. (2009). Roseococcus suduntuyensis sp. nov., a new aerobic bacteriochlorophyll a-containing bacterium isolated from a low-mineralized soda lake of Eastern Siberia. Microbiology 78, 92–101. doi: 10.1134/S0026261709010123
Boswell, C. D., Dick, R. E., Eccles, H., and Macaskie, L. E. (2001). Phosphate uptake and release by Acinetobacter johnsonii in continuous culture and coupling of phosphate release to heavy metal accumulation. J. Ind. Microbiol. Biotechnol. 26, 333–340. doi: 10.1038/sj.jim.7000139
Braissant, O., Verrecchia, E. P., and Aragno, M. (2002). Is the contribution of bacteria to terrestrial carbon budget greatly underestimated? Naturwissenschaften 89, 366–370. doi: 10.1007/s00114-002-0340-0
Brankatschk, R., Bodenhausen, N., Zeyer, J., and Burgmann, H. (2012). Simple absolute quantification method correcting for quantitative PCR efficiency variations for microbial community samples. Appl. Environ. Microbiol. 78, 4481–4489. doi: 10.1128/AEM.07878-11
Brettar, I., Christen, R., and Hofle, M. G. (2004). Aquiflexum balticum gen. nov., sp. nov., a novel marine bacterium of the Cytophaga-Flavobacterium-Bacteroides group isolated from surface water of the central Baltic Sea. Int. J. Syst. Evol. Microbiol. 54, 2335–2341. doi: 10.1099/ijs.0.63255-0
Brodie, E. L., DeSantis, T. Z., Joyner, D. C., Baek, S. M., Larsen, J. T., Andersen, G. L., et al. (2006). Application of a high-density oligonucleotide microarray approach to study bacterial population dynamics during uranium reduction and reoxidation. Appl. Environ. Microbiol. 72, 6288–6298. doi: 10.1128/AEM.00246-06
Brown, M. J., and Lester, J. N. (1982). Role of bacterial extracellular polymers in metal uptake in pure bacterial culture and activate sludge II. Effects of mean cell retention time. Water Res. 16, 1549–1560. doi: 10.1016/0043-1354(82)90207-x
Bruhn, D. F., Frank, S. M., Roberto, F. F., Pinhero, P. J., and Johnson, S. G. (2009). Microbial biofilm growth on irradiated, spent nuclear fuel cladding. J. Nucl. Mater. 384, 140–145. doi: 10.1016/j.jnucmat.2008.11.008
Caporaso, J. G., Kucynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E., et al. (2010). QIIME allows analysis of high- Intensity normalization improves color. Nat. Publ. Group 7, 335–336. doi: 10.1038/nmeth0510-335
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108(Suppl. 1), 4516–4522. doi: 10.1073/pnas.1000080107
Chen, C. L., Liu, W. T., Chong, M. L., Wong, M. T., Ong, S. L., Seah, H., et al. (2004). Community structure of microbial biofilms associated with membrane-based water purification processes as revealed using a polyphasic approach. Appl. Microbiol. Biotechnol. 63, 466–473. doi: 10.1007/s00253-003-1286-7
Chicote, E., García, A. M., Moreno, D. A., Sarró, M. I., Lorenzo, P. I., and Montero, F. (2005). Isolation and identification of bacteria from spent nuclear fuel pools. J. Ind. Microbiol. Biotechnol. 32, 155–162. doi: 10.1007/s10295-005-0216-3
Chicote, E., Moreno, D. A., Garcia, A. M., Sarro, M. I., Lorenzo, P. I., and Montero, F. (2004). Biofouling on the walls of a spent nuclear fuel pool with radioactive ultrapure water. Biofouling 20, 35–42. doi: 10.1080/08927010410001662670
Comte, S., Guibaud, G., and Baudu, M. (2008). Biosorption properties of extracellular polymeric substances (EPS) towards Cd, Cu and Pb for different pH values. J. Hazard. Mater. 151, 185–193. doi: 10.1016/j.jhazmat.2007.05.070
Dekker, L., Osborne, T. H., and Santini, J. M. (2014). Isolation and identification of cobalt- and caesium-resistant bacteria from a nuclear fuel storage pond. FEMS Microbiol. Lett. 359, 81–84. doi: 10.1111/1574-6968.12562
DeLong, E. F. (1992). Archaea in coastal marine environments. Proc. Natl. Acad. Sci. U.S.A. 89, 5685–5689. doi: 10.1073/pnas.89.12.5685
Deutch, J. M., Forsberg, C., Kadak, A. C., Kazimi, M. S., Moniz, E. J., and Parsons, J. E. (2009). Future of Nuclear Power, an Interdisciplinary MIY Study. Cambridge, MA: Massachusetts Institute of Technology.
Diesendorf, N., Köhler, S., Geißdörfer, W., Grobecker-Karl, T., Karl, M., and Burkovski, A. (2017). Characterisation of Roseomonas mucosa isolated from the root canal of an infected tooth. BMC Res. Notes 10:212. doi: 10.1186/s13104-017-2538-4
Ding, L., and Yokota, A. (2010). Curvibacter fontana sp. nov., a microaerobic bacteria isolated from well water. J. Gen. Appl. Microbiol. 56, 267–271. doi: 10.2323/jgam.56.267
Dittrich, M., Müller, B., Mavrocordatos, D., and Wehrli, B. (2003). Induced calcite precipitation by cyanobacterium Synechococcus. Acta Hydrochim. Hydrobiol. 31, 162–169. doi: 10.1002/aheh.200300486
Eden, P. A., Schmidt, T. M., Blakemore, R. P., and Pace, N. R. (1991). Phylogenetic analysis of Aquaspirillum magnetotacticum Using Polymerase Chain Reaction-Amplified 16s rRNA-Specific DNA. Int. J. Syst. Bacteriol. 41, 324–325. doi: 10.1201/b10765-4
Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fahy, A., Ball, A. S., Lethbridge, G., Timmis, K. N., and McGenity, T. J. (2008). Isolation of alkali-tolerant benzene-degrading bacteria from a contaminated aquifer. Lett. Appl. Microbiol. 47, 60–66. doi: 10.1111/j.1472-765x.2008.02386.x
Feng, G. D., Yang, S. Z., Wang, Y. H., Zhang, X. X., Zhao, G. Z., Deng, M. R., et al. (2014). Description of a Gram-negative bacterium, Sphingomonas guangdongensis sp. nov. Int. J. Syst. Evol. Microbiol. 64(PART 5), 1697–1702. doi: 10.1099/ijs.0.056853-0
Ferris, F. G., Wiese, R. G., and Fyfe, W. S. (1994). Precipitation of carbonate minerals by microorganisms: implications for silicate weathering and the global carbon dioxide budget. Geomicrobiol. J. 12, 1–13. doi: 10.1080/01490459409377966
Foster, L., Boothman, C., Ruiz-Lopez, S., Boshoff, G., Jenkinson, P., Sigee, D., et al. (2020a). Microbial bloom formation in a high pH spent nuclear fuel pond. Sci. Total Environ. 720:137515. doi: 10.1016/j.scitotenv.2020.137515
Foster, L., Muhamadali, H., Boothman, C., Sigee, D., Pittman, J. K., Goodacre, R., et al. (2020b). Radiation tolerance of Pseudanabaena catenata, a cyanobacterium relevant to the first generation magnox storage pond. Front. Microbiol. 11:515. doi: 10.3389/fmicb.2020.00515
Finneran, K. T., Johnsen, C. V., and Lovley, D. R. (2003). Rhodoferax ferrireducens sp. nov., a psychrotolerant, facultatively anaerobic bacterium that oxidizes acetate with the reduction of Fe(III). Int. J. Syst. Evol. Microbiol. 53, 669–673. doi: 10.1099/ijs.0.02298-0
Francis, A. J. (2012). “Impacts of microorganisms on radionuclides in contaminated environments and waste materials,” in Radionuclide Behaviour in the Natural Environment: Science, Implications and Lessons for the Nuclear Industry, eds C. Poinssot and H. Geckeis (Cambridge: Woodhead Publishing Limited). doi: 10.1533/9780857097194.1.161
Fuerst, J. A., Hawkins, J. A., Holmes, A., Sly, L. I., Moore, C. J., and Stackebrandt, E. (1993). Porphyrobacter neustonensis gen. nov., sp. nov., an aerobic bacteriochlorophyll-synthesizing budding bacterium from fresh water. Int. J. Syst. Bacteriol. 43, 125–134. doi: 10.1099/00207713-43-1-125
Fujita, Y., Redden, G. D., Ingram, J. C., Cortez, M. M., Ferris, F. G., and Smith, R. W. (2004). Strontium incorporation into calcite generated by bacterial ureolysis. Geochim. Cosmochim. Acta 68, 3261–3270. doi: 10.1016/j.gca.2003.12.018
Gadd, G. M. (2009). Biosorption: critical review of scientific rationale, environmental importance and significance for pollution treatment. J. Chem. Technol. Biotechnol. 84, 13–28. doi: 10.1002/jctb.1999
Galès, G., Libert, M. F., Sellier, R., Cournac, L., Chapon, V., and Heulin, T. (2004). Molecular hydrogen from water radiolysis as an energy source for bacterial growth in a basin containing irradiating waste. FEMS Microbiol. Lett. 240, 155–162. doi: 10.1016/j.femsle.2004.09.025
Ghorbanzadeh, M. S., and Mohammad, T. G. P. (2009). Biotechnological potential of Azolla filiculoides for biosorption of Cs and Sr: application of micro-PIXE for measurement of biosorption. Bioresour. Technol. 100, 1915–1921. doi: 10.1016/j.biortech.2008.10.019
Glaring, M. A., Vester, J. K., Lylloff, J. E., Al-Soud, W. A., Soørensen, S. J., and Stougaard, P. (2015). Microbial diversity in a permanently cold and alkaline environment in Greenland. PLoS One 10:e0124863. doi: 10.1371/journal.pone.0124863
Gov UK (2018). Sellafield Ltd. Available online at: https://www.gov.uk/government/organisations/sellafield-ltd/about (accessed July 6, 2018).
Hanada, S., Kawase, Y., Hiraishi, A., Takaichi, S., Matsuura, K., Shimada, K., et al. (1997). Porphyrobacter tepidarius sp. nov., a moderately thermophilic aerobic photosynthetic bacterium isolated from a hot spring. Int. J. Syst. Bacteriol. 47, 408–413. doi: 10.1099/00207713-47-2-408
Howden, M. (1987). Radioactive effluent treatment plant - sellafield reprocessing factory. Proc. Inst. Mech. Eng. A Power Process Eng. 201, 1–15. doi: 10.1243/pime_proc_1987_201_002_02
Jackson, S. F., Monk, S. D., and Riaz, Z. (2014). An investigation towards real time dose rate monitoring, and fuel rod detection in a First Generation Magnox Storage Pond (FGMSP). Appl. Radiat. Isot. 94, 254–259. doi: 10.1016/j.apradiso.2014.08.019
Jonsson, M., Nielsen, F., Roth, O., Ekeroth, E., Nilsson, S., and Hossain, M. M. (2007). Radiation induced spent nuclear fuel dissolution under deep repository conditions. Environm. Sci. Technol. 41, 7087–7093. doi: 10.1021/es070832y
Joshi, N., and Fass, J. (2011). Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files (Version 1.33). Available online at: https://github.com/najoshi/sickle (accessed December 18, 2019)
Kalyuzhnaya, M. G., Beck, D. A. C., Vorobev, A., Smalley, N., Kunkel, D. D., Lidstrom, M. E., et al. (2011). Novel methylotrophic isolates from lake sediment, description of Methylotenera versatilis sp. nov. and emended description of the genus methylotenera. Int. J. Syst. Evol. Microbiol. 62, 106–111. doi: 10.1099/ijs.0.029165-0
Kalyuzhnaya, M. G., Bowerman, S., Lara, J. C., Lidstrom, M. E., and Chistoserdova, L. (2006). Methylotenera mobilis gen. nov., sp. nov., an obligately methelamine-utilizing bacterium within the family Methylophilaceae. Int. J. Syst. Evol. Microbiol. 56, 2819–2823. doi: 10.1099/ijs.0.64191-0
Kampfer, P., Schulze, R., Jackel, U., Malik, K. A., Amann, R., and Spring, S. (2005). Hydrogenophaga defluvii sp. nov and Hydrogenophaga atypica sp. nov., isolated from activated sludge. Int. J. Syst. Evol. Microbiol. 55, 341–344. doi: 10.1099/ijs.0.03041-0
Kang, H., kim, H., Lee, B. I., Joung, Y., and Joh, K. (2014). Sediminibacterium goheungense sp. nov., isolated from a freshwater reservoir. Int. J. Syst. Evol. Microbiol. 64(PART 4), 1328–1333. doi: 10.1099/ijs.0.055137-0
Kang, H., Weerawongwiwat, V., Jung, M. Y., Myung, S. C., and Kim, W. (2013). Algoriphagus chungangensis sp. nov., isolated from a tidal flat sediment. Int. J. Syst. Evol. Microbiol. 63(PART2), 648–653. doi: 10.1099/ijs.0.039214-0
Karley, D., Shukla, S. K., and Rao, T. S. (2018). Isolation and characterization of culturable bacteria present in the spent nuclear fuel pool water. Environ. Sci. Pollut. Res. 25, 20518–20526. doi: 10.1007/s11356-017-0376-5
Kawai, M., Matsutera, E., Kanda, H., Yamaguchi, N., Tani, K., and Nasu, M. (2002). 16S ribosomal DNA-based analysis of bacterial diversity in purified water used in pharmaceutical manufacturing processes by PCR and denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 68, 699–704. doi: 10.1128/AEM.68.2.699-704.2002
Kim, M. S., Baik, K. S., Park, S. C., Rhee, M. S., Oh, H. M., and Seong, C. N. (2009). Roseomonas frigidaquae sp. nov., isolated from a water-cooling system. Int. J. Syst. Evol. Microbiol. 59, 1630–1634. doi: 10.1099/ijs.0.004812-0
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., and Schloss, P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the miseq illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. doi: 10.1128/AEM.01043-13
Kulakov, L. A., McAlister, M. B., Ogden, K. L., Larkin, M. J., and O’Hanlon, J. F. (2002). Analysis of bacteria contaminating ultrapure water in industrial systems. Appl. Environ. Microbiol. 68, 1548–1555. doi: 10.1128/AEM.68.4.1548-1555.2002
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis Version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lambo, A. J., and Patel, T. R. (2006). Isolation and characterization of a biphenyl-utilizing psychrotrophic bacterium, Hydrogenophaga taeniospiralis IA3-A, that cometabolize dichlorobiphenyls and polychlorinated biphenyl congeners in Aroclor 1221. J. Basic Microbiol. 46, 94–107. doi: 10.1002/jobm.200510006
Lane, D. J., Pace, B., Olsen, G. J., Stahl, D. A., Sogin, M. L., and Pace, N. R. (1986). Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc. Natl. Acad. Sci. U.S.A. 83:4792.
Lang, A. R., Engelberg, D. L., Walther, C., Weiss, M., Bosco, H., Jenkins, A., et al. (2019). Cesium and strontium contamination of nuclear plant stainless steel: implications for decommissioning and waste minimization. ACS Omega 4, 14420–14429. doi: 10.1021/acsomega.9b01311
Lawson, P. S., Sterrit, R. M., and Lester, J. N. (1984). Factor affecting the removal of metals during activated sludge wastewater treatment II. The role of mixed liquor biomass. Arch. Environ. Contam. Toxicol. 13, 391–402. doi: 10.1007/bf01056254
Lee, J. S., Shin, Y. K., Yoon, J. H., Takeuchi, M., Pyun, Y. R., and Park, Y. H. (2001). Sphingomonas aquatilis sp. nov., Sphingomonas koreensis sp. nov. and Sphingomonas taejonensis sp. nov., yellow-pigmented bacteria isolated from natural mineral water. Int. J. Syst. Evol. Microbiol. 51, 1491–1498. doi: 10.1099/00207713-51-4-1491
Lee, S. Y., Jung, K. H., Lee, J. E., Lee, K. A., Lee, S. H., Lee, J. Y., et al. (2014). Photosynthetic biomineralization of radioactive Sr via microalgal CO2 absorption. Bioresour. Technol. 172, 449–452. doi: 10.1016/j.biortech.2014.09.023
Leys, N. M. E. J., Ryngaert, A., Bastiaens, L., Verstraete, W., Top, E. M., Springael, D., et al. (2004). Occurrence and phylogenetic diversity of Sphingomonas strains in soils contaminated with polycyclic aromatic hydrocarbons. Appl. Environ. Microbiol. 70, 1944–1955. doi: 10.1128/AEM.70.4.1944-1955.2004
Libert, M., Bildstein, O., Esnault, L., Jullien, M., and Sellier, R. (2011). Molecular hydrogen: an abundant energy source for bacterial activity in nuclear waste repositories. Phys. Chem. Earth 36, 1616–1623. doi: 10.1016/j.pce.2011.10.010
Liu, M., Dong, F., Kang, W., Sun, S., Wei, H., Zhang, W., et al. (2014). Biosorption of strontium from simulated nuclear wastewater by Scenedesmus spinosus under culture conditions: adsorption and bioaccumulation processes and models. Int. J. Environ. Res. Public Health 11, 6099–6118. doi: 10.3390/ijerph110606099
Liu, Q., Wu, Y. H., Cheng, H., Xu, L., Wang, C. S., and Xu, X. W. (2017). Complete genome sequence of bacteriochlorophyll-synthesizing bacterium Porphyrobacter neustonensis DSM 9434. Stand. Genomic Sci. 12:32. doi: 10.1186/s40793-017-0243-5
Lloyd, J. R. (2003). Microbial reduction of metals and radionuclides. FEMS Microbiol. Rev. 27, 411–425. doi: 10.1016/S0168-6445(03)00044-5
Lloyd, J. R., and Macaskie, L. E. (2000). “Bioremediation of radionuclide-containing wastewaters,” in Environmental Microbe-metal Interactions, ed. R. D. Lovley (Washington, DC: ASM Press).
Lloyd, J. R., and Macaskie, L. E. (2002). “Biochemical basis of microbe-radionuclide interactions,” in Interactions of Microorganisms with Radionuclides, eds M. J. Keith-Roach and F. R. Livens (Amsterdam: Elsevier).
Lloyd, J. R., and Renshaw, J. C. (2005). Bioremediation of radioactive waste: radionuclide-microbe interactions in laboratory and field-scale studies. Curr. Opin. Biotechnol. 16, 254–60. doi: 10.1016/j.copbio.2005.04.012
Lorenz, T. C. (2012). Polymerase chain reaction: basic protocol plus troubleshooting and optimization strategies. J. Vis. Exp. 63:e3998. doi: 10.3791/3998
Lovley, D. R., Greening, R. C., and Ferry, J. G. (1984). Rapidly growing rumen methanogenic organism that synthesizes coenzyme M and Has a High Affinity for Formate. Appl. Environ. Microbiol. 48, 81–87. doi: 10.1128/aem.48.1.81-87.1984
Ma, D., Hao, Z., Sun, R., Bartlam, M., and Wang, Y. (2016). Genome sequence of a typical Ultramicrobacterium, Curvibacter sp. Strain PAE-UM, capable of phtalate ester degradation. Genome Announc. 4:e01510-15. doi: 10.1128/genomeA.01510-15.Copyright
Macaskie, L. E., Empson, R. M., Cheetham, A. K., Grey, C. P., and Skarnulis, J. (1992). Uranium Bioaccumulation by a Citrobacter sp as a result of enzymically mediated growth of polycrystalline HUO2PO4. Science 257, 782–784. doi: 10.1126/science.1496397
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.Journal 7, 10–12. doi: 10.14806/ej.17.1.200
Masella, A. P., Bartram, A. K., Truszkowski, J. M., Brown, D. G., and Neufeld, J. D. (2012). PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics 13. doi: 10.1016/s0739-5930(09)79285-2
Masurat, P., Fru, E. C., and Pedersen, K. (2005). Identification of Meiothermus as the dominant genus in a storage system for spent nuclear fuel. J. Appl. Microbiol. 98, 727–740. doi: 10.1111/j.1365-2672.2004.02519.x
MeGraw, V. E., Brown, A. R., Boothman, C., Goodacre, R., Morris, K., Sigee, D., et al. (2018). A novel adaptation mechanism underpinning algal colonization of a nuclear fuel storage pond. mBio 9:e02395-17. doi: 10.1128/mBio.02395-17
Merroun, M. L., and Selenska-Pobell, S. (2008). Bacterial interactions with uranium: an environmental perspective. J. Contam. Hydrol. 102, 285–295. doi: 10.1016/j.jconhyd.2008.09.019
Misal, S. A., Bajoria, V. D., Lingojwar, D. P., and Gawai, K. R. (2013). Purification and characterization of nitroreductase from red alkaliphilic bacterium Aquiflexum sp. DL6. Appl. Biochem. Microbiol. 49, 227–232. doi: 10.1134/S0003683813030125
Mortensen, B. M., Haber, M. J., DeJong, J. T., Caslake, L. F., and Nelson, D. C. (2011). Effects of environmental factors on microbial induced calcium carbonate precipitation. J. Appl. Microbiol. 111, 338–349. doi: 10.1111/j.1365-2672.2011.05065.x
Neidhart, F. C., Bloch, P. L., and Smith, D. F. (1974). Culture medium for enterobacteria. J. Bacteriol. 119, 736–747. doi: 10.1128/jb.119.3.736-747.1974
Newsome, L., Morris, K., and Lloyd, J. R. (2014a). The biogeochemistry and bioremediation of uranium and other priority radionuclides. Chem. Geol. 363, 164–184. doi: 10.1016/j.chemgeo.2013.10.034
Newsome, L., Morris, K., Trivedi, D., Atherton, N., and Lloyd, J. R. (2014b). Microbial reduction of uranium(VI) in sediments of different lithologies collected from Sellafield. Appl. Geochem. 51, 55–64. doi: 10.1016/j.apgeochem.2014.09.008
Niharika, N., Jindal, S., Kaur, J., and Lal, R. (2012). Sphingomonas indica sp. nov., isolated from hexachlorocyclohexane (HCH)-contaminated soil. Int. J. Syst. Evol. Microbiol. 62, 2997–3002. doi: 10.1099/ijs.0.033845-0
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A., Korobeynikov, A., Lapidus, A., et al. (2013). “Assembling genomes and mini-metagenomes from highly chimeric reads,” in Research in Computational Molecular Biology. RECOMB 2013. Lecture Notes in Computer Science, eds M. Deng, R. Jiang, F. Sun, and X. Zhang (Berlin: Springer), 7821, 158–170. doi: 10.1007/978-3-642-37195-0_13
Peschek, G. A. (1999). “Photosynthesis and respiration of cyanobacteria,” in The Phototrophic Prokaryotes, eds G. A. Peschek, W. Loffelhardt, and G. Schmetterer (Boston, MA: Springer).
Pipíška, M., Trajtešová, Z., Horník, M., and Frišták, V. (2018). Evaluation of Mn bioaccumulation and biosorption by bacteria isolated from spent nuclear fuel pools using 54Mn as a radioindicator. Radiochim. Acta 106, 217–228. doi: 10.1515/ract-2017-2836
Proctor, C. R., Edwards, M. A., and Pruden, A. (2015). Microbial composition of purified waters and implications for regrowth control in municipal water systems. Environ. Sci. Water Res. Technol. 1, 882–892. doi: 10.1039/c5ew00134j
Qu, J. H., and Yuan, H. L. (2008). Sediminibacterium salmoneum gen. nov., sp. nov., a member of the phylum Bacteroidetes isolated from sediment of a eutrophic reservoir. Int. J. Syst. Evol. Microbiol. 58, 2191–2194. doi: 10.1099/ijs.0.65514-0
Rajala, P., Bomberg, M., Vepsalainen, M., and Carpen, L. (2017). Microbial fouling and corrosion of carbon steel in deep anoxic alkaline groundwater. Biofouling 33, 195–209. doi: 10.1080/08927014.2017.1285914
Reddy, S. F., Monk, S. D., Nye, D. W., Colling, B. R., and Stanley, S. J. (2012). Proposal to characterize legacy Sellafield ponds using SONAR and RadLine. Appl. Radiat. Isot. 70, 1162–1165. doi: 10.1016/j.apradiso.2012.04.002
Reeder, J. R., Nugent, M., Tait, C. D., Morris, D. E., Heald, S., Beck, K. M., et al. (2001). Coprecipitation of Uranium (VI) with calcite: XAFS, micro XAS, and luminescence characterization. Geochim. Cosmochim. Acta 65, 3491–3503. doi: 10.1016/s0016-7037(01)00647-0
Rivasseau, C., Farhi, E., Compagnon, E., de Gouvion Saint Cyr, D., van Lis, R., Falconet, D., et al. (2016). Coccomyxa actinabiotis sp. nov. (Trebouxiophyceae, Chlorophyta), a new green microalga living in the spent fuel cooling pool of a nuclear reactor. J. Phycol. 52, 689–703. doi: 10.1111/jpy.12442
Risso, C., Sun, J., Zhuang, K., Mahadevan, R., DeBoy, R., Ismail, W., et al. (2009). Genome-scale comparison and constraint-based metabolic reconstruction of the facultative anaerobic Fe(III)-reducer Rhodoferax ferrireducens. BMC Genomics 10:447. doi: 10.1186/1471-2164-10-447
Rizoulis, A. E., Morris, K., and Lloyd, J. R. (2016). Bacterial Diversity in the Hyperalkaline Allas Springs (Cyprus), a Natural Analogue for Cementitious Radioactive Waste Repository. Geomicrobiol. J. 73–84. doi: 10.1080/01490451.2014.961107
Santo Domingo, J. W., Berry, C. J., Summer, M., and Fliermans, C. B. (1998). Microbiology of spent nuclear fuel storage basins. Curr. Microbiol. 37, 387–394. doi: 10.1007/s002849900398
Sarró, M. I., García, A. M., and Moreno, D. A. (2005). Biofilm formation in spent nuclear fuel pools and bioremediation of radioactive water. Int. Microbiol. 8, 223–230. doi: 10.2436/im.v8i3.9529
Schwartz, E., Fritsch, J., and Friedrich, B. (2013). “H2-metabolizing prokaryotes,” in The Prokaryotes: Prokaryotic Physiology and Biochemistry, Vol. 9783642301414, (Berlin: Springer-Verlag), 119–199. doi: 10.1007/978-3-642-30141-4_65
Sellafield Ltd (2011). Sellafield Plan. Available online at: https://www.cumbria.gov.uk/eLibrary/Content/Internet/538/755/1929/17716/17720/17722/41333114920.pdf (accessed December 2, 2015).
Sellafield Ltd (2015). Enablers. https://webarchive.nationalarchives.gov.uk/20170712124354/http://www.sellafieldsites.com/solution/spent-fuel-management/magnox-reprocessing/enablers/ (accessed November 14, 2015)
Sezonov, G., Joseleau-Petit, D., and D’Ari, R. (2007). Escherichia coli physiology in Luria-Bertani broth. J. Bacteriol. 189, 8746–8749. doi: 10.1128/jb.01368-07
Shaw, R. D. (1990). Corrosion prevention and control at Sellafield nuclear fuel reprocessing plant. Br. Corros. J. 25, 97–107. doi: 10.1179/bcj.1990.25.2.97
Shoesmith, D. W. (2000). Fuel corrosion processes under waste disposal conditions. J. Nucl. Mater. 282, 1–31. doi: 10.1016/s0022-3115(00)00392-5
Silva, R., De Almeida, D. M., Cabral, B. C. A., Dias, V. H. G., De Toledo, E., Mello, I. C., et al. (2018). Microbial enrichment and gene functional categories revealed on the walls of a spent fuel pool of a nuclear power plant. PLoS One 13:e0205228. doi: 10.1371/journal.pone.0205228
Singh, P., Kim, Y. J., Hoang, V. A., Farh, M. E. A., and Yang, D. C. (2015). Sphingomonas panacis sp. nov., isolated from rhizosphere of rusty ginseng. Antonie van Leeuwenhoek 108, 711–720. doi: 10.1007/s10482-015-0527-y
Slatko, B. E., Albright, L. M., Tabor, S., and Ju, J. (1999). DNA Sequencing by the Dideoxy Method. Curr. Protoc. Mol. Biol. 47, 7.4A.1–7.4A.39. doi: 10.1002/0471142727.mb0704as47
Springell, R., Rennie, S., Costelle, L., Darnbrough, J., Stitt, C., Cocklin, E., et al. (2014). Water corrosion of spent nuclear fuel: radiolysis driven dissolution at the UO2/water interface. Faraday Discuss. 180, 301–311. doi: 10.1039/c4fd00254g
Sullivan, L. (2017). Hypotehsis Testing for Means and Proportions. Available online at: https://sphweb.bumc.bu.edu/otlt/mph-modules/bs/bs704_hypothesistest-means-proportions/ (accessed September 13, 2020).
Suzuki, S., Kenuen, J. G., Schipper, K., Van de Velde, S., Ishii, S., Sorokin, D. Y., et al. (2014). Physiological and genomic features of highly alkaliphilic hydrogen-utilizing Betaproteobacteria from a continental serpentinizing site. Nat. Commun. 5:3900.
Tiago, I., Chung, A. P., and Veríssimo, A. (2004). Bacterial diversity in a nonsaline alkaline environment: heterotrophic aerobic populations. Appl. Environ. Microbiol. 70, 7378–7387. doi: 10.1128/AEM.70.12.7378-7387.2004
Tišáková, L., Pipíška, M., Godány, A., Horník, M., Vidová, B., and Augustín, J. (2013). Bioaccumulation of 137Cs and 60Co by bacteria isolated from spent nuclear fuel pools. J. Radioanal. Nucl. Chem. 295, 737–748. doi: 10.1007/s10967-012-1932-6
Tomioka, N., Uchiyama, H., and Yagi, O. (1992). Isolation and characterization of cesium-accumulating bacteria. Appl. Environ. Microbiol. 58, 1019–1023. doi: 10.1128/aem.58.3.1019-1023.1992
Turner, S., Miao, V. P., and Palmer, J. D. (1999). Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J. Eukaryot. Microbiol. 46, 327–338. doi: 10.1111/j.1550-7408.1999.tb04612.x
Van Roy, S., Peys, K., Dresselaers, T., and Diels, L. (1997). The use of Alcaligenes eutrophus biofilm in a membrane bioreactor for heavy metal recovery. Res. Microbiol. 148, 526–528. doi: 10.1016/s0923-2508(97)88356-8
Větrovský, T., and Baldrian, P. (2013). The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One 8:e57923. doi: 10.1371/journal.pone.0057923
Wang, C., Deng, S., Liu, X., Yao, L., Shi, C., Jiang, J., et al. (2016). Roseomonas eburnea sp. nov., isolated from activated sludge. Int. J. Syst. Evol. Microbiol. 66, 385–390. doi: 10.1099/ijsem.0.000728
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/aem.00062-07
White, C., and Gadd, G. M. (1990). Biosorption of radionuclides by fungal biomass. J. Chem. Technol. Biotechnol. 49, 331–343. doi: 10.1002/jctb.280490406
White, C., Sharman, A. K., and Gadd, G. M. (1998). An integrated microbial process for the bioremediation of soild contaminated with toxic metals. Nat. Biotechnol. 16, 572–575. doi: 10.1038/nbt0698-572
Willems, A., Busse, J., Goor, M., Pot, B., Falsen, E., Jantzen, E., et al. (1989). Hydrogenophaga, a new genus of hydrogen-oxidizing bacteria that includes Hydrogenophaga flava comb. nov. (formerly Pseudomonas flava), Hydrogenophaga palleronii (formerly Pseudomonas palleronii), Hydrogenophaga pseudoflava (formerly Pseudomonas pseudoflav). Int. J. Syst. Bacteriol. 39, 319–333. doi: 10.1099/00207713-39-3-319
Wingender, J., Neu, T. R., and Flemming, H.-C. (1999). Microbial Extracellular Polymeric Substances?: Characterization, Structure, and Function, eds J. Wingender, T. R. Neu, and H.-C. Flemming (Berlin: Springer).
WNA (2018a). Nuclear Power in the United Kingdom. Available online at: http://www.world-nuclear.org/information-library/country-profiles/countries-t-z/united-kingdom.aspx (accessed February 4, 2019).
WNA (2018b). Radioactive Waste Management. Available online at: http://www.world-nuclear.org/information-library/nuclear-fuel-cycle/nuclear-wastes/radioactive-waste- (accessed May 25, 2019).
Wolfram, J. H., Mizia, R. E., Jex, R., Nelson, L., and Garcia, K. M. (1996). The Impact of Microbially Influenced Corrosion on Spent Nuclear Fuel and Storage Life. Idaho Falls, ID: Idaho National Laboratory.
Yoon, J. H., Lee, M. H., and Oh, T. K. (2004). Porphyrobacter donghaensis sp. nov., isolated from sea water of the East Sea in Korea. Int. J. Syst. Evol. Microbiol. 54, 2231–2235. doi: 10.1099/ijs.0.63226-0
Yoon, J. H., Tsukada, N., Sakai, Y., Ishii, M., Igarashi, Y., and Nishihara, H. (2008). Isolation and characterization of a new facultatively autotrophic hydrogen-oxidizing Betaproteobacterium, Hydrogenophaga sp. AH-24. FEMS Microbiol. Lett. 278, 94–100. doi: 10.1111/j.1574-6968.2007.00983.x
Yu, J. (2018). Fixation of carbon dioxide by a hydrogen-oxidizing bacterium for value-added products. World Microbiol. Technol. 34:89.
Yurkov, V., Stackebrandt, E., Holmes, A., Fuerst, J. A., Hugenholtz, P., Golecki, J., et al. (1994). Phylogenetic positions of novel aerobic, bacteriochlorophyll a-containing bacteria and description of Roseococcus thiosulfatophilus gen. nov., sp. nov., Erythromicrobium ramosum gen. nov., sp. nov., and Erythrobacter litoralis sp. nov. Int. J. Syst. Bacteriol. 44, 427–434. doi: 10.1099/00207713-44-3-427
Yurkov, V. V. (2015). “Roseococcus,” in Bergey’s Manual of Systematics of Archaea and Bacteria, eds V. Yurkov, E. Stackebrandt, A. Holmes, J. A. Fuerst, P. Hugenholtz, J. Golecki, et al. (Hoboken, NJ: John Wiley & Sons, Inc.), 1–4. doi: 10.1002/9781118960608.gbm00887
Keywords: spent nuclear fuel, microbial ecology, Sellafield, Hydrogenophaga, spent nuclear fuel ponds, hydrogen metabolism
Citation: Ruiz-Lopez S, Foster L, Boothman C, Cole N, Morris K and Lloyd JR (2020) Identification of a Stable Hydrogen-Driven Microbiome in a Highly Radioactive Storage Facility on the Sellafield Site. Front. Microbiol. 11:587556. doi: 10.3389/fmicb.2020.587556
Received: 26 July 2020; Accepted: 26 October 2020;
Published: 24 November 2020.
Edited by:
Rakesh Mogul, California State Polytechnic University, United StatesReviewed by:
Cynthia B. Silveira, San Diego State University, United StatesSteven Singer, Lawrence Berkeley National Laboratory, United States
Copyright © 2020 Ruiz-Lopez, Foster, Boothman, Cole, Morris and Lloyd. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sharon Ruiz-Lopez, c2hhcnVsb18yNjhAaG90bWFpbC5jb20=; Jonathan R. Lloyd, Sm9uLkxsb3lkQG1hbmNoZXN0ZXIuYWMudWs=
†Present address: Sharon Ruiz-Lopez, Department of Environmental Microbiology, Manchester Metropolitan University, Manchester, United Kingdom