 Li-Yin Lai1Li-Yu Hsu1Shang-Hui Weng1Shuo-En Chung1Hui-En Ke1Tzu-Lung Lin1Pei-Fang Hsieh1Wei-Ting Lee2,3Hsing-Yuan Tsai2,3Wan-Hsuan Lin2,3
Li-Yin Lai1Li-Yu Hsu1Shang-Hui Weng1Shuo-En Chung1Hui-En Ke1Tzu-Lung Lin1Pei-Fang Hsieh1Wei-Ting Lee2,3Hsing-Yuan Tsai2,3Wan-Hsuan Lin2,3 Ruwen Jou2,3
Ruwen Jou2,3 Jin-Town Wang1,4*
Jin-Town Wang1,4*- 1Department of Microbiology, National Taiwan University College of Medicine, Taipei, Taiwan
- 2Tuberculosis Research Center, Centers for Disease Control, Ministry of Health and Welfare of Taiwan, Taipei, Taiwan
- 3Center for Diagnostics and Vaccine Development, Centers for Disease Control, Ministry of Health and Welfare of Taiwan, Taipei, Taiwan
- 4Department of Internal Medicine, National Taiwan University Hospital, Taipei, Taiwan
Tuberculosis (TB) is an infectious respiratory disease caused by Mycobacterium tuberculosis and one of the top 10 causes of death worldwide. Treating TB is challenging; successful treatment requires a long course of multiple antibiotics. Rifampicin (RIF) is a first-line drug for treating TB, and the development of RIF-resistant M. tuberculosis makes treatment even more difficult. To determine the mechanism of RIF resistance in these strains, we searched for novel mutations by sequencing. Four isolates, CDC-1, CDC-2, CDC-3, and CDC-4, had high-level RIF resistance and unique mutations encoding RpoB G158R, RpoB V168A, RpoB S188P, and RpoB Q432insQ, respectively. To evaluate their correlation with RIF resistance, plasmids carrying rpoB genes encoding these mutant proteins were transfected into the H37Rv reference strain. The plasmid complementation of RpoB indicated that G158R, V168A, and S188P did not affect the MIC of RIF. However, the MIC of RIF was increased in H37Rv carrying RpoB Q432insQ. To confirm the correlation between RIF resistance and Q432insQ, we cloned an rpoB fragment carrying the insertion (encoding RpoB Q432insQ) into H37Rv by homologous recombination using a suicide vector. All replacement mutants expressing RpoB Q432insQ were resistant to RIF (MIC > 1 mg/L). These results indicate that RpoB Q432insQ causes RIF resistance in M. tuberculosis.
Introduction
Mycobacterium tuberculosis is an important human pathogen that causes tuberculosis (TB). It was first isolated by Robert Koch in 1882, and numerous TB drugs have been developed since the 1930s, although the first effective anti-TB drug was not discovered until 1944 (Vilcheze and Jacobs, 2014; Islam et al., 2017; Long et al., 2019; Mabhula and Singh, 2019). TB is a common, chronic infectious respiratory disease that affects nearly one-third of the world’s population (Long et al., 2019; Yong et al., 2019) and is transmitted via aerosol (e.g., cough) from infected persons. According to the World Health Organization, TB is one of the top 10 causes of death worldwide. In 2018, there were approximately 10 million people with TB and 1.4 million deaths due to TB worldwide (World Health Organization [WHO], 2019). Approximately 500,000 new cases were found to be resistant to the most effective first-line drug, rifampicin (RIF), of which 78% had multidrug-resistant TB (resistant to at least isoniazid and RIF) (World Health Organization [WHO], 2019). According to the Taiwan Centers for Disease Control, there were 9,179 new cases of TB in Taiwan in 2018, and 294 of these were RIF-resistant. Over the period of long-term anti-TB therapy, M. tuberculosis is exposed to the appropriate drug concentration, which might lead to the development of drug-resistant TB and increase the risk of transmission (Yong et al., 2019). It takes at least 6 months to successfully treat TB, and the development of drug resistance makes therapy even more difficult and is a threat to public health. The treatment of drug-resistant TB requires the administration of more than five drugs for more than 9 months (Vekemans et al., 2020). In addition, RIF-resistant TB is frequently not adequately treated because of a delay in the diagnosis of drug resistance. Such delayed treatment not only has a poor therapeutic effect on the infected patient but also increases the risk of transmission (Boyd et al., 2017; Yong et al., 2019). First-line RIF acts by binding the β-subunit of RNA polymerase, blocking RNA synthesis, and inducing hydroxyl radical formation, which likely contributes to its killing effect (Piccaro et al., 2014; Lohrasbi et al., 2018). It has been reported that drug resistance develops when mutations in rpoB block RIF binding to the β-subunit. Most point mutations causing RIF resistance occur in an 81-nucleotide region (codons 426–452 in M. tuberculosis or codons 507–533 in the Escherichia coli codon numbering system) of rpoB that is called the RIF resistance-determining region (RRDR) (Pang et al., 2013; Chikaonda et al., 2017). According to previous studies, >90% of RIF-resistant TB strains have mutations in the RRDR within codon 435, 445, and 450 (codon 516, 526, and 531 in E. coli) (Pang et al., 2013; Swain et al., 2020). Drug resistance is a major challenge for TB control. Due to the slow growth rate of M. tuberculosis, conventional drug susceptibility testing takes several weeks. Therefore, sequencing drug resistance-related mutations can be used to quickly detect drug-resistant TB, significantly reducing the duration of therapy, and avoiding treatment delays. Thus, we examined the effects of novel mutations in known target gene regions and mutations outside of the target region on RIF resistance in M. tuberculosis with the aim to expand upon known RIF resistance-causing mutations for use in clinical molecular diagnostics.
Materials and Methods
Bacteria Strains
Clinical isolates were obtained from the Reference Laboratory of Mycobacteriology of the Taiwan Centers for Disease Control. M. tuberculosis H37Rv was used as the reference strain. M. tuberculosis H37Rv and clinical isolates were cultured in DifcoTM Middlebrook 7H9 medium (BD REF: 271310; MD, NJ, United States) supplemented with 10% oleic acid/albumin/dextrose/catalase (OADC), 0.5% glycerol, and 0.05% tween-80 at 37°C (Tan et al., 2006). E. coli DH10B was grown in Luria broth (LBL405.1; BioShop, Burlington, Canada). All experiments involving M. tuberculosis strains were conducted at the Biosafety level 3 lab in the National Taiwan University College of Medicine and followed institutional biosafety procedures.
Isolation of Clinical Strains and Drug Susceptibility Testing
Clinical isolates of M. tuberculosis were tested in the Reference Laboratory of Mycobacteriology of the Taiwan Centers for Disease Control. Each isolate was initially obtained from a patient and was inoculated in solid and liquid culture. The minimum inhibitory concentration (MIC) of RIF was determined using a SensititreTM Mycobacterium tuberculosis MIC Plate (MYCOTB; Thermo Fisher Scientific, Cleveland, OH, United States) according to the manufacturer’s instructions. The bacterial culture was adjusted to a 0.5 McFarland standard and then added to the SensititreTM plate, which was covered with an adhesive plastic seal. After incubation at 37°C, the results were recorded with a SensititreTM VizionTM Digital MIC Viewing System. The critical concentration of RIF was 1 mg/L.
Microplate Alamar Blue Assays (MABAs)
Microplate alamar blue assays were performed as previously described with minor modifications (Burke et al., 2017). Briefly, M. tuberculosis strains were cultured in Middlebrook 7H9 medium, and then the OD600nm was adjusted to 0.1. A 200-μL aliquot of each prepared bacterial suspension was placed in a 96-well sterile plate (LabServ, Singapore) and incubated at 37°C for 14 days with and without RIF (0.03–512 mg/L). After 14 days of incubation, 50 μL of a freshly prepared 1:1 mixture of Alamar blue (alamarBlue®; BioRad, Hercules, CA, United States) and 10% tween 80 was added to each well. Then, the plate was incubated at 37°C for 48 h, and the color was recorded. Blue indicates no growth, and pink indicates growth. During incubation and staining, the plate was sealed with an adhesive plate seal.
RpoB Expression Plasmids
The plasmid pMN437 was used to express RpoB in M. tuberculosis (Steinhauer et al., 2010). Derivative plasmids carrying resistance-associated rpoB mutations were generated using the QuikChange II Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA, United States). The primers used are shown in Supplementary Table 1. M. tuberculosis was transformed by electroporation at 2,500 V, 1,000 Ω, and 25 μF and recovered in 10 mL of Middlebrook 7H9 medium supplemented with 10% OADC and 0.5% glycerol for 24 h (Larsen et al., 2007; Lai et al., 2018). Recovered cells were then plated on BBLTM 7H11 solid agar (BD REF: 212203; BD, MD, NJ, United States) containing 50 mg/L hygromycin and incubated at 37°C for 4 weeks.
Site-Directed Mutagenesis
The putative resistance-associated rpoB mutations were amplified by PCR from the genomic DNA of the resistant strains as a template and then cloned into the ScaI site of the suicide plasmid pGOAL19 (Addgene; MA, United States) (Parish and Stoker, 2000). The primers used are shown in Supplementary Table 1. The resulting plasmids were transformed into M. tuberculosis H37Rv, and point mutants were selected after two rounds of homologous recombination, as previously described (Parish and Stoker, 2000; Larsen et al., 2007).
DNA Sequencing
Mycobacterium tuberculosis isolates were lysed with InstantGetTM DNA Extraction Solution (Catalog Number: 17001; HNG, Taipei, Taiwan) and heat-killed (100°C for 15 min). Then, the supernatants of heat-killed M. tuberculosis isolates were added to the PCR reagent containing KOD XtremeTM Hot Start DNA Polymerase (Catalog Number: 71976; Novagen, CA, United States) and gene-specific primers, rpoB out F1 and rpoB-out-R (Supplementary Table 1), to amplify the rpoB fragment. The PCR products were sent for sequencing (Mission Biotech, Taipei, Taiwan) and were verified by capillary electrophoresis (ABI 3730xl; Thermo Fisher Scientific, Cleveland, OH, United States). The rpoB sequences of clinical strains CDC-1, CDC-2, and CDC-4 were submitted to NCBI (GenBank accession numbers: MT774526, MT774527, and MT774528, respectively).
Results
Identification of Novel rpoB Mutations in RIF-Resistant Clinical Isolates
Four clinical isolates, CDC-1, CDC-2, CDC-3, and CDC-4, had high-level RIF resistance and different novel mutations within the rpoB gene. The RIF resistance levels of clinical strains were determined using a SensititreTM Mycobacterium tuberculosis MIC Plate. The MIC of RIF for CDC-1 was 8 mg/L, and the MICs of RIF for CDC-2, CDC-3, and CDC-4 were >16 mg/L (Table 1). Sequencing showed that each of these four clinical isolates harbored genes encoding two amino acid changes in RpoB. When compared to the sequence of the reference strain H37Rv, in CDC-1, the G at codon 158 was replaced with R (G158R) and the V at codon 170 was replaced with F (V170F). In CDC-2, the V at codon 168 was replaced with A (V168A) and the V at codon 170 was replaced with F (V170F). In CDC-3, the V at codon 170 of RpoB was substituted with F (V170F), and the S at codon 188 was replaced with P (S188P). In CDC-4, the S at codon 431 was replaced with G (S431G) and a Q was inserted at codon 432 (Q432insQ). However, G158R, V168A, S188P, and Q432insQ had not previously been confirmed to be related to drug resistance.
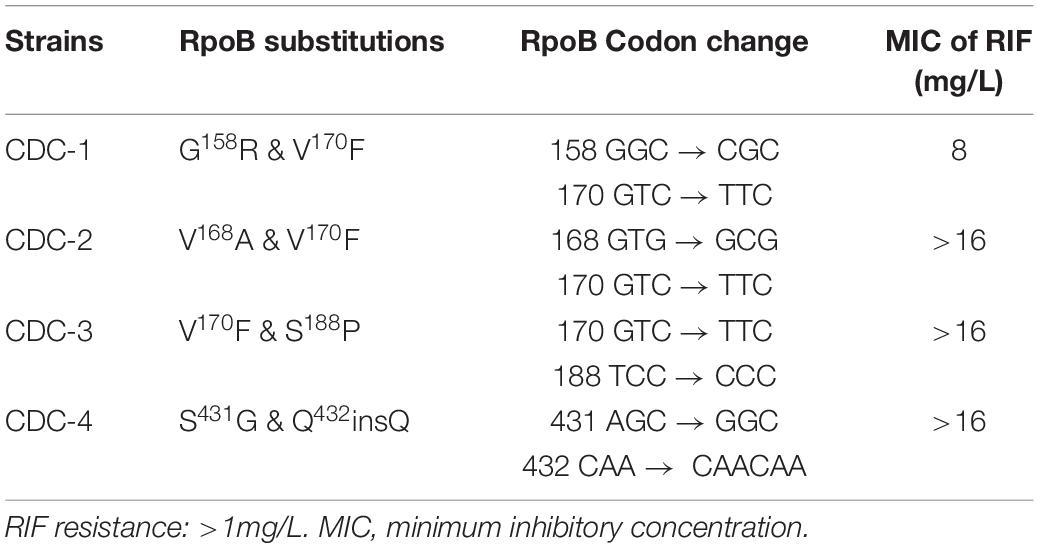
Table 1. Rifampicin (RIF) resistance levels of clinical strains as measured by SensititreTM Mycobacterium tuberculosis MIC Plate.
An RpoB Q432insQ Expression Plasmid Increases the MIC of RIF in H37Rv
Clinical isolates CDC-1, CDC-2, CDC-3, and CDC-4 had high-level RIF resistance and harbored unique RpoB amino acid changes (G158R, V168A, S188P, and Q432insQ, respectively). To evaluate the association between these unique amino acid changes and the RIF resistance of the clinical isolates, plasmids (pMN437-derived) expressing these RpoB proteins were transformed into H37Rv (Table 2), and RIF susceptibility was measured in these strains by a MABA. The results showed that the MICs of RIF for the wild-type strains, H37Rv and H37Rv:pMN437, were 0.03–0.125 mg/L. The MIC of RIF for clinical strain CDC-1 was 4–32 mg/L, that for clinical strain CDC-2 was 256–512 mg/L, and that for clinical strain CDC-4 was 128–256 mg/L. These results were consistent with the MIC data obtained with the SensititreTM Mycobacterium tuberculosis MIC Plate. However, the MIC of RIF for clinical strain CDC-3 could not be determined by the MABA because it could not be cultured. The MIC of RIF for H37Rv:pMN437-RpoB G158R, H37Rv:pMN437-RpoB V168A, and H37Rv:pMN437-RpoB S188P was 0.03–0.25 mg/L, that for H37Rv:pMN437-CDC-4 was 128–256 mg/L, and that for H37Rv:pMN437-RpoB Q432insQ was 64–128 mg/L (Tables 2, 3, Figure 1). These results showed that H37Rv:pMN437-RpoB G158R, H37Rv:pMN437-RpoB V168A, and H37Rv:pMN437-RpoB S188P were not resistant to RIF (MIC < 1 mg/L). The MIC of RIF for H37Rv:pMN437-CDC-4 was the same as that of clinical strain CDC-4, and the MIC of RIF for H37Rv:pMN437-RpoB Q432insQ was two-fold lower than that for H37Rv:pMN437-CDC-4 and clinical strain CDC-4. This suggests that RpoB G158R, RpoB V168A, and RpoB S188P likely do not contribute to RIF resistance, whereas RpoB Q432insQ contributes to high-level RIF resistance in M. tuberculosis.
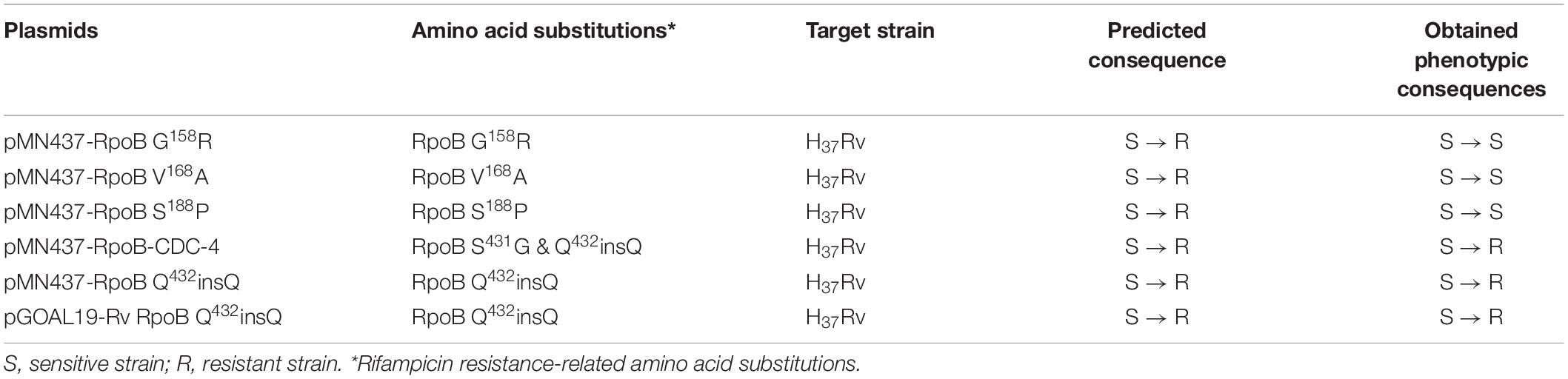
Table 2. Plasmids used in this study.
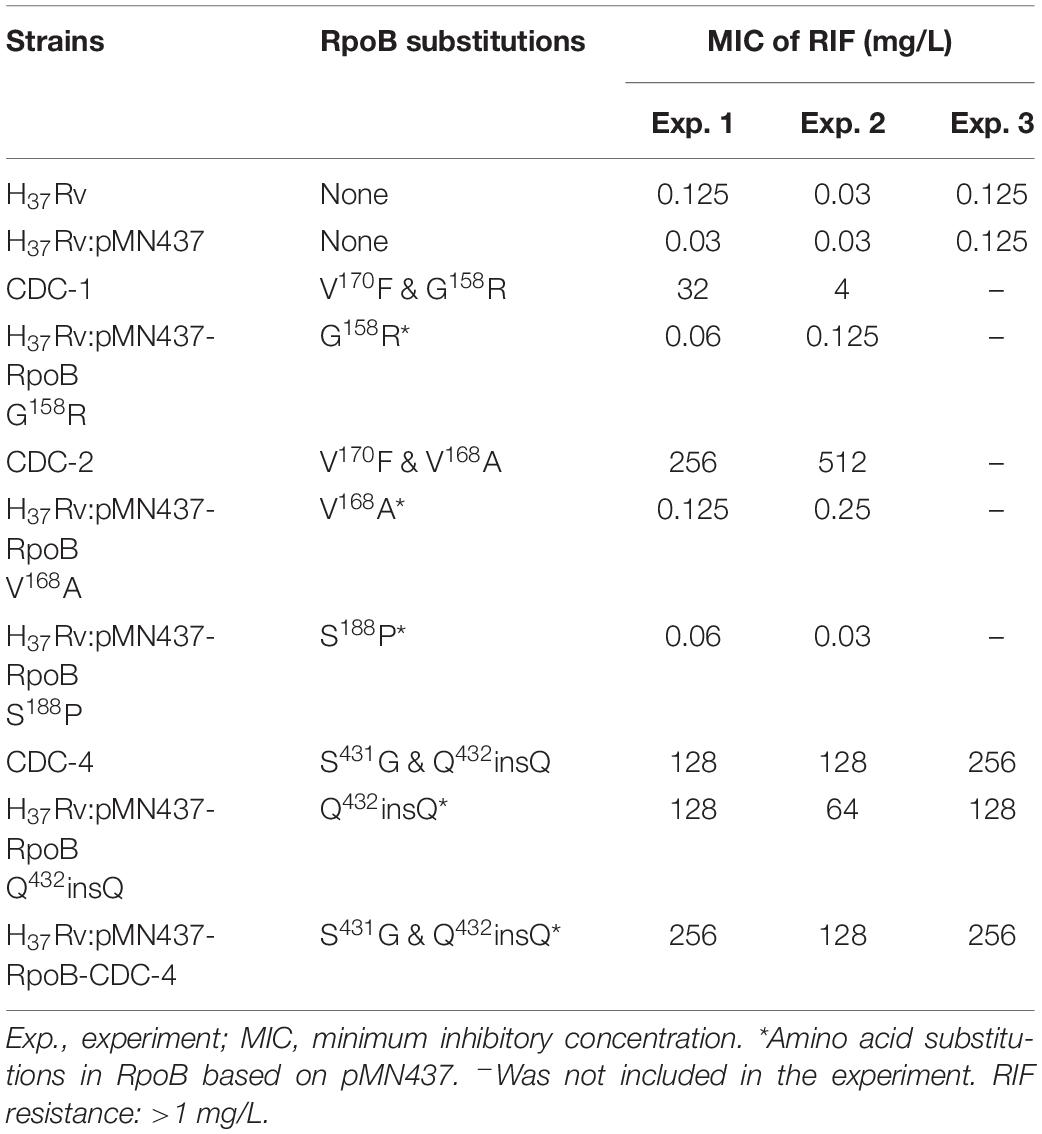
Table 3. Susceptibility test results of clinical isolates and H37Rv strains carrying RpoB expression plasmids as measured by microplate Alamar blue assays.

Figure 1. Rifampicin (RIF) susceptibility test for Mycobacterium tuberculosis strains expressing wild-type and mutant RpoB as measured by microplate Alamar blue assays (MABAs). The MIC of RIF for M. tuberculosis strains carrying RpoB expression plasmids was measured by MABA. M. tuberculosis was cultured in 7H9 broth containing different concentrations of RIF for 2 weeks and then stained with Alamar blue for 2 days. CDC-1, CDC-2, and CDC-4 are RIF-resistant clinical strains that harbored RpoB amino acid changes (G158R & V170F, V168A & V170F, and S431G & Q432insQ, respectively). Red indicates bacterial growth, and blue indicates no growth. The critical concentration for RIF resistance was1.0 mg/L. All experiments were performed in duplicate to confirm reproducibility and repeated at least twice with similar results (Table 3).
RpoB Q432insQ Confers RIF Resistance to M. tuberculosis H37Rv
Among the tested RpoB expression plasmids, only RpoB Q432insQ increased the MIC of RIF in H37Rv. To further confirm that the RpoB Q432insQ was correlated with RIF resistance, unmarked mutants of H37Rv expressing RpoB Q432insQ were generated. The rpoB fragment with the CAA insertion was subcloned from pMN437-RpoB Q432insQ into the ScaI site of the pGOAL19 plasmid, and the resulting construct, pGOAL19-Rv RpoB Q432insQ (Table 2) was checked by sequencing. Then, the plasmid was transformed into M. tuberculosis H37Rv, and the unmarked replacement mutant (RpoB Q432insQ) was selected after two rounds of homologous recombination. DNA sequencing confirmed that seven strains (#19, #112, #132, #133, #135, #136, and #137) had the CAA insertion in codon 432 of rpoB, which leads to a Q insertion (Figure 2). All tested strains were resistant to RIF, with an MIC >1 mg/L.
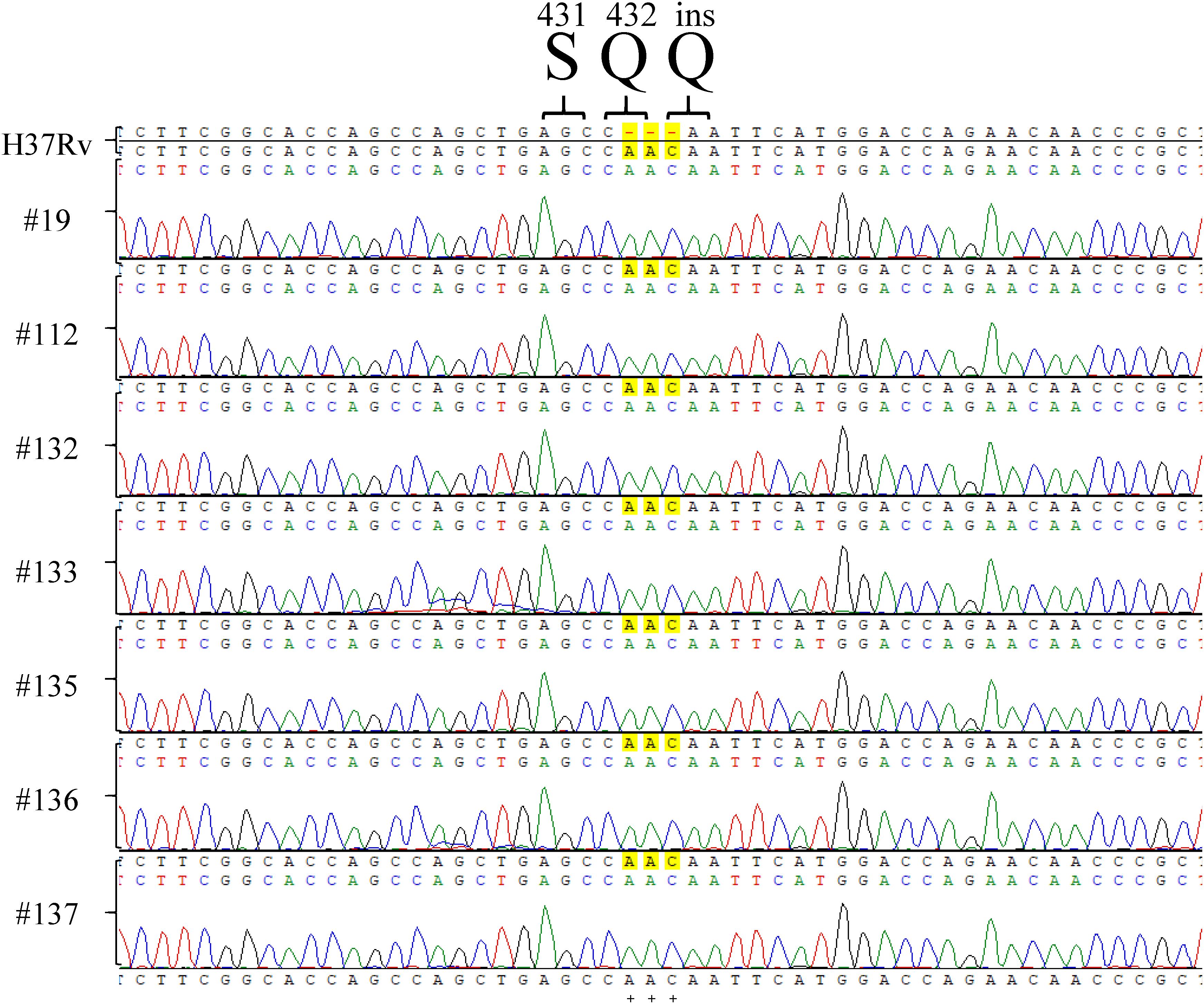
Figure 2. rpoB sequencing results of Mycobacterium tuberculosis H37Rv RpoB Q432insQ strains generated by site-directed mutagenesis. Sequencing of rpoB gene fragments of the strains generated by site-directed mutagenesis, specifically #9, #112, #132, #133, #135, #136, and #137, showed that RpoB was successfully substituted with RpoB Q432insQ in M. tuberculosis H37Rv-derived strains. H37Rv: M. tuberculosis H37Rv. Green pick: adenine, A. Red pick: thymine, T. Blue pick: cytosine, C. Black pick: guanine, G.
Discussion
Clinical strains CDC-1, CDC-2, and CDC-3 have high-level RIF resistance and unique rpoB gene mutations. All of them also encoded V170F, which is a well-known RIF-resistance associated mutation, as it has been reported that M. tuberculosis isolates encoding the V170F mutation have high-level resistance to RIF (8–32 mg/L) (Heep et al., 2000). According to our data, G158R, V168A, and S188P mutations likely did not contribute to RIF resistance in these strains. However, strain CDC-2 had a RIF MIC of 256–512 mg/L, which was 32–64 fold higher than the MIC previously reported for a strain encoding RpoB V170F (Heep et al., 2000), suggesting that strain CDC-2 might have another drug-resistance mechanism not involving RpoB. Because we only sequenced the rpoB genes of RIF-resistant strains, resistance mechanisms outside rpoB could not be detected. Mutations causing other antibiotic resistance could be missed too. Chromosome mutations comprise the major mechanism causing drug resistance in M. tuberculosis (Miotto et al., 2018). Drug resistance in strains could be caused by other resistance mechanisms such as antibiotic modifications or neutralization, augmented efflux pumps, porin alterations, and the downregulation of cell-wall permeability (Mnyambwa et al., 2017; Sharma et al., 2018).
Codons 431 and 432 of rpoB are located in the RRDR (Friehs, 2004), and mutations in this region are related to RIF resistance (Sandgren et al., 2009). Mutations at codon 431 (S431T, S431I, S431R, and S431G) were previously reported to be associated with RIF resistance (Kapur et al., 1994; Sajduda et al., 2004; Chan et al., 2007). Multiple mutations including codon 431 or 432 of RpoB resulted in higher-level RIF resistance (MIC > 100 mg/L) (Bahrmand et al., 2009). We also demonstrated that plasmid complementation or chromosomal mutagenesis of Q432insQ alone could cause RIF resistance, whereas S431G plus Q432insQ complementation resulted in a two-fold higher MIC of RIF (Tables 2, 3, Figure 1).
RIF interacts with the β-subunit of RpoB, and most RIF resistance in M. tuberculosis is caused by mutations in the RRDR of RpoB (Tupin et al., 2010). The domain structure of RpoB was previously deduced by analyzing the crystal structure, which showed that RpoB directly interacts with RIF via 12 hydrogen bonds (Nusrath Unissa et al., 2016). Molecular docking experiments showed stronger RIF binding by wild-type RpoB than by mutant RpoB proteins (Nusrath Unissa et al., 2016). Q432 is an energetically favorable binding site and is considered part of the active site that is involved in ligand binding (Campbell et al., 2001; Nusrath Unissa et al., 2016). RpoB Q432insQ might influence the binding enthalpy to weaken the molecular interaction between RpoB and RIF, which could result in RIF resistance.
We observed that CDC-4 grew slower compared to the H37Rv strain. Therefore, mutants survived better only in the presence of RIF. Multiple mutations in rpoB have been reported, shown to result in increased drug resistance (Bahrmand et al., 2009). The occurrence of multiple mutations could be accumulated due to continuous RIF usage after a first mutation or they could occur simultaneously in high RIF environments.
Rifampicin is an effective drug used to treat most cases of drug-susceptible TB (Long et al., 2019). However, cases of RIF resistance have been reported since the early 1990s, leading to problems in TB control (Zaw et al., 2018). In M. tuberculosis, drug resistance is mostly caused by genetic changes rather than gene transfer from other bacteria (Tupin et al., 2010; Zaw et al., 2018). Thus, sequencing well-known mutation sites is important to detect drug-resistant M. tuberculosis. Several molecular diagnostic methods have been developed for the rapid detection of drug resistance in M. tuberculosis (Yong et al., 2019). Nevertheless, more comprehensive information on drug resistance-associated mutations must be established to improve the diagnosis and treatment of TB.
Conclusion
In summary, we studied four isolates with high-level RIF resistance and unique mutations encoding RpoB G158R, RpoB V168A, RpoB S188P, and RpoB Q432insQ. Results of plasmid complementation of RpoB indicated that G158R, V168A, and S188P of RpoB do not affect the MIC of RIF. However, the transfer of pMN437-RpoB Q432insQ plasmids to M. tuberculosis H37Rv or chromosomal mutagenesis generating RpoB Q432insQ turned sensitive strains into RIF-resistant strains. Therefore, RpoB Q432insQ confers RIF resistance in M. tuberculosis.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
J-TW designed the study. L-YL, P-FH, T-LL, and J-TW discussed the results and revised the manuscript. W-TL, H-YT, W-HL, and RJ provided and analyzed the clinical strains. L-YL, S-HW, S-EC, L-YH, and H-EK prepared materials and performed experiments. L-YL and P-FH analyzed the data. L-YL wrote the manuscript. All authors reviewed and approved the final version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by the Ministry of Science and Technology, the Excellent Translational Medicine Research Projects of National Taiwan University College of Medicine and National Taiwan University Hospital, the Liver Disease Prevention and Treatment Research Foundation of Taiwan, and “Center of Precision Medicine” from the Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.583194/full#supplementary-material
References
Bahrmand, A. R., Titov, L. P., Tasbiti, A. H., Yari, S., and Graviss, E. A. (2009). High-level rifampin resistance correlates with multiple mutations in the rpoB gene of pulmonary tuberculosis isolates from the Afghanistan border of Iran. J. Clin. Microbiol. 47, 2744–2750. doi: 10.1128/jcm.r00548-09
Boyd, R., Ford, N., Padgen, P., and Cox, H. (2017). Time to treatment for rifampicin-resistant tuberculosis: systematic review and meta-analysis. Int. J. Tuberc. Lung Dis. 21, 1173–1180. doi: 10.5588/ijtld.17.0230
Burke, R. M., Coronel, J., and Moore, D. (2017). Minimum inhibitory concentration distributions for first- and second-line antimicrobials against Mycobacterium tuberculosis. J. Med. Microbiol. 66, 1023–1026. doi: 10.1099/jmm.0.000534
Campbell, E. A., Korzheva, N., Mustaev, A., Murakami, K., Nair, S., Goldfarb, A., et al. (2001). Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell 104, 901–912. doi: 10.1016/s0092-8674(01)00286-0
Chan, R. C., Hui, M., Chan, E. W., Au, T. K., Chin, M. L., Yip, C. K., et al. (2007). Genetic and phenotypic characterization of drug-resistant Mycobacterium tuberculosis isolates in Hong Kong. J. Antimicrob. Chemother. 59, 866–873. doi: 10.1093/jac/dkm054
Chikaonda, T., Ketseoglou, I., Nguluwe, N., Krysiak, R., Thengolose, I., Nyakwawa, F., et al. (2017). Molecular characterisation of rifampicin-resistant Mycobacterium tuberculosis strains from Malawi. Afr. J. Lab. Med. 6:463.
Friehs, K. (2004). Plasmid copy number and plasmid stability. Adv. Biochem. Eng. Biotechnol. 86, 47–82.
Heep, M., Rieger, U., Beck, D., and Lehn, N. (2000). Mutations in the beginning of the rpoB gene can induce resistance to rifamycins in both Helicobacter pylori and Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 44, 1075–1077. doi: 10.1128/aac.44.4.1075-1077.2000
Islam, M. M., Hameed, H. M. A., Mugweru, J., Chhotaray, C., Wang, C., Tan, Y., et al. (2017). Drug resistance mechanisms and novel drug targets for tuberculosis therapy. J. Genet. Genom. 44, 21–37. doi: 10.1016/j.jgg.2016.10.002
Kapur, V., Li, L. L., Iordanescu, S., Hamrick, M. R., Wanger, A., Kreiswirth, B. N., et al. (1994). Characterization by automated DNA sequencing of mutations in the gene (rpoB) encoding the RNA polymerase beta subunit in rifampin-resistant Mycobacterium tuberculosis strains from New York City and Texas. J. Clin. Microbiol. 32, 1095–1098. doi: 10.1128/jcm.32.4.1095-1098.1994
Lai, L. Y., Lin, T. L., Chen, Y. Y., Hsieh, P. F., and Wang, J. T. (2018). Role of the Mycobacterium marinum ESX-1 secretion system in sliding motility and biofilm formation. Front. Microbiol. 9:1160. doi: 10.3389/fmicb.2018.01160
Larsen, M. H., Biermann, K., Tandberg, S., Hsu, T., and Jacobs, W. R. Jr. (2007). Genetic manipulation of Mycobacterium tuberculosis. Curr. Protoc. Microbiol. Chap. 10:Unit 10A 12.
Lohrasbi, V., Talebi, M., Bialvaei, A. Z., Fattorini, L., Drancourt, M., Heidary, M., et al. (2018). Trends in the discovery of new drugs for Mycobacterium tuberculosis therapy with a glance at resistance. Tuberculosis 109, 17–27. doi: 10.1016/j.tube.2017.12.002
Long, B., Liang, S. Y., Koyfman, A., and Gottlieb, M. (2019). Tuberculosis: a focused review for the emergency medicine clinician. Am. J. Emerg. Med. 38, 1014–1022. doi: 10.1016/j.ajem.2019.12.040
Mabhula, A., and Singh, V. (2019). Drug-resistance in Mycobacterium tuberculosis: where we stand. Med. Chem. Commun. 10, 1342–1360. doi: 10.1039/c9md00057g
Miotto, P., Zhang, Y., Cirillo, D. M., and Yam, W. C. (2018). Drug resistance mechanisms and drug susceptibility testing for tuberculosis. Respirology 23, 1098–1113. doi: 10.1111/resp.13393
Mnyambwa, N. P., Kim, D. J., Ngadaya, E. S., Kazwala, R., Petrucka, P., and Mfinanga, S. G. (2017). Clinical implication of novel drug resistance-conferring mutations in resistant tuberculosis. Eur. J. Clin. Microbiol. Infect. Dis. 36, 2021–2028. doi: 10.1007/s10096-017-3027-3
Nusrath Unissa, A., Hassan, S., Indira Kumari, V., Revathy, R., and Hanna, L. E. (2016). Insights into RpoB clinical mutants in mediating rifampicin resistance in Mycobacterium tuberculosis. J. Mol. Graph. Model. 67, 20–32. doi: 10.1016/j.jmgm.2016.04.005
Pang, Y., Lu, J., Wang, Y., Song, Y., Wang, S., and Zhao, Y. (2013). Study of the rifampin monoresistance mechanism in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 57, 893–900. doi: 10.1128/aac.01024-12
Parish, T., and Stoker, N. G. (2000). Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology 146(Pt 8), 1969–1975. doi: 10.1099/00221287-146-8-1969
Piccaro, G., Pietraforte, D., Giannoni, F., Mustazzolu, A., and Fattorini, L. (2014). Rifampin induces hydroxyl radical formation in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58, 7527–7533. doi: 10.1128/aac.03169-14
Sajduda, A., Brzostek, A., Poplawska, M., Augustynowicz-Kopec, E., Zwolska, Z., Niemann, S., et al. (2004). Molecular characterization of rifampin- and isoniazid-resistant Mycobacterium tuberculosis strains isolated in Poland. J. Clin. Microbiol. 42, 2425–2431. doi: 10.1128/jcm.42.6.2425-2431.2004
Sandgren, A., Strong, M., Muthukrishnan, P., Weiner, B. K., Church, G. M., and Murray, M. B. (2009). Tuberculosis drug resistance mutation database. PLoS Med. 6:e1000002. doi: 10.1371/journal.pmed.1000002
Sharma, D., Bisht, D., and Khan, A. U. (2018). Potential alternative strategy against drug resistant tuberculosis: a proteomics prospect. Proteomes 6:26. doi: 10.3390/proteomes6020026
Steinhauer, K., Eschenbacher, I., Radischat, N., Detsch, C., Niederweis, M., and Goroncy-Bermes, P. (2010). Rapid evaluation of the mycobactericidal efficacy of disinfectants in the quantitative carrier test EN 14563 by using fluorescent Mycobacterium terrae. Appl. Environ. Microbiol. 76, 546–554. doi: 10.1128/aem.01660-09
Swain, S. S., Sharma, D., Hussain, T., and Pati, S. (2020). Molecular mechanisms of underlying genetic factors and associated mutations for drug resistance in Mycobacterium tuberculosis. Emerg. Microb. Infect. 9, 1651–1663. doi: 10.1080/22221751.2020.1785334
Tan, T., Lee, W. L., Alexander, D. C., Grinstein, S., and Liu, J. (2006). The ESAT-6/CFP-10 secretion system of Mycobacterium marinum modulates phagosome maturation. Cell Microbiol. 8, 1417–1429. doi: 10.1111/j.1462-5822.2006.00721.x
Tupin, A., Gualtieri, M., Roquet-Baneres, F., Morichaud, Z., Brodolin, K., and Leonetti, J. P. (2010). Resistance to rifampicin: at the crossroads between ecological, genomic and medical concerns. Int. J. Antimicrob. Agents 35, 519–523. doi: 10.1016/j.ijantimicag.2009.12.017
Vekemans, J., Brennan, M. J., Hatherill, M., Schrager, L., Fritzell, B., Rutkowski, K., et al. (2020). Preferred product characteristics for therapeutic vaccines to improve tuberculosis treatment outcomes: key considerations from World Health Organization consultations. Vaccine 38, 135–142. doi: 10.1016/j.vaccine.2019.10.072
Vilcheze, C., and Jacobs, W. R. Jr. (2014). Resistance to isoniazid and ethionamide in Mycobacterium tuberculosis: genes, mutations, and causalities. Microbiol. Spectr. 2:MGM2-0014-2013. doi: 10.1128/microbiolspec.MGM2-0014-2013
World Health Organization [WHO] (2019). Global Tuberculosis Report 2019. Geneva: World Health Organization.
Yong, Y. K., Tan, H. Y., Saeidi, A., Wong, W. F., Vignesh, R., Velu, V., et al. (2019). Immune biomarkers for diagnosis and treatment monitoring of tuberculosis: current developments and future prospects. Front. Microbiol. 10:2789. doi: 10.3389/fmicb.2018.02789
Keywords: Mycobacterium tuberculosis, drug resistance, rifampicin, rpoB, codon 432 of RpoB
Citation: Lai L-Y, Hsu L-Y, Weng S-H, Chung S-E, Ke H-E, Lin T-L, Hsieh P-F, Lee W-T, Tsai H-Y, Lin W-H, Jou R and Wang J-T (2020) A Glutamine Insertion at Codon 432 of RpoB Confers Rifampicin Resistance in Mycobacterium tuberculosis. Front. Microbiol. 11:583194. doi: 10.3389/fmicb.2020.583194
Received: 14 July 2020; Accepted: 23 September 2020;
Published: 19 October 2020.
Edited by:
Maria Rosalia Pasca, University of Pavia, ItalyReviewed by:
Divakar Sharma, Indian Institute of Technology Delhi, IndiaYu Pang, Beijing Chest Hospital, Capital Medical University, China
Copyright © 2020 Lai, Hsu, Weng, Chung, Ke, Lin, Hsieh, Lee, Tsai, Lin, Jou and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin-Town Wang, d2FuZ2p0QG50dS5lZHUudHc=