 Belinda Loh
Belinda Loh Jiayuan Chen
Jiayuan Chen Prasanth Manohar
Prasanth Manohar Yunsong Yu2
Yunsong Yu2 Xiaoting Hua
Xiaoting Hua Sebastian Leptihn
Sebastian Leptihn
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 03 December 2020
Sec. Infectious Agents and Disease
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.579802
This article is part of the Research Topic Bacterial Pathogen Genomics: Recent Achievements, Current Applications and Future Challenges View all 16 articles
Acinetobacter baumannii is of major clinical importance as the bacterial pathogen often causes hospital acquired infections, further complicated by the high prevalence of antibiotic resistant strains. Aside from natural tolerance to certain antibiotic classes, resistance is often acquired by the exchange of genetic information via conjugation but also by the high natural competence exhibited by A. baumannii. In addition, bacteriophages are able to introduce resistance genes but also toxins and virulence factors via phage mediated transduction. In this work, we analyzed the complete genomes of 177 A. baumannii strains for the occurrence of prophages, and analyzed their taxonomy, size and positions of insertion. Among all the prophages that were detected, Siphoviridae and Myoviridae were the two most commonly found families, while the average genome size was determined to be approximately 4 Mbp. Our data shows the wide variation in the number of prophages in A. baumannii genomes and the prevalence of certain prophages within strains that are most “successful” or potentially beneficial to the host. Our study also revealed that only two specific sites of insertion within the genome of the host bacterium are being used, with few exceptions only. Lastly, we analyzed the existence of genes that are encoded in the prophages, which may confer antimicrobial resistance (AMR). Several phages carry AMR genes, including OXA-23 and NDM-1, illustrating the importance of lysogenic phages in the acquisition of resistance genes.
The opportunistic pathogen Acinetobacter baumannii is the causative agent for bloodstream infections, meningitis and urinary tract infections, and is responsible for 2–10% of all Gram-negative hospital-acquired infections (Joly-Guillou, 2005). Such infections include ventilator-associated pneumonia and bacteremia with a mortality rate of 35–52% (Dijkshoorn et al., 2007; Kempf et al., 2013; Antunes et al., 2014). As a multitude of strains cause nosocomial infections, A. baumannii has become an important pathogen in hospital care and is of global concern. Many clinical isolates have acquired genes coding for virulence factors, such as toxins or efflux pumps, through various genetic uptake mechanisms (Morris et al., 2019). While A. baumannii easily acquires genetic material by conjugation, natural transformation is also widespread as many strains are highly naturally competent (Hu et al., 2019). Such mechanisms ultimately give rise to an increasing number of strains that display high levels of antimicrobial resistance (AMR), against which antibiotics show little or no effect. Genetic information for AMR genes is often embedded in genetic elements such as transposons or plasmids (Partridge et al., 2018; Rozwandowicz et al., 2018). In addition, bacteriophages (or phages) are able to transfer non-viral genetic information through a process called transduction, which can include genes coding for toxins or antimicrobial resistance (Wagner and Waldor, 2002; Derbise et al., 2007; Wachino et al., 2019). Therefore, phages play an important role in the development of AMR.
Regardless of their morphology or infection mechanism, phages can be divided into two types based on their life cycle: Lytic phages and lysogenic phages (sometimes also called temperate). Both eventually kill the host cell by lysis, employing various enzymes that create holes in the membrane and disintegrate the bacterial cell envelope, to allow the release of phage progeny. Few exceptions exist, such as the filamentous phages that are assembled in the membrane and secreted from the host while the bacterium continues to grow and divide (Loh et al., 2017, 2019; Kuhn and Leptihn, 2019). Nonetheless, phages that destroy the host by lysis upon completion of their life cycle, either start their viral replication immediately after entry (lytic phages) or integrate their genome into that of the host first (lysogenic). Lysogenic phages can remain “dormant” without replicating their genome or initiating phage coat protein synthesis. This way, lysogenic phages are being inherited by daughter cells, and might only replicate to form phage particles after many generations. The trigger for phage replication and synthesis is usually a stress signal produced by the host, such as a SOS response after DNA damage (Howard-Varona et al., 2017). However, if the host resides under favorable conditions, lysogenic phages continue their passive co-existence as so-called prophages embedded inside the DNA of the host.
Prophages are a major source of new genes for bacteria, occupying up to 20% of bacterial chromosomes and therefore may provide new functions to its host (Brüssow et al., 2004; Brüssow, 2007; Cortez et al., 2009; Fortier and Sekulovic, 2013; Wang and Wood, 2016). These functions include virulence factors and drug resistance mechanisms which include extracellular toxins and effector proteins involved in adhesion factors, enzymes, super antigens and invasion (Tinsley and Khan, 2006; Wang and Wood, 2016; Argov et al., 2017; Fortier, 2017). In some cases, the acquisition of virulence genes allows non-virulent bacteria to become a virulent pathogen. The most prominent example is that of the CTXΦ cholera toxin, encoded by a filamentous phage, making Vibrio cholerae the clinical pathogen that poses a substantial socio-economic burden on developing countries with poor hygiene due to frequent cholera outbreaks (Davis and Waldor, 2003). Another example is the Shiga toxin-encoding prophages found in highly virulent Escherichia coli strains, causing food-borne infections across the world (Gamage et al., 2004; Tozzoli et al., 2014). As part of the bacteriophage life cycle, prophages of lytic phages are a double edged sword; while they provide the advantage of increasing chances of survival in challenging environments, they could also lead to the killing of the host through the release of progeny at the end of the phage life cycle. As our relationship with microorganisms is a complex and vital one, from the important role of gut microbiota to the increase in mortality due to virulent microorganisms, understanding bacterial genomes is crucial. Yet in order to obtain detailed insight, we also need to be able to identify viral genes and to comprehend the impact of these genes on its host. As part of the bacterial genome, prophages are subjected to the general effects of mutation, recombination and deletion events. For some phages it has been clearly established that prophage genes have an influence on the host, such as motility or biofilm formation, both important aspects with regards to virulence. However for many other prophages, it is less well understood. Previous work on A. baumannii prophages have identified putative virulence factors and antibiotic resistance genes in host genomes deposited on GenBank (Costa et al., 2018; López-Leal et al., 2020). The work presented here analyses clinical A. baumannii strain genomes in search of possible prophages. We describe the identification of active prophages, analyzed their taxonomy, size and detail the regions in the bacterial genomes where these prophages have been found. Our data reveal a wide variation in the number of prophages in A. baumannii genomes and the prevalence of certain prophages within strains. From an evolutionary perspective, these might represent the “most successful” phages, or the ones that bring a benefit to the host. Our data analysis also allowed us to identify two major sites of insertion within the genome of the host bacterium, with most phage genomes inserting in these two regions, while only a few exceptions are being observed. In addition, our study indicates two distinct genome size distributions of prophages, as we observe a bimodal distribution when analyzing all prophage genomes. Furthermore, we describe genes coding for virulence factors, in particular for antimicrobial resistance in the prophages.
A. baumannii genomes: Complete genome sequences of A. baumannii only were selected for this study. Detailed information of each A. baumannii strain used in this study is disclosed in supplementary material (Supplementary Table S1). For the characterization of genome lengths of A. baumannii, the following values were obtained: mean, median, mode, the smallest and the largest genome. The distribution of genome lengths was plotted using the geom_density function provided in the ggplot2 package in R (Wickham, 2016).
Alignment of A. baumannii genomes: All strains were aligned so that their starting position is identical, with the gene dnaA defined as the start. BLAST Scoring Parameters provided by NCBI (Gertz et al., 2005; available at https://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to blast the locations of dnaA and the adjustment of genome sequences was achieved by SnapGene software (from Insightful Science; available at snapgene.com).
Identification of Prophage Genes: The tool used to identify prophages in A. baumannii genomes was Prophage Hunter (Song et al., 2019; available at https://pro-hunter.bgi.com/). Here, we obtained data on the start, end, length, score, category, and the name of the closest phage. Phaster (Arndt et al., 2016; available at http://phaster.ca/) was used to further confirm some conflicting results. According to the algorithm created by the authors of Prophage Hunter, an “active” prophage is defined by a score close to 1, while the probability decreases the lower the score gets. This means that an active prophage region received a scoring of higher that 0.8 while 0.5–0.8 is defined as “ambiguous,” and a score lower than 0.5 as “inactive.”
Prophage number analysis: Calculations of the mean, median, and mode on the prophage number (total, only active, and only ambiguous) were performed after removing the overlaps of the same prophage in the same strain. The 10 strains with the fewest and with the largest numbers of total, only active, and only ambiguous prophages were selected to show the 2 extremes, while the density plot achieved through geom_density function provided by ggplot2 package in R were conducted to describe the general distribution. The boxplot produced by R reflected the relationship between total, only active, and only ambiguous prophage number and A. baumannii genome length.
Phylogenetic analysis of host strains: Prokka v1.13 (Seemann, 2014) was used to generate the gff files for the genome sequences of 177 A. baumannii strains. The core genome alignment was constructed with Roary v3.12.0 (Page et al., 2015). A maximum-likelihood phylogenetic tree was created using FastTree v2.1.10 (Price et al., 2010). The tree was annotated and visualized with ggtree.
Prophage classification and phylogeny analysis: Prophage classification presented was provided by the program Prophage Hunter which was based on the NCBI’s database. Different orders and families were taken into consideration. The prophage number in different families and their proportions were revealed by histograms and pie charts generated in Microsoft Excel, respectively. Different bacterial hosts were referred to in the calculation of the number of prophages and the number of phage species they had. Based on the species of prophages, 10 most common ones for total, only active, and only ambiguous were selected, with a heatmap which was completed through geom_tile provided by ggplot2 package in R showing their number with the activity value in different strains. The phylogeny of phages was mapped according to their sequences. The alignment of the phage sequences were performed using Multiple Alignment using Fast Fourier Transform (MAFFT) (Katoh and Standley, 2013) with default options. Maximum-likelihood phylogenetic trees were created using FastTree v2.1.10. The tree was annotated and visualized using ggtree.
Prophage location analysis: The positions of all prophages were first showed in a stacked bar chart in Microsoft Excel. Considering the overlaps of different prophages in the same strain, all the prophage starts and ends were mapped in a density plot created by ggplot2 geom_density and aes functions in R. The stacked bar charts of different prophage species were used to estimate their preference of insertion which were then summarized in tables.
Prophage length analysis: The use of ggplot2 geom_density function in R facilitated the creation of density plots of prophage length. With the help of aes function, the mapping of prophage categories (active and ambiguous) and families were achieved in the density plots.
Identification of virulence factors and antibiotic resistance genes: No program is currently available that allows the search of virulence genes which are embedded in prophage sequences within bacterial genomes. Thus, we identified prophages first followed by manually correlating genomic positions of virulence genes with those that were also identified to belong to prophage genes.
Mapping of prophage-encoded antimicrobial resistance genes (ARG): To search for the specific virulence genes we identified (above), we first downloaded all available 4,128 A. baumannii Illumina sequencing reads from the Sequence Read Archive (SRA) with the cut-off date for deposited sequences on 2019/11/17. The raw Illumina sequencing reads were mapped against the ARG prophage sequences employing BWA-MEM v0.7.17 (80% coverage cutoff) (Li, 2013).
Our first aim was to analyze how frequently prophages occur in the genomes of A. baumannii strains. We randomly chose 177 genome sequences of A. baumannii strains, many of them clinical isolates. For the subsequent analyses, we aligned all sequences so that their starting positions are identical. To this end, we defined the gene dnaA as the start, which codes for a replication initiation factor that facilitates DNA replication in bacteria. From the sequence alignments, we observed a large variation in genome sizes. The average length of the genomes was 3,981,579 bp with a median value of 4,001,318 bp; the smallest genome had a length of 3,072,399 bp (22.9% shorter than average), and the largest genome displayed a size of 4,389,990 bp (10.25% larger than average) (Figure 1A).
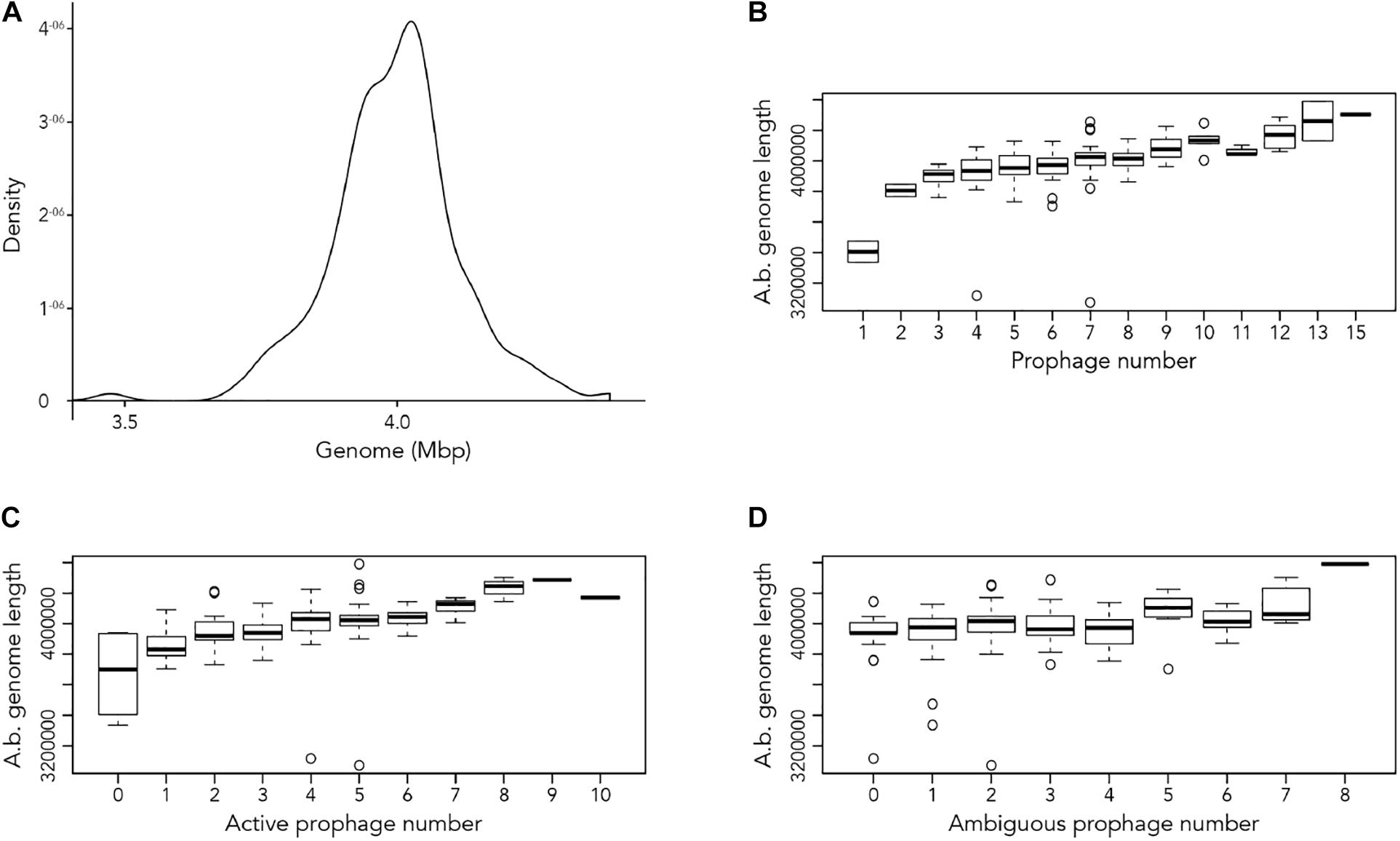
Figure 1. Length of A. baumannii genomes and its correlation with number of prophages present. (A) Density graph of 177 genome sequences of A. baumannii strains, indicating the distribution of the lengths of genome sequences analyzed. (B–D) Correlation between A. baumannii genome length and number of (B) all prophages, (C) active prophages, and (D) ambiguous prophages identified in genomes.
Next, we used the online platform Prophage Hunter (Song et al., 2019) to identify active and ambiguous prophage genomes. The algorithm provides an output value for each prophage identified, which allows the researcher to establish whether a sequence contains an “active” or an ambiguous prophage. While “active” prophages exhibit the complete genomic sequence of a prophage, and are therefore likely to allow the production of phage particles, ambiguous prophage sequences are truncated, mutated or otherwise incomplete, and unlikely to be able to form infectious phages. Among the 177 A. baumannii genomes analyzed, we identified 1,156 prophages, with 459 of them being defined as “ambiguous” while the remaining 697 sequences were labeled as “active” according to the program (Supplementary Table S2). To determine the prevalence of prophages in the A. baumannii genomes, we analyzed the number of prophages per genome. Using a heatmap to illustrate our results, we found that while some prophages are rarely found, others are quite common in the genomes of A. baumannii isolates (Figure 2). One phage that was only found once, for example, is a prophage with high sequence similarity to the Yersinia Podovirus fHe-Yen3-01, while the Acinetobacter phage Bphi-B1251, a Siphovirus, has been found in 79.1% (140 in 177) of all analyzed A. baumannii genomes. Such high prevalence observed by prophages such as Bphi-B1251 could indicate high infectivity and wide host range of the active phage particle. Table 1 shows 10 strains with the fewest and with the largest numbers of prophages identified (Table 1). We found that the average prophage number in an A. baumannii genome is 6.53, with some bacterial genomes containing only one (n = 2) prophage sequence, such as in case of the A. baumannii strains DS002 and VB1190. In contrast to these, other strains have been found to contain as many as 10 prophage sequences, such as in strain 9201 (n = 1), that were labeled “active” by Prophage Hunter; additionally this strain contains two prophage sequences that were defined as ambiguous. The highest prophage number was found in the strain AF-401 which contains 15 prophages, however, only 8 were defined as active. Our results show that prophage sequences are relatively common and that most A. baumannii strains show a median of seven and a mode of eight prophages per genome. Additional genome analysis of the clinical isolates illustrates a possible relationship between prophages and host strains (Supplementary Figure S1). Prophages, such as Bacillus phage PfEFR 4 and Enterobacteria phage CUS 3 were observed more frequently in strains whose genomes are in the same clade or are closely related, indicating a narrow host range.
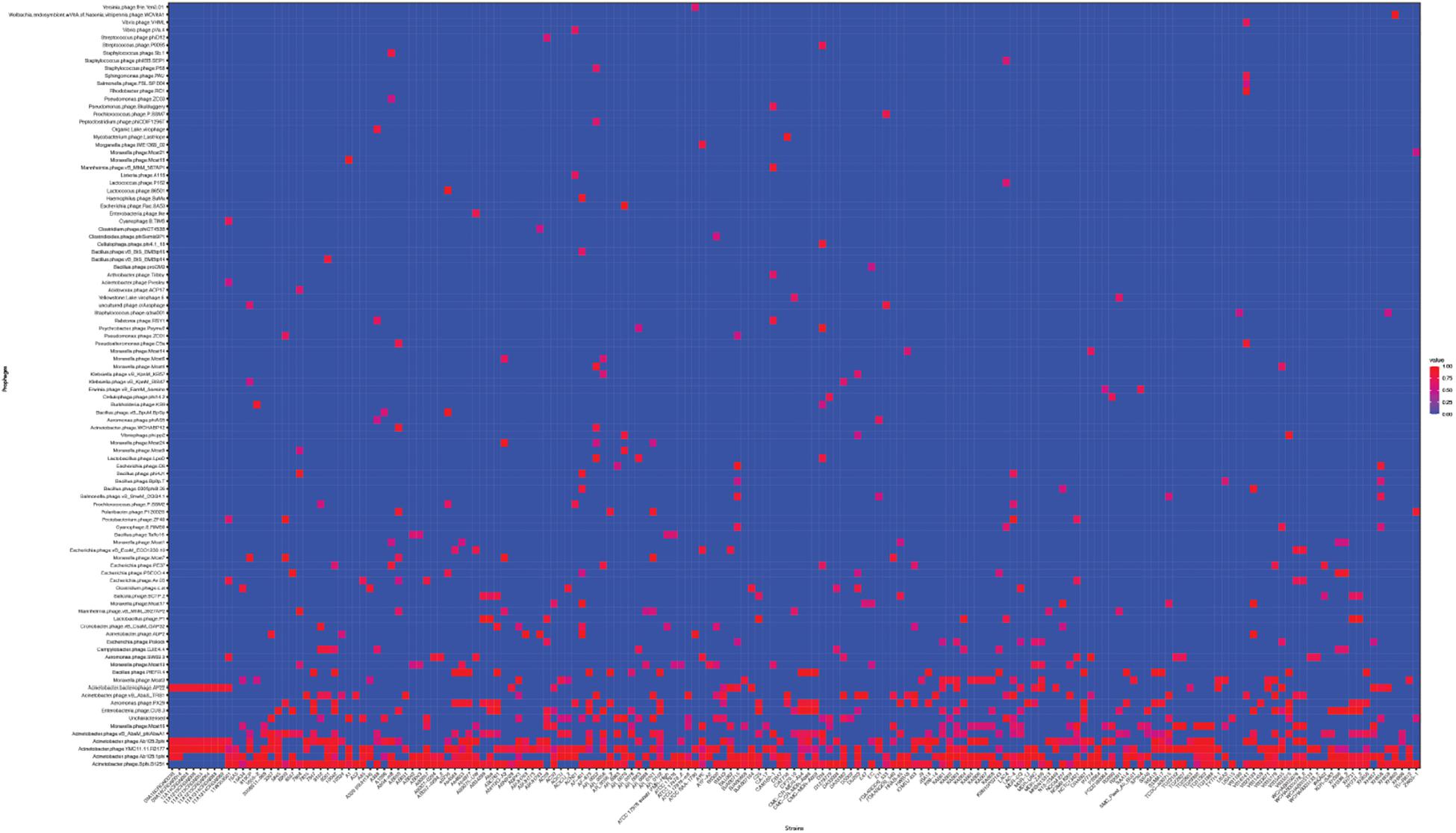
Figure 2. The prevalence of prophages analyzed. Heat map of prophages found in all A. baumannii strains analyzed. Prophages (y-axis) are plotted against each A. baumannii strain (x-axis). Red squares indicate the presence of the indicated prophage. Blue squares indicate the lack thereof. Please refer to the PDF of the figure and use the zoom function to identify names of strains and phages.
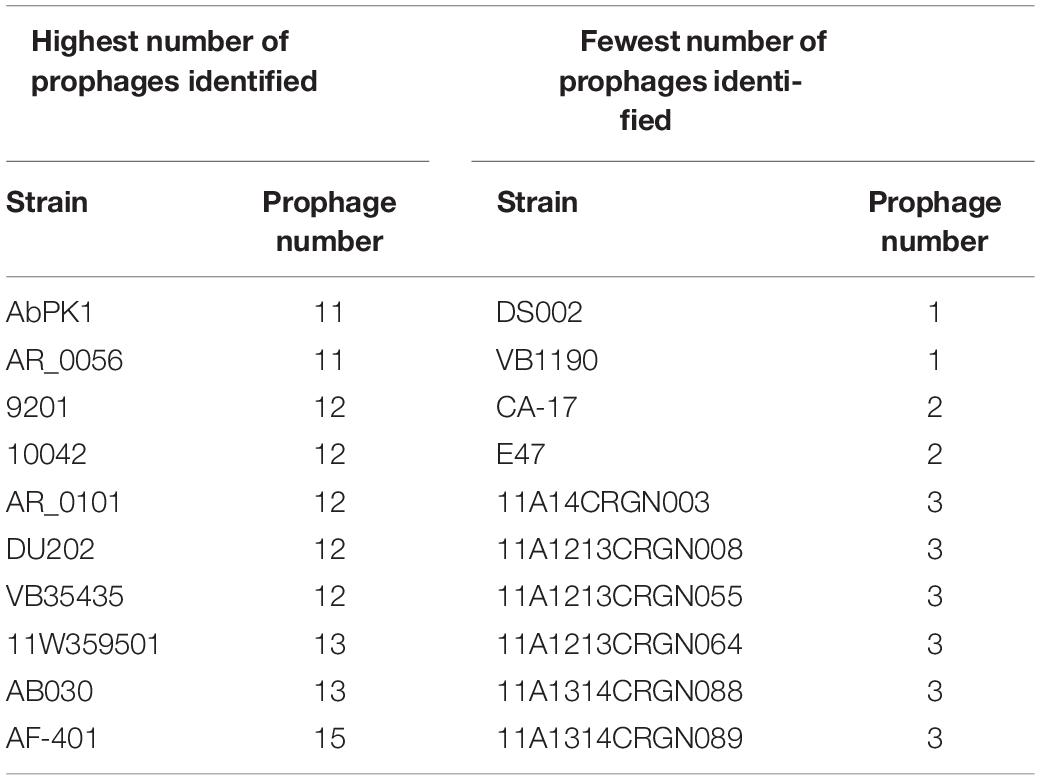
Table 1. A. baumannii stains with the highest and the fewest number of prophages identified.
We next correlated host genome length with number of prophages identified to determine if there is a relationship between the two variables. Perhaps unsurprisingly, the length of A. baumannii genomes increased as more prophages are identified, disregarding whether the prophage genomes are “active” or “ambiguous” (Figure 1B). However, when prophages are classified, the correlation between host genome length and number of prophage genomes identified were less distinct (Figures 1C,D).
We analyzed the relationship of all prophages we identified and created a phylogenetic tree (Supplementary Figure S2). Phage phylogeny is very complex. While bacteria share many common genes, microbial viruses are less related to each other creating large phylogenetic distances. The phylogenetic analysis shows that in some instances the phylogenetic clustering does not necessarily result in grouping of the phages according to their classes. This is, however, not surprising as phages often display diversity by “mosaicity of their genomes” (Dion et al., 2020). After identifying all prophage sequences in the genomes of the 177 A. baumannii isolates, we set out to analyze the most prevalent classes of phages present. Our analysis of all prophage sequences (n = 1,156) revealed that the majority of them, ∼57% (n = 660) of the prophages, belong to the Siphoviridae group (Figure 3A). The Siphoviridae is a class of head-and-tail phages, with the best known representative being phage lambda, that exhibit long, non-contractile but comparably flexible tail structures (Nobrega et al., 2018). The second most commonly found prophages are Myoviridae, with a percentage of ∼33% (n = 385) (Figure 3A). With the best-known Myovirus, the E. coli phage T4, these phages have a stiff, contractible tail that allows the active penetration of the bacterial host envelope (Hu et al., 2015). Together, these two phage classes make up 90% of all prophage genomes. The third most common class, albeit only 4.7% (n = 55) of all prophage genomes, belongs to Podoviridae. The best known Podovirus is probably T7, which has a short, stubby tail and internal core proteins that get ejected for the formation of a DNA-translocating channel across the bacterial cell envelope (Guo et al., 2014; Lupo et al., 2015; Leptihn et al., 2016). Prophages that could not be conclusively classified to a viral group accounted for 3.3% (n = 38). Siphoviridae, Myoviridae and Podoviridae all belong to the order Caudovirales, phages that exhibit a head-and-tail structure. Within the two phage classes we found several phages that were most successful, i.e., most common. Examples are the A. baumannii phages Bphi-B1251 and YMC11/11/R3177, which both belong to the Siphoviridae (Table 2A). The most common Myovirus was Ab105-1phi. Not only are these the most common prophages found, they are also the most common active prophages identified (Table 2B). In addition, the distribution of the classes was similar if only active prophages were analyzed. Here, 62% (432/697) belonged to the Siphoviridae and 32% (223/697) to the Myoviridae (Figure 3B).
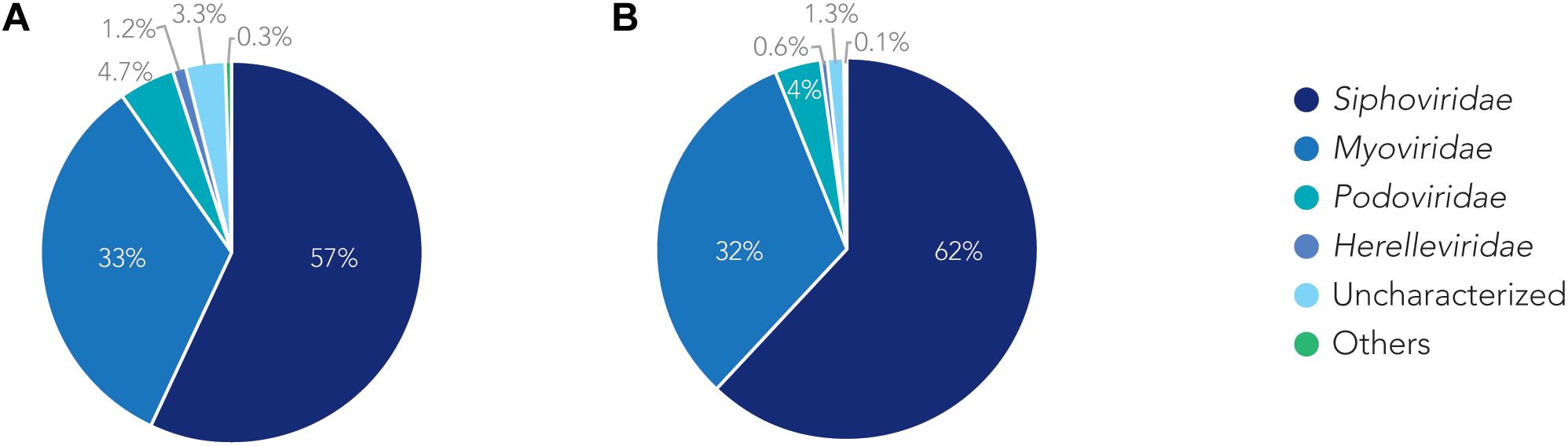
Figure 3. The families of prophages found. Pie charts of prophages identified showing the percentage make up of each family. (A) Classification for all prophages. (B) Classification of active prophages only.
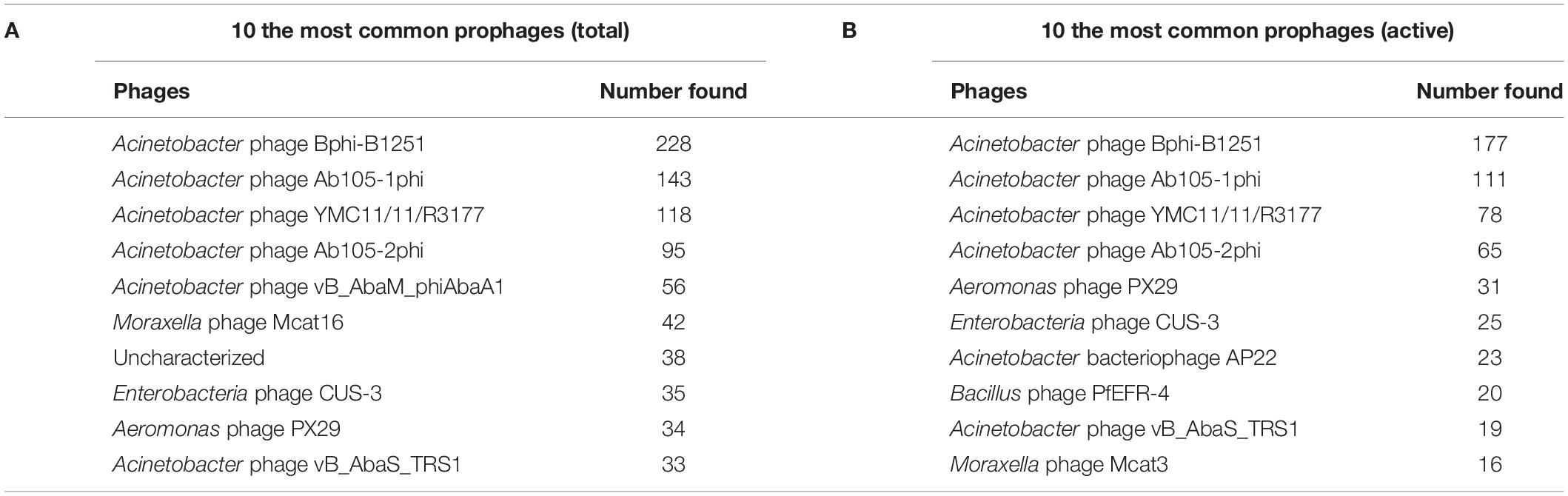
Table 2. The most common prophages identified in A. baumannii stains.
To determine where all prophages -regardless of their class- are found in the bacterial genome, or if they are possibly distributed at random within the host DNA, we visualized the position of the prophages in all 177 genomes (Figure 4A). We then plotted all prophage positions found in all genomes against the position in the bacterial host genome sequence. Surprisingly, we observed a bimodal distribution, with two clear peaks in the position of prophages (Figure 4B), indicating that there are two main sites of attachment for prophages and their genomic insertion. While this reflects the situation for all prophages, we then analyzed the position of several individual prophages within the bacterial genome. First, we assessed one of the most commonly found phages YMC11/11/R3177. The position of this phage reflects the overall distribution of all phages in the analyzed genomes, with two main areas of insertion. However, in some cases the position is outside the main area of insertion, possibly due to recombination of the bacterial genome (Figure 5A). The second phage we analyzed was phage vB_AbauS_TRS1. Here, the distribution of the phage within the bacterial genome seems to be more random as compared to the overall distribution (Figure 5B). In case of the Aeromonas phage PX29, insertion seems to be very “strict,” i.e., only observed in one location within the genome (Figure 5C). The observations made when analyzing the prophage positions show that the insertion of phages could be described as “directed,” and less random, indicating that attachment sites, if they are required, are found more commonly in certain positions of the bacterial genome.
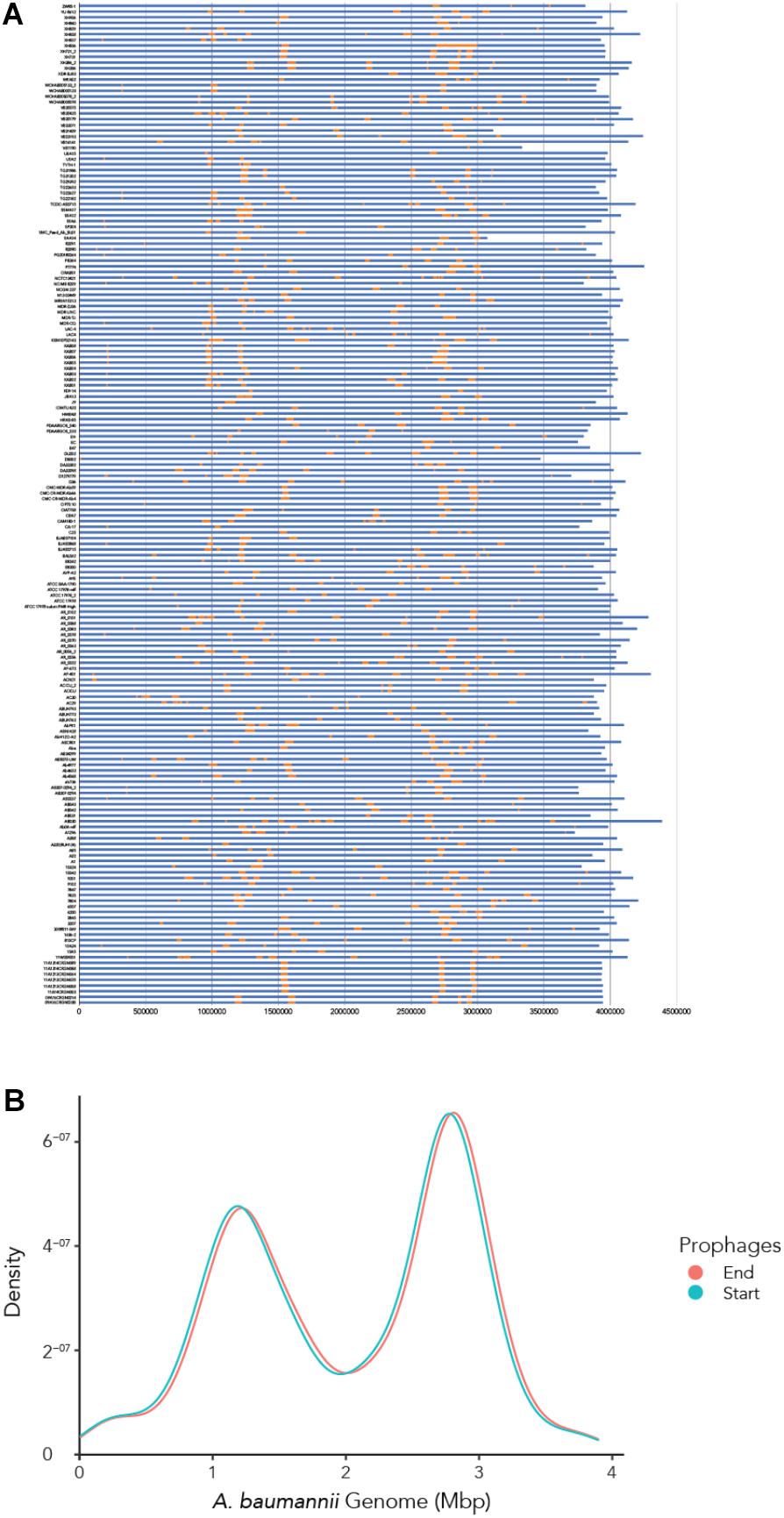
Figure 4. The location of prophages found in each A. baumannii genome. (A) Stacked bar chart of each bacterial strain (y-axis). Yellow segments indicate prophage sequences identified on the genome (in blue). (B) Density graph compiled from the stacked bar chart. Please refer to the PDF of the figure and use the zoom function to identify names of strains and phages.
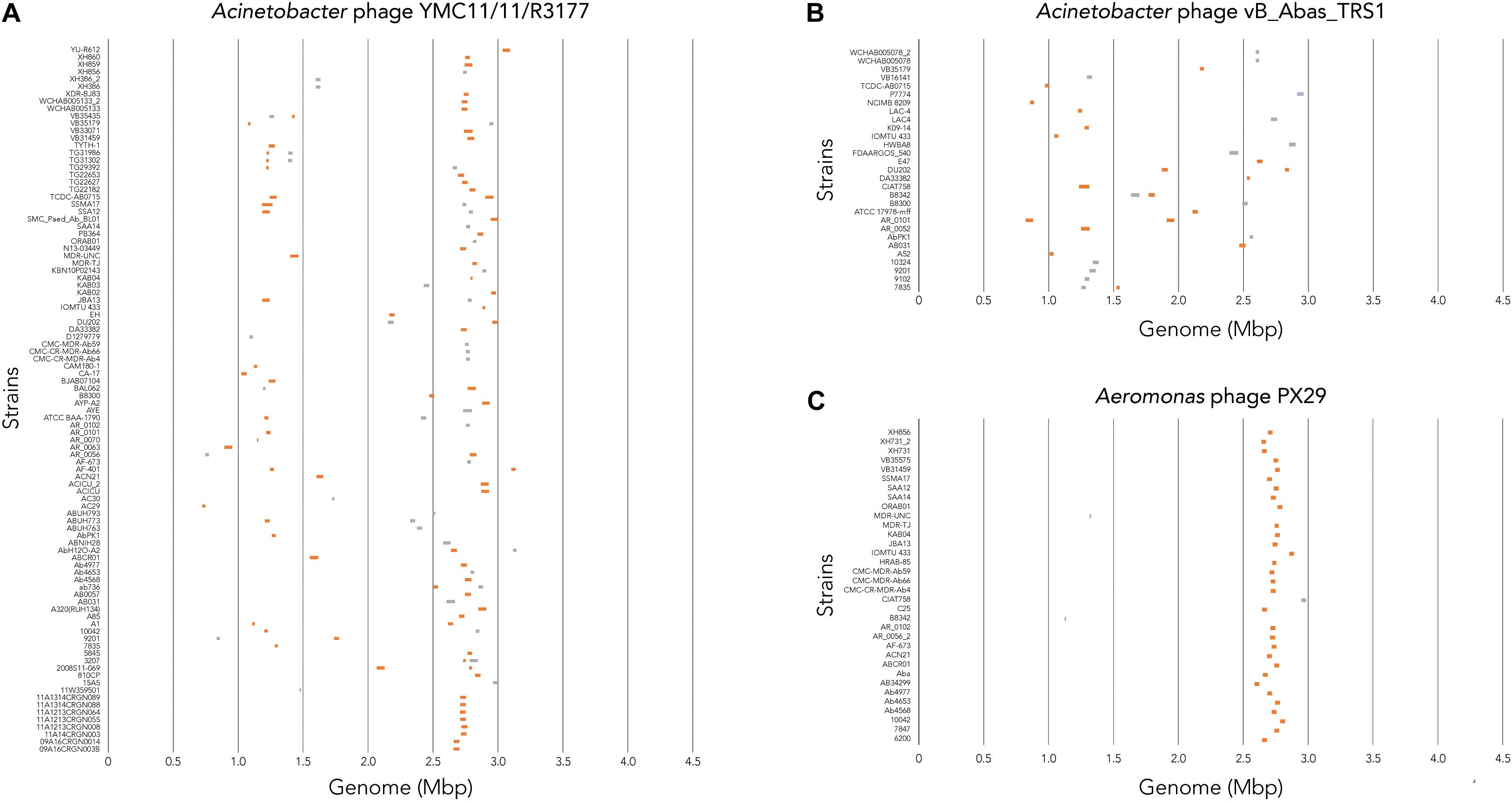
Figure 5. Location of prophage genome insertion differs between phages. (A) Comparison of phage location for Acinetobacter phage YMC11/11/R3177. (B) Comparison of insertion locations for Acinetobacter phage vB_Abas_TRS1. (C) Comparison of prophage insertion sites for Aeromonas phage PX29. Boxes in orange indicate active prophages identified. Gray boxes indicate ambiguous prophage sequences. Please refer to the PDF of the figure and use the zoom function to identify names of strains and phages.
Using the data provided by the program Prophage Hunter, we were interested in evaluating the size distribution of prophage genomes. Therefore, we plotted sizes against the frequency of prophages present in the bacterial genomes and calculated average prophage genome sizes. In the case of ambiguous prophages, a main population at 15 kb became visible followed by a minor peak of substantial size at approximately 60 kb, leaving the average and median length of ambiguous prophage genomes at 29.2 and 25.8 kb, respectively (Figure 6A and Table 3). In contrast to this, two main peaks were observed when analyzing only active prophages. Here, one peak is observed at around 17 kb while the other at around 36 kb was observed (Figure 6A). The average genome length of active prophages is 34 kb (Table 3). As these peaks include all phage categories, we re- analyzed the genomic length of the active prophages according to their classes: Siphoviridae, Myoviridae and Podoviridae, which together constitutes almost 95% of all prophages (see Figure 3A). When analyzing the length of all Siphoviridae sequences, we observed two main populations, one sharp peak at around 20 kb and one broad peak with a shoulder containing larger sequences from 18 to 56 kb (Figure 6B). The average prophage length of Siphoviridae is 36.7 kb (Table 3). Myoviridae sequences similarly exhibited two sharp peaks (17 and 36 kb), with a third minor one of around 60 kb (Figure 6C). The average prophage genome size of active Myoviridae is 32.4 kb (Table 3). The Podoviridae showed several minor peaks with a large sharp peak at about 12 kb and the average prophage genome length is calculated to be 17.4 kb (Figure 6D and Table 3). The results of these analyses show that there are distinct distributions of bacteriophage genome sizes. Two clearly separated groups of prophages can be observed just based on size, in the case of Myoviridae. In the case of Siphoviridae, we saw a less defined area with possibly multiple species within the broad distribution.
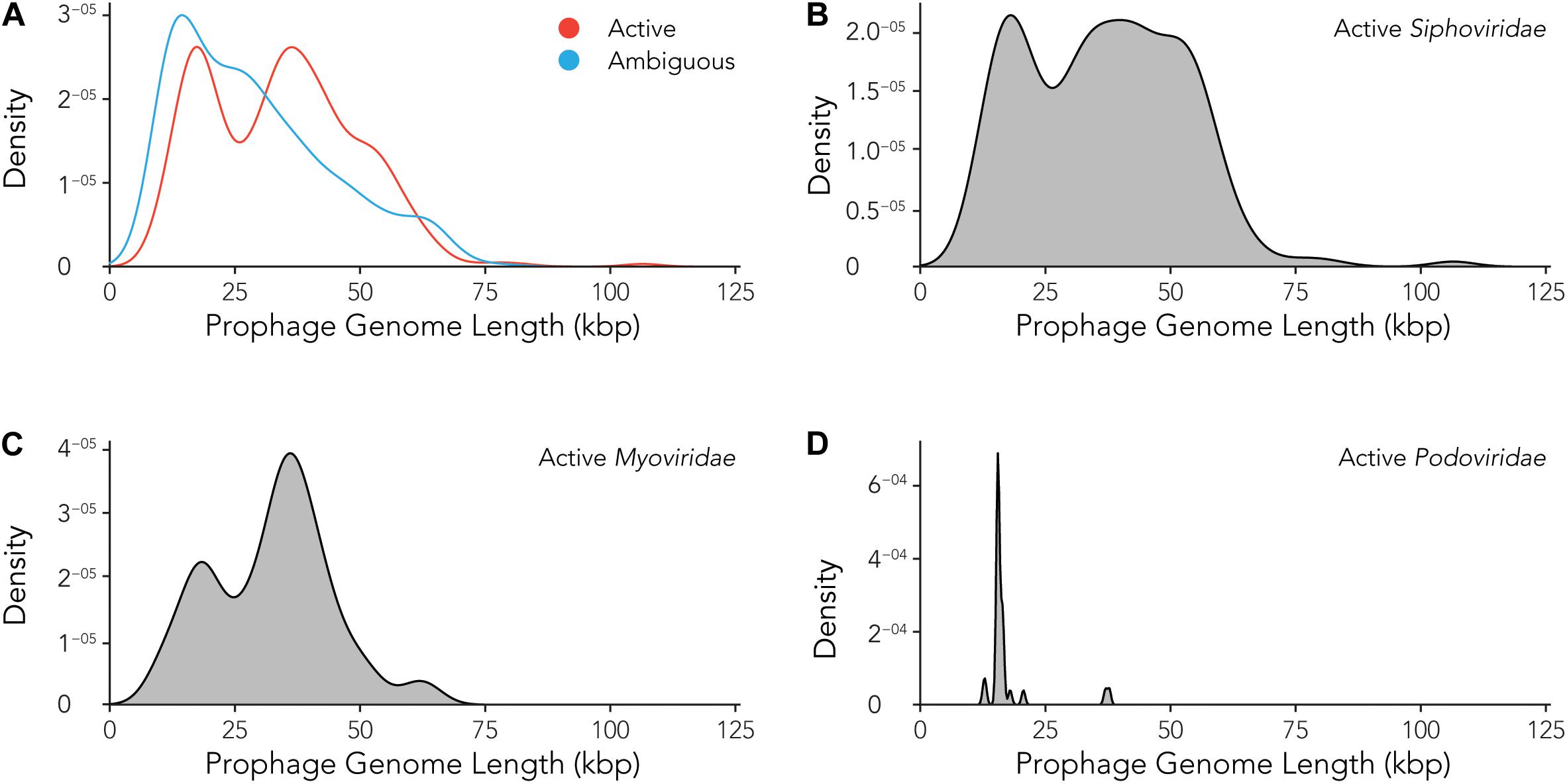
Figure 6. Active prophages categorized by prophage lengths. (A) Comparison of prophage lengths between active and ambiguous prophages. (B–D) Distribution of prophage length for active Siphoviridae (B), Myoviridae (C), and Podoviridae (D).
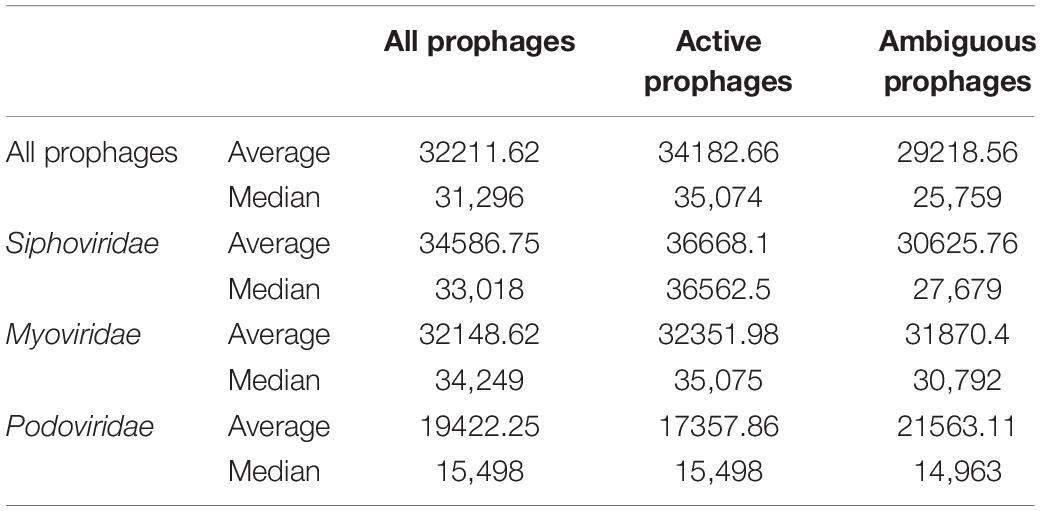
Table 3. Prophage genome lengths for active and ambiguous prophages, among the three major families in the order of Caudovirales.
As prophages are able to encode genes that might allow its host to become more virulent and therefore more evolutionary successful, we aimed to analyze prophage-encoded virulence factors. However, in contrast to e.g., E. coli, a databank for A. baumannii virulence factors currently does not exist. We therefore searched for prophage sequences that contain genes that contribute to antibiotic resistance. Table 4 lists the start and end of the genes that are encoded within a respective prophage. Among others, we found AMR genes for OXA-23 and NDM-1. OXA-23 is the most widespread carbapenem resistance gene globally (Hamidian and Nigro, 2019). NDM-1 encodes a carbapenemase, a beta-lactamase enzyme with a broad substrate specificity capable of hydrolyzing penicillins, carbapenems, cephalosporins, and monobactams. Other beta-lactamase genes were blaADC–5, blaOXA–67, blaOXA–115, and blaTEM–12. In addition, we were able to identify genes coding for N-Acetyltransferases(aac(3)-I, aac(3)-Id, aacA16), Aminoglycoside phosphotransferases (aph(3′)-Ia, aph(3′)-VI, aph(6)-Id, aph(3″)-Ib), both groups mediating aminoglycoside resistance. Other genes that contribute to antibiotic resistance were sulfonamide resistance gene (sul2), and the macrolide-resistance conferring genes msr(E), encoding an efflux pump, and mph(E), coding for a macrolide-inactivating phosphotransferase.
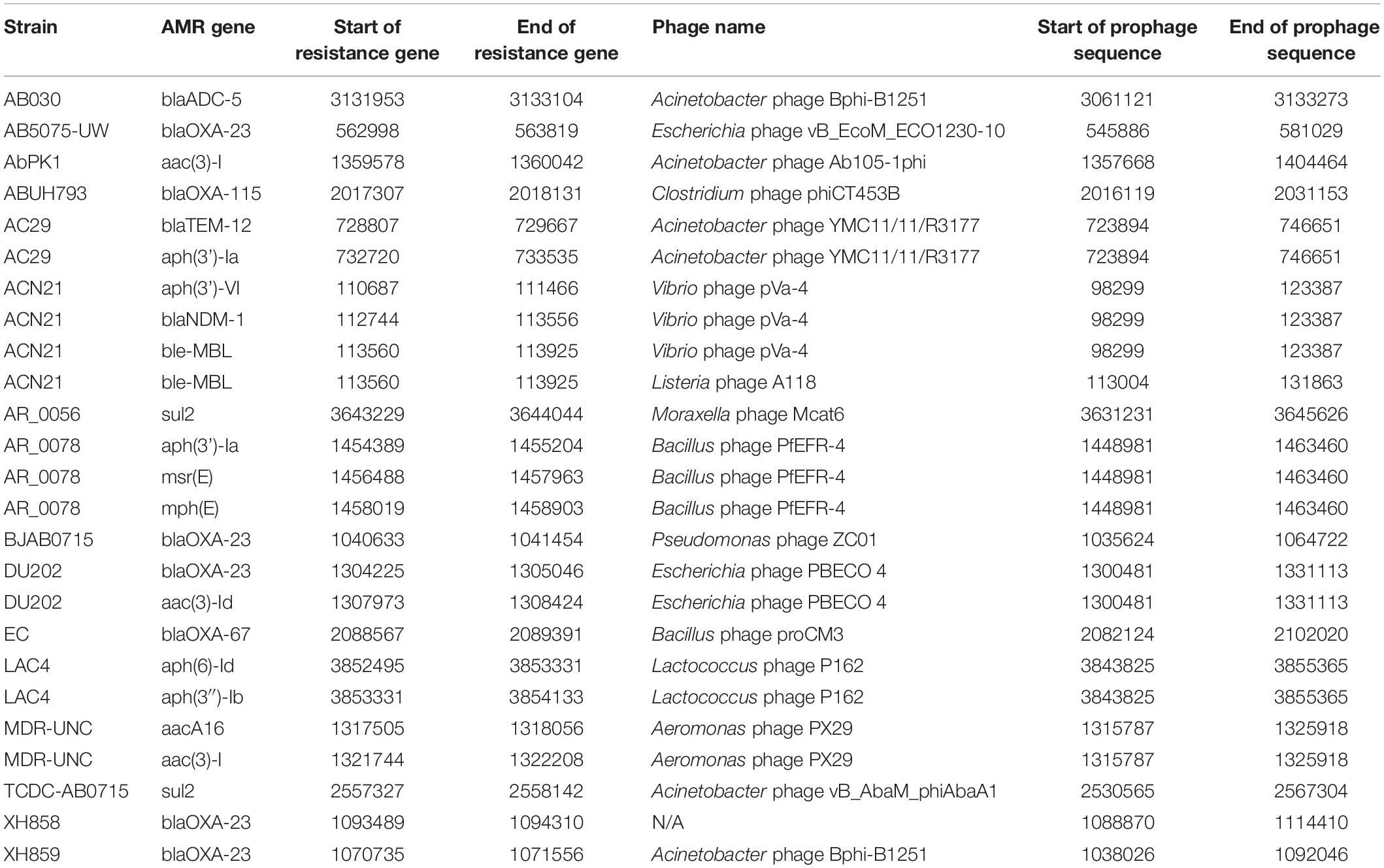
Table 4. Antimicrobial resistance genes found in prophages embedded in A. baumannii stains.
Interestingly, one A. baumannii strain, ACN21, contains a prophage which encodes three antibiotic resistance genes [aph(3’)-VI, blaNDM–1, bleMBL]. The phage is most closely related to Vibrio phage pVa-4, a Myovirus that infects V. alginolyticus (Kim et al., 2019). In contrast to its relative, phage pVa-4 is a lytic phage was grouped to be part of the phiKZ-like phages (Phikzviruses), which are considered as “jumbo” phages. A second A. baumannii strain, AR_0078, contains a prophage sequence that shares a high degree of similarity to the Bacillus phage PfEFR-4, a Siphovirus with a prolate head, in contrast to its E. coli relative lambda (Geng et al., 2017). The prophage encodes three antimicrobial resistance genes that confer macrolide-resistance, msr(E), encoding an efflux pump, and mph(E), coding for a macrolide-inactivating phosphotransferase. The third gene encodes the Aminoglycoside phosphotransferases [aph(3’)-Ia], inactivating aminoglycoside antibiotics.
A. baumannii strain DU202 contains a prophage sequence that is related to the lytic E. coli myovirus PBCO 4 (Kim et al., 2013). Within the genome sequence, two antimicrobial resistance genes are encoded: aac(3)-Id codes for an N-Acetyltransferase mediating aminoglycoside resistance and OXA-23, which is encodes the most widespread resistance mechanism toward the β-lactamase inhibitor sulbactam. The gene was also embedded in the prophage sequences of two phages in the A. baumannii strain XH859; here, the A. baumannii phage Bphi-B1251 was found to be the most closely related phage, a lytic Podovirus, that was previously shown to be able to infect and lyse an OXA-23- harboring A. baumannii isolate from a septic patient (Jeon et al., 2012).
To determine how prevalent the prophage encoded antimicrobial resistance genes are in other genomes, we mapped all available 4,128 A. baumannii Illumina sequence reads that were accessible by 2019/11/17 on the Sequence Read Archive (SRA) to the AMR prophage sequences using an 80% cutoff for the coverage. We identified 174 A. baumannii genomes that contain prophage sequences, or about 4.2% of all available reads, not including the genomes we used for the initial analysis. Supplementary Figure S3 illustrates the prevalence of the prophage-encoded AMR genes (ARGs) and their respective prophages. Fairly “successful,” i.e., widely distributed, were two prophages: ABUH793 and AR_0056. ABUH793 is a close relative of the Clostridium phage phiCT453B, containing the resistance gene blaOXA–115. The second prophage sequence that was found often in A. baumannii genomes in comparison to other prophages encoding ARGs, was AR_0056, a relative of the Moraxella phage Mcat6, encoding the ARG sul2. While the prophage does not necessarily render the host antibiotic resistant as genetic regulators might be missing, prophages containing ARGs can present an evolutionary advantage for the host (Wendling et al., 2020).
Our finding demonstrates the importance of phages in the acquisition of antimicrobial resistance; the above described genes may confer the ability to grow in the presence of antibiotics when the bacterial host is infected by a phage that encodes not only the information for its own replication but also genes that inactivate or remove antibiotic compounds.
Our search for prophages in the genomes of A. baumannii strains revealed several interesting findings. One surprising observation was the positions of the prophages within the genome of the bacterial host. When analyzing the prophage positions one might expect that the insertion of phages would be less directed, and more random. However, we found that the majority of phages inserted into two locations as seen by a bimodal density plot with a sharp separation between the two peaks. Prophage genome integration can either be a site-specific recombination event at so-called att sites or occurs in a non-directed manner by transposition into random sites (Ramisetty and Sudhakari, 2019). Our data could indicate that the two areas in the genome contain most of the attachment sites for the majority of phages. A previously published analysis of Salmonella and E. coli genomes found a large number of distinctive phage integration loci; in the case of Salmonella, 24 loci were shared among 102 Salmonella phages, amounting to four phages statistically sharing one integration site. In case of E. coli, 58 distinctive integration loci were identified for 369 phages, with statistically 6.6 phages per site (Bobay et al., 2013). It might be reasonable to assume that A. baumannii contains similar numbers of attachment sites, although we have not analyzed potential sites in the genomes we investigated. However, regardless of whether a phage inserts via one of the various attachment sites or randomly via transposition, only two “hot spots” were observed in our study. In addition to the explanation that attachment sites might be more frequent in these two sections of the bacterial genome, prophage insertion into segments crucial for e.g., over-all gene regulation, or into household genes, would be an evolutionary disadvantage and might therefore be less commonly found.
Bacteriophages are classified into 12 families (ICTV, 2019). Pioneering work in taxonomy divided tailed phages into three classes based on the morphology of the phages; Myoviridae have long contractile tails, Siphoviridae have long non contractile tails, and Podoviridae have short tails. Recently, the International Committee on Taxonomy of Viruses (ICTV) expanded the order Caudovirales, describing tailed bacteriophages to include six additional families, i.e.,: Ackermannviridae, Autographiviridae, Chaseviridae, Demerecviridae, Drexlerviridae, and Herelleviridae, taking additional characteristics into consideration such as genome sequence, gene content, protein homology and the host (Adriaenssens et al., 2020). When analyzing the families of prophages in this population of A. baumannii strains, we observed a prevalence of Siphoviridae which constituted 57% of all identified prophages. Together with the next family of phages, Myoviridae, which consists 1/3 of all prophages, the two groups make up 90% of all prophages identified. Among the remaining 10%, the largest group belongs to Podoviridae. These ratios are very similar to the ones that have been reported in other studies, and also the ratio of the most commonly found phages in nature (Costa et al., 2018).
Interestingly, the ratio between ambiguous to active prophages in case of the ones that have been identified as Siphoviridae, 0.654, markedly differs from the ratio calculated for the prophages that belong to Myoviridae, 0.579. It is unlikely that Siphoviridae prophage sequences are less prone to mutations, as they should be occurring at random. However, a mechanism that would specifically “protect” Siphoviridae prophages might be the case if daughter cells, where mutations in the prophages occur, would have an evolutionary disadvantage. This would imply that prophages influence the host behavior positively, which has previously been shown in some cases (Bondy-Denomy and Davidson, 2014; Nanda et al., 2015; Loh et al., 2019). Could the most likely scenario be that the genomes of Myoviridae are possibly larger than those of the Siphoviridae, making them more prone to random mutations and deletions? However, the size comparisons of the genomic sequences of the prophages that we identified, does not support this possible explanation: The Siphoviridae sequences display distribution with one clear peak at around 20 kb followed by a fairly broad peak with a plateau and a shoulder toward larger genome sizes, ranging from 28 to 65 kb (Figure 6B). In contrast to this, the genomic size distributions of Myoviridae showed two peaks, one around 20 kb, the second around 42 kb (Figure 6C). The size estimations are corroborated by the findings of a previous study which estimated the genome sizes of Siphoviridae to be approximately 50 kb in average, with a broad distribution between 24 and 101 kb, while Myoviridae display smaller genomes or around 34 kb (Costa et al., 2018).
One question we could pose is why there is no broad distribution of prophage sizes, and why do we observe “peaks”? Can we conclude from this data that certain genome sizes are advantageous from an evolutionary standpoint? The arch-Myovirus might have had a certain size that proved to be sufficient for the successful persistence during the course of evolution. Only smaller increases or decreases of the genome allowed evolutionary success, and no gradual increase or decrease in genome size occurred. However, an evolutionary leap or jump might have happened at some point, which might have led to a major increase of genomic size, creating a new, second type of a Myovirus class which is represented in the second, larger peak. Starting from this size, again only smaller changes, decreases or increases with regards to the genomic size, may have occurred, preserving the sharp separation of each peak. It would be interesting to investigate if the smaller Myovirus display a prolate head as does T4. The increase volume of this geometry allows the packing of a larger genome, which might explain the possible separation in two sizes. To test this hypothesis, smaller Myoviruses should have non-prolate heads. Viral classification is a complex topic. Possibly the genome sizes might help to contribute to classifying of microbial viruses in the future.
While Prophage Hunter extracts prophage genomes from bacterial genomes, the platform is a web-based tool that also distinguishes between “active” and “ambiguous” prophage genomes (Song et al., 2019). The developers of Prophage Hunter have used experimental data and conducted induction experiments with mitomycin C, to validate the program’s output, showing its ability to hunt for “active,” inducible prophages. Yet, conclusions should not be hastily drawn to assume that all “active” prophages can definitively excise from the host genome to commence the bacteriophage lytic life cycle; false positives may still exist. In this regard, induction experiments should be conducted to confirm that “active” prophages can indeed produce active particles.
Prophages are an important source for acquiring new genetic information, including antibiotic resistance genes, for their bacterial host. Phage-mediated transfer of genes from donor to recipient cells, also called transduction, has been shown to be instrumental in the spread of AMR genes both in vitro and in vivo (Haaber et al., 2016). In our study, we also investigated AMR genes that are embedded in prophage sequences. Previous studies on prophage diversity in A. baumannii had found AMR genes (also called: ARGs) in many prophages that were analyzed (Costa et al., 2018; López-Leal et al., 2020). Yet despite this, it remains to be shown whether prophages confer antimicrobial resistance to its host A. baumannii. Our observation illustrates that phages might represent important contributors in the process of AMR acquisition. However, it remains to be said that we found less than 5% of a publicly available, deposited sequence reads to contain the prophage-encoded ARGs we initially identified, arguing that phage transduction is possibly not the prevalent mode of AMR acquisition but is second to other mechanisms such as plasmid uptake via conjugation. Interestingly, despite viruses in general showing highly condensed genomes trying to pack essential information in small volumes, bacterial viruses seem to have co-evolved with their hosts and carry genes that are not directly required for the virus but are beneficial to the host and thus also to the prophage.
Our study attempts to take an inventory of prophages in the important nosocomial pathogen A. baumannii. We have analyzed the phylogeny of the prophages, their position in the host genome and characterized their lengths, identifying “successful,” i.e., widely distributed phages, and the dominant families, Myoviridae and Siphoviridae.
Several prophage sequences contained genes coding for antimicrobial resistance genes. By mapping these genes in all deposited illumina A. baummannii sequence reads, we found that less than 5% of all available host sequences contain such prophage-embedded genes, indicating that transduction may not be the major contributor to the emergence of antimicrobial resistance.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
SL and XH devised this study. JC, BL, XH, and SL performed the work. BL created the figures. BL, PM, XH, YY, and SL wrote the manuscript. All authors contributed to the article and approved the submitted version.
We thank the National Science Foundation of China for supporting this work (NSFC 32011530116). BL was supported by the China Postdoctoral Science Foundation (530000-X91902).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank the reviewers who made very valuable suggestions that helped to improve the work. A preprint of this article has been published on BioRxiv (Loh et al., 2020).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.579802/full#supplementary-material
Supplementary Figure 1 | In silico detection and distribution of prophage sequences in A. baumannii strains. Unrooted core phylogenetic tree of 177 A. baumannii genomes from clinical isolates. Presence of prophage sequences are indicated by red squares. Green squares indicate the lack thereof. Please refer to the PDF of the figure and use the zoom function to identify names of strains and phages.
Supplementary Figure 2 | Phylogeny of prophages found in A. baumannii genomes.
Supplementary Figure 3 | Prevalence of prophages carrying AMR genes in A. baumannii strains.
Supplementary Table 1 | Metadata of all A. baumannii genomes used in this study.
Supplementary Table 2 | Details of detected active and ambiguous prophage in all 177 A. baumannii strains analyzed in this study.
Adriaenssens, E. M., Sullivan, M. B., Knezevic, P., van Zyl, L. J., Sarkar, B. L., Dutilh, B. E., et al. (2020). Taxonomy of prokaryotic viruses: 2018-2019 update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 165, 1253–1260. doi: 10.1007/s00705-020-04577-8
Antunes, L. C., Visca, P., and Towner, K. J. (2014). Acinetobacter baumannii: evolution of a global pathogen. Pathog. Dis. 71, 292–301. doi: 10.1111/2049-632X.12125
Argov, T., Rabinovich, L., Sigal, N., and Herskovits, A. A. (2017). An effective counterselection system for Listeria monocytogenes and its use to characterize the monocin genomic region of strain 10403S. Appl. Environ. Microbiol. 83:e02927-16. doi: 10.1128/AEM.02927-16
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Bobay, L. M., Rocha, E. P., and Touchon, M. (2013). The adaptation of temperate bacteriophages to their host genomes. Mol. Biol. Evol. 30, 737–751. doi: 10.1093/molbev/mss279
Bondy-Denomy, J., and Davidson, A. R. (2014). When a virus is not a parasite: the beneficial effects of prophages on bacterial fitness. J. Microbiol. 52, 235–242. doi: 10.1007/s12275-014-4083-3
Brüssow, H. (2007). Bacteria between protists and phages: from antipredation strategies to the evolution of pathogenicity. Mol. Microbiol. 65, 583–589. doi: 10.1111/j.1365-2958.2007.05826.x
Brüssow, H., Canchaya, C., and Hardt, W. D. (2004). Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 68, 560–602. doi: 10.1128/MMBR.68.3.560-602.2004
Cortez, D., Forterre, P., and Gribaldo, S. (2009). A hidden reservoir of integrative elements is the major source of recently acquired foreign genes and ORFans in archaeal and bacterial genomes. Genome Biol. 10:R65. doi: 10.1186/gb-2009-10-6-r65
Costa, A. R., Monteiro, R., and Azeredo, J. (2018). Genomic analysis of Acinetobacter baumannii prophages reveals remarkable diversity and suggests profound impact on bacterial virulence and fitness. Sci. Rep. 8:15346. doi: 10.1038/s41598-018-33800-5
Davis, B. M., and Waldor, M. K. (2003). Filamentous phages linked to virulence of Vibrio cholerae. Curr. Opin. Microbiol. 6, 35–42. doi: 10.1016/s1369-5274(02)00005-x
Derbise, A., Chenal-Francisque, V., Pouillot, F., Fayolle, C., Prévost, M. C., Médigue, C., et al. (2007). A horizontally acquired filamentous phage contributes to the pathogenicity of the plague bacillus. Mol. Microbiol. 63, 1145–1157. doi: 10.1111/j.1365-2958.2006.05570.x
Dijkshoorn, L., Nemec, A., and Seifert, H. (2007). An increasing threat in hospitals: multidrug-resistant Acinetobacter baumannii. Nat. Rev. Microbiol. 5, 939–951. doi: 10.1038/nrmicro1789
Dion, M. B., Oechslin, F., and Moineau, S. (2020). Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 18, 125–138. doi: 10.1038/s41579-019-0311-5
Fortier, L. C. (2017). The Contribution of Bacteriophages to the biology and virulence of pathogenic clostridia. Adv. Appl. Microbiol. 101, 169–200. doi: 10.1016/bs.aambs.2017.05.002
Fortier, L. C., and Sekulovic, O. (2013). Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 4, 354–365. doi: 10.4161/viru.24498
Gamage, S. D., Patton, A. K., Hanson, J. F., and Weiss, A. A. (2004). Diversity and host range of Shiga toxin-encoding phage. Infect. Immun. 72, 7131–7139. doi: 10.1128/IAI.72.12.7131-7139.2004
Geng, P., Tian, S., Yuan, Z., and Hu, X. (2017). Identification and genomic comparison of temperate bacteriophages derived from emetic Bacillus cereus. PLoS One 12:e0184572. doi: 10.1371/journal.pone.0184572
Gertz, J., Riles, L., Turnbaugh, P., Ho, S. W., and Cohen, B. A. (2005). Discovery, validation, and genetic dissection of transcription factor binding sites by comparative and functional genomics. Genome Res. 15, 1145–1152. doi: 10.1101/gr.3859605
Guo, F., Liu, Z., Fang, P. A., Zhang, Q., Wright, E. T., Wu, W., et al. (2014). Capsid expansion mechanism of bacteriophage T7 revealed by multistate atomic models derived from cryo-EM reconstructions. Proc. Natl. Acad. Sci. U.S.A. 111, E4606–E4614. doi: 10.1073/pnas.1407020111
Haaber, J., Leisner, J. J., Cohn, M. T., Catalan-Moreno, A., Nielsen, J. B., Westh, H., et al. (2016). Bacterial viruses enable their host to acquire antibiotic resistance genes from neighbouring cells. Nat. Commun. 7:13333. doi: 10.1038/ncomms13333
Hamidian, M., and Nigro, S. J. (2019). Emergence, molecular mechanisms and global spread of carbapenem-resistant Acinetobacter baumannii. Microb. Genom. 5:e000306. doi: 10.1099/mgen.0.000306
Howard-Varona, C., Hargreaves, K. R., Abedon, S. T., and Sullivan, M. B. (2017). Lysogeny in nature: mechanisms, impact and ecology of temperate phages. ISME J. 11, 1511–1520. doi: 10.1038/ismej.2017.16
Hu, B., Margolin, W., Molineux, I. J., and Liu, J. (2015). Structural remodeling of bacteriophage T4 and host membranes during infection initiation. Proc. Natl. Acad. Sci. U.S.A. 112, E4919–E4928. doi: 10.1073/pnas.1501064112
Hu, Y., He, L., Tao, X., Meng, F., and Zhang, J. (2019). High DNA uptake capacity of international clone II Acinetobacter baumannii detected by a novel planktonic natural transformation assay. Front. Microbiol. 10:2165. doi: 10.3389/fmicb.2019.02165
ICTV (2019). International Committee on Taxonomy of Viruses: New Taxonomy Release. Available online at: https://talk.ictvonline.org/taxonomy/ (accessed June 03, 2020).
Jeon, J., Kim, J. W., Yong, D., Lee, K., and Chong, Y. (2012). Complete genome sequence of the podoviral bacteriophage YMC/09/02/B1251 ABA BP, which causes the lysis of an OXA-23-producing carbapenem-resistant Acinetobacter baumannii isolate from a septic patient. J. Virol. 86, 12437–12438. doi: 10.1128/JVI.02132-12
Joly-Guillou, M. L. (2005). Clinical impact and pathogenicity of acinetobacter. Clin. Microbiol. Infect. 11, 868–873. doi: 10.1111/j.1469-0691.2005.01227.x
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kempf, M., Rolain, J. M., Azza, S., Diene, S., Joly-Guillou, M. L., Dubourg, G., et al. (2013). Investigation of Acinetobacter baumannii resistance to carbapenems in Marseille hospitals, south of France: a transition from an epidemic to an endemic situation. APMIS 121, 64–71. doi: 10.1111/j.1600-0463.2012.02935.x
Kim, M. S., Hong, S. S., Park, K., and Myung, H. (2013). Genomic analysis of bacteriophage PBECO4 infecting Escherichia coli O157:H7. Arch. Virol. 158, 2399–2403. doi: 10.1007/s00705-013-1718-3
Kim, S. G., Jun, J. W., Giri, S. S., Yun, S., Kim, H. J., Kim, S. W., et al. (2019). Isolation and characterisation of pVa-21, a giant bacteriophage with anti-biofilm potential against Vibrio alginolyticus. Sci. Rep. 9:6284. doi: 10.1038/s41598-019-42681-1
Kuhn, A., and Leptihn, S. (2019). “Helical and filamentous phages,” in Reference Module in Life Sciences, ed. M. G. Feiss (Amsterdam: Elsevier), doi: 10.1016/B978-0-12-809633-8.20986-2
Leptihn, S., Gottschalk, J., and Kuhn, A. (2016). T7 ejectosome assembly: a story unfolds. Bacteriophage 6:e1128513. doi: 10.1080/21597081.2015.1128513
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [Preprint]. Available online at: https://arxiv.org/abs/1303.3997 (accessed November 20, 2020).
Loh, B., Haase, M., Mueller, L., Kuhn, A., and Leptihn, S. (2017). The transmembrane morphogenesis protein gp1 of filamentous phages contains Walker A and Walker B motifs essential for phage assembly. Viruses 9:73. doi: 10.3390/v9040073
Loh, B., Kuhn, A., and Leptihn, S. (2019). The fascinating biology behind phage display: filamentous phage assembly. Mol. Microbiol. 111, 1132–1138. doi: 10.1111/mmi.14187
Loh, B., Chen, J., Manohar, P., Yu, Y., Hua, X., Leptihn, S. A. et al. (2020). Biological inventory of prophages in A. baumannii genomes reveal distinct distributions in classes, length and genomic positions. bioRxiv. doi: 10.1101/2020.10.26.355222
López-Leal, G., Santamaria, R. I., Cevallos, M. Á, Gonzalez, V., and Castillo-Ramírez, S. (2020). Prophages encode antibiotic resistance genes in Acinetobacter baumannii. Microb. Drug Resist. 26, 1275–1277. doi: 10.1089/mdr.2019.0362
Lupo, D., Leptihn, S., Nagler, G., Haase, M., Molineux, J. I., and Kuhn, A. (2015). The T7 ejection nanomachine components gp15-gp16 form a spiral ring complex that binds DNA and a lipid membrane. Virology 486, 263–271. doi: 10.1016/j.virol.2015.09.022
Morris, F. C., Dexter, C., Kostoulias, X., Uddin, M. I., and Peleg, A. Y. (2019). The mechanisms of disease caused by Acinetobacter baumannii. Front. Microbiol. 10:1601. doi: 10.3389/fmicb.2019.01601
Nanda, A. M., Thormann, K., and Frunzke, J. (2015). Impact of spontaneous prophage induction on the fitness of bacterial populations and host-microbe interactions. J. Bacteriol. 197, 410–419. doi: 10.1128/JB.02230-14
Nobrega, F. L., Vlot, M., de Jonge, P. A., Dreesens, L. L., Beaumont, H. J. E., Lavigne, R., et al. (2018). Targeting mechanisms of tailed bacteriophages. Nat. Rev. Microbiol. 16, 760–773. doi: 10.1038/s41579-018-0070-8
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Partridge, S. R., Kwong, S. M., Firth, N., and Jensen, S. O. (2018). Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 31:e0088-17. doi: 10.1128/CMR.00088-17
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Ramisetty, B. C. M., and Sudhakari, P. A. (2019). Bacterial ‘Grounded’ prophages: hotspots for genetic renovation and innovation. Front. Genet. 10:65. doi: 10.3389/fgene.2019.00065
Rozwandowicz, M., Brouwer, M. S. M., Fischer, J., Wagenaar, J. A., Gonzalez-Zorn, B., Guerra, B., et al. (2018). Plasmids carrying antimicrobial resistance genes in Enterobacteriaceae. J. Antimicrob. Chemother. 73, 1121–1137. doi: 10.1093/jac/dkx488
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Song, W., Sun, H. X., Zhang, C., Cheng, L., Peng, Y., Deng, Z., et al. (2019). Prophage Hunter: an integrative hunting tool for active prophages. Nucleic Acids Res. 47, W74–W80. doi: 10.1093/nar/gkz380
Tinsley, E., and Khan, S. A. (2006). A novel FtsZ-like protein is involved in replication of the anthrax toxin-encoding pXO1 plasmid in Bacillus anthracis. J. Bacteriol. 188, 2829–2835. doi: 10.1128/JB.188.8.2829-2835.2006
Tozzoli, R., Grande, L., Michelacci, V., Ranieri, P., Maugliani, A., Caprioli, A., et al. (2014). Shiga toxin-converting phages and the emergence of new pathogenic Escherichia coli: a world in motion. Front. Cell Infect. Microbiol. 4:80. doi: 10.3389/fcimb.2014.00080
Wachino, J. I., Jin, W., Kimura, K., and Arakawa, Y. (2019). Intercellular transfer of chromosomal antimicrobial resistance genes between Acinetobacter baumannii strains mediated by prophages. Antimicrob. Agents Chemother. 63:e00334-19. doi: 10.1128/AAC.00334-19
Wagner, P. L., and Waldor, M. K. (2002). Bacteriophage control of bacterial virulence. Infect. Immun. 70, 3985–3993. doi: 10.1128/iai.70.8.3985-3993.2002
Wang, X., and Wood, T. K. (2016). Cryptic prophages as targets for drug development. Drug Resist. Updat. 27, 30–38. doi: 10.1016/j.drup.2016.06.001
Wendling, C. C., Refardt, D., and Hall, A. R. (2020). Fitness benefits to bacteria of carrying prophages and prophage-encoded antibiotic-resistance genes peak in different environments. bioRxiv [Preprint]. doi: 10.1101/2020.03.13.990044
Keywords: bacteriophage, prophage, A. baumannii, horizontal gene transfer, evolution, viral classification, antimicrobial resistance genes, phage genomes
Citation: Loh B, Chen J, Manohar P, Yu Y, Hua X and Leptihn S (2020) A Biological Inventory of Prophages in A. baumannii Genomes Reveal Distinct Distributions in Classes, Length, and Genomic Positions. Front. Microbiol. 11:579802. doi: 10.3389/fmicb.2020.579802
Received: 03 July 2020; Accepted: 10 November 2020;
Published: 03 December 2020.
Edited by:
Se-Ran Jun, University of Arkansas for Medical Sciences, United StatesReviewed by:
Li-Kuang Chen, Tzu Chi University, TaiwanCopyright © 2020 Loh, Chen, Manohar, Yu, Hua and Leptihn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoting Hua, eGlhb3RpbmdodWFAemp1LmVkdS5jbg==; Sebastian Leptihn, TGVwdGlobkBpbnRsLnpqdS5lZHUuY24=; c2ViYWxlcEB5YWhvby5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.