 Gajraj Singh Kushwaha
Gajraj Singh Kushwaha Anupam Patra
Anupam Patra Neel Sarovar Bhavesh
Neel Sarovar Bhavesh
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 05 November 2020
Sec. Microbial Physiology and Metabolism
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.575041
This article is part of the Research Topic (p)ppGpp and Its Homologs: Enzymatic and Mechanistic Diversity Among the Microbes View all 19 articles
(p)ppGpp, highly phosphorylated guanosine, are global regulatory nucleotides that modulate several biochemical events in bacterial physiology ranging from core central dogma to various metabolic pathways. Conventionally, (p)ppGpp collectively refers to two nucleotides, ppGpp, and pppGpp in the literature. Initially, (p)ppGpp has been discovered as a transcription regulatory molecule as it binds to RNA polymerase and regulates transcriptional gene regulation. During the past decade, several other target proteins of (p)ppGpp have been discovered and as of now, more than 30 proteins have been reported to be regulated by the binding of these two signaling nucleotides. The regulation of diverse biochemical activities by (p)ppGpp requires fine-tuned molecular interactions with various classes of proteins so that it can moderate varied functions. Here we report a structural dynamics of (p)ppGpp in the unbound state using well-defined computational tools and its interactions with target proteins to understand the differential regulation by (p)ppGpp at the molecular level. We carried out replica exchange molecular dynamics simulation studies to enhance sampling of conformations during (p)ppGpp simulation. The detailed comparative analysis of torsion angle conformation of ribose sugar of unbound (p)ppGpp and bound states of (p)ppGpp was carried out. The structural dynamics shows that two linear phosphate chains provide plasticity to (p)ppGpp nucleotides for the binding to diverse proteins. Moreover, the intermolecular interactions between (p)ppGpp and target proteins were characterized through various physicochemical parameters including, hydrogen bonds, van der Waal’s interactions, aromatic stacking, and side chains of interacting residues of proteins. Surprisingly, we observed that interactions of (p)ppGpp to target protein have a consensus binding pattern for a particular functional class of enzymes. For example, the binding of (p)ppGpp to RNA polymerase is significantly different from the binding of (p)ppGpp to the proteins involved in the ribosome biogenesis pathway. Whereas, (p)ppGpp binding to enzymes involved in nucleotide metabolism facilitates the functional regulation through oligomerization. Analysis of these datasets revealed that guanine base-specific contacts are key determinants to discriminate functional class of protein. Altogether, our studies provide significant information to understand the differential interaction pattern of (p)ppGpp to its target and this information may be useful to design antibacterial compounds based on (p)ppGpp analogs.
Bacterial physiology is regulated by various types of secondary messenger nucleotides and these nucleotides regulate almost all major biochemical events (Hengge et al., 2019). These nucleotides are key players of the signaling network intended to responsive cellular behavior to various environmental conditions. The stringent response is such a pleiotropic and global regulatory process which modulates at least one-third of bacterial physiological processes (Cashel and Gallant, 1969; Cashel et al., 1996; Hauryliuk et al., 2015). It is meticulously regulated by the synthesis of two signaling nucleotides namely, pppGpp and ppGpp (together called (p)ppGpp) (Potrykus and Cashel, 2008; Hauryliuk et al., 2015; Steinchen and Bange, 2016). (p)ppGpp messenger nucleotides are highly phosphorylated and bind to several protein targets to regulate biochemical events (Dalebroux and Swanson, 2012; Kanjee et al., 2012; Zhang et al., 2018). Historically, these nucleotides were discovered by two-dimensional thin-layer chromatography of samples from amino acid starved Escherichia coli culture. The concentration of these nucleotides was enhanced drastically in E coli cells during amino acid starvation and these nucleotides were inhibiting rRNA synthesis (Cashel and Gallant, 1969). Initially, the functional role of (p)ppGpp was discovered as transcriptional regulatory as it binds to bacterial RNA polymerase to down-regulate rRNA gene expression (Cashel and Gallant, 1969; Ross et al., 2013; Zuo et al., 2013). Later on, several enzymes from various pathways were identified which are regulated by binding of (p)ppGpp nucleotides. Most of those proteins are part of the core process of molecular machinery such as replication, transcription, translation, and cellular metabolism (Rojas et al., 1984; Wang et al., 2007, 2019; Srivatsan and Wang, 2008; Kriel et al., 2012; Bærentsen et al., 2019; Kushwaha et al., 2019b). The resultant effects of (p)ppGpp mediated regulation has been shown in virulence, host invasion, biofilm formation, persistence, long term survival, pathogenesis, antibiotic resistance, and antibiotic tolerance (Primm et al., 2000; Abranches et al., 2009; Dalebroux et al., 2010; Kudrin et al., 2017; Prusa et al., 2018). Therefore, being a modulator of several processes, (p)ppGpp has been considered as a master regulator for the survival of bacteria during unfavorable conditions.
Conventionally, term (p)ppGpp is used for two nucleotides, guanosine 5′-diphosphate-3′-diphosphate (ppGpp) and guanosine 5′-triphosphate-3′-diphosphate (pppGpp). These nucleotides are synthesized by (p)ppGpp synthetase by transferring pyrophosphate groups from ATP to GDP/GTP to form ppGpp and pppGpp, respectively (Mechold et al., 2002; Hogg et al., 2004; Tamman et al., 2020). There are primarily two types of (p)ppGpp synthetases that have been identified, multi-domain long RelA type and small alarmone synthetases. The long-form (p)ppGpp synthetases are found in two forms; mono functional comprises active synthetase and inactive hydrolase domain while bifunctional (p)ppGpp synthetases have both synthetase and hydrolase active domain. These are classified and named as RelA/SpoT Homolog (RSH) proteins (Atkinson et al., 2011; Hauryliuk et al., 2015). Several regulatory mechanisms have been proposed to explain the activation of (p)ppGpp synthetase enzymes (Hogg et al., 2004; Shyp et al., 2012; Steinchen et al., 2015; Beljantseva et al., 2017; Hauryliuk and Atkinson, 2017; Winther et al., 2018; Kushwaha et al., 2019a; Ronneau and Hallez, 2019). Subsequently, these nucleotide binds to various proteins to modulate the functional activity of respective biochemical reactions, hence, the structural information at the molecular level is essential to understand the fundamental differences associated with these biomolecular interactions. Although the structural studies on (p)ppGpp have been carried out in the bound form as complexes with its binding protein, the structural conformation and dynamics on (p)ppGpp in unbound states have not been reported so far. The computational methods have been an efficient choice to understand the structural dynamics of secondary messenger nucleotides (Stern et al., 2010; Wang et al., 2017). Here, we report a detailed structural analysis on (p)ppGpp in the unbound state using extensive conformation sampling molecular dynamics simulation along with their comparison with structural conformation in bound states. Additionally, a systematic analysis of the interactions between (p)ppGpp and binding proteins has been carried out to probe the particular binding pattern of these interactions for various types of proteins.
The molecular dynamics simulation studies provide structural dynamic information in the solution state of a molecule. Although, glycosidic bond and ribose sugar in the nucleotide structure exhibit conformational flexibility and classical molecular dynamics simulation on nucleotides is challenging because of energy barriers and sampling of conformations are limited. Therefore, Replica Exchange Molecular Dynamics (REMD) simulation was carried out on ppGpp and pppGpp nucleotides in solution states. In REMD, several identical replicas run in parallel at different temperatures and these allow enhanced sampling of high energy conformations. Moreover, these replicas are allowed to swap their states based on Boltzmann-weighted probability at neighboring temperature state. This process is repeated iteratively during the simulation and subsequently enhanced sampling of conformations is achieved at various temperatures.
The structural coordinates of ppGpp and pppGpp were extracted from Protein Data Bank (Berman et al., 2000) and were prepared for simulation using the Maestro program from Schrodinger suite (Figures 1A–D). LigPrep tool was used to retain original chirality and biological pH 7.0 ± 2.0 of the (p)ppGpp nucleotides. For each nucleotide, the lowest energy conformer was used for the simulation process. The simulation system was built using the system builder tool in Maestro. The explicit solvent model TIP3P (Jorgensen et al., 1983)was included in an orthorhombic periodic boundary condition (PBC) computational box. The initial absolute box volume was 1000 Å3 for both molecules and upon addition of buffer, the box volume was expanded to 145262 and 150766 Å3 for ppGpp and pppGpp, respectively. Negative charges, due to phosphates groups of (p)ppGpp molecule, were neutralized by the addition of 20 mM Mg2+ in the simulation system. The Optimized Potential for Liquid Simulations 3 enhanced (OPLS3e) force field was selected for the simulation (Roos et al., 2019). Next, the prepared systems of ppGpp and pppGpp were loaded to Desmond workspace for energy minimization and replica-exchange simulation. The system was energy minimized before running the simulations. The replica-exchange parameters were set in a replica-exchange panel in Desmond. A tempering method was selected with nine replicas covering of temperature range from 273 to 373 K. The simulation time was fixed as 200 ns with a recording interval of 200 ps trajectory with energy of 1.2. The ensemble was selected as NPT for replica exchange. Finally, both REMD simulations were carried out for 200 ns. The REMD simulation result statistics were analyzed using the Desmond simulation interaction diagram report. The molecular properties of simulation trajectory were plotted using root-mean-square fluctuation (RMSF), root-mean-square deviation (RMSD), radius of gyration (rGyr), intramolecular hydrogen bonds (intraHB), molecular surface area (MolSA), solvent-accessible surface area (SASA), and polar surface area (PSA) parameters.
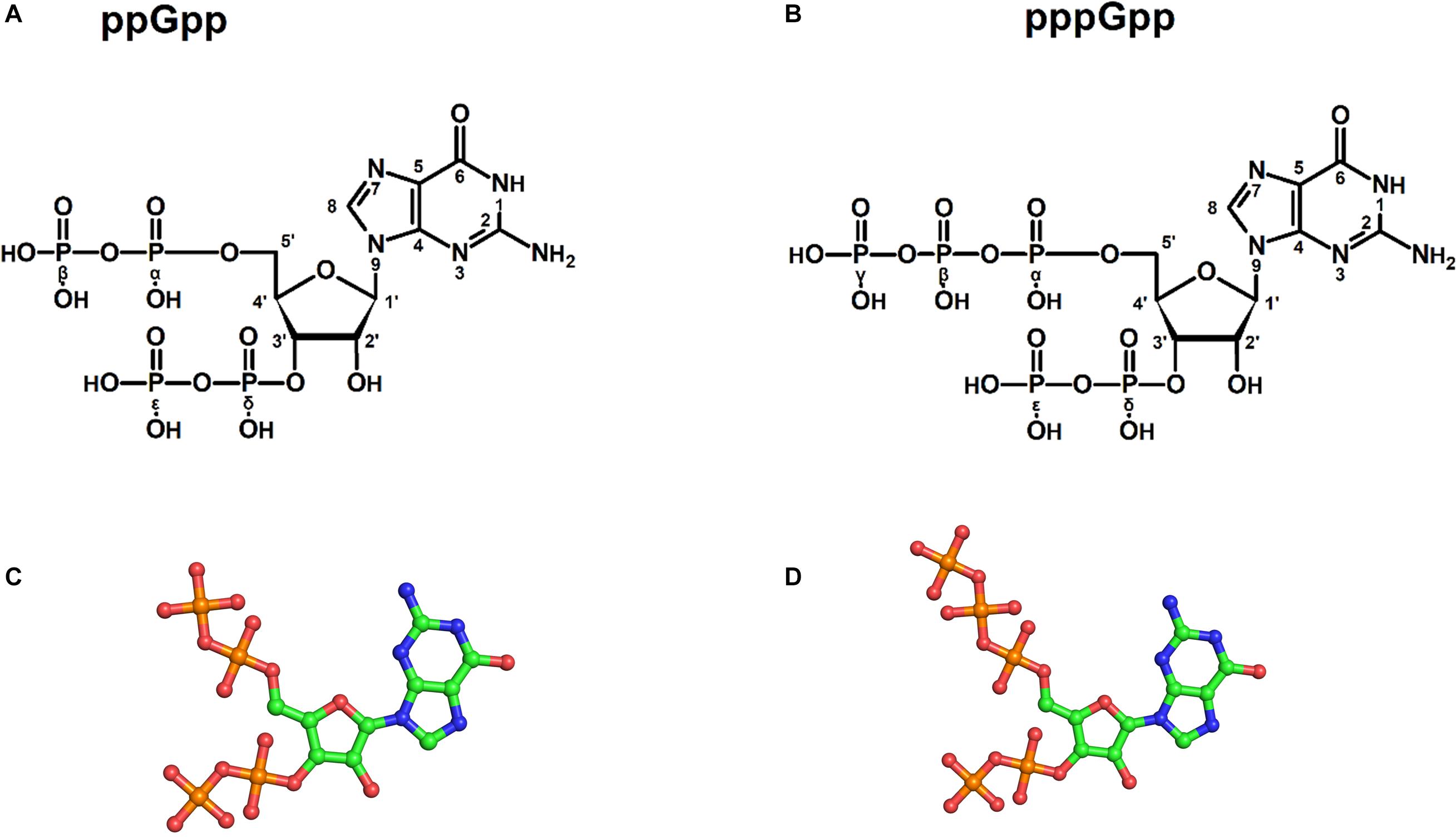
Figure 1. Structures of (p)ppGpp signaling nucleotides: a two-dimensional chemical sketch of ppGpp (A), and pppGpp (B). Ball and stick model of ppGpp (C), and pppGpp (D). The color coding for atoms in ball stick model is displayed as follows; green-carbon, blue-nitrogen, red-oxygen, and orange-phosphorous atoms.
The energy minimized three-dimensional structural coordinates of ppGpp and pppGpp were compared with coordinates extracted from crystal structures of (p)ppGpp-protein complexes. The structural alignment was carried out in PyMol using an atom alignment algorithm. The distribution frequency of glycosidic bonds in unbound states of ppGpp and pppGpp were plotted at 300 K simulation pose in Schrodinger. The torsion angle and phase angle values were calculated from Pseudo-Rotational Online Service and Interactive Tool (PROSIT) (Sun et al., 2004, 2005).
The structures of (p)ppGpp-protein complexes were downloaded from Protein Data Bank (PDB). The PDB ligand code G4P for ppGpp, 0o2, and C1Z for pppGpp was used as a search term to obtain structures of ppGpp-protein and pppGpp-protein complexes, respectively. As shown in Table 1, there were 26 structures of ppGpp-protein while eight structures of pppGpp-protein complexes were found in the database. Four crystal structures of ppGpp-riboswitch complex were also found. The protein structures in complex with ppGpp and pppGpp were uploaded to the Arpeggio server (Jubb et al., 2017). Arpeggio server, based on Python, extract the interaction data between atoms located within 5 Å radial cutoff. The interaction results of each structures were downloaded from the server and converted to tabular form. The columns containing interactions involving proximal, clashes, covalent bond, halogen bonds, hydrophobic, carbonyl were removed before calculations. The nine interaction parameters were included in the interaction calculations including, van der Waal’s clashes, van der Waal’s interactions, hydrogen bonds, weak hydrogen bonds, ionic, metal complex, aromatic, polar, and weak polar interactions. The stereochemical parameters for the definition of interatomic interactions in the Arpeggio program are employed from the CREDO database (Marcou and Rognan, 2007; Schreyer and Blundell, 2009). The hydrogen bonds were considered as the distance at 2.8 to 3.5 Å and the angle between 120 and 180° while weak hydrogen bonds were shorter than 2.8 Å and having an angle less than 120°(Marcou and Rognan, 2007). The van der Waal’s interactions are defined as interactions between two non-hydrogen bonding atoms which are present in their van der Waal’s radii of corresponding atoms. The distance parameters for van der Waal’s interactions and clashes in the Arpeggio program are taken from Open Babel. All these interactions for 33 complexes of (p)ppGpp-protein are provided in the table (Supplementary Table S1). The occurrence of these non-covalent interactions between (p)ppGpp and functional class of protein was plotted to identify the binding pattern of (p)ppGpp to respective class. For the sake of clarity, the two-dimensional interaction diagrams of each representative (p)ppGpp-protein complex were also plotted using the PoseView tool from the Protein Plus server (Stierand et al., 2006; Fährrolfes et al., 2017).
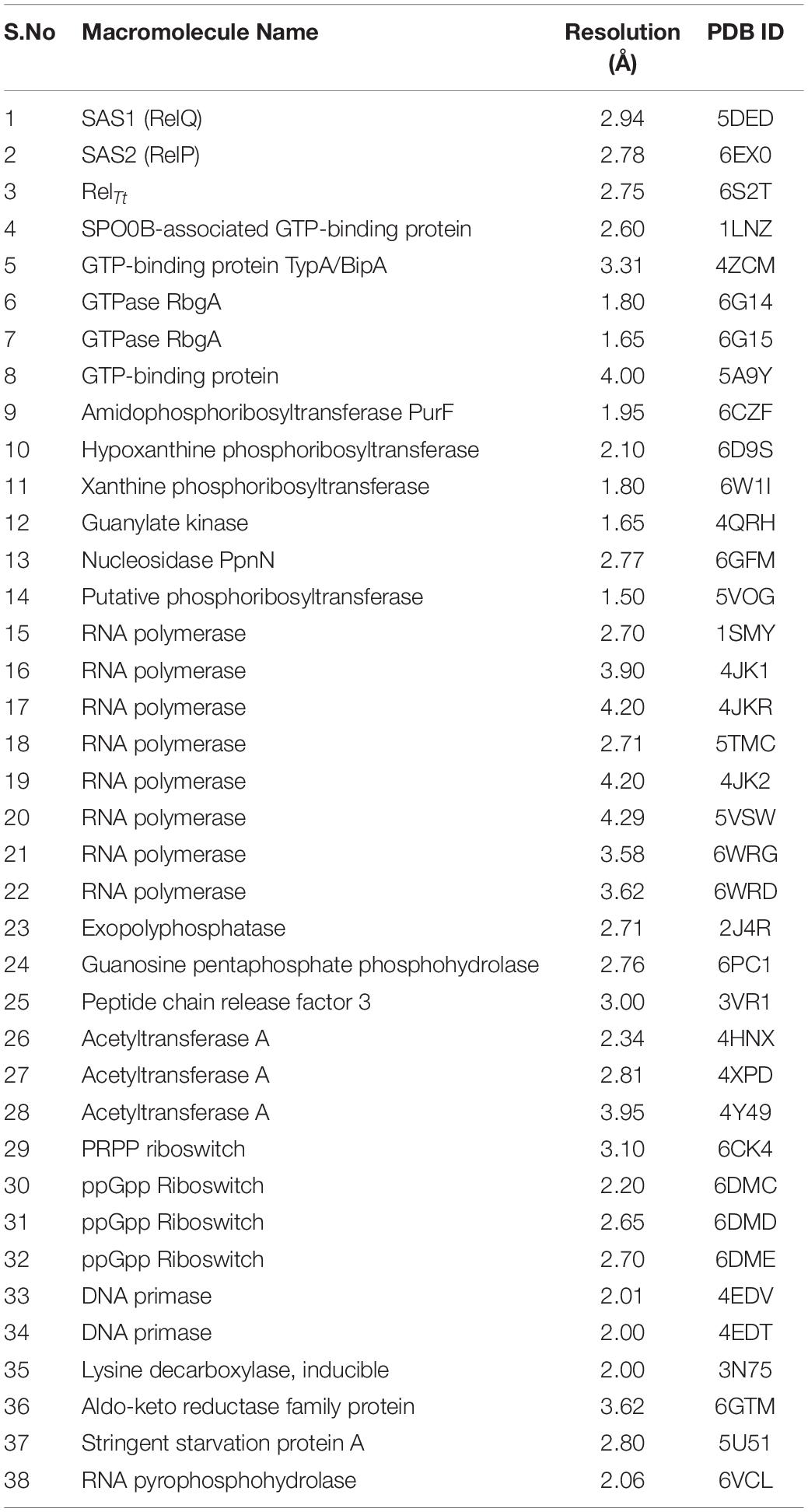
Table 1. List of available structures in complex with (p)ppGpp (ligand code G4P C1Z, and 0o2) in Protein Data Bank. The references are provided within the main text.
Molecular dynamics simulation is one of the most commonly used approaches to understand structural dynamics of biomolecules in solution state (Karplus and Petsko, 1990; Karplus and McCammon, 2002; Hollingsworth and Dror, 2018). It has been an efficient choice for the characterization of structural dynamics of free nucleotides and nucleic acid (Stern et al., 2010; Kobayashi et al., 2013; Šponer et al., 2014; Wang et al., 2017; Mlýnský and Bussi, 2018; Cassone et al., 2019). However, classical MD simulations studies on small molecules such as free nucleotides are challenging due to the presence of high energy barriers of the glycosidic bond between nucleotide base and ribose sugar as well as conformational flexibility of ribose moiety (Wang et al., 2017; Wang and Berne, 2018; Yang et al., 2019). To overcome the sampling issue, the replica-exchange method has been explored previously on small biomolecules to enhance sampling to cover more conformation space (Smith et al., 2016; Wang and Berne, 2018). Recently, cyclic nucleotides and small nucleic acid have been characterized for structural dynamics using REMD enhanced sampling methods (Šponer et al., 2014; Wang et al., 2017; Mlýnský and Bussi, 2018).
We used a well-defined protocol for REMD simulation to obtain the structural dynamics information of unbound ppGpp and pppGpp nucleotides in the solution state. A total of eight replicas simulation were exchanged at the 273, 285, 298, 310, 323, 335, 348, 360, 373 K during simulation covering temperature range from 273 to 373 K (Supplementary Figure S1). As shown in the graph, the RMSF, a parameter for displacement measurement of an atom in a molecular simulation trajectory in comparison with reference position, indicates that the major structural changes in (p)ppGpp molecules are observed (Figures 2A,B). There are 18 torsion bonds in pppGpp and 15 torsion bonds in ppGpp which can rotate therefore higher RMSF values are observed in these corresponding atoms. Although the negatively charged phosphate groups were neutralized by two Mg2+ ions during the simulation, yet higher flexibility of phosphate atoms was observed in the RMSF plot. In contrast to (p)ppGpp, the phosphate groups in cyclic messenger nucleotides show lesser flexibility due to the unavailability of free phosphate chains (Stern et al., 2010; Wang et al., 2017; Cassone et al., 2019). Based on our results, we propose that the dynamics in two free phosphate chains may provide greater flexibility to (p)ppGpp nucleotides for binding to diverse proteins. Surprisingly, the higher value of RMSF was observed in the NH2 group of guanine ring of (p)ppGpp in REMD simulation. The detailed analysis of molecular properties shows the overall quality and conformational dynamics of (p)ppGpp during the simulation (Figures 3A,B). These graphs show that molecular properties in ppGpp and pppGpp are similar, however, there are minor differences in these values that may be because of additional phosphate group present in pppGpp. The higher RMSD and rGyr show the conformational flexibility which indicates that (p)ppGpp nucleotides show dynamics in the unbound states which is necessary to accommodate in the various types of the binding site. The other important parameter of (p)ppGpp nucleotides is the SASA which depicts the surface area of molecules accessible by the water molecules. The higher SASA values of (p)ppGpp contribute water-mediated interactions to the proteins and it is consistent with structures of (p)ppGpp-protein-complexes. The molecular parameters including rGyr, intraHB, MolSA, SASA, and PSA exhibit slightly higher value for pppGpp as compare to ppGpp because of an additional γ phosphate group present in pppGpp. In addition to inter-molecular non-covalent interactions, water-mediated hydrogen bonds play a significant role in stabilizing small molecules in the binding site of the protein.
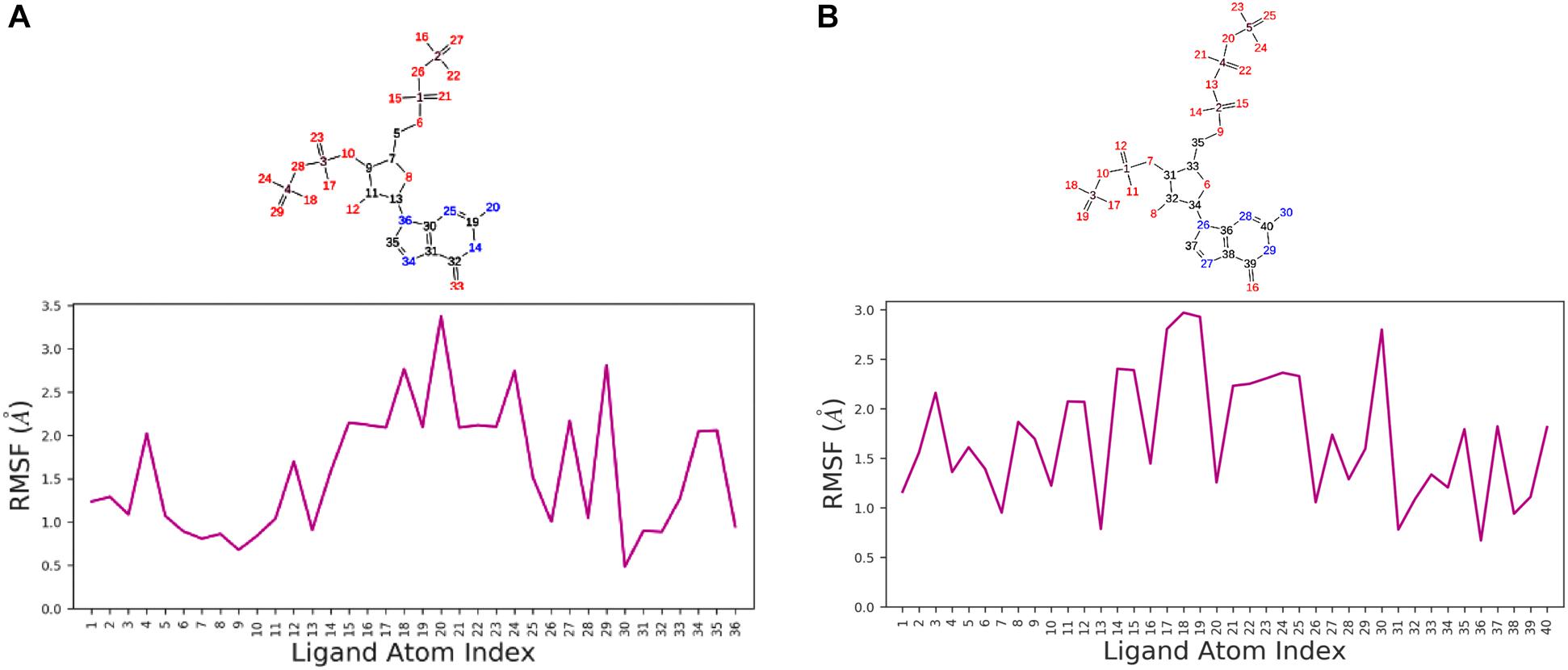
Figure 2. Replica exchange molecular dynamics simulation analysis. Atom numbering at top panel and root-mean-square fluctuation (RMSF) plot for ppGpp (A) and pppGpp (B) displays the fluctuation in individual atoms with reference to an initial state.
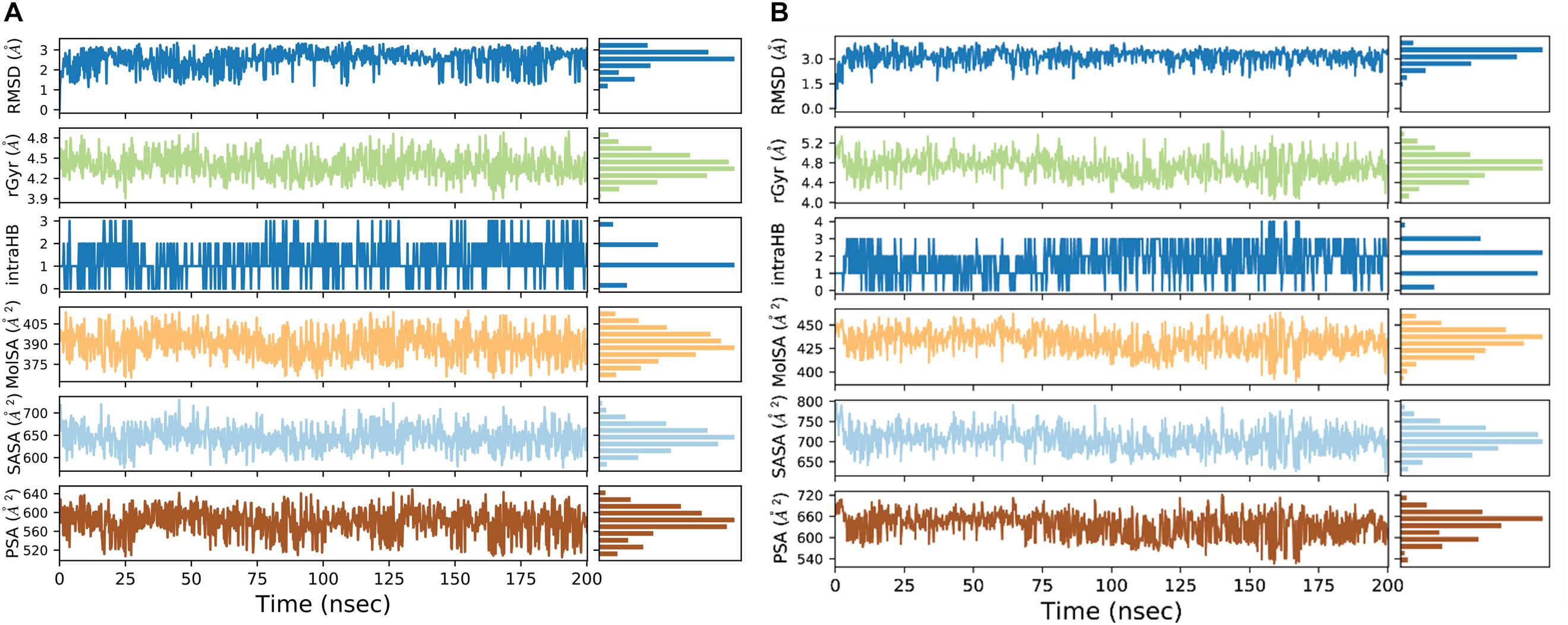
Figure 3. Molecular properties graph for the description of various parameters of ppGpp (A), and pppGpp (B) during simulation run.
The three-dimensional structures of unbound (p)ppGpp were obtained by energy minimization in Maestro. The structural coordinates of bound (p)ppGpp were extracted from structures of (p)ppGpp-protein complexes. The structural alignment of unbound state and bound state of (p)ppGpp shows substantial similarity in the guanine ring region with RMSD of atoms less than one. However, the phosphate chain shows divergence upon structural alignment which is in agreement with our simulation RMSF plot.
(p)ppGpp nucleotides exhibit a substantial extent of conformational flexibility because of various rotatable torsion angles present in the structure particularly a ribose ring (Figure 4). Therefore, we analyzed the glycosidic bond conformation of (p)ppGpp in unbound and bound states. The glycosidic conformation signifies the orientation of base and sugar in nucleotides and is measured as a torsion angle (χ) between O4’-C1’-N9-C4 of guanine. As shown in Figure 5, the distribution frequency of glycosidic bond conformation in both nucleotides observed during REMD was mostly in syn conformation, however, some occurrences were observed in anti conformation. Our results on the frequency distribution of glycosidic conformation in unbound states are similar to those observed in the bound states in the crystal structure. As most of the structures of (p)ppGpp-protein complexes have anti conformation of glycosidic bond except in few cases it was found in syn conformation such as nucleosidase (6GFM) (Zhang et al., 2019), lysine decarboxylase (3N75) (Kanjee et al., 2011). The sugar pucker in (p)ppGpp nucleotides in the bound state is observed majorly in endo conformation (Table 2). Whereas, the sugar pucker conformation energy minimized unbound states are found as exo for ppGpp and pppGpp (Table 2). The five endocyclic torsion angles, ν0, ν1, ν2, ν3, and ν4, of backbone atoms, define the conformation description of the ribose sugar (Figure 4). The ribose sugar of (p)ppGpp nucleotides shows substantial conformational flexibility in unbound and bound states (Table 2). Additional parameters to characterize ribose sugar conformation are pseudorotational phase angle (P) and maximum puckering amplitude (νmax) which show that ribose sugar adopts north conformation in both states (Sun et al., 2004). The comparative description of these torsion angles is given in the Table 2 for unbound states and few bound states of (p)ppGpp. All these parameters indicate that ribose moiety in (p)ppGpp behave dynamically in both unbound and bound states.
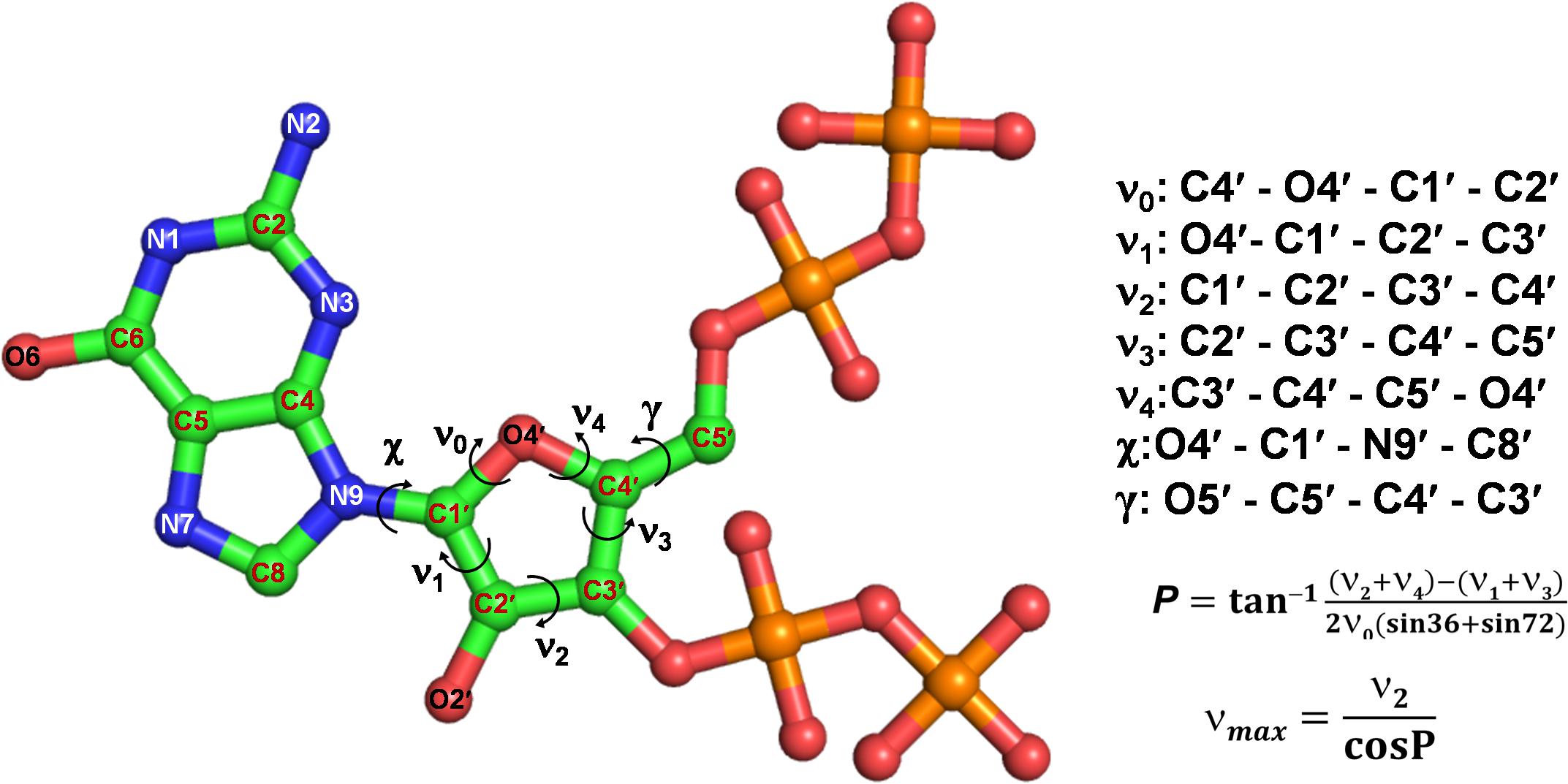
Figure 4. Different torsion angles in the structure of (p)ppGpp. The glycosidic torsion angle (χ) depicts the rotation between guanine and ribose ring, the angle γ denotes rotation between ribose ring and C4 branch of ribose sugar. The torsion angles of ribose rings are denoted fromν0 to ν4.
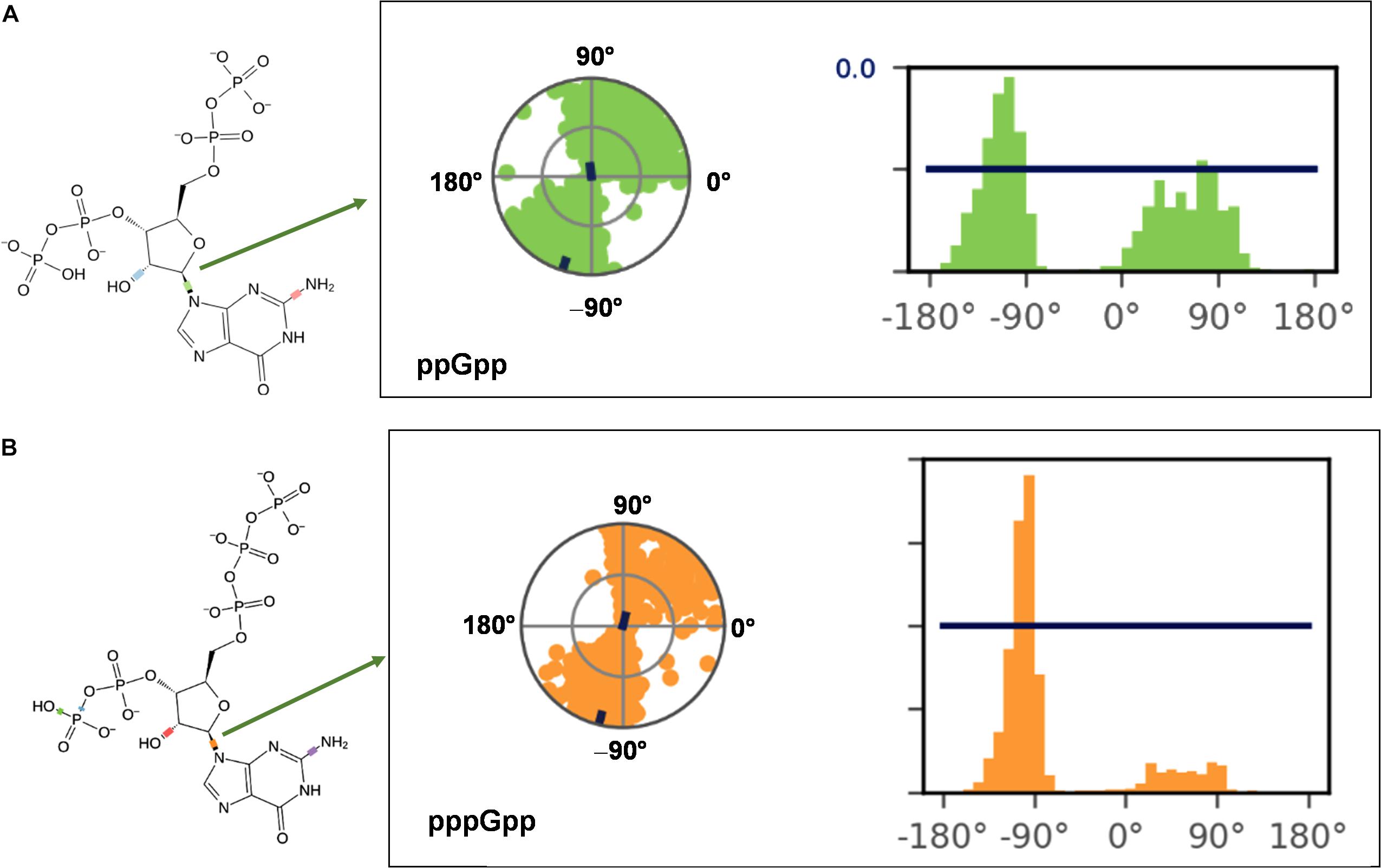
Figure 5. Probable frequency distribution of glycosidic torsion angle (χ) conformation in ppGpp (A) and pppGpp (B) during REMD simulation. The radial plot denotes the conformation of glycosidic bond throughout the simulation time course. The beginning of the simulation started at the center of a circle and radially outward portrays time evolution during the simulation run. The bar diagram representation of the distribution data of torsion angle. X-axis denotes the angle of glycosidic bond and Y-axis shows the number of times the bond was observed at this angle i.e., percentage frequency distribution. The value of χ falls into ranges of +90 to +180° (or 180 to 270°) corresponds to anti conformation while the value χ founds in the ranges of –90 to +90° refers to syn conformation of glycosidic bond.
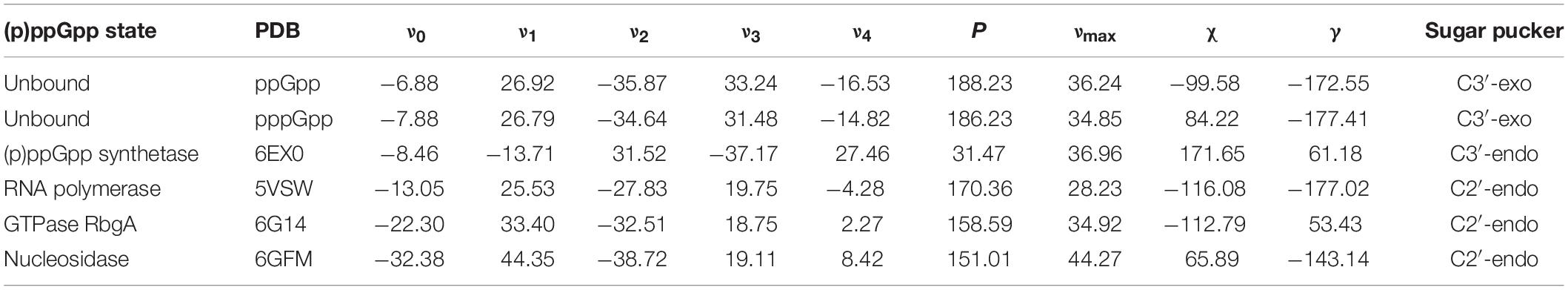
Table 2. Pseudorotational phase angle and puckering amplitude for (p)ppGpp nucleotides.
The structural diversity of secondary messenger nucleotides deliver generous adaptability for the binding to the cognate proteins. Based on their structural architecture, these nucleotides may be divided into two classes; linear and cyclic secondary nucleotides. The interaction analysis of cyclic secondary nucleotides with their binding protein complexes have been characterized extensively (Moodie and Thornton, 1993; Wang et al., 2017; Cassone et al., 2019; He et al., 2020). These studies provide significant information about the binding mode and interaction pattern of cyclic nucleotides and their protein complexes (He et al., 2020). Similarly, linear secondary messenger nucleotides, (p)ppGpp binds to a diverse class of proteins and should exhibit a particular binding mode for each functional class of protein. We have examined these interaction patterns using various computational tools to assess the binding stereochemistry between (p)ppGpp and respective protein. A detailed survey has been carried out on available structures of (p)ppGpp-protein complexes and the non-covalent interactions were quantified (Figure 6A). In addition to conventional interatomic interactions, various other types of molecular interaction fingerprints were included in the study which provides additional information for binding stability of ligand (Marcou and Rognan, 2007). Other types of non-conventional interactions that were included in the analysis are weak polar and weak hydrogen bonds that denote the hydrogen bonds without considering angles. A total of five major types of bonding parameters were included in the analysis interactions between (p)ppGpp and proteins. Among these interactions, hydrogen bonds contribute most as five nitrogen and 17 oxygen atoms in the (p)ppGpp have properties to make hydrogen bonds (Figure 6B). In the structures of many complexes, the phosphate group of (p)ppGpp nucleotide is observed as making interaction to protein through Mg2+ ions. As seen by the hydrogen bonds histogram, the major interatomic polar interactions were observed in guanine ring and phosphate group atoms. The aromatic ring of guanine hadπ-π base stacking interactions with the side chain of tyrosine in the structures of (p)ppGpp synthetases and nucleotide metabolic enzymes. The ribose moiety of (p)ppGpp makes comparatively fewer interactions with protein atoms which may be the consequence of the structural restrain of the ribose ring. Similarly, van der Waal’s interactions were found majorly in the region of the guanine ring of (p)ppGpp (Figure 6C). The available structures of the complexes between (p)ppGpp and protein span several functional classes. Here the characterization of (p)ppGpp-protein interactions focuses only on four major functional classes.
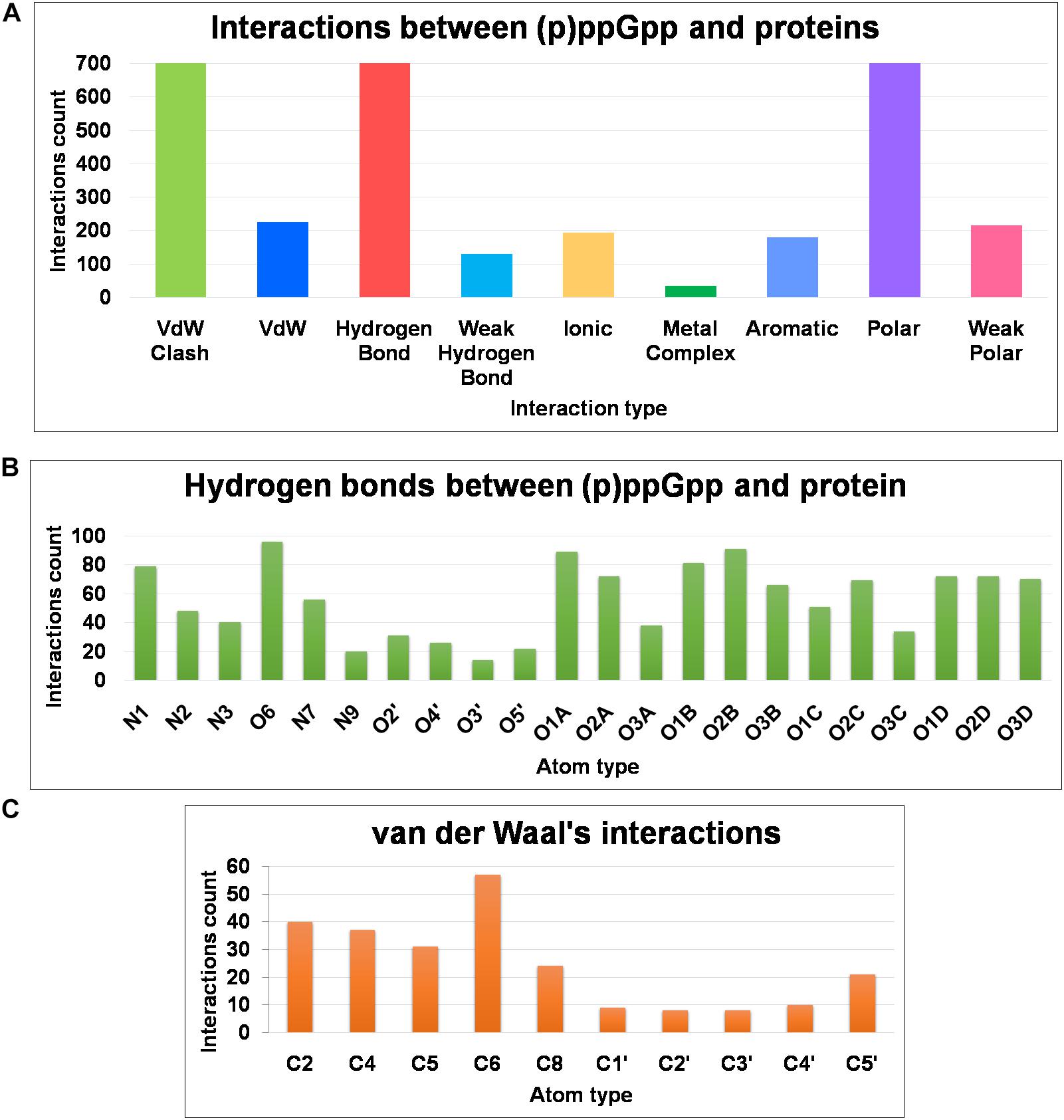
Figure 6. Frequency distribution of non-covalent interactions, between (p)ppGpp and protein, extracted from available crystal structures of (p)ppGpp-protein complexes. (A) Various types of inter-molecular interactions and their occurrences. (B) Atom-level distribution frequency of hydrogen bonds found in the (p)ppGpp-protein complexes. (C) Atom-level distribution frequency of van der Waal’s interactions found between (p)ppGpp and protein.
The (p)ppGpp synthetases are apparent (p)ppGpp binding proteins as these enzymes synthesize ppGpp and pppGpp from ATP and GDP/GTP, respectively. However, most of the structures determined so far are in complexes with substrate analogs as these complexes explain the catalytic mechanism. A brief structural review on (p)ppGpp synthetase proteins has been described previously (Kushwaha et al., 2019b) hence current study focus only on (p)ppGpp and (p)ppGpp synthetase complexes. There are three structures of (p)ppGpp synthetases available in the Protein Data Bank in complex with (p)ppGpp. It includes two single domain (p)ppGpp synthetases, small alarmone synthetase-1 (SAS1) from Bacillus subtilis (Steinchen et al., 2015); small alarmone synthetase-2 (SAS2) from Staphylococcus aureus (Manav et al., 2018) and one long (p)ppGpp synthetase RelTt from Thermus thermophiles (Tamman et al., 2020). The (p)ppGpp binding site is primarily comprised of polar residues which hold the (p)ppGpp strongly by making several hydrogen bonds. The most distinguishing feature of these complexes is aromatic stacking with the guanine ring which is stacked by the side chain-ring of tyrosine. The negatively charged phosphate groups are stabilized by ionic interactions with the guanidinium group of several arginine residues. The Mg2+ ions also contribute to stabilizing these phosphate groups. As shown the Figure 7A, the nitrogen and oxygen atoms of the guanine ring significantly contribute to the hydrogen bond interactions with (p)ppGpp synthetase proteins. This type of binding pattern of (p)ppGpp may be considered as an optimum binding pattern for (p)ppGpp interactions with proteins.
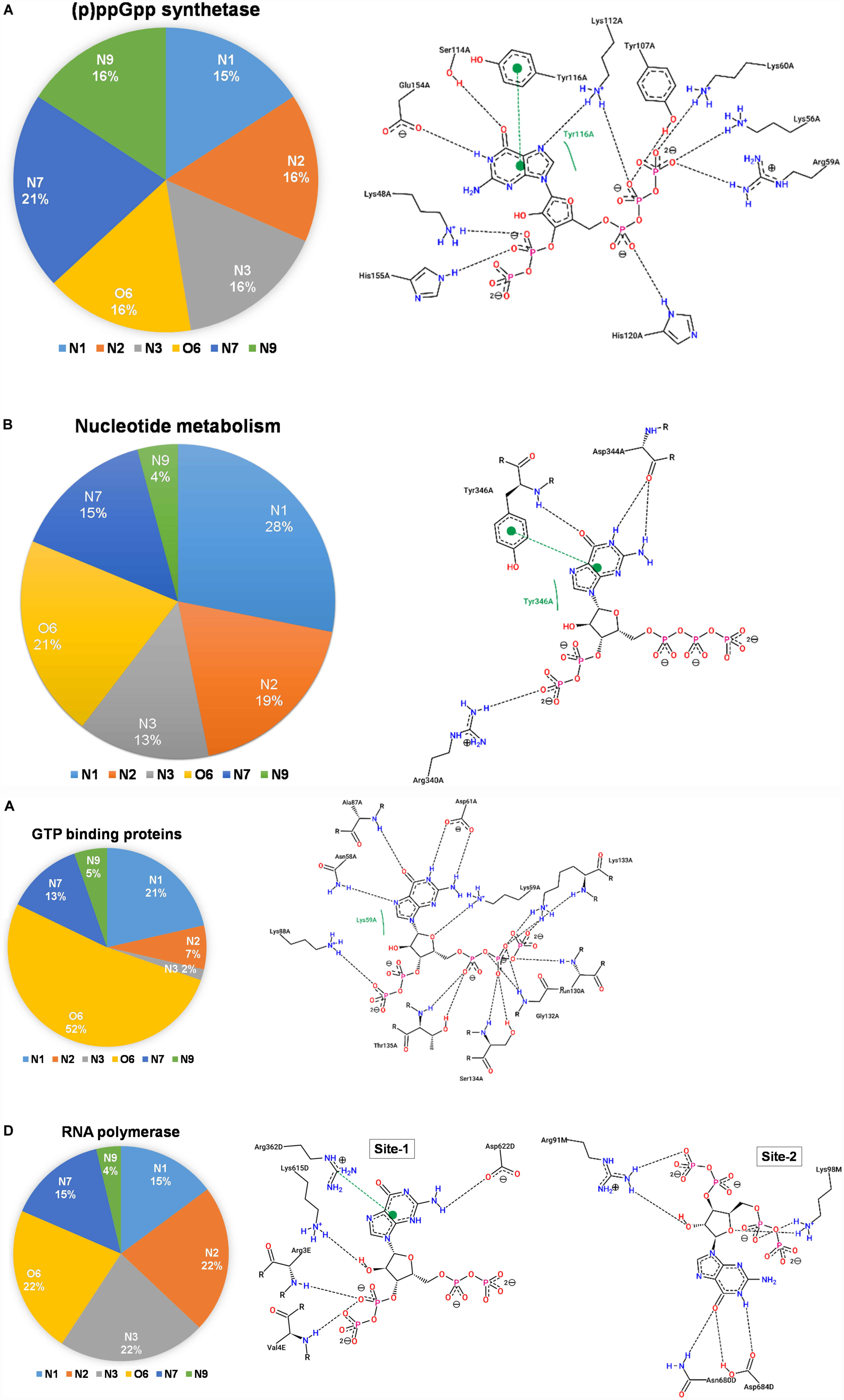
Figure 7. Interactions between (p)ppGpp atoms and protein are identified in the crystal structures of (p)ppGpp-protein complexes. The left panel of each figure displays the frequency distribution of interactions formed by guanine atoms of (p)ppGpp with protein atoms. The right panel of each figure indicates the two-dimensional interaction diagram between (p)ppGpp and protein. The interaction plots, generated by PoseView tool, show hydrogen bonds between (p)ppGpp and protein. (A) (p)ppGpp synthetase (5DED), (B) nucleotide metabolic enzymes (6GFM), (C) GTPase (6G15), and (D) RNA polymerase (5VSW).
There are six crystal structures of (p)ppGpp and nucleotide metabolic enzymes complexes available in the Protein Data Bank which includes Xanthine phosphoribosyltransferase (XPRT) (Anderson et al., 2020), Pyrimidine/purine nucleotide 5′-monophosphate nucleosidase (nucleosidase, PpnN) (Zhang et al., 2019), Hypoxanthine phosphoribosyltransferase (HPRT) (Anderson et al., 2019), Amidophosphoribosyltransferase (PurF) (Wang et al., 2019), Guanylate kinase (Liu et al., 2015; Table 1). A most striking feature of the structures of these enzyme-(p)ppGpp complexes are oligomeric stoichiometry in these complexes. The (p)ppGpp nucleotides facilitates the oligomeric cooperation through binding at an oligomeric interface. Similar to (p)ppGpp synthetases, (p)ppGpp binding site in these enzymes is primarily comprised of residues containing side chains with the polar groups for hydrogen bonding and aromatic ring for stacking interactions. The characteristic feature of (p)ppGpp binding to enzymes involved in nucleotide metabolism is aromatic stacking interaction between guanine base of (p)ppGpp and the aromatic ring-containing side chain of residues of corresponding binding proteins (Figure 7B). For example, Phe126 in XPRT, Trp153 in XGPRT, Tyr346 in PpnN, Phe152 in HPRT, Tyr83 in guanylate kinase makes stacking interactions. However, the aromatic stacking was not observed in the structure of PurF-ppGpp complex. The ribose ring generally interacted with protein though water-mediated hydrogen bonds interactions. The negatively charged phosphate groups of (p)ppGpp nucleotides are stabilized by mostly guanidine moiety of arginine side chains and Mg2+ ions. Therefore, these interactions are considered as ionic interactions. The observed binding mode of (p)ppGpp to the nucleotide enzymes is similar due to an analogy of substrate nucleotide structure to the (p)ppGpp. As shown in the graph, the N1, N2, N7, and O6 atoms of the aromatic guanine ring of (p)ppGpp is a major contributor to hydrogen bond interaction with protein residues.
There are five crystal structures of GTP binding GTPase proteins in complex with (p)ppGpp available till now. These include Obg GTP binding protein (Buglino et al., 2002), GTPase BipA/TypA (Fan et al., 2015), GTPase BipA (Kumar et al., 2015), and GTPase RbgA (Pausch et al., 2018). The (p)ppGpp binding site in GTPases proteins are shallower in comparison to (p)ppGpp synthetases and nucleotide metabolic enzymes. In contrast to (p)ppGpp synthetases, and nucleotide metabolic enzymes, aromatic stacking and guanidinium moiety was not observed in the structures of GTPases in complexes with (p)ppGpp. Interestingly, the major interaction of (p)ppGpp guanine ring was contributed by the O6 atom of (p)ppGpp (Figure 7C). The metal ions were also not observed to neutralize the negatively charged phosphate group of (p)ppGpp. Therefore, the major forces for the stabilization of (p)ppGpp are hydrogen bonds and van der Waal’s interactions.
RNA polymerase was the first protein complex reported to regulate its activity by binding of (p)ppGpp (Potrykus and Cashel, 2008). There are two (p)ppGpp binding sites that have been reported in the RNA polymerase which modulates allosteric transcription regulation (Molodtsov et al., 2018). Site 1 is located at the interface formed by β′ and ω subunits of RNA polymerase. This site is comparatively shallower and the guanine ring of (p)ppGpp interacts with the side chain of arginine, isoleucine, histidine, and aspartic acid residues. The second (p)ppGpp binding site is found at the secondary channel and it acts synergistically by the binding of a transcription regulator protein, DksA. Therefore, complete binding of (p)ppGpp to site 2 is accomplished by the interactions between (p)ppGpp and residues of β′ rim and DksA. In the site 2, the guanine ring of (p)ppGpp is stabilized by interactions between (p)ppGpp and side chains of aspartic acid, tyrosine, asparagine, and isoleucine while negatively charged phosphate group is neutralized by basic residues such as lysine and arginine (Figure 7D). The binding of (p)ppGpp to site 2, exhibit a allosteric change in the corresponding areas of RNAP and DksA to facilitate transcription regulation.
There are several crystal structures of various other functional class of (p)ppGpp binding proteins reported including translation peptide chain release factor 3 (PDB: 3VR1) (Kihira et al., 2012), acetyltransferase A (PDB: 4HNX, 4XPD, 4Y49), PPX/GppA phosphatases (PDB: 2J4R, 6PC1) (Kristensen et al., 2008; Song et al., 2020), DNA primase (PDB: 4EDV, 4EDT) (Rymer et al., 2012), lysine decarboxylase (PDB: 3N75) (Kanjee et al., 2011), aldo-keto reductase (PDB: 6GTM)and RNA pyrophosphohydrolase (PDB: 6VCL) (Gao et al., 2020). The PPX/GppA phosphatases are pppGpp hydrolyzing enzymes that remove the γ-phosphate group from pppGpp to make ppGpp nucleotide, therefore, pppGpp serves as a substrate for these enzymes. DnaG is a DNA dependent RNA polymerase primase, responsible for primer synthesis during DNA replication. DnaG primase binds to various nucleotides including (p)ppGpp nucleotides. Lysine decarboxylase is an acid response protein that catalyzes the decarboxylation of L-lysine. (p)ppGpp binds to Ldcl and inhibits its enzymatic activity. RppH is Nudix hydrolase enzyme involved in RNA processing. It hydrolyzes various nucleotides including (p)ppGpp alarmone. The detailed interactions analysis and binding pattern of these (p)ppGpp-complexes are given in Supplementary Figure S2.
Magnesium ion (Mg2+) plays a significant role in the structural stability of nucleic acid and nucleotides by neutralizing highly negative charged phosphate groups (Black et al., 1994; Pechlaner and Sigel, 2012; Leonarski et al., 2017). Interestingly, highly phosphorylated (p)ppGpp has two linear chains of phosphate groups therefore Mg2+ ions assist in the specific interaction of (p)ppGpp to their target protein. As shown in the graph (Figure 6), we have observed several metal interactions in the structures of (p)ppGpp-protein complexes. In (p)ppGpp synthetases structures, Mg2+ ions provide robust physical support to the flexible phosphate chains. In the case of SAS1, one Mg2+ ion firmly stabilizes two phosphate chains with the side chain of Lys 32 while in the case of long-form of (p)ppGpp synthetase, RelTt, it binds to only one chain of phosphate (Tamman et al., 2020). In addition to physical stability, Mg2+ ions assist in the deprotonation of 3′ OH of GDP/GTP by acidic residues in RelSeq and SAS1 (Hogg et al., 2004; Steinchen et al., 2015). Similarly, in the case of nucleotide metabolic enzymes, Mg2+ ions provide not only physical stability to one chain of (p)ppGpp but it also mediates interactions between (p)ppGpp and protein side chains. Each phosphate chain of (p)ppGpp is making interactions with one Mg2+ ion in the complex of RNA polymerase-(p)ppGpp. Altogether, we have observed Mg2+ ions in several other complexes of (p)ppGpp-protein structures except for few complexes which may be the limitation of electron density interpretation as discussed earlier (Leonarski et al., 2017).
Secondary messenger nucleotides are key signaling molecules that modulate several cellular functions particularly in response to environmental changes. These nucleotides bind to several enzymes involve in various functional activities. The versatility of the binding mode of secondary nucleotides is facilitated by the conformation flexibility of glycosidic bond, ribose sugar puckering. In contrast to the cyclic nucleotides, (p)ppGpp have linear phosphate chains that provide additional flexibility to adapt various conformations according to the stereochemistry of the binding site of a respective target protein. Overall, our results support the hypothesis that the conformation flexibility of glycosidic bond, ribose sugar puckering, and phosphate groups provide structural plasticity to the (p)ppGpp for binding to the various functional class of proteins. The structures of unbound ppGpp and pppGpp in solution states obtained by MD simulation are similar to that observed in the structures in bound form, hence, these structures resemble biologically active conformations of (p)ppGpp. The gyration conformation profile of glycosidic bond is in agreement with the conformation of (p)ppGpp observed in the bound state. The analysis of (p)ppGpp-protein interactions reveals that the binding pattern of these nucleotides governs the regulation for a particular class of target proteins.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
GK and NB conceptualized the study. GK and AP performed the study. All authors analyzed the data, wrote and edited the manuscript, and agreed on the final version of the manuscript.
This study was supported by Science and Engineering Research Board (SERB), Government of India (SB/YS/LS-176/2014), and ICGEB, New Delhi core funds. GK was supported by grant no. BT/IC-06/003/91-Flagship proposal, from the DBT, Government of India. AP is a recipient of Council of Scientific and Industrial Research (CSIR) Senior Research Fellowship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.575041/full#supplementary-material
Abranches, J., Martinez, A. R., Kajfasz, J. K., Chávez, V., Garsin, D. A., and Lemos, J. A. (2009). The molecular alarmone (p)ppGpp mediates stress responses, vancomycin tolerance, and virulence in Enterococcus faecalis. J. Bacteriol. 191, 2248–2256. doi: 10.1128/JB.01726-08
Anderson, B. W., Hao, A., Satyshur, K. A., Keck, J. L., and Wang, J. D. (2020). Molecular mechanism of regulation of the purine salvage enzyme XPRT by the AlarmonespppGpp, ppGpp, and pGpp. J. Mol. Biol. 432, 4108–4126. doi: 10.1016/j.jmb.2020.05.013
Anderson, B. W., Liu, K., Wolak, C., Dubiel, K., She, F., Satyshur, K. A., et al. (2019). Evolution of (P)ppGpp-HPRT regulation through diversification of an allosteric oligomeric interaction. Elife 8:e47534. doi: 10.7554/eLife.47534
Atkinson, G. C., Tenson, T., and Hauryliuk, V. (2011). The RelA/SpoT Homolog (RSH) superfamily: distribution and functional evolution of ppGppSynthetases and hydrolases across the tree of life. PLoS One 6:e23479. doi: 10.1371/journal.pone.0023479
Beljantseva, J., Kudrin, P., Andresen, L., Shingler, V., Atkinson, G. C., Tenson, T., et al. (2017). Negative allosteric regulation of Enterococcus faecalis small alarmonesynthetaseRelQ by single-stranded RNA. Proc. Natl. Acad. Sci. U. S. A. 114, 3726–3731. doi: 10.1073/pnas.1617868114
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The protein data bank. Nucleic Acids Res. 28, 235–242. doi: 10.1093/nar/28.1.235
Black, C. B., Huang, H. W., and Cowan, J. A. (1994). Biological coordination chemistry of magnesium, sodium, and potassium ions.Protein and nucleotide binding sites.Coord. Chem. Rev. 135–136, 165–202. doi: 10.1016/0010-8545(94)80068-5
Buglino, J., Shen, V., Hakimian, P., and Lima, C. D. (2002). Structural and biochemical analysis of the Obg GTP binding protein. Structure 10, 1581–1592. doi: 10.1016/S0969-2126(02)00882-1
Bærentsen, R. L., Brodersen, D. E., and Zhang, Y. E. (2019). Evolution of the bacterial nucleosidasePpnN and its relation to the stringent response. Microb. Cell 6, 450–453. doi: 10.15698/mic2019.09.692
Cashel, M., and Gallant, J. (1969). Two compounds implicated in the function of the RC gene of Escherichia coli. Nature 221, 838–841.
Cashel, M., Gentry, D., Hernandez, V. J., and Vinella, D. (1996). “The stringent response,” in Escherichia coli and Salmonella: Cellular and Molecular Biology, ed. F. C. Neidhardt (Washington, DC: ASM Press), 1458–1496.
Cassone, G., Kruse, H., and Sponer, J. (2019). Interactions between cyclic nucleotides and common cations: an: ab initio molecular dynamics study. Phys. Chem. Chem. Phys. 21, 8121–8132. doi: 10.1039/c8cp07492e
Dalebroux, Z. D., Svensson, S. L., Gaynor, E. C., and Swanson, M. S. (2010). ppGpp conjures bacterial virulence. Microbiol. Mol. Biol. Rev. 74, 171–199. doi: 10.1128/MMBR.00046-09
Dalebroux, Z. D., and Swanson, M. S. (2012). ppGpp: magic beyond RNA polymerase. Nat. Rev. Microbiol. 10, 203–212. doi: 10.1038/nrmicro2720
Fährrolfes, R., Bietz, S., Flachsenberg, F., Meyder, A., Nittinger, E., Otto, T., et al. (2017). Proteins plus: a web portal for structure analysis of macromolecules. Nucleic Acids Res. 45, W337–W343. doi: 10.1093/nar/gkx333
Fan, H., Hahm, J., Diggs, S., Perry, J. J. P., and Blaha, G. (2015). Structural and functional analysis of BipA, a regulator of virulence in enteropathogenic Escherichia coli. J. Biol. Chem. 290, 20856–20864. doi: 10.1074/jbc.M115.659136
Gao, A., Vasilyev, N., Kaushik, A., Duan, W., and Serganov, A. (2020). Principles of RNA and nucleotide discrimination by the RNA processing enzyme RppH. Nucleic Acids Res. 48, 3776–3788. doi: 10.1093/nar/gkaa024
Hauryliuk, V., and Atkinson, G. C. (2017). Small alarmone synthetases as novel bacterial RNA-binding proteins. RNA Biol. 14, 1695–1699. doi: 10.1080/15476286.2017.1367889
Hauryliuk, V., Atkinson, G. C., Murakami, K. S., Tenson, T., and Gerdes, K. (2015). Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat. Rev. Microbiol. 13, 298–309. doi: 10.1038/nrmicro3448
He, J., Yin, W., Galperin, M. Y., and Chou, S. H. (2020). Cyclic di-AMP, a second messenger of primary importance: tertiary structures and binding mechanisms. Nucleic Acids Res. 48, 2807–2829. doi: 10.1093/nar/gkaa112
Hengge, R., Häussler, S., Pruteanu, M., Stülke, J., Tschowri, N., and Turgay, K. (2019). Recent advances and current trends in nucleotide second messenger signaling in bacteria. J. Mol. Biol. 431, 908–927. doi: 10.1016/j.jmb.2019.01.014
Hogg, T., Mechold, U., Malke, H., Cashel, M., and Hilgenfeld, R. (2004). Conformational antagonism between opposing active sites in a bifunctionalRelA/SpoT homolog modulates (p)ppGpp metabolism during the stringent response. Cell 117, 57–68. doi: 10.1016/S0092-8674(04)00260-0
Hollingsworth, S. A., and Dror, R. O. (2018). Molecular dynamics simulation for all. Neuron 99, 1129–1143. doi: 10.1016/j.neuron.2018.08.011
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., and Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. doi: 10.1063/1.445869
Jubb, H. C., Higueruelo, A. P., Ochoa-Montaño, B., Pitt, W. R., Ascher, D. B., and Blundell, T. L. (2017). Arpeggio: a web server for calculating and visualising interatomic interactions in protein structures. J. Mol. Biol. 429, 365–371. doi: 10.1016/j.jmb.2016.12.004
Kanjee, U., Gutsche, I., Alexopoulos, E., Zhao, B., El Bakkouri, M., Thibault, G., et al. (2011). Linkage between the bacterial acid stress and stringent responses: the structure of the inducible lysine decarboxylase. EMBO J. 30, 931–944. doi: 10.1038/emboj.2011.5
Kanjee, U., Ogata, K., and Houry, W. A. (2012). Direct binding targets of the stringent response alarmone (p)ppGpp. Mol. Microbiol. 85, 1029–1043. doi: 10.1111/j.1365-2958.2012.08177.x
Karplus, M., and McCammon, J. A. (2002). Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 9, 646–652. doi: 10.1038/nsb0902-646
Karplus, M., and Petsko, G. A. (1990). Molecular dynamics simulations in biology. Nature 347, 631–639. doi: 10.1038/347631a0
Kihira, K., Shimizu, Y., Shomura, Y., Shibata, N., Kitamura, M., Nakagawa, A., et al. (2012). Crystal structure analysis of the translation factor RF3 (release factor 3). FEBS Lett. 586, 3705–3709. doi: 10.1016/j.febslet.2012.08.029
Kobayashi, E., Yura, K., and Nagai, Y. (2013). Distinct conformation of ATP molecule in solution and on protein. Biophys. 9, 1–12. doi: 10.2142/biophysics.9.1
Kriel, A., Bittner, A. N., Kim, S. H., Liu, K., Tehranchi, A. K., Zou, W. Y., et al. (2012). Direct regulation of GTP homeostasis by (p)ppGpp: a critical component of viability and stress resistance. Mol. Cell 48, 231–241. doi: 10.1016/j.molcel.2012.08.009
Kristensen, O., Ross, B., and Gajhede, M. (2008). Structure of the PPX/GPPA phosphatase from aquifexaeolicus in complex with the AlarmoneppGpp. J. Mol. Biol. 375, 1469–1476. doi: 10.1016/j.jmb.2007.11.073
Kudrin, P., Varik, V., Oliveira, S. R. A., Beljantseva, J., Del Peso Santos, T., Dzhygyr, I., et al. (2017). Subinhibitory concentrations of bacteriostatic antibiotics induce relA -dependent and relA -independent tolerance to β-Lactams. Antimicrob. Agents Chemother. 61:e2173-16. doi: 10.1128/AAC.02173-16
Kumar, V., Chen, Y., Ero, R., Ahmed, T., Tan, J., Li, Z., et al. (2015). Structure of BipA in GTP form bound to the ratcheted ribosome. Proc. Natl. Acad. Sci. U. S. A. 112, 10944–10949. doi: 10.1073/pnas.1513216112
Kushwaha, G. S., Bange, G., and Bhavesh, N. S. (2019a). Interaction studies on bacterial stringent response protein RelA with uncharged tRNA provide evidence for its prerequisite complex for ribosome binding. Curr. Genet. 65, 1173–1184. doi: 10.1007/s00294-019-00966-y
Kushwaha, G. S., Oyeyemi, B. F., and Bhavesh, N. S. (2019b). Stringent response protein as a potential target to intervene persistent bacterial infection. Biochimie 165, 67–75. doi: 10.1016/j.biochi.2019.07.006
Leonarski, F., D’Ascenzo, L., and Auffinger, P. (2017). Mg2+ ions: do they bind to nucleobasenitrogens? Nucleic Acids Res. 45, 987–1004. doi: 10.1093/nar/gkw1175
Liu, K., Myers, A. R., Pisithkul, T., Claas, K. R., Satyshur, K. A., Amador-Noguez, D., et al. (2015). Molecular mechanism and evolution of guanylate kinase regulation by (p)ppGpp. Mol. Cell 57, 735–749. doi: 10.1016/j.molcel.2014.12.037
Manav, M. C., Beljantseva, J., Bojer, M. S., Tenson, T., Ingmer, H., Hauryliuk, V., et al. (2018). Structural basis for (p)ppGpp synthesis by the Staphylococcus aureus small alarmonesynthetaseRelP. J. Biol. Chem. 293, 3254–3264. doi: 10.1074/jbc.RA117.001374
Marcou, G., and Rognan, D. (2007). Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. J. Chem. Inf. Model. 47, 195–207. doi: 10.1021/ci600342e
Mechold, U., Murphy, H., Brown, L., and Cashel, M. (2002). Intramolecular regulation of the opposing (p)ppGpp catalytic activities of Rel(Seq), the Rel/Spo enzyme from Streptococcus equisimilis. J. Bacteriol. 184, 2878–2888. doi: 10.1128/JB.184.11.2878-2888.2002
Mlýnský, V., and Bussi, G. (2018). Exploring RNA structure and dynamics through enhanced sampling simulations. Curr. Opin. Struct. Biol. 49, 63–71. doi: 10.1016/j.sbi.2018.01.004
Molodtsov, V., Sineva, E., Zhang, L., Huang, X., Cashel, M., Ades, S. E., et al. (2018). Allosteric effector ppGppPotentiates the inhibition of transcript initiation by DksA. Mol. Cell 69, 828.e5–839.e5. doi: 10.1016/J.MOLCEL.2018.01.035
Moodie, S. L., and Thornton, J. M. (1993). A study into the effects of protein binding on nucleotide conformation. Nucleic Acids Res. 21, 1369–1380. doi: 10.1093/nar/21.6.1369
Pausch, P., Steinchen, W., Wieland, M., Klaus, T., Freibert, S. A., Altegoer, F., et al. (2018). Structural basis for (p)ppGpp-mediated inhibition of the GTPaseRbgA. J. Biol. Chem. 293, 19699–19709. doi: 10.1074/jbc.RA118.003070
Pechlaner, M., and Sigel, R. K. O. (2012). Characterization of metal ion-nucleic acid interactions in solution. Met. Ions Life Sci. 10, 1–42. doi: 10.1007/978-94-007-2172-2-1
Potrykus, K., and Cashel, M. (2008). (p)ppGpp: still magical? Annu. Rev. Microbiol. 62, 35–51. doi: 10.1146/annurev.micro.62.081307.162903
Primm, T. P., Andersen, S. J., Mizrahi, V., Avarbock, D., Rubin, H., Barry, C. E., et al. (2000). The stringent response of Mycobacterium tuberculosis is required for long-term survival. J. Bacteriol. 182, 4889–4898.
Prusa, J., Zhu, D. X., and Stallings, C. L. (2018). The stringent response and Mycobacterium tuberculosis pathogenesis. Pathog. Dis. 1:fty054. doi: 10.1093/femspd/fty054
Rojas, A. M., Ehrenberg, M., Andersson, S. G., and Kurland, C. G. (1984). ppGpp inhibition of elongation factors Tu, G and Ts during polypeptide synthesis. Mol. Gen. Genet. 197, 36–45.
Ronneau, S., and Hallez, R. (2019). Make and break the alarmone: regulation of (p)ppGppsynthetase/hydrolase enzymes in bacteria. FEMS Microbiol. Rev. 43, 389–400. doi: 10.1093/femsre/fuz009
Roos, K., Wu, C., Damm, W., Reboul, M., Stevenson, J. M., Lu, C., et al. (2019). OPLS3e: extending force field coverage for drug-like small molecules. J. Chem. Theory Comput. 15, 1863–1874. doi: 10.1021/acs.jctc.8b01026
Ross, W., Vrentas, C. E., Sanchez-Vazquez, P., Gaal, T., and Gourse, R. L. (2013). The magic spot: a ppgpp binding site on E. coli RNA polymerase responsible for regulation of transcription initiation. Mol. Cell 50, 420–429. doi: 10.1016/J.MOLCEL.2013.03.021
Rymer, R. U., Solorio, F. A., Tehranchi, A. K., Chu, C., Corn, J. E., Keck, J. L., et al. (2012). Binding mechanism of metalṡNTP substrates and stringent-response alarmones to bacterial DnaG-type primases. Structure 20, 1478–1489. doi: 10.1016/j.str.2012.05.017
Schreyer, A., and Blundell, T. (2009). CREDO: a protein-ligand interaction database for drug discovery. Chem. Biol. Drug Des. 73, 157–167. doi: 10.1111/j.1747-0285.2008.00762.x
Shyp, V., Tankov, S., Ermakov, A., Kudrin, P., English, B. P., Ehrenberg, M., et al. (2012). Positive allosteric feedback regulation of the stringent response enzyme RelA by its product. EMBO Rep. 13, 835–839. doi: 10.1038/embor.2012.106
Smith, A. K., Lockhart, C., and Klimov, D. K. (2016). Does replica exchange with solute tempering efficiently sample Aβ peptide conformational ensembles? J. Chem. Theory Comput. 12, 5201–5214. doi: 10.1021/acs.jctc.6b00660
Song, H., Dharmasena, M. N., Wang, C., Shaw, G. X., Cherry, S., Tropea, J. E., et al. (2020). Structure and activity of PPX/GppA homologs from Escherichia coli and Helicobacter pylori. FEBS J. 287, 1865–1885. doi: 10.1111/febs.15120
Šponer, J., Banáš, P., Jurečka, P., Zgarbová, M., Kührová, P., Havrila, M., et al. (2014). Molecular dynamics simulations of nucleic acids. From Tetranucleotides to the ribosome. J. Phys. Chem. Lett. 5, 1771–1782. doi: 10.1021/jz500557y
Srivatsan, A., and Wang, J. D. (2008). Control of bacterial transcription, translation and replication by (p)ppGpp. Curr. Opin. Microbiol. 11, 100–105. doi: 10.1016/j.mib.2008.02.001
Steinchen, W., and Bange, G. (2016). The magic dance of the alarmones (p)ppGpp. Mol. Microbiol. 101, 531–544. doi: 10.1111/mmi.13412
Steinchen, W., Schuhmacher, J. S., Altegoer, F., Fage, C. D., Srinivasan, V., Linne, U., et al. (2015). Catalytic mechanism and allosteric regulation of an oligomeric (p)ppGppsynthetase by an alarmone. Proc. Natl. Acad. Sci. U. S. A. 112, 13348–13353. doi: 10.1073/pnas.1505271112
Stern, N., Major, D. T., Gottlieb, H. E., Weizman, D., and Fischer, B. (2010). What is the conformation of physiologically-activedinucleoside polyphosphates in solution? Conformational analysis of free dinucleoside polyphosphates by NMR and molecular dynamics simulations. Org. Biomol. Chem. 8, 4637–4652. doi: 10.1039/c005122e
Stierand, K., Maaß, P. C., and Rarey, M. (2006). Molecular complexes at a glance: automated generation of two-dimensional complex diagrams. Bioinformatics 22, 1710–1716. doi: 10.1093/bioinformatics/btl150
Sun, G., Voigt, J. H., Filippov, I. V., Marquez, V. E., and Nicklaus, M. C. (2004). PROSIT: pseudo-rotational online service and interactive tool, applied to a conformational survey of nucleosides and nucleotides. J. Chem. Inf. Comput. Sci. 44, 1752–1762. doi: 10.1021/ci049881
Sun, G., Voigt, J. H., Marquez, V. E., and Nicklaus, M. C. (2005). Prosit, an online service to calculate pseudorotational parameters of nucleosides and nucleotides. Nucleos. Nucleot. Nucleic Acids. 24, 1029–1032. doi: 10.1081/NCN-200059757
Tamman, H., Van Nerom, K., Takada, H., Vandenberk, N., Scholl, D., Polikanov, Y., et al. (2020). A nucleotide-switch mechanism mediates opposing catalytic activities of Rel enzymes. Nat. Chem. Biol. 16, 834–840. doi: 10.1038/s41589-020-0520-2
Wang, B., Dai, P., Ding, D., Del Rosario, A., Grant, R. A., Pentelute, B. L., et al. (2019). Affinity-based capture and identification of protein effectors of the growth regulator ppGpp. Nat. Chem. Biol. 15, 141–150. doi: 10.1038/s41589-018-0183-4
Wang, B., Wang, Z., Javornik, U., Xi, Z., and Plavec, J. (2017). Computational and NMR spectroscopy insights into the conformation of cyclic di-nucleotides. Sci. Rep. 7:16550. doi: 10.1038/s41598-017-16794-4
Wang, J. D., Sanders, G. M., and Grossman, A. D. (2007). Nutritional control of elongation of DNA replication by (p)ppGpp. Cell 128, 865–875. doi: 10.1016/j.cell.2006.12.043
Wang, L., and Berne, B. J. (2018). Efficient sampling of puckering states of monosaccharides through replica exchange with solute tempering and bond softening. J. Chem. Phys. 149:072306. doi: 10.1063/1.5024389
Winther, K. S., Roghanian, M., and Gerdes, K. (2018). Activation of the stringent response by loading of RelA-tRNA complexes at the ribosomal a-site. Mol. Cell 70, 95.e4–105.e4. doi: 10.1016/j.molcel.2018.02.033
Yang, Y. I., Shao, Q., Zhang, J., Yang, L., and Gao, Y. Q. (2019). Enhanced sampling in molecular dynamics. J. Chem. Phys. 151:070902. doi: 10.1063/1.5109531
Zhang, Y., Zbornikova, E., Rejman, D., and Gerdes, K. (2018). Novel (p)ppGpp binding and metabolizing proteins of Escherichiacoli. mBio 9:e2188-17. doi: 10.1128/mBio.02188-17
Zhang, Y. E., Bærentsen, R. L., Fuhrer, T., Sauer, U., Gerdes, K., and Brodersen, D. E. (2019). (p)ppGpp Regulates a bacterial nucleosidase by an allosteric two-domain switch. Mol. Cell 74, 1239.e4–1249.e4. doi: 10.1016/j.molcel.2019.03.035
Keywords: stringent response, (p)ppGpp, secondary messenger nucleotide, interaction analysis, structural dynamics, (p)ppGpp synthetase
Citation: Kushwaha GS, Patra A and Bhavesh NS (2020) Structural Analysis of (p)ppGpp Reveals Its Versatile Binding Pattern for Diverse Types of Target Proteins. Front. Microbiol. 11:575041. doi: 10.3389/fmicb.2020.575041
Received: 22 June 2020; Accepted: 06 October 2020;
Published: 05 November 2020.
Edited by:
Katarzyna Potrykus, University of Gdańsk, PolandReviewed by:
Kenneth A. Satyshur, University of Wisconsin–Madison, United StatesCopyright © 2020 Kushwaha, Patra and Bhavesh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gajraj Singh Kushwaha, Z2FqcmFqc2tAZ21haWwuY29t; Neel Sarovar Bhavesh, bmVlbHNiQGljZ2ViLnJlcy5pbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.