 Vincent Cattoir1,2,3*
Vincent Cattoir1,2,3* Annabelle Pourbaix4Mélanie Magnan4Françoise Chau4Victoire de Lastours4,5Brice Felden3Bruno Fantin4,5François Guérin6,7
Annabelle Pourbaix4Mélanie Magnan4Françoise Chau4Victoire de Lastours4,5Brice Felden3Bruno Fantin4,5François Guérin6,7- 1CHU de Rennes, Service de Bactériologie-Hygiène Hospitalière, Rennes, France
- 2Centre National de Référence sur la Résistance aux Antibiotiques (laboratoire associé ‘Entérocoques’), Rennes, France
- 3Inserm, Bacterial Regulatory RNAs and Medicine - UMR_S 1230, Rennes, France
- 4IAME, UMR-1137, Inserm and Université de Paris Diderot, Paris, France
- 5Service de Médecine Interne, Hôpital Beaujon, Assistance Publique – Hôpitaux de Paris, Paris, France
- 6CHU de Caen, Service de Microbiologie, Caen, France
- 7Université de Caen Normandie, EA4655, Caen, France
Fosfomycin resistance in Escherichia coli results from chromosomal mutations or acquisition of plasmid-mediated genes. Because these mechanisms may be absent in some resistant isolates, we aimed at decipher the genetic basis of fosfomycin resistance in E. coli. Different groups of isolates were studied: fosfomycin-resistant mutants selected in vitro from E. coli CFT073 (MIC = 1 mg/L) and two groups (wildtype and non-wildtype) of E. coli clinical isolates. Single-nucleotide allelic replacement was performed to confirm the implication of novel mutations into resistance. Induction of uhpT expression by glucose-6-phosphate (G6P) was assessed by RT-qPCR. The genome of all clinical isolates was sequenced by MiSeq (Illumina). Two first-step mutants were obtained in vitro from CFT073 (MICs, 128 mg/L) with single mutations: G469R in uhpB (M3); F384L in uhpC (M4). Second-step mutants (MICs, 256 mg/L) presented additional mutations: R282V in galU (M7 from M3); Q558∗ in lon (M8 from M4). Introduction of uhpB or uhpC mutations by site-directed mutagenesis conferred a 128-fold increase in fosfomycin MICs, whereas single mutations in galU or lon were only responsible for a 2-fold increase. Also, these mutations abolished the induction of uhpT expression by G6P. All 14 fosfomycin-susceptible clinical isolates (MICs, 0.5–8 mg/L) were devoid of any mutation. At least one genetic change was detected in all but one fosfomycin-resistant clinical isolates (MICs, 32 – >256 mg/L) including 8, 17, 18, 5, and 8 in uhpA, uhpB, uhpC, uhpT, and glpT genes, respectively. In conclusion, novel mutations in uhpB and uhpC are associated with fosfomycin resistance in E. coli clinical isolates.
Introduction
Fosfomycin, a phosphonic acid derivative discovered in 1969, has become the first-choice antibiotic for the ‘single-dose’ oral treatment of uncomplicated urinary tract infections (UTIs) (Falagas et al., 2016). It is a bactericidal antibiotic with a broad spectrum of activity that interferes with the first step of peptidoglycan synthesis in both Gram-positive and Gram-negative bacteria (Castaneda-Garcia et al., 2013; Falagas et al., 2016). As a phosphoenolpyruvate analog, fosfomycin inhibits the cytosolic UDP-N-acetylglucosamine enolpyruvyltransferase (also named MurA) by covalent binding to key residue C115 of the enzyme active site, preventing the formation of N-acetylmuramic acid (Kahan et al., 1974). This low-molecular-weight antibiotic enters into the bacterial cell via two transport uptake systems: the glycerol-3-phosphate permease (encoded by glpT) constitutively expressed and, the hexose phosphate uptake transporter (encoded by uhpT) inducible by extracellular glucose-6-phosphate (G6P) (Castaneda-Garcia et al., 2013). While transcription of glpT and uhpT is regulated by glpR and uhpABC, respectively, their expression also requires high levels of cyclic AMP (cAMP) combined with, as a complex, the cAMP receptor protein (CRP) (Castaneda-Garcia et al., 2013). cAMP levels depend on the activity of CyaA adenyl cyclase and are regulated by the phosphotransferase enzyme PtsI (Castaneda-Garcia et al., 2013).
Despite its widespread clinical use for many years in several countries, the prevalence of fosfomycin resistance is still low among E. coli clinical isolates, usually below 3% (4,5). Concerning multi-drug-resistant (MDR) isolates as ESBL-producing E. coli, levels of susceptibility to fosfomycin remain as high as 80% (Falagas et al., 2016, 2019; Aghamali et al., 2019). By contrast, the selection of fosfomycin-resistant mutants is much easier under in vitro conditions at high mutation frequencies (ca. 10–8–10–7) (Karageorgopoulos et al., 2012). This paradox is partially due to a significant resistance-associated fitness cost with decrease in vitro rate and attenuated virulence in vivo (Marchese et al., 2003; Nilsson et al., 2003; Pourbaix et al., 2017), and higher fosfomycin activity under urinary tract physiological conditions (i.e., urine acidification and anaerobiosis counterbalanced by negligible amounts of urinary G6P) that enhance expression of GlpT and UhpT (Martin-Gutierrez et al., 2018; Pourbaix et al., 2019).
Due to the unique mechanism of action of fosfomycin, there are no cross-resistances with other antibacterial agents (Falagas et al., 2016; Silver, 2017). However, three specific mechanisms of fosfomycin resistance were described in E. coli: impaired drug uptake, enzymatic drug inactivation and target modification (Cattoir and Guérin, 2018). Reduced drug uptake is the most frequent resistance mechanism for in in vitro mutants and clinical isolates. It results from chromosomal mutations that alter the function or expression of GlpT and/or UhpT transporters. These mutations (mutations, insertions, deletions) can arise either in structural genes (i.e., glpT and uhpT) or in genes coding for regulators (i.e., uhpA, cyaA, and ptsI) (Castaneda-Garcia et al., 2013; Silver, 2017). More recently, there is the emergence of plasmid-mediated metallo-dependent enzymes (including FosA, FosB, and FosX) that inactivate the drug, of which FosA3 is, by far, the most frequently variant in E. coli (Yang et al., 2019). Much more uncommon, fosfomycin resistance can be mediated by qualitative and/or quantitative modifications of MurA (Silver, 2017).
The aim of this study was to (1) investigate the genetic basis of fosfomycin resistance in E. coli mutants selected in vitro that had no mutations in genes previously reported to be involved in resistance (i.e., glpT, uhpT, uhpA, murA, cyaA, and ptsI), (2) demonstrate experimentally the role of novel mutations identified in four different genes, and (3) determine their prevalence among a collection fosfomycin-resistant E. coli clinical isolates recently collected in France.
Materials and Methods
Bacterial Strains
Three different groups of E. coli strains were used in this study (Tables 1, 2). The first group consisted of fosfomycin-resistant mutants (CFT073_M3 to CFT073_M8) obtained from the parental strain E. coli CFT073 (uropathogenic strain belonging to phylogroup B2) (Welch et al., 2002) after serial passages on Mueller–Hinton (MH) medium (Difco, Becton Dickinson, Rungis, France) containing increased concentrations of fosfomycin (from 32 to 128 mg/L) in the presence of G6P (25 mg/L). The two other groups consisted of E. coli epidemiologically unrelated clinical isolates (wildtype and non-wildtype phenotype of resistance to fosfomycin, according to the epidemiological cut-off established at 8 mg/L) responsible for UTIs in patients hospitalized in two French university hospitals between 2012 and 2017.
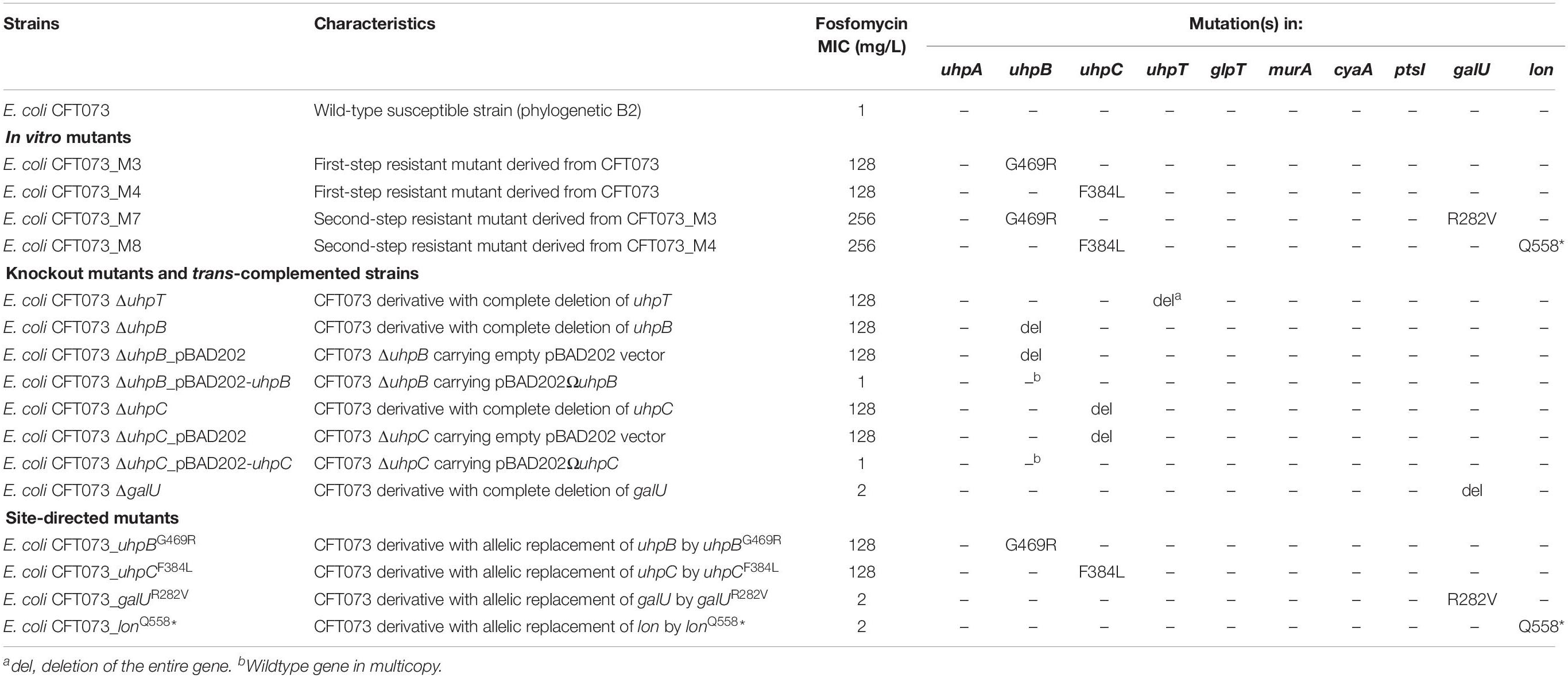
Table 1. Genotypic and phenotypic characteristics and susceptibility to fosfomycin of isogenic strains derived from E. coli CFT073.
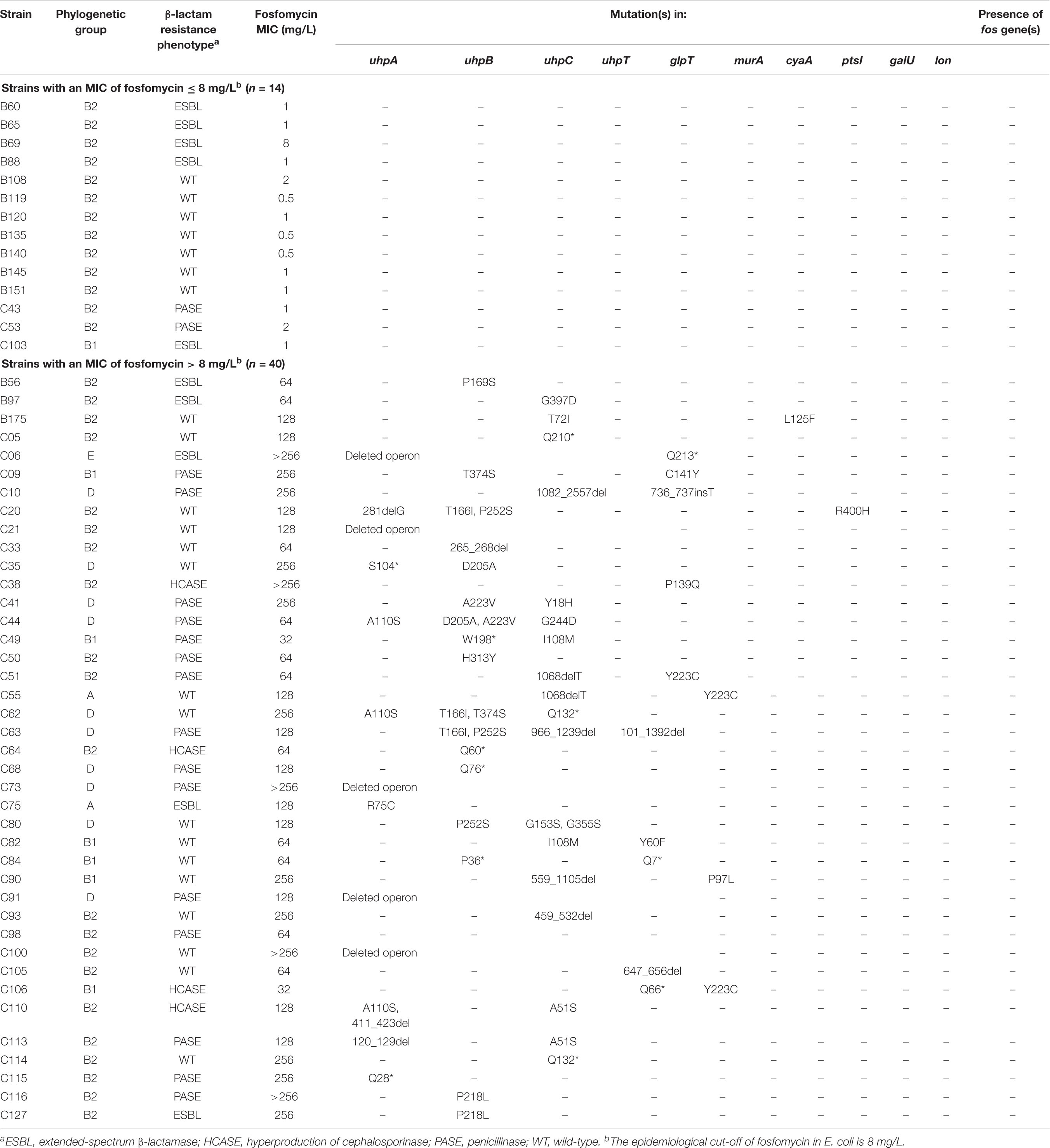
Table 2. Phenotypic and genotypic characteristics of clinical isolates.
Bacterial strains were routinely grown at 35°C in Luria–Bertani (LB) broth or agar supplemented with appropriate antibiotics, unless otherwise specified. When required, E. coli were grown on media supplemented with 100 mg/L ampicillin, 40 mg/L kanamycin or 25 mg/L chloramphenicol.
Antimicrobial Susceptibility Testing
MICs of fosfomycin were determined by using the agar dilution reference method described by the European Committee on Antimicrobial Susceptibility Testing1. Briefly, bacterial suspension was prepared to match the turbidity of the 0.5 McFarland in sterile physiological water (ca. 108 CFU/mL). Agar dilution was performed using MH agar plates containing 25 mg/L G6P. Cell suspensions were further diluted in MH broth and were delivered onto plates using a Steer replicator, which delivered ca. 104 CFU for each isolate. Concentrations tested ranged from 256 to 0.125 mg/L. E. coli ATCC 25922 and Pseudomonas aeruginosa ATCC 25923 were used as control strains and were run in parallel with every experiment. Each MIC determination was performed at least three times. The current susceptibility breakpoint of fosfomycin for Enterobacteriaceae is a MIC ≤ 32 mg/L according to the EUCAST guidelines (see text footnote 1).
In vitro Bacterial Growth Rate
Growth rates at 35°C were measured in Luria–Bertani (LB) broth and Nutrient broth (NB) at pH 5 or 7 as well as in sterile-filtered pooled human male urine (pH = 6.5). The bacteria were grown aerobically overnight at 35°C and approximately 105 colony-forming units (CFUs) were inoculated into 200 μL of growth medium on a bioscreen plate and the optical density at 600 nm was read each 5 min for 24 h with a multimode reader Infinite 200 Pro® (Tecan, Männendorf, Switzerland). Maximal growth rate (MGR) of each strain was calculated as the inflexion point of first by-product of the curve of growth. For each strain and condition, MGR was measured in duplicate in three separate experiments.
Construction of the Knockout Mutants
The disruption of the genes coding for putative transporters (glpT and uhpT) and their regulators (uhpA, uhpB, and uhpC) were performed using the method previously described, with some modifications, using the Red helper plasmid pKOBEG (Datsenko and Wanner, 2000; Derbise et al., 2003). This vector is a low-copy-number plasmid that contains a gene for chloramphenicol resistance selection, a temperature-sensitive origin of replication, and a gene encoding a recombinase. Briefly, pKOBEG was first introduced into CFT073 competent cells by electroporation, and transformants were selected on LB agar with chloramphenicol (25 mg/L) after incubation for 24 h at 30°C. A selectable kanamycin resistance cassette (flanked by flippase recognition target [FRT] sequences) was amplified by PCR using DNA of pKD4 plasmid as the template. The primers used included 5′ extensions with homology for the candidate genes (around 50 bases) (Table 3). The PCR product was introduced into the pKOBEG-harboring CFT073 by electroporation, and after homologous recombination, the disruption of the candidate gene was obtained. Selected clones were cured for the pKOBEG plasmid following a heat shock, creating the kanamycin-resistant variant. In order to have deletion mutants free of the antibiotic marker, strains then were transformed with the pCP20_Gm plasmid, which is able to express the FLP nuclease that recognizes the FRT sequences present on either side of the kan gene (Doublet et al., 2008). Lastly, the mutants were verified by Sanger sequencing.
Table 3. Deoxynucleotide primers used in the study.
Construction of Trans-Complemented Strains
The uhpB and uhpC wildtype genes were amplified by PCR using specific primers (Table 3) and each amplicon was TA cloned into the overexpression plasmid, pBAD202 directional TOPO (Invitrogen, Courtaboeuf, France). E. coli TOP10 cells (Invitrogen) carrying pBAD202 recombinants containing correctly oriented inserts were selected on LB plates with 40 mg/L kanamycin. After purification, recombinant plasmids pBAD202ΩuhpB and pBAD202ΩuhpC were used to transform by electroporation ΔuhpB and ΔuhpC mutants, respectively.
Site-Directed Mutagenesis
Single-nucleotide allelic replacement was carried out using the suicide vector pDS132 in order to confirm the role of novel mutations (Philippe et al., 2004). The cloning steps of the desired gene alleles into pDS132 were performed in E. coli DH5αλpir strain to allow replication of the plasmid. The recombinant plasmids were then purified and introduced in E. coli CFT073 by electro-transformation. The first step of allelic exchange was selection of plasmid integration into the recipient chromosome by plating cells on chloramphenicol-containing LB plates. After overnight growth at 35°C, one colony was picked, diluted in 10 mM MgSO4 solution, and serial dilutions were plated on LB agar plates with 5% sucrose and without NaCl. This plating step allowed selection of plasmid excision from the chromosome by a second cross-over. After overnight incubation at 35°C, 100 clones were streaked on chloramphenicol-containing LB agar plates and on LB agar with 5% sucrose and without NaCl. Several clones were screened by PCR-sequencing in order to identify those carrying the desired allele.
RNA Extraction and RT-qPCR
The levels of expression of uhpT were determined by RT-qPCR using specific primers (Table 3). E. coli cells were grown for 24 h in LB broth, and the cells were harvested and washed twice with M9 minimum salt solution as previously described (Ohkoshi et al., 2017). The suspended cells were used to inoculate to M9 minimum salt solution with or without 0.2% G6P supplementation and incubated for 30 min at 35°C. Total RNAs were extracted from all clinical isolates using the Direct-zol RNA miniprep kit (Zymo Research, Irvine, CA, United States). Residual chromosomal DNA was removed by treating samples with the Turbo DNA-free kit (Life Technologies, Saint-Aubin, France). Samples were quantified using the BioSpec-nano spectrophotometer (Shimadzu, Noisiel, France), and the integrity was assessed using the Agilent 2100 bioanalyzer according to the manufacturer’s instructions. cDNA was synthesized from total RNA (∼25 ng) using the QuantiFast SYBR green RT-PCR kit (Qiagen), and transcript levels were determined by the ΔΔ threshold cycle (ΔΔCt) method using the rrsA (16S rRNA) gene as a housekeeping control gene (Table 3).
WGS and Bioinformatic Analysis
Genomic DNA was isolated using the using the Quick-DNA fungal/bacterial miniprep kit (Zymo Research, Irvine, CA, United States). DNA libraries were prepared using the NEBNext Ultra DNA library prep kit for Illumina (New England Biolabs, Ipswich, MA, United States) and sequenced as paired-end reads (2 × 300 bp) using an Illumina MiSeq platform and the MiSeq reagent kit version 3. The Illumina reads were assembled using the CLC Genomics Workbench software (Qiagen). The annotation of chromosome and plasmids was performed using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP)2. The nucleotide sequences were also submitted to ResFinder server3 (version 3.1) to identify fosfomycin resistance mutations and acquired genes. Raw and processed data generated in this study were deposited in GenBank as bioproject no. PRJNA625505.
Results
In vitro Fosfomycin-Resistant Mutants of E. coli CFT073
Four different mutants were selected in vitro from the parental strain E. coli CFT073, including two single-step and two second-step mutants (Table 1). The two first-step mutants harbored only one mutation each: CFT073_M3 possessed a non-synonymous mutation in uhpB (leading to the substitution G469R) and CFT073_M4 had a non-synonymous mutation in uhpC (leading to the substitution F384L). Both mutations were associated with a 128-fold increase in fosfomycin MICs (Table 1), and mutants were categorized as resistant according to the EUCAST breakpoints. Concerning the two-step mutants, CFT073_M7 and CFT073_M8 were obtained on agar plates supplemented with 128 mg/L of fosfomycin from CFT073_M3 and CFT073_M4, respectively. Both exhibited a two-fold increase in fosfomycin MICs (256 mg/L), and harbored one more mutation each: a non-synonymous mutation in galU (leading to the substitution R282V) in CFT073_M7, and a nonsense mutation in lon (leading to Q558∗) in CFT073_M8 (Table 1).
Overall, bacterial growth rates were reduced as the pH was lower and exhibited their lowest levels in urine (Figure 1). MGR of CFT073_M4 was significantly decreased as compared to that of CFT073 (P < 0.05, unpaired t test), except in urine (Figure 1). Interestingly, CFT073_M7 had a significant decreased MGR as compared with CFT073 in LB and NB at pH 5 (P < 0.01, unpaired t-test) and also in urine at pH 6.5 (P < 0.001, unpaired t test) (Figure 1). There was no difference in MGRs for CFT073_M3 and CFT073_M8 (Figure 1).
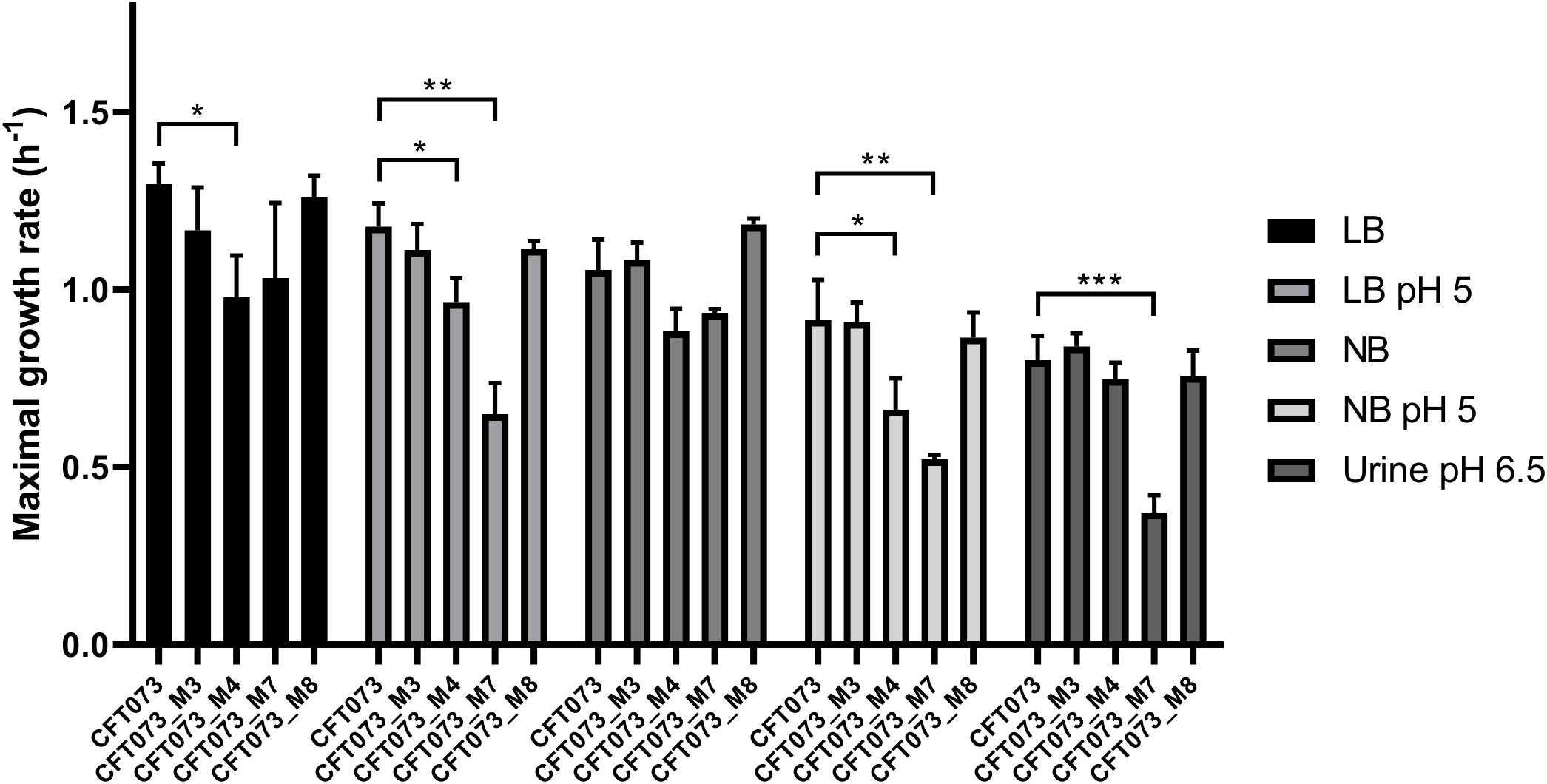
Figure 1. Maximal growth rates of CFT073 and in vitro mutants in Luria–Bertani (LB), LB pH 5, Nutrient broth (NB), NB pH 5 and urine (pH 6.5). Statistical comparison was performed using the unpaired t-test. *P < 0.05; **P < 0.01; ***P < 0.001.
Role of the Novel Mutations Into Fosfomycin Resistance
To confirm the role of uhpB, uhpC, galU, and lon and their corresponding mutations in fosfomycin resistance, several approaches were used. First, knockout mutants were constructed, as well as their corresponding trans-complemented strains. Both ΔuhpB and ΔuhpC mutants were resistant to fosfomycin, with MICs at 128 mg/L (Table 1). As expected, the trans-complementation of ΔuhpB and ΔuhpC mutants with their respective isogenic copies restored the fosfomycin susceptibility, with MICs at 1 mg/L (Table 2). Whereas we failed to construct a Δlon mutant, a deleted mutant was obtained for galU that only exhibited a two-fold increase in MICs of fosfomycin (Table 2).
Second, we constructed site-directed mutants of CFT073 by single-nucleotide allelic replacement, to introduce the same mutations than those observed in mutants obtained in vitro by antibiotic selection. The introduction of a unique mutation in uhpB (G469R) or in uhpC (F384L) was responsible for a significant increase in MICs (from 1 to 128 mg/L) in both cases (Table 1), confirming experimentally their role into fosfomycin resistance. The unique mutation in galU (R282V) conferred a two-fold increase in MIC of fosfomycin as did the sole mutation in lon (Q558∗) (Table 1). The latter results are consistent with the increase of MICs of fosfomycin in second-step mutants as compared to single-step mutants (256 vs. 128 mg/L, respectively).
To understand the mechanism(s) by which these mutations confer higher fosfomycin MICs, we compared by RT-qPCR the differential expression of uhpT in the absence or presence of 0.2% G6P. After induction, uhpT expression was strongly enhanced (244-fold ± 47) in the CFT073 parental strain, as expected, whereas it was significantly lower in all mutants M3 (1.1-fold ± 0.4), M4 (1.4-fold ± 0.1), M7 (1.2-fold ± 0.2), and M8 (1.5-fold ± 0.1) (all P < 0.007 by an unpaired t) (Figure 2). This lack of induction by G6P was also observed with deleted and site-directed mutants for uhpB and uhpC (Figure 2). The deletion of glpT in CFT073 had no significant effect on G6P-mediated induction of uhpT expression, as expected, and it was also the case in CFT073_galUR282V and CFT073_lonQ558* mutants (Figure 2). Surprisingly, the change in uhpT expression after G6P induction was significantly higher in galU-deleted mutant than in the parental strain (407 ± 33 vs. 244-fold ± 47; P = 0.0082) (Figure 2).
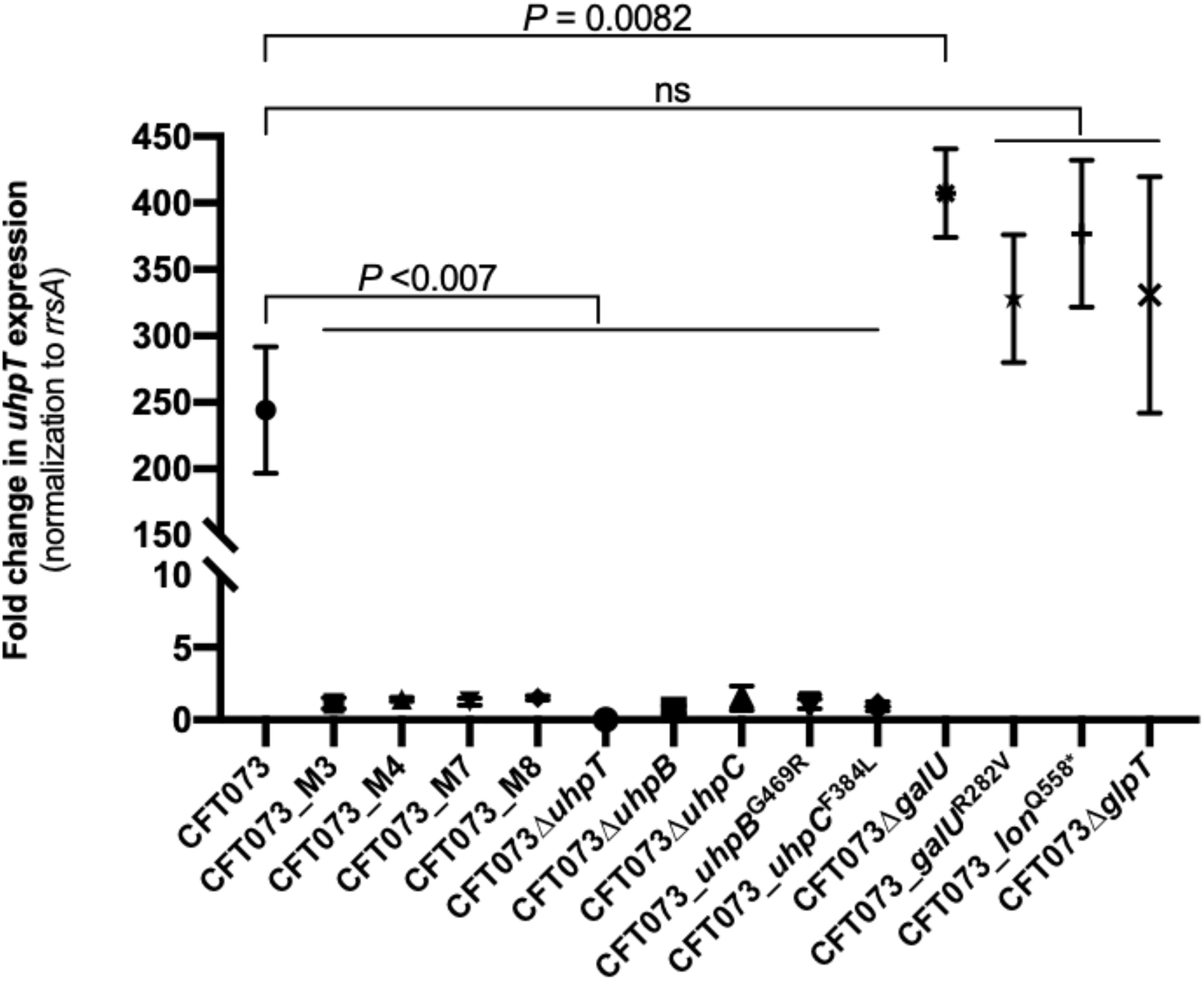
Figure 2. Changes in uhpT expression after induction with 0.2% of G6P in E. coli CFT073 parental strain and its derivative mutants. Transcript levels of uhpT are shown as relative values compared to those of rrsA (16S rRNA) gene. Data plotted correspond to the means and SDs of three biological replicates.
Prevalence of Novel Mutations in Fosfomycin-Resistant E. coli Clinical Isolates
To know if these mutations have been underestimated until now, we assessed their prevalence in a panel of 40 unrelated non-wildtype (MICs > 8 mg/L) E. coli clinical isolates (Table 2). We also studied a collection of 14 wildtype (MICs ≤ 8 mg/L) clinical isolates in which we verified the absence of mutations, as expected (Table 2).
Of the 40 non-wildtype isolates, no plasmid-mediated fosfomycin resistance genes (especially fosA3) were detected (Table 2). By contrast, at least one mutation/insertion/deletion was identified in almost all (n = 39) isolates, whereas only one isolate (C98, MIC at 64 mg/L) did not possess any change in uhpA, uhpB, uhpC, uhpT, glpT, murA, cyaA, ptsI, galU or lon genes (Table 2). Only two isolates (C49 and C106) were categorized as susceptible to fosfomycin (MICs at the susceptibility breakpoint, 32 mg/L) and harbored two mutations each (Table 2). Five isolates had a full deletion of the uhp operon, including one with one additional non-sense mutation in glpT (Q213∗). Besides these five cases, a genetic change was identified in uhpA, uhpB, uhpC, uhpT, glpT, cyaA, and ptsI in 8, 17, 18, 5, 8, 1, and 1 isolates, respectively (Table 2). Even though half of isolates presented several mutations in up to three genes, some unique mutations were sufficient to confer fosfomycin resistance (MICs ranging from 64 to >256 mg/L) such as in uhpB (Q60∗, Q76∗, 265_268del, P169S, P218L, and H313Y), uhpC (459_532del, Q132∗, Q210∗ and G397D), uhpA (Q28∗ and R75C), uhpT (647_656del), and glpT (P139Q) (Table 2). Finally, no mutations were detected in galU and lon genes among the 40 clinical isolates tested.
Discussion
The Uhp hexose phosphate transport pathway and its regulation are well described in E. coli (Kadner, 1973; Kadner and Winkler, 1973; Kadner and Shattuck-Eidens, 1983; Weston and Kadner, 1987, 1988; Island et al., 1992; Island and Kadner, 1993; Wright et al., 2000; Verhamme et al., 2001, 2002). UhpT is a member of the Major Facilitator Superfamily (MFS) containing 12 transmembrane protein segments, and it is responsible for the accumulation of G6P into the bacterial cells. The UhpT system is tightly controlled by the UhpABC phosphorelay system UhpABC, which is necessary for high-level expression of uhpT. UhpC is also an MFS member that shares approximately 30% amino acid sequence identity with UhpT. UhpC is a membrane-bound protein that senses external G6P in the periplasm and interacts with UhpB, stimulating its kinase activity. UhpB is a membrane-bound histidine kinase (HK) in a two-component system that possesses eight predicted transmembrane helices and a C-terminal cytoplasmic domain containing the conserved sequence elements common to HK proteins (i.e., the H-box around the phosphorylated histidine, the N-box, and the G-box comprising the ATP-binding and phosphate transfer region) (Parkinson and Kofoid, 1992). Upon interaction with UhpC, UhpB autophosphorylates the conserved histidine residue (His313), with subsequent phosphorylation at Asp54 of its cognate response regulator UhpA. Phosphorylated UhpA increases the affinity for its specific DNA binding sites, hence promoting transcription of uphT.
Many mutants defective in the hexose phosphate transport were isolated between 1970s and 1990s, but shortcomings can be found in these old studies, such as the imprecise position of the mutation/deletion/insertion due to the poor annotation of the uhp region sequence, the absence of determination of fosfomycin MICs, and the ‘artificial nature’ of many in vitro mutants that were obtained by transposon insertion (Mu, Tn10), or a resistance cassette (Kadner, 1973; Kadner and Shattuck-Eidens, 1983; Weston and Kadner, 1987, 1988; Island et al., 1992). Also, mutations/insertions can have different impacts on fosfomycin susceptibility since some of them do not impair uhpT expression and others confer constitutive expression (Weston and Kadner, 1987; Island and Kadner, 1993). Deleted mutants with a kanamycin resistance cassette in uhpA, uhpB, or uhpC from the E. coli BW25113 parental strain only conferred a modest increase in fosfomycin MICs to 8, 8, and 4 mg/L (Castaneda-Garcia et al., 2009), respectively, which is different from our findings. Altogether, it suggests that ‘artificial’ insertional mutants do not represent systematically how bacteria develop fosfomycin resistance.
Unexpectedly, we found here novel mutations in uhpB and uhpC in mutants, which are not often detected in fosfomycin-resistant clinical isolates. Indeed, fosfomycin resistance in E. coli clinical isolates is usually due to chromosomal mutations in uhpT, uhpA, glpT, murA, cyaA, and ptsI genes (Nilsson et al., 2003; Oteo et al., 2009; Takahata et al., 2010; Li et al., 2015b; Tseng et al., 2015; Ohkoshi et al., 2017; Lucas et al., 2018; Seok et al., 2020), and little is known about the involvement of mutations in other genes, especially those in uhpB and uhpC that have been exceptionally reported (Castaneda-Garcia et al., 2013).
Recently, mutations in uhpB or uhpC were described in E. coli BW25133-derived laboratory mutants ΔcyaA, ΔglpT-cyaA, ΔglpT-ptsI, and ΔptsI-cyaA recovered in vitro after time-kill experiments with fosfomycin (Ballestero-Tellez et al., 2017) and in two E. coli clinical isolates (Martin-Gutierrez et al., 2018). All the mutants were resistant to high levels to fosfomycin (MICs > 1,024 mg/L) and possessed the following mutations one or two mutations in uhpB (48del, W181∗, L255∗, and Q262∗) and uhpC (T27∗, T72P and 541_548del) (Ballestero-Tellez et al., 2017). In the two clinical isolates, one uhpB mutation (D205A) and three uhpC mutations (Y18H, G282D, T435A) were found in the first while two uhpC mutations (I14M, Q17Y) were found in the second (Martin-Gutierrez et al., 2018). We found here two mutations at the exact same position (D205A in UhpB and T72 in UhpC) of these previous studies (Figure 3), which is in favor of their role in fosfomycin resistance. In our study, uhpB mutations were distributed all along the 500-amino-acid-long protein in either periplasmic (n = 3), transmembrane (n = 5), or cytoplasmic (n = 6) regions, including one in the autophosphorylation H-box (H313Y) and another in the conserved G-box (G469R) that part of the ATP-binding domain (Figure 3). Concerning uhpC mutations, they were more frequently detected within the transmembrane segments (8/15) than into the cytoplasm (n = 6) or periplasm (n = 1) portions of the 439-amino-acid protein, suggesting that it could impair external G6P sensing through the membrane (Figure 3). Among fosfomycin-resistant clinical isolates, five had a full deletion of the uhp region (uhpA-uhpB-uhpC-uhpT), as reported (Lucas et al., 2018).
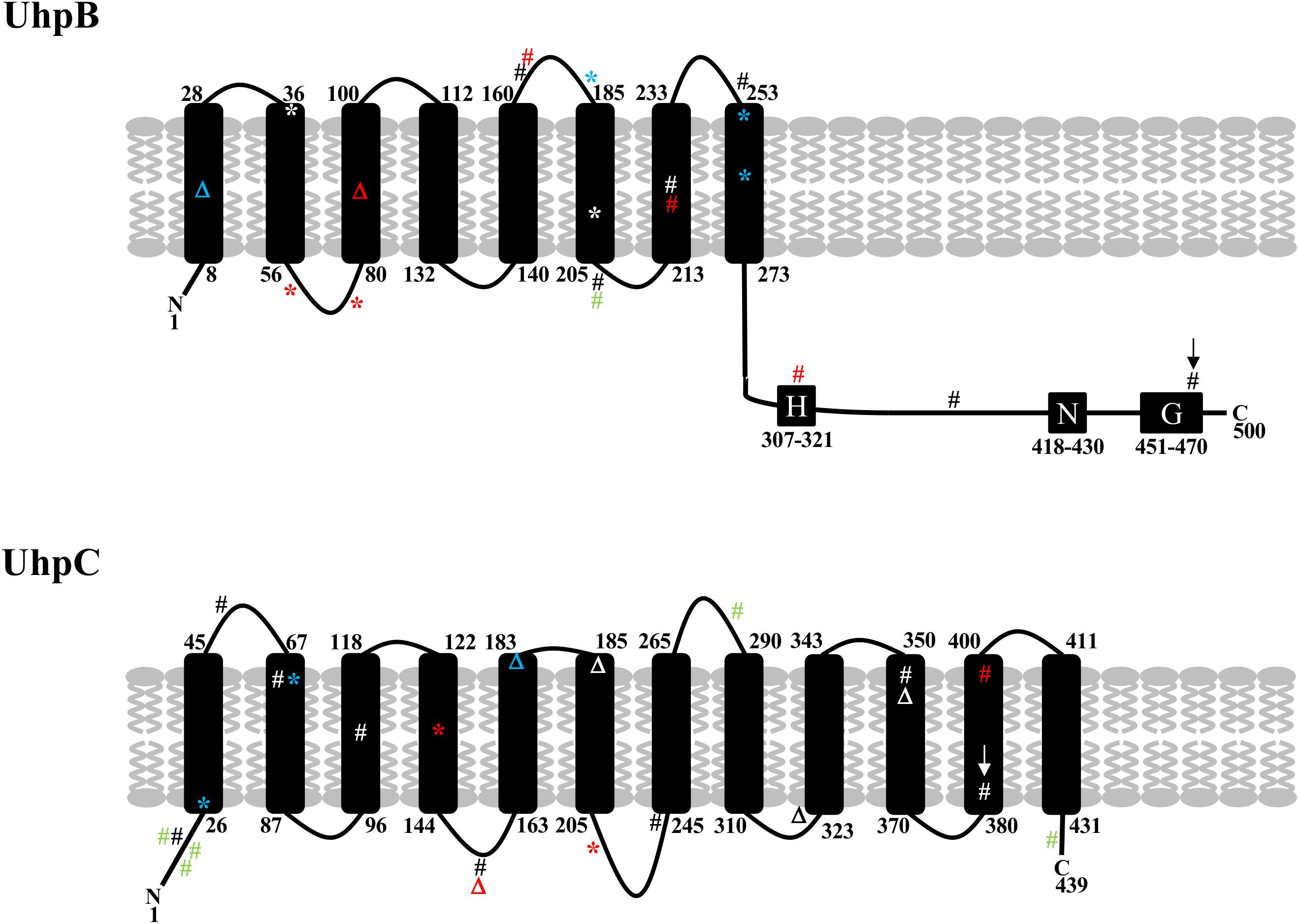
Figure 3. Schematic representation of the structure of UhpB and UhpC proteins with position of mutations identified in our study (in black and red) and previously described by Ballestero-Téllez (34) (in blue) and by Martin-Gutiérrez (10) (in green). Mutations in red correspond to single mutations associated with fosfomycin resistance in clinical isolates. Mutations with arrows correspond to single mutations (G469R in UhpB and F384L in UhpC) demonstrated experimentally to be responsible for fosfomycin resistance. Amino acids of transmembrane segments are indicated for UhpB (500 amino acids) and UhpC (439 amino acids). For UhpB, the putative conserved H-, N-, and G-boxes are also represented. #, non-synonymous mutation; *, non-sense mutation; Δ, deletion.
A majority of clinical isolates harbored uhpB and uhpC mutations that were widely distributed over the protein sequences. It is likely that all these mutations impact fosfomycin susceptibility differently, as described for insertion mutants exhibiting variable Uhp phenotypes (Weston and Kadner, 1988; Island et al., 1992; Island and Kadner, 1993). We also demonstrated that deletions and mutations in uhpB and uhpC were responsible for an absence of induction by G6P of uhpT expression, as described in several ΔuhpA, ΔuhpB, and ΔuhpC laboratory mutants and one clinical isolate with a truncated UhpA (Weston and Kadner, 1988; Island et al., 1992; Island and Kadner, 1993; Wright et al., 2000; Lucas et al., 2018). This confirms the role of UhpB and UhpC as G6P-response regulators required for the induction of uhpT expression. In addition, it appears that the mutation in uhpC (leading to the substitution F384L) also alters in vitro bacterial growth rate in LB and NB (regardless the pH) but not in urine, suggesting that it may occur in vivo.
Besides uhpB and uhpC mutations, two novel mutations were also identified in the two-step in vitro mutants. The first mutation occurred in galU that codes for a 302-amino-acid-long protein named UTP-glucose-1-phosphate uridylyltransferase, which catalyzes synthesis of UDP-D-glucose from UTP and α-D-glucose 1-phosphate (Weissborn et al., 1994). It is a central precursor for synthesis of cell surface carbohydrates, colanic acid, trehalose, cellulose, capsule- and membrane-derived oligosaccharides, and also has a major role in galactose metabolism (Ebrecht et al., 2015). Then, the deletion of galU has many consequences on different carbon metabolic pathways: for instance, they are unable to ferment galactose and fail to incorporate glucose and galactose into bacterial cell membranes, resulting in the incomplete synthesis of lipopolysaccharides (Fukasawa et al., 1962; Sundararajan et al., 1962). Also, the absence of galU leads to a reduced level of TolC into the outer membrane (Sharma et al., 2009), which might be related to antibiotic susceptibility. Here, we identified a non-synonymous mutation (R282V) in the C-terminal region of GalU that is outside the enzyme active site formed by the key residues T20, R21, and K202 (Ebrecht et al., 2015). Then, it is difficult to explain the implication of R282V mutation into the fosfomycin MIC two-fold increase. Note that it seems that this mutation also impacts on bacterial fitness when grown in acidic pH or in urine, suggesting that it may be difficult to develop in vivo.
The second mutation appeared in lon coding for an ATP-dependent serine protease that plays a major role in protein quality control, degrading incorrect proteins, and has an important role into many biological processes in bacteria (Tsilibaris et al., 2006). It degrades abnormal and misfolded proteins, but has also regulatory proteins as substrates, such as MarA and SoxS (Griffith et al., 2004). Here, we identified a lon mutation giving rise to a premature stop codon (Q558∗), and then a truncated protein, probably not functional. Indeed, with a length of 784 amino acids in E. coli, a large part of the C-terminal domain is lacking (Amerik et al., 1991). Therefore, we can assume that this truncated protein is inactive since the Ser679-Lys722 catalytic dyad is absent (Botos et al., 2004). Interestingly, it was demonstrated that mutations in lon were implicated in the development of multiple antibiotic resistance phenotype related to the efflux pump system AcrAB-TolC, and to the OmpF porin (Nicoloff et al., 2006, 2007; Duval et al., 2009; Nicoloff and Andersson, 2013; Bhaskarla et al., 2016). MarA, SoxS, and Rob, positively control the expression of acrAB, tolC, and micF, and micF regulatory RNA post-transcriptionally represses the translation of ompF mRNA (Li et al., 2015a). In a lon mutant, the accumulation of MarA and SoxS could enhance the micF-mediated inhibition of the OmpF production, that could impact on fosfomycin activity since OmpF can facilitate the spontaneous diffusion of the antibiotic across the outer membrane (Golla et al., 2019).
In conclusion, we demonstrated here experimentally the role of novel mutations in four genes implicated in fosfomycin resistance, and the prevalence of uhpB and uhpC mutations among fosfomycin-resistant E. coli clinical isolates.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
VC and FG conceptualized the study. VC, AP, MM, FC, VL, BF, and FG contributed to methodology. VC and FG provided the formal analysis and visualization. VC, AP, MM, FC, VL, BF, BF, and FG carried out the investigation. VC, BF, and FG were responsible for the resources. VC, BF, BF, and FG wrote the manuscript. All authors read and approved the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We warmly thank Michel Auzou, Sébastien Galopin, and Mamadou Godet for technical assistance.
Footnotes
- ^ www.eucast.org
- ^ www.ncbi.nlm.nih.gov/genome/annotation_prok/
- ^ https://cge.cbs.dtu.dk/services/ResFinder/
References
Aghamali, M., Sedighi, M., Zahedi Bialvaei, A., Mohammadzadeh, N., Abbasian, S., Ghafouri, Z., et al. (2019). Fosfomycin: mechanisms and the increasing prevalence of resistance. J. Med. Microbiol. 68, 11–25. doi: 10.1099/jmm.0.000874
Amerik, A., Antonov, V. K., Gorbalenya, A. E., Kotova, S. A., Rotanova, T. V., and Shimbarevich, E. V. (1991). Site-directed mutagenesis of La protease. a catalytically active serine residue. FEBS Lett. 287, 211–214. doi: 10.1016/0014-5793(91)80053-6
Ballestero-Tellez, M., Docobo-Perez, F., Portillo-Calderon, I., Rodriguez-Martinez, J. M., Racero, L., Ramos-Guelfo, M. S., et al. (2017). Molecular insights into fosfomycin resistance in Escherichia coli. J. Antimicrob Chemother 72, 1303–1309.
Bhaskarla, C., Das, M., Verma, T., Kumar, A., Mahadevan, S., and Nandi, D. (2016). Roles of Lon protease and its substrate MarA during sodium salicylate-mediated growth reduction and antibiotic resistance in Escherichia coli. Microbiology 162, 764–776. doi: 10.1099/mic.0.000271
Botos, I., Melnikov, E. E., Cherry, S., Tropea, J. E., Khalatova, A. G., Rasulova, F., et al. (2004). The catalytic domain of Escherichia coli Lon protease has a unique fold and a Ser-Lys dyad in the active site. J. Biol. Chem. 279, 8140–8148. doi: 10.1074/jbc.m312243200
Castaneda-Garcia, A., Blazquez, J., and Rodriguez-Rojas, A. (2013). Molecular mechanisms and clinical impact of acquired and intrinsic fosfomycin resistance. Antibiotics (Basel) 2, 217–236. doi: 10.3390/antibiotics2020217
Castaneda-Garcia, A., Rodriguez-Rojas, A., Guelfo, J. R., and Blazquez, J. (2009). The glycerol-3-phosphate permease GlpT is the only fosfomycin transporter in Pseudomonas aeruginosa. J. Bacteriol. 191, 6968–6974. doi: 10.1128/jb.00748-09
Cattoir, V., and Guérin, F. (2018). How is fosfomycin resistance developed in Escherichia coli? Fut. Microbiol. 13, 1693–1696. doi: 10.2217/fmb-2018-0294
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645. doi: 10.1073/pnas.120163297
Derbise, A., Lesic, B., Dacheux, D., Ghigo, J. M., and Carniel, E. (2003). A rapid and simple method for inactivating chromosomal genes in Yersinia. FEMS Immunol. Med. Microbiol. 38, 113–116. doi: 10.1016/s0928-8244(03)00181-0
Doublet, B., Douard, G., Targant, H., Meunier, D., Madec, J. Y., and Cloeckaert, A. (2008). Antibiotic marker modifications of lambda Red and FLP helper plasmids, pKD46 and pCP20, for inactivation of chromosomal genes using PCR products in multidrug-resistant strains. J. Microbiol. Methods 75, 359–361. doi: 10.1016/j.mimet.2008.06.010
Duval, V., Nicoloff, H., and Levy, S. B. (2009). Combined inactivation of lon and ycgE decreases multidrug susceptibility by reducing the amount of OmpF porin in Escherichia coli. Antimicrob Agents Chemother 53, 4944–4948. doi: 10.1128/aac.00787-09
Ebrecht, A. C., Orlof, A. M., Sasoni, N., Figueroa, C. M., Iglesias, A. A., and Ballicora, M. A. (2015). On the ancestral UDP-glucose pyrophosphorylase activity of Galf from Escherichia coli. Front. Microbiol. 6:1253. doi: 10.3389/fmicb.2015.01253
Falagas, M. E., Athanasaki, F., Voulgaris, G. L., Triarides, N. A., and Vardakas, K. Z. (2019). Resistance to fosfomycin: mechanisms, frequency and clinical consequences. Int. J. Antimicrob Agents 53, 22–28. doi: 10.1016/j.ijantimicag.2018.09.013
Falagas, M. E., Vouloumanou, E. K., Samonis, G., and Vardakas, K. Z. (2016). Fosfomycin. Clin. Microbiol. Rev. 29, 321–347.
Fukasawa, T., Jokura, K., and Kurahashi, K. (1962). A new enzymic defect of galactose metabolism in Escherichia coli K-12 mutants. Biochem. Biophys. Res. Commun. 7, 121–125. doi: 10.1016/0006-291x(62)90158-4
Golla, V. K., Sans-Serramitjana, E., Pothula, K. R., Benier, L., Bafna, J. A., Winterhalter, M., et al. (2019). Fosfomycin permeation through the outer membrane porin OmpF. Biophys. J. 116, 258–269. doi: 10.1016/j.bpj.2018.12.002
Griffith, K. L., Shah, I. M., and Wolf, R. E. Jr. (2004). Proteolytic degradation of Escherichia coli transcription activators SoxS and MarA as the mechanism for reversing the induction of the superoxide (SoxRS) and multiple antibiotic resistance (Mar) regulons. Mol. Microbiol. 51, 1801–1816. doi: 10.1046/j.1365-2958.2003.03952.x
Island, M. D., Wei, B. Y., and Kadner, R. J. (1992). Structure and function of the uhp genes for the sugar phosphate transport system in Escherichia coli and Salmonella typhimurium. J. Bacteriol. 174, 2754–2762. doi: 10.1128/jb.174.9.2754-2762.1992
Island, M. D., and Kadner, R. J. (1993). Interplay between the membrane-associated UhpB and UhpC regulatory proteins. J. Bacteriol. 175, 5028–5034. doi: 10.1128/jb.175.16.5028-5034.1993
Kadner, R. J. (1973). Genetic control of the transport of hexose phosphates in Escherichia coli: mapping of the UHP locus. J. Bacteriol. 116, 764–770. doi: 10.1128/jb.116.2.764-770.1973
Kadner, R. J., and Shattuck-Eidens, D. M. (1983). Genetic control of the hexose phosphate transport system of Escherichia coli: mapping of deletion and insertion mutations in the uhp region. J. Bacteriol. 155, 1052–1061. doi: 10.1128/jb.155.3.1052-1061.1983
Kadner, R. J., and Winkler, H. H. (1973). Isolation and characterization of mutations affecting the transport of hexose phosphates in Escherichia coli. J. Bacteriol. 113, 895–900. doi: 10.1128/jb.113.2.895-900.1973
Kahan, F. M., Kahan, J. S., Cassidy, P. J., and Kropp, H. (1974). The mechanism of action of fosfomycin (phosphonomycin). Ann. N. Y. Acad. Sci. 235, 364–386. doi: 10.1111/j.1749-6632.1974.tb43277.x
Karageorgopoulos, D. E., Wang, R., Yu, X. H., and Falagas, M. E. (2012). Fosfomycin: evaluation of the published evidence on the emergence of antimicrobial resistance in Gram-negative pathogens. J. Antimicrob Chemother 67, 255–268. doi: 10.1093/jac/dkr466
Li, X. Z., Plesiat, P., and Nikaido, H. (2015a). The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 28, 337–418. doi: 10.1128/cmr.00117-14
Li, Y., Zheng, B., Li, Y., Zhu, S., Xue, F., and Liu, J. (2015b). Antimicrobial susceptibility and molecular mechanisms of fosfomycin resistance in clinical Escherichia coli Isolates in mainland China. PLoS One 10:e0135269. doi: 10.1371/journal.pone.0135269
Lucas, A. E., Ito, R., Mustapha, M. M., Mcelheny, C. L., Mettus, R. T., Bowler, S. L., et al. (2018). Frequency and mechanisms of spontaneous fosfomycin nonsusceptibility observed upon disk diffusion testing of Escherichia coli. J. Clin. Microbiol. 56:e01368-17.
Marchese, A., Gualco, L., Debbia, E. A., Schito, G. C., and Schito, A. M. (2003). In vitro activity of fosfomycin against gram-negative urinary pathogens and the biological cost of fosfomycin resistance. Int. J. Antimicrob Agents 22, (Suppl. 2), 53–59. doi: 10.1016/s0924-8579(03)00230-9
Martin-Gutierrez, G., Docobo-Perez, F., Rodriguez-Beltran, J., Rodriguez-Martinez, J. M., Aznar, J., Pascual, A., et al. (2018). Urinary tract conditions affect fosfomycin activity against Escherichia coli strains harboring chromosomal mutations involved in fosfomycin uptake. Antimicrob Agents Chemother 62:e01899-17.
Nicoloff, H., and Andersson, D. I. (2013). Lon protease inactivation, or translocation of the lon gene, potentiate bacterial evolution to antibiotic resistance. Mol. Microbiol. 90, 1233–1248. doi: 10.1111/mmi.12429
Nicoloff, H., Perreten, V., and Levy, S. B. (2007). Increased genome instability in Escherichia coli lon mutants: relation to emergence of multiple-antibiotic-resistant (Mar) mutants caused by insertion sequence elements and large tandem genomic amplifications. Antimicrob Agents Chemother 51, 1293–1303. doi: 10.1128/aac.01128-06
Nicoloff, H., Perreten, V., Mcmurry, L. M., and Levy, S. B. (2006). Role for tandem duplication and lon protease in AcrAB-TolC- dependent multiple antibiotic resistance (Mar) in an Escherichia coli mutant without mutations in marRAB or acrRAB. J. Bacteriol. 188, 4413–4423. doi: 10.1128/jb.01502-05
Nilsson, A. I., Berg, O. G., Aspevall, O., Kahlmeter, G., and Andersson, D. I. (2003). Biological costs and mechanisms of fosfomycin resistance in Escherichia coli. Antimicrob Agents Chemother 47, 2850–2858. doi: 10.1128/aac.47.9.2850-2858.2003
Ohkoshi, Y., Sato, T., Suzuki, Y., Yamamoto, S., Shiraishi, T., Ogasawara, N., et al. (2017). Mechanism of reduced susceptibility to fosfomycin in Escherichia coli clinical isolates. Biomed. Res. Int. 2017:5470241.
Oteo, J., Orden, B., Bautista, V., Cuevas, O., Arroyo, M., Martinez-Ruiz, R., et al. (2009). CTX-M-15-producing urinary Escherichia coli O25b-ST131-phylogroup B2 has acquired resistance to fosfomycin. J. Antimicrob Chemother 64, 712–717. doi: 10.1093/jac/dkp288
Parkinson, J. S., and Kofoid, E. C. (1992). Communication modules in bacterial signaling proteins. Annu. Rev. Genet. 26, 71–112. doi: 10.1146/annurev.ge.26.120192.000443
Peng, S., Stephan, R., Hummerjohann, J., and Tasara, T. (2014). Evaluation of three reference genes of Escherichia coli for mRNA expression level normalization in view of salt and organic acid stress exposure in food. FEMS Microbiol. Lett. 355, 78–82. doi: 10.1111/1574-6968.12447.
Philippe, N., Alcaraz, J. P., Coursange, E., Geiselmann, J., and Schneider, D. (2004). Improvement of pCVD442, a suicide plasmid for gene allele exchange in bacteria. Plasmid 51, 246–255. doi: 10.1016/j.plasmid.2004.02.003
Pourbaix, A., Guerin, F., Burdet, C., Massias, L., Chau, F., Cattoir, V., et al. (2019). Unexpected activity of oral fosfomycin against resistant strains of Escherichia coli in murine pyelonephritis. Antimicrob Agents Chemother 63, e903–e919.
Pourbaix, A., Guerin, F., Lastours, V., Chau, F., Auzou, M., Boulley, E., et al. (2017). Biological cost of fosfomycin resistance in Escherichia coli in a murine model of urinary tract infection. Int. J. Med. Microbiol. 307, 452–459. doi: 10.1016/j.ijmm.2017.09.019
Seok, H., Choi, J. Y., Wi, Y. M., Park, D. W., Peck, K. R., and Ko, K. S. (2020). Fosfomycin resistance in Escherichia coli isolates from south korea and in vitro activity of fosfomycin alone and in combination with other antibiotics. Antibiotics (Basel) 9:112. doi: 10.3390/antibiotics9030112
Sharma, O., Datsenko, K. A., Ess, S. C., Zhalnina, M. V., Wanner, B. L., and Cramer, W. A. (2009). Genome-wide screens: novel mechanisms in colicin import and cytotoxicity. Mol. Microbiol. 73, 571–585. doi: 10.1111/j.1365-2958.2009.06788.x
Silver, L. L. (2017). Fosfomycin: mechanism and resistance. Cold Spring Harb. Perspect. Med. 7:a025262. doi: 10.1101/cshperspect.a025262
Sundararajan, T. A., Rapin, A. M., and Kalckar, H. M. (1962). Biochemical observations on E. coli mutants defective in uridine diphosphoglucose. Proc. Natl. Acad. Sci. U.S.A. 48, 2187–2193. doi: 10.1073/pnas.48.12.2187
Takahata, S., Ida, T., Hiraishi, T., Sakakibara, S., Maebashi, K., Terada, S., et al. (2010). Molecular mechanisms of fosfomycin resistance in clinical isolates of Escherichia coli. Int. J. Antimicrob Agents 35, 333–337.
Tseng, S. P., Wang, S. F., Kuo, C. Y., Huang, J. W., Hung, W. C., Ke, G. M., et al. (2015). Characterization of fosfomycin resistant extended-spectrum beta-lactamase-producing Escherichia coli isolates from human and pig in Taiwan. PLoS One 10:e0135864. doi: 10.1371/journal.pone.0135864
Tsilibaris, V., Maenhaut-Michel, G., and Van Melderen, L. (2006). Biological roles of the Lon ATP-dependent protease. Res. Microbiol. 157, 701–713.
Verhamme, D. T., Arents, J. C., Postma, P. W., Crielaard, W., and Hellingwerf, K. J. (2001). Glucose-6-phosphate-dependent phosphoryl flow through the Uhp two-component regulatory system. Microbiology 147, 3345–3352.
Verhamme, D. T., Postma, P. W., Crielaard, W., and Hellingwerf, K. J. (2002). Cooperativity in signal transfer through the Uhp system of Escherichia coli. J. Bacteriol. 184, 4205–4210.
Weissborn, A. C., Liu, Q., Rumley, M. K., and Kennedy, E. P. (1994). UTP: alpha-D-glucose-1-phosphate uridylyltransferase of Escherichia coli: isolation and DNA sequence of the galU gene and purification of the enzyme. J. Bacteriol. 176, 2611–2618.
Welch, R. A., Burland, V., Plunkett, G. III, Redford, P., Roesch, P., Rasko, D., et al. (2002). Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 99, 17020–17024.
Weston, L. A., and Kadner, R. J. (1987). Identification of uhp polypeptides and evidence for their role in exogenous induction of the sugar phosphate transport system of Escherichia coli K-12. J. Bacteriol. 169, 3546–3555.
Weston, L. A., and Kadner, R. J. (1988). Role of uhp genes in expression of the Escherichia coli sugar-phosphate transport system. J. Bacteriol. 170, 3375–3383.
Wright, J. S., Olekhnovich, I. N., Touchie, G., and Kadner, R. J. (2000). The histidine kinase domain of UhpB inhibits UhpA action at the Escherichia coli uhpT promoter. J. Bacteriol. 182, 6279–6286.
Keywords: E. coli, fosfomycin-resistant, uhpB, uhpC, galU, lon
Citation: Cattoir V, Pourbaix A, Magnan M, Chau F, de Lastours V, Felden B, Fantin B and Guérin F (2020) Novel Chromosomal Mutations Responsible for Fosfomycin Resistance in Escherichia coli. Front. Microbiol. 11:575031. doi: 10.3389/fmicb.2020.575031
Received: 22 June 2020; Accepted: 29 September 2020;
Published: 20 October 2020.
Edited by:
Etienne Giraud, Institut National de la Recherche Agronomique de Toulouse, FranceCopyright © 2020 Cattoir, Pourbaix, Magnan, Chau, de Lastours, Felden, Fantin and Guérin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincent Cattoir, dmluY2VudC5jYXR0b2lyQGNodS1yZW5uZXMuZnI=; dmluY2VudC5jYXR0b2lyQHVuaXYtcmVubmVzMS5mcg==