
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 01 December 2020
Sec. Systems Microbiology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.573679
 Arif Luqman1,2
Arif Luqman1,2 Susanne Zabel3
Susanne Zabel3 Samane Rahmdel4
Samane Rahmdel4 Britta Merz5
Britta Merz5 Nicole Gruenheit5
Nicole Gruenheit5 Johannes Harter5
Johannes Harter5 Kay Nieselt3
Kay Nieselt3 Friedrich Götz1*
Friedrich Götz1*Trace amines (TA) are endogenously produced in mammals, have a low concentration in the central nervous system (CNS), but trigger a variety of neurological effects and intervene in host cell communication. It emerged that neurotransmitters and TA are produced also by the microbiota. As it has been shown that TA contribute to wound healing, we examined the skin microbiome of probands using shotgun metagenomics. The phyla Actinobacteria, Proteobacteria, Firmicutes, and Bacteroidetes were predominant. Since SadA is a highly promiscuous TA-producing decarboxylase in Firmicutes, the skin microbiome was specifically examined for the presence of sadA-homologous genes. By mapping the reads of certain genes, we found that, although there were less reads mapping to sadA than to ubiquitous housekeeping genes (arcC and mutS), normalized reads counts were still >1000 times higher than those of rare control genes (icaA, icaB, and epiA). At protein sequence level SadA homologs were found in at least 7 phyla: Firmicutes, Actinobacteria, Proteobacteria, Bacteroidetes, Acidobacteria, Chloroflexi, and Cyanobacteria, and in 23 genera of the phylum Firmicutes. A high proportion of the genera that have a SadA homolog belong to the classical skin and intestinal microbiota. The distribution of sadA in so many different phyla illustrates the importance of horizontal gene transfer (HGT). We show that the sadA gene is widely distributed in the human skin microbiome. When comparing the sadA read counts in the probands, there was no correlation between age and gender, but an enormous difference in the sadA read counts in the microbiome of the individuals. Since sadA is involved in TA synthesis, it is likely that the TA content of the skin is correlated with the amount of TA producing bacteria in the microbiome. In this way, the microbiome-generated TA could influence signal transmission in the epithelial and nervous system.
The term “trace amine” (TA) has been coined in the early 1970s by Alan Boulton and his colleagues to distinguish a group of endogenous vertebrate monoamines from their more abundant structural relatives, the catecholamine and indoleamine neurotransmitters (Boulton, 1974; Gainetdinov et al., 2018). TA are stored in nerve terminals with classical neurotransmitters such as dopamine (DOP), norepinephrine, or serotonin, and are released together with these classical neurotransmitters (Dewar et al., 1988; Premont et al., 2001). Despite their low abundance (Berry, 2004), there is evidence on the crucial physiological roles of TA in the neuromodulation of synaptic transmission in mammalian brains (Burchett and Hicks, 2006; Gainetdinov et al., 2018).
TA are produced by a wide range of organisms from bacteria to plants and vertebrates. In vertebrates, TA can be formed directly by the action of aromatic L-amino acid decarboxylase (AADC) on L-phenylalanine, L-tyrosine, and L-tryptophan, (Boulton and Wu, 1972; Saavedra, 1974; Silkaitis and Mosnaim, 1976; Dyck et al., 1983). TA production in bacteria has been mainly studied in food microorganisms, such as enterococci, lactobacilli, streptococci, lactococci, pediococci, and oenococci which represents the main producers of biogenic amines (Marcobal et al., 2006; Irsfeld et al., 2013; Williams et al., 2014; Barbieri et al., 2019).
Although there is some evidence on the low abundance of amine production in food-associated staphylococci (Rahmdel et al., 2018), the investigation of TA production in this genus has provided information about TA formation by the Staphylococcus species (Luqman et al., 2018). In this genus only some species are capable of TA production which can mainly be attributed to the presence of the gene sadA encoding staphylococcal aromatic amino acid decarboxylase. SadA decarboxylates tryptophan, tyrosine, and phenylalanine to tryptamine (TRY), tyramine (TYM), and phenethylamine (PEA), in a pyridoxalphosphate (PLP)-dependent reaction. It also decarboxylates dihydroxy phenylalanine (L-DOPA) and 5-hydroxytryptophan (5-HTP) to the neurotransmitters DOP and serotonin (Luqman et al., 2018). TA producing staphylococci triggered the internalization into human colon adenocarcinoma cells by activation of the α2-adrenergic receptor (α2-AR) (Luqman et al., 2018, 2019).
A study of the human intestinal microflora revealed that TA-producing staphylococci are present in the majority of human probands, suggesting a selective advantage. Moreover, a sadA deletion mutant of the animal pathogen Staphylococcus pseudintermedius showed a lower internalization rate than the parent strain in the presence of aromatic amino acids (AAAs). This may reinforce the hypothesis that the excreted TA interfere with host communication to improve the survival and colonization of the bacteria (Luqman et al., 2018, 2019). More recently it has been shown that TA-producing Staphylococcus epidermidis strains expressing SadA are predominant on human skin and that TA accelerate wound healing by antagonizing the β2-adrenergic receptor (β2-AR) in keratinocytes (Luqman et al., 2020b).
In mammalians, TA are synthesized by aromatic L-amino acid decarboxylases (AADC; EC 4.1.1.28) (Boulton and Wu, 1972; Snodgrass and Iversen, 1974; Silkaitis and Mosnaim, 1976; Dyck et al., 1983). Although AADC is widely accepted as the vertebrate synthetic enzyme for PEA, TYM, and TRY, the precursor amino acids are in fact extremely poor substrates for AADC (Christenson et al., 1970; Juorio and Yu, 1985; Gainetdinov et al., 2018). This raises the question whether the endogenous synthesis of TA plays a major role at all, or whether the uptake of TA from food and the production of TA by the microbiota are not as such decisive for exerting effects in mammalians. This assumption is supported by the relatively high concentrations of AAAs and TA present on human skin (10 and 5 μg/100 cm2, respectively) (Luqman et al., 2020b). Substantial TA concentrations have also been reported in the human gut in the decreasing order of TYM (from 7.6 to 621 μg g–1 of stool sample, depending on the subject), DOP, TRY, serotonin, and PEA (Luqman et al., 2018). These relatively high TA levels on the skin and in the intestine indicate that they are not endogenous but can be of microbial origin.
The PLP-dependent Trp decarboxylases of the common gut Firmicutes Clostridium sporogenes and Ruminococcus gnavus have been enzymatically and structurally characterized (Williams et al., 2014). However, they assumed that such activities are extremely rare in bacteria. On the other hand, the staphylococcal aromatic amino acid decarboxylase, SadA, is a highly promiscuous enzyme. It decarboxylates all AAAs to TAs, and also dihydroxylated phenylalanine and 5-HTP to the neurotransmitters DOP and serotonin (Luqman et al., 2018). SadA producing staphylococci are prevalent in the gut and the human skin (Luqman et al., 2018, 2019, 2020b).
TA are neuromodulators that may have an impact on the well-being of mammals. Therefore, the answer to the question whether SadA is widespread in the human microbiome or whether it is an exotic exception in some Staphylococcus species, is of great importance. The metagenomic profiling of the microbiome allows the analysis of the phylogenetic distribution of individual genes. In this study we investigated the occurrence of SadA homologs in the human skin microbiome. We could show that the microbiome of each volunteer contained sadA homologous genes, with large variations from person to person. In addition, SadA homologs are widely distributed throughout almost the entire bacterial kingdom, especially among representatives of the human microbiota, suggesting that the skin microbiota may be able to produce significant amounts of TA to influence host signaling and physiology.
We collected skin swab samples from 27 healthy people (age 14–78; 18 males and 10 females) by swabbing the antecubital fossa using sterile swab sample collectors. The swabs were resuspended in sterile phosphate buffer saline (PBS). We incubated the resuspended skin swabs with lysostaphin for 1 h and isolated genomic DNA using the Quick-DNA Microprep Kit (Zymo Research).
Genomic DNA was quality controlled and then used as input material for the preparation of sequencing libraries. Library preparation was carried out using the Nextera XT DNA Library Preparation Kit (Illumina). After library preparation, all libraries were pooled and sequenced with a read length of 2 × 100 bp on an Ilumina NovaseqTM 6000 system.
Ten million of the adapter-trimmed raw forward reads were aligned to the RefSeq protein database (version 94) using Diamond in BLASTX mode (Buchfink et al., 2015). Taxonomic placement was performed using the lowest common ancestor (LCA) algorithm implemented in MEGAN6 Ultimate Edition (version 6.15.2) (Huson et al., 2016). Only Taxa with relative sequence abundances above 0.001% were considered. Functional classification was carried out in MEGAN6 Ultimate Edition (version 6.15.2) (Huson et al., 2016) by assigning the reads to KEGG, SEED, VFDB, and Interpro identifiers.
Sequences of the genes [arcC (NC_004461.1), mutS (NC_004461.1), sadA (NP_763667.1), epiA (CUY02274.1), icaA (AAC06117.1), and icaB (AAC06118.1)] were downloaded from the NCBI database. All reads of all samples were mapped against these sequences using BWA (Li and Durbin, 2009) and the number of mapped reads extracted from the resulting samfiles using samtools (Li et al., 2009). Read counts were then normalized for the number of sequenced reads and the length of the respective genes. Therefore, normalized read counts are comparable between samples and between genes.
We performed a phylogenetic analysis of SadA homologs on four different taxonomic rank levels: within the species S. epidermidis, the genus Staphylococcus, the phylum Firmicutes and within the whole Bacteria domain. Homologs of SadA (WP_014612792) from S. pseudintermedius (Luqman et al., 2018) were identified using different flavor of NCBI’s Microbial BLAST and appropriate databases depending on the taxonomic rank the search was restricted to. To find homologs on the strain level within the species S. epidermidis tblastn was used and all complete as well as draft genomes of S. epidermidis were used as a database. Within the genus Staphylococcus, the phylum Firmicutes, and the domain Bacteria homologs of SadA were identified using blastp. The search within the bacterial domain was restricted to proteins of the RefSeq database (O’Leary et al., 2016) due to the large number of hits. Since several species have more than one putative homolog of SadA, we restricted the hits for each species in all blast runs to the best scoring protein. Based on the results of each individual blast run we computed separate phylogenetic trees of the SadA homologs. All best hits of the tblastn run were translated into protein sequences which were subsequently aligned using Clustal Omega (version 1.2.1) (Sievers et al., 2011). Also, the best hits of the three other blast runs were independently aligned using Clustal Omega. Based on the respective resulting multiple sequence alignment, a phylogenetic tree was constructed using maximum likelihood (ML) analysis using RaxML (version8.2.9) (Stamatakis, 2014). The GAMMA Model of rate heterogeneity was used and all model parameters were estimated by RaxML. LG with empirical base frequencies was used as the protein substitution model. Results were assessed using 100 bootstrap replicates. The best tree of each run was visualized using Interactive Tree Of Life (iTOL, version 4.4.2) (Letunic and Bork., 2019).
The pyridoxal phosphate (PLP)-dependent aspartate aminotransferase superfamily (fold I) domain (accession cd06450) of SadA was selected and its multiple sequence alignment within the genus Staphylococcus was visualized using Jalview 2.11 (Waterhouse et al., 2009).
We modeled the 3D structure of SadA homologs using SWISS-MODEL (31).
Topographically, the human skin can be divided into many regions and each region represents its own micro-environment (Grice and Segre, 2011). In this study, we performed metagenomic profiling on skin swabs collected from the forearm (antecubital fossa) of 27 probands. This approach enabled us to gain insights into the microbial composition and the phylogenetic distribution of individual genes of the skin microbiome. The most abundant bacterial phyla on the forearm of all probands were Actinobacteria, Proteobacteria, and Firmicutes (Figure 1A). As the genus Staphylococcus has been reported to be a very prominent component of the commensal skin microbiota (Kloos, 1980) we specifically focused on the richness and relative sequence abundances of staphylococcal species. In the current study, 28 different staphylococcal species were identified. The most abundant species found in all probands included S. aureus, S. epidermidis, S. capitis, S. hominis, and S. simulans, followed by S. saccharolyticus, S. haemolyticus, S. pseudintermedius, S. xylosus, and S. warneri (Figure 1B).
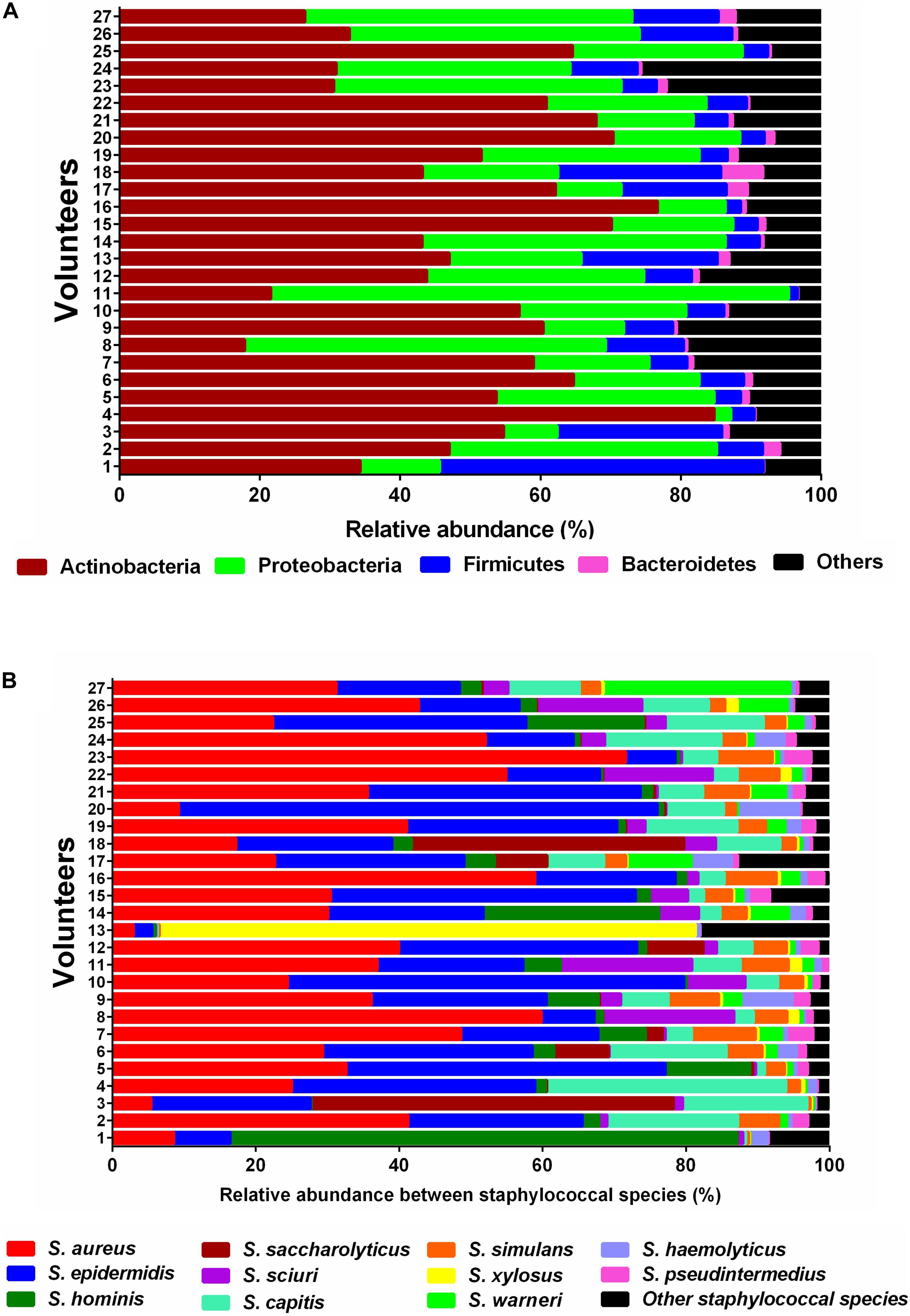
Figure 1. Relative sequence abundances of bacterial phyla and Staphylococcus species in skin samples. (A) Stacked bar plot of relative sequence abundances of the phyla present in each skin swab sample (n = 27). Relative sequence abundances were derived for all phyla present in any sample and the samples sorted by Bray-Curtis similarity. The predominant phyla were Actinobacteria, Proteobacteria, Firmicutes, and Bacteroidetes. (B) Stacked bar plot of relative sequence abundances of the species present in each skin swab sample (n = 27). Relative abundances were derived for all species present in any sample and the samples sorted by Bray-Curtis similarity. The predominant species were S. aureus, S. epidermidis, S. hominis, S. saccharolyticus, S. sciuri, and S. capitis. The black bars represent the other species with very low relative sequence abundances.
One of the main goals of this study was to determine how widespread the SadA gene and protein is in the skin microbiome. To investigate the abundance of sadA homologs in the skin microbiome, the sadA gene sequence from S. pseudintermedius (WP_014612792) was used as a reference to analyze the reads obtained from all skin swab samples. As a control, we also determined the normalized read counts of two housekeeping genes arcC (Shikimate dehydrogenase) and mutS (DNA mismatch repair), which occur almost in all bacteria (Thomas et al., 2007), as well as the less frequently occurring genes epiA [representing the structural gene of epidermin biosynthesis (Schnell et al., 1992)] and icaA and icaB [involved in the biosynthesis of polysaccharide intercellular adhesin (PIA) (Heilmann et al., 1996)]. Normalized read counts of sadA were about 400-times lower than those of the housekeeping genes (arcC and mutS), but more than 1,000-fold higher than the read counts of the rare genes (icaA, icaB, and epiA) (Figure 2). Based on the number of reads mapping to the sadA gene, there are more copies of this gene in the skin microbiota than there are copies of the rare genes. Therefore, it is highly likely that there are more bacteria whose genomes contain this gene than the rare genes.
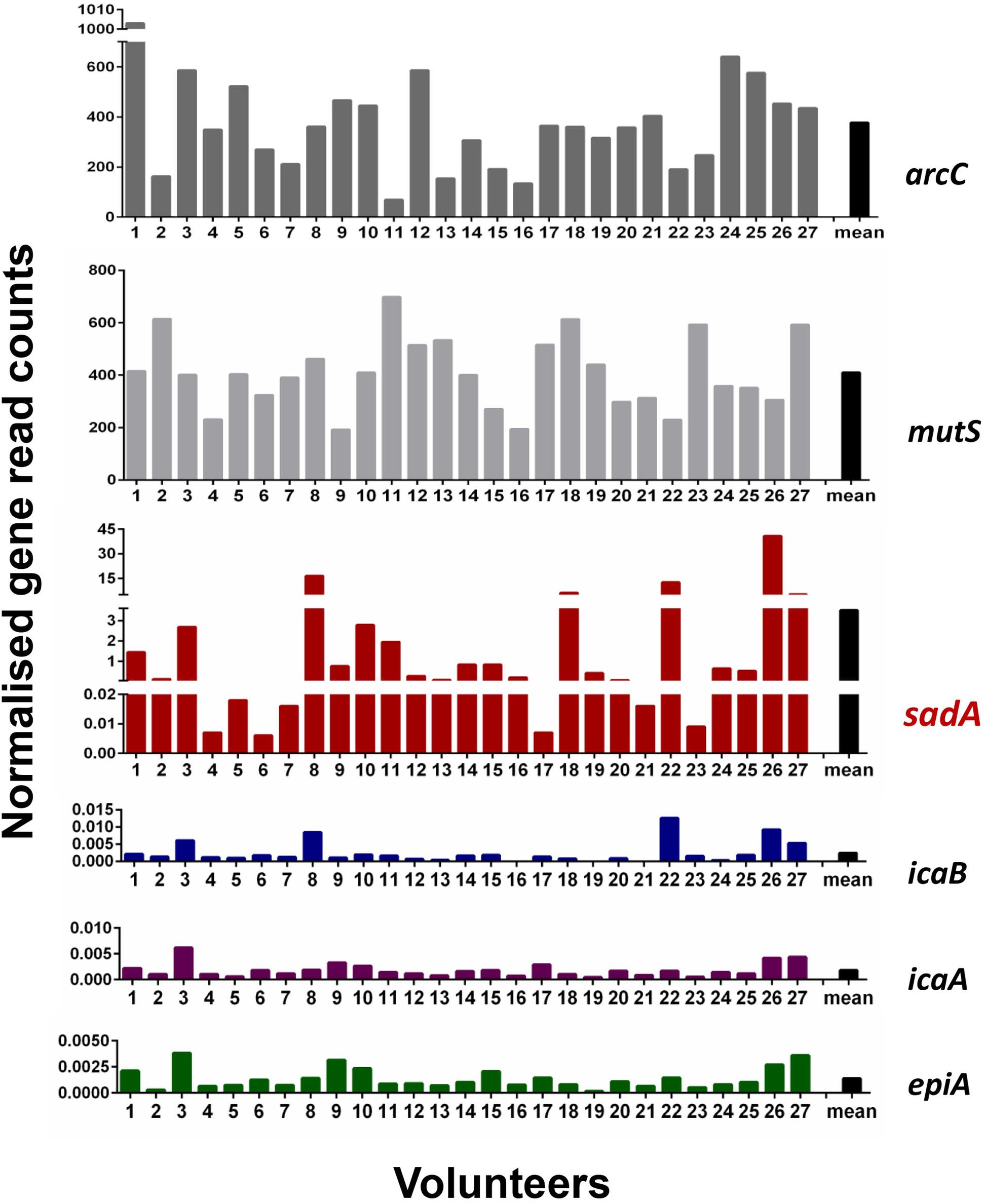
Figure 2. Normalized read counts for arcC, mutS, sadA, icaB, icaA, and epiA genes in skin samples. Reads were mapped against the sequences of the six genes and the obtained read counts normalized for the number of reads per sample and the length of the genes. Paired reads are counted as one.
To determine the presence and absence of the SadA homologs at protein level in all sequenced bacterial genomes we used BLASTP and sequences corresponding to the best hits were then aligned. We then constructed phylogenetic trees based on the alignments. Within the domain of Bacteria, SadA homologs can be found in at least 7 phyla: Firmicutes, Actinobacteria, Proteobacteria, Bacteroidetes, Acidobacteria, Chloroflexi, and Cyanobacteria (Figure 3). This analysis shows that SadA homologs are also widely distributed within bacteria that have been isolated from the skin. In fact, there is a high number of bacterial species that possess SadA homologs (Figure 3 and Supplementary Table 1).
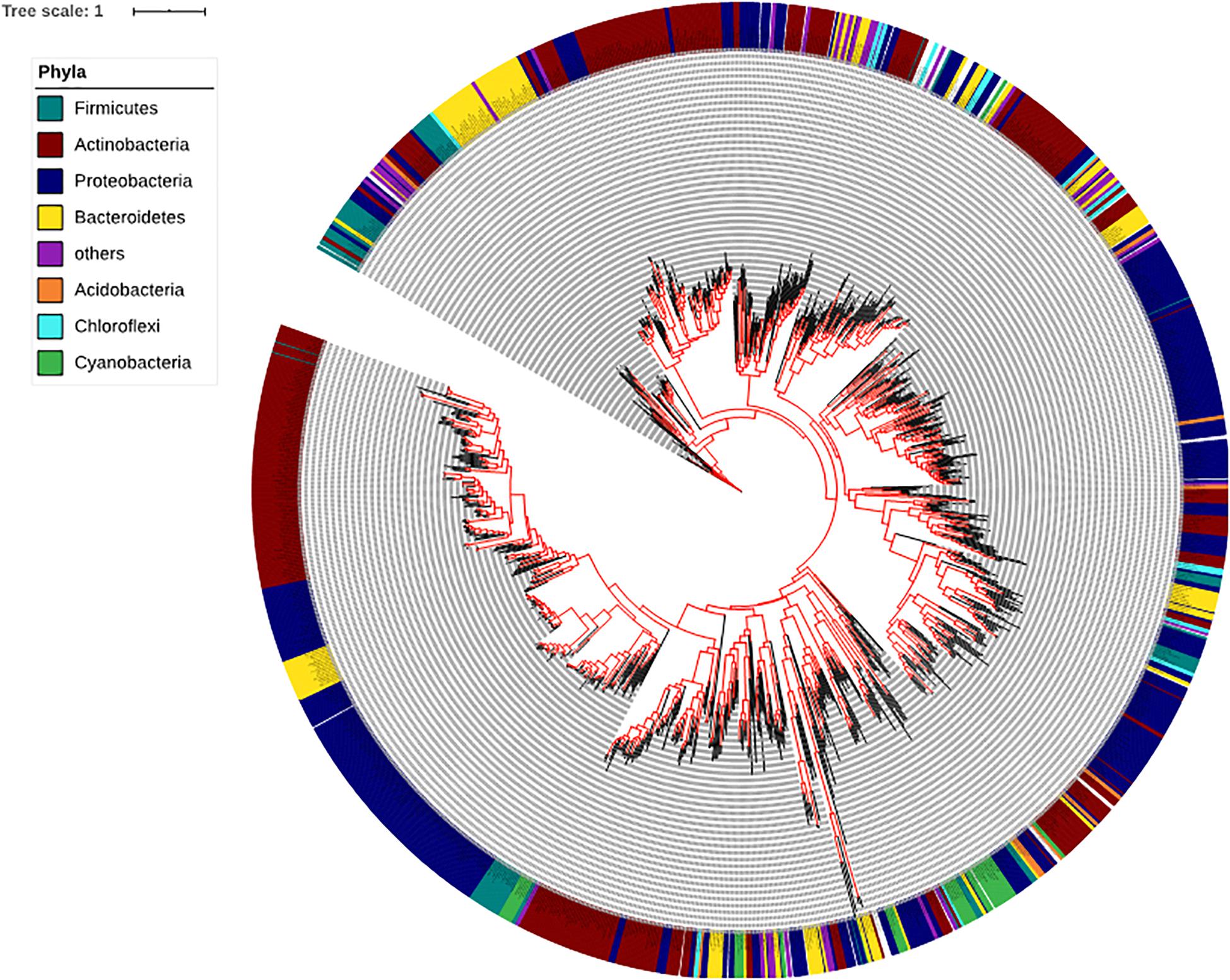
Figure 3. Phylogenetic tree of the bacteria based on the SadA sequence. The SadA sequence from S. pseudintermedius ED99 (WP_014612792) was used as a reference to identify homologs within the domain of Bacteria using BLASTP. All best hits were used to construct a phylogenetic tree, using the Randomized Axelerated maximum-likelihood (RaxML) method (version8.2.9) (Sievers et al., 2011). SadA was widespread among bacteria and could be found in at least seven phyla. Based on the SadA sequences, phyla did not cluster perfectly, but the subtrees often consisted of one phylum. The list of bacterial species is shown in Supplementary Table 1.
Since families belonging to Firmicutes play an important role on our skin, we have given special consideration to corresponding genera. SadA homologs were also found in 23 genera of the phylum Firmicutes (Figure 4), which is comprised of the “classic” low-GC Gram-positive bacteria. Together with the phylum Bacteroidetes, they represent 90% of the gut microbiota (Rinninella et al., 2019). Interestingly, many of the genera that possess SadA homologs like Clostridiaceae, Enterococcaceae, Lactobacillaceae, or Ruminococcaceae also belong to the elementary components of the human intestine. (Figure 4 and Supplementary Table 2).
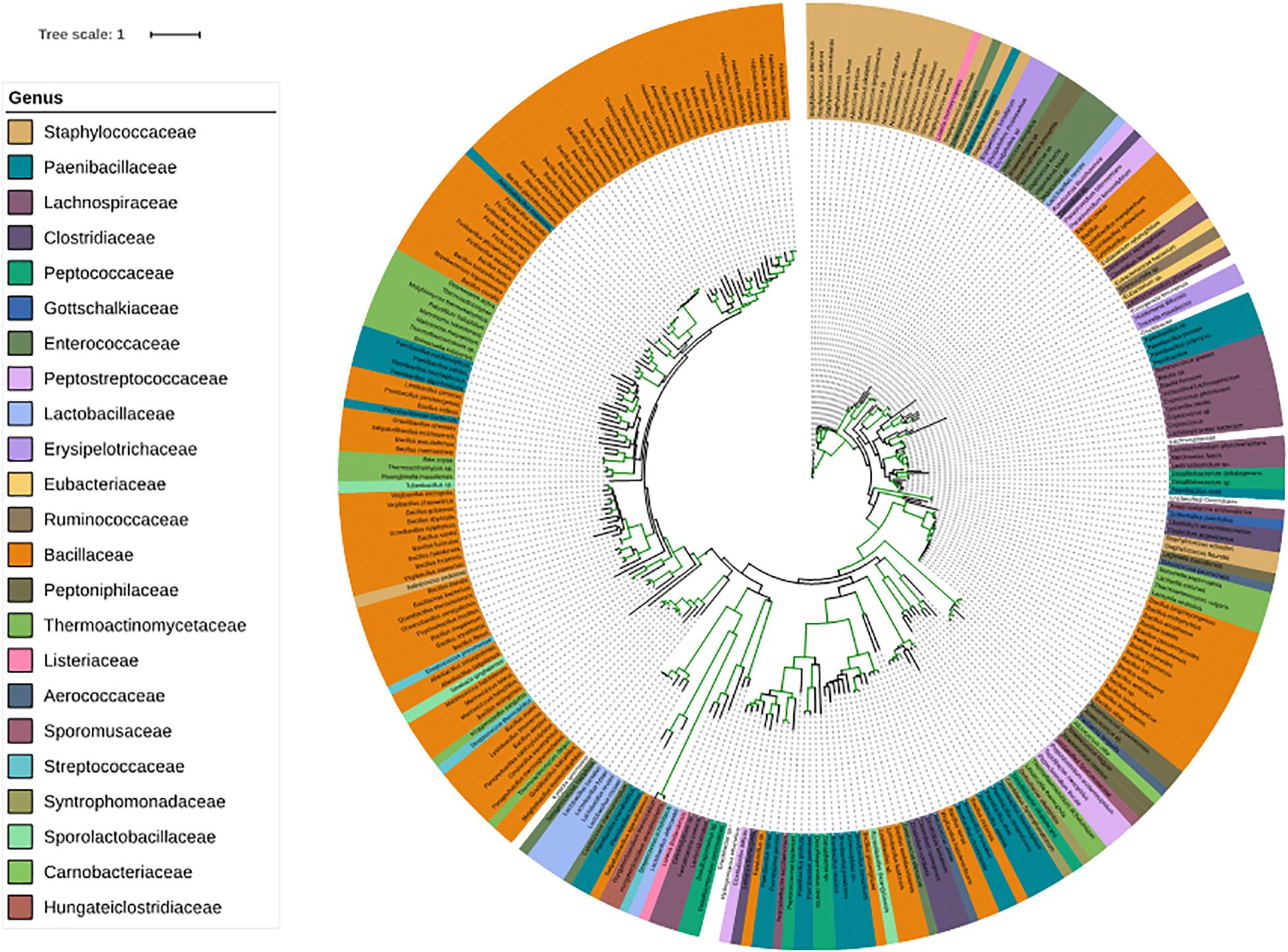
Figure 4. Phylogenetic tree of the SadA sequences within the Firmicutes. The SadA sequence from S. pseudintermedius ED99 (WP_014612792) was used as a reference to identify homologs within the Firmicutes phylum using BLASTP. All best hits were used to construct a phylogenetic tree, using the Randomized Axelerated maximum-likelihood (RaxML) method (version8.2.9) (Sievers et al., 2011). SadA was found in many genera of Firmicutes. Based on the SadA sequences, genera were not clustered perfectly, but the subtrees often consisted of one genus. The list of bacterial species is shown in Supplementary Table 2.
We also analyzed the correlation between the sadA read counts and the age of the probands using Pearson correlation; the p-value was 0.306. We categorized the sadA read counts based on the gender of the subjects and analyzed the difference between the genders using the Student’s t-test. This test yielded a p-value of 0.868, indicating that intrinsic factors (age and gender) do not show a significant correlation with the sadA read counts (Table 1).
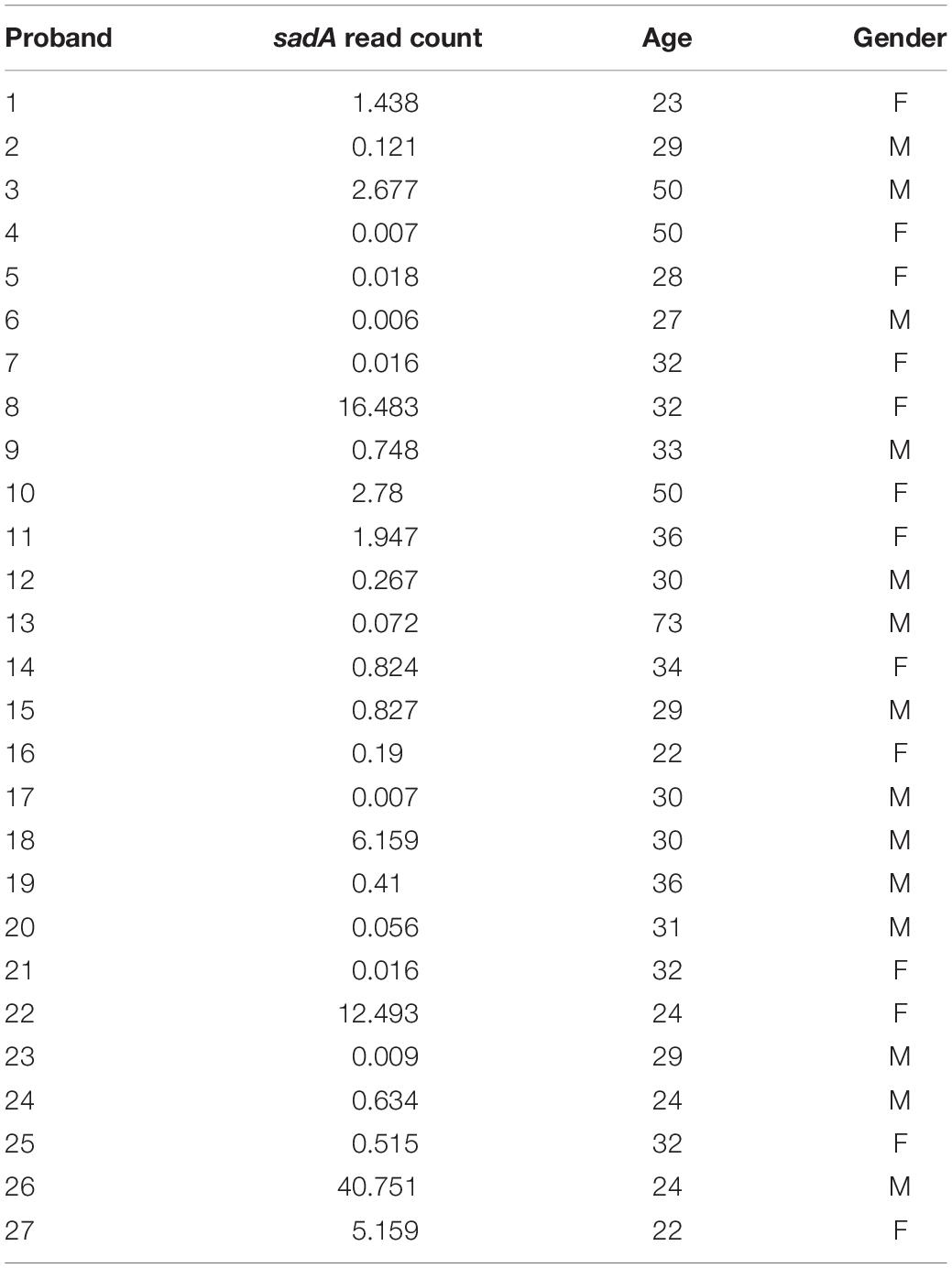
Table 1. sadA read counts in correlation with age and gender of probands.
To further substantiate the results, we aligned staphylococcal SadA and marked amino acids (aa) involved in PLP binding and the active site lysine (Figure 5A). The knowledge about the active site aa is derived from structural analysis of Trp decarboxylase (RUMGNA_01526) from Ruminococcus gnavus, an anaerobic Gram-positive gut bacterium of the Clostridiales family (Williams et al., 2014). This enzyme is 65% similar (44% identity) to SadA, making it currently the most closely related enzyme (Luqman et al., 2018). In all staphylococcal SadA homologs, the pyridoxal-dependent decarboxylase (PLP)-binding domain and the catalytic lysine in the active site were highly conserved. They also had a very similar protein length with about 470 aa.
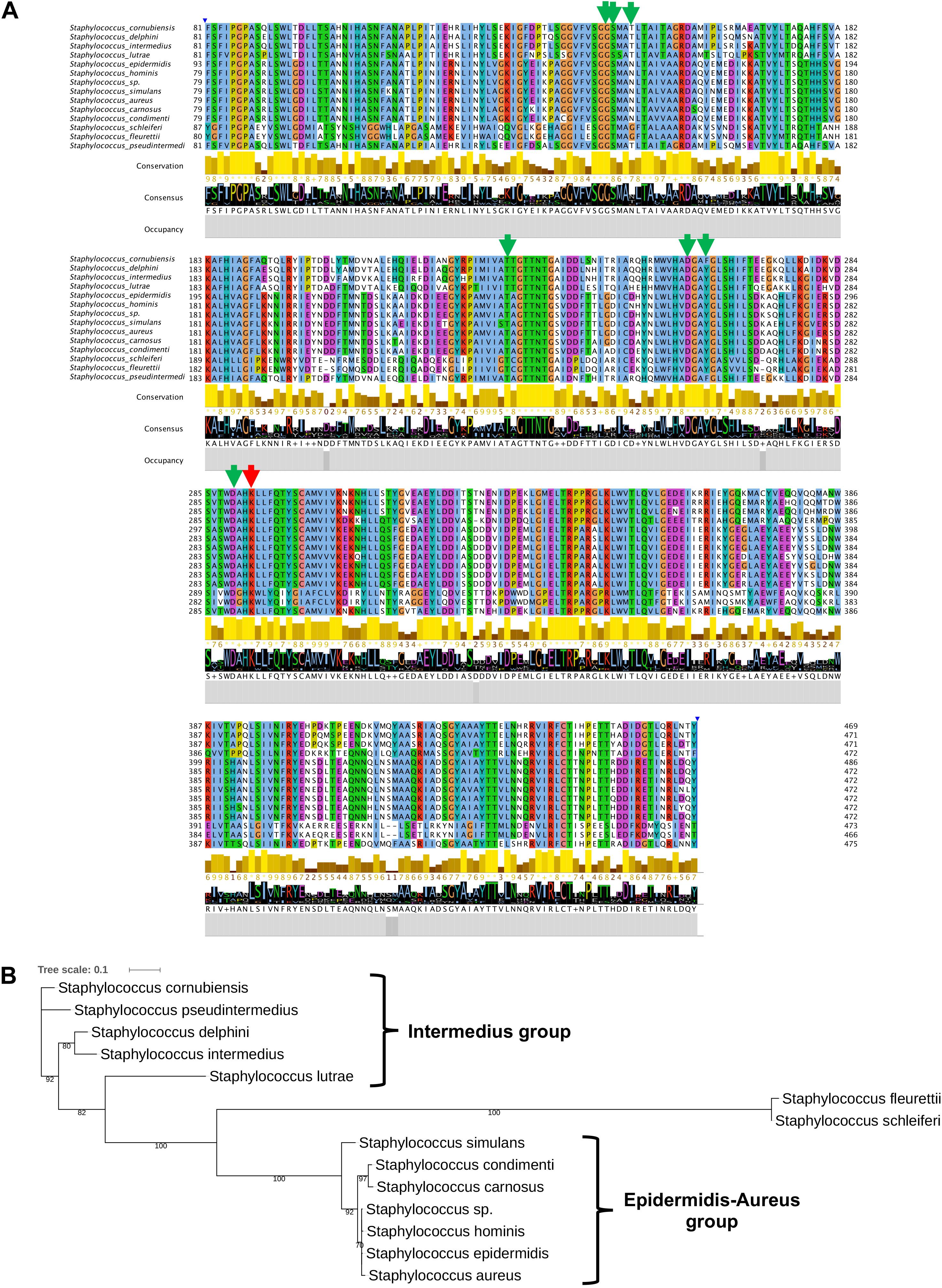
Figure 5. SadA in TA-producing staphylococci is highly conserved. (A) We aligned multiple sequences of the pyridoxal phosphate (PLP)-dependent aspartate aminotransferase superfamily (fold I) domain (accession cd06450) of SadA from species within the genus Staphylococcus and visualized using Jalview 2.11 (Letunic and Bork., 2019). The proposed PLP-binding domain (green) of SadA and the catalytic site Lysine (red) are highly conserved. (B) The 475 aa SadA sequence from S. pseudintermedius ED99 (WP_014612792) was used as a reference to identify homologs within the Staphylococcus genus using BLASTP. All best hits were used to construct the phylogenetic tree, using the Randomized Axelerated maximum-likelihood (RaxML) method (version8.2.9) (Sievers et al., 2011). Node support is indicated by bootstrap values from 100 resamplings of the alignment.
As shown previously, SadA is not present in all staphylococcal species (Luqman et al., 2018). Using SadA (475 aa) from S. pseudintermedius (WP_014612792) as a reference, we constructed the maximum-likelihood phylogenetic tree (Figure 5B). The clustering obtained is broadly consistent with the published phylogram with six species groups and 15 clusters (Lamers et al., 2012). All the species were correctly clustered into the expected groups except S. schleiferi which belongs to the intermedius group. S. fleurettii, belonging to the oxidase-positive sciuri species group is only distantly related with the other staphylococcal species and formed a separate small cluster.
To substantiate the taxonomic presence/absence pattern, we compared SadA with corresponding homologs of representatives of unrelated genera (Figure 6A) and predicted the 3D protein structure of the aa sequences (Figure 6B). The protein length varies from 350–520 aa and the active site amino acid Lysine as well as the proposed PLP-binding sites were highly conserved. The 3D structure models of the homolog proteins show similar folding as SadA from S. pseudintermedius. Some of the homologs are annotated as aromatic amino acid decarboxylase, PLP-dependent decarboxylase, or annotated as the domain: Fold Type I–aspartate aminotransferase family with lysine in the active site (Schneider et al., 2000; Milano et al., 2013). The annotation and 3D structure not only confirm the metagenomic analyses but also show that SadA homologs are represented in Lactobacilli, Bacteroides, Pseudomonas, Acetobacter, or Dermacocci, which all belong to the human microbiota. A comparison of SadA homologs between the genera Pseudomonas and Corynebacterium showed sequence similarity over most of their length with an average identity of about 27% (Supplementary Table 3).
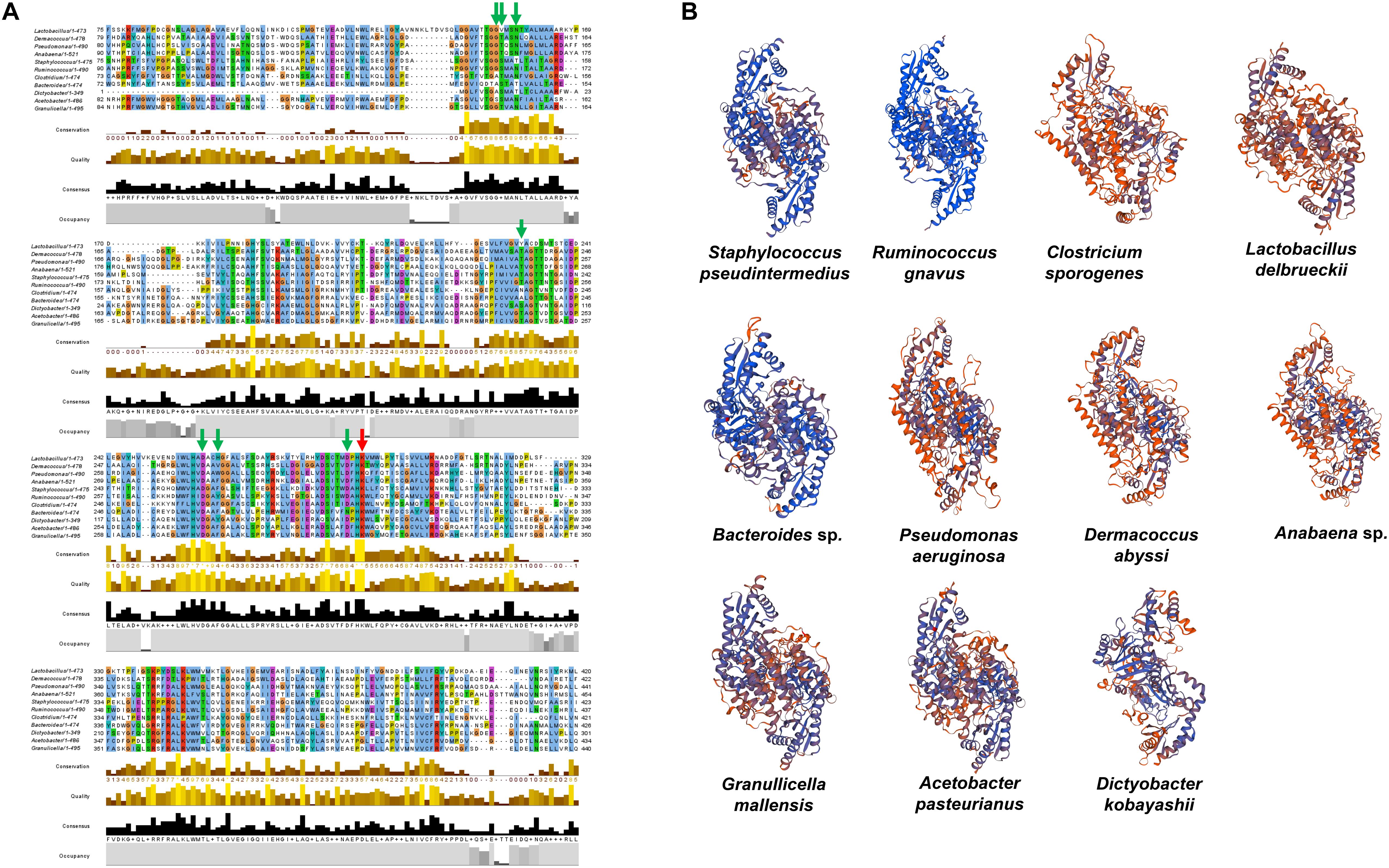
Figure 6. SadA homologs of several bacterial species from different phyla. (A) We aligned multiple sequences of SadA homologs of different species from different phyla. Jalview 2.11 was used for visualization (Letunic and Bork., 2019). The PLP-binding motifs are marked with green arrows and the active site lysine with a red arrow. (B) We also predicted the 3D structure of SadA homologs using SWISS-MODEL (31). Staphylococcus pseudintermedius, Clostridium sporogenes, Lactobacillus delbrueckii, and Ruminococcus gnavus represent Firmicutes; Dermacoccus abyssi represents Actinobacteria; Pseudomonas aeruginosa and Acetobacter pasteurianus represent Proteobacteria; Anabaena sp. represents Cyanobacteria; Bacteroides sp. represents Bacteroidetes; Dictyobacter kobayashii represents Chloroflexi; and Granulicella mallensis represents Acidobacteria. Accession numbers of the SadA homologs used for the alignment: Staphylococcus pseudintermedius WP_014612792, Ruminococcus gnavus A7B1V0.1, Clostridium sporogenes WP_058007894.1, Lactobacillus delbrueckii WP_130183735.1, Pseudomonas aeruginosa MXH35303.1, Dermacoccus abyssi QEH92696.1, Acetobacter pasteurianus GAB29948.1, Bacteroides sp. KPL16553.1, Granulicella mallensis WP_014266912.1, Dictyobacter kobayashii GCE20849.1, and Anabaena sp. HFS08142.1.
The promiscuous activity of SadA was the reason why its gene sequence was used to analyze its prevalence in the human skin microbiome. On the human skin, on the forearm (antecubital fossa), the phyla Actinobacteria, Proteobacteria, Firmicutes, and Bacteroidetes predominate (Figure 1A) which is consistent with the previous reports (Grice et al., 2008). Among Firmicutes, staphylococci belong to the classic skin bacteria and play an ambivalent role as typical commensals or opportunistic pathogens. Within this phylum with a focus on the genus Staphylococcus, eleven different species were found to be predominant (Figure 1B). Strikingly, S. aureus was almost as abundant as S. epidermidis in the healthy probands although its primary habitat is the nasal cavity. However, the metagenomic shotgun analysis used (as opposed to a 16S analysis) is able to reliably assign reads down to the species level.
The next question was: to what extent are staphylococcal SadA homologs represented in the entire skin microbiome? Since the gene sequence cannot provide reliable data due to the different GC content in the different phyla, this study was carried out at the protein sequence level. This allowed us to control the active site, the PLP binding site as well as the approximate length of the SadA homologs. We then constructed a phylogenetic tree based on the SadA protein sequence (Figure 3). To verify the metagenomic analyses, SadA homologs from completely unrelated family representatives were aligned and the similarity compared. As shown in Figure 6, the enzymes had a comparable length and the active site amino acids and PLP binding domain were conserved. Therefore, it can be assumed that these are SadA-like decarboxylases. However, nothing can be deduced about their substrate specificity.
Surprisingly, SadA homologs were much more widespread in the bacterial kingdom than expected. SadA homologs were found in at least seven phyla of Gram-positive and -negative bacteria. It is worth mentioning that many families and genera of the Firmicutes, Actinobacteria, Proteobacteria or Bacteroidetes belong to the predominant skin and intestinal microbiota. In depth analysis of the phylum Firmicutes reveals that SadA homologs are represented in 23 genera, many of which belong to the elementary components of the human skin and/or intestine such as Clostridiaceae, Enterococcaceae, Lactobacillaceae, or Ruminococcaceae (Figure 4). Interestingly, we did not find SadA homologs in cuti- and propionibacteria, which also belong to the skin microbiota.
From the phylogenetic trees of SadA homologs (Figures 3, 4), one can see that both the phyla and the genus are not perfectly clustered. Furthermore, the subtrees often consist of only one branch and of one genus, which suggests that SadA might undergo lateral gene transfer in the process of evolution. The seemingly arbitrary distribution of sadA within a bacterial family suggests horizontal gene transfer (HGT). Since in Gram-positive bacteria phages and prophages are widely distributed phage transduction is the most likely mechanism for lateral sadA spreading (Desiere et al., 2002; Novick et al., 2010; Alexeeva et al., 2018; van Zyl et al., 2018). In a metagenomic study on evolutionary trajectory and functional distribution of S. epidermidis skin isolates it was reported that they coalesce into multiple founder lineages rather than a single colonizer (Zhou et al., 2020). This is in accordance with our results as only about half of the skin S. epidermidis isolates possess the sadA gene. However, when we compared the sadA synteny in different staphylococcal species, we saw that its position differs within the different staphylococcal species-clusters suggesting transmission via mobile genetic elements (MGEs); but in no case did we see phage-specific flanking genes as relics of a phage transduction, or sadA as part of a genomic island (Luqman et al., 2018). Nevertheless, since in Firmicutes and other Gram-positive bacteria phage transduction is more common than conjugation or transformation, we still think that phages play a crucial role in spreading of sadA. Perhaps traces of sadA acquisition in the genome of staphylococci have become obscured in the course of evolution. However, such traces could possibly be found in other bacteria families.
Our metagenomic analysis represents only a snapshot in the evolution of sadA. The question is therefore, is sadA in a phase of degression or further expansion. Since sadA has no obvious benefit for the growth of bacteria, at least in staphylococci (Luqman et al., 2018), one could imagine a degression of sadA. On the other hand, for all bacteria that live together with host organisms sadA might be advantageous as it can contribute to colonization and internalization. Given the benefit of sadA for host interaction and communication we believe that there is an advantage in obtaining and keeping the gene, at least for such bacterial species whose living is strongly associated with a host; therefore, it is likely that sadA spreading is expansive.
What we know so far about SadA and its role for the host is maybe only the tip of the iceberg. We believe that SadA has many other activities regarding the coexistence of bacteria with the host waiting to be revealed. Burchett and Hicks have described TA as “the mysterious TA” (Burchett and Hicks, 2006). In low (“trace”) concentrations, they can interact with TAARs, which play an important role in the coordination of synaptic physiology by potentiating the activity of other neurotransmitters, especially DOP and serotonin. At high concentrations, they have well-characterized presynaptic “amphetamine-like” effects. We show that the intestinal (Luqman et al., 2018) and skin microbiota is quite capable of increasing the concentration of TA, and that the increased TA concentration causes new activities and interactions with receptors. It has been shown already that TA interact with α2- and β-AR and the biological consequences (Luqman et al., 2018, 2019, 2020b). But these are probably not the only receptors with which TA interact. Only recently it has been shown that TA have a high binding activity to the human α1-AR and are able to antagonize the epinephrine effect (Luqman et al., 2020a). α1-AR is a G protein-coupled receptor in the central and peripheral nervous system; its activation by epinephrine causes among other activities also vasoconstriction. The finding that TA acts as an α1-AR antagonist opens completely new perspectives. For example it was found that α1-AR antagonists prevent acute respiratory distress syndrome and death from cytokine storm syndrome (Vogelstein et al., 2020), prevent cytokine storm syndrome in COVID-19 (Konig et al., 2020), are used for treatment of lower urinary tract symptoms (Kim et al., 2019), or used for treatments of patients with nightmares after posttraumatic stress disorder and/or borderline personality disorders (Roepke et al., 2017).
When we compare the sadA read counts in the probands we see no correlation between age and gender, however, we see an enormous difference in the sadA prevalence in the microbiome of the probands (Table 1). Could this difference be related to the unique personality structure and psyche of each individual? The manifold influence of TA on the signaling cascade of the nervous system, the physiology, the behavior, gives an idea of the effects that skin and intestinal microbiota can have on our well-being.
The original contributions presented in the study are publicly available. This data can be found in the NCBI database using bioproject accession number PRJNA641765. Further information about the associated raw sequence data is shown in Supplementary Table 4.
The studies involving human participants were reviewed and approved by Ethic Commission of the University of Tübingen. The patients/participants provided their written informed consent to participate in this study.
FG and AL designed the study. AL, SZ, SR, BM, NG, JH, KN, and FG designed the experiments. AL collected the samples, isolated genomic DNA, and carried out 3D structure modeling. BM, NG, and JH performed the metagenomic analysis. SZ and KN done the phylogenetic analysis. FG and AL wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the DFG, German Research Foundation, SFB766 and SFB/TRR34 to FG and by DFG, Germany’s Excellence Strategy–EXC 2124–390838134 “Controlling Microbes to Fight Infections.”
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.573679/full#supplementary-material
Supplementary Figure 1 | Illustration of the sample collection and analysis workflow. We collected skin swab samples from 27 probands, isolated genomic DNA, prepared sequencing libraries and then sequenced these using. The libraries were subjected to next generation sequencing (NGS) analysis.
Supplementary Table 1 | List of species in Figure 3.
Supplementary Table 2 | List of species in Figure 4.
Supplementary Table 3 | SadA homologs found in Pseudomonas and Corynebacterium.
Supplementary Table 4 | Metadata.
Alexeeva, S., Guerra Martinez, J. A., Spus, M., and Smid, E. J. (2018). Spontaneously induced prophages are abundant in a naturally evolved bacterial starter culture and deliver competitive advantage to the host. Bmc Microbiol. 18:120. doi: 10.1186/s12866-018-1229-1
Barbieri, F., Montanari, C., Gardini, F., and Tabanelli, G. (2019). Biogenic amine production by lactic acid bacteria: a review. Foods 8:17. doi: 10.3390/foods8010017
Berry, M. D. (2004). Mammalian central nervous system trace amines. Pharmacologic amphetamines, physiologic neuromodulators. J. Neurochem. 90, 257–271. doi: 10.1111/j.1471-4159.2004.02501.x
Boulton, A. A. (1974). Letter: amines and theories in psychiatry. Lancet 2, 52–53. doi: 10.1016/s0140-6736(74)91390-7
Boulton, A. A., and Wu, P. H. (1972). Biosynthesis of cerebral phenolic amines. I. In vivo formation of p-tyramine, octopamine, and synephrine. Can. J. Biochem. 50, 261–267. doi: 10.1139/o72-037
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Burchett, S. A., and Hicks, T. P. (2006). The mysterious trace amines: protean neuromodulators of synaptic transmission in mammalian brain. Prog. Neurobiol. 79, 223–246. doi: 10.1016/j.pneurobio.2006.07.003
Christenson, J. G., Dairman, W., and Udenfriend, S. (1970). Preparation and properties of a homogeneous aromatic L-amino acid decarboxylase from hog kidney. Arch. Biochem. Biophys. 141, 356–367. doi: 10.1016/0003-9861(70)90144-x
Desiere, F., Lucchini, S., Canchaya, C., Ventura, M., and Brussow, H. (2002). Comparative genomics of phages and prophages in lactic acid bacteria. Antonie Van Leeuwenhoek 82, 73–91. doi: 10.1007/978-94-017-2029-8_5
Dewar, K. M., Dyck, L. E., Durden, D. A., and Boulton, A. A. (1988). Involvement of brain trace amines in the behavioural effects of phenelzine. Neurochem. Res. 13, 113–119. doi: 10.1007/bf00973322
Dyck, L. E., Yang, C. R., and Boulton, A. A. (1983). The biosynthesis of p-tyramine, m-tyramine, and beta-phenylethylamine by rat striatal slices. J. Neurosci. Res. 10, 211–220. doi: 10.1002/jnr.490100209
Gainetdinov, R. R., Hoener, M. C., and Berry, M. D. (2018). Trace amines and their receptors. Pharmacol. Rev. 70, 549–620. doi: 10.1124/pr.117.015305
Grice, E. A., Kong, H. H., Renaud, G., Young, A. C., Program, N. C. S., Bouffard, G. G., et al. (2008). A diversity profile of the human skin microbiota. Genome Res. 18, 1043–1050. doi: 10.1101/gr.075549.107
Grice, E. A., and Segre, J. A. (2011). The skin microbiome. Nat. Rev. Microbiol. 9, 244–253. doi: 10.1038/nrmicro2537
Heilmann, C., Schweitzer, O., Gerke, C., Vanittanakom, N., Mack, D., and Götz, F. (1996). Molecular basis of intercellular adhesion in the biofilm-forming Staphylococcus epidermidis. Mol. Microbiol. 20, 1083–1091. doi: 10.1111/j.1365-2958.1996.tb02548.x
Huson, D. H., Beier, S., Flade, I., Gorska, A., El-Hadidi, M., Mitra, S., et al. (2016). MEGAN community edition – interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 12:e1004957. doi: 10.1371/journal.pcbi.1004957
Irsfeld, M., Spadafore, M., and Pruss, B. M. (2013). Beta-phenylethylamine, a small molecule with a large impact. Webmedcentral 4:4409.
Juorio, A. V., and Yu, P. H. (1985). Effects of benzene and other organic solvents on the decarboxylation of some brain aromatic-L-amino acids. Biochem. Pharmacol. 34, 1381–1387. doi: 10.1016/0006-2952(85)90674-4
Kim, D. K., Lee, J. Y., Jung, J. H., Kim, J. H., Hah, Y. S., Hong, C. H., et al. (2019). Alpha-1 adrenergic receptor blockers for the treatment of lower urinary tract symptoms in women: a systematic review and meta-analysis. Int. Neurourol. J. 23, 56–68. doi: 10.5213/inj.1836188.094
Kloos, W. E. (1980). Natural populations of the genus Staphylococcus. Annu. Rev. Microbiol. 34, 559–592. doi: 10.1146/annurev.mi.34.100180.003015
Konig, M. F., Powell, M., Staedtke, V., Bai, R. Y., Thomas, D. L., Fischer, N., et al. (2020). Preventing cytokine storm syndrome in COVID-19 using alpha-1 adrenergic receptor antagonists. J. Clin. Invest. 130, 3345–3347. doi: 10.1172/JCI139642
Lamers, R. P., Muthukrishnan, G., Castoe, T. A., Tafur, S., Cole, A. M., and Parkinson, C. L. (2012). Phylogenetic relationships among Staphylococcus species and refinement of cluster groups based on multilocus data. BMC Evol. Biol. 12:171. doi: 10.1186/1471-2148-12-171
Letunic, I., and Bork., P. (2019). Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Luqman, A., Ebner, P., Reichert, S., Sass, P., Kabagema-Bilan, C., Heilmann, C., et al. (2019). A new host cell internalisation pathway for SadA-expressing staphylococci triggered by excreted neurochemicals. Cell. Microbiol. 21:e13044. doi: 10.1111/cmi.13044
Luqman, A., Kharisma, V. D., Ruiz, R. A., and Götz, F. (2020a). In silico and in vitro study of trace amines (TA) and dopamine (DOP) interaction with human alpha 1-adrenergic receptor and the bacterial adrenergic receptor QseC. Cell. Physiol. Biochem. 54, 888–898. doi: 10.33594/000000276
Luqman, A., Muttaqin, M. Z., Yulaipi, S., Ebner, P., Matsuo, M., Zabel, S., et al. (2020b). Trace amines produced by skin bacteria accelerate wound healing in mice. Commun. Biol. 3:277. doi: 10.1038/s42003-020-1000-7
Luqman, A., Nega, M., Nguyen, M. T., Ebner, P., and Götz, F. (2018). SadA-expressing staphylococci in the human gut show increased cell adherence and internalization. Cell Rep. 22, 535–545. doi: 10.1016/j.celrep.2017.12.058
Marcobal, A., de las Rivas, B., and Munoz, R. (2006). First genetic characterization of a bacterial beta-phenylethylamine biosynthetic enzyme in Enterococcus faecium RM58. FEMS Microbiol. Lett. 258, 144–149. doi: 10.1111/j.1574-6968.2006.00206.x
Milano, T., Paiardini, A., Grgurina, I., and Pascarella, S. (2013). Type I pyridoxal 5’-phosphate dependent enzymatic domains embedded within multimodular nonribosomal peptide synthetase and polyketide synthase assembly lines. BMC Struct. Biol. 13:26. doi: 10.1186/1472-6807-13-26
Novick, R. P., Christie, G. E., and Penades, J. R. (2010). The phage-related chromosomal islands of Gram-positive bacteria. Nat. Rev. Microbiol. 8, 541–551. doi: 10.1038/nrmicro2393
O’Leary, N. A., Wright, M. W., Brister, J. R., Ciufo, S., McVeigh, D. H. R., Rajput, B., et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. doi: 10.1093/nar/gkv1189
Premont, R. T., Gainetdinov, R. R., and Caron, M. G. (2001). Following the trace of elusive amines. Proc. Natl. Acad. Sci. U.S.A. 98, 9474–9475. doi: 10.1073/pnas.181356198
Rahmdel, S., Hosseinzadeh, S., Shekarforoush, S. S., Torriani, S., Gatto, V. and Pashangeh, S. (2018). Safety hazards in bacteriocinogenic Staphylococcus strains isolated from goat and sheep milk. Microb. Pathog. 116, 100–108. doi: 10.1016/j.micpath.2018.01.016
Rinninella, E., Raoul, P., Cintoni, M., Franceschi, F., Miggiano, G. A. D., Gasbarrini, A., et al. (2019). What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms 7:14. doi: 10.3390/microorganisms7010014
Roepke, S., Danker-Hopfe, H., Repantis, D., Behnia, B., Bernard, F., Hansen, M. L., et al. (2017). Doxazosin, an alpha-1-adrenergic-receptor antagonist, for nightmares in patients with posttraumatic stress disorder and/or borderline personality disorder: a chart review. Pharmacopsychiatry 50, 26–31. doi: 10.1055/s-0042-107794
Saavedra, J. M. (1974). Enzymatic isotopic assay for and presence of beta-phenylethylamine in brain. J. Neurochem. 22, 211–216. doi: 10.1111/j.1471-4159.1974.tb11581.x
Schneider, G., Kack, H., and Lindqvist, Y. (2000). The manifold of vitamin B6 dependent enzymes. Structure 8, R1–R6. doi: 10.1016/s0969-2126(00)00085-x
Schnell, N., Egelke, G., Augustin, J., Rosenstein, R., Ungermann, V., Götz, F., et al. (1992). Analysis of genes involved in the biosynthesis of lantibiotic epidermin. Eur. J. Biochem. 204, 57–68. doi: 10.1111/j.1432-1033.1992.tb16605.x
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W. Z., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Silkaitis, R. P., and Mosnaim, A. D. (1976). Pathways linking L-phenylalanine and 2-phenylethylamine with p-tyramine in rabbit brain. Brain Res. 114, 105–115. doi: 10.1016/0006-8993(76)91010-6
Snodgrass, S. R., and Iversen, L. L. (1974). Formation and release of 3H-tryptamine from 3H-tryptophan in rat spinal cord slices. Adv. Biochem. Psychopharmacol. 10, 141–150.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Thomas, J. C., Vargas, M. R., Miragaia, M., Peacock, S. J., Archer, G. L., and Enright, M. C. (2007). Improved multilocus sequence typing scheme for Staphylococcus epidermidis. J. Clin. Microbiol. 45, 616–619. doi: 10.1128/JCM.01934-06
van Zyl, L. J., Abrahams, Y., Stander, E. A., Kirby-McCollough, B., Jourdain, R., Clavaud, C., et al. (2018). Novel phages of healthy skin metaviromes from South Africa. Sci. Rep. 8:12265. doi: 10.1038/s41598-018-30705-1
Vogelstein, J. T., Powell, M., Koenecke, A., Xiong, R., Fischer, N., Huq, S., et al. (2020). Alpha-1 adrenergic receptor antagonists for preventing acute respiratory distress syndrome and death from cytokine storm syndrome. arXiv [Preprint].
Waterhouse, A. M., Procter, J. B., Martin, D. M. A., Clamp, M., and Barton, G. J. (2009). Jalview version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. doi: 10.1093/bioinformatics/btp033
Williams, B. B., Van Benschoten, A. H., Cimermancic, P., Donia, M. S., Zimmermann, M., Taketani, M., et al. (2014). Discovery and characterization of gut microbiota decarboxylases that can produce the neurotransmitter tryptamine. Cell Host Microbe 16, 495–503. doi: 10.1016/j.chom.2014.09.001
Keywords: metagenomic profiling, microbiota, microbiome, skin, trace amines
Citation: Luqman A, Zabel S, Rahmdel S, Merz B, Gruenheit N, Harter J, Nieselt K and Götz F (2020) The Neuromodulator-Encoding sadA Gene Is Widely Distributed in the Human Skin Microbiome. Front. Microbiol. 11:573679. doi: 10.3389/fmicb.2020.573679
Received: 17 June 2020; Accepted: 09 November 2020;
Published: 01 December 2020.
Edited by:
George Tsiamis, University of Patras, GreeceReviewed by:
Zhigang Zhou, Feed Research Institute (CAAS), ChinaCopyright © 2020 Luqman, Zabel, Rahmdel, Merz, Gruenheit, Harter, Nieselt and Götz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Friedrich Götz, ZnJpZWRyaWNoLmdvZXR6QHVuaS10dWViaW5nZW4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.