 Vitaly V. Kadnikov
Vitaly V. Kadnikov Andrey V. Mardanov
Andrey V. Mardanov Alexey V. Beletsky
Alexey V. Beletsky Olga V. Karnachuk
Olga V. Karnachuk Nikolai V. Ravin
Nikolai V. Ravin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 03 September 2020
Sec. Extreme Microbiology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.572252
To get insights into microbial diversity and biogeochemical processes in the terrestrial deep subsurface aquifer, we sequenced the metagenome of artesian water collected at a 2.8 km deep oil exploration borehole 5P in Western Siberia, Russia. We obtained 71 metagenome-assembled genomes (MAGs), altogether comprising 93% of the metagenome. Methanogenic archaea accounted for about 20% of the community and mostly belonged to hydrogenotrophic Methanobacteriaceae; acetoclastic and methylotrophic lineages were less abundant. ANME archaea were not found. The most numerous bacteria were the Firmicutes, Ignavibacteriae, Deltaproteobacteria, Chloroflexi, and Armatimonadetes. Most of the community was composed of anaerobic heterotrophs. Only six MAGs belonged to sulfate reducers. These MAGs accounted for 5% of the metagenome and were assigned to the Firmicutes, Deltaproteobacteria, Candidatus Kapabacteria, and Nitrospirae. Organotrophic bacteria carrying cytochrome c oxidase genes and presumably capable of aerobic respiration mostly belonged to the Chloroflexi, Ignavibacteriae, and Armatimonadetes. They accounted for 13% of the community. The first complete closed genomes were obtained for members of the Ignavibacteriae SJA-28 lineage and the candidate phylum Kapabacteria. Metabolic reconstruction of the SJA-28 bacterium, designated Candidatus Tepidiaquacella proteinivora, predicted that it is an anaerobe growing on proteinaceous substrates by fermentation or anaerobic respiration. The Ca. Kapabacteria genome contained both the sulfate reduction pathway and cytochrome c oxidase. Presumably, the availability of buried organic matter of Mesozoic marine sediments, long-term recharge of the aquifer with meteoric waters and its spatial heterogeneity provided the conditions for the development of microbial communities, taxonomically and functionally more diverse than those found in oligotrophic underground ecosystems.
Deep subsurface environments provide the largest habitats for prokaryotes in terms of size (Whitman et al., 1998). According to recent estimates (Magnabosco et al., 2018), the deep subsurface biosphere contains from 12 to 20% of the total biomass of microorganisms on Earth, mostly in the continental subsurface. Both the deep sub-seafloor and terrestrial subsurface habitats contain a variety of functionally active microbial communities, the existence of which is limited only by temperature increase with depth and is possible at depths of up to 3–5 km (Ghiorse and Wilson, 1988; Parkes et al., 2000; Takai et al., 2001; Teske, 2005; Itävaara et al., 2011; Lomstein et al., 2012; Bomberg et al., 2015). Such microbial communities may be independent of the supply of organic matter from the surface and exist autonomously for hundreds of millions of years (Chivian et al., 2008; Edwards et al., 2012). Depending on the type of rocks, underground microbial communities may be lithoautotrophic or organotrophic (Fredrickson and Hicks, 1987). In the lithoautotrophic communities characteristic of igneous rocks, the main source of energy is molecular hydrogen of abiotic origin, and the content of organic carbon is extremely low. Such microbial communities are usually characterized by low diversity and contain sulfate-reducing or methanogenic microorganisms (Moser et al., 2005; Chivian et al., 2008). In sedimentary rocks, organic matter buried since their formation provides energy sources and organic carbon, which enable the development of various organotrophic microorganisms. The best-studied example of such ecosystems is the formation waters of oil reservoirs, which harbor diverse communities of bacteria and archaea (Orphan et al., 2003; Lewin et al., 2014; Hu et al., 2016). The microbial communities of deep terrestrial subsurface ecosystems have been investigated in a number of studies (Miettinen et al., 2015; Hubalek et al., 2016; Magnabosco et al., 2016; Wu et al., 2016; Colman et al., 2017; Momper et al., 2017). Deep subsurface ecosystems can be accessed through drilled boreholes penetrating deep strata. Particularly, oil-exploration boreholes, drilled to a depth of several kilometers, provide a unique opportunity to access microbial communities of deep subsurface aquifers. The Western Siberian megabasin, formed from marine sediments of the Mesozoic era, is one of the largest oil and gas reserves in the world (Vyssotski et al., 2006). This region also harbors huge underground waterbodies located at depths of 1–3 km (Novikov and Sukhorukova, 2015). A number of oil-exploration boreholes have been drilled here since the 1950s. Some of them remain open and are currently used as sources of artesian thermal waters, thus providing a unique opportunity for sampling microbial communities of subsurface aquifers. Three such deep subsurface aquifers have been studied by molecular and cultivation approaches. The microbial community of an aquifer broached by the 3P borehole in the Parabel district (Tomsk region, Russia) was characterized by 16S rRNA profiling and cultivation; it was found to consist mostly of chemolithoautotrophs, – sulfate-reducing Firmicutes and hydrogenotrophic methanogenic archaea (Frank et al., 2016). The microbial community of the second aquifer, accessed via the 1-R borehole in the Byelii Yar district, has been analyzed by metagenomics. This community was more complex: it lacked methanogens and comprised sulfate-reducing Firmicutes and Deltaproteobacteria, as well as various presumably organotrophic members of the phyla Chlorofexi, Ignavibacteriae, Ca. Aminicenantes, Ca. Riflebacteria and BRC1 (Kadnikov et al., 2018). The metagenomic data enabled genome-based characterization of members of Ca. Aminicenantes, Ca. Riflebacteria and BRC1 (Kadnikov et al., 2018, 2019a,b) and cultivation of an enigmatic uncultured firmicute, Ca. Desulforudis audaxviator (Karnachuk et al., 2019), previously known by its genome retrieved from a deep gold mine in South Africa, where it formed a single-species hydrogen-driven ecosystem (Chivian et al., 2008).
The third site, borehole 5P in the Chazhemto district, was analyzed by 16S rRNA profiling (Kadnikov et al., 2017a,b). It harbored a rather different microbial community, consisting of various lineages of the phyla Firmicutes, Ignavibacteria, Chloroflexi, Bacteroidetes, and Proteobacteria, phylogenetically distant from cultured species, and methanogenic archaea of the genera Methanothermobacter and Methanosaeta. Known sulfate reducers were not identified. However, the lack of metagenomics data prevents us from predicting the functional roles of the majority of this community represented by uncultured lineages and obtaining insights into the ecology of this subsurface aquifer.
Here we report a metagenomics analysis of the deep subsurface aquifer accessed via the 5P borehole. We successfully recovered high-quality metagenome-assembled genomes of the vast majority of community members that enabled us to perform an accurate metabolic reconstruction and propose an ecological model of the deep subsurface aquifer.
The oil-exploration borehole 5P is located near the village of Chazhemto in the Tomsk region of Russia (58.077481N, 82.836091E). The borehole was drilled in the 1950s to a depth of 2.8 km. It penetrated Quaternary sediments near the surface followed by Paleogene, Cretaceous and Jurassic sediments to a depth of ∼2.5 km. Upon passing Jurassic sedimentary rocks, the borehole entered the Palaeozoic basement (Banks et al., 2014). According to oil exploration practices of the 1950s, the borehole was tested by a bottom-up casing perforation method. This method involves perforating the walls of the pipe and testing for the outflow. After testing, the section was sealed with cement, and then the next section was tested in the same way. The last tested section is assumed to be located at a depth of more than 2 km. Therefore, the water is expected to originate from an aquifer system located at a depth of 2–2.5 km in the Mesozoic sedimentary rocks. The artesian water flows out spontaneously under natural pressure with the rate of about 11.5 m3 per day and is used by local populations as a source of mineral water through a closed wellhead with a set of connectors.
Water samples were collected at the wellhead on April 30, 2016. Water temperature, pH and Eh were determined on site using an HI 8314 pH meter (Hanna Instruments, Germany). For chemical analysis, the water samples were filtered through a 0.2-μm sterile filter (Merck Millipore, Germany) and analyzed by ICP-MS and ion chromatography as reported previously (Kadnikov et al., 2017a). Gas content was determined using a Kristall-5000.1 gas chromatograph (Russia) equipped with a katharometer detector. Microbial biomass from 25 L of groundwater was collected by filtration on 0.22 μm cellulose nitrate membranes (Sartorius, Germany) using a Sartorius filtration unit. The filters were frozen in liquid nitrogen and then ground and melted with TE buffer in a water bath at 37°C. Total DNA was extracted using a Power Soil DNA isolation kit (MO BIO Laboratories, Inc., Carlsbad, CA, United States). About 1 μg of DNA was isolated.
Sequencing of a paired-end (2 × 250 bp) TruSeq DNA library using an Illumina HiSeq2500 platform generated 57,579,354 read pairs (Kadnikov et al., 2017c). Adapters were trimmed using Cutadapt v.1.14 (Martin, 2011). Sequencing reads were preprocessed by trimming low-quality sequences (Q < 33) using Sickle version 1.331. Trimmed reads were merged with FLASH v.1.2.11 (Magoć and Salzberg, 2011). Resulting merged and unmerged reads (about 16.9 Gbp in total) were de novo assembled using metaSPAdes version 3.7.1 (Bankevich et al., 2012) into 185,598 contigs longer than 500 bp.
Contigs longer than 2000 bp were binned into MAGs using the program CONCOCT v.0.4.1 (Alneberg et al., 2014). The completeness of obtained MAGs and their possible contamination were estimated using CheckM v.1.05 (Parks et al., 2015) with lineage-specific marker genes. The assembled MAGs were taxonomically assigned using the GTDB-Tk version 0.1.3 tool and Genome Taxonomy database (GTDB) (Parks et al., 2018).
The 16S rRNA genes in the contigs were identified using CheckM. The search for continuations of contigs representing fragments of 16S rRNA genes using Bandage v.0.8.0 program (Wick et al., 2015) allowed linkage of 16S rRNA genes to most of the genome bins where 16S rRNA carrying contigs were missing.
Metagenomic DNA was additionally sequenced on a MinION system (Oxford Nanopore, United Kingdom) using the 1D Genomic DNA by ligation protocol. Sequencing generated 1,418,419 reads of a total length of about 1.54 Gbp. These long reads were used to assemble the Illumina contigs of a given MAG into longer sequences. For this purpose, the MinION reads exhibiting high sequence similarity to the contigs of a given MAG were selected using BWA v.0.7.15 (Li and Durbin, 2010). Then the contigs were merged using the npScarf program (Cao et al., 2017). The consensus sequence was polished using Pilon v.1.22 (Walker et al., 2014) with mapping of Illumina reads back to the assembled sequence using Bowtie 2 (Langmead and Salzberg, 2012).
Gene search and annotation of MAGs was performed using the RAST server 2.0 (Brettin et al., 2015), followed by manual correction of the annotation by the comparison of predicted protein sequences with the National Center for Biotechnology Information (NCBI) databases. Signal peptides were predicted by Signal P v.4.12 and PRED-TAT3, and the presence of transmembrane helices was predicted by TMHMM v. 2.04.
The average amino acid identity (AAI) between the selected genomes was calculated using the aai.rb script from the Enveomics Collection (Rodriguez-R and Konstantinidis, 2016).
The GTDB-Tk v.0.1.3 tool was used to find single-copy marker genes in the assembled MAGs and to construct a multiple alignment of concatenated single-copy gene sequences, comprising those from a given MAG and all species from the GTDB. In some cases, genomes of related organisms missing in GTDB were additionally included in this analysis. A selected part of the multiple alignment built into GTDB-Tk was used to construct a phylogenetic tree in PhyML v. 3.3 (Guindon et al., 2010) with default parameters. The level of support for internal branches was assessed using the Bayesian test in PhyML.
The metagenomic sequences obtained in this study have been deposited in the NCBI Sequence Read Archive under the accession numbers SRR6186653 (Illumina reads) and SRR11854357 (MinION reads). The annotated sequences of high-quality MAGs have been deposited in the GenBank database under the BioProject PRJNA414521.
The physical and chemical characteristics of the water collected at the 5P borehole in April 2016 are shown in Table 1. The water temperature was surprisingly low (19.8°C), considering a typical thermal gradient of 20–30°C per km and an expected 2 km depth of the productive horizon. However, the water may cool when passing through the borehole, and it should be noted that over the past 15 years of observation, the water temperature has decreased by about 10°C, and the flow rate up the well has also decreased, which may be due to clogging of the well pipe. The water had a near neutral pH (about 7.5) and was highly reduced (Eh – 329 mV). The ionic content of the water was dominated by sodium and chloride, with subsidiary calcium. The total mineralization of the water (about 6 g L–1), estimated by summing the concentrations of ions, was only 17% of marine salinity. Therefore, most of the water is expected to be derived from meteoric recharge. The magnesium/strontium mass ratio was very low (0.06), suggesting that strontium accumulated in water during the prolonged residence time. The concentration of barium, a key indicator of sulfate removal by sufate reduction, was rather high (3.9 mg L–1). The concentration of sulfate was higher than in waters collected at the 3P Parabel (Frank et al., 2016) and 1-R Byelii Yar boreholes (Kadnikov et al., 2018) and varied from year to year (90.4 mg L–1 in 2016 and 23.3 mg L–1 in 2015). The water contained dissolved hydrogen sulfide at concentrations of 15.7 mg L–1 in 2016 and 7.6 mg L–1 in 2015.
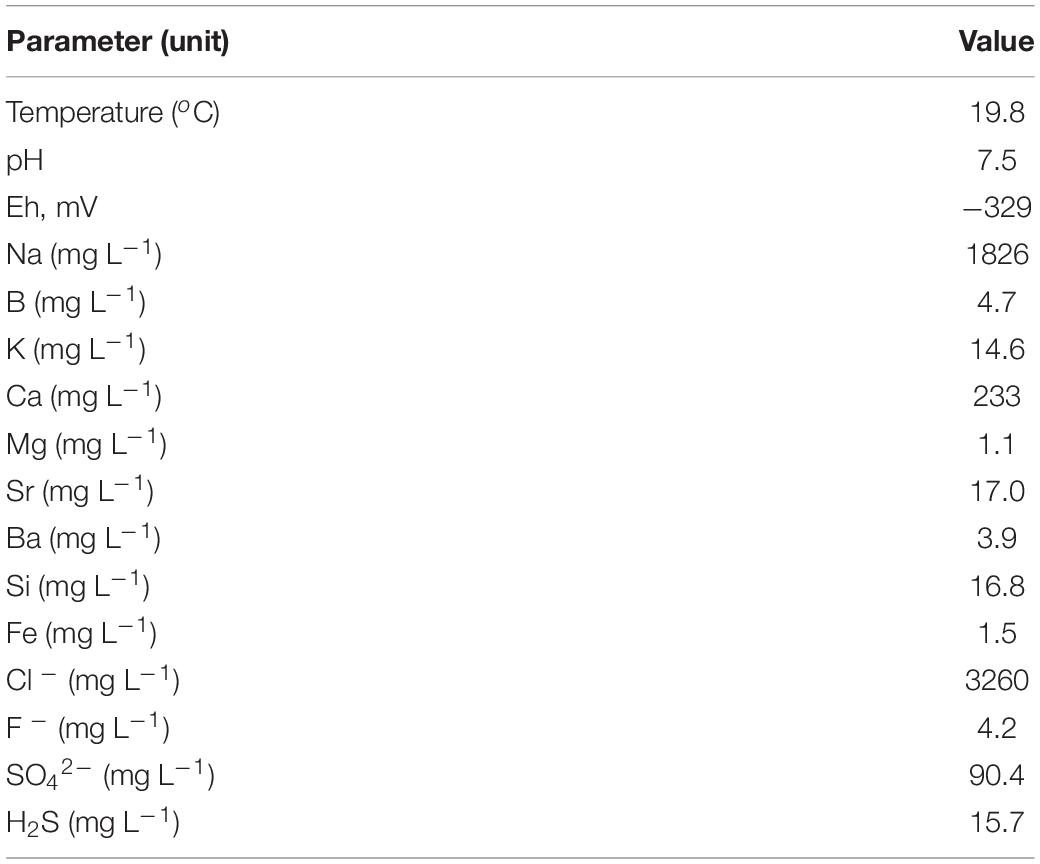
Table 1. Physical and chemical characteristics of the water at 5P borehole.
The water flowing out from the borehole exsolved gas bubbles under atmospheric pressure. The gas contained mostly methane (81.1%) followed by nitrogen (16.3%) and oxygen (2.5%). The isotopic composition of methane (δ13Cav. = −59.87–60.03‰) is consistent with its mostly biogenic origin (Kadnikov et al., 2017a).
In order to assemble the composite genomes of the most abundant members of the microbial community, we sequenced the metagenome of a water sample using the Illumina HiSeq2500 platform. A total of 16.9 Gbp metagenomic sequences were assembled into contigs, which were clustered into 136 genome bins. Seventy one bins representing MAGs with more than 85% completeness altogether comprised 93.3% of the whole metagenome (Table 2 and Supplementary Table S1). Analysis of the presence of a set of single-copy conserved genes revealed that 45 of these 71 bins could be classified as high-quality MAGs with more than 90% completeness and less than 5% contamination, as proposed by Bowers et al. (2017).
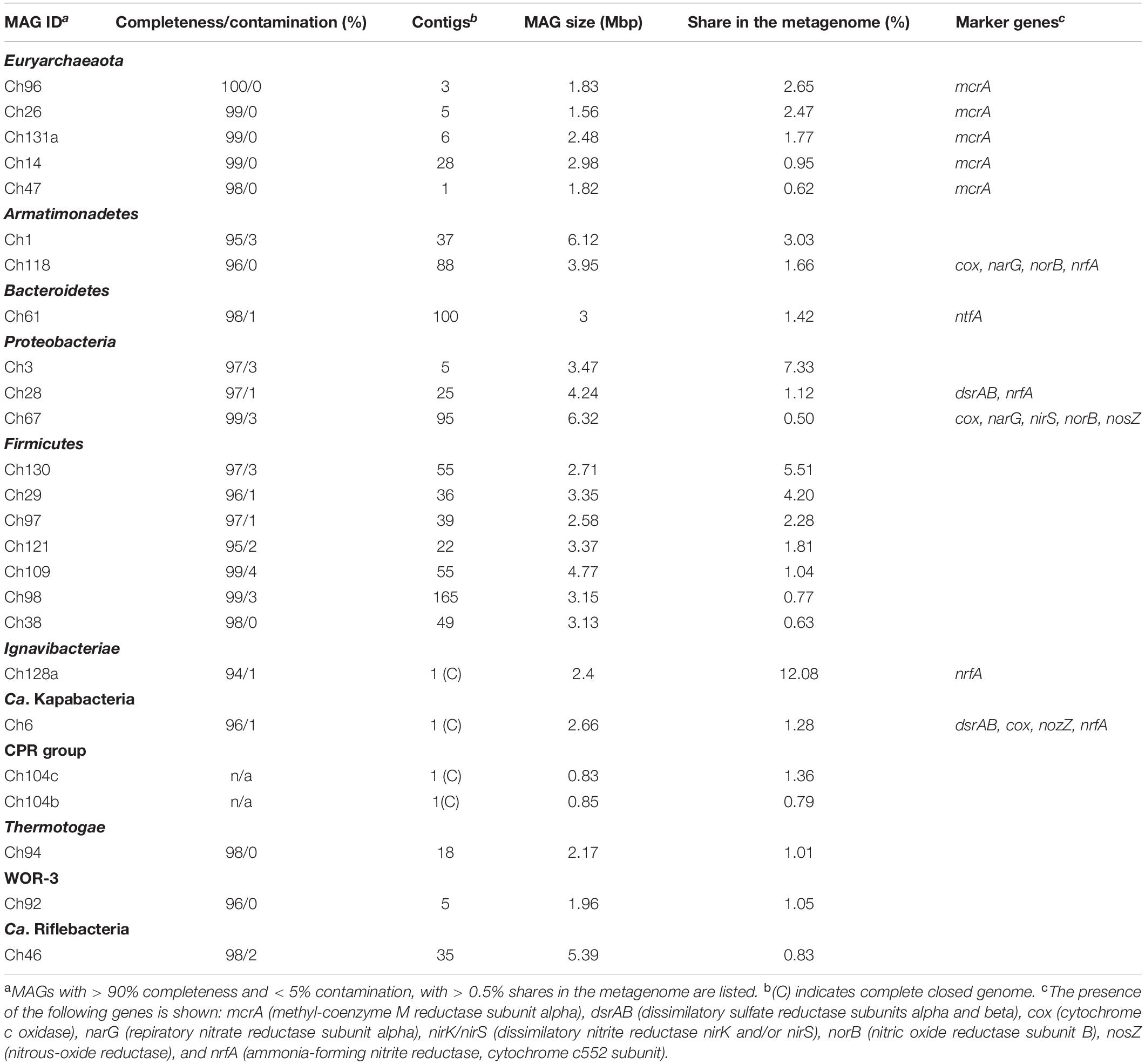
Table 2. General characteristics of high quality MAGs.
Phylogenetic identification of MAGs based on searches against the GTDB (Parks et al., 2018) revealed the same major prokaryotic lineages that were detected by 16S rRNA profiling (Kadnikov et al., 2017a): Euryarchaeota (20.4% of the whole metagenome), Firmicutes (24.1%), Ignavibacteriae (14.3%), Proteobacteria (9.2%), Chloroflexi (7.9%), Armatimonadetes (5.0%), Bacteroidetes (2.0%), Thermotogae (1.0%), the candidate phyla Kapabacteria (1.3%), and WOR-3 (1.1%) (Figure 1). Four MAGs assigned to the Candidate Phyla Radiation (CPR), a lineage comprising symbiotic or parasitic bacteria with a small cell size and reduced genomes (Castelle et al., 2018), accounted for about 2.8% of the metagenome. The actual relative abundance of CPR bacteria may be higher, because a 0.2-μm filter was used to collect the biomass, and small cells could pass through it. The shares of other identified phylum-level lineages, Acidobacteria, Actinobacteria, Ca. Aminicenantes, Ca. Atribacteria, Ca. Bipolaricaulota, Coprothermobacterota, Nitrospirae, Ca. Omnitrophica, Ca. Riflebacteria, Spirochaetes, and Ca. Verstraetearchaeota were less than 1% each (Supplementary Table S1).
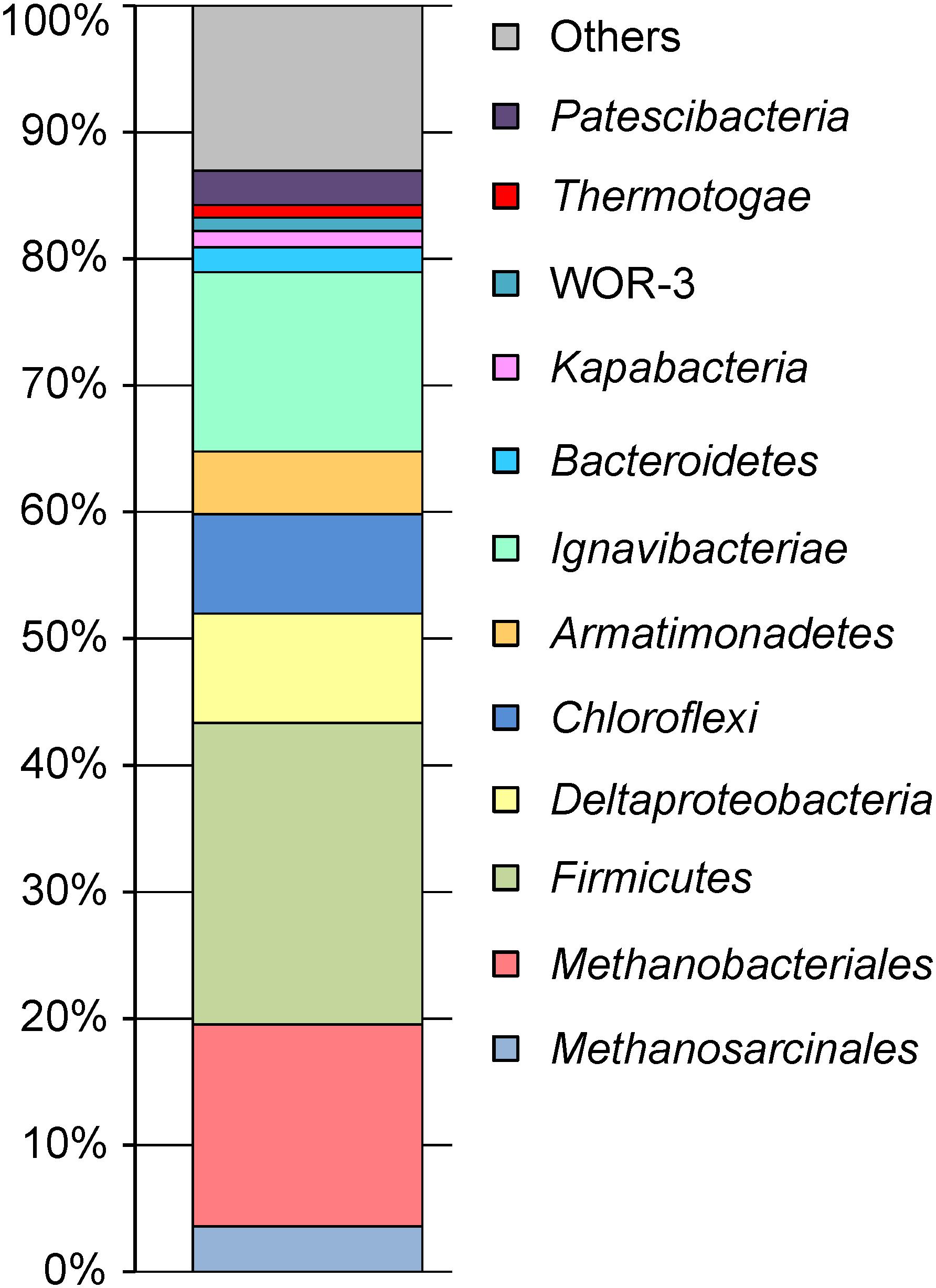
Figure 1. The relative abundance of taxonomic groups of microorganisms in the metagenome.
All archaeal MAGs belonged to known methanogenic lineages, namely Methanobacteriales (Ch26, Ch35, Ch131a, and Ch131b), Methanosarcinales (Methanosaetaceae, Ch14 and Ch96), Methanomassiliicoccales (Ch47), Ca. Methanofastidiosales (Ch133), and Ca. Verstraetearchaeota (Ch88). Therefore, all three types of methanogens, hydrogenotrophic, acetoclastic and methyl-reducing, were present in the groundwater. Hydrogenotrophic methanogens predominated (16.1% of metagenome) and mostly belonged to the genus Methanothermobacter (Ch35 and Ch26 with average AAIs of 96.4 and 66.3% to Methanothermobacter marburgensis str. Marburg). According to the AAI thresholds proposed by Konstantinidis et al. (2017) for uncultivated microorganisms (45–65% for the same family, 65–95% for the same genus and 95–100% for the same species), Ch35 is a distinct strain of M. marburgensis, while Ch26 represented another species in this genus. The second most numerous group (3.6%) were acetoclastic methanogens of the family Methanosaetaceae, represented by Methanosaeta thermophila (Ch96–99.0% AAI to Methanosaeta thermophila PT) and a novel genus-level lineage (Ch14–61.7% AAI to M. thermophila). Another group of methanogens, members of the order of Methanomassiliicoccales that produces methane by reducing methanol and other methylated compounds with hydrogen as an electron donor (Lang et al., 2015), were less abundant (0.52%). Methanomassiliicoccales were represented by a single MAG Ch47 phylogenetically distant from known species of this order (47–59% AAI). Two other archaeal lineages present in minor amounts are likely characterized by a similar type of methanogenesis through reduction of methylated compounds. Ch133 belongs to the candidate genus Methanofastidiosum (81% AAI to Arc I group archaeon ADurb1213_Bin02801) of the euryarchaeal order Ca. Methanofastidiosales (Nobu et al., 2016) also known as WSA2 or Arc1. The second MAG, Ch88, belongs to the genus Ca. Methanosuratus (88.7% AAI to Ca. Methanosuratus petracarbonis) within the candidate phylum Verstraetearchaeota (Vanwonterghem et al., 2016). Notably, we found no organisms related to known anaerobic methane oxidizers (ANME-1, 2, 3).
The most abundant phylum, Firmicutes, accounted for 24.6% of the community and was represented by 21 MAGs. Only two of these MAGs could be assigned to known genera, while most others were phylogenetically distant from known lineages (Supplementary Table S1). Ch97 belongs to the genus Pelotomaculum, sharing 98.21% AAI with P. thermopropionicum, and Ch98 belongs to Syntrophothermus, sharing 95.7% AAI with Syntrophothermus lipocalidus DSM 12680. Members of these genera are mostly syntrophs growing in association with methanogens or anaerobic respiring organisms and capable of oxidizing propionate, fatty acids, alcohols or aromatic compounds (Sekiguchi et al., 2000; Imachi et al., 2002; Hidalgo-Ahumada et al., 2018).
The most abundant Firmicutes genotype, Ch130, accounting for 5.5% of the whole metagenome, may also have syntrophic metabolism. This MAG has an average AAI of 65.1% with Thermacetogenium phaeum DSM 12270, an anaerobic thermophilic bacterium oxidizing acetate in syntrophic association with a methanogenic partner (Hattori et al., 2000; Oehler et al., 2012). The presence of the Wood–Ljungdahl pathway used in the reverse direction for anaerobic acetate oxidation in T. phaeum in the Ch130 MAG indicates that this bacterium could have the same metabolism. Firmicutes phylogenetically related to Thermacetogenium, along with methanogenic archaea, were previously detected in another Western Siberian subsurface aquifer accessed via a 3P Parabel borehole (Frank et al., 2016).
Since sulfate-reducing members of the Firmicutes are common to the deep subsurface (e.g., Moser et al., 2005; Magnabosco et al., 2016; Jungbluth et al., 2017; Escudero et al., 2018) we searched for the presence of dissimilatory sulfate reduction pathways in all obtained Firmicutes MAGs. Surprisingly, only two MAGs, Ch2 and Ch87, whose shares in the genome were 1.61 and 0.24%, respectively, encoded the necessary set of enzymes and could be sulfate reducers. The presence of NrfAH ammonia-forming cytochrome c nitrite reductases in the MAGs Ch2 and Ch87 suggested that nitrite could also be used as an electron acceptor in anaerobic respiration. Both these MAGs are phylogenetically distant from cultured Firmicutes and belong to the candidate class DTU015 proposed in the GTDB taxonomy (Parks et al., 2018).
Ch19, assigned to the order Limnochordales (Watanabe et al., 2015) and accounting for about 0.37% of the metagenome, was the only Firmicutes MAG encoding cytochrome c oxidase, indicating the capacity for aerobic respiration. This bacterium also possesses a complete set of genes coding for dissimilatory nitrite reduction pathways that could operate under anaerobic conditions, including cytochrome c nitrite reductase NrfAH, NO-forming nitrite reductase NirK, nitric oxide reductase (Nor) and nitrous oxide (Nos) reductase. All other Firmicutes MAGs lacked known enzymes for dissimilatory reduction of oxygen, sulfate, nitrate and nitrite and are probably limited to anaerobic fermentative metabolism.
MAG Ch108, sharing 63.4% AAI with Coprothermobacter proteolyticus DSM 5265, could be assigned to the family Coprothermobacteraceae. Coprothermobacter species are proteolytic anaerobic fermentative bacteria (Etchebehere et al., 1998) phylogenetically distant from other Firmicutes and recently proposed to be considered as a distinct phylum Coprothermobacterota (Pavan et al., 2018).
About 7.9% of the metagenome was assigned to the phylum Chloroflexi. All 12 Chloroflexi MAGs were assigned to the class Anaerolinea, members of which have been identified from diverse environments, including marine and freshwater sediments and deep subsurface aquifers (Yamada and Sekiguchi, 2009). They were also found in the Western Siberian deep subsurface (Kadnikov et al., 2018). Only one MAG, Ch72, was classified at the genus level, being assigned to the Thermanaerothrix, sharing 83.69% AAI with the only cultured species of this genus, thermophilic anaerobic bacterium Thermanaerothrix daxensis, isolated from a deep hot aquifer in France (Grégoire et al., 2011).
Anaerolinea are metabolically versatile heterotrophs capable of growing on various carbohydrates by fermentation, as well as aerobic and anaerobic respiration (Yamada et al., 2006). The search for respiratory pathways revealed the presence of cytochrome c oxidases in six MAGs, cytochrome c nitrite reductase NrfAH in four MAGs, NarGHI-type nitrate reductase in one MAG, NirK/NirS nitrite reductases in three MAGs and nitrous oxide (Nos) reductase in one MAG (Supplementary Table S1). Interestingly, contrary to T. daxensis, described as an anaerobic chemoorganotroph fermenting various carbohydrates, Ch72 contained cytochrome c oxidase and dissimilatory nitrite reductases and can probably obtain energy through respiration. Five Chloroflexi MAGs (Ch5, Ch9, Ch43, Ch80, and Ch93) do not contain any of the above-mentioned reductases. They also do not possess the complete set of genes of the TCA cycle and therefore are likely limited to a fermentative lifestyle.
Deltaproteobacteria accounted for 8.7% of the metagenome. The two most abundant genotypes, Ch3 (7.3%) and Ch 28 (1.1%), shared 54–55% AAI with Syntrophus aciditrophicus SB and were assigned to the family Syntrophaceae (recognized as the order Syntrophales in the GTDB taxonomy).
Cultured members of Syntrophacea are syntrophic organisms that degrade intermediates of organic decomposition, such as short-chain fatty and aromatic acids, to acetate, formate and hydrogen in co-culture with hydrogen-/formate-consuming methanogens or sulfate reducers (Elshahed et al., 2001; McInerney et al., 2007). Interestingly, these two MAGs have drastically different respiratory capacities. The more abundant organism, Ch3, appeared to lack known enzymes that enable dissimilatory reduction of sulfate, sulfite, nitrate and nitrite, as well as cytochrome c oxidases. Analysis of the Ch3 genome revealed the complete pathway for beta-oxidation of fatty acids, as well as enzymes involved in the utilization of butyrate and propionate. Hydrogen and formate could be formed as fermentation products, as evidenced by the presence of formate dehydrogenase and hydrogenase. The search for potentially secreted hydrolytic enzymes revealed the absence of signal peptide-containing glycosyl hydrolases and proteases. Probably, Ch3 bacterium is devoted to fermentation of low-molecular-weight fatty acids, presumably in syntrophic association with methanogenic archaea and/or sulfate reducers. The presence of multiple transporters for amino acids and peptides indicates the ability of Ch3 bacterium to ferment these substrates.
The genome of the second member of the Deltaproteobacteria, Ch28, was found to encode most of the enzymes of the dissimilatory sulfate reduction pathway, namely, sulfate adenylyltransferase, dissimilatory sulfite reductase (DsrAB and DsrC), sulfite reduction-associated complex DsrMKJOP, and the heterodisulfide reductase complex. However, genes for adenosine 5’-phosphosulfate reductase (AprAB) were not found. It is possible that Ch28 can reduce only sulfite rather than sulfate, or the aprAB genes were missed in the assembly. The presence of a [NiFe] group 1b hydrogenase that enables respiratory H2 uptake further supports the possibility of anaerobic respiration. Cytochrome c oxidases were not identified, but cytochrome c nitrite reductase NrfAH was encoded. Like the Ch3 bacterium, the Ch28 genome encodes the enzymes of the beta-oxidation pathway, indicating the possibility of utilization of fatty acids. The presence of an ABC transporter for the import of carbohydrates and amino acids/peptides likely indicates a broader substrate range of the Ch28 bacterium.
The third species, Ch74, assigned to the family Syntrophobacteraceae, represented a minor part of the community (0.26% of metagenome). Cultured members of this family are strictly anaerobic bacteria, having a respiratory or fermentative type of metabolism; some species form a syntrophic association with hydrogen/formate-utilizing partners (Kuever, 2014). Syntrophobacteraceae species are major acetate- and propionate-degrading sulfate reducers in paddy soil (Liu et al., 2018; Liu and Conrad, 2017). Analysis of the Ch74 genome revealed the apr and dsr genes of the sulfate reduction pathway, as well as nrfAH nitrite reductase and norB nitric oxide reductase.
The phylum Armatimonadetes was represented by three MAGs, and together they accounted for about 5% of the metagenome. Members of this bacterial phylum, initially found in Obsidian Pool, Yellowstone National Park and designated as OP10 (Hugenholtz et al., 1998), have been detected in numerous 16S rRNA gene surveys and metagenomic studies from various environments. A few cultured members of this phylum are aerobic heterotrophs growing on various sugars, including complex polysaccharides (Lee et al., 2011, 2014; Im et al., 2012; Li et al., 2019).
One MAG, Ch118, was assigned to the class Chthonomonadetes, comprising a single cultured member, Chthonomonas calidirosea, a thermophilic motile aerobic bacterium capable of utilizing a wide range of carbohydrates, including starch, carboxymethyl cellulose and xylan (Lee et al., 2011). Although Ch118 is phylogenetically rather distant from this species (47.5% AAI), its genome also encoded flagellar motility, an aerobic respiratory chain with cytochrome c oxidase and numerous carbohydrate-active enzymes and sugar transporters. Anaerobic respiration could be enabled by dissimilatory nitrate, nitrite, nitric-oxide, and tetrathionate reductases.
The most abundant Armatimonadetes MAG (3.3% of the metagenome), Ch1, was assigned to the candidate class UBA5377 in the GTDB taxonomy, still lacking cultured members. The third Armatimonadetes MAG, Ch33, belonged to the same class. Analysis of both these genomes revealed the absence of an aerobic respiratory chain, as well as pathways for dissimilatory reduction of sulfate and nitrate. Presumably, these two organisms are anaerobic fermenters, unlike all cultured Armatimonadetes species.
The recently proposed candidate phylum Kapabacteria (Kantor et al., 2015), also known as the OPB56 clade within the Bacteroidetes/Chlorobi/Ignavibacteriae superphylum, accounted for 1.29% of the metagenome and comprised a single genotype, Ch6. The genome of this bacterium was assembled as a single circular 2,660,650 bp long chromosome; this is the first known complete genome of Ca. Kapabacteria. It was predicted to contain 2275 protein-coding genes, 47 tRNA genes and a single 16S-23S-5S rRNA operon.
Analysis of the Ch6 genome revealed the presence of a complete set of genes required for dissimilatory sulfate reduction, including sulfate adenyltransferase, adenisine-5’-phisphate reductase, dissimilatory sulfite reductase and related redox complexes Qmo and DsrMKJOP. These results are consistent with the finding of a sulfate reduction pathway in the partial genomes of another member of Ca. Kapabacteria, Ca. Thermonerobacter thiotrophicus, obtained from hot springs in United States and Japan (Thiel et al., 2019). Taking into account 96.81% AAI between Ch6 and Ca. T. thiotrophicus MAG Naka2016_bin-10 (GenBank GCA_003731655.1) from Japan, these two bacteria probably represent a single species. Like the latter genome, Ch6 MAG contained genes for NrfAH-type nitrite reductase, and nitrous oxide reductase (NosZ), cytochrome bd ubiquinol oxidase and an aerobic respiratory pathway with a terminal aa3-type cytochrome c oxidase. Additionally, the Ch6 genome encodes ArrAB-type respiratory arsenate reductase, absent in the Naka2016_bin-10 genome assembly. The presence of both dsr and cox genes is unusual since sulfate-reducing bacteria are unable to derive energy from aerobic respiration. Nevertheless, cytochrome c oxidase genes were found in Desulfovibrio genomes, and the ability of a mutant strain of Desulfovibrio vulgaris to grow with energy derived from oxidative phosphorylation linked to oxygen reduction was recently demonstrated (Schoeffler et al., 2019). Therefore, it is possible that the Ch6 bacterium is metabolically versatile and adapted to fluctuating concentrations of oxygen and alternative electron acceptors.
Analysis of the Ch6 genome revealed a very limited potential of this bacterium to utilize organic substrates. No presumably secreted carbohydrate-active and proteolytic enzymes were identified. We found no transporters for the import of sugars and only a few transporters of peptides and amino acids. A chemolithoautotrophic lifestyle is unlikely, since the genome lacked the Wood–Ljungdahl pathway typically used by sulfate reducers for carbon fixation, as well as other CO2 fixation pathways. Therefore, the Ch6 bacterium is probably a highly specialized organism with a narrow substrate range. The presence of acetyl-CoA synthetase (ADP-forming) suggests that the Ch6 bacterium could assimilate acetate and oxidize it in the course of aerobic or anaerobic respiration.
The Ch6 genome contained genes required for flagellar motility and chemotaxis, indicating that this bacterium is motile, like members of the Bacteroidetes and Ignavibacteriae. However, genes encoding the type IV pili for twitching motility were not found. The type IV pili enable attachment of the cells to insoluble substrates, and their absence is consistent with the predicted inability of Ch6 to hydrolyse polysaccharides.
The phylum Ignavibacteriae was the second most numerous lineage in the microbial community of groundwater. It was represented by two MAGs, Ch128a, and Ch128b, accounting for 12.08 and 2.22% of the whole metagenome, respectively. Both MAGs were only distantly related to cultured species of this phylum, Ignavibacterium album (Iino et al., 2010) and Melioribacter roseus (Podosokorskaya et al., 2013).
The genome of the Ch128a bacterium was sequenced to 764x average coverage and assembled into a single circular chromosome with a length of 2,403,876 bp. Taking into account the abundance of this lineage, we took advantage of availability of a complete genome sequence for detailed analysis of the phylogenetic position and metabolic potential of the Ch128a bacterium.
As a result of genome annotation, 2,067 potential protein-coding genes were identified, and the functions of 1,017 (49%) of them were tentatively assigned. A single ribosomal RNA operon comprising the 16S-23S-5S rRNA genes and 44 transfer RNA (tRNA) genes were identified in the genome. The Ch128a cells were predicted to be rod-shaped, based on the identification of genes encoding the rod-shape determining proteins MreBCD, RodA and peptidoglycan D,D-transpeptidase MrdA. The bacterium is likely non-motile because it lacks flagellar machinery and chemotaxis genes.
The Ch128a 16S rRNA gene shares only 86% sequence identity with the closest cultured bacterium, Ignavibacterium album. Several 16S rRNA sequences with about 90% identity (AY344403, RPQH01000040, QEVB01000012, MWSW01000018, etc.) were detected in freshwater lakes and bioreactor sludge. To determine the phylogenetic position of the Ch128a bacterium, a phylogenetic tree based on concatenated sequences of conservative marker genes of bacteria assigned to the Bacteroidetes/Chlorobi group (the phylum Bacteroidota in the GTDB taxonomy) was constructed. The results confirmed that Ch128a belongs to Ignavibacteriae and placed it within the candidate order SJA-28 (also known as Chlorobi lineage 4), as defined by the GTDB taxonomy (Figure 2). The AAI values between Ch128a and other SJA-28 genomes, OLB4, OLB5, UTCHB1, UBA2330 and UBA6667 was in the range of 48–53%. The taxonomic status of the SJA-28 lineage, as well as Ignavibacteriae, remained unclear. Ignavibacteriae have been described as a distinct phylum in the Bacteroidetes/Chlorobi group, along with Bacteroidetes and Chlorobi (Podosokorskaya et al., 2013), and such classification is currently used in the NCBI taxonomy database (Federhen, 2012). The GTDB taxonomy considers Ignaviibacteria as a class (comprising orders Ignavibacteriales and SJA-28) in the phylum Bacteroidota, along with the classes Bacteroidia, Chlorobi, Rhodothermia, Ca. Kapabacteria, UBA10030, and Ca. Kryptonia. Our phylogenetic analysis showed that SJA-28 is a distinct lineage within a clade comprising Ignavibacteriales, UBA10030 and Ca. Kryptonia (Figure 2). This whole group is separated from the Chlorobi, Ca. Kapabacteria and Bacteroidetes/Rhodothermia clades and thus could be considered as the phylum Ignavibacteriae or as the class Ignaviibacteria of the larger phylum Bacteroidota.
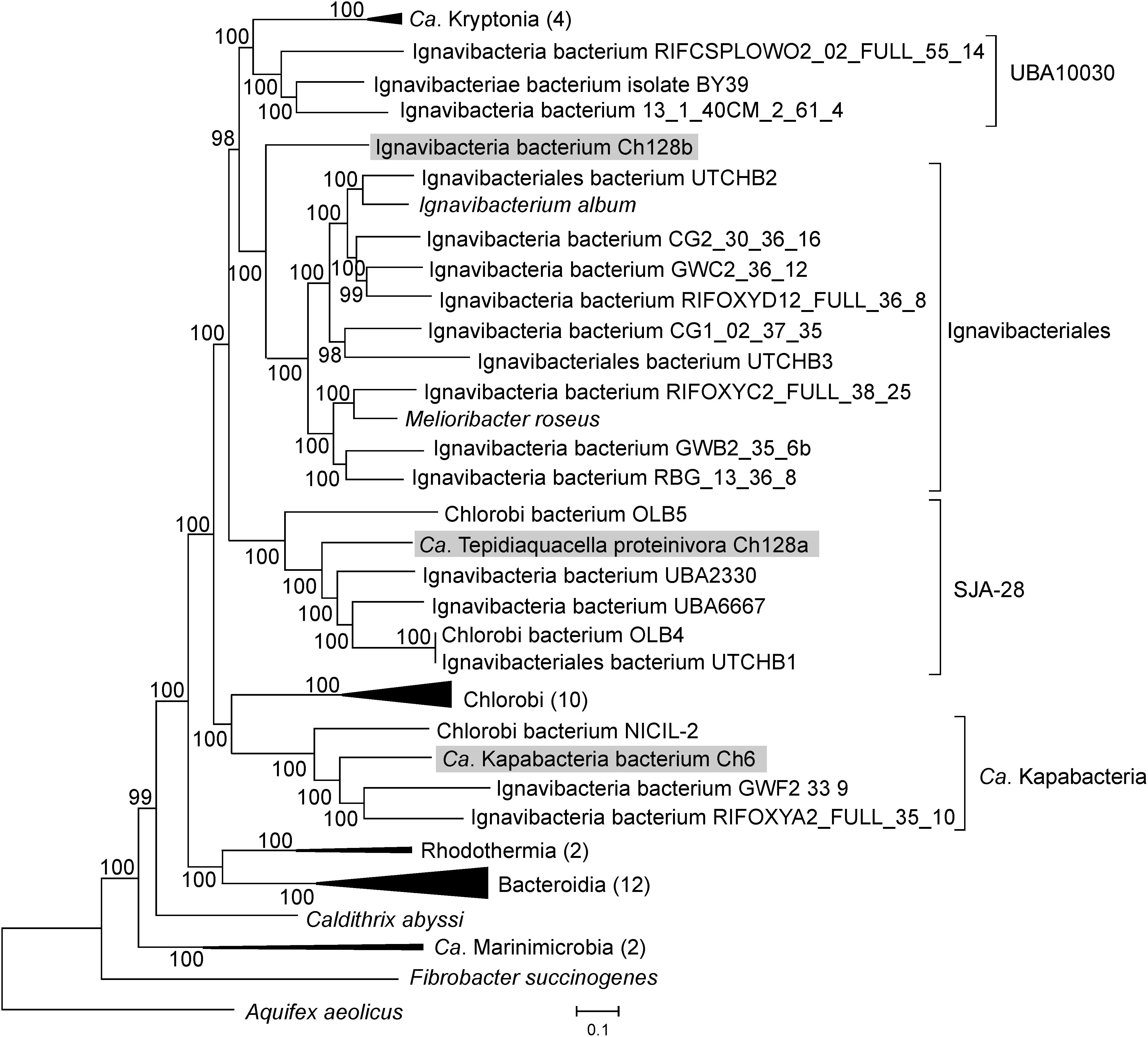
Figure 2. Phylogenomic placement of the Ch6, Ch128a, and Ch128b genomes in the maximum likelihood concatenated protein phylogeny of the Bacteroidetes/Chlorobi group. The level of support for internal branches was assessed using the Bayesian test in PhyML. The numbers of genomes used for the analysis in the collapsed clusters is shown after the name of the taxon. Taxonomy is shown according to the GTDB.
Analysis of the Ch128a genome revealed that this microorganism has a limited capacity to degrade complex organic substrates. Contrary to cultured members of Ignavibacteriae, M. roseus and I. album, metabolically versatile facultative anaerobes growing on various carbohydrates (Liu et al., 2012; Kadnikov et al., 2013), the Ch128a bacterium lacked secreted carbohydrate-active enzymes and thus was predicted to be unable to utilize polysaccharides. A few encoded glycoside hydrolases lacked N-terminal secretion signals and are likely involved in the synthesis and degradation of a storage polysaccharide, glycogen. Consistently, the Ch128a genome encodes a complete set of genes for the Embden-Meyerhof pathway of glycolysis and gluconeogenesis, as well as the non-oxidative branch of the pentose phosphate pathway (Figure 3). The ability of the Ch128a bacterium to utilize proteinaceous substrates is indicated by the presence of five signal-peptide-containing proteases belonging to the M3, M13, M16, M28, and C1 families, and amino acid transport systems. The imported amino acids can then be deaminated by a number of aminotransferases in glutamate dehydrogenase coupled reactions, followed by oxidation to generate the corresponding coenzyme A (CoA) derivatives (Figure 3). This step could be performed by ferredoxin-dependent oxidoreductases with different substrate specificities, including pyruvate: ferredoxin oxidoreductase, indolepyruvate: ferredoxin oxidoreductase and 2-oxoglutarate ferredoxin oxidoreductase. Pyruvate could also be used by pyruvate formate lyase, which catalyses the conversion of pyruvate and CoA to formate and acetyl-CoA. The acyl-CoA derivatives in Ch128a could be oxidized to the corresponding acids by acetyl-CoA synthetase and succinyl-CoA synthetase with concomitant generation of ATP. The presence of aldehyde and alcohol dehydrogenases suggested that alcohols could be produced as fermentation products. NADH and reduced ferredoxin, generated in fermentation pathways, could be re-oxidized by cytoplasmic [FeFe] group A hydrogenase and group 3c [NiFe] heterodisulfide reductase-linked hydrogenase (MvhADG/HdrABC) that bifurcates electrons from H2 to heterodisulfide and ferredoxin (Kaster et al., 2011).
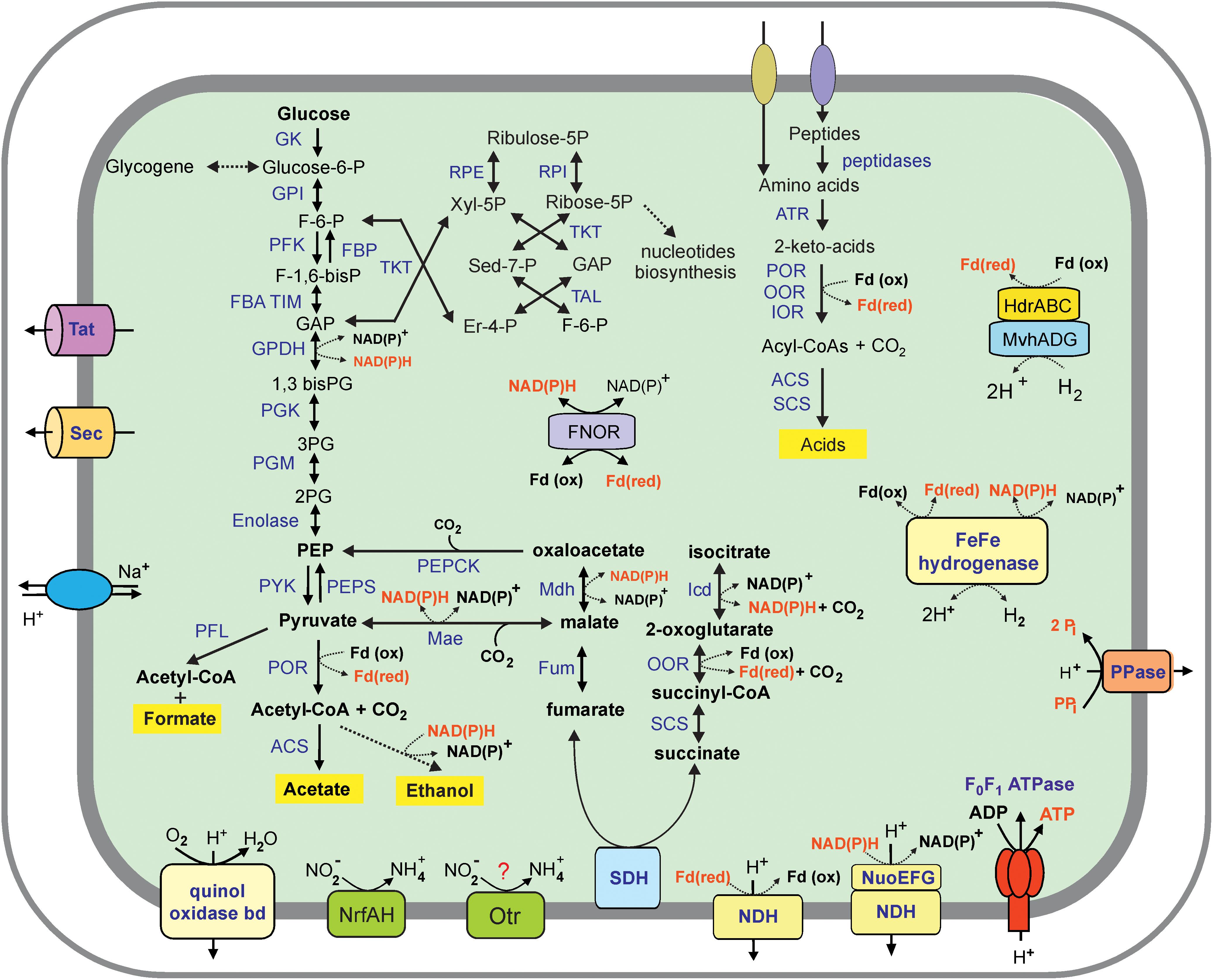
Figure 3. An overview of the metabolism of Ignavibacteriae bacterium Ch128a (Candidatus Tepidiaquacella proteinivora) reconstructed from its genome. Enzyme abbreviations: GK, glukokinase; GPI, glucose-6-phosphate isomerase; PFK, 6-phosphofructokinase; FBA, fructose-bisphosphate aldolase; TIM, triosephosphate isomerase; GPDH, glyceraldehyde 3-phosphate dehydrogenase; PGK, phosphoglycerate kinase; PGM, phosphoglycerate mutase; PYK, pyruvate kinase; PEPS, phosphoenolpyruvate synthase; FBP, fructose-1,6-bisphosphatase; PEPCK, phosphoenolpyruvate carboxykinase; RPE, ribulose-phosphate 3-epimerase; RPI, ribose 5-phosphate isomerase; TKT, transketolase, TAL, transaldolase, PFL, pyruvate formate lyase; POR, pyruvate ferredoxin oxidoreductase; ACS, acetyl-CoA synthetase, Mdh, malate dehydrogenase; Mae, malic enzyme; Fum, fumarate hydratase; SDH, succinate dehydrogenase; SCS, succinyl-CoA ligase; OOR, 2-oxoglutarate ferredoxin oxidoreductase; Icd, isocitrate dehydrogenase; ATR, aminotransferase; IOR, indolepyruvate ferredoxin oxidoreductase; FNOR, ferredoxin-NAD(P) + reductase; PPase, pyrophosphatase; NDH, NADH dehydrogenase (subunits NuoABCDHIJKLMN); Otr, octaheme tetrathionate reductase. Other abbreviations: F-6-P, fructose 6-phosphate; F-1,6-bisP, fructose-1,6-bisphosphate; GAP, glyceraldehyde-3 phosphate; 1,3–bis–PG, 1,3–biphosphoglycerate; 3–PG, 3–phosphoglycerate; 2–PG, 2–phosphoglycerate; PEP, phosphoenolpyruvate; Xyl-5P, xylulose 5-phosphate; Er-4-P, erythrose 4-phosphate; Sed–7-P, sedoheptulose–7–phosphate; Pi, phosphate; PPi, pyrophosphate.
The key enzymes of autotrophic carbon fixation pathways, the Calvin-Benson-Bassham, 3-hydroxypropionate and Wood–Ljungdahl pathways were not found. The tricarboxylic acid cycle is incomplete due to the lack of citrate synthase and aconitate hydratase and is likely used only for biosynthetic purposes.
Analysis of the Ch128a genome reveal the presence of subunits of the proton-translocating NADH-dehydrogenase complex, while the cytochrome bc1 complex, quinol-oxidizing alternative complex III, and terminal cytochrome c oxidases were not found, suggesting the inability of the Ch128a bacterium to grow by aerobic respiration. The only terminal reductase, the quinol oxidase bd complex, is probably involved in oxygen detoxification rather than respiration, as in most anaerobic bacteria. The Ch128a genome contained several clusters and separate genes encoding a full set of 14 NADH dehydrogenase subunits: ndhABCDHIJKLMN, ndhABCDEF, ndhKL, ndhG, ndhH, ndhJ, ndhM, and ndhAB. However, the absence of nuoEFG genes encoding the NADH-interacting module (Sazanov, 2007) in the first cluster suggests that it could encode an alternative proton-transporting complex that accepts electrons from reduced ferredoxin rather than NADH, as suggested for M. roseus (Kadnikov et al., 2013). The generated transmembrane ion gradient can be used for ATP generation by membrane F0F1-type ATP synthase (Figure 3).
Genome analysis revealed only two potential terminal reductases of anaerobic respiratory pathways. The first one is an ammonia-forming cytochrome c nitrite reductase of the NrfAH-type. The second gene cluster encodes the octaheme tetrathionate reductase (Otr) and cytochrome b561 protein. The Otr enzymes are responsible for the reduction of tetrathionate to thiosulfate but could also be involved in nitrite reduction (Simon et al., 2011). Both predicted reductases contain subunits with transmembrane helices and are probably linked to the cytoplasmic membrane where they could accept electrons from the quinone pool. The presence of this enzyme can enable the use of nitrite and sulfur compounds under anaerobic conditions. The genome of Ch128a is the first complete genome of a member of the SJA-28 lineage of the phylum Ignavibacteriae, and we propose the following taxonomic names for the novel genus and species of Ch128a:
Description of Candidatus Tepidiaquacella gen. nov.
Candidatus Tepidiaquacella (Te.pi.di.a.qua.cel’la, L. adj. tepidus warm; L. n. aqua, water; L. fem. n. cella, cell; N.L. fem. n. Tepidiaquacella, a cell from warm water).
Description of Candidatus Tepidiaquacella proteinivora sp. nov.
Tepidiaquacella proteinivora (pro.te.i.ni’vo.ra N.L. neut. n. proteinum protein; L. fem. suff. vora devouring; N.L. fem. adj. proteinivora devouring proteins).
Not cultivated. Inferred to be a rod-shaped, non-motile, anaerobic, obligate organotroph. Obtains energy by fermentation or respiration with nitrite and is able to use proteinaceous substrates for growth. Represented by the complete genome (GenBank CP054675) obtained from metagenome of a deep subsurface thermal aquifer in Western Siberia, Russia.
On this basis, we propose the following names for the order and family:
Candidatus Tepidiaquacellales ord. nov.
Candidatus Tepidiaquacellaceae fam. nov.
Assuming the results of phylogenetic analysis, and because of the appearance of a representative with known complete genome sequence, the SJA-28 lineage is proposed to be named as Candidatus Tepidiaquacellales ord. nov. within the class Ignavibacteria in the phylum Ignavibacteriae (Podosokorskaya et al., 2013). The order Candidatus Tepidiaquacellales is defined on a phylogenetic basis by comparative genome sequence analysis of Candidatus Tepidiaquacella proteinivora Ch128a, OLB4, OLB5, UTCHB1, UBA2330, and UBA6667.
Metagenomic analysis of the underground water reservoir revealed a diverse microbial community. First, it should be emphasized that the analyzed aquifer is located in Mesozoic sedimentary rocks, formed from sediments of marine origin. It is these deposits that became the basis for the formation of oil and gas fields in Western Siberia. This buried and completely or partly altered organic matter can support the development of organotrophic microbial communities that differ from the lithoautotrophic communities of oligotrophic subsurface ecosystems in which molecular hydrogen of abiotic origin is the main source of energy (Takai et al., 2004; Chivian et al., 2008; Pedersen, 2012).
Metagenomic analysis revealed four main metabolic groups of microorganisms (Figure 4). The first group is methanogenic archaea, which accounted for about 20% of the community. The most numerous among them were hydrogenotrophic methanogens of the order Methanobacteriales, which are widespread in various subsurface ecosystems, including deep underground aquifers in Western Siberia (Frank et al., 2016). The presence of acetoclastic and methyl-reducing methanogens indicates the use of not only hydrogen (which can be of both biogenic and abiogenic origin), but also products of microbial metabolism for methanogenesis. The second important functional group is sulfate reducers. Like methanogens, they are typical members of both lithoautotrophic and organotrophic microbial communities of the deep subsurface ecosystems, including Western Siberia (Moser et al., 2005; Chivian et al., 2008; Frank et al., 2016; Kadnikov et al., 2018). The sulfate reduction pathway was revealed in two genomes of Firmicutes (Ch2 and Ch87) and in the members of Deltaproteobacteria (Ch28 and Ch74), Ca. Kapabacteria (Ch6), and Nitrospirae (Thermodesulfovibrio aggregans, Ch24). In total, sulfate reducers accounted for only about 5% of the community, which is a much smaller proportion than in the microbial communities of aquifers accessible through boreholes 3P Parabel (∼50%) and 1-R Byelii Yar (∼30%). Considering that the concentration of sulfate in 5P water is several times higher than at these two boreholes (<5 mg L–1, Frank et al., 2016; Kadnikov et al., 2018), a lower relative abundance of sulfate reducers is unexpected, but this may be explained by a higher total concentration of microorganisms in the water collected at the 5P borehole. Alternatively, a higher concentration of sulfate in 5P water could be a consequence of lower abundance of sulfate-reducers and an overall low rate of the sulfur cycle.
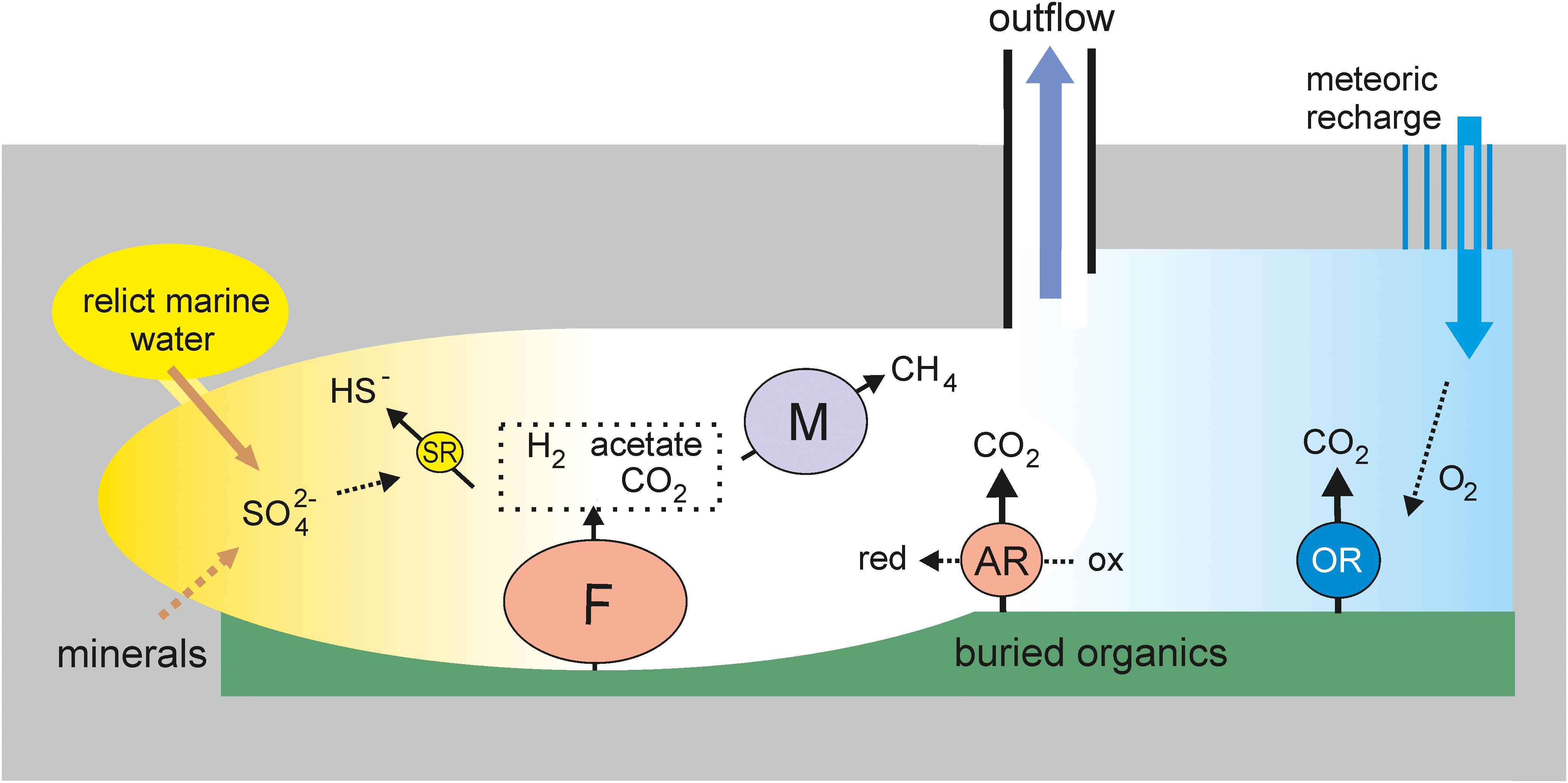
Figure 4. An ecological model of the deep subsurface aquifer. OR, aerobic respiration; F, fermentation; SR, sulfate reduction; AR, anaerobic respiration (other than SR); M, methanogenesis. The sizes of the circles roughly reflect the relative abundance of the respective metabolic groups in the metagenome.
The third major functional group is organotrophs with aerobic respiratory pathways, marked by the presence of cytochrome c oxidase. These include MAGs from the phyla Chloroflexi (Ch21, Ch27, Ch132, Ch71, Ch72, and Ch58), Firmicutes (Ch19), Ignavibacteriae (Ch128b), Gammaproteobacteria (Ch67), Armatimonadetes (Ch118), Bacteroidetes (Ch62), Ca. Omnitrophica (Ch114), and Acidobacteria (Ch122). The cytochrome c oxidase genes are also present in sulfate reducers assigned to Ca. Kapabacteria (Ch6). Some of these genomes also contain genes for enzymes involved in the oxidation of sulfur compounds. In total, organotrophs potentially capable of aerobic respiration accounted for 13% of the community.
Most of the community is composed of anaerobic, presumably heterotrophic microorganisms with a fermentative type of metabolism; the genomes of many of them also contain enzymes for the dissimilatory reduction of nitrite, nitrate, Fe (III), thiosulfate, tetrathionate, polysulfides, etc. This metabolic group includes most Firmicutes, some Chloroflexi, the most abundant lineages of Ignavibacteriae (Ch128a), Deltaproteobacteria (Ch3), Armatimonadetes (Ch1 and Ch33), and Bacteroidetes (Ch61), as well as a number of less abundant phyla (WOR-3, Thermotogae, Ca. Riflebacteria, Spirochaetes, Ca. Bipolaricaulota, Actinobacteria, Ca. Aminicenantes, and Ca. Atribacteria). The genomes of some anaerobic members of Firmicutes and Chloroflexi encoded a Wood–Ljungdahl pathway that can be used for autotrophic carbon fixation. These microorganisms can produce acetate and hydrogen consumed by sulfate reducers and methanogens. Fermentative Firmicutes and Deltaproteobacteria capable of oxidizing low-molecular-weight organics could be involved in syntrophic associations with methanogens or anaerobic respiring organisms. An important role for syntropy in deep subsurface environments has been reported (Gray et al., 2010; Kimura et al., 2010; Matsushita et al., 2020).
Some of the functional groups of microorganisms described above, for example, methanogens and aerobic heterotrophs, have mutually exclusive requirements regarding environmental conditions, which indicates the heterogeneity of conditions in the deep subsurface aquifer (Figure 4). The capacity for aerobic respiration is a rather unexpected property for microorganisms of the deep underground biosphere, which is considered as a strictly anaerobic habitat (Teske, 2005). However, their presence is consistent with the presence of several percent oxygen in dissolved gas. Oxygen was also found in gas samples taken over several years from 3P Parabel and 1-R Byelii Yar boreholes (Frank et al., 2016; Kadnikov et al., 2018). It can be proposed that oxygen and aerobic microorganisms were delivered to the reservoir from the surface, with meteoric recharge waters. Favorable conditions for the development of aerobes can be formed locally in the zones of entry of such waters into the aquifer, and sulfide oxidation with the formation of sulfate can also occur in them. In other parts of the underground aquifer system, strictly anaerobic conditions favorable to fermentative microorganisms, as well as sulfate reducers and methanogens consuming their metabolic products, can be maintained. Such respiratory versatile bacteria as Ch6 Kapabacteria, predicted to be able to use both oxygen and sulfate for respiration, could grow successfully at the oxic-anoxic interface. Sulfate reducers and methanogens probably develop in spatially separated parts of the aquifer, respectively, those rich and poor in sulfate. Sulfate could originate from the oxidation of sulfide minerals or the dissolution of sulfates, such as gypsum (calcium sulfate). The sulfate moiety from sedimentary low-soluble sulfates, such as barite (barium sulfate) and celectine (strontium sulfate), can also be used by sulfate reducing bacteria as an electron acceptor (Karnachuk et al., 2002). Sulfate could also remain in relict waters of marine origin. The observed temporary fluctuations in the sulfate content in the water collected at the borehole may reflect the flow of water from different parts of the reservoir.
It should be mentioned that the above four functional groups of prokaryotes were also detected in two other previously characterized deep subsurface aquifers in Western Siberia, albeit in various combinations. Methanogens, sulfate reducers and a small fraction of anaerobic heterotrophs were found in the 3P Parabel borehole, while potential aerobes were missing (Frank et al., 2016). Sulfate reducers, as well as aerobic and anaerobic heterotrophs, were found in the 1-R Byelii Yar borehole, but methanogens were nearly absent (Kadnikov et al., 2018). Apparently, the composition of the aquifer community as a whole is determined by the rate of meteoric recharge, which delivers oxygen, and the availability of sulfate. Thus, the water from the 3P Parabel borehole had the highest salinity (13–14 g L–1), which indicates the minimal inflow of oxygenated water from the surface, and a predominantly anaerobic chemolithoautotrophic community of sulfate reducers and methanogens became formed.
Overall, the availability of buried organic matter of marine sediments of the Mesozoic era and the spatial heterogeneity of the underground aquifer create the conditions for the development of microbial communities, taxonomically and functionally much more diverse than those found in oligotrophic underground ecosystems.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, SRR6186653; https://www.ncbi.nlm.nih.gov/genbank/, SRR11854357; and https://www.ncbi.nlm.nih.gov/genbank/, PRJNA414521.
NR designed the research project and drafted the manuscript. VK and AM performed metegenome sequencing. VK, AB, OK, and NR analyzed the data. All authors read and approved the manuscript.
This work was partly supported by the Russian Science Foundation (grant no. 19-14-00245 to NR).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was performed using the scientific equipment of the Core Research Facility “Bioengineering” (Research Center of Biotechnology RAS).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.572252/full#supplementary-material
Alneberg, J., Bjarnason, B. S., de Bruijn, I., Schirmer, M., Quick, J., Ijaz, U. Z., et al. (2014). Binning metagenomics contigs by coverage and composition. Nat. Methods 11, 1144–1146. doi: 10.1038/nmeth.3103
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Banks, D., Frank, Y. A., Kadnikov, V. V., Watts, M., Boyce, A., and Frengstad, B. S. (2014). Hydrochemical Data Report from Sampling of Two Deep Abandoned Hydrocarbon Exploration Wells: Byelii Yar and Parabel’, Tomsk oblast’, Western Siberia, Russian Federation. NGU Report No. 2014.034, Trondheim: Geological Survey of Norway.
Bomberg, M., Nyyssönen, M., Pitkänen, P., Lehtinen, A., and Itävaara, M. (2015). Active microbial communities inhabit sulphate-methane interphase in deep bedrock fracture fluids in Olkiluoto, Finland. Biomed. Res. Int. 2015:979530.
Bowers, R. M., Kyrpides, N. C., Stepanauskas, R., Harmon-Smith, M., Doud, D., Reddy, T. B. K., et al. (2017). Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725–731.
Brettin, T., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Olsen, G. J., et al. (2015). RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5:8365.
Cao, M. D., Nguyen, S. H., Ganesamoorthy, D., Elliott, A. G., Cooper, M. A., and Coin, L. J. (2017). Scaffolding and completing genome assemblies in real-time with nanopore sequencing. Nat. Commun. 8:14515.
Castelle, C. J., Brown, C. T., Anantharaman, K., Probst, A. J., Huang, R. H., and Banfield, J. F. (2018). Biosynthetic capacity, metabolic variety and unusual biology in the CPR and DPANN radiations. Nat. Rev. Microbiol. 16, 629–645. doi: 10.1038/s41579-018-0076-2
Chivian, D., Brodie, E. L., Alm, E. J., Culley, D. E., Dehal, P. S., DeSantis, T. Z., et al. (2008). Environmental genomics reveals a single-species ecosystem deep within Earth. Science 322, 275–278. doi: 10.1126/science.1155495
Colman, D. R., Poudel, S., Stamps, B. W., Boyd, E. S., and Spear, J. R. (2017). The deep, hot biosphere: twenty-five years of retrospection. Proc. Natl. Acad. Sci. U.S.A. 114, 6895–6903.
Edwards, K. J., Becker, K., and Colwell, F. (2012). The deep, dark energy biosphere: intraterrestrial life on earth. Annu. Rev. Earth Planet. Sci. 40, 551–568. doi: 10.1146/annurev-earth-042711-105500
Elshahed, M. S., Bhupathiraju, V. K., Wofford, N. Q., Nanny, M. A., and McInerney, M. J. (2001). Metabolism of benzoate, cyclohex-1-ene carboxylate, and cyclohexane carboxylate by “Syntrophus aciditrophicus” strain SB in syntrophic association with H(2)-using microorganisms. Appl. Environ. Microbiol. 67, 1728–1738. doi: 10.1128/aem.67.4.1728-1738.2001
Escudero, C., Oggerin, M., and Amils, R. (2018). The deep continental subsurface: the dark biosphere. Int. Microbiol. 21, 3–14. doi: 10.1007/s10123-018-0009-y
Etchebehere, C., Pavan, M. E., Zorzópulos, J., Soubes, M., and Muxí, L. (1998). Coprothermobacter platensis sp. nov., a new anaerobic proteolytic thermophilic bacterium isolated from an anaerobic mesophilic sludge. Int. J. Syst. Bacteriol. 48(Pt. 4), 1297–1304. doi: 10.1099/00207713-48-4-1297
Frank, Y. A., Kadnikov, V. V., Gavrilov, S. N., Banks, D., Gerasimchuk, A. L., Podosokorskaya, O. A., et al. (2016). Stable and variable parts of microbial community in siberian deep subsurface thermal aquifer system revealed in a long-term monitoring study. Front. Microbiol. 7:2101. doi: 10.3389/fmicb.2016.02101
Fredrickson, J. K., and Hicks, R. J. (1987). Probing reveals many microbes beneath Earth’s surface. A.S.M. News 53, 78–79.
Ghiorse, W. C., and Wilson, J. T. (1988). Microbial ecology of the terrestrial subsurface. Adv. Appl. Microbiol. 33, 107–173.
Gray, N. D., Sherry, A., Hubert, C., Dolfing, J., and Head, I. M. (2010). Methanogenic degradation of petroleum hydrocarbons in subsurface environments remediation, heavy oil formation, and energy recovery. Adv. Appl. Microbiol. 72, 137–161. doi: 10.1016/s0065-2164(10)72005-0
Grégoire, P., Fardeau, M. L., Joseph, M., Guasco, S., Hamaide, F., Biasutti, S., et al. (2011). Isolation and characterization of Thermanaerothrix daxensis gen. nov., sp. nov., a thermophilic anaerobic bacterium pertaining to the phylum “Chloroflexi”, isolated from a deep hot aquifer in the Aquitaine Basin. Syst. Appl. Microbiol. 34, 494–497. doi: 10.1016/j.syapm.2011.02.004
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hattori, S., Kamagata, Y., Hanada, S., and Shoun, H. (2000). Thermacetogenium phaeum gen. nov., sp. nov., a strictly anaerobic, thermophilic, syntrophic acetate-oxidizing bacterium. Int. J. Syst. Evol. Microbiol. 50(Pt. 4). 1601–1609. doi: 10.1099/00207713-50-4-1601
Hidalgo-Ahumada, C. A. P., Nobu, M. K., Narihiro, T., Tamaki, H., Liu, W. T., and Kamagata, Y, et al. (2018). Novel energy conservation strategies and behaviour of Pelotomaculum schinkii driving syntrophic propionate catabolism. Environ. Microbiol. 20, 4503–4511. doi: 10.1111/1462-2920.14388
Hu, P., Tom, L., Singh, A., Thomas, B. C., Baker, B. J., Piceno, Y. M., et al. (2016). Genome-resolved metagenomic analysis reveals roles for candidate phyla and other microbial community members in biogeochemical transformations in oil reservoirs. mBio 7:e01669-15.
Hubalek, V., Wu, X., Eiler, A., Buck, M., Heim, C., Dopson, M., et al. (2016). Connectivity to the surface determines diversity patterns in subsurface aquifers of the Fennoscandian shield. ISME J. 10, 2447–2458. doi: 10.1038/ismej.2016.36
Hugenholtz, P., Pitulle, C., Hershberger, K. L., and Pace, N. R. (1998). Novel division level bacterial diversity in a Yellowstone hot spring. J. Bacteriol. 180, 366–376. doi: 10.1128/jb.180.2.366-376.1998
Iino, T., Mori, K., Uchino, Y., Nakagawa, T., Harayama, S., and Suzuki, K. (2010). Ignavibacterium album gen. nov., sp. nov., amoderately thermophilic anaerobic bacterium isolated frommicrobial mats at a terrestrial hot spring and proposal of Ignavibacteria classis nov., for a novel lineage at the periphery of green sulfur bacteria. Int. J. Syst. Evol. Microbiol. 60, 1376–1382. doi: 10.1099/ijs.0.012484-0
Im, W. T., Hu, Z. Y., Kim, K. H., Rhee, S. K., Meng, H., Lee, S. T., et al. (2012). Description of Fimbriimonas ginsengisoli gen. nov., sp. nov. within the Fimbriimonadia class nov., of the phylum Armatimonadetes. Antonie Van Leeuwenhoek. 102, 307–317. doi: 10.1007/s10482-012-9739-6
Imachi, H., Sekiguchi, Y., Kamagata, Y., Hanada, S., Ohashi, A., and Harada, H. (2002). Pelotomaculum thermopropionicum gen. nov., sp. nov., an anaerobic, thermophilic, syntrophic propionate-oxidizing bacterium. Int. J. Syst. Evol. Microbiol. 52(Pt. 5), 1729–1735. doi: 10.1099/ijs.0.02212-0
Itävaara, M., Nyyssönen, M., Kapanen, A., Nousiainen, A., Ahonen, L., and Kukkonen, I. (2011). Characterization of bacterial diversity to a depth of 1500 m in the Outokumpu deep borehole, Fennoscandian Shield. FEMS Microbiol. Ecol. 77, 295–309. doi: 10.1111/j.1574-6941.2011.01111.x
Jungbluth, S. P., Glavina Del Rio, T., Tringe, S. G., Stepanauskas, R., and Rappé, M. S. (2017). Genomic comparisons of a bacterial lineage that inhabits both marine and terrestrial deep subsurface systems. PeerJ 5:e3134.
Kadnikov, V. V., Mardanov, A. V., Beletsky, A. V., Banks, D., Pimenov, N. V., Frank, Y. A., et al. (2018). A metagenomic window into the 2-km-deep terrestrial subsurface aquifer revealed multiple pathways of organic matter decomposition. FEMS Microbiol. Ecol. 94:fiy152.
Kadnikov, V. V., Mardanov, A. V., Beletsky, A. V., Karnachuk, O. V., and Ravin, N. V. (2019a). Genome of the candidate phylum Aminicenantes bacterium from a deep subsurface thermal aquifer revealed its fermentative saccharolytic lifestyle. Extremophiles 23, 189–200. doi: 10.1007/s00792-018-01073-5
Kadnikov, V. V., Mardanov, A. V., Beletsky, A. V., Rakitin, A. L., Frank, Y. A., Karnachuk, O. V., et al. (2019b). Phylogeny and physiology of candidate phylum BRC1 inferred from the first complete metagenome-assembled genome obtained from deep subsurface aquifer. Syst. Appl. Microbiol. 42, 67–76. doi: 10.1016/j.syapm.2018.08.013
Kadnikov, V. V., Mardanov, A. V., Podosokorskaya, O. A., Gavrilov, S. N., Kublanov, I. V., Beletsky, A. V., et al. (2013). Genomic analysis of Melioribacter roseus, facultatively anaerobic organotrophic bacterium representing a novel deep lineage within Bacteriodetes/Chlorobi group. PLoS One 8:e53047. doi: 10.1371/journal.pone.0053047
Kadnikov, V. V., Frank, Y. A., Mardanov, A. V., Beletskii, A. V., Ivasenko, D. A., Pimenov, N. V., et al. (2017a). Uncultured bacteria and methanogenic archaea predominate in the microbial community of Western Siberian deep subsurface aquifer. Microbiology 86, 412–415. doi: 10.1134/S0026261717030079
Kadnikov, V. V., Frank, Y. A., Mardanov, A. V., Beletsky, A. V., Ivasenko, D. A., Pimenov, N. V., et al. (2017b). Variability of the composition of the microbial community of the deep subsurface thermal aquifer in Western Siberia. Microbiology 86, 765–772. doi: 10.1134/S002626171706008X
Kadnikov, V. V., Frank, Y. A., Mardanov, A. V., Beletsky, A. V., Karnachuk, O. V., and Ravin, N. V. (2017c). Metagenome of the Siberian underground water reservoir. Genome Announc. 5:e01317-17. doi: 10.1128/genomeA.01317-17
Kantor, R. S., van Zyl, A. W., van Hille, R. P., Thomas, B. C., Harrison, S. T., and Banfield, J. F. (2015). Bioreactor microbial ecosystems for thiocyanate and cyanide degradation unravelled with genome-resolved metagenomics. Environ. Microbiol. 17, 4929–4941. doi: 10.1111/1462-2920.12936
Karnachuk, O. V., Kurochkina, S. Y., and Tuovinen, O. H. (2002). Growth of sulfate-reducing bacteria with solid-phase electron acceptors. Appl. Microbiol. Biotechnol. 58, 482–486. doi: 10.1007/s00253-001-0914-3
Karnachuk, O. V., Frank, Y. A., Lukina, A. P., Kadnikov, V. V., Beletsky, A. V, Mardanov, A. V., et al. (2019). Domestication of previously uncultivated Candidatus Desulforudis audaxviator from a deep aquifer in Siberia sheds light on its physiology and evolution. ISME J. 13, 1947–1959. doi: 10.1038/s41396-019-0402-3
Kaster, A. K., Moll, J., Parey, K., and Thauer, R. K. (2011). Coupling of ferredoxin and heterodisulfide reduction via electron bifurcation in hydrogenotrophic methanogenic archaea. Proc. Natl. Acad. Sci. U.S.A. 108, 2981–2986. doi: 10.1073/pnas.1016761108
Kimura, H., Nashimoto, H., Shimizu, M., Hattori, S., Yamada, K., Koba, K., et al. (2010). Microbial methane production in deep aquifer associated with the accretionary prism in Japan. ISME J. 4, 531–541. doi: 10.1038/ismej.2009.132
Konstantinidis, K. T., Rosselló-Móra, R., and Amann, R. (2017). Uncultivated microbes in need of their own taxonomy. ISME J. 11, 2399–2406. doi: 10.1038/ismej.2017.113
Kuever, J. (2014). “The Family Syntrophobacteraceae,” in The Prokaryotes, eds E. Rosenberg, E. F DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin: Springer,).
Lang, K., Schuldes, J., Klingl, A., Poehlein, A., Daniel, R., and Brunea, A. (2015). New mode of energy metabolism in the seventh order of methanogens as revealed by comparative genome analysis of “Candidatus Methanoplasma termitum”. Appl. Environ. Microbiol. 81, 1338–1352. doi: 10.1128/aem.03389-14
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2.Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lee, K. C., Dunfield, P. F., Morgan, X. C., Crowe, M. A., Houghton, K. M., Vyssotski, M., et al. (2011). Chthonomonas calidirosea gen. nov., sp. nov., an aerobic, pigmented, thermophilic micro-organism of a novel bacterial class, Chthonomonadetes classis nov., of the newly described phylum Armatimonadetes originally designated candidate division OP10. Int. J. Syst. Evol. Microbiol. 61(Pt. 10), 2482–2490. doi: 10.1099/ijs.0.027235-0
Lee, K. C., Morgan, X. C., Dunfield, P. F., Tamas, I., McDonald, I. R., and Stott, M. B. (2014). Genomic analysis of Chthonomonas calidirosea, the first sequenced isolate of the phylum Armatimonadetes. ISME J. 8, 1522–1533. doi: 10.1038/ismej.2013.251
Lewin, A., Johansen, J., Wentzel, A., Kotlar, H. K., Drabløs, F., and Valla, S. (2014). The microbial communities in two apparently physically separated deep subsurface oil reservoirs show extensive DNA sequence similarities. Environ. Microbiol. 16, 545–558. doi: 10.1111/1462-2920.12181
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, J., Kudo, C., and Tonouchi, A. (2019). Capsulimonas corticalis gen. nov., sp. nov., an aerobic capsulated bacterium, of a novel bacterial order, Capsulimonadales ord. nov., of the class Armatimonadia of the phylum Armatimonadetes. Int. J. Syst. Evol. Microbiol. 69, 220–226. doi: 10.1099/ijsem.0.003135
Liu, P., and Conrad, R. (2017). Syntrophobacteraceae-affiliated species are major propionate-degrading sulfate reducers in paddy soil. Environ. Microbiol. 19, 1669–1686. doi: 10.1111/1462-2920.13698
Liu, Z., Frigaard, N. U., Vogl, K, Iino, T., Ohkuma, M., Overmann, J., et al. (2012). Complete genome of Ignavibacterium album, a metabolically versatile, flagellated, facultative anaerobe from the phylum Chlorobi. Front. Microbiol. 3:185. doi: 10.3389/fmicb.2012.00185
Liu, P., Pommerenke, B., and Conrad, R. (2018). Identification of Syntrophobacteraceae as major acetate-degrading sulfate reducing bacteria in Italian paddy soil. Environ. Microbiol. 20, 337–354. doi: 10.1111/1462-2920.14001
Lomstein, B. A., Langerhuus, A. T., D’Hondt, S., Jørgensen, B. B., and Spivack, A. J. (2012). Endospore abundance, microbial growth and necromass turnover in deep sub-seafloor sediment. Nature 484, 101–104.
Magnabosco, C., Lin, L. H., Dong, H., Bomberg, M., Ghiorse, W., Stan-Lotter, H., et al. (2018). The biomass and biodiversity of the continental subsurface. Nat. Geosci. 11, 707–717. doi: 10.1038/s41561-018-0221-6
Magnabosco, C., Ryan, K., Lau, M. C., Kuloyo, O., Sherwood Lollar, B., Kieft, T. L., et al. (2016). A metagenomic window into carbon metabolism at 3 km depth in Precambrian continental crust. ISME J. 10, 730–741. doi: 10.1038/ismej.2015.150
Magoć, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17, 10–12.
Matsushita, M., Ishikawa, S., Magara, K., Sato, Y., and Kimura, H. (2020). The potential for CH4 production by syntrophic microbial communities in diverse deep aquifers associated with an accretionary prism and its overlying sedimentary layers. Microbes Environ. 35, 1–10.
McInerney, M. J., Rohlin, L., Mouttaki, H., Kim, U., Krupp, R. S., Rios-Hernandez, L., et al. (2007). The genome of Syntrophus aciditrophicus: life at the thermodynamic limit of microbial growth. Proc. Natl. Acad. Sci. U.S.A. 104, 7600–7605. doi: 10.1073/pnas.0610456104
Miettinen, H., Kietäväinen, R., Sohlberg, E., Numminen, M., Ahonen, L., and Itävaara, M. (2015). Microbiome composition and geochemical characteristics of deep subsurface high-pressure environment, Pyhasalmi mine Finland. Front. Microbiol. 6:1203. doi: 10.3389/fmicb.2015.01203
Momper, L., Kiel Reese, B., Zinke, L., Wanger, G., Osburn, M. R., Moser, D., et al. (2017). Amend JP, major phylum-level differences between porefluid and host rock bacterial communities in the terrestrial deep subsurface. Environ. Microbiol. Rep. 9, 501–511. doi: 10.1111/1758-2229.12563
Moser, D. P., Gihring, T. M., Brockman, F. J., Fredrickson, J. K., Balkwill, D. L., Dollhopf, M. E., et al. (2005). Desulfotomaculum and Methanobacterium spp. dominate a 4- to 5-kilometer-deep fault. Appl. Environ. Microbiol. 71, 8773–8783. doi: 10.1128/aem.71.12.8773-8783.2005
Novikov, D. A., and Sukhorukova, A. F. (2015). Hydrogeology of the northwestern margin of the West Siberian Artesian Basin. Arab. J. Geosci. 8, 8703–8719. doi: 10.1007/s12517-015-1832-5,
Nobu, M. K., Narihiro, T., Kuroda, K., Mei, R., and Liu, W. T. (2016). Chasing the elusive Euryarchaeota class WSA2: genomes reveal a uniquely fastidious methyl-reducing methanogen. ISME J. 10, 2478–2487. doi: 10.1038/ismej.2016.33
Oehler, D., Poehlein, A., Leimbach, A., Müller, N., Daniel, R., Gottschalk, G., et al. (2012). Genome-guided analysis of physiological and morphological traits of the fermentative acetate oxidizer Thermacetogenium phaeum. BMC Genomics 13:723. doi: 10.1186/1471-2164-13-723
Orphan, V. J., Goffredi, S. K., and DeLong, E. F. (2003). Geochemical influence on diversity and microbial processes in high temperature oil reservoirs. Geomicrobiol. J. 20, 295–311. doi: 10.1080/01490450303898
Parkes, R. J., Cragg, B. A., and Wellsbury, P. (2000). Recent studies on bacterial populations and processes in subseafloor sediments: a review. Hydrogeol. J. 8, 11–28. doi: 10.1007/PL00010971
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P. A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Pavan, M. E., Pavan, E. E., Glaeser, S. P., Etchebehere, C., Kämpfer, P., Pettinari, M. J., et al. (2018). Proposal for a new classification of a deep branching bacterial phylogenetic lineage: transfer of Coprothermobacter proteolyticus and Coprothermobacter platensis to Coprothermobacteraceae fam. nov., within Coprothermobacterales ord. nov., Coprothermobacteria classis nov. and Coprothermobacterota phyl. nov. and emended description of the family Thermodesulfobiaceae. Int. J. Syst. Evol. Microbiol. 68, 1627–1632. doi: 10.1099/ijsem.0.002720
Pedersen, K. (2012). Subterranean microbial populations metabolize hydrogen and acetate under in situ conditions in granitic groundwater at 450 m depth in the Äspö Hard Rock Laboratory, Sweden. FEMS Microbiol. Ecol. 81, 217–229. doi: 10.1111/j.1574-6941.2012.01370.x
Podosokorskaya, O. A., Kadnikov, V. V., Gavrilov, S. N., Mardanov, A. V., Merkel, A. Y., Karnachuk, O. V., et al. (2013). Characterization of Melioribacter roseus gen. nov., sp. nov., a novel facultatively anaerobic thermophilic cellulolytic bacterium from the class Ignavibacteria, and a proposal of a novel bacterial phylum Ignavibacteriae. Environ. Microbiol. 15, 1759–1771. doi: 10.1111/1462-2920.12067
Rodriguez-R, L. M., and Konstantinidis, K. T. (2016). The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ 4:e1900v1
Sazanov, L. A. (2007). Respiratory complex I: mechanistic and structural insights provided by the crystal structure of the hydrophilic domain. Biochemistry 46, 2275–2288. doi: 10.1021/bi602508x
Schoeffler, M., Gaudin, A. L., Ramel, F., Valette, O., Denis, Y., Hania, W. B., et al. (2019). Growth of an anaerobic sulfate-reducing bacterium sustained by oxygen respiratory energy conservation after O2 -driven experimental evolution. Environ. Microbiol. 21, 360–373. doi: 10.1111/1462-2920.14466
Sekiguchi, Y., Kamagata, Y., Nakamura, K., Ohashi, A., and Harada, H. (2000). Syntrophothermus lipocalidus gen. nov., sp. nov., a novel thermophilic, syntrophic, fatty-acid-oxidizing anaerobe which utilizes isobutyrate. Int. J. Syst. Evol. Microbiol. 50(Pt 2), 771–779. doi: 10.1099/00207713-50-2-771
Simon, J., Kern, M., Hermann, B., Einsle, O., and Butt, J. N. (2011). Physiological function and catalytic versatility of bacterial multihaem cytochromes c involved in nitrogen and sulfur cycling. Biochem. Soc. Trans. 39, 1864–1870. doi: 10.1042/bst20110713
Takai, K., Gamo, T., Tsunogai, U., Nakayama, N., Hirayama, H., Nealson, K. H., et al. (2004). Geochemical and microbiological evidence for a hydrogen-based, hyperthermophilic subsurface lithoautotrophic microbial ecosystem (HyperSLiME) beneath an active deep-sea hydrothermal field. Extremophiles 8, 269-282.
Takai, K., Moser, D. P., DeFlaun, M., Onstott, T. C., and Fredrickson, J. K. (2001). Archaeal diversity in waters from deep South African gold mines. Appl. Environ. Microbiol. 67, 750–760.
Teske, A. P. (2005). The deep subsurface biosphere is alive and well. Trends Microbiol. 13, 402–404. doi: 10.1016/j.tim.2005.07.004
Thiel, V., Garcia Costas, A. M., Fortney, N. W., Martinez, J. N., Tank, M., Roden, E. E., et al. (2019). “Candidatus Thermonerobacter thiotrophicus,” A Non-phototrophic Member of the Bacteroidetes/Chlorobi With Dissimilatory Sulfur Metabolism in Hot Spring Mat Communities. Front. Microbiol. 9:3159. doi: 10.3389/fmicb.2018.03159
Vanwonterghem, I., Evans, P. N., Parks, D. H., Jensen, P. D., Woodcroft, B. J., Hugenholtz, P., et al. (2016). Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nat. Microbiol. 1:16170.
Vyssotski, A. V., Vyssotski, V. N., and Nezhdanov, A. A. (2006). Evolution of the west siberian basin. Mar. Pet. Geol. 23, 93–126. doi: 10.1016/j.marpetgeo.2005.03.002
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Watanabe, M., Kojima, H., and Fukui, M. (2015). Limnochorda pilosa gen. nov., sp. nov., a moderately thermophilic, facultatively anaerobic, pleomorphic bacterium and proposal of Limnochordaceae fam. nov., Limnochordales ord. nov. and Limnochordia classis nov. in the phylum Firmicutes. Int. J. Syst. Evol. Microbiol. 65, 2378–2384. doi: 10.1099/ijs.0.000267
Whitman, W. B., Coleman, D. C., and Wiebe, W. J. (1998). Prokaryotes: the unseen majority. Proc. Natl. Acad. Sci. U.S.A. 95. 6578–6583. doi: 10.1073/pnas.95.12.6578
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wu, X., Holmfeldt, K., Hubalek, V., Lundin, D., Åström, M., Bertilsson, S., et al. (2016). Microbial metagenomes from three aquifers in the Fennoscandian shield terrestrial deep biosphere reveal metabolic partitioning among populations. ISME J. 10, 1192–1203 doi: 10.1038/ismej.2015.185
Yamada, T., and Sekiguchi, Y. (2009). Cultivation of uncultured chloroflexi subphyla: significance and ecophysiology of formerly uncultured chloroflexi ‘subphylum i’ with natural and biotechnological relevance. Microbes Environ. 24, 205–216. doi: 10.1264/jsme2.me09151s
Yamada, T., Sekiguchi, Y., Hanada, S., Imachi, H., Ohashi, A., Harada, H., et al. (2006). Anaerolinea thermolimosa sp. nov., Levilinea saccharolytica gen. nov., sp. nov. and Leptolinea tardivitalis gen. nov., sp. nov., novel filamentous anaerobes, and description of the new classes Anaerolineae classis nov. and Caldilineae classis nov. in the bacterial phylum Chloroflexi. Int. J. Syst. Evol. Microbiol. 56(Pt. 6), 1331–1340. doi: 10.1099/ijs.0.64169-0
Keywords: deep subsurface, metagenome, uncultivable bacteria, candidate phylum, microbial diversity
Citation: Kadnikov VV, Mardanov AV, Beletsky AV, Karnachuk OV and Ravin NV (2020) Microbial Life in the Deep Subsurface Aquifer Illuminated by Metagenomics. Front. Microbiol. 11:572252. doi: 10.3389/fmicb.2020.572252
Received: 13 June 2020; Accepted: 13 August 2020;
Published: 03 September 2020.
Edited by:
Nils-Kaare Birkeland, University of Bergen, NorwayReviewed by:
Mircea Podar, Oak Ridge National Laboratory (DOE), United StatesCopyright © 2020 Kadnikov, Mardanov, Beletsky, Karnachuk and Ravin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nikolai V. Ravin, bnJhdmluQGJpZW5naS5hYy5ydQ==; bnJhdmluQG1haWwucnU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.