
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 12 November 2020
Sec. Evolutionary and Genomic Microbiology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.560667
 Mohammad Reza Rahbar1Mahboubeh Zarei1
Mohammad Reza Rahbar1Mahboubeh Zarei1 Abolfazl Jahangiri2Saeed Khalili3
Abolfazl Jahangiri2Saeed Khalili3 Navid Nezafat1,4Manica Negahdaripour1,4Yaser Fattahian5Amir Savardashtaki6
Navid Nezafat1,4Manica Negahdaripour1,4Yaser Fattahian5Amir Savardashtaki6 Younes Ghasemi1,4*
Younes Ghasemi1,4*Brucella species are Gram-negative, facultative intracellular pathogens. They are the main cause of brucellosis, which has led to a global health burden. Adherence of the pathogen to the host cells is the first step in the infection process. The bacteria can adhere to various biotic and abiotic surfaces using their outer membrane proteins. Trimeric autotransporter adhesins (TAAs) are modular homotrimers of various length and domain complexity. They are a diverse, and widespread gene family constituting the type Vc secretion pathway. These adhesins have been established as virulence factors in Brucellaceae. To date, no comprehensive and exhaustive study has been performed on the trimeric autotransporter family in the genus. In the present study, various bioinformatics tools were used to provide a novel evolutionary insight into the sequence and structure of this protein family in Brucellaceae. To this end, a dataset of all trimeric autotransporters from the Brucella genomes was built. Analyses included but were not limited to sequence alignment, phylogenetic tree constructions, codon-based test for selection, clustering of the sequences, and structure (primary to quaternary) predictions. Batch analyzes of the dataset suggested the existence of a few structural domains within the whole population. BatA from the B. abortus 2308 genome was selected as a reference to describe the features of these structural domains. Furthermore, we examined the structural basis for the observed rigidity and resiliency of the protein structure through a molecular dynamics evaluation, which led us to deduce that the random drift results in the non-adaptive evolution of the trimeric autotransporter genes in the Brucella genus. Notably, the modifications have occurred across the genus without interference of gene transmission.
Members of the genus Brucella are Gram-negative, facultative intracellular pathogens of the alpha-subclass of Proteobacteria phylum. Brucella species cause brucellosis, a damaging zoonotic disease, in a broad range of mammalian hosts. Brucellosis imposes a significant health burden, particularly in developing countries and in the livestock dependent economies (Gopalakrishnan et al., 2016). The ingestion of contaminated dairy products and direct contact with the infected animals could transmit the infection to humans, whereas infection in animals occurs through the environmental transmission of bacteria (Gopalakrishnan et al., 2016).
The typical species within the Brucellae genus share a high level of genomic identity. This observation initially led to the conclusion that Brucella is a monospecific genus (B. melitensis), which also contained six biovars based on their host prevalence. However, recent molecular analyses suggest that B. abortus, B. melitensis, B. neotomae, and B. ovis are four related clones of one organism; whereas, B. suis forms a separate cluster (Foster et al., 2009; Whatmore, 2009). Currently, at least 12 Brucella species are identified (El-Sayed and Awad, 2018). The most economically significant species are B. abortus, B. melitensis, and B. suis.
The Brucella ancestor was a free-living bacterium that became an animal parasite (El-Sayed and Awad, 2018). Some species have only one chromosome. Most species, by contrast, harbor two chromosomes; one larger in size and a smaller one originating from a plasmid. Interestingly, some species such as B. suis have also kept their ancestor accessory genes, responsible for exploiting plant-derived nutrients. B. suis chromosome 1 and the genome of Mesorhizobium loti, a plant symbiont, show a high level of gene synteny. Their metabolic activities are similar to those of soil-plant associated bacteria, suggesting an evolutionary relationship between Brucellae and the plant pathogens and symbionts (Paulsen et al., 2002; Ficht, 2010).
The bacterial attachment to some molecules on the host cells triggers a multistep infection process, which is followed by internalization, replication, and survival within macrophages. A delay in an antibiotic-based treatment following the initial stages of the infection could ultimately lead to the settlement of Brucellae in various organs and tissues (Corbel, 1997). The progress in the course of infection demands several mechanistic determinants and regulatory systems. Professional and non-professional phagocytic cells provide a replication niche for the bacterium; they can also carry the pathogen through the mucosal surfaces into the lymph nodes (Roop et al., 2009).
The secretome and membrane proteome of the pathogens are an integral part of interaction between bacterial cells and their environment. Bacteria can sense stress, adhere to various biotic and abiotic surfaces, and establish a cell to cell communication by means of some secreted proteins (Sankarasubramanian et al., 2016a).
Adherence to the host cells governs the cellular and tissue tropism, entrance site, host specificity, and replicative niche of the pathogen (Czibener and Ugalde, 2012). The pathogen’s outer membrane proteins play pivotal roles in this process (Edmonds et al., 2001). Several open reading frames, coding for different adhesin proteins, have been found in Brucella genomes (Rocha-Gracia et al., 2002; Castañeda-Roldán et al., 2006; Backert et al., 2008; Posadas et al., 2012; Czibener et al., 2016; Sankarasubramanian et al., 2016a). A pathogenicity island harboring an immunoglobulin-like domain protein was also identified in the Brucellae genomes. This domain involves the attachment and internalization of the bacteria into the host cells (Czibener and Ugalde, 2012). More recently, in Brucella, a collagen-binding adhesin (major outer membrane protein, Bp26) was described by enzyme-linked immunosorbent assay (ELISA) and bio-layer interferometry. The protein was shown to bind to type I collagen, vitronectin, and soluble fibronectin (ElTahir et al., 2019).
Two members of trimeric autotransporter adhesins (TAAs, also known as type Vc secretion pathway) namely, BtaE (Ruiz-Ranwez et al., 2013b), and BtaF (Ruiz-Ranwez et al., 2013a) were identified in B. suis. Both adhesins involve the attachment of bacteria to the extracellular matrix. Functional studies using genetic knockouts of the adhesin proteins reduce the virulence of B. suis.
The comparative genome analysis of B. abortus 2308 and Brucella S19, a spontaneously attenuated vaccine strain, resulted in the identification of a set of virulence genes. One of the candidates for virulence is BAB1_0069 (Crasta et al., 2008), which, encodes an ortholog of BtaE. An adherence assay suggested that this adhesin participates in the interaction of B. abortus with the host cell, similar to its ortholog in B. suis 1330 (Ruiz-Ranwez et al., 2013b). This ortholog also assists the initial attachment of the bacteria to the HeLa cells (Sieira et al., 2017). The corresponding locus in S19 has undergone a 1,695-nucleotide deletion. Hence, 565 amino acids were removed from the mature protein during the deletion, so that the attenuated S19 strain comprises a 768 aa long adhesin. Similar deletions were also observed in the same open reading frame of other virulent species. In B. melitensis, the locus contains three deletions: the first causes a 512 amino acid deletion, second and third are single nucleotide deletions leading to a frameshift. As a result, the strain contains only the 365 C-terminal amino acids. In B. suis genome, the related open reading frames are predicted to encode a 740 amino acid long protein because of two deletions. The position and length of these open reading frames are identical in the attenuated S19 strain and the virulent B. suis. The difference is related to 133 amino acid repeat module. These deletions and similar ones in other virulent species have ruled out the association of deletions with the lack of virulence in the S19 strain (Crasta et al., 2008). Therefore, the trimeric autotransporters are putative virulence factors in Brucellae and not essential or key virulence factors.
Trimeric autotransporter adhesins are a diverse, simple, and widespread type Vc secretion pathway (Dautin and Bernstein, 2007; Bernstein, 2019; Kiessling et al., 2020). TAAs are modular homotrimers with various lengths and domain complexity. The structure of these adhesins obeys the simple pattern of (from the N-terminal) signal peptide, passenger, and membrane anchoring domain (Linke et al., 2006). The passenger is a complex of structurally-conserved analogous blocks, which immediately forms a trimeric quaternary structure upon passing through the barrel made by membrane anchoring domain (Linke et al., 2006). The rearrangement of these blocks has resulted in considerable diversity among family members.
The TAAs are well-worthy of study as potential candidates to design novel vaccines, disease biomarkers, and novel anti-virulence drugs (Qin et al., 2015). Understanding the intricacies of their structure, architecture, and evolutionary insights are critical for developing novel therapeutic approaches and to advance the field further.
In recent years, several members of TAAs have been investigated in extensive detail. As the crystal structures of various domains of TAAs are solved, it becomes feasible to identify the domains of new members via a homology modeling-based approach. The examples of solved crystal structures are the crystal structure of Burkholderia pseudomallei antigen Bpsl2063 under Protein Data Bank (PDB) accession number 4USX (Gourlay et al., 2015), Acinetobacter species (sp.) Tol 5 AtaA C-terminal stalk (PDB ID: 3WPA; Koiwai et al., 2016), Salmonella enterica SadA coiled-coil domain (PDB ID: 2WPQ; Hartmann et al., 2009), crystal structure of Hia binding domain from Haemophilus influenzae genome (PDB ID:1S7M; Yeo et al., 2004); structure of the head of the Bartonella adhesin BadA (PDB ID: 3D9X; Szczesny et al., 2008); and the left-handed parallel beta-roll collagen-binding domain of Yersinia enterocolitica adhesin YadA (PDB ID: 1P9H; Nummelin et al., 2004).
In the present study, a wide variety of in silico tools were employed to determine the properties of trimeric autotransporters in Brucella species (BTAA). This paper provides comprehensive information, regarding the distribution of TAAs in the genus, the architecture of BTAAs, and a hypothesis as to the evolution of the BTAAs. BTAA sequences were extracted from databases to make a dataset. Batch analyzes of the dataset suggested the existence of a few structural domains. Focusing on TAA from the B. abortus 2308 genome enabled us to describe some features of BTAAs. Henceforth, these proteins were described as BatA (Brucella abortus trimeric autotransporter adhesin). Following homology modeling, the sequence and structural features of the proteins were annotated. The evolutionary analysis were also performed and showed that these domains are repeated by different frequencies within BTAA sequences. We finally concluded that the random drift results in the non-adaptive evolution of TAA genes in the Brucella genus.
The phylogenetic tree of Brucellaceae, the related genomes, protein sequences, and protein-coding genes were obtained from the Pathosystems Resource Integration Center (PATRIC ver. 3.5.39; Wattam et al., 2016). The BLAST databases were built separately from genomes, proteomes, and protein-coding genes of all members of the Brucellaceae family using the makeblastdb application version 2.9.0 (Altschul et al., 1997) in the UGENE environment version 1.32 (Okonechnikov et al., 2012). For all BLAST search strategies, the consensus sequence of YadA like C-terminal domain (IPR005594) was queried. The significant BLAST hits were mapped to related genomes on the phylogenetic tree of the Brucellaceae family.
The evolutionary analyzes including disparity index test, detecting duplication events, alignments, and phylogenetic tree constructions were performed in MEGA 7 software (Kumar et al., 2016). All analyses were based on the nucleotide sequences of the highly conserved membrane anchoring region of BTAAs; and the sequences were grouped based on related species. The best evolutionary model for analyzing the DNA dataset was estimated by the model selection function of MEGA 7.
Possible recombination events were tracked by the Genetic Algorithm for Recombination Detection (GARD; Kosakovsky Pond et al., 2006) as provided by the Datamonkey server (Weaver et al., 2018) at www.datamonkey.org. The analysis was performed on the complete nucleotide sequences of BTAA encoding genes of Brucellaceae.
Codon bias within the nucleotide sequences of BTAAs was inferred by MEGA 7. Codon usage of each representative sequence was compared to the species codon table by the graphical codon usage analyzer (Fuhrmann et al., 2004) as provided by http://www.gcua.schoedl.de. The codon usage frequency was converted into relative adaptiveness values. Contrary to the codon usage frequency, the relative adaptiveness takes into account the number of codons which code for the respective amino acid. For each amino acid, the codon with the highest frequency value was set to 100% relative adaptiveness. All other codons for the same amino acid were scaled accordingly. The codon usage table of each representative genome was obtained from the codon usage database at https://www.kazusa.or.jp (Bzhalava et al., 2018). The nucleotide sequence of Omp31, a species-specific gene (Herman and De Ridder, 1992; Romero et al., 1995; Singh et al., 2013), was used as control.
The probability of rejecting the null hypothesis of strict-neutrality (dN = dS) in favor of the alternative hypotheses [positive (dN > dS) and purifying (dN < dS) selections] was calculated on membrane anchoring domain encoding portion of the genes in MEGA 7 based on the Z-test and Fisher’s exact test of neutrality (Zhang et al., 1997). Values of P less than 0.05 were considered significant. The variance of the difference was computed using 1000 bootstrap replicates. The analyses were conducted using the Pamilo-Bianchi-Li method (Pamilo and Bianchi, 1993) (dN: non-synonymous substitutions per non-synonymous site; dS: synonymous substitutions per synonymous site).
An all-against-all approach was employed for clustering the amino acid sequences of all TAAs from Brucella genomes by CLANS software as provided by the MPI Bioinformatics Toolkit (Zimmermann et al., 2018) at https://toolkit.tuebingen.mpg.de (Alva et al., 2016). The pairwise attraction values based on the P values of the high scoring segment pairs (HSPs) were calculated. The BLAST analysis of each BTAA sequence against all other BTAA sequences based on the P values of high-scoring segment pairs was performed and enabled the three-dimensional visualization of pair-wise sequence similarities. The resulting file was visualized and analyzed by CLANS stand-alone software (Frickey and Lupas, 2004). To keep the sequences from collapsing into one point, the P values above 10–6 were discarded. Furthermore, the distribution of HSPs was collected and mapped onto the BatA sequence as a prototype and visualized in a graph.
The domain architectures of proteins were viewed at the InterPro database (Finn et al., 2017) and Pfam (Finn et al., 2016).
The presence of signal peptides (SP) was assessed by Phobius (Käll et al., 2004) at http://phobius.sbc.su.se/ (Käll et al., 2007).
The location of signal peptides was predicted based on the SignalP ver. 5 (Petersen et al., 2011) server at http://www.cbs.dtu.dk, using deep neural network [taking advantage of the algorithm ability to differentiate the “standard” signal peptidase I-cleaved SPs translocated by the Sec translocon (Sec/SPI) and two other types of SPs in prokaryotes, namely lipoprotein SPs cleaved by signal peptidase II (Sec/SPII) and Twin-Arginine Translocation (Tat) SPs translocated by the Tat translocon (Tat/SPI)].
The presence and location of Twin-arginine signal peptide cleavage sites in BTAA sequences were predicted by TatP ver 1 server (Bendtsen et al., 2005) at http://www.cbs.dtu.dk using a combination of two artificial neural networks. Pred-Tat (Bagos et al., 2010), at http://www.compgen.org, was employed for predicting the signal peptides based on the hidden Markov model. The Signal BLAST server (Frank and Sippl, 2008) at http://sigpep.services.came.sbg.ac.at was also employed for comparing the signal peptide against the Uniprot database (Consortium, 2017).
The repeat modules within the primary sequences were identified by generating a dot-plot matrix at https://myhits.isb-sib.ch by Dotlet software (Junier and Pagni, 2000). The Xstream software (Newman and Cooper, 2007) available at http://jimcooperlab.mcdb.ucsb.edu and the T-REKS standalone software (Jorda and Kajava, 2009) were also employed for defining the repeat modules within the entire protein dataset.
The existence of coiled-coil regions was predicted by the Wagga Wagga (Simm et al., 2015) server at https://waggawagga.motorprotein.de. The server employs six external tools to determine the position of coiled-coil regions.
Consensus patterns were searched across the primary amino acid sequences by the ScanProsite tool at https://prosite.expasy.org (De Castro et al., 2006). The consensus patterns and motives were extracted from the literature (Szczesny et al., 2008; Leo et al., 2011; Bassler et al., 2015).
The gene order in the vicinity of BTAAs was assessed by the SyntTax server (Oberto, 2013) implementable at http://archaea.u-psud.fr. The 60 C-terminal residues related to the membrane anchoring region were queried to perform the analysis.
The existence of BTAAs in association with other genes to build an operon was assessed at http://www.microbesonline.org and http://operondb.cbcb.umd.edu by querying the locus tags of the protein-encoding genes.
The transmembrane beta-barrel topology of the BTAAs was predicted by BOCTOPUS at http://boctopus.bioinfo.se/pred/, PRED TMBB at http://bioinformatics.biol.uoa.gr/PRED-TMBB/input.jsp and PRED TMBB2 at http://www.compgen.org/tools/PRED-TMBB2.
The 3-state secondary structures of the proteins were predicted at RaptorX server (Wang et al., 2016) at http://raptorx.uchicago.edu, based on an emerging machine learning model (Wang et al., 2015). The accuracy of RaptorX for 3-state secondary structure prediction is 84%. The secondary structures were also predicted by PSIPRED (McGuffin et al., 2000), available by the UCL department of computational biology at http://www.bioinf.cs.ucl.ac.uk.
Considering the importance of B. abortus 2308 and the aforesaid facts, we prompted to model the tertiary structures and forecast some other properties of these building blocks. To this end, we chose a sequence (1333 amino acid in length) from B. abortus 2308 (locus tag: BAB1_0069) as a prototype; the sequence is called BatA (Brucella abortus trimeric autotransporter adhesin) hereafter.
To achieve the best template for the 3D-structure prediction, four approaches were employed. (i) the amino acid sequence of BatA was subjected to PSI-BALST against the PDB with five iterations. The structures with lower E-Values (lower than 10–4), higher identities (more than 40%), and better query coverages (more than 40%) were selected. (ii) Searching for templates, which is also a part of tertiary structure predictor algorithms using different methods and even external softwares, such as Phyre2 (Kelley et al., 2015), SwissModel (Waterhouse et al., 2018), and I-TASSER (Yang et al., 2015). (iii) Additionally, a pairwise comparison of hidden Markov models was applied for the BatA sequence to find the remote homologs by HHpred software at www.toolkit.tuebingen.mpg.de (Söding et al., 2005). This method is more sensitive than the BLAST search for finding the remote homologous structures. HHpred can detect homologous associations far beyond the twilight zone (below 20% sequence identity). in this method, the estimated probability of the template to be homolog to the query is the most important principle for choosing the template. (iv) The models generated by the tertiary structure predictors (Phyre2, SwissModel, and I-Tasser) were also added to the collection of templates.
The structures with satisfactory results in terms of sequence identity and similarity to each part of BatA were used as the templates for homology modeling by Modeler (Webb and Sali, 2014). The approach initiated by the alignment of the templates, aligning templates with the query, then generating 10 models for each query.
In all stages, the generated models (modeled with the online software tools or Modeler) were evaluated in terms of quality and agreement between the secondary structures of the 3D models and the predicted ones. The quality of the predicted models was evaluated by MolProbity (Chen et al., 2010) at http://molprobity.biochem.duke.edu.
To obtain the quaternary structures of each part, the predicted qualified models were submitted to GalaxyHomomer (Baek et al., 2017), embedded in http://galaxy.seoklab.org server. The predicted models underwent ab initio docking, and after refinement, final trimers were generated. The additional refinements were done at the Galaxy server, if needed.
The analysis of the membrane channel of the protein was based on Caver ver. 3 (Chovancova et al., 2012), embedded as a plug-in in the Pymol software (DeLano, 2002). ChexVis (Masood et al., 2015), at https://vgl.csa.iisc.ac.in, was also employed for analyzing the channel properties. The probe radius was set at zero angstroms to consider channels of any width as feasible.
The flexibility and toughness of the modeled structures were assessed via simulation of protein dynamics by the CABS-Flex tool (Jamroz et al., 2013). The restraints were generated for all residues by selecting the “All” mode. The minimum distance along the protein chain for the two residues to be bound was set at 2, meaning that each residue cannot be restrained with contiguous residues. Restraints were automatically generated for residues within the minimal and maximal distances of 3.8 to 8 Angstrom (default setting length of restraints). The number of cycles was set at 100, and the temperature of the simulation was 1.4.
While cavities in the coiled-coils are assumed to play role in the bending properties of TAAs (Leo et al., 2011), the presence and volume of the cavities in the C-terminal coiled-coils were evaluated and measured by the CASTp server (Tian et al., 2018) at http://sts.bioe.uic.edu/castp. A similar assessment was also conducted by CavityPlus (Xu et al., 2018) at http://repharma.pku.edu.cn/cavityplus; and the overlapping results of both servers were presented. Furthermore, to better understand the effect of cavities, the volume data was compared to similar data derived from the structure of the 527–665 fragment of UspA1 protein from Moraxella catarrhalis (Conners et al., 2008; PDB ID: 2QIH) coiled-coil region of Escherichia coli EibD (PDB ID: 2XZR; Leo et al., 2011), as well as a conserved coiled-coil segment of TAA of Y. enterocolitica (Alvarez et al., 2010; PDB ID: 3H7X).
To obtain the predicted model representation in a natural-like environment, the model was embedded within a membrane patch. VMD v1.9.2 software was used to perform all steps of the process, while the Tcl scripting language was used to perform all of the external manipulations. The membrane builder tool of the VMD software was used to build a membrane patch (X length of 80 and Y length of 80). The protein was aligned according to its center of mass within the lipid bilayer. Setting ten residues of membrane barrel as the center of mass, the protein was located at a proper orientation. Moreover, the matrix was set to rotate about the X, Y, and Z-axis by 0, −90, and 0. All of the overlapping lipid molecules with the protein stem were removed for the proper accommodation of the protein within the membrane. Water molecules were also added to the protein and membrane structure using the VMD solvate tool.
To observe the location of BTAA encoding genes and their neighbors, a gene synteny analysis was performed. In all Brucella species, the TAA encoding genes are located in chromosome 1; the immediate neighbor of the TAA encoding genes is the invasion-associated locus B [in the case of B. abortus 2308, TAA (BatA, Locus tag: BAB1_0069), which is located 209 nucleotide pairs downstream of the invasion-associated locus B (Ialb, locus tag: BAB1_0070)] (Figure 1). This pair of genes are located in the same direction, making a set of consecutive genes on the same DNA strand.
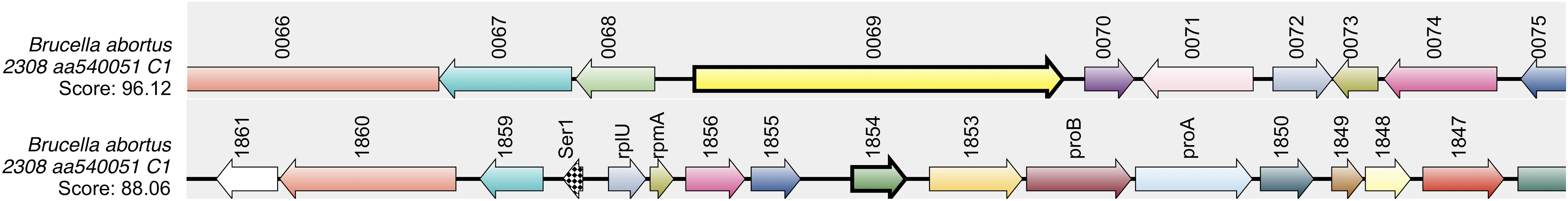
Figure 1. The location of the BatA encoding gene in the genome of B. abortus 2308. Different loci are presented with arrows of different colors. The locus tags are at the top of each arrow. BAB1_0069 and BAB1_1854 are trimeric autotransporter encoding genes.
To determine whether batA is associated with other genes, the operon databases were searched for related locus tags. Based on the operon database, the probability of existence of batA and its downstream neighbor in the same operon is estimated as 0.80 (average). This indicates that these genes are likely to be parts of the same operon. Values near to 1 or 0 are confident predictions of being in the same operon or not, respectively, while values near 0.5 are low-confidence predictions. Searching the operon database also indicated that these two genes could be found near each other in the genomes of few genera and species including Bartonella. The confidence was approximately 42 percent, which is a relatively low score. The conservation of this nucleotide pair across multiple genomes was estimated as 0.75, based on Microbesonline Ortholog Groups Dataset. The conservation score here is typically ranged from 0 (not conserved) to 1 (100% conserved). Based on these results, BTAAs encoding genes and their neighbors do not arrange as an operon.
The tBLASTn results revealed that there are two genes in the genome of B. abortus 2308 that could encode proteins, containing the YadA-like membrane anchoring domain (YadA-anchor: Pfam: PF03895, InterPro: IPR005594): a 3999-base pair protein identified by locus tag “BAB1_0069,” “UniProtKB/TrEMBL:Q2YPR0: 1333 amino acids,” and a 591 base-pair one identified by locus tag “BAB1_1854,” “UniProtKB/TrEMBL:Q2YLM1: 197 amino acids.” The pairwise alignment of their amino acid sequences showed that similarities are limited to two portions (Figure 2A). The longer sequence (under acc. no. of Q2YPR0) is a more complex adhesin (BatA); therefore, it was the focus of this study.
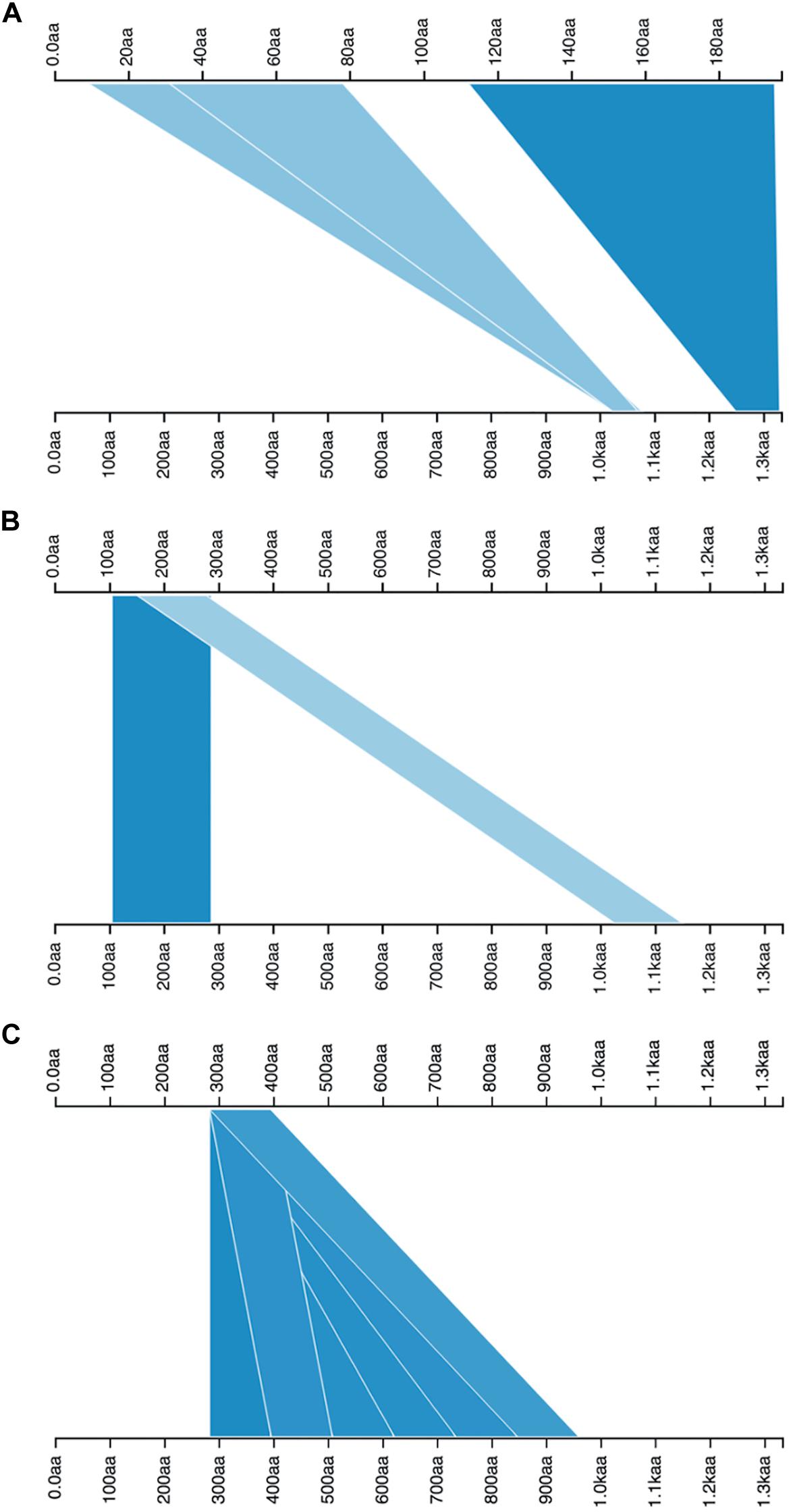
Figure 2. Visualization of pairwise alignments. The alignments between queries (top axes) and subjects (bottom axes) are illustrated by blue triangles or ribbons; a higher density of blue color shows a higher score. For each query subject pairing, there is more than one alignment. (A) the alignment of two TAAs in the proteome of B. abortus 2308, the top axis is representative of shorter TAA, and the bottom axis is BatA. The alignment hits include the head domains (light blue triangles) and membrane anchoring region (dark blue triangle). (B) Both axes are the BatA sequence, the positions of head domains are demarcated by blue ribbons. (C) The plot shows the presence of a repeat module, which is repeated six times along the sequence of BatA; both axes are BatA sequence. The pairwise BLAST results are visualized by Kablammo at http://kablammo.wasmuthlab.org/.
To define the domain architecture of BatA, the location of the signal peptide was predicted by multiple servers initially.
The sequence is initiated by a signal peptide at N-terminus. The cleavage site is located after residue Ala39 (Supplementary Data 1 and Supplementary Figure 1). Specific BLAST search to confirm the existence of the signal peptide showed that the most significant BLAST hit belongs to BtaE signal peptide, which is a trimeric autotransporter from B. suis genome (Score = 75.1 bits, E-value = 9e-15).
The definition of repeat modules allowed us to perform the domain annotation of BatA as a prototype of the BTAAs. By getting clues from the dot-plot matrix results and repeat module predictors, the locations of repeated blocks were determined. The pairwise alignment function of the BALSTP tool at NCBI and visualization of the BLASTP results finalized the repeat module definition. The approach was initiated by aligning the BatA sequence with itself. The sequence contains two repetitive modules, one of which is repeated two times (Figure 2), which is a low complexity region (based on dot-plot matrix results; Figure 3), and the second one is perfectly and tandemly repeated with six repeats of the same sequence (the core domain of tandem repeats, Figures 2C, 3).

Figure 3. The dot-plot matrix, alignment of BatA against itself. The plot displays the alignment of the BatA amino acid sequence against itself. The horizontal and vertical axes are both the BatA amino acid sequence. The plot shows how the sequence matches to itself, therefore represents the repeat modules and low complexity regions. Each pixel corresponds to a residue in the horizontal and vertical sequences. The pixel shading represents the score of matching the residues; the darker the pixel, the higher the score. In the middle of the sequence, there is a core domain of tandem repeats, which is repeated six times within the sequence (282–950). The low complexity regions stand out as black squares.
As expected, the protein ends with a potential membrane anchoring region. The transmembrane barrel is connected to the passenger by a coiled-coil domain (1208–1251) containing the period of six heptads. Short coiled-coils also mediate the connection of other segments to each other.
Several attempts were made for collecting the proper templates for each domain block of BatA. Due to the high sequence variability of TAAs, sequence identity was not an appropriate criterion to measure the relationship. Thus, to achieve more reliable results, a Hidden Markov model profiling was utilized. A search against alignment databases such as Pfam and SMART by HHpred provides an alternative approach to identify the proper structural templates. HHpred introduces the homologous structures in a more informative manner than the BLAST search. The templates with a probability larger than 95% were selected as the final templates for homology modeling. This criterion suggests that the homology is nearly certain.
The template library included some irrelevant members, suggesting that artificial intelligence was not sufficient for template selection. Therefore, a knowledge-based approach was adapted to select the templates; it means that: (i) the template should be biologically suggestive or reasonable. The organismic origin and function of the templates were also taken into account. (ii) the secondary structures of the templates should be in good agreement with those of the query. (iii) the templates should be the members of the same superfamily. This approach considerably ruled out the irrelevant hits. The existence of famous conserved motives (the result of the primary sequence analysis) was also considered (The identified patterns with their descriptions are summarized in Table 1). Taken these criteria into account, the spurious templates were filtered out, and a library of the templates that was proportional to each part of the protein was built (Table 2). In general, the alignments of templates with different parts of the protein showed a low level of sequence identity (Supplementary Data 1 and Supplementary Figure 2).

Table 1. Consensus patterns found in the primary sequence of BatA.
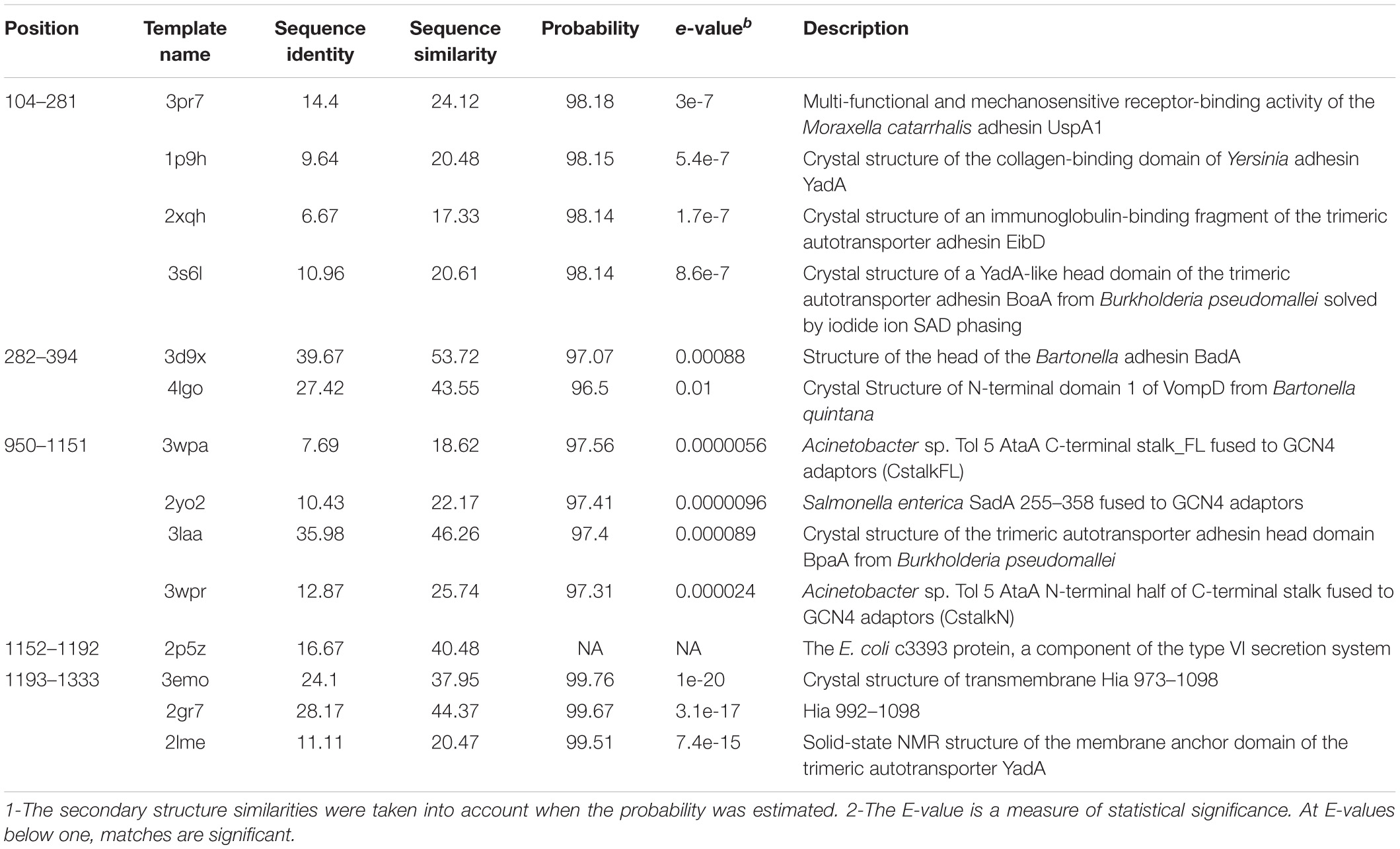
Table 2. Selected templates for building tertiary structures of each block.
Since many templates were found for different parts of the protein, the homology modeling approach was exploited to build the monomeric structures with various computational methods. Multiple tertiary structures were built for each block of the protein. Amongst, the best-qualified models -the ones that matched the consensus patterns and secondary structures-were selected. The secondary structure components of BatA were predicted to be 11% helix, 27% extended strand, and 60% coil (Supplementary Data 1 and Supplementary Figure 3). All the qualified monomers were built by a modeler software.
These structures satisfied all the criteria considered to be essential for quaternary structure assembly (favored rotamers, Ramachandran favored, bonds, and angles). Hence, they were submitted to Galaxyhomomer to build their trimeric complexes. The resulting library was evaluated in the term of overall quality (Table 3). All building blocks of the protein were built (the PDB file of the modeled structures are provided as a compressed Supplementary File).
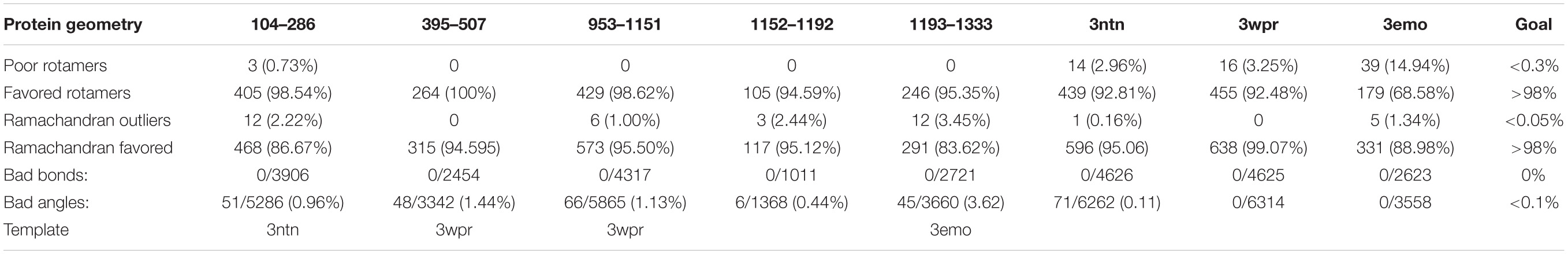
Table 3. Quality assessment of the built structures.
To define the domain architecture of BatA, a structural comparison was made (including superimposition and visual inspection).
The abundant domains were head, GIN, TrpRing, and coiled-coils. Details on the identified domains are provided in Supplementary Data 1. Briefly, low complexity regions were found at the N-terminus YadA like head and the C-terminus YadA like head, which are left-handed beta rolls, common in TAA architectures. Both the head domains at the N-terminus contain an inserted motif called HIM (Head Insert Motif), which extend downwards over the neck to connect the heads to a short coiled-coil region. The N-terminus head is connected to the TrpRing; and the C- terminus head is connected to a coiled-coil and FGG.
A core domain of repeated modules is an evident architecture within the sequence, which is related to the tripartite architecture of the TrpRing-GIN-Neck domain (this arrangement is appeared to be the crucial repeated modular units). This sequence is perfectly and tandemly repeated six times within the sequence of BatA. TrpRing is an interleaved head domain, parallel to the fiber axis. GIN domain is located after the TrpRing domain. GIN is a traversal head perpendicular to the fiber axis. The domain is connected to a short coiled-coil region by a neck connector.
Following the tandem repeats, there is a region composed of FGG-HANS- YLHead –HIM, which is connected to a narrower coiled-coil by a neck connector domain. FGG is a 3-stranded beta meander inserted into the coiled-coil region, and HANS is a short beta hairpin interacting with the YLHead.
At the C-terminus, the protein ends with a membrane anchoring region. The membrane channel is embedded in the membrane by a β-barrel. The membrane anchoring domain virtually represents a similar structure in all TAAs and forms a β-barrel pore containing 12 transmembrane beta strands (each subunit includes four beta strands). The BatA β-barrel has an 18.19 Angstrom-diameter central channel. The length of the barrel is estimated as 37.86 angstroms (Supplementary Data 1 and Supplementary Figure 4). The β-barrel is traversed by three N-terminal α-helices, one from each subunit.
The coiled-coils are bundles of alpha helices; typically, hydrophobic residues organized in a reiterating pattern of seven residues. The position of coiled-coils represents abcdefg (Figure 4). Moreover, the NXYTD motif, the site for transition of the right-handed coiled-coil to the left handed one, is present in C-terminal coiled-coil domain (Figure 4 and Table 1).
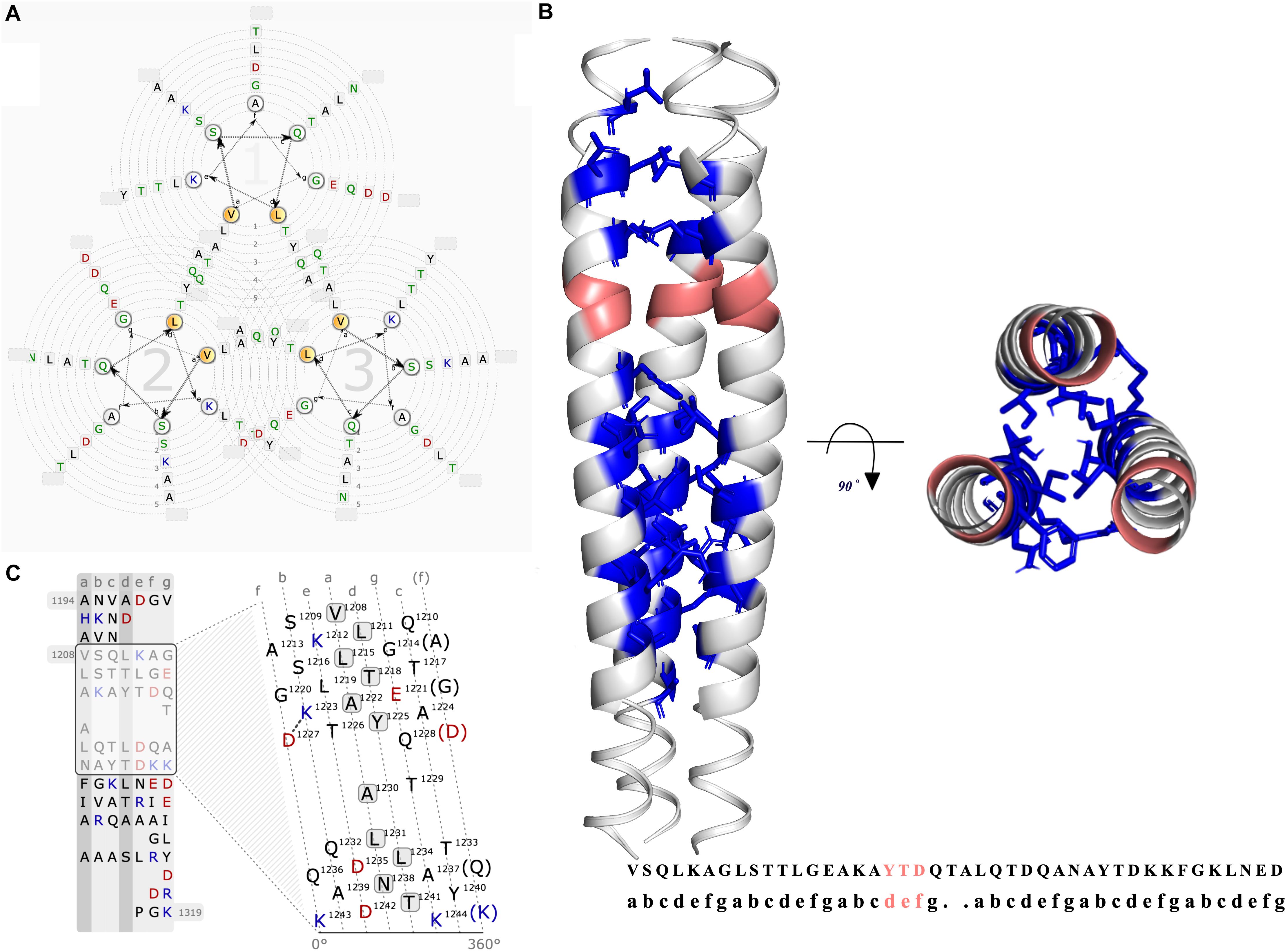
Figure 4. Graphical representation of the coiled-coil region of BatA. (A) The helical wheel view of the coiled-coil region is presented. The possible residue interactions inside a predicted coiled-coil are visualized. Each helix is represented by a wheel that is made up of seven groups (a,b,c,d,e,f,g) with the residues sorted according to the predicted heptad pattern. The sequence starts in the most inner circle of the wheel-spiral and follows in an arrow-indicated fashion to the outside. The heptad positions a and d are highlighted in orange and are oriented to the opposing wheel, representing the inner side of a coiled-coil helix. (B) The coiled-coil region located at the C-terminal of the protein is presented by three different colors for each chain. The location of YDD and YTD motifs (transition site of the right-handed coiled-coil segment to the left-handed coiled-coil) are colored pinkish-orange (deep salmon). The quaternary interaction of the three alpha-helices are presented with the Knob-into-holes and colored in blue. (C) The lower left graph is the illustration of the interaction net of an open-cut single alpha helix. In this view, one helix is cut and flattened. The flattened helix is made along the surface in the direction of the helix axis. The heptad repeat pattern, as a basis to divide the residues, is presented in strict columns, the cut is made along the column. Solid and dashed connections represent the strong (black lines) and weak (dashed gray) types of interactions between the residues.
The passenger domain in this protein is composed of heads, connectors, and coiled-coils. The mature protein is the result of wrapping three identical chains around an axis to build a ∼85.4-nanometer nano-fiber (Figure 5).
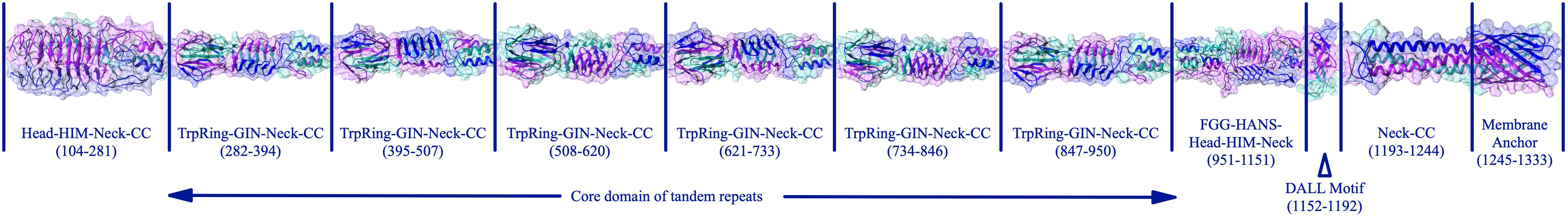
Figure 5. The complete assembly of the BatA structure. The complete structure of BatA is shown as cartoons with transplant surfaces. The position of each domain within the amino acid sequence of the protein is given at the bottom of each domain.
To gain a better insight into the characteristics of TAAs in the Brucellaceae family, an all-against-all approach was employed to cluster the BTAA amino acid sequences. Based on all-against-all pairwise similarities, TAAs were clustered into three distinct and clear clusters and few scattered sequences (Figure 6A). The amino acid length of sequences ranged from 86 (locus tag: BSPT1_II1279, B. suis bv. 2 strain PT09143) to 1559 (locus tag: P050_01136, B. abortus 90-12178). The first cluster included the longest sequences and involved all species except for B. pinnipedialis. A 1333 amino acid sequence was the most abundant sequence in the population (see Supplementary Data 2, CLANS tab, for details of different clusters). In general, the sequence lengths of BTAAs are heterogeneous [compare it with Acinetobacter, which is highly heterogenous (Rahbar et al., 2019)]. BatA is a member of the cluster 1. Moreover, two well-defined BTAAs, viz. BtaE and BtaF, are members of the cluster 1 and the cluster 2, respectively.
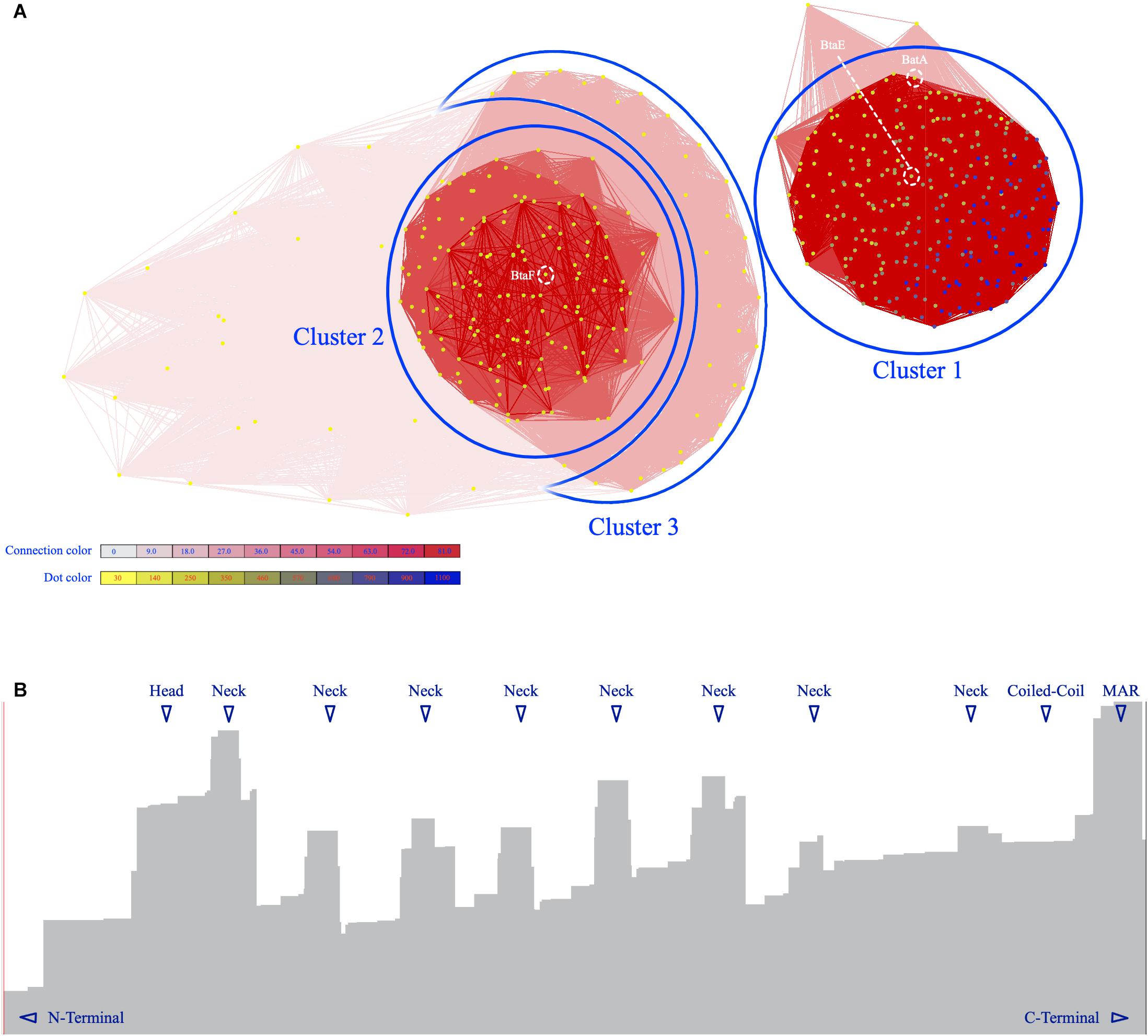
Figure 6. The graphical layout of 490 BTAA amino acid sequences. (A) The Fruchterman–Reingold graph layout algorithm to visualize pairwise sequence similarities. Sequences are represented by vertices in the graph, BLAST high scoring segment pairs (HSPs) are shown as edges connecting vertices and provide attractive forces proportional to the negative logarithm of the HSP’s P value (P value cutoff: 10– 6), different clusters are indicated by numbers. BatA and BtaE are members of cluster 1; BtaF in member of cluster 2. The vertices are colored based on the sequence length. The redness intensity of connecting edges is proportional to attraction forces. (B) The distribution of HSPs over the BatA sequence. The X-axis is the amino acid sequence and the Y-axis is the level of distribution. The highest levels are indicated by triangles.
The results of topology prediction indicated that the BatA protein is an outer membrane beta-barrel protein. The position for transmembrane strands (TM) was predicted to be: TM1: 70–79, TM2: 83–92, TM3: 97–106, and TM4: 110–120 by BOCTOPUS, TM1: 68–78, TM2: 97–105, and TM3: 110–120 by PRED TMBB and TM1: 52–59, TM2: 71–79, TM3: 84–92, and TM4: 112–122 by PRED TMBB2. The predictions of the BOCTOPUS and PRED TMBB2 are highly concordant in regards to the number of transmembrane β-strands, however, the PRED TMBB predicted an outer membrane strand spanning the amino acids 79–96. The globular beta barrel-shaped domain of the molecule I is located within the membrane, while the helical stems of the molecule are sprouted out of the membrane. Collectively, the topology predictions and membrane alignment studies could be considered as a reliable source for the prediction BatA structure. Figure 7 shows the BatA structure is embedded within the lipid membrane in the presence of water molecules.
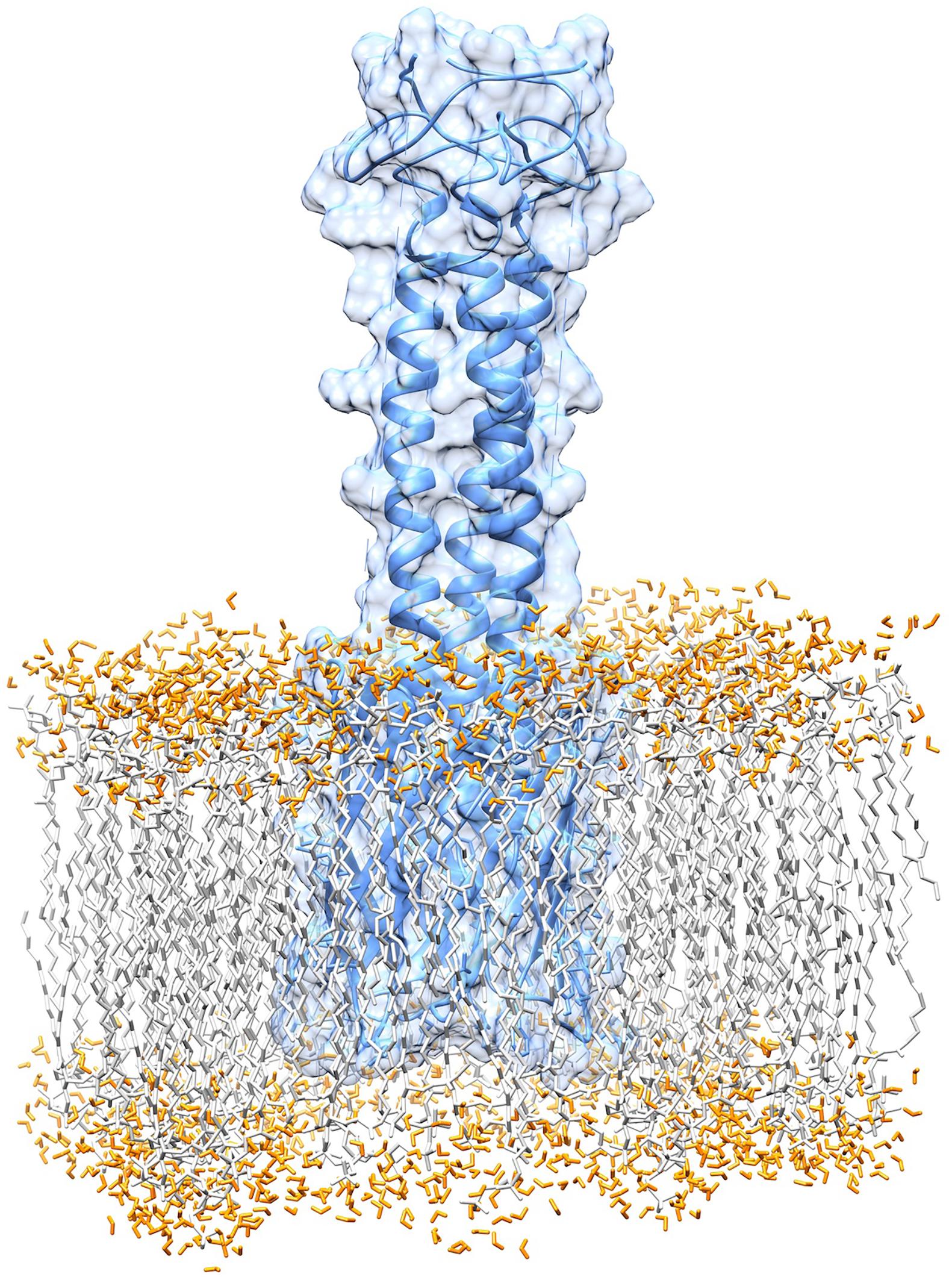
Figure 7. Graphical representation of the membrane anchoring domain of BatA. The alignment of the membrane anchoring region with the lipid bilayer of the membrane is provided.
To estimate the resiliency and toughness of the structures, a coarse-grained dynamics approach was employed, at which the high root mean square fluctuation (RMSF) value is indicative of high residue flexibility. The highest flexibility was associated with the HANS, HIM, and neck connectors, respectively, (Figure 8). Conversely, the beta hairpins represented lower RMSF values. The residues of YLHead domains and the membrane barrel had far lower RMSF values, representing their less flexibility.
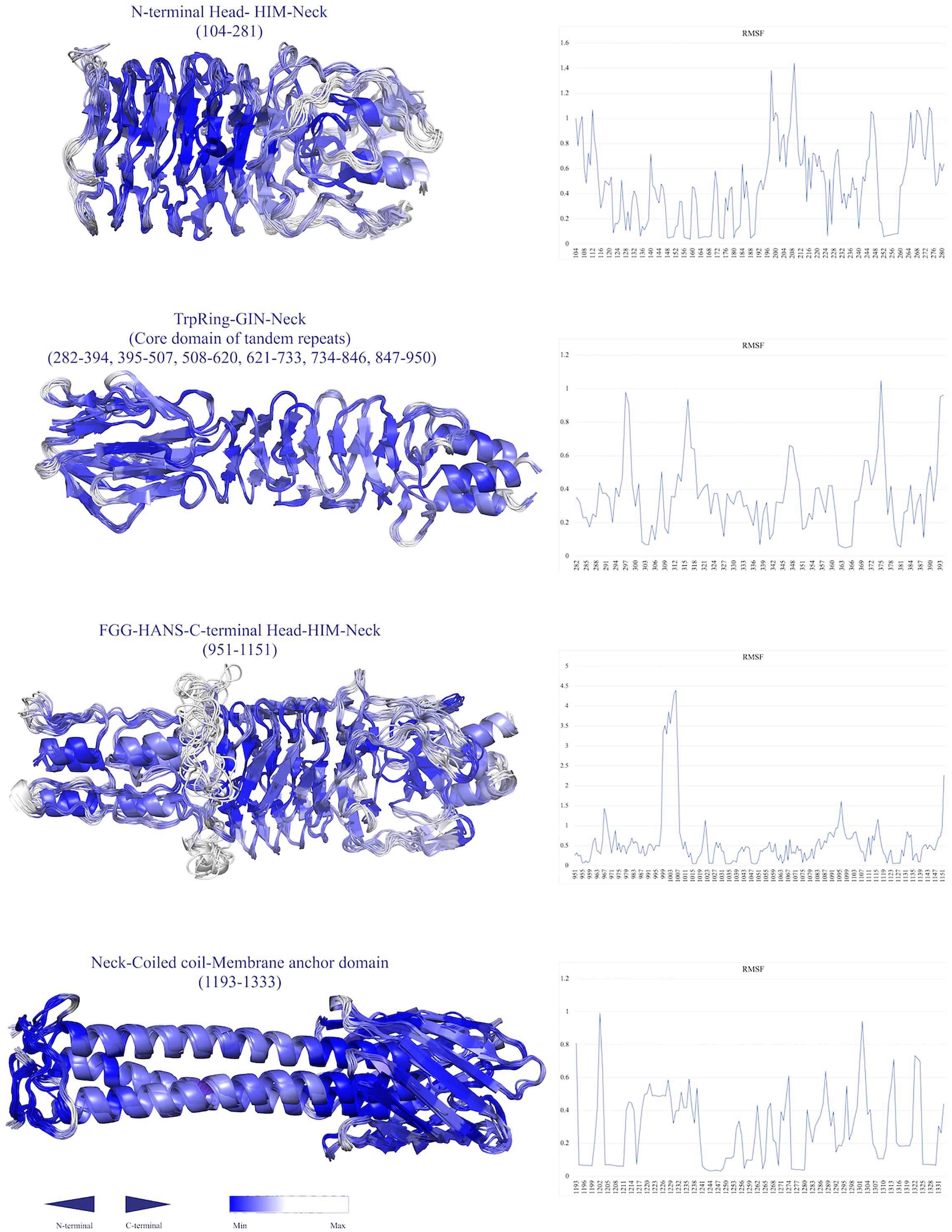
Figure 8. Simulations of the conformational flexibility of the structures by a Monte Carlo-based method (CABS dynamics). The residue fluctuation profiles are shown on the right and the ensemble fluctuation models are displayed on the left. The structures are labeled by their name and position. The residues are colored by a gradient of blue color (the higher the RMSF, the lower the density of blue color.
To examine the possible effect of cavities within the coiled-coil region of BatA, the existence and volume of cavities were assessed and measured. The largest cavities measured by the two servers (see “Materials and Methods” section), were presented in Figure 9. The numerical data representing the volume and surface of cavities are summarized in Table 4. As is evident in the table, the cavities of BatA coiled-coil are comparable to the other three examined molecules in term of volume. Moreover, the surrounding residues are similar to the conserved coiled-coil segment of TAA of Y. enterocolitica. The second large cavity in the coiled-coil structure of BatA is located in the transition site of this section (right-handed coiled-coil to left-handed coiled-coil; also see Figure 4).
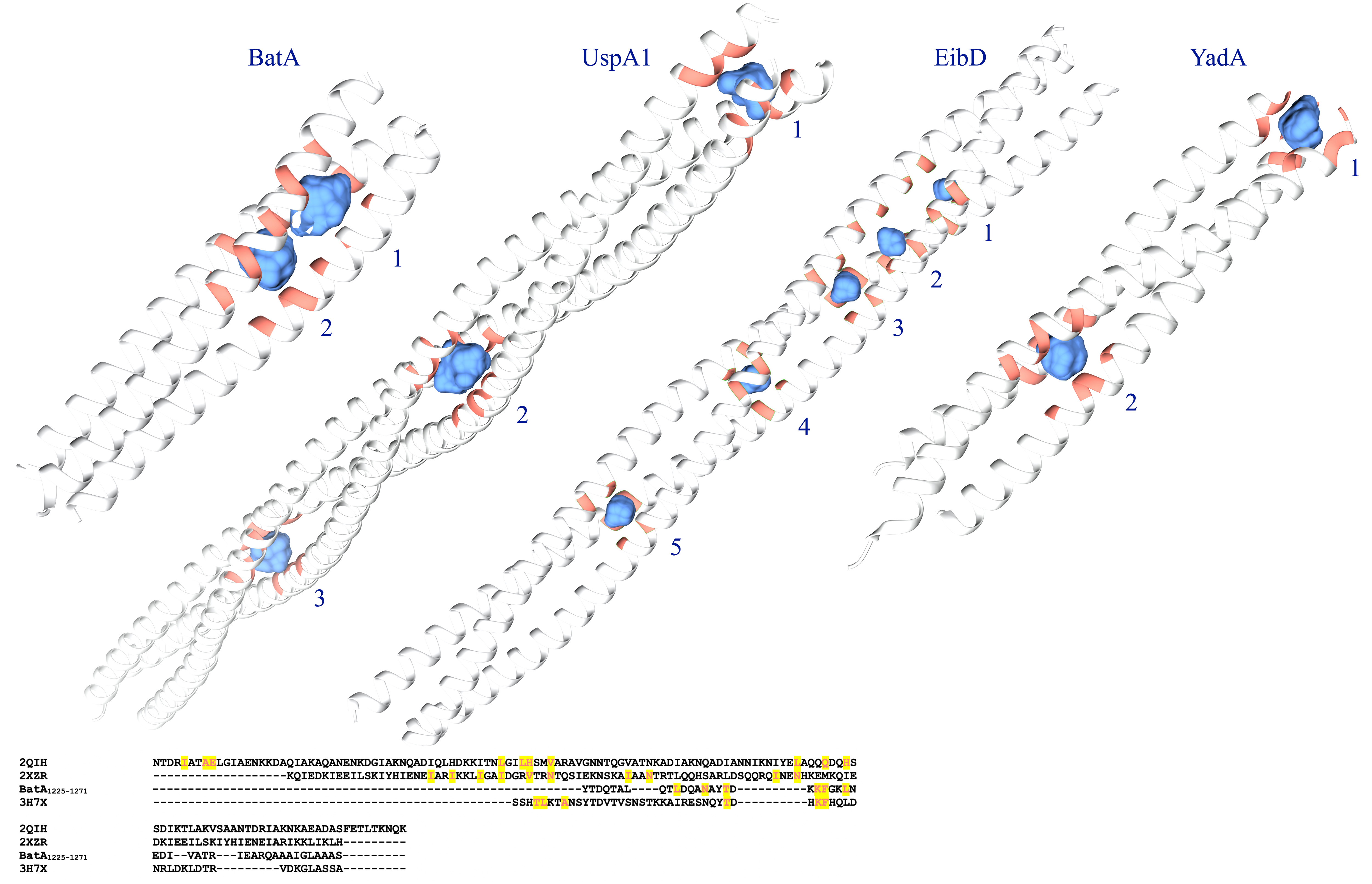
Figure 9. Graphical visualization of the major cavities. The cavities within the coiled-coil structure of BatA and the three well defined TAA coiled-coil structures are presented by blue surfaces. The structures are labeled by protein names; cavities are labeled by the Arabic numbers; details of the volumes are provided in Table 5. The lower panel shows the sequence alignment of these coiled-coil structures; the residues that surround the cavities are highlighted.
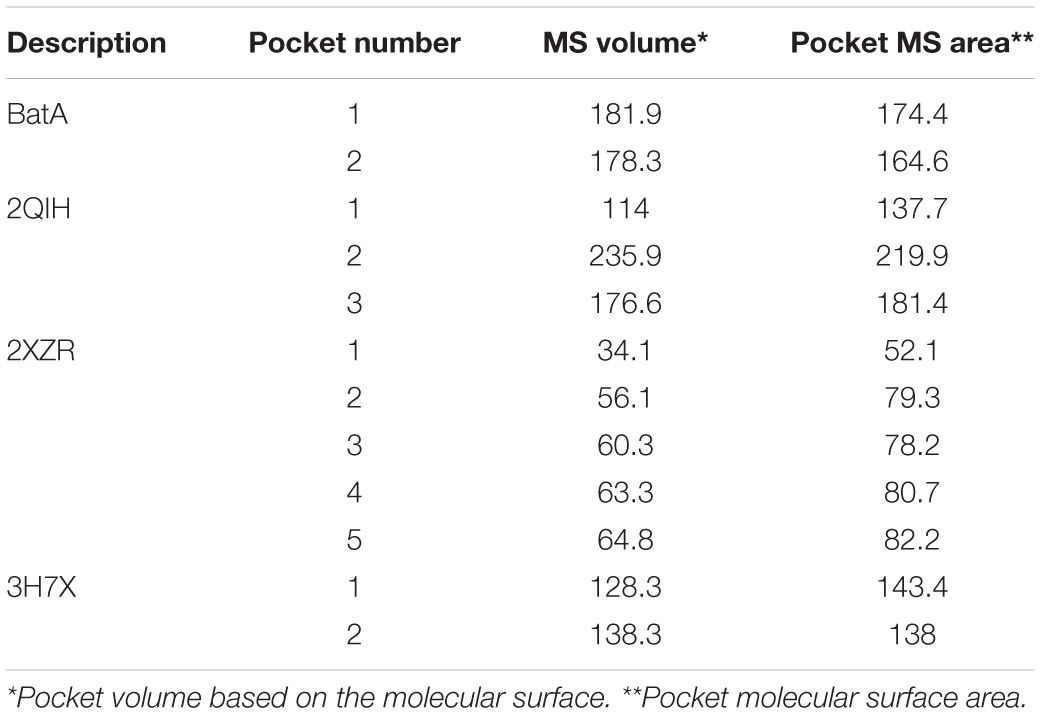
Table 4. The volume and surface area of the interior coiled-coil cavities of four TAAs.
To determine the distribution of TAA encoding genes in Brucellaceae, a tBLASTn search against all genomes of the family members was conducted by querying the consensus amino acid sequence of the membrane anchoring region of TAAs. All significant BLAST hits (E-value < 10–6) were mapped to the phylogenetic tree of Brucellaceae. The results showed that the TAA encoding genes were present in all 300 leaves of the tree; suggesting that all species share a common ancestor (Supplementary Data 1 and Supplementary Figure 4). Moreover, 201 leaves contained two TAA encoding genes (orange leaves in Supplementary Figure 4).
For estimating the best evolutionary model, the dataset involving 490 nucleotide sequences was analyzed by the model selection function of MEGA 7. Codon positions included were 1st + 2nd + 3rd + Non-coding. The positions containing gaps and missing data were eliminated. The best model for a proper description of the substitution pattern of nucleotide collection of BTAA membrane anchoring region (it is the model with the lowest Bayesian information criterion; BIC), was Kimura 2 parameter (Kimura, 1980; BIC: 12063.54 versus higher values of other models, Supplementary Data 2).
To examine whether the pattern of nucleotide substitution was homogenous, the disparity index tests of substitution patterns were performed for each pair of BTAA nucleotide sequences. In the disparity index (ID) test using all (1st + 2nd + 3rd) codons at the 5% significance level, the null hypothesis of neutrality could not be rejected for membrane anchoring region (Supplementary Data 2). Therefore, all sequences were retained for further analysis.
No significant bias was observed in the codon usage of the BTAA encoding genes. The mean differences between adaptiveness values ranged from 20.45% to 35.88%. The highest value attributed to the TAA genes of B. pinnipedialis (versus that of Omp31: 28.14%; Supplementary Data 1 and Supplementary Figure 5). The G + C content of TAA encoding genes was ∼54% (versus G + C content of Brucellaceae: 57.2%; Sankarasubramanian et al., 2016b).
To trace possible duplication events, an unrooted gene tree was built based on the nucleotide sequences of the membrane anchoring region. The search for duplication events was performed by finding the placement of the root of branches that produced the minimum number of duplication events. There were at least 14 gene duplication events within the tree (data not shown).
To define the recombination events, the whole sequences of BTAA encoding genes were analyzed by GARD. Substantial evidence for recombination breakpoints was found. The alignment contained 4691 potential breakpoints, translating into a search space of 11005086 models with up to two breakpoints, of which, 0.01% was explored by the genetic algorithm. The AICc [Akaike Information Criterion (AIC; Sugiura, 1978)] score allows for different topologies between segments (26929.4), the model then assumes a similar number of trees for all the partitions inferred by GARD, but allows for different branch lengths between partitions (28750.0). Comparing the AICc score of the best fitting GARD model suggested that the multiple tree model is preferred over the single tree model (based on an evidence ratio of greater than 100). At least one of the breakpoints reflects a true topological incongruence.
In order to measure the diversity and distances within BTAAs, all nucleotide sequences of the membrane anchoring region of BTAAs were grouped by their related species. The highest divergence was found between B. pinnipedialis and other species (Table 5). The number of base substitutions per site was calculated by averaging the substitution rate over all the sequence pairs within groups (Table 5). Analyses were conducted using the Kimura 2-parameter model. The included codon positions were 1st + 2nd + 3rd + Non-coding. All positions containing gaps and missing data were eliminated.
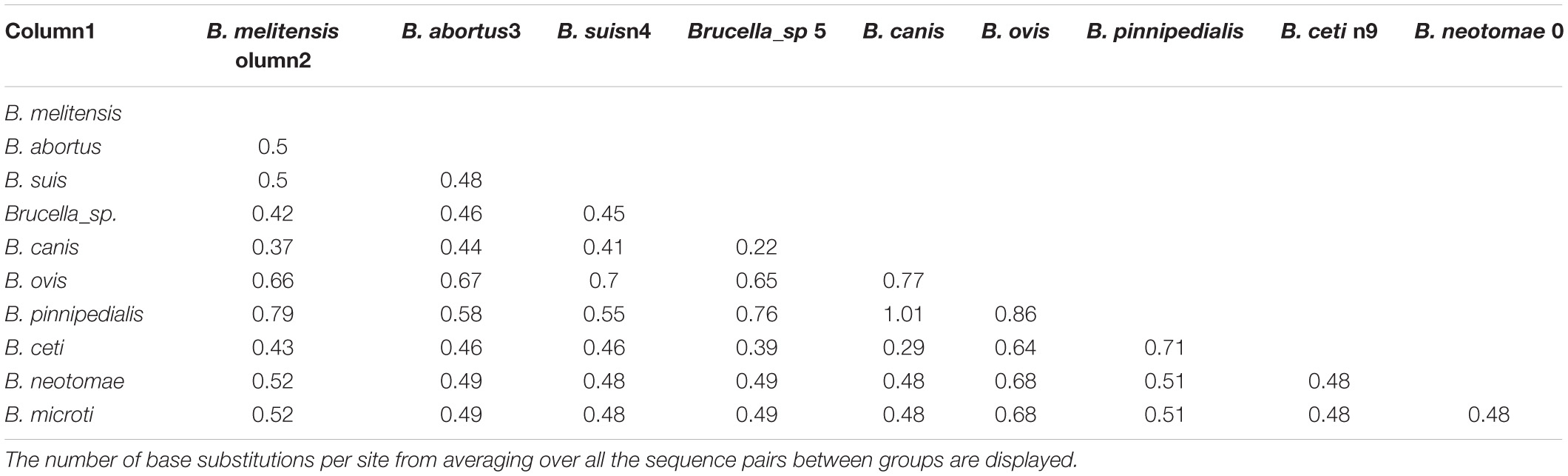
Table 5. Estimates of evolutionary divergence over sequence pairs between groups.
The Z-test for codon-based selection showed no evidence of positive or purifying selection in BTAA encoding genes (Supplementary Data 2), so the null hypothesis of neutral selection could not be rejected in favor of the alternative hypotheses of positive and purifying selection. Moreover, Fisher’s exact test of neutrality was not significant in rejecting the neutral selection hypothesis in favor of positive selection (Supplementary Data 2). These data suggest that mutations are neutral rather than beneficial.
This section contemplates the overall architecture of BTAAs. The batch analysis of all sequences revealed that the Sec signal peptide is predicted to exist in 17% of the sequences; 48% contain Tat signal peptide, and the remaining sequences had no detectable signal peptides. Interestingly, no correlation was observed between sequence length and the existence of signal peptides. The lengthiest sequences belong to B. abortus and B. melitensis (Supplementary Data 1 and Supplementary Table 1). As is summarized in Supplementary Table 1, the presence of identical sequences is common in the Brucella genus, consistent with the high level of genome identity in this genus.
The sequences are rich in coiled-coil regions. Moreover, 55% of the dataset contains repeat modules. The repeat modules include a tripartite module of TrpRing-GIN-Neck, which is repeated multiple times in many sequences, such as the BTAAs from B. abortus, B. melitensis, B. ceti, B. microti, and B. neotomae genomes, or is present in one copy in the TAAs of B. canis, B. ovis, B. suis, or is completely absent in the TAAs of B. pinnipedialis. The other common repeat modules are short sequences within the head domains. The head domains followed by HIM and Neck connectors are present in both the N-terminal and C-terminal regions of many BTAAs. Moreover, truncated heads and TrpRings can be observed in BTAA architectures (Figure 10).
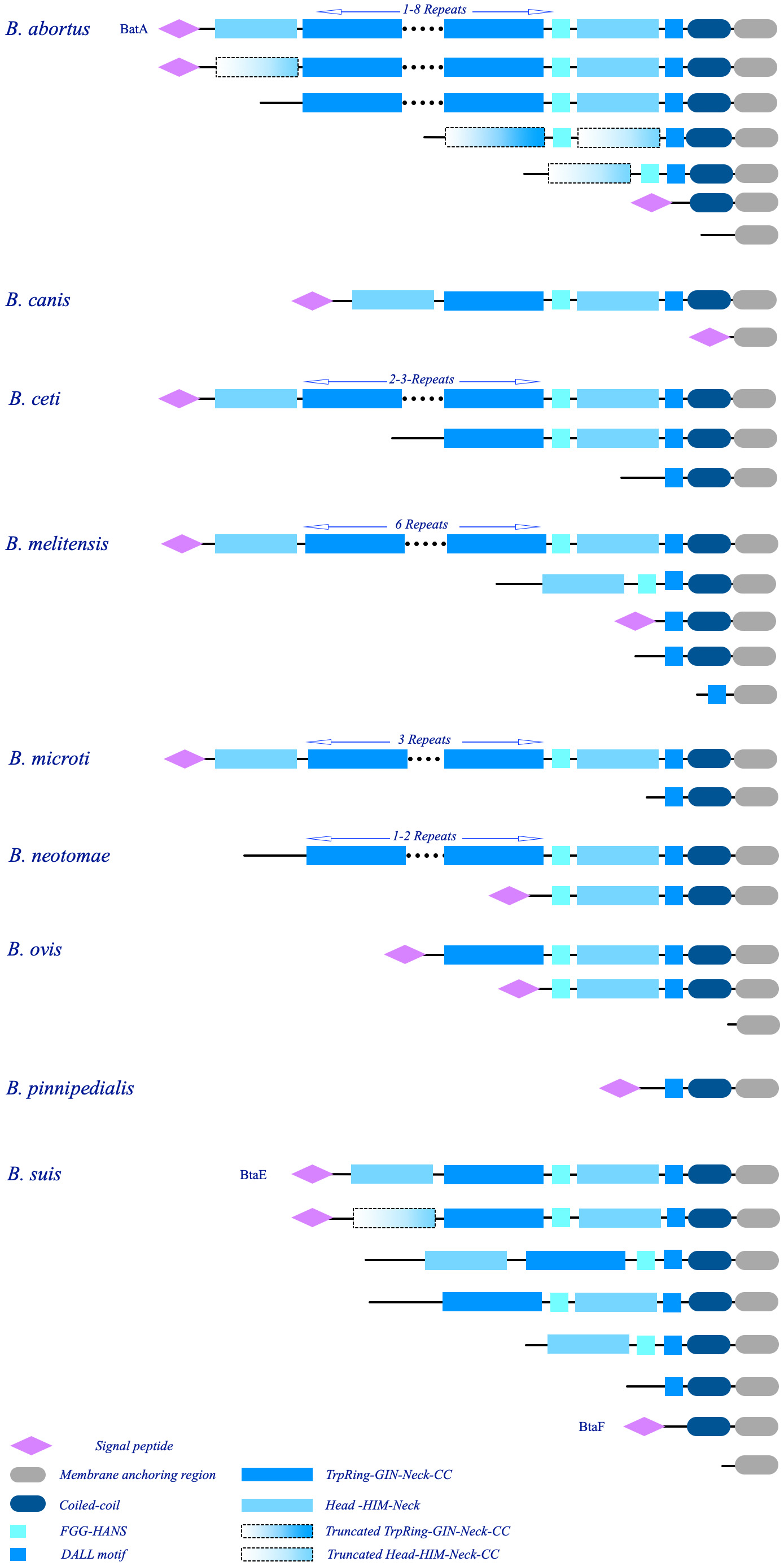
Figure 10. Schematic illustration of the major architectures of BTAAs. The observed architectures of BTAAs are presented; each group is labeled by the hosted species. The number of the repeat modules are shown at the top of the respective structures. The architectures of BatA, BtaE, and BtaF are assigned by protein names; notably, similar architectures to these three proteins can be found in the library (Supplementary Data 2). The schematic domains are identified at the bottom.
This paper defined the BatA as a prototypical TAA in the Brucella genomes. The architecture of BatA is a combination of all observed domains and motifs; however, although the exact architecture is not presented in all genomes (except for B. melitensis), similar architecture is presented in other species (except for B. pinnipedialis). As is evident in Figure 10, truncated heads and TrpRings are obvious in B. abortus and B. suis. Further, small TAAs including a single membrane anchoring region is presented in all species (except for B. suis).
The architectures of the two previously defined BTAAs, BtaE, and BtaF, are as follows: BtaF: Signal-peptide-N-terminal head-TrpRing-GIN-Neck-FGG-C-terminal Head-DALL-Coiled-coil-Membrane anchoring region; BtaE: Signal-Peptide-extended coiled-coil-Membrane anchoring region. The relatively small sequences of approximately 200 amino acid long, contain extended coiled-coils. Details of BTAAs are provided in Supplementary Data 2.
Duplicated genes are further apart in one chromosome and often in both forward and reverse strands. Therefore, both orthologous, and paralogs TAA genes are present in the Brucella genomes. The details of BTAA are provided in Supplementary Data 2 and Figure 10.
The present study was aimed at identifying and characterizing the TAAs in Brucellae via in silico approach. Computational tools provide a means to attain structural data (Negahdaripour et al., 2016) which is, by itself, a gateway to access a massive plethora of biological information (Negahdaripour et al., 2017). End conclusions are context-dependent, yet are generally derived from the broad concept of homology.
It appears that the few domains shared by the whole Brucellae population are responsible for the observed heterogeneity in the sequence length of BTAAs. Significant differences were also found in the arrangement and repeat frequencies of TAA domains in Brucella species. Paralogs of different sequence lengths are common within the species, which is indicative of duplication events; this conclusion is also supported by the evolutionary analyzes (see the evolutionary analysis). However, in comparison with Gammaproteobacterial TAAs, such as Acinetobacter species TAAs (Rahbar et al., 2019), the length of the sequences is shorter and less heterogeneous.
Moreover, the distribution of BLAST HSPs across the sequences revealed the existence of a few structural building blocks in the entire population (Figure 6B). Differences in the structural blocks of BTAAs are limited to the repeat frequencies of these blocks. These differences may play a variety of roles in host preferences and tissue tropism. This hypothesis was previously suggested by Chain et al. (2005) and Ruiz-Ranwez et al. (2013b) as well. The result of the disparity index test supported the homogeneity of membrane anchoring sequences, suggesting that TAA genes have evolved via the same or similar evolutionary processes. Moreover, the presence of the TAA-encoding genes in all members of the family implies that these genes share a common ancestor. This conclusion is further supported by a lack of evidence for conjugation (no plasmid is identified in Brucellaceae, except for the Ochrobacteria, which harbors TAA encoding plasmids), as well as similar G + C contents and the paucity of codon biases.
The differences in sequence length, domain complexity, and the existence of paralogs could be explained by recombination and duplication events. It can be deduced that all sequences are derived from a common ancestor, which can, in turn, provide insight into the existence of population-wise domains. The observed heterogeneity could be the consequence of genetic events such as the duplication and recombination events that occurred intraspecies. This conclusion is consistent with the evolutionary history of Brucellaceae.
According to a previous genomic comparison study, 95% DNA identity was revealed across the genomes of B. abortus, B melitensis, B. canis, B. neotomae, and B. ovis, which led to the assumption that they have all diverged from a common ancestor that was most likely very close to B. ovis (Foster et al., 2009).
Introducing a prototype sequence is commonly used to define a protein family (Andreeva et al., 2013). This approach is more pronounced for TAAs, which share common structural and functional properties (Linke et al., 2006). As is the case here, it seems rational to analyze a prototype sequence and make comparisons to define the entire members of the gene family.
It has been shown that due to the TAA sequence diversity, alignment and tree-based analyzes are not suitable to investigate the properties of TAAs (Pina et al., 2009; Fialho and Mil-Homens, 2011). Hence, all-against-all BLAST was harnessed here, as an appropriate alternative to handle the massive data, including very disparate sequences (Rahbar et al., 2019). Furthermore, it is difficult to annotate the domains of TAAs due to their considerable sequence diversity, the various patterns of domain arrangements, and the limited coverage of sequences by known structures (Meng et al., 2006; Bassler et al., 2015). This was evident in our study following the alignment and observation of the low level of sequence similarity. Due to the same reasons, general domain annotators, such as Pfam, perform poorly in recognizing the TAA domains (Szczesny and Lupas, 2008). Given these circumstances, invoking an innovative and integrated approach could be of value. The solution could be an structural analysis, especially when the homologous structures are available via the process of homology modeling (Montgomerie et al., 2008; Steinegger et al., 2019; Zheng et al., 2019).
Brucella abortus is the main causative agent of brucellosis in cattle leading to abortion, infertility, and decreased milk production (Neta et al., 2010). Brucella abortus 2308, a highly virulent strain isolated from an aborted fetus of a cow (Jones et al., 1965), is wildly used as a reference strain in many brucellosis research (Suárez-Esquivel et al., 2016). A 1559 amino acid long TAA (locus tag: P050_01136, B. abortus 90–12178) from this bacteria was the largest TAA found in our dataset. The difference between BatA and the latter (P050_01136) is limited to the frequency of core repeat modules. Among the architectures defined for various TAAs, BatA has a relatively low level of complexity and the protein architecture matched to 104 similar protein architectures in the Interpro database.
Homology modeling based on the known homologous structures is the most successful approach for protein structure prediction. The phenomenon is rooted in the fact that structures diverge much more slowly than sequences, even when their sequences have diverged out of a distinguishing range (Kinch and Grishin, 2002). Sensitivity is crucial for success in homology detection, since many proteins have only remote relatives in the structure database (Söding et al., 2005). TAAs do not share global sequence similarities, although they perform common functions (Linke et al., 2006). Despite the differences in their sequences, structural similarities highlight the important role of structure in these proteins. On the other hand, it implies that various evolutionary events have manipulated the primary sequence and domain arrangements of these proteins.
By searching the databases with HHpred, the best templates were selected to generate tertiary structures. Comparing the built structures with that of the resolved structures allowed us to annotate the domain architecture of the protein.
The null hypothesis of homogeneity of N-terminal of BTAAs was rejected (data not shown); revealing a diversity in this region (the 160 nucleotides of N-terminal of BTAAs is considered to be the coding region the signal peptide).
The signal peptide in TAAs generally delivers the unfolded proteins to the Sec secretion machinery (Navarro-Garcia et al., 2004). However, in our dataset, inclusion of the N-terminus region of BTAA encoding genes resulted in a significant level of diversity. Both Tat and Sec signal peptides were predicted in the population, though many sequences did not comprise any detectable signal peptide. Therefore, while the secretion of some BTAAs is under debate, the microarray data have shown that both TAAs from the B. abortus 2308 genome are expressed (Kleinman et al., 2017). Although based on the prediction of interaction among secretion systems in the Brucella genus (Sankarasubramanian et al., 2016a), both Sec and Tat pathway proteins interact with T4SS and T5SS proteins, and it seems unlikely that Tat pathway participates in BTAAs translocation. This notion could be rooted in two facts: (i) Sec signal peptide is the most common signal peptide in TAAs in different genera; and (ii) TAAs are known to be secreted in unfolded forms (Navarro-Garcia et al., 2004; Cotter et al., 2005; Linke et al., 2006; Łyskowski et al., 2011), which is not consistent with the Tat system (Natale et al., 2008). Thus, the predicted Tat signal could probably be a computational error due to the high similarity between the targeting signals (Natale et al., 2008).
A trimeric lipoprotein in Enterobacteria is involved in the biogenesis of TAA (Grin et al., 2014) and arranged as an operon with TAAs in Enterobacteria. A similar operon has been also introduced in Acinetobacter Tol5 (AtaA and TpgA; Ishikawa et al., 2016). The BatA encoding gene and its downstream gene (IalB, 182 aa) are stated to constitute a bicistronic unit (Kleinman et al., 2017). IalB was predicted to be localized in the periplasmic region or the inner-membrane of Brucellae (in contrast to the trimeric lipoprotein of Enterobacteria and TpgA, which are outer membrane proteins). IalB has been also introduced in Bartonella bacilliformis as a major virulence factor with a direct role in human erythrocyte parasitism (Coleman and Minnick, 2001). The structural alignments of IalB and TpgA (TCoffee expresso package (Armougom et al., 2006); the server combines the structural information with sequence data to build alignment; the alignment is built on sequences) showed some levels of similarity (data not shown). However, our method did not provide sufficient data to interpret the relationship between BatA and IalB. Kleinman et al. (2017) proposed that this bicistronic unit is under the regulatory control of VjbR. A combinatorial control system following the association of HutC and MdrA regulatory proteins was also observed (Sieira et al., 2017). The MucR was reported to be another regulatory element for this open reading frame (Caswell et al., 2013). It seems that operon configuration is not the most likely explanation for this genomic proximity between BTAAs and IalB encoding genes. The exact relationship of these two neighbor genes (if any) is remained to be explored.
Residue fluctuation profile of the BatA domains suggests that coils were generally responsible for the highest RMSF values. The most flexible regions with the highest RMSF (coil-containing structures including HANS, HIM, and neck connectors) provide the bending sites for the nano-fiber. This property was previously noted for the HANS motif (Hartmann et al., 2012). The C-terminal coiled-coil region exhibited lower levels of flexibility, which is consistent with the fact that coiled-coils stabilize the structure of the protein (Meng et al., 2006). However, as previously ascribed to some TAAs, such as the UspA1 protein from Moraxella catarrhalis (Conners et al., 2008), the coiled-coil region of the E. coli EibD (Leo et al., 2011); and a conserved coiled-coil segment of TAA of Y. enterocolitica (Alvarez et al., 2010), interior cavities are the regions of deformation to allow bending the stalk. Attribution of such a role to the cavities within the C-terminal coiled-coils is not ruled out. Because, cavities of the similar volume comparable to the aforesaid proteins were observed in BTAAs.
Globular heads and beta rolls also represented a low RMSF value implying the rigidity of these domains. While both the aforesaid domains contain structurally repetitive units, it seems that repetitive structures [mostly cross beta-prisms (Roche et al., 2018)] may confer an overall toughness to the protein. This is obvious in the membrane anchoring region, which is composed of repetitive up and down beta hair-pins structures (Roche et al., 2018). These strands were in a lower RMSF state.
The primary requirement for such an adhesin to deliver its functions is its structural length. It should be long enough to pass through the surface elements of the bacterial cell and reach the receptors on the host cell. Moreover, it has to be flexible to form multiple binding sites and ensure a high affinity. The core domain of repeat modules (consisted of TrpRing), which is suspected to bind to fibronectin (Meng et al., 2008; Szczesny et al., 2008; Qin et al., 2016), along with the proper flexibility of the specific domains, provide opportunities for the adhesin to bind from multiple sites (six Trp-ring-GIN strung together by neck connectors). Thus, it could be suggested that despite the low affinity of TrpRing to fibronectin, binding from multiple sites might enhance the overall affinity and enable the protein to overcome the mechanical forces. Given the approximate length of 85 nm (BatA), it can pass through the surface components of the bacterial cells. Moreover, trimerization provides three identical faces that expose multivalent binding sites upon adhesion (Cotter et al., 2006).
It has been shown that trimerization is essential for the full functionality of TAAs (Cotter et al., 2006; Schütz et al., 2010). The main structural reason for such trimerization is hydrophobic interactions especially head domain, whose cores are extremely hydrophobic (Nummelin et al., 2004). Furthermore, the collocation of three alpha-helical coils makes a hydrophilic region (due to polar side chains) that sequesters the ions into the coiled-coils (Hartmann et al., 2009). C-terminal coiled-coils are available in almost all BTAAs. Therefore, it can be speculated that all BTAAs have minimal requirements for trimerization. However, lacking functional domains (perpendicular and interleaved heads) in many members would cast doubt on the extent of functionality of all BTAAs.
It has been proved that many TAAs are involved in biofilm formation. Brucellae has long been considered as a facultative intracellular pathogen in most references. However, they were re-designated as a facultative extracellular-intracellular pathogen due to their evolutionary relationship to other alpha-proteobacteria (El-Sayed and Awad, 2018). The biofilm lifestyle of Brucella has already been confirmed (Uzureau et al., 2007; Godefroid et al., 2010; Almirón et al., 2013). Therefore, the possible relationship of TAA with the biofilm lifestyle of Brucellae could be the objective of future studies.
Contrary to other genera such as Acinetobacter, Burkholderia, and Haemophilus, the absence of any plasmid or phage in the Brucella genus has ruled out the possibility for transfer of TAA genes by conjugation.
Brucellae coevolved with animals in a pathogenic manner. Given their broad mammalian host range, Brucella species are faced with different environmental conditions (phagocytic and non-phagocytic cells such as fibroblasts and epithelial cells) and consequently various stressful conditions. However, the genetic diversity in the species is relatively low. The bacterium required several components to survive within a mammalian host cell. Here, it seems that TAA is not a critical factor for the bacterial survival. The mechanism for the expression of most genes is debatable due to the deletion of relative signal peptides. Moreover, the functionality of several members is also questionable due to the lack of functional domains (i.e., various sorts of head domains). The relative abundance of synonymous and non-synonymous substitutions that have occurred in the gene sequences has been compared in the present study. The data implies that substitutions are neutral. TAA harboring phenotype is not an optimum phenotype for survival. Therefore, TAA encoding genes are not under positive selection. On the other hand, the virulence of pathogenic species was weakened by mutations in the TAA gene. These mutations did not end with the complete abolishment of virulence, which indicates that these sorts of adhesins are not critical to establish the infection. It can be assumed that the adhesion to the host cells takes place by other components. Thus, positive or purifying selection does not affect the TAA genes in Brucellaceae. Presumably, due to neutral mutations in the population, the non-adaptive evolution would be a well-fitted theory. Since the model does not invoke a positive selection as the driving force of fixation, the BTAA encoding genes may have evolved through a non-adaptive evolution.
Brucella species, as zoonotic intracellular pathogens, harness an arsenal of membrane components to penetrate the host cells. The trimeric autotransporters are among the many virulence factors of Brucellaceae, which have evolved through a non-adaptive evolution process. The evolutionary events, sequence diversity, and structural complexity are dictated partly by the repetitive nature of these adhesins. The modifications include alterations within the repeat frequency of a few structural blocks of a common ancestral gene. The events have occurred among species without the interference of foreign sequences. The issue is traceable by computational tools. The in silico approach used here holds the potential for handling such sets of disparate sequences to investigate the great regularity of the living systems, especially at a molecular level. However, the proposed model herein does not rule out the existence of any other evolutionary event. Thus, other possible processes could also be identified through further studies.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
MR proposed and designed the idea and the study, contributed to the writing of the manuscript, and discussed the results. MZ, AJ, and SK collected, processed, and analyzed the data, and were involved in the study design. NN, MN, and YF contributed to the writing of the manuscript and revised the final version. MN also edited the final version. AS contributed to the writing of the manuscript and discussed the results. YG proposed and designed the idea and the study, provided the facilities, funding, commented, and revised the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors wish to thanks the Pharmaceutical Science Research Center, Shiraz University of Medical Science. They also would like to thank Dr. Mohammad Hajdezfulian for language editing and proofreading the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.560667/full#supplementary-material
Almirón, M. A., Roset, M. S., and Sanjuan, N. (2013). The aggregation of Brucella abortus occurs under microaerobic conditions and promotes desiccation tolerance and biofilm formation. Open Microbiol. J. 7.87–91. doi: 10.2174/1874285801307010087
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl. Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Alva, V., Nam, S.-Z., Söding, J., and Lupas, A. N. (2016). The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucl Acids Res. 44, W410–W415.
Alvarez, B. H., Gruber, M., Ursinus, A., Dunin-Horkawicz, S., Lupas, A. N., and Zeth, K. (2010). A transition from strong right-handed to canonical left-handed supercoiling in a conserved coiled-coil segment of trimeric autotransporter adhesins. J. Struct. Biol. 170, 236–245. doi: 10.1016/j.jsb.2010.02.009
Andreeva, A., Howorth, D., Chothia, C., Kulesha, E., and Murzin, A. G. (2013). SCOP2 prototype: a new approach to protein structure mining. Nucl. Acids Res. 42, D310–D314.
Armougom, F., Moretti, S., Poirot, O., Audic, S., Dumas, P., Schaeli, B., et al. (2006). Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucl. Acids Res. 34, W604–W608.
Backert, S., Fronzes, R., and Waksman, G. (2008). VirB2 and VirB5 proteins: specialized adhesins in bacterial type-IV secretion systems? Trends Microbiol. 16, 409–413. doi: 10.1016/j.tim.2008.07.001
Baek, M., Park, T., Heo, L., Park, C., and Seok, C. (2017). GalaxyHomomer: a web server for protein homo-oligomer structure prediction from a monomer sequence or structure. Nucl. Acids Res. 45, W320-W324.
Bagos, P. G., Nikolaou, E. P., Liakopoulos, T. D., and Tsirigos, K. D. (2010). Combined prediction of Tat and Sec signal peptides with hidden Markov models. Bioinformatics 26, 2811–2817. doi: 10.1093/bioinformatics/btq530
Bassler, J., Alvarez, B. H., Hartmann, M. D., and Lupas, A. N. (2015). A domain dictionary of trimeric autotransporter adhesins. Int. J. Med. Microbiol. 305, 265–275. doi: 10.1016/j.ijmm.2014.12.010
Bendtsen, J. D., Nielsen, H., Widdick, D., Palmer, T., and Brunak, S. (2005). Prediction of twin-arginine signal peptides. BMC Bioinformatics 6:167. doi: 10.1186/1471-2105-6-167
Bernstein, H. D. (2019). Type V secretion in gram−negative bacteria. EcoSal Plus 8:10.1128/ecosalplus.ESP-0031-2018.
Bzhalava, Z., Tampuu, A., Bała, P., Vicente, R., and Dillner, J. (2018). Machine Learning for detection of viral sequences in human metagenomic datasets. BMC Bioinformatics 19:336. doi: 10.1186/s12859-018-2340-x
Castañeda-Roldán, E. I., Ouahrani-Bettache, S., Saldaña, Z., Avelino, F., Rendón, M. A., Dornand, J., et al. (2006). Characterization of SP41, a surface protein of Brucella associated with adherence and invasion of host epithelial cells. Cell. Microbiol. 8, 1877–1887. doi: 10.1111/j.1462-5822.2006.00754.x
Caswell, C. C., Elhassanny, A. E., Planchin, E. E., Roux, C. M., Weeks-Gorospe, J. N., Ficht, T. A., et al. (2013). Diverse genetic regulon of the virulence-associated transcriptional regulator MucR in Brucella abortus 2308. Infect. Immun. 81, 1040–1051. doi: 10.1128/iai.01097-12
Chain, P. S., Comerci, D. J., Tolmasky, M. E., Larimer, F. W., Malfatti, S. A., Vergez, L. M., et al. (2005). Whole-genome analyses of speciation events in pathogenic Brucellae. Infect. Immun. 73, 8353–8361. doi: 10.1128/iai.73.12.8353-8361.2005
Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., et al. (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sec. D. Biol. Crystallogr. 66, 12–21. doi: 10.1107/s0907444909042073
Chovancova, E., Pavelka, A., Benes, P., Strnad, O., Brezovsky, J., Kozlikova, B., et al. (2012). CAVER 3.0: a tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol. 8:e1002708. doi: 10.1371/journal.pcbi.1002708
Coleman, S. A., and Minnick, M. F. (2001). Establishing a direct role for the Bartonella bacilliformis invasion-associated locus B (IalB) protein in human erythrocyte parasitism. Infect. Immun. 69, 4373–4381. doi: 10.1128/iai.69.7.4373-4381.2001
Conners, R., Hill, D. J., Borodina, E., Agnew, C., Daniell, S. J., Burton, N. M., et al. (2008). The Moraxella adhesin UspA1 binds to its human CEACAM1 receptor by a deformable trimeric coiled-coil. EMBO J. 27, 1779–1789. doi: 10.1038/emboj.2008.101
Consortium, U. (2017). UniProt: the universal protein knowledgebase. Nucl. Acids Res. 45, D158–D169.
Corbel, M. J. (1997). Brucellosis: an overview. Emerg. Infect. Dis. 3:213. doi: 10.3201/eid0302.970219
Cotter, S. E., Surana, N. K., and Geme, J. W. S. III (2005). Trimeric autotransporters: a distinct subfamily of autotransporter proteins. Trends Microbiol. 13, 199–205. doi: 10.1016/j.tim.2005.03.004
Cotter, S. E., Surana, N. K., Grass, S., and Geme, J. W. S. (2006). Trimeric autotransporters require trimerization of the passenger domain for stability and adhesive activity. J. Bacteriol. 188, 5400–5407. doi: 10.1128/jb.00164-06
Crasta, O. R., Folkerts, O., Fei, Z., Mane, S. P., Evans, C., Martino-Catt, S., et al. (2008). Genome sequence of Brucella abortus vaccine strain S19 compared to virulent strains yields candidate virulence genes. PLoS One 3:e2193. doi: 10.1371/journal.pone.0002193
Czibener, C., Merwaiss, F., Guaimas, F., Del Giudice, M. G., Serantes, D. A. R., Spera, J. M., et al. (2016). BigA is a novel adhesin of Brucella that mediates adhesion to epithelial cells. Cell. Microbiol. 18, 500–513. doi: 10.1111/cmi.12526
Czibener, C., and Ugalde, J. E. (2012). Identification of a unique gene cluster of Brucella spp. that mediates adhesion to host cells. Microb. Infect. 14, 79–85. doi: 10.1016/j.micinf.2011.08.012
Dautin, N., and Bernstein, H. D. (2007). Protein secretion in gram-negative bacteria via the autotransporter pathway. Annu. Rev. Microbiol. 61, 89–112. doi: 10.1146/annurev.micro.61.080706.093233
De Castro, E., Sigrist, C. J., Gattiker, A., Bulliard, V., Langendijk-Genevaux, P. S., Gasteiger, E., et al. (2006). ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucl. Acids Res. 34, W362–W365.
Edmonds, M. D., Cloeckaert, A., Booth, N. J., Fulton, W. T., Hagius, S. D., Walker, J. V., et al. (2001). Attenuation of a Brucella abortus mutant lacking a major 25 kDa outer membrane protein in cattle. Am. J. Vet. Res. 62, 1461–1466. doi: 10.2460/ajvr.2001.62.1461
El-Sayed, A., and Awad, W. (2018). Brucellosis: Evolution and expected comeback. Int. J. Vet. Sci. Med. 6, S31–S35.
ElTahir, Y., Al-Araimi, A., Nair, R. R., Autio, K. J., Tu, H., Leo, J. C., et al. (2019). Binding of Brucella protein, Bp26, to select extracellular matrix molecules. BMC Mole Cell Biol. 20:1–12. doi: 10.1186/s12860-020-00263-4
Fialho, A. M., and Mil-Homens, D. (2011). Trimeric autotransporter adhesins in members of the Burkholderia cepacia complex: a multifunctional family of proteins implicated in virulence. Front. Cell. Infect. Microbiol. 1:13. doi: 10.3389/fcimb.2011.00013
Finn, R. D., Attwood, T. K., Babbitt, P. C., Bateman, A., Bork, P., Bridge, A. J., et al. (2017). InterPro in 2017—beyond protein family and domain annotations. Nucl. Acids Res. 45, D190–D199.
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., Mitchell, A. L., et al. (2016). The Pfam protein families database: towards a more sustainable future. Nucl. Acids Res. 44, D279–D285.
Foster, J. T., Beckstrom-Sternberg, S. M., Pearson, T., Beckstrom-Sternberg, J. S., Chain, P. S., Roberto, F. F., et al. (2009). Whole-genome-based phylogeny and divergence of the genus Brucella. J. Bacteriol. 191, 2864–2870. doi: 10.1128/jb.01581-08
Frank, K., and Sippl, M. J. (2008). High-performance signal peptide prediction based on sequence alignment techniques. Bioinformatics 24, 2172–2176. doi: 10.1093/bioinformatics/btn422
Frickey, T., and Lupas, A. (2004). CLANS: a Java application for visualizing protein families based on pairwise similarity. Bioinformatics 20, 3702–3704. doi: 10.1093/bioinformatics/bth444
Fuhrmann, M., Hausherr, A., Ferbitz, L., Schödl, T., Heitzer, M., and Hegemann, P. (2004). Monitoring dynamic expression of nuclear genes in Chlamydomonas reinhardtii by using a synthetic luciferase reporter gene. Plant Mole. Biol. 55, 869–881. doi: 10.1007/s11103-005-2150-1
Godefroid, M., Svensson, M. V., Cambier, P., Uzureau, S., Mirabella, A., De Bolle, X., et al. (2010). Brucella melitensis 16M produces a mannan and other extracellular matrix components typical of a biofilm. FEMS Immun. Med. Microbiol. 59, 364–377. doi: 10.1111/j.1574-695x.2010.00689.x
Gopalakrishnan, A., Dimri, U., Saminathan, M., Yatoo, M., Priya, G. B., Gopinath, D., et al. (2016). Virulence factors, intracellular survivability and mechanism of evasion from host immune response by brucella: an overview. JAPS J. Anim. Plant Sci. 26, 1542-1555.
Gourlay, L. J., Peano, C., Deantonio, C., Perletti, L., Pietrelli, A., Villa, R., et al. (2015). Selecting soluble/foldable protein domains through single-gene or genomic ORF filtering: structure of the head domain of Burkholderia pseudomallei antigen BPSL2063. Acta Crystallogr. Sec. D. Biol. Crystallogr 71, 2227–2235. doi: 10.1107/s1399004715015680
Grin, I., Hartmann, M. D., Sauer, G., Alvarez, B. H., Schütz, M., Wagner, S., et al. (2014). A trimeric lipoprotein assists in trimeric autotransporter biogenesis in enterobacteria. J. Biol. Chem. 289, 7388–7398. doi: 10.1074/jbc.m113.513275
Hartmann, M. D., Grin, I., Dunin-Horkawicz, S., Deiss, S., Linke, D., Lupas, A. N., et al. (2012). Complete fiber structures of complex trimeric autotransporter adhesins conserved in enterobacteria. Proc. Natl. Acad. Sci. 109, 20907–20912. doi: 10.1073/pnas.1211872110
Hartmann, M. D., Ridderbusch, O., Zeth, K., Albrecht, R., Testa, O., Woolfson, D. N., et al. (2009). A coiled-coil motif that sequesters ions to the hydrophobic core. Proc. Natl. Acad. Sci. 106, 16950–16955. doi: 10.1073/pnas.0907256106
Herman, L., and De Ridder, H. (1992). Identification of Brucella spp. by using the polymerase chain reaction. Appl. Environ. Microbiol. 58, 2099–2101. doi: 10.1128/aem.58.6.2099-2101.1992
Ishikawa, M., Yoshimoto, S., Hayashi, A., Kanie, J., and Hori, K. (2016). Discovery of a novel periplasmic protein that forms a complex with a trimeric autotransporter adhesin and peptidoglycan. Mole. Microbiol. 101, 394–410. doi: 10.1111/mmi.13398
Jamroz, M., Kolinski, A., and Kmiecik, S. (2013). CABS-flex: server for fast simulation of protein structure fluctuations. Nucl. Acids Res. 41, W427–W431.
Jones, L. M., Montgomery, V., and Wilson, J. (1965). Characteristics of carbon dioxide-independent cultures of Brucella abortus isolated from cattle vaccinated with strain 19. J. Infect. Dis. 115, 312–320. doi: 10.1093/infdis/115.3.312
Jorda, J., and Kajava, A. V. (2009). T-REKS: identification of Tandem REpeats in sequences with a K-meanS based algorithm. Bioinformatics 25, 2632–2638. doi: 10.1093/bioinformatics/btp482
Junier, T., and Pagni, M. (2000). Dotlet: diagonal plots in a web browser. Bioinformatics 16, 178–179. doi: 10.1093/bioinformatics/16.2.178
Käll, L., Krogh, A., and Sonnhammer, E. L. (2004). A combined transmembrane topology and signal peptide prediction method. J. Mole. Biol. 338, 1027–1036. doi: 10.1016/j.jmb.2004.03.016
Käll, L., Krogh, A., and Sonnhammer, E. L. (2007). Advantages of combined transmembrane topology and signal peptide prediction—the Phobius web server. Nucl. Acids Res. 35, W429–W432. doi: 10.1093/nar/gkm256
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Prot. 10, 845–858. doi: 10.1038/nprot.2015.053
Kiessling, A. R., Malik, A., and Goldman, A. (2020). Recent advances in the understanding of trimeric autotransporter adhesins. Med. Microbiol. Immun. 209, 233–242. doi: 10.1007/s00430-019-00652-3
Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mole. Evol. 16, 111–120. doi: 10.1007/bf01731581
Kinch, L. N., and Grishin, N. V. (2002). Evolution of protein structures and functions. Curr. Opin. Struct. Biol. 12, 400–408.
Kleinman, C. L., Sycz, G., Bonomi, H. R., Rodríguez, R. M., Zorreguieta, A., and Sieira, R. (2017). ChIP-seq analysis of the LuxR-type regulator VjbR reveals novel insights into the Brucella virulence gene expression network. Nucl. Acids Res. 45, 5757–5769. doi: 10.1093/nar/gkx165
Koiwai, K., Hartmann, M. D., Linke, D., Lupas, A. N., and Hori, K. (2016). Structural basis for toughness and flexibility in the C-terminal passenger domain of an Acinetobacter trimeric autotransporter adhesin. J. Biol. Chem. 291, 3705–3724. doi: 10.1074/jbc.m115.701698
Kosakovsky Pond, S. L., Posada, D., Gravenor, M. B., Woelk, C. H., and Frost, S. D. (2006). Automated phylogenetic detection of recombination using a genetic algorithm. Mole. Biol. Evol. 23, 1891–1901. doi: 10.1093/molbev/msl051
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mole. Bio. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Leo, J. C., Lyskowski, A., Hattula, K., Hartmann, M. D., Schwarz, H., Butcher, S. J., et al. (2011). The structure of E. coli IgG-binding protein D suggests a general model for bending and binding in trimeric autotransporter adhesins. Structure 19, 1021–1030. doi: 10.1016/j.str.2011.03.021
Linke, D., Riess, T., Autenrieth, I. B., Lupas, A., and Kempf, V. A. (2006). Trimeric autotransporter adhesins: variable structure, common function. Trends Microbiol. 14, 264–270. doi: 10.1016/j.tim.2006.04.005
Łyskowski, A., Leo, J. C., and Goldman, A. (2011). Structure and biology of trimeric autotransporter adhesins. Adv. Exp. Med. Biol. 715, 143–158. doi: 10.1007/978-94-007-0940-9_9
Masood, T. B., Sandhya, S., Chandra, N., and Natarajan, V. (2015). CHEXVIS: a tool for molecular channel extraction and visualization. BMC Bioinformatics 16:119. doi: 10.1186/s12859-015-0545-9
McGuffin, L. J., Bryson, K., and Jones, D. T. (2000). The PSIPRED protein structure prediction server. Bioinformatics 16, 404–405. doi: 10.1093/bioinformatics/16.4.404
Meng, G., Geme, J. W. S. III, and Waksman, G. (2008). Repetitive architecture of the Haemophilus influenzae Hia trimeric autotransporter. J. Mole. Biol. 384, 824–836. doi: 10.1016/j.jmb.2008.09.085
Meng, G., Surana, N. K., St Geme, J. W., and Waksman, G. (2006). Structure of the outer membrane translocator domain of the Haemophilus influenzae Hia trimeric autotransporter. EMBO J. 25, 2297–2304. doi: 10.1038/sj.emboj.7601132
Montgomerie, S., Cruz, J. A., Shrivastava, S., Arndt, D., Berjanskii, M., and Wishart, D. S. (2008). PROTEUS2: a web server for comprehensive protein structure prediction and structure-based annotation. Nucl. Acids Res. 36, W202–W209.
Mühlenkamp, M., Oberhettinger, P., Leo, J. C., Linke, D., and Schütz, M. S. (2015). Yersinia adhesin A (YadA)–beauty & beast. Int. J. Med. Microbiol. 305, 252–258. doi: 10.1016/j.ijmm.2014.12.008
Natale, P., Brüser, T., and Driessen, A. J. (2008). Sec-and Tat-mediated protein secretion across the bacterial cytoplasmic membrane—distinct translocases and mechanisms. Biochim. Biophys. Acta Biomembr. 1778, 1735–1756. doi: 10.1016/j.bbamem.2007.07.015
Navarro-Garcia, F., Ala’aldeen, D., Desvaux, M., Fernandez, R. C., and Henderson, I. R. (2004). Type V protein secretion pathway: the autotransporter story. Microbiol. Mole. Biol. Rev. 68, 692–744. doi: 10.1128/mmbr.68.4.692-744.2004
Negahdaripour, M., Nezafat, N., and Ghasemi, Y. (2016). A panoramic review and in silico analysis of IL-11 structure and function. Cytokine Growth Factor Rev. 32, 41–61. doi: 10.1016/j.cytogfr.2016.06.002
Negahdaripour, M., Nezafat, N., Hajighahramani, N., Rahmatabadi, S. S., and Ghasemi, Y. (2017). Investigating CRISPR-Cas systems in Clostridium botulinum via bioinformatics tools. Infect. Genet. Evol. 54, 355–373. doi: 10.1016/j.meegid.2017.06.027
Neta, A. V. C., Mol, J. P., Xavier, M. N., Paixão, T. A., Lage, A. P., and Santos, R. L. (2010). Pathogenesis of bovine brucellosis. Vet. J. 184, 146–155. doi: 10.1016/j.tvjl.2009.04.010
Newman, A. M., and Cooper, J. B. (2007). XSTREAM: a practical algorithm for identification and architecture modeling of tandem repeats in protein sequences. BMC Bioinformatics 8:1. doi: 10.1186/1471-2105-8-38
Nummelin, H., Merckel, M. C., Leo, J. C., Lankinen, H., Skurnik, M., and Goldman, A. (2004). The Yersinia adhesin YadA collagen-binding domain structure is a novel left-handed parallel β-roll. EMBO J. 23, 701–711. doi: 10.1038/sj.emboj.7600100
Oberto, J. (2013). SyntTax: a web server linking synteny to prokaryotic taxonomy. BMC Bioinformatics 14:4. doi: 10.1186/1471-2105-14-4
Okonechnikov, K., Golosova, O., Fursov, M., and Team, U. (2012). Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167. doi: 10.1093/bioinformatics/bts091
Pamilo, P., and Bianchi, N. O. (1993). Evolution of the Zfx and Zfy genes: rates and interdependence between the genes. Mole. Biol. Evol. 10, 271–281.
Paulsen, I. T., Seshadri, R., Nelson, K. E., Eisen, J. A., Heidelberg, J. F., Read, T. D., et al. (2002). The Brucella suis genome reveals fundamental similarities between animal and plant pathogens and symbionts. Proc. Natl. Acad. Sci. 99, 13148–13153. doi: 10.1073/pnas.192319099
Petersen, T. N., Brunak, S., Von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Pina, S., Olvera, A., Barceló, A., and Bensaid, A. (2009). Trimeric autotransporters of Haemophilus parasuis: generation of an extensive passenger domain repertoire specific for pathogenic strains. J. Bacteriol. 191, 576–587. doi: 10.1128/jb.00703-08
Posadas, D. M., Ruiz-Ranwez, V., Bonomi, H. R., Martín, F. A., and Zorreguieta, A. (2012). BmaC, a novel autotransporter of Brucella suis, is involved in bacterial adhesion to host cells. Cell. Microbiol. 14, 965–982. doi: 10.1111/j.1462-5822.2012.01771.x
Qin, W., Wang, L., and Lei, L. (2015). New findings on the function and potential applications of the trimeric autotransporter adhesin. Antonie van Leeuwenhoek 108, 1–14. doi: 10.1007/s10482-015-0477-4
Qin, W., Wang, L., Zhai, R., Ma, Q., Liu, J., Bao, C., et al. (2016). Trimeric autotransporter adhesins contribute to Actinobacillus pleuropneumoniae pathogenicity in mice and regulate bacterial gene expression during interactions between bacteria and porcine primary alveolar macrophages. Antonie van Leeuwenhoek 109, 51–70. doi: 10.1007/s10482-015-0609-x
Rahbar, M. R., Zarei, M., Jahangiri, A., Khalili, S., Nezafat, N., Negahdaripour, M., et al. (2019). Trimeric autotransporter adhesins in Acinetobacter baumannii, coincidental evolution at work. Infect. Genet. Evol. 71, 116-127.
Rocha-Gracia, R. D. C., Castañeda-Roldán, E. I., Giono-Cerezo, S., and Girón, J. A. (2002). Brucella sp. bind to sialic acid residues on human and animal red blood cells. FEMS Microbiol. Lett. 213, 219–224. doi: 10.1016/s0378-1097(02)00817-0
Roche, D. B., Do Viet, P., Bakulina, A., Hirsh, L., Tosatto, S. C., and Kajava, A. V. (2018). Classification of β-hairpin repeat proteins. J. Struct. Biol. 201, 130–138. doi: 10.1016/j.jsb.2017.10.001
Romero, C., Gamazo, C., Pardo, M., and Lopez-Goñi, I. (1995). Specific detection of Brucella DNA by PCR. J. Clin. Microbiol. 33, 615–617. doi: 10.1128/jcm.33.3.615-617.1995
Roop, R. M., Gaines, J. M., Anderson, E. S., Caswell, C. C., and Martin, D. W. (2009). Survival of the fittest: how Brucella strains adapt to their intracellular niche in the host. Med. Microbiol. Immun. 198, 221–238. doi: 10.1007/s00430-009-0123-8
Ruiz-Ranwez, V., Posadas, D. M., Estein, S. M., Abdian, P. L., Martin, F. A., and Zorreguieta, A. (2013a). The BtaF trimeric autotransporter of Brucella suis is involved in attachment to various surfaces, resistance to serum and virulence. PloS One 8:e79770. doi: 10.1371/journal.pone.0079770
Ruiz-Ranwez, V., Posadas, D. M., Van Der Henst, C., Estein, S. M., Arocena, G. M., Abdian, P. L., et al. (2013b). BtaE, an adhesin that belongs to the trimeric autotransporter family, is required for full virulence and defines a specific adhesive pole of Brucella suis. Infect. Immun. 81, 996–1007. doi: 10.1128/iai.01241-12
Sankarasubramanian, J., Vishnu, U. S., Dinakaran, V., Sridhar, J., Gunasekaran, P., and Rajendhran, J. (2016a). Computational prediction of secretion systems and secretomes of Brucella: identification of novel type IV effectors and their interaction with the host. Mole. BioSys. 12, 178–190. doi: 10.1039/c5mb00607d
Sankarasubramanian, J., Vishnu, U. S., Khader, L. A., Sridhar, J., Gunasekaran, P., and Rajendhran, J. (2016b). BrucellaBase: genome information resource. Infect., Genet. Evol. 43, 38–42. doi: 10.1016/j.meegid.2016.05.006
Schütz, M., Weiss, E.-M., Schindler, M., Hallström, T., Zipfel, P., Linke, D., et al. (2010). Trimer stability of YadA is critical for virulence of Yersinia enterocolitica. Infect. Immun. 78, 2677–2690. doi: 10.1128/iai.01350-09
Sieira, R., Bialer, M. G., Roset, M. S., Ruiz-Ranwez, V., Langer, T., Arocena, G. M., et al. (2017). Combinatorial control of adhesion of B rucella abortus 2308 to host cells by transcriptional rewiring of the trimeric autotransporter bta E gene. Mole. Microbiol. 103, 553–565. doi: 10.1111/mmi.13576
Simm, D., Hatje, K., and Kollmar, M. (2015). Waggawagga: comparative visualization of coiled-coil predictions and detection of stable single α-helices (SAH domains). Bioinformatics 31, 767–769. doi: 10.1093/bioinformatics/btu700
Singh, A., Gupta, V. K., Kumar, A., Singh, V. K., and Nayakwadi, S. (2013). 16S rRNA and omp31 gene based molecular characterization of field strains of B. melitensis from aborted foetus of goats in India. Sci. World J. 2013:160376
Söding, J., Biegert, A., and Lupas, A. N. (2005). The HHpred interactive server for protein homology detection and structure prediction. Nucl. Acids Res. 33, W244–W248.
Steinegger, M., Meier, M., Mirdita, M., Vöhringer, H., Haunsberger, S. J., and Söding, J. (2019). HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics 20:1–15. doi: 10.1186/s12859-019-3019-7
Suárez-Esquivel, M., Ruiz-Villalobos, N., Castillo-Zeledón, A., Jiménez-Rojas, C., Roop, I., Martin, R., et al. (2016). Brucella abortus strain 2308 Wisconsin genome: importance of the definition of reference strains. Front. Microbiol. 7:1557. doi: 10.3389/fmicb.2016.01557
Sugiura, N. (1978). Further analysts of the data by akaike’s information criterion and the finite corrections: Further analysts of the data by akaike’s. Commun. Statist. Theory Methods 7, 13–26. doi: 10.1080/03610927808827599
Szczesny, P., Linke, D., Ursinus, A., Bär, K., Schwarz, H., Riess, T. M., et al. (2008). Structure of the head of the Bartonella adhesin BadA. PLoS Pathog. 4:e1000119. doi: 10.1371/journal.ppat.1000119
Szczesny, P., and Lupas, A. (2008). Domain annotation of trimeric autotransporter adhesins—daTAA. Bioinformatics 24, 1251–1256. doi: 10.1093/bioinformatics/btn118
Tian, W., Chen, C., Lei, X., Zhao, J., and Liang, J. (2018). CASTp 3.0: computed atlas of surface topography of proteins. Nucl. Acids Res. 46, W363–W367.
Uzureau, S., Godefroid, M., Deschamps, C., Lemaire, J., De Bolle, X., and Letesson, J.-J. (2007). Mutations of the quorum sensing-dependent regulator VjbR lead to drastic surface modifications in Brucella melitensis. J. Bacteriol. 189, 6035–6047. doi: 10.1128/jb.00265-07
Wang, S., Li, W., Liu, S., and Xu, J. (2016). RaptorX-Property: a web server for protein structure property prediction. Nucl. Acids Res. 44, W430–W435.
Wang, S., Peng, J., Ma, J., and Xu, J. (2015). Protein secondary structure prediction using deep convolutional neural fields. Sci. Rep. 6:18962.
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucl. Acids Res. 46, W296–W303.
Wattam, A. R., Davis, J. J., Assaf, R., Boisvert, S., Brettin, T., Bun, C., et al. (2016). Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucl. Acids Res. 45, D535–D542.
Weaver, S., Shank, S. D., Spielman, S. J., Li, M., Muse, S. V., and Kosakovsky Pond, S. L. (2018). Datamonkey 2.0: a modern web application for characterizing selective and other evolutionary processes. Mole. Biol. Evol. 35, 773–777. doi: 10.1093/molbev/msx335
Webb, B., and Sali, A. (2014). Protein structure modeling with MODELLER. Prot. Struct. Predict. 1137, 1–15. doi: 10.1007/978-1-4939-0366-5_1
Whatmore, A. M. (2009). Current understanding of the genetic diversity of Brucella, an expanding genus of zoonotic pathogens. Infect. Genet. Evol. 9, 1168–1184. doi: 10.1016/j.meegid.2009.07.001
Xu, Y., Wang, S., Hu, Q., Gao, S., Ma, X., Zhang, W., et al. (2018). CavityPlus: a web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucl. Acids Res. 46, W374–W379.
Yang, J., Yan, R., Roy, A., Xu, D., Poisson, J., and Zhang, Y. (2015). The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8. doi: 10.1038/nmeth.3213
Yeo, H. J., Cotter, S. E., Laarmann, S., Juehne, T., St Geme, J. W. III, and Waksman, G. (2004). Structural basis for host recognition by the Haemophilus influenzae Hia autotransporter. EMBO J. 23, 1245–1256. doi: 10.1038/sj.emboj.7600142
Zhang, J., Kumar, S., and Nei, M. (1997). Small-sample tests of episodic adaptive evolution: a case study of primate lysozymes. Mole. Biol. Evol. 14, 1335–1338. doi: 10.1093/oxfordjournals.molbev.a025743
Zheng, W., Zhang, C., Wuyun, Q., Pearce, R., Li, Y., and Zhang, Y. (2019). LOMETS2: improved meta-threading server for fold-recognition and structure-based function annotation for distant-homology proteins. Nucl. Acids Res. 47, W429–W436.
Keywords: in silico, bioinformatics, TAA, BatA, Brucellaceae, adhesin
Citation: Rahbar MR, Zarei M, Jahangiri A, Khalili S, Nezafat N, Negahdaripour M, Fattahian Y, Savardashtaki A and Ghasemi Y (2020) Non-adaptive Evolution of Trimeric Autotransporters in Brucellaceae. Front. Microbiol. 11:560667. doi: 10.3389/fmicb.2020.560667
Received: 09 May 2020; Accepted: 05 October 2020;
Published: 12 November 2020.
Edited by:
John R. Battista, Louisiana State University, United StatesReviewed by:
Jack Christopher Leo, Nottingham Trent University, United KingdomCopyright © 2020 Rahbar, Zarei, Jahangiri, Khalili, Nezafat, Negahdaripour, Fattahian, Savardashtaki and Ghasemi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Younes Ghasemi, Z2hhc2VtaXlAc3Vtcy5hYy5pcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.