 Abhishek Srivastava1,2
Abhishek Srivastava1,2 Jayaseelan Murugaiyan3,4
Jayaseelan Murugaiyan3,4 Juan A. L. Garcia2
Juan A. L. Garcia2 Daniele De Corte5Matthias Hoetzinger6
Daniele De Corte5Matthias Hoetzinger6 Murat Eravci7Christoph Weise7Yadhu Kumar8
Murat Eravci7Christoph Weise7Yadhu Kumar8 Uwe Roesler3
Uwe Roesler3 Martin W. Hahn9
Martin W. Hahn9 Hans-Peter Grossart1,10*
Hans-Peter Grossart1,10*- 1Leibniz-Institute of Freshwater Ecology and Inland Fisheries, Stechlin, Germany
- 2Department of Functional and Evolutionary Ecology, University of Vienna, Vienna, Austria
- 3Centre for Infectious Medicine, Institute for Animal Health and Environmental Hygiene, Freie Universität Berlin, Berlin, Germany
- 4Department of Biotechnology, SRM University-AP, Guntur, India
- 5Research and Development Center for Marine Biosciences, Japan Agency for Marine-Earth Science and Technology, Yokosuka, Japan
- 6Department of Biology and Environmental Science, Linnaeus University, Kalmar, Sweden
- 7Institute of Chemistry and Biochemistry, Freie Universität Berlin, Berlin, Germany
- 8Eurofins Genomics Europe Sequencing GmbH, Konstanz, Germany
- 9Research Department for Limnology, University of Innsbruck, Mondsee, Austria
- 10Institute for Biochemistry and Biology, Potsdam University, Potsdam, Germany
Polynucleobacter asymbioticus strain QLW-P1DMWA-1T represents a group of highly successful heterotrophic ultramicrobacteria that is frequently very abundant (up to 70% of total bacterioplankton) in freshwater habitats across all seven continents. This strain was originally isolated from a shallow Alpine pond characterized by rapid changes in water temperature and elevated UV radiation due to its location at an altitude of 1300 m. To elucidate the strain’s adjustment to fluctuating environmental conditions, we recorded changes occurring in its transcriptomic and proteomic profiles under contrasting experimental conditions by simulating thermal conditions in winter and summer as well as high UV irradiation. To analyze the potential connection between gene expression and regulation via methyl group modification of the genome, we also analyzed its methylome. The methylation pattern differed between the three treatments, pointing to its potential role in differential gene expression. An adaptive process due to evolutionary pressure in the genus was deduced by calculating the ratios of non-synonymous to synonymous substitution rates for 20 Polynucleobacter spp. genomes obtained from geographically diverse isolates. The results indicate purifying selection.
Introduction
The model bacterium Polynucleobacter asymbioticus strain QLW-P1DMWA-1T (Meincke et al., 2012) belongs to the class Gammaproteobacteria (Parks et al., 2018). The genus Polynucleobacter mainly represents a group of highly successful heterotrophic planktonic bacteria (Jezberová et al., 2010) inhabiting freshwater ecosystems (lakes, ponds, and streams) across all climatic zones and across all continents (Hahn et al., 2015). The genus also includes some obligate endosymbionts of ciliates, which represent evolutionarily derived stages (Boscaro et al., 2013). The strain investigated here persistently inhabits a small and shallow acidic Alpine pond in Austria (Hahn et al., 2005, 2012, 2016; Hoetzinger et al., 2017) that is covered by ice for 6 months of the year and, during the warmer season, undergoes pronounced diurnal fluctuations in thermal and irradiation conditions (Hahn et al., 2012).
Previous investigations revealed that the global ubiquity of Polynucleobacter in freshwater ecosystems results from ecological diversification (Jezbera et al., 2011; Hahn et al., 2012). Moreover, the small cell size of the ultramicrobacteria related to the genus Polynucleobacter is reportedly an advantage: flagellate predation was weak if not completely absent (Boenigk et al., 2004). Ultramicrobacteria (cell volume < 0.1 μm3) in general are numerically dominant organisms in many freshwater ecosystems (Proctor et al., 2018). They display extreme (e.g., LD12 Alphaproteobacteria and acI Actinobacteria) or moderate genome streamlining (e.g., genus Polynucleobacter) (Hahn et al., 2005; Newton et al., 2007; Salcher et al., 2011; Neuenschwander et al., 2017). Finally, this strain exhibits a non-motile planktonic lifestyle with limited metabolic plasticity: its members lack motility and quorum sensing genes with a very low number of signal transduction-related genes (Hahn et al., 2012). Our expectation therefore was that only minimal metabolic adjustment occurs in this strain. We strove to cover the majority of the molecular cause and effects in this strain’s adjustment to varying stress scenarios. A standard transcriptome and proteome study was designed to determine the underlying regulatory mechanisms and metabolic changes. At the same time, the presence of several methyltransferases in the genome1 prompted us to test whether any one of them could enable DNA modification that would possibly be involved in gene expression followed by metabolic adjustment.
DNA methylation is the most common epigenetic modification in nearly all life-forms, whereby the information is passed on from the parent cells to their daughter cells in addition to the genetic makeup (Jeltsch, 2002). Such modifications help bacteria defend their genome against viral infection using restriction-modification (R-M) systems, control DNA damage by mismatch-repair, coordinate gene regulation, control transposition events, and mediate host-pathogen associations (Wion and Casadesus, 2006; Sánchez-Romero et al., 2015). Many bacterial methylomes have been resolved at a single base resolution, but the efforts have largely been directed toward discovering methylated motifs and describing restriction-modification systems (Fang et al., 2012; Murray et al., 2012; Krebes et al., 2013; Lluch-Senar et al., 2013; Lee et al., 2015; Blow et al., 2016; Hiraoka et al., 2019). We, however, focus on testing the co-occurrence of methylation on the bacterial genome and gene expression. This approach enables us to assess whether the DNA methylation patterns play a role for the cell’s flexibility in manifesting important traits under different environmental circumstances.
Our study therefore included pinpointing molecular candidates responsible for the strain’s acclimatization in its natural habitat. We chose distinct experimental parameters for this strain, i.e., incubation scenarios at 4°C, at 26°C, and at 26°C with UV irradiation (hereafter denoted as 26°C∗). These experimental conditions reflect the abiotic factors common in the strain’s Alpine habitat (Hahn et al., 2012).
Materials and Methods
Investigated Strains and Genomes
The strain QLW-P1DMWA-1T was used for stressor-related experiments as detailed below. Twenty other genomes of freshwater strains belonging to Polynucleobacter spp. were also used for this study. See Supplementary Table S1 for more details. Their genomes were used to assess the natural selection process operating in all these strains.
Background Information on the Criteria for Selecting the Experimental Conditions and Details of the Bacterial Growth Conditions
The species Polynucleobacter asymbioticus typically inhabits shallow bog ponds (Hahn et al., 2012), which are characterized by rather unstable environmental conditions (Figures 1A,B), with strong diurnal changes in the upper water layers (<50 cm) and more stable conditions in deeper layers (>50 cm). This habitat type can be characterized as polymictic, slightly acidic (typically pH 5–6), shallow (max. depths usually 1–1.5 m) freshwater systems. We compared the temperature parameters of a high altitude (1300 m, Pond-1) and a low altitude (450 m, Schönramer Moor) Alpine habitat of Polynucleobacter bacteria that are 35 km apart. During calm and sunny days, the upper water layers quickly warm up (frequently by up to 5–8°C) and thermally stratify. Although, upper layers of Pond-1 (strain’s home habitat) can reach up to 26°C, the temperature is frequently higher in the lower altitude ponds during the summer where this species is also found (Figures 1A,B). Nocturnal cooling causes mixing of the upper water layers, but such mixing usually does not extend down to the lower layers. Thunderstorms with strong winds or cool weather periods cause complete mixing of these water bodies. Such complete mixing events take place several times during spring to late fall. The unstable environmental conditions result in sudden changes in growth conditions for the bacteria in the water columns of such ponds. Beyond the changes in water temperature, sudden strong changes in UV intensities also occur during the diurnal mixing cycles (Hahn et al., 2012).
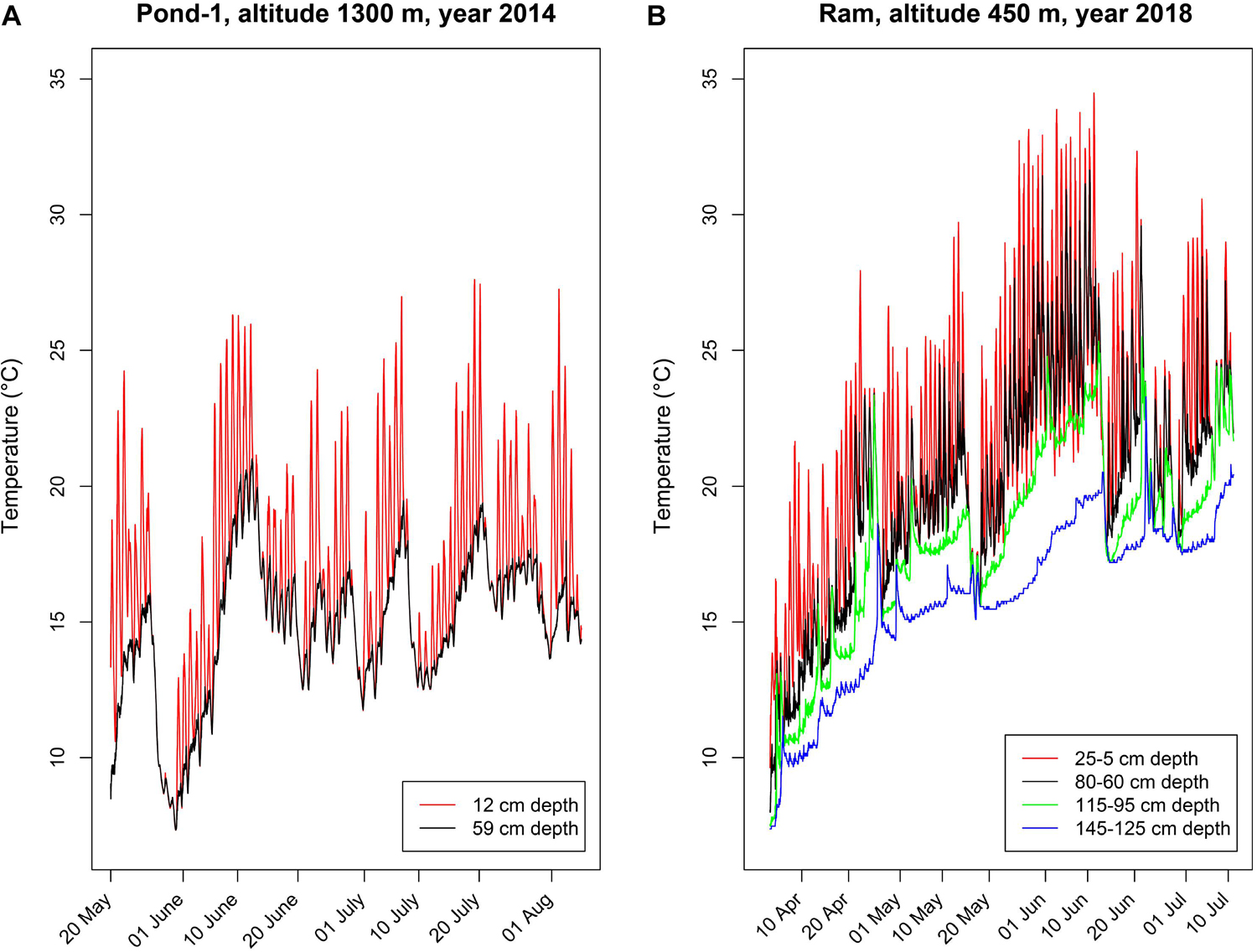
Figure 1. Depth-specific time series of water temperatures in two shallow freshwater systems inhabited by Polynucleobacter asymbioticus. Loggers deployed in different water depths measured temperatures in 30-min intervals. (A) Water temperatures measured in Pond-1, the home habitat of strain QLW-P1DMWA-1T. This shallow pond (maximum depth ∼ 1.2 m) is located at an altitude of 1300 m. (B) Water temperatures measured in four different water depths in a shallow (maximum water depth 1.5 m) bog pond located in Schönramer Moor. Designated here as Ram, this pond is located at an altitude of 450 m and 35 km away from Pond-1. The water level dropped by 20 cm during the investigation period in summer, which changed the water depths in which the loggers were moored. Note the diurnal changes in water temperature in the upper water layers, which frequently show amplitudes of more than 5°C of warming and nocturnal cooling with mixing.
The bacterial samples were cultured in 500 ml R2A liquid media in sterile UV-permeable polyethylene bags (∼35% permeability; Whirl-Pak®, Nasco). Experiments were performed in triplicates. Initial bacterial inoculum was transferred from pre-culture to the 500 ml main culture in the nine bags, which were kept on constant rotation of 200 rpm at 26°C. All bacterial samples were initially allowed to grow in an incubator maintained at 26°C. During this time the optical density (OD) at a wavelength of 575 nm approached 0.10. Afterward, the bags were distributed according to their planned treatment, i.e., (a) 26°C incubation continued for three bags, (b) three other bags were transferred to the 4°C incubator at 200 rpm, (c) radiation (UV) experiment was conducted with the remaining three bags, which were also maintained at 26°C (26°C∗). Humic substances stain the water of the home habitat Pond-1, thus the UV intensity is already reduced in deeper water layers (probably at 10–50 cm). Due to wind-driven mixing, the cells may cycle through different water depths with different UV intensities. Accordingly, only during a calm day with strong thermal stratification of the water column would cells in the top centimeters (i.e., a small fraction of the population) experience the natural UV intensity over a period of a whole day. We therefore opted for a shorter UV exposure duration. For the UV irradiation experiment, all three bags containing bacterial samples were transferred from the 26°C incubator to a position that was 40 cm from five parallel 100W ARIMED® B UV lamps (Cosmedico®, Germany) for 30 min of exposure two times a day. At this distance, the spectral irradiance readings of UV lamps by the spectro-radiometer were 3 and 97 Wm–2 for the UVB and UVA range, respectively. Samples were harvested when the optical density (OD) values of the media (at wavelength 575 nm) approached 0.2 (late logarithmic/early stationary phase). Harvesting at this OD best resembles the state in which they are found in their natural setting, where they live a non-motile planktonic lifestyle (Hahn et al., 2012). 500 μl of UV-treated bacterial sample was spread on an R2A agar-media-containing plate. This produced viable bacterial colonies in 5 days at room temperature, confirming that radiation was not provided at lethal dose. Every experimental bag yielded 3 × 150 ml of bacterial samples that were harvested for the methylome, transcriptome and proteome studies.
Sequencing and Methylome Analysis
Sample preparation and sequencing were performed at GATC Biotech (Constance, Germany) using the PacBio RSII system (Pacific Biosciences, San Diego). PacBio libraries with 8–12 kb insert size were prepared and sequenced on 1–2 SMRT (Single Molecule, Real-Time) cells with 120-min movie length and MagBead loading. For each sample, about 100x average coverage per genome was achieved (minimum >25× per strand). Depth coverage varied (∼54–∼70) in the three samples (Supplementary Figure S1). The base modification was analyzed with SMRT Portal 2.1 (Pacific Biosciences, San Diego).
SMRT sequencing reads from 4°C, 26°C, and 26°C plus UV-treated samples (26°C∗) were mapped against the Polynucleobacter asymbioticus reference genome down-loaded from NCBI (Accession Nr. NC_009379) using the BLASR mapper2 and the Pacific Biosciences SMRTAnalysis pipeline3 using the standard mapping protocol. Interpulse durations (IPDs) were measured as described by Flusberg et al. (2010) for all pulses aligned to each position in the reference sequence. IPD is used to determine if a DNA nucleobase is modified. This is achieved by monitoring a real time progression of DNA polymerase while it incorporates fluorescently labeled nucleotides. This is followed by detecting a fluorescence pulse and obtaining the kinetics of polymerase translocating toward the next base on a DNA template. The pulse extinguishes when the fluorophore attached to the terminal phosphate of a nucleotide is clipped away by the progressing polymerase. Modified bases affect the kinetics of DNA polymerase, and IPD increases when the base is methylated because the time needed for next base incorporation into the active site of the enzyme differs from the unmethylated site (Flusberg et al., 2010). Delayed incorporation of nucleotide when a methylated base is encountered eventually helps calculate the IPD ratio. IPDs were normalized by calculating the ratio of the IPD in the 4°C, 26°C, and 26°C∗ samples to the IPD of a control (the ‘IPD ratio’).
Transcriptomic Study
An Agilent 60-mer microarray containing the P. asymbioticus str. QLW-P1DMWA-1T genome was designed by the company Source BioScience GmbH. The 8 × 15K microarrays have 2123 genes of P. asymbioticus str. QLW-P1DMWA-1T represented by seven 60-mer oligonucleotide probes on average. Additionally, 536 Agilent control probes were spotted onto the array for intra-array reproducibility measurements and against 10 different mRNA spike-in control transcripts. For the design, the Agilent web-based design-tool e-array was used, and the arrays were printed with Agilent SurePrint® technology. The RNA was extracted using the NucleoSpin® RNA kit (Macherey-Nagel GmbH & Co. KG; Düren, Germany) according to the manufacturer’s recommendations. The extracted RNA was quantified using a Nanodrop-ND-1000 (PEQLAB Biotechnologie GmbH, Erlangen, Germany), and RNA integrity was evaluated with the Agilent 2100 Bioanalyzer and RNA 6000 Nano Kit (Agilent Technologies, Böblingen, Germany). Cyanine-3 (Cy3-) labeled cRNA was prepared from 0.2 μg total RNA using the Quick Amp Labeling Kit, One-Color (Agilent Technologies, Böblingen, Germany) according to the manufacturer’s instructions. The full spectrum Multistart Primer (BioCat GmbH, Heidelberg, Germany) was used for the RNA isolated from the Gram-negative bacteria instead of the oligo-dT primer. Samples were purified with the RNeasy Mini Kit (QIAGEN, Hilden, Germany), and the cRNA yield and the Cy3 incorporation were determined with the Nanodrop-ND-1000 (PEQLAB Biotechnologie GmbH, Erlangen, Germany). 625 ng of cy-labeled cRNA was fragmented at 60°C for exactly 30 min in a volume of 25 μl containing 1 × Agilent fragmentation buffer and 2 × Agilent blocking agent. After incubation, 25 μl 2 × Agilent hybridization buffer was added to the sample and 40 μl were hybridized on the Agilent P. asymbioticus 8 × 15K microarray for 17 h at 65°C and at 10 rpm in an Agilent hybridization oven. After hybridization the microarray was dissembled at room temperature in Gene Expression Wash Buffer 1 (Agilent Technologies, Böblingen, Germany) and then washed for 1 min at room temperature with Gene Expression Wash Buffer 1, and for 1 min at 37°C with Gene Expression Wash Buffer 2. Acetonitrile was used to dry the array. The microarray was scanned with the Agilent DNA Microarray scanner (G2565CA) using the following settings: scan area 61 × 21.6 mm, scan resolution 3 μm, Dye Channel Green, and PMT 100%. The scanned array was extracted with the Feature Extraction software (version 10.5.1.1), and group comparisons were performed with the analysis pipeline of the company Source Bioscience GmbH to detect those genes that are significantly differentially expressed. Raw data were analyzed, normalized, and finally evaluated with t-tests (unequal variance), principal component analysis (PCA), and hierarchical clustering. The P-value cut-off was set to 0.05, and values less than that were considered significant in our study.
Proteomic Study
We used an Orbitrap mass analyzer to identify the proteins present in our three experimental samples, i.e., 26°C, 26°C∗, and 4°C-incubated bacterial culture. The cells were harvested and the whole cell proteins were extracted as described elsewhere (Murugaiyan et al., 2016). The cell pellet was reconstituted with 500 μl of 20 mM HEPES (pH 7.4) and subjected to sonication on ice for 1 min (cycle, 1.0; amplitude, 100%; UP100H; Hielscher Ultrasound Technology, Teltow, Germany). The suspension was centrifuged at 11,290 × g for 5 min at 4°C and the clear supernatant was collected. The protein content was estimated using a modified Bradford’s method (Bio-Rad, Munich, Germany). The protein estimation consistency was verified using a volume of protein extraction containing 5 μg of protein mixing with 10 μL sample loading buffer, heated for 5 min at 60°C and subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (stacking gel 4%, and separating gel 12%). The protein bands were visualized using Coomassie Brilliant Blue staining (Candiano et al., 2004).
In-Solution Trypsin Digestion and Mass Spectrometry (MS) Analysis
In-solution trypsin digestion was carried out with incubation steps at room temperature under gentle shaking. 10 μg of bacterial proteins were precipitated with acetone and reconstituted in 20 μl of denaturation buffer (6 M urea/2 M thiourea in 10 mM HEPES, pH 8.0). 0.2 μl of 10 mM dithiothreitol were added in 50 mM of ammonium bicarbonate (NH4HCO3) and incubated for 30 min. Thereafter, 0.4 μl of 55 mM iodoacetamide were added in NH4HCO3 and then incubated for 20 min. 0.4 μl of Lys-C protease (Sigma-Aldrich, Germany) solution (0.5 μg/μl in NH4HCO3) were added and incubated overnight. The urea concentration was diluted by adding 75 μl of NH4HCO3, whereby 0.4 μl of 0.5 μg/μL trypsin protease were added in 50 mM NH4HCO3. Following overnight incubation, trypsin digestion was stopped by adding 100 μl of 5% acetonitrile in 3% trifluroacetic acid. Overnight trypsin treatment was performed to obtain a better sequence coverage of all digested proteins.
After digestion, peptide samples were desalted by solid phase extraction (SPE) using C18 EmporeTM disks Stagetips (Supelco, Germany). Desalted peptide mixtures were separated by reversed phase chromatography using the Dionex UltiMate 3000 Nano LC on in-house manufactured 25 cm fritless silica microcolumns with an inner diameter of 100 μm. Columns were packed with ReproSil-Pur C18-AQ 3 μm resin. Peptides were separated on a 5–60% acetonitrile gradient (90 min) with 0.1% formic acid at a flow rate of 350 nl/min. Eluting peptides were ionized online by electrospray ionization and transferred into a Thermo ScientificTM LTQ-Orbitrap VelosTM mass spectrometer (Thermo Fisher Scientific, Germany).
The LTQ-Orbitrap was operated in the positive mode to record full scan MS spectra (from m/z 300–1700) in the Orbitrap mass analyzer at a resolution of R = 60,000. This was followed by isolation and fragmentation of the 20 most intense ions in the LTQ part by collision-induced dissociation. The MaxQuant (version 1.3.0.5) software suit was used to process the raw MS files. The search engine ANDROMEDA (Cox et al., 2011) was utilized to search the peak list files against forward and backward protein sequences of P. asymbioticus down-loaded from the Uniprot database and 248 frequently observed laboratory contaminants. Initial maximum precursor and fragment mass deviations were set to 7 ppm and 0.5 Da, respectively. Methionine oxidation/acetylation of peptide N-termini and cysteine carbamidomethylation were set as variable and fixed modification, respectively, for the search. Furthermore, enzyme specificity was set to trypsin and a maximum of two missed cleavages was allowed for searching. The target-decoy-based false discovery rate (FDR) for peptide and protein identification was set to 1% for peptides and proteins, and the minimum peptide length was set to six amino acids. Precursor mass tolerance was set to 20 ppm. The mass tolerance for fragment ions was set to 0.5 Da. Protein identification was based on the detection of more than one unique peptide specific for a protein. MS-Quantification of proteins was performed using the label-free quantification algorithm of the MaxQuant software package. The freely available software Perseus (version 1.4.1.3, Max-Planck-Institute of Biochemistry, Martinsried, Germany) was used to compare the peak intensity across the whole set of measurements to obtain the quantitative measurements for all of the peptides in the sample. The normalized protein intensities from the MaxQuant analysis were imported and transformed into logarithmic scale with base two. The missing values were replaced with the value of the lowest intensity. The proteins were quantified and statistical significance determined using two-sample Student t-test and FDR using the method of Benjamini–Hochberg (Benjamini and Hochberg, 1995). For further visualization, heat-map and principal component analysis (PCA) were computed.
Melting Temperature Calculation
The melting temperature (Tm) calculation was adopted from SantaLucia’s study on unified nearest-neighbor thermodynamic stability parameters (ΔG°37 or Gibb’s free energy at 37°C) of Watson–Crick base pairs in 1 M sodium chloride solution (SantaLucia, 1998) with 100-kb window size. Data were plotted to prepare circular maps using Circos software package v0.694.
Non-synonymous Substitution Rate (Ka) and Synonymous Substitution Rate (Ks) Calculation
Ka/Ks values were calculated with the KaKs calculator 2.0 (Wang et al., 2010) using both an approximate method (γ-MYN) and a maximum-likelihood method based on a substitution model selected from a set of candidate models (MS). Gene pairs with less than three substitutions and KaKs values with p-values higher than 0.05 were removed from our study.
The GEO accession number corresponding to microarray data is GSE98129.
The mass spectrometry proteomics data were deposited at the ProteomeXchange Consortium (Perez-Riverol et al., 2019) via the PRIDE partner repository with the dataset identifier PXD020103.
Results and Discussion
Transcriptome and Proteome Analyses
4°C-Incubated Sample
When incubated at 4°C (winter conditions), only 10% of the total 2088 protein-coding genes were up-regulated (Figure 2A, Supplementary Figure S2, and Supplementary Tables S2a,b). Bacterial responses to cold temperature have been investigated and summarized in detail elsewhere (Barria et al., 2013), and our findings agree with earlier reports on maintaining membrane fluidity, efficient transcription or translation, enriched chaperones, oxidative stress compensation, and cell shape maintenance. Several key features are summarized below that might be helpful for strain QLW-P1DMWA-1T to survive in alpine winter.
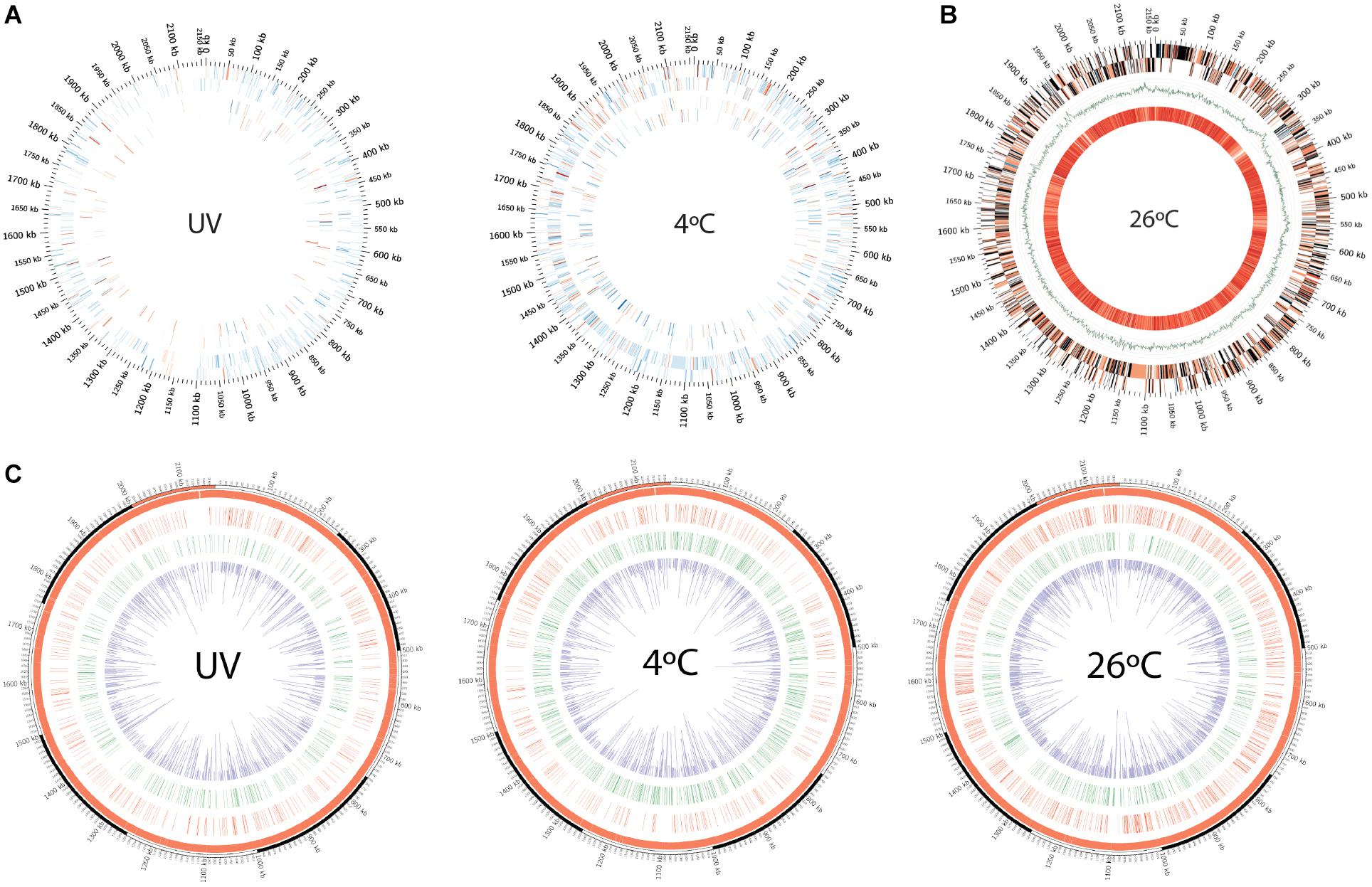
Figure 2. (A) Circos plot depiction of differentially expressed genes and proteins in cold treatment and UV-irradiated bacterial samples compared against 26°C treatment, as revealed by microarray and LC-MS. Outermost tick marks: bacterial genome of 2.15-Mb. Concentric tracks from outer to inner: sense and antisense strand, respectively. (B) Circos plot representation of differentially expressed genes in bacteria incubated at 26°C (in relative comparison to 4°C-incubated cells) and averaged melting energy distribution in bacterial genome. The outer two concentric tracks represent sense and antisense strands. Color-coding: red (up-regulated genes) with transient color (expressed transcript gradients) toward blue (down-regulated genes), black genes are undetected in microarray analysis. Green GC skew curve is then followed with the innermost track, representing average negative DNA melting energy with 100-kb scanning window size (bright orange to pale yellow represent high to low values). (C) Circos plots showing the kinetic variation measured as IPD ratio recorded across the Polynucleobacter asymbioticus strain QLW-P1DMWA-1T genome. Red and green tracks represent the genome locations where 4mC and 6mA modification motifs were recorded, respectively. Inner (purple) track: IPD ratios of the corresponding modifications recorded across the genome.
General Low Temperature Anti-stress Proteins
Cold treatment induced a nine-fold higher expression of cold-shock protein (CSP) (p = 9.4 × 10–6) (Table 1 and Supplementary Table S3). Low temperatures can impose secondary folding of the mRNA, and CSP can therefore efficiently counteract such folding, which impedes ribosomal access to the mRNA (Giuliodori et al., 2004). Moreover, we observed that the fatty acid desaturase-encoding gene was moderately over expressed (+1.0 fold; p = 0.001) in the cold-treated samples. Such a feature may help bacteria to acclimatize in near-freezing lakes during the winter season.

Table 1. Quantitative proteome analysis. (A) Tabulation of 50 strongest up-regulated proteins obtained from 4°C-incubated cells. (B) Tabulation of 50 strongest down-regulated proteins obtained from 4°C-incubated cells. (C) Tabulation of 50 strongest up-regulated proteins obtained from UV irradiated (26°C-incubated) cells (26°C*). (D) Tabulation of 50 strongest down-regulated proteins obtained from UV irradiated (26°C-incubated) cells (26°C*).
The upregulation of several chaperones, co-chaperones, and anti-oxidative stress factors indicates the necessity of keeping the cell and its molecular machineries operational at stressful low temperature. For example, chaperone HscA and co-chaperone HscB, which participate in iron–sulfur-cluster biosynthesis, were enriched (+0.93; p = 0.006 and +2.19; p = 0.001, respectively) (Vickery and Cupp-Vickery, 2007). We also observed that antioxidant-related genes are also up-regulated, e.g., superoxide dismutase Pnuc_1626 (+3.9 fold; p = 0.04), catalase Pnuc_2053 (+0.7 fold; p = 0.03), and alkyl hydroperoxide reductase Pnuc_1534 (+0.7 fold; p = 0.002). These antioxidant genes, amongst many located in this strain’s genome, might be cold induced (Smirnova et al., 2001). Similarly, lipoic acid is a cofactor for enzymes that are multicomponent dehydrogenases. Due to its solubility in lipid- or aqueous solutions, it functions as a wide-range antioxidant that can quench free radicals (Spalding and Prigge, 2010). Cold treatments led to a +2.2 fold (p = 0.001) higher detection of lipoyl synthase compared to warmer incubation of bacterial cells. The antioxidant peroxiredoxin (Pnuc_0173) was also enriched (+1.87 fold; p = 7 × 10–4) in cold-treated samples.
Another class of protein folding chaperones consists of peptidyl-prolyl isomerase (PPIase), which catalyses the isomerization reaction of the cis–trans configuration in peptide bonds (Shaw, 2002). Proteins generally contain trans configuration peptide linkages except proline amino acid, which exists in a cis configuration (Joseph et al., 2012). Therefore, prolyl isomerization is a rate-limiting step during protein folding, which is additionally affected by extreme temperatures leading to potential impaired folding during cold (Feller, 2018). Such mis-folding is avoided by PPIases, which help microbes to adapt to low temperature (Budiman et al., 2011). Our dataset clearly indicates that cold treatment induced PPIases in the cell: Pnuc_0379 - PpiC-type (+7.74 fold; p = 1 × 10–8), Pnuc_1732 - FKBP-type (+2.53 fold; p = 5.4 × 10–5), Pnuc_1869 - PpiC-type SurA (+1.98 fold; p = 3.3 × 10–5). Note, however, that a consecutive run of proline residues in an elongating polypeptide may cause increased stalling of ribosomes during the translation process. Such stalling is prevented by the elongation factor P (EF-P), which facilitates peptide-bond formation (Doerfel et al., 2013). Interestingly, we detected an elevated EF-P signal (+1.43 fold; p = 4 × 10–4) in our proteomic analysis, suggesting its important role in rapid protein biosynthesis.
Cell Shape Maintenance
Bacterial cell wall integrity is crucial in cell shape maintenance and viability (Dörr et al., 2019). Importantly, an additional role of the cell wall in psychrophilic bacteria is protection against cold, as revealed by transcriptomic analyses in Exiguobacterium sibiricum (Rodrigues et al., 2008). Increased expression of peptidoglycan biosynthetic transcripts during cold treatment was explained by cell wall thickening, which possibly contributes to cell protection in the event of ice formation and related cell disruption (Rodrigues et al., 2008). We recorded moderately increased protein expression related to the cell wall biosynthetic pathway, which supports the cell protection notion [UDP-MurNAc-tripeptide synthetase (+1.79 fold; p = 2.3 × 10–4), UDP-N-acetylmuramoyl-tripeptide–D-alanyl-D-alanine ligase (+1.04 fold; p = 0.02), UDP-N-acetylmuramoyl-L-alanyl-D-glutamate synthetase (+0.56 fold; p = 0.01), D-alanylalanine synthetase (+1.59 fold; p = 0.01)]. Similarly, the structural protein MreB was enriched, which would help maintain bacterial cell shape (+1.3 fold; p = 1.6 × 10–5) at 4°C. This was also reported for cold-shock treated Vibrio parahaemolyticus (Chiu et al., 2008).
GTP Homeostasis-Related Cellular Processes
The GTPase family proteins act as a sensor for the cellular GDP/GTP pools by cycling between the inactive GDP- and active GTP-bound state (Verstraeten et al., 2011). GTPase proteins also participate in diverse cellular processes that affect cell physiology (Verstraeten et al., 2011). For example, the GTPase Obg in Escherichia coli is involved in chromosomal partitioning (Kobayashi et al., 2001). Similarly, the GTPase Era is required in septum initiation and acts as a critical checkpoint regulator in the E. coli cell cycle (Britton et al., 1998). The GTPase FtsZ multimerization at the future cell division site forms a ring where other cytokinesis molecular machinery also assembles (Arjes et al., 2015). The GTPase TypA (tyrosine phosphorylation protein A) acts as a global regulator in bacteria and has been shown to be important in the survival and growth of Sinorhizobium meliloti and E. coli at low temperatures (Pfennig and Flower, 2001; Kiss et al., 2004). Interestingly, we observed increased protein signals of the above-mentioned GTPases in the cold-treated bacterial samples: Obg (+1.87 fold; p = 3 × 10–4), Era (+1.23 fold; p = 2.9 × 10–3), FtsZ (+1.04 fold; p = 3.2 × 10–3), TypA (+2.1 fold; p = 1.7 × 10–5).
In contrast, one GTPase, namely LepA (or, elongation factor 4) transcripts, were under-expressed (Pnuc_0404 fold; −1.3 fold, p = 0.02). The protein signal of LepA was also under-detected in the cold-treated sample, although non-significantly (−0.53 fold; p = 0.2). Interesting in this respect is that LepA translocates the ribosome complex toward one codon backward and helps to increase translation fidelity at low concentration only (≤0.3 LepA molecules per 70S ribosome subunits). Higher cellular concentrations of LepA, however, cause a non-productive translation process (Qin et al., 2006). Collectively, GTPases may act as a molecular switch that monitors intracellular GDP/GTP, and our study highlights their potential roles during bacterial cold stress.
It is also interesting to note that the relA (Pnuc_0828) transcript is down-regulated (−0.65 fold; p = 0.002) in cold treatment but remains nominally enriched at the protein level (+0.53 fold; p = 0.01). Such an expression mismatch might be due to post-transcriptional regulation of relA. Potential regulation of relA under-expression is discussed in the section “Methylation Features.” relA encodes for the stress responsive alarmone guanosine pentaphosphate [(p)ppGpp]. This is a nucleotide second messenger and its accumulation is associated with stress survival. Such a signaling molecule can bind directly to RNA polymerase and influence numerous metabolic reactions, thereby possibly helping bacteria to survive cold periods. Regulation of the GTP pool in the cell is one outcome of the (p)ppGpp availability (Gaca et al., 2015).
Translational Machinery Assembly and Functioning at Low Temperature
Low temperature causes instability in bacterial 70S ribosomal subunits (Bayles et al., 2000). The 70S ribosome is composed of 30S and 50S subunits. Our expression data show that assemblies of ribosomal subunits are prioritized in the cold-treated samples. The GTPase Era (+1.23 fold; p = 2.9 × 10–3), the maturation factor RimM (+1.33 fold; p = 0.01), and the maturation factor RimP (+5.5 fold; p = 0.003) involved in 30S subunit assembly were up-regulated. Similarly, the GTPase Der is a 50S ribosomal subunit-associated factor that helps in maturing and stabilizing the large subunit (Hwang and Inouye, 2006). Cold-induced samples contained a moderately over-represented Der protein signal (+0.9 fold; p = 8.4 × 10–5). The ATP-dependent RNA helicase RhlE was also enriched (+3.5 fold; p = 1.3 × 10–5) and may contribute toward 50S subunit assembly by catalyzing unwinding of RNA under cold induction (Owttrim, 2013). Thus, RhlE overexpression indicates a promotion of the cold-specific ribosomal assembly pathway (Owttrim, 2013). Higher expression of the sua5 gene (+1.3 fold; p = 0.03) at cold temperature indicates threonylcarbamoyl group modification on adenine of tRNA to recognize ANN codons. Such modification helps accurate translation by stabilizing the tRNA codon interaction with the ribosome (El Yacoubi et al., 2009).
Storage Reserves and Metabolic Adaptation
The lower detection of transcript and proteome signals of the respiratory chain and Krebs cycle process at 4°C indicates a slowing down of key metabolic processes (Supplementary Table S4). This is apparently crucial for cost-effectiveness of the operating cellular machinery during cold periods. Upregulation of the amino acid ABC transporter subunit gene (+3.69 fold; p = 0.03) underlines the importance of amino acid utilization for protein biosynthesis or in replenishing TCA cycle intermediates. Moreover, moderate upregulation of cyanophycin synthetase (+1.30 fold; p = 0.04) also suggests that storage of polypeptides in the form of cyanophycin is an important step to ensure sufficient nutrients for the winter period. Similarly, carbon and energy storage biopolymers such as polyhydroxyalkanoate (PHA) and the associated stabilizer phasin protein are moderately stimulated at colder temperature, i.e., poly(R)-hydroxyalkanoic acid synthase (+1.34 fold; p = 0.00002), phasin (+2.64 fold; p = 0.01). PHA biosynthesis is known to increase fitness in the Antarctic bacterium Pseudomonas sp. 14-3 during cold shock (Ayub et al., 2009). Polymeric PHA is actually an esterified (hydroxy-) fatty acid that represents a lipophilic carbon storage compound. Accumulating PHA might be advantageous during nutrient-scarce long winter scenarios until sufficient nutrients again become available. Interestingly, we detected several lipases and esterases in the cold-treated bacterial samples. The signal peptide-bearing lipase (Pnuc_1435) and carboxylesterase (Pnuc_0443) were overexpressed: +6.0 fold; p = 7.6 × 10–5 and +7.56 fold; p = 8 × 10–6, respectively. Intracellular predicted esterases (Pnuc_0531, Pnuc_0959) were also overexpressed: +1.21 fold; p = 0.002 and +0.78 fold; p = 0.04, respectively. Lipases and esterases can directly modulate the turnover of lipids and long/short chain triacylglycerols (Röttiga and Steinbüchel, 2013) and possibly participate in membrane recycling.
UV-Irradiated and 26°C-Incubated Sample
Stress-inducing ultraviolet doses prompt bacterial cells to assume a defensive stance, and survival becomes the priority. Cells harvested from irradiated bacteria grown at 26°C∗ showed an upregulation of only 4% of the total genes (Figure 2A, Supplementary Figure S2, and Supplementary Tables S2c,d). UV-irradiated bacterial cells displayed over-expression of repair- and defense-related genes, a feature often reported since the 1950s (Beukers et al., 2008). We therefore outlined important features and expression profiles that may help strain QLW-P1DMWA-1T cope with UV radiation.
Effect of Radiation on Nucleotides and Transcription
UV radiation causes pyrimidine dimer photoproducts on the DNA strand that could prematurely terminate transcription (Michalke and Bremer, 1969). In this treatment, the xthA gene encoding for exodeoxyribonuclease III for base excision repair was up-regulated +1.95 fold (p = 0.0008). The upregulation of the pyrH gene (+2.55 fold; p = 0.04) encoding for uridylate kinase points to an enhanced supply of pyrimidine bases (thymine and cytosine) because this gene participates in de novo pyrimidine biosynthesis (Bucurenci et al., 1998).
Beyond de novo nucleotide biosynthesis, RNAse G overexpression (+1.58 fold, p = 0.001) suggests RNA processing and degradation of transcripts due to its endoribonuclease activity toward the 5′ end of rRNA and mRNA, respectively (Aït-Bara and Carpousis, 2015). Similarly, RhlE RNA helicase has been reported to facilitate RNA degradation by PNPase, i.e., polynucleotide phosphorylase (Khemici et al., 2004). Both genes (Pnuc_1342 and Pnuc_1222) were up-regulated (+2.19 fold; p = 0.0003, +3.84 fold; p = 1.78934 × 10–5, respectively), suggesting nucleotide recycling by complementing RNAse G function. Furthermore, HrpA RNA helicase was down-regulated (−4.99 fold; p = 4.45 × 10–6), which could otherwise enhance transcript stability (Salman-Dilgimen et al., 2013). If the RNA polymerase is blocked during the transcription process, it may be rescued by UvrD helicase (+1.29 fold; p = 0.001), which can backtrack the polymerase enzyme (Epshtein, 2015).
Defense Against Oxidative Stress
Cell protection is apparently a high priority for UV-treated cells because periplasmic catalase and cytosolic rubrerythrin were significantly enriched (+5.25 fold; p = 1.7 × 10–6 and +6.52 fold; p = 0.008, respectively) (Table 1 and Supplementary Table S3). The upregulation of sod, encoding for the antioxidant superoxide dismutase, also signifies the superoxide-combating capability of a cell; such superoxide compounds could otherwise damage DNA bases and iron–sulfur clusters in proteins (Keyer and Imlay, 1996). The engagement of cells in guarding the iron–sulfur cluster under radiation has been described for Deinococcus radiodurans (Slade and Radman, 2011). Additionally, three PFAM predicted Ahp (alkyl hydroperoxide reductase) enzymes were detected that were mildly up-regulated, i.e., Pnuc_0173, Pnuc_0429, and Pnuc_1534 (+1.34; p = 0.003, +0.65; p = 0.019, +0.77; p = 0.0002, respectively). Multiple hydrogen peroxide- (H2O2) scavenging enzymes are not uncommon in bacteria (Mishra and Imlay, 2012). Based on that review, we found eleven H2O2-scavenging enzyme-encoding genes in the genome of the present bacterium (Pnuc_0173, Pnuc_0212, Pnuc_0429, Pnuc_1344, Pnuc_1376, Pnuc_1512, Pnuc_1534, Pnuc_1930, Pnuc_1970, Pnuc_2044, Pnuc_2053). Interestingly, the proteins expressed by six of these genes contain predicted signal peptide sequences, suggesting their active role in periplasmic space. Hydrogen peroxide can originate intracellularly or exogenously (Mishra and Imlay, 2012). Moreover, humic matter and chromophoric dissolved organic substances are important sources of H2O2, which is produced photochemically (Glaeser et al., 2010; Zhang et al., 2012). This makes it very relevant for this strain to maintain several anti-oxidative enzymes because the natural, humic-matter-rich habitat is shallow and strongly exposed to sunlight in summer (Hahn et al., 2012). Other than catalase (+5.25 fold; p = 1.76 × 10–6), we also detected glutathione-S-transferase (+3.40 fold; p = 0.005), which could protect the cell under a UV-induced oxidative damage scenario (Rastogi et al., 2014).
Oxidative stress can seriously impair bacterial cells at the macromolecular level, e.g., oxidative species can directly target the bacterial translation machinery (Katz and Orellana, 2012). Another feature we observed was the reassessment of correct protein synthesis at the level of translation (Supplementary Table S3). Slight enrichment of tmRNA (+0.6 fold; p = 0.03) indicates the importance of recycling stalled ribosomes that are stuck during incomplete protein biosynthesis and that have attached a tag on incomplete nascent polypeptides for further proteolysis (Janssen and Hayes, 2012).
We recorded another up-regulated candidate (Pnuc_0887, tRNA 5-hydroxyuridine modification protein YegQ, +1.1 fold; p = 0.003) for efficient translation in the UV-treated sample. YegQ is a peptidase U32 family protein involved in modification (hydroxylation) of uridine nucleoside in tRNA (Lauhon, 2019). Base pairing between the mRNA codon and tRNA anticodon is flexible at the third codon base position, which is known as wobble base pairing (Crick, 1966). Moreover, post-transcriptional modification of tRNA’s anticodon loop nucleotide allows expanded base-pairing possibilities (Agris, 1991). Hence, we expect reading and expanded decoding of degenerate mRNA codons by modified tRNAs when the cells are under defensive mode.
In the UV irradiation experiment, we detected slight upregulation of polyphosphate kinase 2 (Pnuc_1582, +1.11 fold; p = 0.0002), which utilizes polyP to produce GTP and could also inversely synthesize polyP chains by using either GTP or ATP (Achbergerová and Nahálka, 2011). Poly P accumulation in the cell is associated with stress endurance, and ppk (polyphosphate kinase) gene disruption is responsible for UV sensitivity in Pseudomonas putida KT2440 (Nikel et al., 2013). Poly P could also act as a chaperone by avoiding damaged protein aggregation such as heat-shock chaperones (Gray et al., 2014). dnaK encoding for the Hsp70 molecular chaperone and groE encoding for the Hsp60 GroEL chaperone were also up-regulated (+2.1 fold; p = 0.006 and +2.4 fold; p = 0.03, respectively). This affirms the smooth operation of protein-folding processes when the translation has been successfully completed (Castanié-Cornet et al., 2014). Poly P forms a complex with the ATP-dependent protease Lon, which could eventually degrade ribosomal proteins (Kuroda et al., 2001). Incidentally, we detected S16: Lon domain peptidase (+2.22 fold; p = 1.7 × 10–5), which is known to increase tolerance for UV (Tsilibaris et al., 2006). Combined, these processes will ultimately make free amino acids available for immediate use in the cell. Complimentarily, the signal-peptide-containing dipeptidyl aminopeptidase (Pnuc_1435) is +6.24 fold (p = 5.64 × 10–7) was up-regulated, apparently in an effort to obtain extracellularly available amino acids instead of biosynthesizing them.
Cell Structure Maintenance
UV radiation may cause bacterial cell wall damage and cytoplasmic leakage (Krishnamurthy et al., 2008). Furthermore, Jagger (1983) proposed that the cell wall may deflect near-UV photons, reducing the overall dosage received by the cellular components. Moderate increase of peptidoglycan-related protein signals may indicate protective measures adapted by irradiated bacterial cells [UDP-MurNAc-tripeptide synthetase (+1.71 fold; p = 2.4 × 10–5), D-alanylalanine synthetase (+1.38 fold; p = 1.9 × 10–3), murein peptide ligase (+0.41 fold; p = 9 × 10–3), UDP-N-acetylmuramate dehydrogenase (+3.47 fold; p = 0.2), anhydro-N-acetylmuramic acid kinase (+0.5 fold; p = 5.8 × 10–3), D-alanyl-D-alanine carboxypeptidase/D-alanyl-D-alanine-endopeptidase (+4.7 fold; p = 1.8 × 10–4)].
Metabolic Adaptation of the Cell
We also observed a lower abundance of the pyruvate dehydrogenase subunit gene and citrate synthase gene, which may hinder reactions running toward the citric acid cycle. Moreover, a pirin interaction with pyruvate dehydrogenase inhibits conversion of pyruvate into acetyl-coA. Enrichment of the pirin-encoding gene (+3.4 fold; p = 0.03) suggests that glycolytic pathway may not proceed into the citric acid cycle because pirin interacts with pyruvate dehydrogenase and inhibits its activity. Krebs cycle-related enzymes were poorly expressed.
The operation of the glyoxylate shunt under radiation toxicity has been reviewed elsewhere for Deinococcus radiodurans (Slade and Radman, 2011). This pathway required a closer examination in our irradiated samples. The glyoxylate shunt is a two-step bypass pathway in which decarboxylation of the Krebs cycle is avoided by the conversion of isocitrate into glyoxylate (with succinate) and further into malate (Dolan and Welch, 2018). Both steps are catalyzed by isocitrate lyase and malate synthase, respectively. In our case, however, the iclR (isocitrate lyase repressor) gene was slightly down-regulated (−0.50 fold; p = 0.04), which may explain the higher expression of isocitrate lyase (+1.25 fold; p = 0002) for triggering the glyoxylate shunt under the radiation-based stress scenario. Interestingly, the transcript of glcB (Pnuc_1280, malate synthase G) is overexpressed under UV treatment (+1.75 fold; p = 0.04). The corresponding enzyme abundance, however, was under-represented (−0.23 fold; p = 0.05). This raised a dilemma as to whether the processing of glyoxylate is low in the cell via the glyoxylate shunt and, if yes, what is the fate of non-processed glyoxylate? We did detect alanine dehydrogenase (Pnuc_0666) (+1.54 fold; p = 1.8 × 10–5), which can possibly catalyze glyoxylate amination to glycine (other than conversion of pyruvate to L-alanine) (Giffin et al., 2012). Alternatively, glyoxylate also behaves as a chromophore that can absorb light under 400 nm (Bersenkowitsch et al., 2018). Therefore, we cannot rule out a protective role of glyoxylate against UV radiation. Furthermore, Eugene et al. (2016) showed experimentally that glyoxylate could be photolyzed into glyoxal and that a total of 8 h of irradiation can lead to the formation of low molecular weight photoproducts, e.g., formic acid, oxalic acid, tartaric acid, and carbon dioxide. This potential should be experimentally tested to verify a protective and nutritional role of glyoxylate under UV radiation to the stressed cells.
Most cells harvested at 26°C appeared to be dividing because DNA replication, segregation, and cell division genes were positively expressed (Figure 2B and Supplementary Table S5). Upregulation of TRAP dicarboxylate transporter subunits of DctP and TAXI families (Pnuc_0468, Pnuc_0528, Pnuc_0628, Pnuc_1105, Pnuc_1145, Pnuc_1509, Pnuc_1539) at warmer temperature supports the previous interpretation by Hahn et al. (2012) that this strain mainly utilizes low-molecular-weight photooxidation products of humic substances in the lake.
All pathways are interpreted based upon our experiments tabulated in Supplementary Tables S2, S3. Importantly, we measured only transcript and protein changes, which provide an indication of the possible functions being carried out in the cells under varying stress scenarios. This leaves future scope for further experimental validation of our observations.
Methylation Features in the Bacterial Genome Under Different Treatments
We found that ∼21%, ∼25%, and ∼16% of the strain’s genes were methylated at 4°C, 26°C, and 26°C∗ conditions, respectively (Figure 2C and Supplementary Table S6). Methylation in the prokaryotic world is widespread and participates in phage defense as well as epigenetic gene regulation (Adhikari and Curtis, 2016). In fact, the steps involved in the bacterial gene expression process (until protein biosynthesis) include various levels of regulation: DNA supercoiling, cis elements and trans regulatory factors, DNA methylation, post-transcriptional regulatory mechanisms, mRNA conformation and modification, and post-translational modifications (Willenbrock and Ussery, 2004; Casadesus and Low, 2006; Richards et al., 2008; Balleza et al., 2009; Geissmann et al., 2009; Grangeasse et al., 2015). This section covers only potential methylation-associated gene expression in the strain QLW-P1DMWA-1T. Nearly half of the detected methylations (on adenine and cytosine together) in our three treatments was found inside the gene body and their 500-bp upstream region, i.e., 49%, 52%, and 47% in 4°C-, 26°C-, and 26°C∗-incubated samples, respectively (Figures 3A,B). The remaining half of the methylations was detected in other intergenic regions of the genome. The presence of methylation at sites other than the gene body and promoter elements is not uncommon (Suzuki and Bird, 2008). For example, intergenic methylation has been cited in the termination of transcription by limiting any aberrant read-through in Arabidopsis thaliana (Yan et al., 2016). Moreover, the role of such broadly distributed base modifications no doubt goes beyond gene expression regulation to include phage defense. It may also help avoid unintended transcription initiation or even accidental elongation processes at non-specific sites. The promoter region is known to contribute < 2% of a bacterial genome, and RNA polymerase may therefore have to conduct random walk along DNA strands or three-dimensional diffusion in order to form promoter-bound complexes (Wang et al., 2013). Accordingly, methylation within the coding region could be an additional prevention mechanism in the event of leaky gene expression due to aberrant transcription initiation of supposedly turned-off genes.
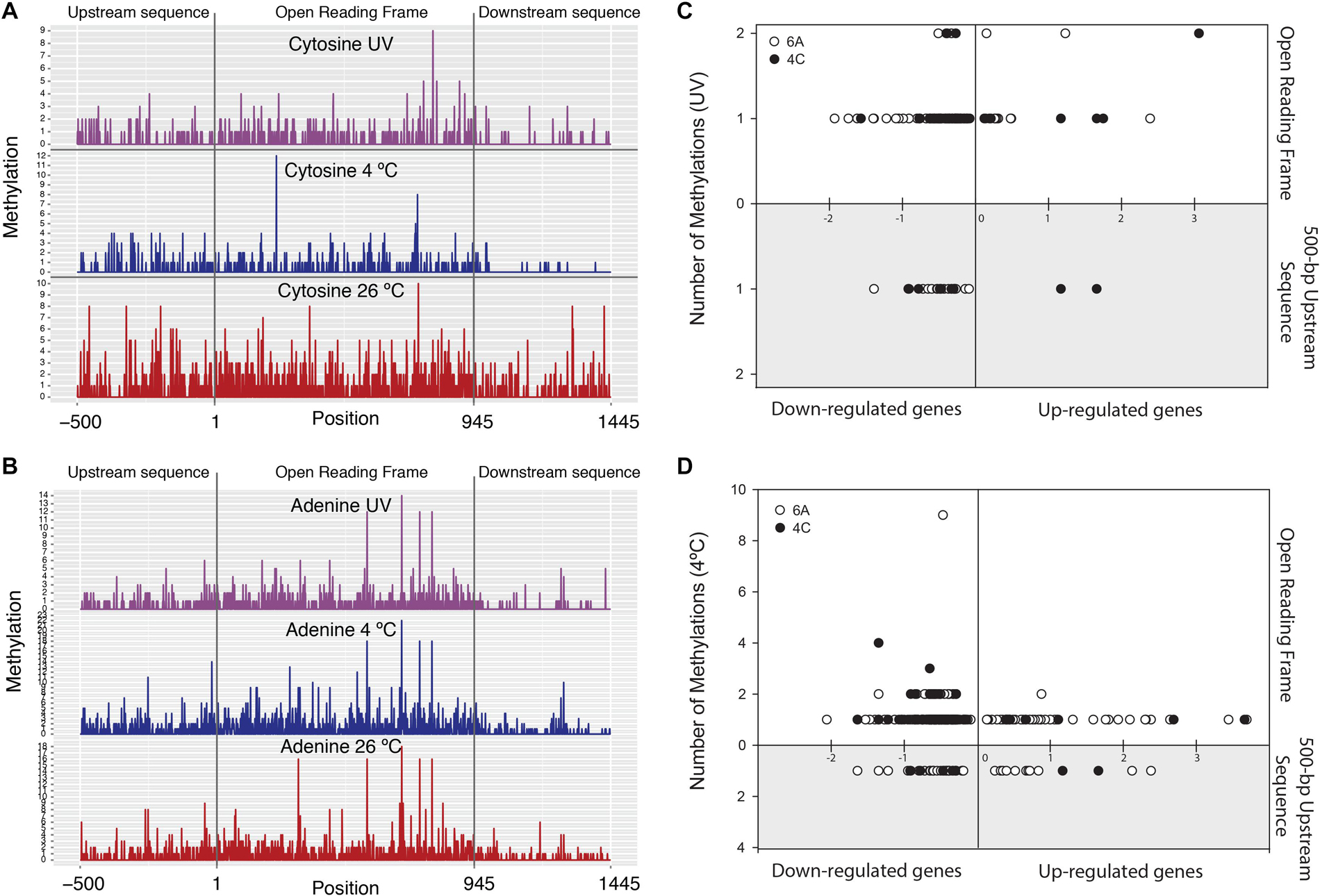
Figure 3. Left panel plots show number of methylations on: (A) cytosine and (B) adenine nucleotides belonging to average length of all genes in the genome (with averaged 500-bp up- and downstream sequences) in three different treatments. Right panel plots show total number of methylations coinciding with transcriptional profiles in: (C) UV-irradiated sample and (D) 4°C-incubated sample. X-axis: up-regulated genes (positive) and down-regulated genes (negative). Y-axis: number of adenine (o) and cytosine (•) methylations. Upper panel X-axis: methylation count within the genes. Lower, gray-shaded panel: number of methylations in 500-bp upstream region of genes.
In our study, ∼13% and ∼6% of the genes were down-regulated when adenine (m6A) and cytosine (m4C) sites were methylated within genes or on +500-bp upstream gene sequences in the 4°C and 26°C∗ cultures (considering the 26°C treatment as control), respectively. Very small sets of genes (∼3% and 1% in 4°C and 26°C∗ cultures, respectively) were also up-regulated under methylation conditions (Figures 3C,D, Supplementary Figures S3a,b, and Supplementary Tables S7a,b). Clearly, the accepted paradigm that methylation represses genes does not hold true in some cases. For example, genes are generally repressed (when the gene-body or its promoter is methylated), but methylation may also be involved in gene activation as described in Escherichia coli, Salmonella enterica, and Caulobacter crescentus (Braun and Wright, 1986; Balbontin et al., 2006; Fioravanti et al., 2013). Most of the gene expressions associated with methylation states were subtle (i.e., lower expression level), an observation noted by Adhikari and Curtis (2016) in a review article covering many dam (DNA adenine methyltransferase) mutant studies in bacteria. Lastly, in our study, the expression link between methylation state, transcription, and proteome status suggested that only ∼5% and ∼1.7% of the genes were differentially expressed (in the 4°C and 26°C∗ cultures, respectively) and could be traced at all three levels (Figure 4 and Supplementary Table S8). Therefore, summary figure (Figure 5) describing major metabolic pathways are broadly based upon transcript and protein expression profiles, and with few methylation-associated expressed genes (Supplementary Tables S5, S8). The fact that we found a trend within our small set of genes that were detected at the three levels indicates that methylation (mainly adenine modification) at low temperature is associated with down-regulated genes (78%) (Table 2A). In contrast, the majority of down-regulated genes (83%) under UV radiation treatment had less association with adenine and cytosine methylation when compared to the 26°C treatment (Table 2A). We examined gene expression because it can be directly affected by methylation, whereas the protein profile might be influenced by post-transcriptional and translational regulations.
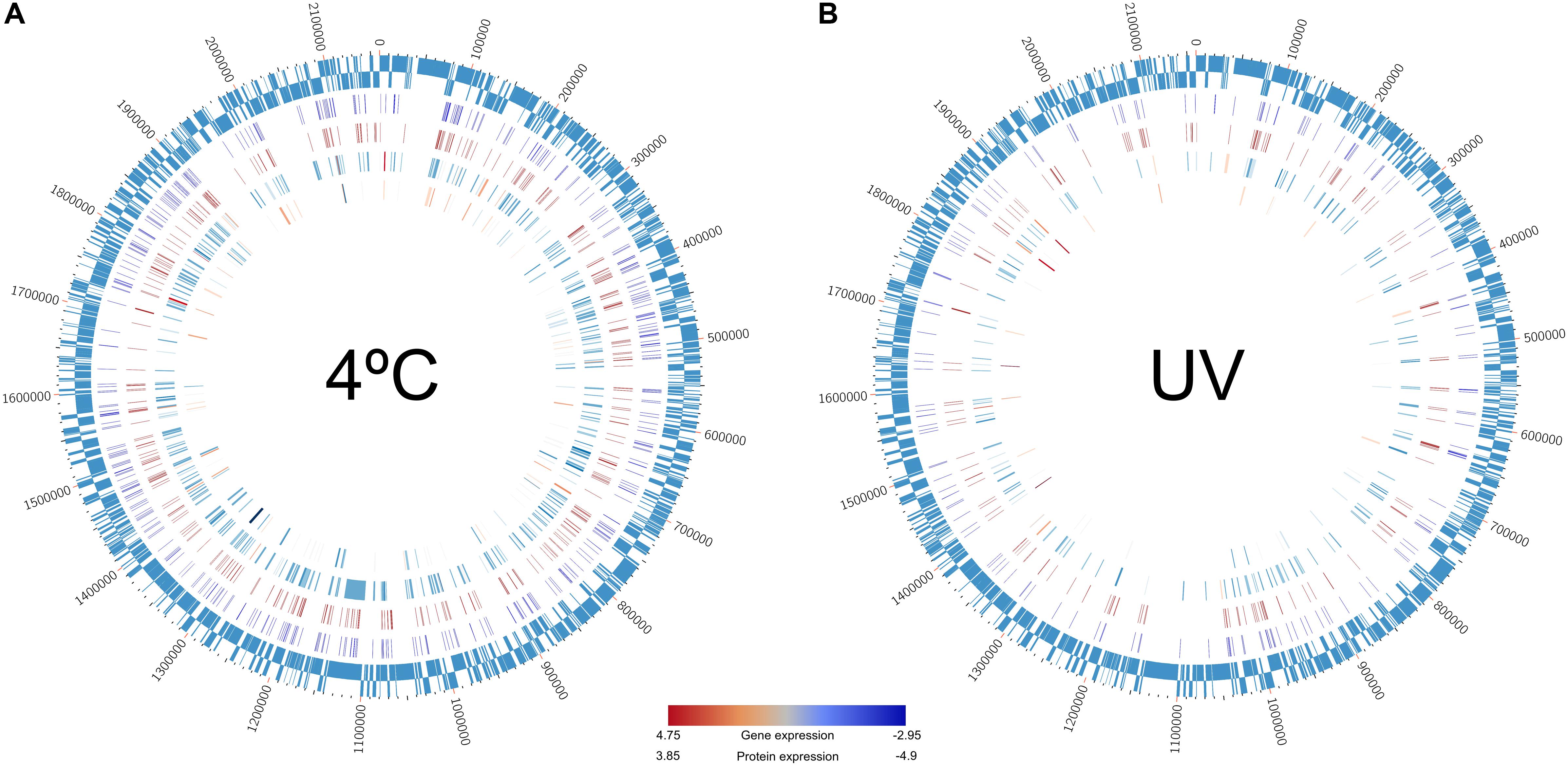
Figure 4. Circos plots showing the direct link between methylome (IPD ratio), transcriptome, and proteome data recorded in the Polynucleobacter asymbioticus strain QLW-P1DMWA-1T. The remaining data have been omitted to highlight only one-to-one relationships across the three levels. Genes painted in the plots are enlisted in Supplementary Table S8. Outermost ring: the 2.15-Mb genome of this strain. Second internal ring: methylation pattern recorded at (A) 4°C and (B) 26°C*-incubation. Third ring: methylation recorded at 26°C-incubation. Here, methylations only within the open reading frame and 500-bp upstream of the genes are shown. Fourth ring: the transcribed genes in (A) 4°C and (B) 26°C*-incubation. Fifth internal ring: protein expression profile in (A) 4°C and (B) 26°C*-incubation. Color gradient bar: red = higher expression, blue = lower expression at transcriptome and proteome levels.
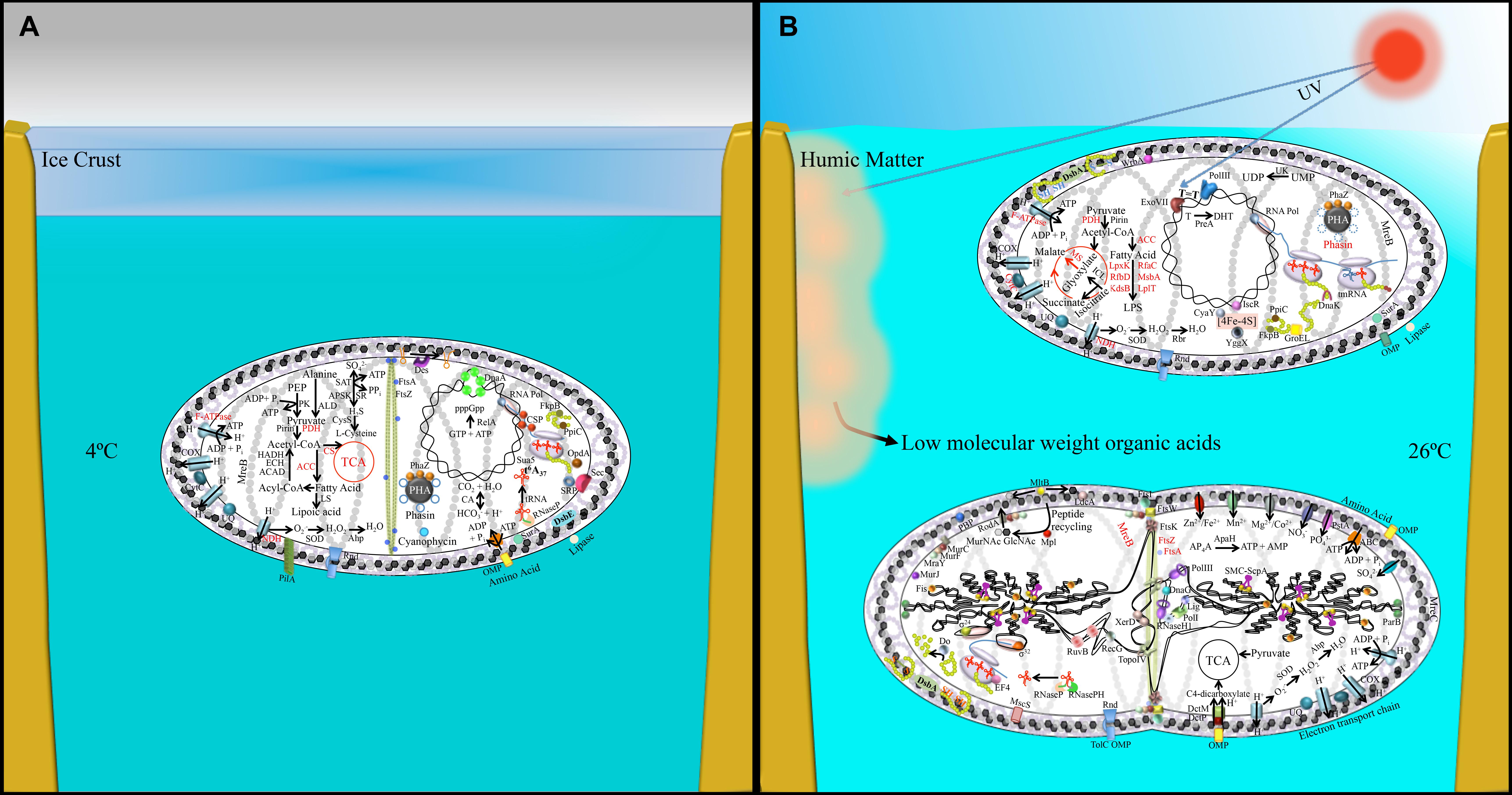
Figure 5. Pictorial representation of transcriptomic and proteomic data obtained from (A) 4°C-incubated bacterial sample and (B) UV-irradiated (upper cell), 26°C-incubated bacterial sample (lower dividing cell). This figure is a lifestyle inference and shows possible physiological changes together with the involved genetics in P. asymbioticus strain QLW-P1DMWA-1T during winter and summer. Black font: up-regulated genes/protein; red font: down-regulated genes/proteins. Abbreviations explained in Supplementary Table S5. Model of humic matter photolysis into low-molecular-weight photo-oxidation products and its utilization by these bacteria has been modified after Hahn et al. (2012).
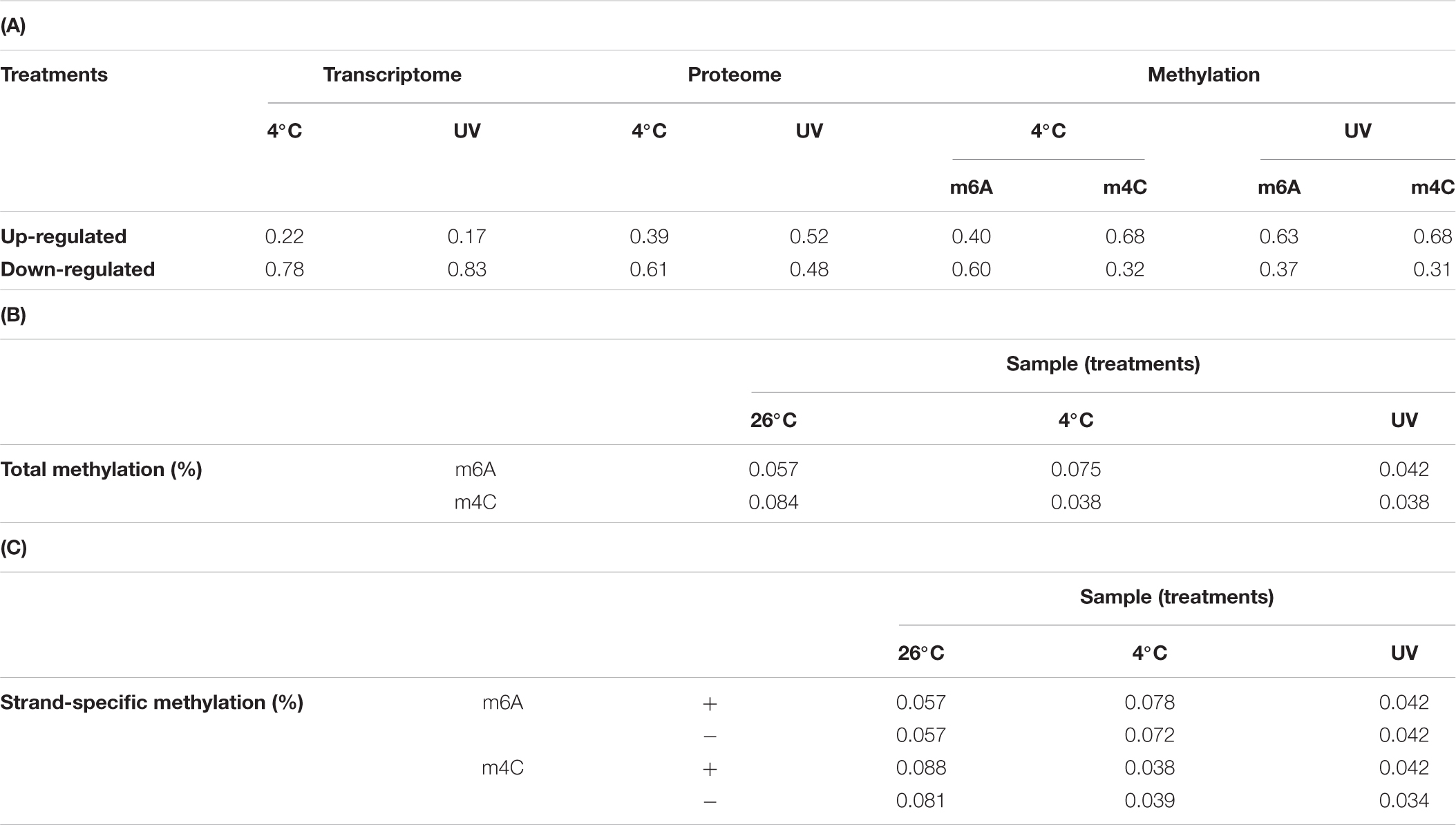
Table 2. Table showing relative variation in the transcriptome, proteome, and in the adenine and cytosine methylation counts as compared to the control (26°C incubation); (B) Tabulation of adenine and cytosine methylation counts in the genome (as percentage) in three treatments (i.e., warm, cold, and UV irradiation, i.e., 26°C*); (C) adenine and cytosine methylation counts as percentage but on sense and antisense strands in three treatments.
In the Polynucleobacter asymbioticus genome, 0.069% (1504 sites) of the adenine plus cytosine sites were methylated at 26°C, 0.058% (1273 sites) at 4°C, and a mere 0.040% (873 sites) at 26°C∗ (Figure 2C and Supplementary Table S6). These values lie in the lower range of recorded methylated bases in tested bacterial genomes: earlier findings revealed a genome methylation range between 0.03% and ∼4% (Fang et al., 2012; Murray et al., 2012; Krebes et al., 2013; Lluch-Senar et al., 2013; Lee et al., 2015; Hiraoka et al., 2019) (Supplementary Table S9). The restriction enzymes database REBASE® lists PacBio-based sequencing information on the genomes of a large number of organisms5 and suggests genomes devoid of methylated motifs. This raised the question whether a low methylation level can still participate in gene regulation. Our detailed analysis revealed that the m4C count dropped to about half when the culture was transferred from 26°C to 4°C or exposed to UV-irradiation. In the m6A count, however, we observed ∼25% more modifications when shifting the sample from warmer to cold temperature (4°C) (Figure 2C and Supplementary Table S6). The N6-adenine-specific DNA methylase enzyme (Pnuc_1133) was marginally enriched (+1.15-fold; p = 0.0004) at cold incubation, suggesting its role in the DNA methylation process. We detected two ‘16 nucleotides long’ conserved motifs (CTAYNNNNNNNNTRTC and GAYANNNNNNNNRTAG) that are complementary to each other (Supplementary Figure S4 and Supplementary Table S6). Bold letter adenine residues in the motifs are modified with a methyl group. We detected 196 motifs of both kinds in the sequenced genome of strain QLW-P1DMWA-1T. Only 92% of the motifs were methylated when the bacteria were incubated at 26°C∗, but this value approached 97% at 26°C and 4°C. Note that we also observed DNA methylation at sites other than identifiable motifs. The presence of non-specific DNA methyltransferase (for adenine residue modification) in E. coli O104:H4 C227-11 might be helpful against phage infection (Murray et al., 2018). Furthermore, we detected only one copy of the type I restriction modification (R-M) system in the genome (Supplementary Figure S5) and two candidate genes (Pnuc_0029, Pnuc_0929) possibly involved in the type III restriction modification process. Many other prokaryotes are known to deploy type I and type III R-M systems to defend their genome against phage infection (Ershova et al., 2015).
At the gene expression level, down-regulated genes encoding for the MscS mechanosensitive ion channel (−1.53 fold; p = 0.05) and ComE DNA binding protein (−1.22 fold; p = 0.02) had methyl group modifications within the gene body on adenine bases at 4°C, but such a methylation pattern was missing from the samples recovered from the 26°C treatment (Figure 2C and Supplementary Table S6). The methylation status of the relA gene is also remarkable considering that this gene is down-regulated in cold treatment: the 26°C sample shows no methylation but becomes methylated (two adenine residues) during 4°C treatment. It is tempting to speculate that the co-occurrence of methylation at 4°C may epigenetically control the expression of the relA gene and consequently many other metabolic pathways. Methylation might be involved in the expression of genes, and such observations need to be verified by mutational analysis in the future. Moreover, experiments with methyltransferase such as gene cloning, overexpression, and enzyme purification are required to confirm the actual binding sites on DNA by electrophoretic mobility shift assays. Such experiments are necessary because variation in the DNA polymerase kinetics signal during SMRT sequencing and a weak signal-to-noise ratio (mainly for cytosine modifications) may introduce false positive modifications being overcalled by the PacBio program (Feng et al., 2013).
In terms of base modification symmetry on DNA strands, when the bacterium was shifted from its optimal growth condition (26°C) to either 4°C or 26°C∗, the m6A and m4C counts remained at similar ratios between both chromosomal strands. This, however, is not evident when considering only total count (Tables 2B,C). One interpretation is that the bacteria try to maintain a balanced methylation pattern on both strands regardless of experimental conditions. No such observation (and therefore no explanation) has ever been reported before in any system. Nonetheless, human epigenetic studies have demonstrated that a balance between methylation and demethylation process maintains regulatory features. At the same time, over- or under-methylation may cause developmental defects or genetic instability, respectively (Martin and Fry, 2018). This might also hold true in a simpler bacterial model system, and the balance of the methylation pattern at both strands appears to reflect an equilibrium between a cell’s methylating and demethylating enzymatic processes.
Natural Selection Process
Earlier studies on Polynucleobacter bacteria have already revealed their relationship with the environment they inhabit (Hahn et al., 2012, 2015). Our study went one step further by investigating the metabolic and genetic potentials of the strain in question. Moreover, apart from the three-tier study (covering methylation, transcriptomic, and proteomics), we conducted additional analyses on the adaptive process operating due to evolutionary pressure.
To investigate which selection processes generally operate in this group of bacteria, we expanded our analysis to include twenty other genomes of Polynucleobacter spp. (Hoetzinger et al., 2017) (Figure 6A and Supplementary Table S1). To study molecular evolution at the population genetics level, we used a standard method that compares the numbers of non-synonymous and synonymous substitution rates in protein-coding genes (Ina, 1996). Twenty highly expressed genes from the transcriptome or proteome analysis and ten housekeeping genes were selected to estimate the selection imposed upon them. The ratios of non-synonymous to synonymous substitution rates (Ka/Ks) revealed that all tested genes underwent purifying selection, i.e., the values were <1 (Figure 6B and Supplementary Tables S10–S12). This indicates that deleterious amino acid replacements have been removed via negative selection (Hurst, 2002). Purifying selection could help explain the moderately streamlined genomes present in all studied Polynucleobacter spp. Genome streamlining due to purifying selection followed by ecological niche specialization in the marine free-living picocyanobacteria Prochlorococcus have already been reported as important reasons for their global dominance (Sun and Blanchard, 2014). The habitats or previous environments of currently studied freshwater strains and their common ancestors remain undefined. Therefore, the nucleotide sequence divergence that we observe reveals – at best – the operation of purifying selection. However, the signals of any potential positive selection in the past will be masked by the dominance of purifying selection.
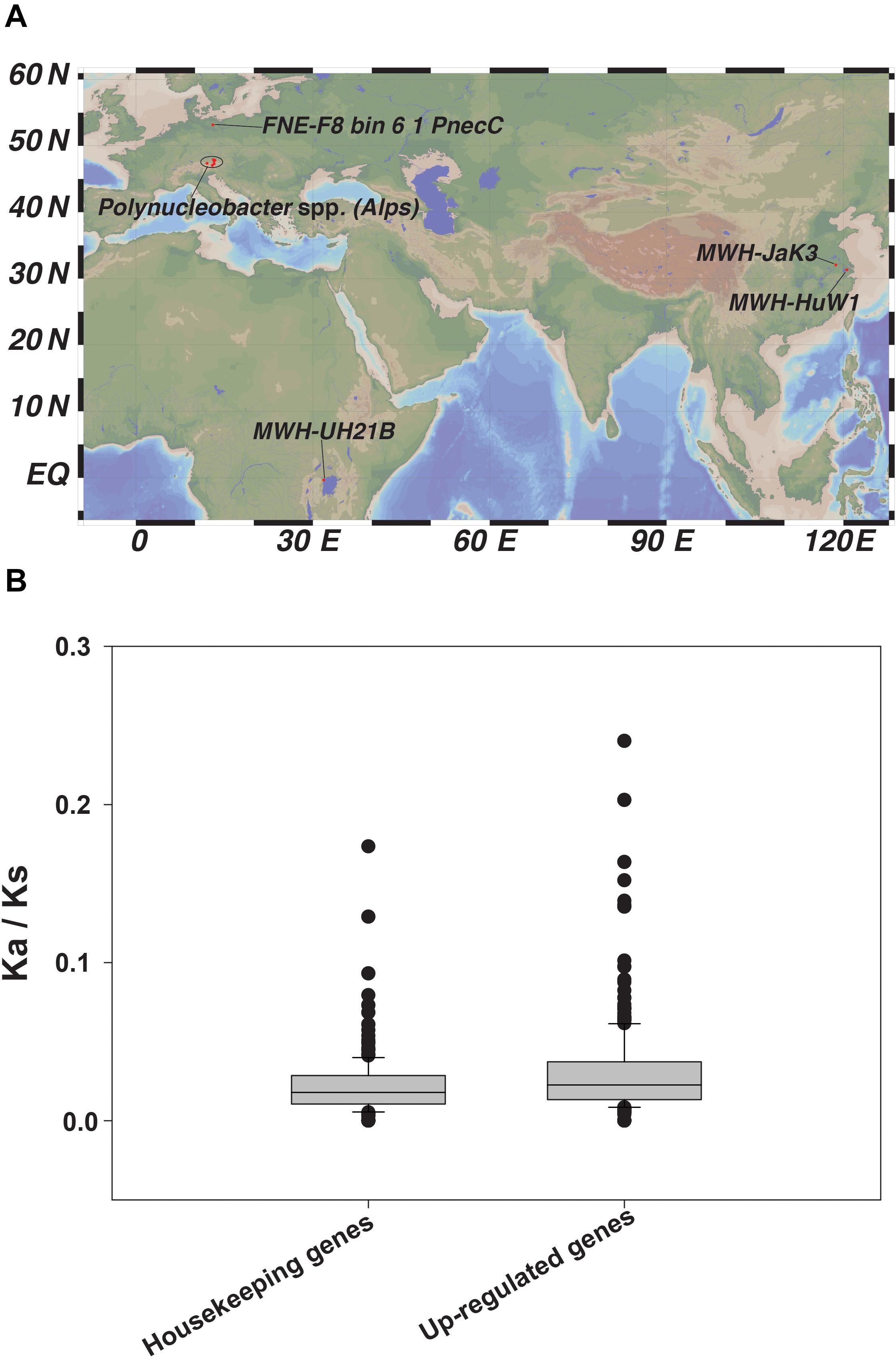
Figure 6. (A) Geographical distribution of the 20 Polynucleobacter spp. used in our study and (B) Ratio of non-synonymous (Ka) to synonymous (Ks) nucleotide substitution represented as box-plot for selected housekeeping genes and other selected up-regulated genes chosen from our -omics dataset.
Populations of a particular species living in different habitats are naturally selected, leading to local adaptation and genetic divergence (Barton, 2010). Furthermore, strong purifying selection may prevail in species inhabiting diverse geographical locations, as reported for several wild varieties of South American tomatoes (Tellier et al., 2011). Importantly, the genomes of enteric bacteria such as Escherichia coli and Salmonella enterica that reside in similar habitats exhibit strong positive selection (Chattopadhyay et al., 2012). Exploiting the environmental situations and sustaining large effective population sizes by maintaining the functional sequences in the genome may have promoted adaptive diversification among various Polynucleobacter spp. occupying distinct biogeographical regions.
Conclusion
UV irradiation and temperature were the only distinguishing aspects of our experimental setups. Expectedly, strain QLW-P1DMWA-1T senses thermal disparity in its immediate environment and expresses a multitude of genes accordingly. Small cells with a streamlined genome appear to follow a trend of small-scale changes in gene expression and methylation, significantly contributing to bacterial acclimatization. Bacterial and mammalian epigenetic studies report methylation-induced repression and gene activation (Casadesus and Low, 2006; Udali et al., 2015). Here, we document that methylation patterns in the bacterial genome are constantly revised in response to rapid environmental changes, possibly via de novo methylation (Figures 2C, 3A–D and Supplementary Figure S3). This strategy, along with the gene regulatory network, might significantly improve the strain’s survival ability. A direct mechanistic relationship between gene expression and methylation status cannot be determined by conducting mutational analysis in the DNA methylase-encoding gene. Nonetheless, the signals obtained for m6A and m4C were consistent with reports elsewhere (Clark et al., 2012). The simultaneous presence or absence of the methyl group signature patterns of various genes and intergenic regions (Figure 3, Supplementary Figure S3, and Supplementary Table S6) indicates their potential in rapid acclimatization to changing environmental conditions. Moreover, local regulatory factors also contribute in organismal responses to environmental disturbances. We have summarized bacterial acclimatization to changing stressors (Figure 5). This summary includes low-temperature-related stress to the cells, which is well known. However, the increased expression of myriad GTPases suggested regulation of wide-ranging global to local processes. Finally, preserving energy reserves appears to be an important feature for withstanding stressed cold phases. In contrast, UV irradiated cells face oxidative stress and become defensive. Not only do they reduce key energy-generating pathways, but they also focus on protein production for important repair systems. This involves enhanced accuracy at the translational level and expanding mRNA decoding with limited sets of tRNAs.
Our proposal is that overall regulatory changes, together with programming of the DNA methylome in parental cells living in a fluctuating environment, are equally important for daughter cells. This is because the latter cells possibly inherit the exact epigenomic information as an epigenetic memory as proposed in other model system (Gaydos et al., 2014). Although not tested here, this strategy potentially gives bacterial cells a head start in a given natural setting despite undergoing natural selection. In this vein, two recent papers have indicated that E. coli daughter cells can be pre-adapted based upon the long-term epigenetic memory they retain upon previous exposure of their parental cells to a particular environment (Ronin et al., 2017; Govers et al., 2018). Determining whether our findings are generally valid across successful species with streamlined genome and smaller cell volumes calls for conducting similar experiments with other environmental bacterial isolates. Some examples to initiate testing would be small-sized prokaryotes with streamlined genomes that are highly abundant in freshwater and marine systems, including more strains of the genus Polynucleobacter, acI Actinobacteria, and Prochlorococcus. Here, epigenetic modifications may play a greater role in cellular functioning than previously recognized.
The strain tested here in the three-tier study (methylome, transcriptome, proteome) is not the complete picture of the success of Polynucleobacter spp. in freshwater systems. It does, however, provide insight into various possibilities that might contribute to rapid adjustments in fluctuating environments. Our natural selection pressure study points toward purifying selection in all 20 P. asymbioticus strains, and the process is associated with deleterious mutation removal from the genome. Combined, we can conclude that exploiting the environmental situations to maintain effective population size is the best way to compensate for such deleterious mutation load. The extinction episodes of other species are compelling examples of a reduced efficiency of purifying selection that led to the accumulation of detrimental mutations in their genomes. The result was that their populations could not be sustained over the long term and declined (Sánchez-Quinto and Lalueza-Fox, 2015; Rogers and Slatkin, 2017).
We believe that conducting experiments in the natural habitat might disclose more shades of epigenetic and regulatory networks than shown in this report using artificial media and an incubator-based set-up. Moreover, diurnal temperature dynamics are more pronounced in small and shallow ponds. It will be interesting to compare the methylation response of shallow pond-dwellers against the methylome pattern obtained from large and deeper lake inhabitants, i.e., from less dynamic systems. We also foresee that epigenetics studies in aquatic microbial ecology can be used as an indicator of environmental fluctuations reflected in microbial epigenome changes.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
AS conceived and designed the details of the project, performed the experiments, analyzed the data, and wrote the manuscript. H-PG designed the overall project, provided mentorship and intellectual input. JM designed the proteomics study, and collected and analyzed data. ME and CW conducted MS analyses. UR contributed with proteomics study designing. JG and YK performed the bioinformatics analyses. DD and MH contributed with statistical analysis. MWH provided the time series data and contributed intellectually. All authors contributed to the article and approved the submitted version.
Funding
AS and H-PG were supported by the European Science Foundation (ESF) via the EUROCORE (PAK 518) project FREDI (DFG: GR 1540/17-1). MH and MWH were supported by the Austrian Science Fund (FWF) project I482-B09 and the European Science Foundation (ESF) project FREDI. We acknowledge the support of the Deutsche Forschungsgemeinschaft and Open Access Publishing Fund of the University of Potsdam.
Conflict of Interest
YK is employed at GATC Biotech AG.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to thank the MIBI team for their support and discussions, in particular Elke Mach and Solvig Pinnow for their practical help. The Leibniz society is thanked for its financial support. We are thankful to Nadine Genzel for her guidance with the transcriptomic study and data submission. We also wish to thank Darren P. Giling, Jason Woodhouse and Michael Stachowitsch for critical feedback. For mass spectrometry, we would like to acknowledge the assistance of the Core Facility BioSupraMol supported by the Deutsche Forschungsgemeinschaft (DFG).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.544785/full#supplementary-material
Footnotes
- ^ https://www.ncbi.nlm.nih.gov/nuccore/CP000655
- ^ http://www.pacbiodevnet.com/SMRT-Analysis/Algorithms/BLASR
- ^ http://www.smrtcommunity.com/SMRT-Analysis/Software/SMRT-Analysis
- ^ http://circos.ca
- ^ http://rebase.neb.com/cgi-bin/pacbiolist?0
References
Achbergerová, L., and Nahálka, J. (2011). Polyphosphate – an ancient energy source and active metabolic regulator. Microb. Cell Fact. 10:63. doi: 10.1186/1475-2859-10-63
Adhikari, S., and Curtis, P. D. (2016). DNA methyltransferases and epigenetic regulation in bacteria. FEMS Microbiol. Rev. 40, 575–591. doi: 10.1093/femsre/fuw023
Agris, P. F. (1991). Wobble position modified nucleosides evolved to select transfer RNA codon recognition: a modified-wobble hypothesis. Biochimie 73, 1345–1349. doi: 10.1016/0300-9084(91)90163-u
Aït-Bara, S., and Carpousis, A. J. (2015). RNA degradosomes in bacteria and chloroplasts: classification, distribution and evolution of RNase E homologs. Mol. Microbiol. 97, 1021–1135. doi: 10.1111/mmi.13095
Arjes, H. A., Lai, B., Emelue, E., Steinbach, A., and Levin, P. A. (2015). Mutations in the bacterial cell division protein FtsZ highlight the role of GTP binding and longitudinal subunit interactions in assembly and function. BMC Microbiol. 15:209. doi: 10.1186/s12866-015-0544-z
Ayub, N. D., Tribelli, P. M., and López, N. I. (2009). Polyhydroxyalkanoates are essential for maintenance of redox state in the Antarctic bacterium Pseudomonas sp. 14-3 during low temperature adaptation. Extremophiles 13, 59–66. doi: 10.1007/s00792-008-0197-z
Balbontin, R., Rowley, G., Pucciarelli, M. G., Lopez-Garrido, J., Wormstone, Y., Lucchini, S., et al. (2006). DNA adenine methylation regulates virulence gene expression in Salmonella enterica serovar Typhimurium. J. Bacteriol. 188, 8160–8168. doi: 10.1128/jb.00847-06
Balleza, E., López-Bojorquez, L. N., Martínez-Antonio, A., Resendis-Antonio, O., Lozada-Chávez, I., Balderas-Martínez, Y. I., et al. (2009). Regulation by transcription factors in bacteria: beyond description. FEMS Microbiol. Rev. 33, 133–151. doi: 10.1111/j.1574-6976.2008.00145.x
Barria, C., Malecki, M., and Arraiano, C. M. (2013). Bacterial adaptation to cold. Microbiology 159, 2437–2443. doi: 10.1099/mic.0.052209-0
Barton, N. H. (2010). What role does natural selection play in speciation? Philos. Trans. R. Soc. Lond. B. Biol. Sci. 365, 1825–1840. doi: 10.1098/rstb.2010.0001
Bayles, D. O., Tunick, M. H., Foglia, T. A., and Miller, A. J. (2000). Cold shock and its effect on ribosomes and thermal tolerance in Listeria monocytogenes. Appl. Environ. Microbiol. 66, 4351–4355. doi: 10.1128/aem.66.10.4351-4355.2000
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate—a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Bersenkowitsch, N. K., Ončák, M., van der Linde, C., Herburger, A., and Beyer, M. K. (2018). Photochemistry of glyoxylate embedded in sodium chloride clusters, a laboratory model for tropospheric sea-salt aerosols. Phys. Chem. Chem. Phys. 20, 8143–8151. doi: 10.1039/c8cp00399h
Beukers, R., Eker, A. P. M., and Lohman, P. H. M. (2008). 50 years thymine dimer. DNA Repair 7, 530–543. doi: 10.1016/j.dnarep.2007.11.010
Blow, M. J., Clark, T. A., Daum, C. G., Deutschbauer, A. M., Fomenkov, A., Fries, R., et al. (2016). The epigenomic landscape of prokaryotes. PLoS Genet. 12:e1005854. doi: 10.1371/journal.pgen.1005854
Boenigk, J., Stadler, P., Wiedlroither, A., and Hahn, M. W. (2004). Strain-specific differences in the grazing sensitivities of closely related ultramicrobacteria affiliated with the Polynucleobacter cluster. Appl. Environ. Microbiol. 70, 5787–5793. doi: 10.1128/aem.70.10.5787-5793.2004
Boscaro, V., Felletti, M., Vannini, C., Ackerman, M. S., Chain, P. S., Malfatti, S., et al. (2013). Polynucleobacter necessarius, a model for genome reduction in both free-living and symbiotic bacteria. Proc. Natl. Acad. Sci. U.S.A. 110, 18590–18595. doi: 10.1073/pnas.1316687110
Braun, R. W., and Wright, A. (1986). DNA methylation differentially enhances the expression of one of the two E. Coli dnaA promoters in vivo and in vitro. Mol. Gen. Genet. 202, 246–250. doi: 10.1007/bf00331644
Britton, R. A., Powell, B. S., Dasgupta, S., Sun, Q., Margolin, W., Lupski, J. R., et al. (1998). Cell cycle arrest in Era GTPase mutants: a potential growth rate-regulated checkpoint in Escherichia coli. Mol. Microbiol. 27, 739–750. doi: 10.1046/j.1365-2958.1998.00719.x
Bucurenci, N., Serina, L., Zaharia, C., Landais, S., Danchin, A., and Bârzu, O. (1998). Mutational analysis of UMP kinase from Escherichia coli. J. Bacteriol. 180, 473–477. doi: 10.1128/jb.180.3.473-477.1998
Budiman, C., Koga, Y., Takano, K., and Kanaya, S. (2011). FK506-binding protein 22 from a psychrophilic bacterium, a cold shock-inducible peptidyl prolyl isomerase with the ability to assist in protein folding. Int. J. Mol. Sci. 12, 5261–5284. doi: 10.3390/ijms12085261
Candiano, G., Bruschi, M., Musante, L., Santucci, L., Ghiggeri, G. M., Carnemolla, B., et al. (2004). Blue silver: a very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis 25, 1327–1333. doi: 10.1002/elps.200305844
Casadesus, J., and Low, D. (2006). Epigenetic gene regulation in the bacterial world. Microbiol. Mol. Biol. Rev. 70, 830–856. doi: 10.1128/mmbr.00016-06
Castanié-Cornet, M. P., Bruel, N., and Genevaux, P. (2014). Chaperone networking facilitates protein targeting to the bacterial cytoplasmic membrane. Biochim. Biophys. Acta 1843, 1442–1456. doi: 10.1016/j.bbamcr.2013.11.007
Chattopadhyay, S., Paul, S., Kisiela, D. I., Linardopoulou, E. V., and Sokurenko, E. V. (2012). Convergent molecular evolution of genomic cores in Salmonella enterica and Escherichia coli. J. Bacteriol. 194, 5002–5011. doi: 10.1128/jb.00552-12
Chiu, S. W., Chen, S. Y., and Wong, H. C. (2008). Localization and expression of MreB in Vibrio parahaemolyticus under different stresses. Appl. Environ. Microbiol. 74, 7016–7022. doi: 10.1128/aem.01020-08
Clark, T. A., Murray, I. A., Morgan, R. D., Kislyuk, A. O., Spittle, K. E., Boitano, M., et al. (2012). Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res. 40:e29. doi: 10.1093/nar/gkr1146
Cox, J., Neuhauser, N., Michalski, A., Scheltema, R. A., Olsen, J. V., and Mann, M. (2011). Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805. doi: 10.1021/pr101065j
Crick, F. H. C. (1966). Codon—anticodon pairing: the wobble hypothesis. J. Mol. Biol. 19, 548–555. doi: 10.1016/s0022-2836(66)80022-0
Doerfel, L. K., Wohlgemuth, I., Kothe, C., Peske, F., Urlaub, H., and Rodnina, M. V. (2013). EF-P is essential for rapid synthesis of proteins containing consecutive proline residues. Science 339, 85–88. doi: 10.1126/science.1229017
Dolan, S. K., and Welch, M. (2018). The glyoxylate shunt, 60 years on. Annu. Rev. Microbiol. 72, 309–330. doi: 10.1146/annurev-micro-090817-062257
Dörr, T., Moynihan, P. J., and Mayer, C. (2019). Editorial: bacterial cell wall structure and dynamics. Front. Microbiol. 10:2051. doi: 10.3389/fmicb.2019.02051
El Yacoubi, B., Lyons, B., Cruz, Y., Reddy, R., Nordin, B., Agnelli, F., et al. (2009). The universal YrdC/Sua5 family is required for the formation of threonylcarbamoyladenosine in tRNA. Nucleic Acids Res. 37, 2894–2909. doi: 10.1093/nar/gkp152
Epshtein, V. (2015). UvrD helicase: an old dog with a new trick: how one step backward leads to many steps forward. Bioessays 37, 12–19. doi: 10.1002/bies.201400106
Ershova, A. S., Rusinov, I. S., Spirin, S. A., Karyagina, A. S., and Alexeevski, A. V. (2015). Role of restriction-modification systems in prokaryotic evolution and ecology. Biochemistry 80, 1373–1386. doi: 10.1134/s0006297915100193
Eugene, A. J., Xia, S.-S., and Guzman, M. I. (2016). Aqueous photochemistry of glyoxylic acid. J. Phys. Chem. A 120, 3817–3826. doi: 10.1021/acs.jpca.6b00225
Fang, G., Munera, D., Friedman, D. I., Mandlik, A., Chao, M. C., Banerjee, O., et al. (2012). Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat. Biotechnol. 30, 1232–1239. doi: 10.1038/nbt.2432
Feller, G. (2018). Protein folding at extreme temperatures: Current issues. Semin. Cell. Dev. Biol. 108, 30125–30128. doi: 10.1016/j.semcdb.2017.09.003
Feng, Z., Fang, G., Korlach, J., Clark, T., Luong, K., Zhang, X., et al. (2013). Detecting DNA modifications from SMRT sequencing data by modeling sequence context dependence of polymerase kinetic. PLoS Comput. Biol. 9:e1002935. doi: 10.1371/journal.pcbi.1002935
Fioravanti, A., Fumeaux, C., Mohapatra, S. S., Bompard, C., Brilli, M., Frandi, A., et al. (2013). DNA binding of the cell cycle transcriptional regulator GcrA depends on N6-adenosine methylation in Caulobacter crescentus and other Alphaproteobacteria. PLoS Genet. 9:e1003541. doi: 10.1371/journal.pgen.1003541
Flusberg, B. A., Webster, D. R., Lee, J. H., Travers, K. J., Olivares, E. C., Clark, T. A., et al. (2010). Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 7, 461–465. doi: 10.1038/nmeth.1459
Gaca, A. O., Colomer-Winter, C., and Lemos, J. A. (2015). Many means to a common end: the intricacies of (p)ppGpp metabolism and its control of bacterial homeostasis. J. Bacteriol. 197, 1146–1156. doi: 10.1128/JB.02577-14
Gaydos, L. J., Wang, W., and Strome, S. (2014). H3K27me and PRC2 transmit a memory of repression across generations and during development. Science 345, 1515–1518. doi: 10.1126/science.1255023
Geissmann, T., Marzi, S., and Romby, P. (2009). The role of mRNA structure in translational control in bacteria. RNA Biol. 6, 153–160. doi: 10.4161/rna.6.2.8047
Giffin, M. M., Modesti, L., Raab, R. W., Wayne, L. G., and Sohaskey, C. D. (2012). ald of Mycobacterium tuberculosis encodes both the alanine dehydrogenase and the putative glycine dehydrogenase. J. Bacteriol. 194, 1045–1054. doi: 10.1128/jb.05914-11
Giuliodori, A. M., Brandi, A., Gualerzi, C. O., and Pon, C. L. (2004). Preferential translation of cold-shock mRNAs during cold adaptation. RNA 10, 265–276. doi: 10.1261/rna.5164904
Glaeser, S. P., Grossart, H.-P., and Glaeser, J. (2010). Singlet oxygen, a neglected but important environmental factor: short-term and long-term effects on bacterioplankton composition in a humic lake. Environ. Microbiol. 12, 3124–3136. doi: 10.1111/j.1462-2920.2010.02285.x
Govers, S. K., Mortier, J., Adam, A., and Aertsen, A. (2018). Protein aggregates encode epigenetic memory of stressful encounters in individual Escherichia coli cells. PLoS Biol. 16:e2003853. doi: 10.1371/journal.pbio.2003853
Grangeasse, C., Stülke, J., and Mijakovic, I. (2015). Regulatory potential of post-translational modifications in bacteria. Front. Microbiol. 6:500. doi: 10.3389/fmicb.2015.00500
Gray, M. J., Wholey, W. Y., Wagner, N. O., Cremers, C. M., Mueller-Schickert, A., Hock, N. T., et al. (2014). Polyphosphate is a primordial chaperone. Mol. Cell 53, 689–699. doi: 10.1016/j.molcel.2014.01.012
Hahn, M. W., Jezberová, J., Koll, U., Saueressig-Beck, T., and Schmidt, J. (2016). Complete ecological isolation and cryptic diversity in Polynucleobacter bacteria not resolved by 16S rRNA gene sequences. ISME J. 10, 1642–1655. doi: 10.1038/ismej.2015.237
Hahn, M. W., Koll, U., Jezberova, J., and Camacho, A. (2015). Global phylogeography of pelagic Polynucleobacter bacteria: restricted geographic distribution of subgroups, isolation by distance, and influence of climate. Environ. Microbiol. 17, 829–840. doi: 10.1111/1462-2920.12532
Hahn, M. W., Pockl, M., and Wu, Q. L. (2005). Low intraspecific diversity in a Polynucleobacter subcluster population numerically dominating bacterioplankton of a freshwater pond. Appl. Environ. Microbiol. 71, 4539–4547. doi: 10.1128/aem.71.8.4539-4547.2005
Hahn, M. W., Scheuerl, T., Jezberová, J., Koll, U., Jezbera, J., Šimek, K., et al. (2012). The passive yet successful way of planktonic life: genomic and experimental analysis of the ecology of a free-living Polynucleobacter population. PLoS One 7:e32772. doi: 10.1371/journal.pone.0032772
Hiraoka, S., Okazaki, Y., Anda, M., Toyoda, A., Nakano, S., and Iwasaki, W. (2019). Metaepigenomic analysis reveals the unexplored diversity of DNA methylation in an environmental prokaryotic community. Nat. Commun. 10:159. doi: 10.1038/s41467-018-08103-y
Hoetzinger, M., Schmidt, J., Jezberová, J., Koll, U., and Hahn, M. W. (2017). Microdiversification of a pelagic Polynucleobacter species is mainly driven by acquisition of genomic islands from a partially interspecific gene pool. Appl. Environ. Microbiol. 83:e0226-16.
Hurst, L. D. (2002). The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends. Genet. 18, 486–487. doi: 10.1016/s0168-9525(02)02722-1
Hwang, J., and Inouye, M. (2006). The tandem GTPase, Der, is essential for the biogenesis of 50S ribosomal subunits in Escherichia coli. Mol. Microbiol. 61, 1660–1672. doi: 10.1111/j.1365-2958.2006.05348.x
Ina, Y. (1996). Pattern of synonymous and nonsynonymous substitutions: an indicator of mechanisms of molecular evolution. J. Genet. 75, 91–115. doi: 10.1007/bf02931754
Jagger, J. (1983). Physiological effects of near-ultraviolet radiation on bacteria. Photochem. Photobiol. Rev. 7, 1–75. doi: 10.1007/978-1-4684-4505-3_1
Janssen, B. D., and Hayes, C. S. (2012). The tmRNA ribosome rescue system. Adv. Protein Chem. Struct. Biol. 86, 151–191. doi: 10.1016/b978-0-12-386497-0.00005-0
Jeltsch, A. (2002). Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem 3, 274–293. doi: 10.1002/1439-7633(20020402)3:4<274::aid-cbic274>3.0.co;2-s
Jezbera, J., Jezberová, J., Brandt, U., and Hahn, M. W. (2011). Ubiquity of Polynucleobacter necessarius subspecies asymbioticus results from ecological diversification. Environ. Microbiol. 13, 922–931. doi: 10.1111/j.1462-2920.2010.02396.x
Jezberová, J., Jezbera, J., Brandt, U., Lindström, E. S., Langenheder, S., and Hahn, M. W. (2010). Ubiquity of Polynucleobacter necessarius ssp. asymbioticus in lentic freshwater habitats of a heterogenous 2000 km2 area. Environ. Microbiol. 12, 658–669. doi: 10.1111/j.1462-2920.2009.02106.x
Joseph, A. P., Srinivasan, N., and de Brevern, A. G. (2012). Cis-trans peptide variations instructurally similar proteins. Amino Acids 43, 1369–1381. doi: 10.1007/s00726-011-1211-9
Katz, A., and Orellana, O. (2012). “Protein Synthesis and the Stress Response,” in Cell-Free Protein Synthesis, ed. M. Biyani (Rijeka: InTech), 111–134.
Keyer, K., and Imlay, J. A. (1996). Superoxide accelerates DNA damage by elevating free-iron levels. Proc. Natl. Acad. Sci. U.S.A. 93, 13635–13640. doi: 10.1073/pnas.93.24.13635
Khemici, V., Toesca, I., Poljak, L., Vanzo, N. F., and Carpousis, A. J. (2004). The RNase E of Escherichia coli has at least two binding sites for DEAD-box RNA helicases: functional replacement of RhlB by RhlE. Mol. Microbiol. 54, 1422–1430. doi: 10.1111/j.1365-2958.2004.04361.x
Kiss, E., Huguet, T., Poinsot, V., and Batut, J. (2004). The typA gene is required for stress adaptation as well as for symbiosis of Sinorhizobium meliloti 1021 with certain Medicago truncatula lines. Mol. Plant Microbe Interact. 17, 235–244. doi: 10.1094/MPMI.2004.17.3.235
Kobayashi, G., Moriya, S., and Wada, C. (2001). Deficiency of essential GTP binding protein ObgE in Escherichia coli inhibits chromosome partition. Mol. Microbiol. 41, 1037–1051. doi: 10.1046/j.1365-2958.2001.02574.x
Krebes, J., Morgan, R. D., Bunk, B., Spröer, C., Luong, K., Parusel, R., et al. (2013). The complex methylome of the human gastric pathogen Helicobacter pylori. Nucleic Acids Res. 42, 2415–2432. doi: 10.1093/nar/gkt1201
Krishnamurthy, K., Tewari, J. C., Irudayaraj, J., and Demirci, A. (2008). Microscopic and spectroscopic evaluation of inactivation of Staphylococcus aureus by pulsed UV light and infrared heating. Food Bioprocess Technol. 3, 93–104. doi: 10.1007/s11947-008-0084-8
Kuroda, A., Nomura, K., Ohtomo, R., Kato, J., Ikeda, T., Takiguchi, N., et al. (2001). Role of inorganic polyphosphate in promoting ribosomal protein degradation by the Lon protease in E. coli. Science 293, 705–708. doi: 10.1126/science.1061315
Lauhon, C. T. (2019). Identification and characterization of genes required for 5-hydroxyuridine synthesis in Bacillus subtilis and Escherichia coli tRNA. J. Bacteriol. 201:e00433-19.
Lee, W. C., Anton, B. P., Wang, S., Baybayan, P., Singh, S., Ashby, M., et al. (2015). The complete methylome of Helicobacter pylori UM032. BMC Genomics 16:424. doi: 10.1186/s12864-015-1585-2
Lluch-Senar, M., Luong, K., Lloréns-Rico, V., Delgado, J., Fang, G., Spittle, K., et al. (2013). Comprehensive methylome characterization of Mycoplasma genitalium and Mycoplasma pneumoniae at single-base resolution. PLoS Genet. 9:e1003191. doi: 10.1371/journal.pgen.1003191
Martin, E. M., and Fry, R. C. (2018). Environmental influences on the epigenome: exposure-associated DNA methylation in human populations. Annu. Rev. Public Health 39, 309–333. doi: 10.1146/annurev-publhealth-040617-014629
Meincke, L., Copeland, A., Lapidus, A., Lucas, S., Berry, K. W., Del Rio, T. G., et al. (2012). Complete genome sequence of Polynucleobacter necessarius subsp. asymbioticus type strain (QLW-P1DMWA-1(T)). Stand. Genomic Sci. 6, 74–83. doi: 10.4056/sigs.2395367
Michalke, H., and Bremer, H. (1969). RNA synthesis in Escherichia coli after irradiation with ultraviolet light. J. Mol. Biol. 41, 1–23. doi: 10.1016/0022-2836(69)90122-3
Mishra, S., and Imlay, J. (2012). Why do bacteria use so many enzymes to scavenge hydrogen peroxide? Arch. Biochem. Biophys. 525, 145–160. doi: 10.1016/j.abb.2012.04.014
Murray, I. A., Clark, T. A., Morgan, R. D., Boitano, M., Anton, B. P., Luong, K., et al. (2012). The methylomes of six bacteria. Nucleic Acids Res. 40, 11450–11462. doi: 10.1093/nar/gks891
Murray, I. A., Morgan, R. D., Luyten, Y., Fomenkov, A., Corrêa, I. R. Jr., Dai, N., et al. (2018). The non-specific adenine DNA methyltransferase M.EcoGII. Nucleic Acids Res. 46, 840–848.
Murugaiyan, J., Eravci, M., Weise, C., and Roesler, U. (2016). Label-free quantitative proteomic analysis of harmless and pathogenic strains of infectious microalgae, Prototheca spp. Int. J. Mol. Sci. 18:59. doi: 10.3390/ijms18010059
Neuenschwander, S. M., Ghai, R., Pernthaler, J., and Salcher, M. M. (2017). Microdiversification in genome-streamlined ubiquitous freshwater Actinobacteria. ISME J. 12, 185–198. doi: 10.1038/ismej.2017.156
Newton, R. J., Jones, S. E., Helmus, M. R., and McMahon, K. D. (2007). Phylogenetic ecology of the freshwater Actinobacteria acI lineage. Appl. Environ. Microbiol. 73, 7169–7176. doi: 10.1128/aem.00794-07
Nikel, P. I., Chavarria, M., Martinez-Garcia, E., Taylor, A. C., and de Lorenzo, V. (2013). Accumulation of inorganic polyphosphate enables stress endurance and catalytic vigour in Pseudomonas putida KT2440. Microb. Cell Fact. 12, 50–64. doi: 10.1186/1475-2859-12-50
Owttrim, G. W. (2013). RNA helicases: diverse roles in prokaryotic response to abiotic stress. RNA Biol. 10, 96–110. doi: 10.4161/rna.22638
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P. A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Perez-Riverol, Y., Csordas, A., Bai, J., Bernal-Llinares, M., Hewapathirana, S., Kundu, D. J., et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450. doi: 10.1093/nar/gky1106
Pfennig, P., and Flower, A. (2001). BipA is required for growth of Escherichia coli K12 at low temperature. Mol. Genet. Genomics 266, 313–317. doi: 10.1007/s004380100559
Proctor, C. R., Besmer, M. D., Langenegger, T., Beck, K., Walser, J.-C., Ackermann, M., et al. (2018). Phylogenetic clustering of small low nucleic acid-content bacteria across diverse freshwater ecosystems. ISME J. 12, 1344–1359. doi: 10.1038/s41396-018-0070-8
Qin, Y., Polacek, N., Vesper, O., Staub, E., Einfeldt, E., Wilson, D. N., et al. (2006). The highly conserved LepA is a ribosomal elongation factor that back-translocates the ribosome. Cell 127, 721–733. doi: 10.1016/j.cell.2006.09.037
Rastogi, R. P., Sinha, R. P., Moh, S. H., Lee, T. K., Kottuparambil, S., Kim, Y. J., et al. (2014). Ultraviolet radiation and cyanobacteria. J. Photochem. Photobiol. B 141, 154–169.
Richards, J., Sundermeier, T., Svetlanov, A., and Karzai, A. W. (2008). Quality control of bacterial mRNA decoding and decay. Biochim. Biophys. Acta 1779, 574–582. doi: 10.1016/j.bbagrm.2008.02.008
Rodrigues, D. F., Ivanova, N., He, Z., Huebner, M., Zhou, J., and Tiedje, J. M. (2008). Architecture of thermal adaptation in an Exiguobacterium sibiricum strain isolated from 3 million year old permafrost: a genome and transcriptome approach. BMC Genomics 9:547. doi: 10.1186/1471-2164-9-547
Rogers, R. L., and Slatkin, M. (2017). Excess of genomic defects in a woolly mammoth on Wrangel island. PLoS Genet. 13:e1006601. doi: 10.1371/journal.pgen.1006601
Ronin, I., Katsowich, N., Rosenshine, I., Balaban, N. Q., Heitmann, V., Niemann, S., et al. (2017). A long-term epigenetic memory switch controls bacterial virulence bimodality. eLife 6:e19599. doi: 10.7554/eLife.19599
Röttiga, A., and Steinbüchel, A. (2013). Acyltransferases in bacteria. Microbiol. Mol. Biol. Rev. 77, 277–321. doi: 10.1128/mmbr.00010-13
Salcher, M. M., Pernthaler, J., and Posch, T. (2011). Seasonal bloom dynamics and ecophysiology of the freshwater sister clade of SAR11 bacteria ‘that rule the waves’ (LD12). ISME J. 5, 1242–1252. doi: 10.1038/ismej.2011.8
Salman-Dilgimen, A., Hardy, P. O., Radolf, J. D., Caimano, M. J., and Chaconas, G. (2013). HrpA, an RNA helicase involved in RNA processing, is required for mouse infectivity and tick transmission of the Lyme disease spirochete. PLoS Pathog. 9:e1003841. doi: 10.1371/journal.ppat.1003841
Sánchez-Quinto, F., and Lalueza-Fox, C. (2015). Almost 20 years of Neanderthal palaeogenetics: adaptation, admixture, diversity, demography and extinction. Phil. Trans. R. Soc. Lond. B Biol. Sci. 370:20130374. doi: 10.1098/rstb.2013.0374
Sánchez-Romero, M. A., Cota, I., and Casadesús, J. (2015). DNA methylation in bacteria: from the methyl group to the methylome. Curr. Opin. Microbiol. 25, 9–16. doi: 10.1016/j.mib.2015.03.004
SantaLucia, J. Jr. (1998). A unified view of polymer, dumbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. U.S.A. 95, 1460–1465. doi: 10.1073/pnas.95.4.1460
Shaw, P. E. (2002). Peptidyl-prolyl isomerases: a new twist to transcription. EMBO Rep. 3, 521–526. doi: 10.1093/embo-reports/kvf118
Slade, D., and Radman, M. (2011). Oxidative stress resistance in Deinococcus radiodurans. Microbiol. Mol. Biol. Rev. 75, 133–191. doi: 10.1128/mmbr.00015-10
Smirnova, G. V., Zakirova, O. N., and Oktyabrskii, O. N. (2001). The role of antioxidant systems in the cold stress response of Escherichia coli. Microbiology 70, 45–50.
Spalding, M. D., and Prigge, S. T. (2010). Lipoic acid metabolism in microbial pathogens. Microbiol. Mol. Biol. Rev. 74, 200–228. doi: 10.1128/mmbr.00008-10
Sun, Z., and Blanchard, J. L. (2014). Strong genome-wide selection early in the evolution of Prochlorococcus resulted in a reduced genome through the loss of a large number of small effect genes. PLoS One 9:e88837. doi: 10.1371/journal.pone.0088837
Suzuki, M. M., and Bird, A. (2008). DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 9, 465–476. doi: 10.1038/nrg2341
Tellier, A., Fischer, I., Merino, C., Xia, H., Camus-Kulandaivelu, L., Städler, T., et al. (2011). Fitness effects of derived deleterious mutations in four closely related wild tomato species with spatial structure. Heredity 107, 189–199. doi: 10.1038/hdy.2010.175
Tsilibaris, V., Maenhaut-Michel, G., and Van Melderen, L. (2006). Biological roles of the Lon ATP-dependent protease. Res. Microbiol. 157, 701–713. doi: 10.1016/j.resmic.2006.05.004
Udali, S., Guarini, P., Ruzzenente, A., Ferrarini, A., Guglielmi, A., Lotto, V., et al. (2015). DNA methylation and gene expression profiles show novel regulatory pathways in hepatocellular carcinoma. Clin. Epigenetics 7:43. doi: 10.1186/s13148-015-0077-1
Verstraeten, N., Fauvart, M., Versées, W., and Michiels, J. (2011). The universally conserved prokaryotic GTPases. Microbiol. Mol. Biol. Rev. 75, 507–542. doi: 10.1128/mmbr.00009-11
Vickery, L. E., and Cupp-Vickery, J. R. (2007). Molecular chaperones HscA/Ssq1 and HscB/Jac1 and their roles in iron-sulfur protein maturation. Crit. Rev. Biochem. Mol. Biol. 42, 95–111. doi: 10.1080/10409230701322298
Wang, D., Zhang, Y., Zhang, Z., Zhu, J., and Yu, J. (2010). KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics 8, 77–80. doi: 10.1016/s1672-0229(10)60008-3
Wang, F., Redding, S., Finkelstein, I. J., Gorman, J., Reichman, D. R., and Greene, E. C. (2013). The promoter-search mechanism of Escherichia coli RNA polymerase is dominated by three-dimensional diffusion. Nat. Struct. Mol. Biol. 20, 174–181. doi: 10.1038/nsmb.2472
Willenbrock, H., and Ussery, D. W. (2004). Chromatin architecture and gene expression in Escherichia coli. Genome Biol. 5:252. doi: 10.1186/gb-2004-5-12-252
Wion, D., and Casadesus, J. (2006). N6-methyl-adenine: an epigenetic signal for DNA-protein interactions. Nat. Rev. Microbiol. 4, 183–192. doi: 10.1038/nrmicro1350
Yan, X., Dong, X., Liu, L., Yang, Y., Lai, J., and Guo, Y. (2016). DNA methylation signature of intergenic region involves in nucleosome remodeler DDM1-mediated repression of aberrant gene transcriptional read-through. J. Genet. Genomics 43, 513–523. doi: 10.1016/j.jgg.2016.03.010
Keywords: DNA modification, gene expression, freshwater heterotrophic bacteria, UV radiation, purifying selection
Citation: Srivastava A, Murugaiyan J, Garcia JAL, De Corte D, Hoetzinger M, Eravci M, Weise C, Kumar Y, Roesler U, Hahn MW and Grossart H-P (2020) Combined Methylome, Transcriptome and Proteome Analyses Document Rapid Acclimatization of a Bacterium to Environmental Changes. Front. Microbiol. 11:544785. doi: 10.3389/fmicb.2020.544785
Received: 22 March 2020; Accepted: 18 August 2020;
Published: 15 September 2020.
Edited by:
Wade H. Jeffrey, University of West Florida, United StatesReviewed by:
Aindrila Mukhopadhyay, Lawrence Berkeley National Laboratory, United StatesBaptiste Leroy, University of Mons, Belgium
Copyright © 2020 Srivastava, Murugaiyan, Garcia, De Corte, Hoetzinger, Eravci, Weise, Kumar, Roesler, Hahn and Grossart. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hans-Peter Grossart, aGdyb3NzYXJ0QGlnYi1iZXJsaW4uZGU=