 Sourav Dutta
Sourav Dutta Nilanjana Das
Nilanjana Das Piyali Mukherjee
Piyali Mukherjee
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Microbiol. , 31 August 2020
Sec. Virology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.01990
This article is part of the Research Topic Cell Organelle Exploitation by Viruses During Infection View all 10 articles
As the world faces the challenge of the COVID-19 pandemic, it has become an urgent need of the hour to understand how our immune system sense and respond to RNA viruses that are often life-threatening. While most vaccine strategies for these viruses are developed around a programmed antibody response, relatively less attention is paid to our innate immune defenses that can determine the outcome of a viral infection via the production of antiviral cytokines like Type I Interferons. However, it is becoming increasingly evident that the “cytokine storm” induced by aberrant activation of the innate immune response against a viral pathogen may sometimes offer replicative advantage to the virus thus promoting disease pathogenesis. Thus, it is important to fine tune the responses of the innate immune network that can be achieved via a deeper insight into the candidate molecules involved. Several pattern recognition receptors (PRRs) like the Toll like receptors (TLRs), NOD-like receptors (NLRs), and the retinoic acid inducible gene-I (RIG-I) like receptors (RLRs) recognize cytosolic RNA viruses and mount an antiviral immune response. RLRs recognize invasive viral RNA produced during infection and mediate the induction of Type I Interferons via the mitochondrial antiviral signaling (MAVS) molecule. It is an intriguing fact that the mitochondrion, one of the cell’s most vital organelle, has evolved to be a central hub in this antiviral defense. However, cytokine responses and interferon signaling via MAVS signalosome at the mitochondria must be tightly regulated to prevent overactivation of the immune responses. This review focuses on our current understanding of the innate immune sensing of the host mitochondria by the RLR-MAVS signalosome and its specificity against some of the emerging/re-emerging RNA viruses like Ebola, Zika, Influenza A virus (IAV), and severe acute respiratory syndrome-coronavirus (SARS-CoV) that may expand our understanding for novel pharmaceutical development.
Mitochondrion, also known as the “powerhouse” of the cell, is critically involved in cellular respiration and ATP synthesis. Apart from its canonical role in cellular energetics, it orchestrates cell fate through the process of apoptosis and mitophagy, thus maintaining cellular homeostasis (Tsujimoto and Shimizu, 2007; Murphy, 2009; Friedman and Nunnari, 2014; Mishra and Chan, 2014; Khan et al., 2015; Sliter et al., 2018). In recent years, several studies have pinpointed the crucial role of mitochondria in stimulating innate immune responses, as well as modulating parts of the adaptive immune response (Walker et al., 2014; Weinberg et al., 2015; Mills et al., 2017). The evolutionary conserved pattern recognition receptors (PRRs), expressed by most immune effector cells recognize conserved sequence within the pathogen and aids in their early detection and containment (Green et al., 2016). The Toll like receptors (TLRs) are a class of PRRs that recognize either dsRNA (TLR3) or ssRNA (TLR7/8) virus (Lester and Li, 2014; Hartmann, 2017; Miyake et al., 2018). The NOD-like receptor (NLR) family of PRRs is cytoplasmic receptors that form a multiprotein complex called “inflammasome” involved in the production of the pro-inflammatory cytokines IL1β and IL18 (He et al., 2016; Hughes and O’Neill, 2018). Another class of PRRs, the RIG-I like receptor (RLR) family involving retinoic acid inducible gene-I (RIG-I), melanoma differentiation-associated protein-5 (MDA-5), and laboratory of genetics and physiology 2 (LGP2) are cytoplasmic sensors of non-self and viral RNA (Vazquez and Horner, 2015; Sadler, 2017; Chow et al., 2018). A few of these receptors have been shown to augment mitochondria mediated antiviral innate immune responses via stimulation of Type I Interferon. Evolutionary conserved signaling intermediate in Toll (ECSIT) pathway, a component of the mitochondrial complex I, has been shown to enhance TLR7 responses via the mitochondrial adaptor protein tumor necrosis factor receptor (TNFR) associated factor 6 (TRAF6; Carneiro et al., 2018). NLRP3 has been shown to form the active inflammasome complex at the mitochondria by associating with the adaptor protein mitochondrial antiviral signaling protein (MAVS; Dorn, 2012; Haneklaus and O’Neill, 2015; Yabal et al., 2019). However, of special interest is the first identified RLR, RIG-I, which recognizes viral RNA that has a triphosphate moiety at its 5' end and has been shown to be targeted by some of the deadliest form of the RNA viruses (Kell and Gale, 2015; Dai et al., 2018). Following viral recognition, RIG-I binds to MAVS located on the outer surface of healthy intact mitochondria leading to interferon production and activation of the NFκB pathway (Kawai and Akira, 2007; Okamoto et al., 2018). This review speculates whether subversion of early viral sensing via the RIG-I/MAVS pathway could determine viral persistence within the host. Further, aberrant activation of the MAVS signalosome by the RLRs could cause hyperstimulation of the inflammatory responses and hence this arm of the innate immune defense could serve as a potential therapeutic target to combat highly communicable infectious RNA viruses.
The “Flu pandemic” over the last century has drawn particular attention to enveloped RNA viruses, a characteristic feature that empowers the virus with greater adaptability and high mutagenic potential, a key strategy in the evasion of host immune response and increased survivability within the host. Here, we systematically review our current understanding of the conserved host RIG-I/MAVS pathway and its regulation in some of the emerging/re-emerging RNA virus infections that include Ebola virus (EBOV) belonging to Filoviridae family, Zika virus (ZIKV) belonging to Flaviviridae family, Influenza A virus (IAV) belonging to Orthomyxoviridae family, and severe acute respiratory syndrome-coronavirus (SARS-CoV) belonging to Coronaviridae family. These viruses have been known to cause deadly outbreaks across the world and it is important to analyze whether key sensors of RNA viruses like the RIG-I/MAVS pathway are important targets of these viruses either to suppress of hyper-activate the immune responses.
Mitochondria play an important role in antiviral immunity by eliciting and maintaining the RLR/MAVS signaling cascade. RLRs are soluble RNA helicase type receptors containing N-terminal tandem of caspase activation and recruitment domains (CARDs) and a DECH-helicase domain required for RNA binding and ATP hydrolysis (Kao et al., 2015; Brisse and Ly, 2019). All the three known RLRs (i.e., RIG-I, MDA-5, and LGP-2) are very efficient in distinguishing between cellular RNAs from those produced by RNA viruses (Züst et al., 2011). Upon recognition of viral RNA, one of the widely studied RLRs, RIG-I, binds to the downstream adaptor protein MAVS (also known as IPS-1, VISA, Cardif) at the mitochondria via CARD-CARD interaction (Liu et al., 2017). MAVS is an integral protein of the mitochondrial outer membrane that binds to the mitochondrial membrane via its C-terminal domain and acts as a key determinant of the antiviral signaling cascade (Xu et al., 2014). Following its interaction with RIG-I, MAVS bind with several kinases and other signaling molecules including TRAF3 and 6, TNFR associated death domain (TRADD), and TRAF associated NF-ĸB activator (TANK1) to form a large multimeric complex called the “MAVS signalosome” (Biacchesi et al., 2009; Vazquez and Horner, 2015). This structure ultimately leads to the activation of the interferon regulatory factor 3 (IRF3) and phosphorylation of IKKε to stimulate the NF-ĸB pathway leading to transcriptional activation of Type I Interferons and other inflammatory cytokines (Pothlichet et al., 2013; Refolo et al., 2020). Interferons in turn stimulate a plethora of interferon stimulated genes (ISGs) that aid in the containment of the viruses as well crosstalk with the adaptive immune response. Thus mitochondrial targeting via the MAVS signalosome by the viral proteins upon their entry appears to be a central executioner of antiviral responses as summarized in Table 1. In a continuous war with the host, viruses have evolved strategies to avoid MAVS mediated innate immune activation. For example, MAVS is expressed only on the surface of intact mitochondria and several studies suggest that RNA viruses alter mitochondrial metabolism and homeostasis that ultimately lead to mitochondrial damage and blocking interferon response via MAVS (Lei et al., 2009; Zhao et al., 2012; Wang et al., 2013; Choi et al., 2017; He et al., 2019). Over centuries, it has been found that enveloped RNA virus causes persistent human infections like the current COVID-19 pandemic (Schoeman and Fielding, 2019). Whether the viral envelope provides additional arsenal to the RNA viruses in the suppression of the protective interferon response via the MAVS signalosome is not known yet.
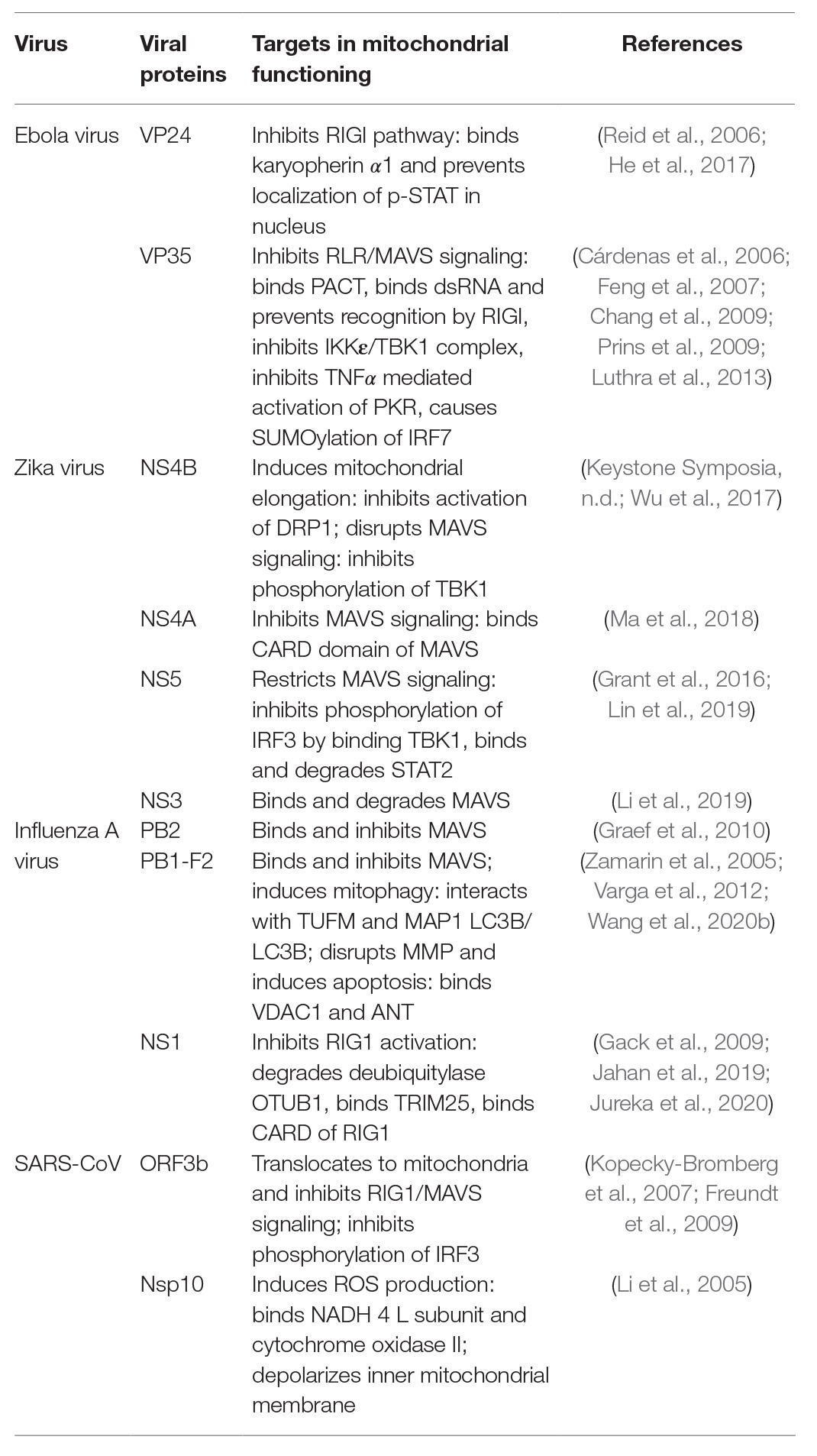
Table 1. Summary of viral proteins and their targets in mitochondria mediated antiviral response.
Following viral infection, our cellular defense machinery systematically induces a number of cytokines (both pro‐ and anti-inflammatory) that, in certain instances, may lead to hyperstimulation of the immune response in a positive feedback loop (Geoghegan et al., 2016; Shrivastava et al., 2016; Orzalli and Kagan, 2017). This leads to a catastrophic damage to the surrounding cells and the side effects of this manifests itself in some of the symptoms like fever, fatigue, nausea along with multiple organ failure (Chen et al., 2020; Hackbart et al., 2020). This has been observed not only in COVID-19 patients but also in case of other strains of the Flu virus, the MERS-CoV, and SARS-CoV1 leading to severe respiratory distress and increased mortality rates (DeDiego et al., 2014; Nieto-Torres et al., 2015; Liang et al., 2020). Hence, the question automatically arises is whether mitochondria can fine tune this response to prevent such overreaction of the immune cells.
Since mitochondria provide the first line of defense against viral infection, signals converging at the mitochondria need to be tightly regulated to prevent bystander tissue damage within the host. One such checkpoint is provided by the NLR, NLRX1 which prevents overactivation of the immune response by its direct competition with RIG-I at its MAVS binding site and antagonizing Type I Interferon responses (Allen et al., 2012; Qin et al., 2017). Further, ubiquitination plays an important immunomodulatory role in the MAVS-signalosome (Gack et al., 2007, 2009). The ubiquitin ligase, tripartite motif containing-25 (TRIM-25), mediates Lys63 polyubiquitination of RIG-I thus, enabling its binding with MAVS for antiviral signaling. It has been shown that TRIM-25 also ubiquitinates MAVS at Lys7 and Lys10 inducing its proteolysis and dissociating it from RIG-I to halt the antiviral signaling cascade (Castanier et al., 2012). Mitophagy induction by reactive oxygen species is another strategy for MAVS degradation at the damaged mitochondria which is sometime adopted by certain viruses to dampen the host immune response (Zhang et al., 2018; He et al., 2019). These observations suggest that stimulators of the MAVS signalosome must work in concert with the negative regulators to strike a balance between activation and deactivation in a timely manner and have been summarized in Figure 1. However, extensive studies are required to find candidate molecules that may act to dampen the overzealous immune activation following an initial protective response via mitochondrial sensing (D’Elia et al., 2013; Cusabio, 2020).
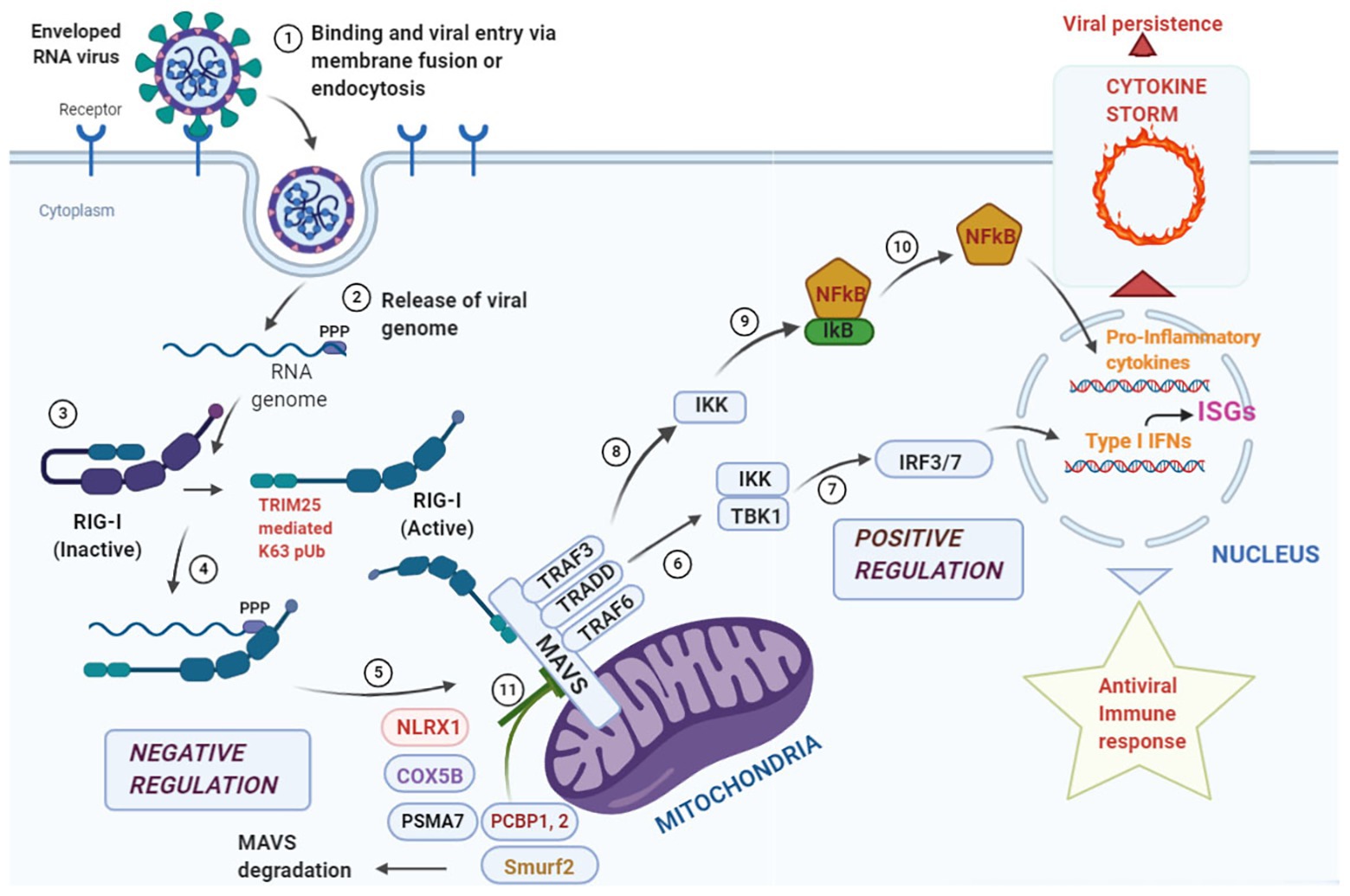
Figure 1. Overview of antiviral response at the mitochondrial antiviral signaling (MAVS) signalosome. Enveloped RNA virus can enter the cell via membrane fusion or endocytosis (Step 1). Following entry, the “ppp” group at the 5' end of the viral genome is recognized by the core domain of the retinoic acid inducible gene-I (RIG-I) like receptor (RLR) helicase RIG-I (Steps 2, 3, and 4). The activated RIG-I molecule binds to MAVS via caspase activation and recruitment domain (CARD)-CARD interaction at the N-terminal domain of MAVS leading to the formation of the “MAVS Signalosome” (Step 5). Two different signals can emanate from this signalosome, the first one leads to the activation of interferon regulatory factor 3/7 (IRF3/7), transcriptional activation of Type I Interferons and stimulation of interferon stimulated genes (ISGs; Steps 6 and 7) leading to a protective antiviral immunity. The second signal phosphorylates IκB via IKK and releases NFκB that translocates to the nucleus and results in the transcriptional activation of pro-inflammatory cytokines (Steps 8, 9, and 10). Events at this step needs to be finely tuned as hyperstimulation of the responses may lead to a “cytokine storm” that aids in viral persistence within the infected cells. To fine tune the hyperstimualtion of the MAVS-signalosome NLRX1 antagonizes IFN signaling by binding to MAVS that may act as a brake on the hyperactive immune response. COX 5B suppress ROS production via suppressing MAVS aggregation and the proteasomal subunit PSMA-7 inhibits MAVS by enhancing its proteasomal degradation. Further, PCBP1, 2 and Smurf2 similarly degrade MAVS via K48 linked polyubiquitination and inhibit Type I Interferon production (Step 11).
The EBOV that causes Ebola Virus Disease (EVD) is an emerging pathogen and has an almost 90% mortality rate. Although it is mostly endemic to West Africa with the Democratic Republic of Congo (DRC) being the hardest hit region over the past decade (2013–2019), it remains a major health concern worldwide due to its high potential to infect other species and the unavailability of a viable therapy till date (Kaner and Schaack, 2016; Chowell et al., 2019; Ebola virus disease, n.d.). EBOV is a non-segmented enveloped (−) single-stranded RNA virus that initially infects the innate immune cells such as macrophages (Structure of Ebola Virus, n.d.). However, the virus has the remarkable ability to infect a wide variety of cells that enables its rapid spread to different tissues. EBOV infection is characterized by hemorrhagic fever accompanied by massive cytokine storm, cytolytic damage, vascular leakage in liver, lungs, and kidneys, and ultimately death (Yu et al., 2012; Falasca et al., 2015).
The role of the viral sensor RIG-I and the subsequent activation of the MAVS pathway in determining the outcome of EBOV infection has not been thoroughly investigated. A study on mouse adapted EBOV (MA-EBOV) infection demonstrated that IFN-dependent and independent MAVS signaling takes place in an organ specific manner, where activation of monocytes and subsequent trafficking to the spleen occurs in a MAVS-dependent manner (Green et al., 2016; Dutta et al., 2017). EBOV mitigates the host immune response by using two viral IFN-antagonists, VP24 and VP35. VP35 has been shown to suppress the IFN-pathway by antagonizing the function of interferon regulatory factor (IRF) activating kinases IKKε and TANK binding kinase-1 (TBK-1; Messaoudi et al., 2015). Further, VP35 has been shown to inhibit RIG-activation via binding to transiently produced dsRNA during EBOV infection, thus preventing viral sensing and also by binding to the RIG-I ATPase activator PACT (Luthra et al., 2013). In an in vitro study, VP24 has been shown to prevent IFN-gene expression by targeting the RIG-I/MAVS pathway. It works downstream of the RIG-I/MAVS pathway by binding karyopherin 𝛼1 and inhibiting p-STAT translocation (Reid et al., 2006). Further studies are required to understand how EBOV suppress early innate immune sensing to develop antiviral strategies.
Zika virus (ZIKV) is a re-emerging mosquito borne pathogen belonging to the genus Flavivirus. However, apart from mosquito-transmission, several other modes of ZIKV infection have been reported and the most striking is the mother to fetus transmission via the transplacental route (Plourde and Bloch, 2016). Although the first document of human infection by ZIKV occurred in 1954 and was associated with mild flu-like symptoms, a recent epidemic in French Polynesia during 2013–2014 that subsequently spread to South and Central America caught the world’s attention with rising symptoms of microcephaly in newborns (Song et al., 2017; Haby et al., 2018). The re-emergence and the rising cases of ZIKV infection with higher infectivity are poorly understood and no clinically approved drug or vaccine is available till date.
ZIKV is a non-segmented enveloped (+) single stranded RNA virus that has shown to co-evolve with the host and strongly antagonizes the host antiviral IFN-responses. Several non-structural proteins of ZIKV like NS1, NS2B/3, NS4A, NS4B, and NS5 have been shown to antagonize IFN-responses (Wu et al., 2017; Ding et al., 2018; Zheng et al., 2018; Lundberg et al., 2019; Zhao et al., 2019). Studies have shown a direct interaction of the ZIKV NS4 with the N-terminal CARD domain of MAVS at the mitochondria (Ma et al., 2018). This prevents binding of RIG-I to MAVS and downstream activation of the interferon responses. It has been further shown that ZIKV NS4 specifically inhibits RIG-I mediated interferon responses and not that mediated by TLRs (Ma et al., 2018; Hu et al., 2019; Schilling et al., 2020). It is also known to disrupt mitochondrial dynamics which aids in infection (Keystone Symposia, n.d.). Further, the non-structural protein NS3 have been shown to target MAVs to proteasomal degradation via K48 linked polyubiquitination and subsequent downregulation of IFNβ pathway (Li et al., 2019). Further, these responses vary among the different ZIKV strains isolated from different geographical locations. ZIKV strains from Brazil and Uganda showed delayed activation of the innate immune responses mediated by RIG-I as compared to the milder Cambodia strain that correlates with their pathological outcomes (Esser-Nobis et al., 2019). Studies using these different strains in lung A549 cells revealed the important role of RIG-I sensing in early innate immune response and induction of Type I Interferon responses (Strottmann et al., 2019). However, the role of this pathway in the host tropism of different isolates of ZIKV is yet to be fully uncovered that may provide a deeper insight into the importance of early viral sensing and productive IFN-response via the MAVS signalosome in ZIKV clearance.
Influenza A virus (IAV) is one of the four types of influenza virus and the only influenza virus sub-type that has been known to cause global pandemic. Based on the presence of two surface proteins, hemagglutinin (HA) and neuraminidase (N), IAV can be sub-categorized into different strains (Bouvier and Palese, 2008). The 1918 Spanish Flu and the pandemic of 2009 were associated with the H1N1 subtype of IAV. H1N1 mostly affect children and young and middle-aged adult contrary to other flu where it affects mostly the older people.
The Influenza virus contains eight segmented (−) single stranded RNA and affects the upper respiratory tract epithelial cells causing the “seasonal” flu or fatal pulmonary disorder in extreme conditions (Shao et al., 2017). The polymeric basic 2 (PB2) subunit of the RNA polymerase complex is a major pathogenic determinant of seasonal IAV (Liu et al., 2019). Further, an intact mitochondrial membrane potential (MMP) is required for MAVS-mediated interferon production and PB2 might indirectly affect MAVS function by altering MMP (Varga et al., 2012). PB2 protein of pdm/09 variant of IAV carrying T5881 mutation has been shown to suppress MAVS-mediated interferon signaling more robustly that could potentially contribute to its increased pathogenicity. It has been demonstrated that PB2 is imported into the mitochondrial matrix and associates with MAVS at the mitochondria that correlates with reduced IFNβ production in vitro (Graef et al., 2010; Long and Fodor, 2016). Besides PB2, other proteins of IAV like the non-structural protein 1 (NS1) can block the RIG-I mediated induction of IFNβ by inhibiting TRIM25 (Gack et al., 2009). Inflammasome formation, a complex of NLRP3/ASC/Caspase-1, that is required for the production the inflammatory cytokine IL1β, is triggered by IFNβ in a positive feedback loop in primary lung epithelial cells and was shown to be mediated by RIG-I via its interaction with MAVS/TRIM2/Riplet (Mibayashi et al., 2007; Pothlichet et al., 2013). TRIM25 and Riplet positively regulates the antiviral responses mediated by RIG-I and the NS1 protein of highly pathogenic 1918 virus binds to RIG-I and TRIM25 to antagonize IFNβ activation (Gack et al., 2009; Koliopoulos et al., 2018). This correlated with the reduced induction of both Type I Interferon, as well as IL1β production by NS1 in IAV infected ferrets. Further RNAse L, a ubiquitous endonuclease for single stranded RNA, enhances NLRP3 activation and complex formation with the DExD/H-box helicase, DHX33, and MAVS in bone marrow derived dendritic cells and THP-1 derived macrophages (Chakrabarti et al., 2015). However, this antiviral response appears to be a double-edged sword as heightened inflammation and production of pro-inflammatory cytokines is often associated with increased morbidity following IAV infection. One of the NLRs, NLRX1 was shown to inhibit the production of antiviral cytokines and reduce lung pathology in IAV virus infected mice via its direct interaction with the RIG-I/MAVS pathway (Allen et al., 2011). Thus, NLRX1 at the mitochondria could provide a brake on the cytokine storm induced by IAV that has often been associated with the higher mortality rates during influenza virus pandemic.
Coronavirus is emerging pathogens that has serious life-threatening impact on human health. Severe acute respiratory syndrome-related coronavirus (SARS-CoV1) caused a major outbreak of respiratory disease in 2002–2004 (SARS | Home | Severe Acute Respiratory Syndrome | SARS-CoV Disease | CDC, n.d.). The current pandemic which has bypassed the death rate of all previous pandemic of the last century is caused by a novel subtype of SARS-CoV and has been named SARS-CoV2 causing corona virus disease 19 (COVID-19). Coming from zoonotic reservoir, SARS-CoV shows extreme adaptivity through species jump and is a major health concern worldwide with the probability of new strains with heightened pathogenic potential emerging every year. SARS-CoV is an enveloped (+) single stranded RNA virus and in human host, it mainly infects the ciliated epithelium and alveolar type II pneumocytes (de Wit et al., 2016). Being mostly asymptomatic in the early stages of infection with flu like symptoms, it can quickly escalate to acute respiratory distress syndrome (ARDS) and multiorgan failure (Cameron et al., 2008; Yin and Wunderink, 2018). A similar manifestation has also been observed in COVID-19 patients, where cytokine storm has been shown to result in ARDS-like symptoms. SARS-CoV-2 can efficiently alter the cytokine profile by promoting the production of pro-inflammatory genes and blocking the stimulation of interferon genes based on their mode of infectivity (i.e., severe or non-severe form of SARS; Mahmudpour et al., 2020; Ratajczak and Kucia, 2020; Wang et al., 2020a; Yap et al., 2020). An arsenal of viral proteins is dedicated for this process and the host mitochondria play a pivotal role in the early response to infection (Maier et al., 2015). SARS-CoV-1 proteins, ORF-3b and nsp-10, show direct mitochondrial association where ORF-3b co-localizes with mitochondria specific markers and nsp-10 specifically interacts with NADH 4 L subunit and cytochrome c oxidase that affects mitochondrial function (Li et al., 2005; Yuan et al., 2006). It can also inhibit the MAVS downstream signaling by directly binding to STAT1 and inhibiting the TBK1/IKK𝛆 signaling. Further, the SARS-CoV-1 envelope protein has been shown to activate inflammasome formation and stimulate the production of pro-inflammatory cytokines like IL6 and TNF which makes it an attractive target for future studies (Nieto-Torres et al., 2014). Hence, studies on SARS-CoV-1 points toward the relevance of mitochondria mediated innate immune signaling pathway that may be further extrapolated to SARS-CoV-2 infection (Singh et al., 2020). No study has reported the role of the mitochondrial innate immune sensing in COVID-19 pathogenesis that may provide effective strategies to limit viral replication within the host and the generation of a protective adaptive immune response.
RNA viruses have become important etiological agents of emerging pathogens in humans constituting a major percentage of all human emerging diseases including those induced by bacteria or parasites. The past decade has seen several cases of pandemics arising due to RNA viruses originating from wild life reservoirs like the Ebola, H1N1 influenza, SARS, and MERS and the recent COVID-19 pandemic. The RNA polymerases of these viruses often lack proofreading activity increasing their mutation rates during the replicative stage of the virus. This comes as a severe challenge in developing vaccine strategies and it is important to understand conserved host immune responses which may help combat a wide range of these RNA viruses.
The innate immune response, which provides the first line of defense against these RNA viruses via the production of Type I Interferon, is often targeted by the viruses for the successful establishment of an infection. However, priming of IFN-responses prior to an infection can be a double edged sword as cytokine storm following hyper-stimulation of the immune responses and the over production of pro-inflammatory cytokines have been shown to be associated with diseases like Ebola, Influenza, and COVID-19 (D’Elia et al., 2013; Infection-cusabio and Topics, 2020) and the mitochondria may act as a central hub in modulating these responses. MAVS dependent pathway at the mitochondria act as a critical factor for limiting virus infection and a detailed understanding of its regulation can help fine tune the host immune responses toward a productive antiviral strategy. Several molecules like NLRX1 and DUBs regulate RIG-I binding to MAVS at the mitochondria or directly target MAVS for degradation, thus acting as a counterbalance to prevent overproduction of Type I Interferons during a persistent viral infection. Further, development of agonists for the RIG-I/MAVS pathways can be used synergistically with antiviral compounds to restrict the replication of viruses at the initial stage and offer prophylactic solution to prevent such deadly outbreaks and rapid spread of RNA-virus induced infection.
PM conceived the work. SD, ND, and PM wrote the manuscript. PM prepared the figures and revised the entire manuscript. All authors contributed to the article and approved the submitted version.
The work was funded by the extramural funding from Dept. of Biotechnology, India (#BT/PR10983/BRB/10/1282/2014) to PM. SD is a recipient of CSIR-NET Fellowship, Govt. of India (File No: 08/155(0085)/2020-EMR-I).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Dr. Amit Sarkar and members of the lab for their insightful comments during the preparation of the manuscript.
Allen, I. C., Moore, C. B., Schneider, M., Lei, Y., Davis, B. K., Scull, M. A., et al. (2011). NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-κB signaling pathways. Immunity 34, 854–865. doi: 10.1016/j.immuni.2011.03.026
Allen, I. C., Moore, C. B., Schneider, M., Lei, Y., Davis, B. K., Scull, A., et al. (2012). NLRX1 protein attenuates inflammatory responses to virus infection by interfering with the RIG-I-MAVS signaling pathway and TRAF6 ubiquitin ligase. Immunity 34, 854–865. doi: 10.1016/j.immuni.2011.03.026.NLRX1
Biacchesi, S., LeBerre, M., Lamoureux, A., Louise, Y., Lauret, E., Boudinot, P., et al. (2009). Mitochondrial antiviral signaling protein plays a major role in induction of the fish innate immune response against RNA and DNA viruses. J. Virol. 83, 7815–7827. doi: 10.1128/JVI.00404-09
Bouvier, N. M., and Palese, P. (2008). The biology of influenza viruses. Vaccine 26, D49–D53. doi: 10.1016/j.vaccine.2008.07.039
Brisse, M., and Ly, H. (2019). Comparative structure and function analysis of the RIG-I-like receptors: RIG-I and MDA5. Front. Immunol. 10:1586. doi: 10.3389/fimmu.2019.01586
Cameron, M. J., Bermejo-Martin, J. F., Danesh, A., Muller, M. P., and Kelvin, D. J. (2008). Human immunopathogenesis of severe acute respiratory syndrome (SARS). Virus Res. 133, 13–19. doi: 10.1016/j.virusres.2007.02.014
Cárdenas, W. B., Loo, Y. -M., Gale, M., Hartman, A. L., Kimberlin, C. R., Martínez-Sobrido, L., et al. (2006). Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J. Virol. 80, 5168–5178. doi: 10.1128/JVI.02199-05
Carneiro, F. R. G., Lepelley, A., Seeley, J. J., Hayden, M. S., and Ghosh, S. (2018). An essential role for ECSIT in mitochondrial complex I assembly and mitophagy in macrophages. Cell Rep. 22, 2654–2666. doi: 10.1016/j.celrep.2018.02.051
Castanier, C., Zemirli, N., Portier, A., Garcin, D., Bidère, N., Vazquez, A., et al. (2012). MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 10:44. doi: 10.1186/1741-7007-10-44
Chakrabarti, A., Banerjee, S., Franchi, L., Loo, Y. M., Gale, M., Núñez, G., et al. (2015). RNase L activates the NLRP3 inflammasome during viral infections. Cell Host Microbe 17, 466–477. doi: 10.1016/j.chom.2015.02.010
Chang, T. H., Kubota, T., Matsuoka, M., Jones, S., Bradfute, S. B., Bray, M., et al. (2009). Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathog. 5:e1000493. doi: 10.1371/journal.ppat.1000493
Chen, G., Wu, D., Guo, W., Cao, Y., Huang, D., Wang, H., et al. (2020). Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 130, 2620–2629. doi: 10.1172/JCI137244
Choi, Y. B., Shembade, N., Parvatiyar, K., Balachandran, S., and Harhaj, E. W. (2017). TAX1BP1 restrains virus-induced apoptosis by facilitating itch-mediated degradation of the mitochondrial adaptor MAVS. Mol. Cell. Biol. 37, e00422–e00416. doi: 10.1128/mcb.00422-16
Chow, K. T., Gale, M., and Loo, Y. -M. (2018). RIG-I and other RNA sensors in antiviral immunity. Annu. Rev. Immunol. 36, 667–694. doi: 10.1146/annurev-immunol-042617-053309
Chowell, G., Tariq, A., and Kiskowski, M. (2019). Vaccination strategies to control Ebola epidemics in the context of variable household inaccessibility levels. PLoS Negl. Trop. Dis. 13:e0007814. doi: 10.1371/journal.pntd.0007814
Dai, T., Wu, L., Wang, S., Wang, J., Xie, F., Zhang, Z., et al. (2018). FAF1 regulates antiviral immunity by inhibiting MAVS but is antagonized by phosphorylation upon viral infection. Cell Host Microbe 24, 776.e5–790.e5. doi: 10.1016/j.chom.2018.10.006
de Wit, E., Van Doremalen, N., Falzarano, D., and Munster, V. J. (2016). SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 14, 523–534. doi: 10.1038/nrmicro.2016.81
DeDiego, M. L., Nieto-Torres, J. L., Regla-Nava, J. A., Jimenez-Guardeno, J. M., Fernandez-Delgado, R., Fett, C., et al. (2014). Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 88, 913–924. doi: 10.1128/jvi.02576-13
D’Elia, R. V., Harrison, K., Oyston, P. C., Lukaszewski, R. A., and Clark, G. C. (2013). Targeting the “Cytokine Storm” for therapeutic benefit. Clin. Vaccine Immunol. 20, 319–327. doi: 10.1128/CVI.00636-12
Ding, Q., Gaska, J. M., Douam, F., Wei, L., Kim, D., Balev, M., et al. (2018). Species-specific disruption of STING-dependent antiviral cellular defenses by the Zika virus NS2B3 protease. Proc. Natl. Acad. Sci. U. S. A. 115, E6310–E6318. doi: 10.1073/pnas.1803406115
Dorn, G. W. (2012). Inflame on!: mitochondrial escape provokes cytokine storms that doom the heart. Circ. Res. 111, 271–273. doi: 10.1161/CIRCRESAHA.112.275867
Dutta, M., Robertson, S. J., Okumura, A., Scott, D. P., Chang, J., Weiss, J. M., et al. (2017). A systems approach reveals MAVS signaling in myeloid cells as critical for resistance to Ebola virus in murine models of infection. Cell Rep. 18, 816–829. doi: 10.1016/j.celrep.2016.12.069
Ebola virus disease (n.d.). Available at: https://www.who.int/health-topics/ebola/#tab=tab_1 (Accessed April 17, 2020).
Esser-Nobis, K., Aarreberg, L. D., Roby, J. A., Fairgrieve, M. R., Green, R., and Gale, M. (2019). Comparative analysis of African and Asian lineage-derived Zika virus strains reveals differences in activation of and sensitivity to antiviral innate immunity. J. Virol. 93, e00640–e00619. doi: 10.1128/JVI.00640-19
Falasca, L., Agrati, C., Petrosillo, N., Di Caro, A., Capobianchi, M. R., Ippolito, G., et al. (2015). Molecular mechanisms of Ebola virus pathogenesis: focus on cell death. Cell Death Differ. 22, 1250–1259. doi: 10.1038/cdd.2015.67
Feng, Z., Cerveny, M., Yan, Z., and He, B. (2007). The VP35 protein of Ebola virus inhibits the antiviral effect mediated by double-stranded RNA-dependent protein kinase PKR. J. Virol. 81, 182–192. doi: 10.1128/JVI.01006-06
Freundt, E. C., Yu, L., Park, E., Lenardo, M. J., and Xu, X. -N. (2009). Molecular determinants for subcellular localization of the severe acute respiratory syndrome coronavirus open reading frame 3b protein. J. Virol. 83, 6631–6640. doi: 10.1128/JVI.00367-09
Friedman, J. R., and Nunnari, J. (2014). Mitochondrial form and function. Nature 505, 335–343. doi: 10.1038/nature12985
Gack, M. U., Albrecht, R. A., Urano, T., Inn, K. S., Huang, I. C., Carnero, E., et al. (2009). Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5, 439–449. doi: 10.1016/j.chom.2009.04.006
Gack, M. U., Shin, Y. C., Joo, C. H., Urano, T., Liang, C., Sun, L., et al. (2007). TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920. doi: 10.1038/nature05732
Geoghegan, J. L., Senior, A. M., Di Giallonardo, F., and Holmes, E. C. (2016). Virological factors that increase the transmissibility of emerging human viruses. Proc. Natl. Acad. Sci. U. S. A. 113, 4170–4175. doi: 10.1073/pnas.1521582113
Graef, K. M., Vreede, F. T., Lau, Y. -F., McCall, A. W., Carr, S. M., Subbarao, K., et al. (2010). The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J. Virol. 84, 8433–8445. doi: 10.1128/JVI.00879-10
Grant, A., Ponia, S. S., Tripathi, S., Balasubramaniam, V., Miorin, L., Sourisseau, M., et al. (2016). Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 19, 882–890. doi: 10.1016/j.chom.2016.05.009
Green, R. R., Wilkins, C., Pattabhi, S., Dong, R., Loo, Y., and Gale, M. (2016). Transcriptional analysis of antiviral small molecule therapeutics as agonists of the RLR pathway. Genom. Data 7, 290–292. doi: 10.1016/j.gdata.2016.01.020
Haby, M. M., Pinart, M., Elias, V., and Reveiz, L. (2018). Prevalence of asymptomatic Zika virus infection: a systematic review. Bull. World Health Organ. 96, 402D–413D. doi: 10.2471/BLT.17.201541
Hackbart, M., Deng, X., and Baker, S. C. (2020). Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc. Natl. Acad. Sci. U. S. A. 117, 8094–8103. doi: 10.1073/pnas.1921485117
Haneklaus, M., and O’Neill, L. A. J. (2015). NLRP3 at the interface of metabolism and inflammation. Immunol. Rev. 265, 53–62. doi: 10.1111/imr.12285
Hartmann, G. (2017). Nucleic acid immunity. Adv. Immunol. 133, 121–169. doi: 10.1016/bs.ai.2016.11.001
He, Y., Hara, H., and Núñez, G. (2016). Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 41, 1012–1021. doi: 10.1016/j.tibs.2016.09.002
He, F., Melén, K., Maljanen, S., Lundberg, R., Jiang, M., Österlund, P., et al. (2017). Ebolavirus protein VP24 interferes with innate immune responses by inhibiting interferon-λ1 gene expression. Virology 509, 23–34. doi: 10.1016/j.virol.2017.06.002
He, X., Zhu, Y., Zhang, Y., Geng, Y., Gong, J., Geng, J., et al. (2019). RNF 34 functions in immunity and selective mitophagy by targeting MAVS for autophagic degradation. EMBO J. 38:e100978. doi: 10.15252/embj.2018100978
Hu, Y., Dong, X., He, Z., Wu, Y., Zhang, S., Lin, J., et al. (2019). Zika virus antagonizes interferon response in patients and disrupts RIG-I-MAVS interaction through its CARD-TM domains. Cell Biosci. 9, 1–15. doi: 10.1186/s13578-019-0308-9
Hughes, M. M., and O’Neill, L. A. J. (2018). Metabolic regulation of NLRP3. Immunol. Rev. 281, 88–98. doi: 10.1111/imr.12608
Infection-cusabio, V., and Topics, O. P. (2020). What you have to know about cytokine storm and virus infection. 1–8.
Jahan, A. S., Biquand, E., Muñoz-Moreno, R., Le Quang, A., Mok, C. K. -P., Wong, H. H., et al. (2019). OTUB1 is a key regulator of RIG-I dependent immune signalling and is targeted for proteasomal degradation by influenza A NS1. Cell Rep. 30, 1570.e6–1584.e6. doi: 10.1016/j.celrep.2020.01.015
Jureka, A. S., Kleinpeter, A. B., Tipper, J. L., Harrod, K. S., and Petit, C. M. (2020). The influenza NS1 protein modulates RIG-I activation via a strain-specific direct interaction with the second CARD of RIG-I. J. Biol. Chem. 295, 1153–1164. doi: 10.1074/jbc.RA119.011410
Kaner, J., and Schaack, S. (2016). Understanding Ebola: the 2014 epidemic. Glob. Health 12:53. doi: 10.1186/s12992-016-0194-4
Kao, W. P., Yang, C. Y., Su, T. W., Wang, Y. T., Lo, Y. C., and Lin, S. C. (2015). The versatile roles of CARDs in regulating apoptosis, inflammation, and NF-κB signaling. Apoptosis 20, 174–195. doi: 10.1007/s10495-014-1062-4
Kawai, T., and Akira, S. (2007). Antiviral signaling through pattern recognition receptors. J. Biochem. 141, 137–145. doi: 10.1093/jb/mvm032
Kell, A. M., and Gale, M. (2015). RIG-I in RNA virus recognition. Virology 479–480, 110–121. doi: 10.1016/j.virol.2015.02.017
Keystone Symposia (n.d.). Available at: https://virtual.keystonesymposia.org/ks/articles/3460/view (Accessed May 17, 2020).
Khan, M., Syed, G. H., Kim, S. J., and Siddiqui, A. (2015). Mitochondrial dynamics and viral infections: a close nexus. Biochim. Biophys. Acta 1853, 2822–2833. doi: 10.1016/j.bbamcr.2014.12.040
Koliopoulos, M. G., Lethier, M., van Der Veen, A. G., Haubrich, K., Hennig, J., Kowalinski, E., et al. (2018). Molecular mechanism of influenza A NS1-mediated TRIM25 recognition and inhibition. Nat. Commun. 9:1820. doi: 10.1038/s41467-018-04214-8
Kopecky-Bromberg, S. A., Martínez-Sobrido, L., Frieman, M., Baric, R. A., and Palese, P. (2007). Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 81, 548–557. doi: 10.1128/JVI.01782-06
Lei, Y., Moore, C. B., Liesman, R. M., O’Connor, B. P., Bergstralh, D. T., Chen, Z. J., et al. (2009). MAVS-mediated apoptosis and its inhibition by viral proteins. PLoS One 4:e5466. doi: 10.1371/journal.pone.0005466
Lester, S. N., and Li, K. (2014). Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 426, 1246–1264. doi: 10.1016/j.jmb.2013.11.024
Li, W., Li, N., Dai, S., Hou, G., Guo, K., Chen, X., et al. (2019). Zika virus circumvents host innate immunity by targeting the adaptor proteins MAVS and MITA. FASEB J. 33, 9929–9944. doi: 10.1096/fj.201900260R
Li, Q., Wang, L., Dong, C., Che, Y., Jiang, L., Liu, L., et al. (2005). The interaction of the SARS coronavirus non-structural protein 10 with the cellular oxido-reductase system causes an extensive cytopathic effect. J. Clin. Virol. 34, 133–139. doi: 10.1016/j.jcv.2004.12.019
Liang, Y., Wang, M. L., Chien, C. S., Yarmishyn, A. A., Yang, Y. P., Lai, W. Y., et al. (2020). Highlight of immune pathogenic response and hematopathologic effect in SARS-CoV, MERS-CoV, and SARS-Cov-2 infection. Front. Immunol. 11:1022. doi: 10.3389/fimmu.2020.01022
Lin, S., Yang, S., He, J., Guest, J. D., Ma, Z., Yang, L., et al. (2019). Zika virus NS5 protein antagonizes type I interferon production via blocking TBK1 activation. Virology 527, 180–187. doi: 10.1016/j.virol.2018.11.009
Liu, Y., Olagnier, D., and Lin, R. (2017). Host and viral modulation of RIG-I-mediated antiviral immunity. Front. Immunol. 7:662. doi: 10.3389/fimmu.2016.00662
Liu, X., Yang, C., Sun, X., Lin, X., Zhao, L., Chen, H., et al. (2019). Evidence for a novel mechanism of influenza A virus host adaptation modulated by PB2-627. FEBS J. 286, 3389–3400. doi: 10.1111/febs.14867
Long, J. C. D., and Fodor, E. (2016). The PB2 subunit of the Influenza A virus RNA polymerase is imported into the mitochondrial matrix. J. Virol. 90, 8729–8738. doi: 10.1128/JVI.01384-16
Lundberg, R., Melén, K., Westenius, V., Jiang, M., Österlund, P., Khan, H., et al. (2019). Zika virus non-structural protein NS5 inhibits the RIG-I pathway and interferon lambda 1 promoter activation by targeting IKK epsilon. Viruses 11:1024. doi: 10.3390/v11111024
Luthra, P., Ramanan, P., Mire, C. E., Weisend, C., Tsuda, Y., Yen, B., et al. (2013). Mutual antagonism between the ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe 14, 74–84. doi: 10.1016/j.chom.2013.06.010
Ma, J., Ketkar, H., Geng, T., Lo, E., Wang, L., Xi, J., et al. (2018). Zika virus non-structural protein 4A blocks the RLR-MAVS signaling. Front. Microbiol. 9:1350. doi: 10.3389/fmicb.2018.01350
Mahmudpour, M., Roozbeh, J., Keshavarz, M., Farrokhi, S., and Nabipour, I. (2020). COVID-19 cytokine storm: the anger of inflammation. Cytokine 133:155151. doi: 10.1016/j.cyto.2020.155151
Maier, H. J., Bickerton, E., and Britton, P. (2015). Coronaviruses: Methods and protocols. Vol. 1282. 1–282.
Messaoudi, I., Amarasinghe, G. K., and Basler, C. F. (2015). Filovirus pathogenesis and immune evasion: insights from Ebola virus and Marburg virus. Nat. Rev. Microbiol. 13, 663–676. doi: 10.1038/nrmicro3524
Mibayashi, M., Martinez-Sobrido, L., Loo, Y. -M., Cardenas, W. B., Gale, M., and Garcia-Sastre, A. (2007). Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of Influenza A virus. J. Virol. 81, 514–524. doi: 10.1128/JVI.01265-06
Mills, E. L., Kelly, B., and O’Neill, L. A. J. (2017). Mitochondria are the powerhouses of immunity. Nat. Immunol. 18, 488–498. doi: 10.1038/ni.3704
Mishra, P., and Chan, D. C. (2014). Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 15, 634–646. doi: 10.1038/nrm3877
Miyake, K., Shibata, T., Ohto, U., Shimizu, T., Saitoh, S. -I., Fukui, R., et al. (2018). Mechanisms controlling nucleic acid-sensing Toll-like receptors. Int. Immunol. 30, 43–51. doi: 10.1093/intimm/dxy016
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/BJ20081386
Nieto-Torres, J. L., DeDiego, M. L., Verdiá-Báguena, C., Jimenez-Guardeño, J. M., Regla-Nava, J. A., Fernandez-Delgado, R., et al. (2014). Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog. 10:e1004077. doi: 10.1371/journal.ppat.1004077
Nieto-Torres, J. L., Verdiá-Báguena, C., Jimenez-Guardeño, J. M., Regla-Nava, J. A., Castaño-Rodriguez, C., Fernandez-Delgado, R., et al. (2015). Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 485, 330–339. doi: 10.1016/j.virol.2015.08.010
Okamoto, M., Kouwaki, T., Fukushima, Y., and Oshiumi, H. (2018). Regulation of RIG-I activation by K63-linked polyubiquitination. Front. Immunol. 8:1942. doi: 10.3389/fimmu.2017.01942
Orzalli, M. H., and Kagan, J. C. (2017). Apoptosis and necroptosis as host defense strategies to prevent viral infection. Trends Cell Biol. 27, 800–809. doi: 10.1016/j.tcb.2017.05.007
Plourde, A. R., and Bloch, E. M. (2016). A literature review of Zika virus. Emerg. Infect. Dis. 22, 1185–1192. doi: 10.3201/eid2207.151990
Pothlichet, J., Meunier, I., Davis, B. K., Ting, J. P. Y., Skamene, E., von Messling, V., et al. (2013). Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in Influenza A virus infected cells. PLoS Pathog. 9:e1003256. doi: 10.1371/journal.ppat.1003256
Prins, K. C., Cardenas, W. B., and Basler, C. F. (2009). Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKK and TBK-1. J. Virol. 83, 3069–3077. doi: 10.1128/JVI.01875-08
Qin, Y., Xue, B., Liu, C., Wang, X., Tian, R., Xie, Q., et al. (2017). NLRX1 mediates MAVS degradation to attenuate the hepatitis C virus-induced innate immune response through PCBP2. J. Virol. 91, e01264–e01217. doi: 10.1128/jvi.01264-17
Ratajczak, M. Z., and Kucia, M. (2020). SARS-CoV-2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine “storm” and risk factor for damage of hematopoietic stem cells. Leukemia 34, 1726–1729. doi: 10.1038/s41375-020-0887-9
Refolo, G., Vescovo, T., Piacentini, M., Fimia, G. M., and Ciccosanti, F. (2020). Mitochondrial interactome: a focus on antiviral signaling pathways. Front. Cell Dev. Biol. 8:8. doi: 10.3389/fcell.2020.00008
Reid, S. P., Leung, L. W., Hartman, A. L., Martinez, O., Shaw, M. L., Carbonnelle, C., et al. (2006). Ebola virus VP24 binds karyopherin α1 and blocks STAT1 nuclear accumulation. J. Virol. 80, 5156–5167. doi: 10.1128/JVI.02349-05
Sadler, A. J. (2017). The role of MDA5 in the development of autoimmune disease. J. Leukoc. Biol. 103, 185–192. doi: 10.1189/jlb.4mr0617-223r
SARS | Home | Severe Acute Respiratory Syndrome | SARS-CoV Disease | CDC (n.d.). Available at: https://www.cdc.gov/sars/index.html (Accessed April 17, 2020).
Schilling, M., Bridgeman, A., Gray, N., Hertzog, J., Hublitz, P., Kohl, A., et al. (2020). RIG-I plays a dominant role in the induction of transcriptional changes in Zika virus-infected cells, which protect from virus-induced cell death. Cells 9:1476. doi: 10.3390/cells9061476
Schoeman, D., and Fielding, B. C. (2019). Coronavirus envelope protein: current knowledge. Virol. J. 16, 1–22. doi: 10.1186/s12985-019-1182-0
Shao, W., Li, X., Goraya, M. U., Wang, S., and Chen, J. L. (2017). Evolution of influenza a virus by mutation and re-assortment. Int. J. Mol. Sci. 18:1650. doi: 10.3390/ijms18081650
Shrivastava, G., León-Juárez, M., García-Cordero, J., Meza-Sánchez, D. E., and Cedillo-Barrón, L. (2016). Inflammasomes and its importance in viral infections. Immunol. Res. 64, 1101–1117. doi: 10.1007/s12026-016-8873-z
Singh, K. K., Chaubey, G., Chen, J. Y., and Suravajhala, P. (2020). Decoding SARS-CoV-2 hijacking of host mitochondria in pathogenesis of COVID-19. Am. J. Physiol. Cell Physiol. 319, C258–C267. doi: 10.1152/ajpcell.00224.2020
Sliter, D. A., Martinez, J., Hao, L., Chen, X., Sun, N., Fischer, T. D., et al. (2018). Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262. doi: 10.1038/s41586-018-0448-9
Song, B. H., Yun, S. I., Woolley, M., and Lee, Y. M. (2017). Zika virus: history, epidemiology, transmission, and clinical presentation. J. Neuroimmunol. 308, 50–64. doi: 10.1016/j.jneuroim.2017.03.001
Strottmann, D. M., Zanluca, C., Mosimann, A. L. P., Koishi, A. C., Auwerter, N. C., Faoro, H., et al. (2019). Genetic and biological characterization of Zika virus isolates from different Brazilian regions. Mem. Inst. Oswaldo Cruz 114:e190150. doi: 10.1590/0074-02760190150
Structure of Ebola Virus (n.d.). Available at: https://microbiologyinfo.com/structure-of-ebola-virus/ (Accessed April 17, 2020).
Tsujimoto, Y., and Shimizu, S. (2007). Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 12, 835–840. doi: 10.1007/s10495-006-0525-7
Varga, Z. T., Grant, A., Manicassamy, B., and Palese, P. (2012). Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J. Virol. 86, 8359–8366. doi: 10.1128/JVI.01122-12
Vazquez, C., and Horner, S. M. (2015). MAVS coordination of antiviral innate immunity. J. Virol. 89, 6974–6977. doi: 10.1128/JVI.01918-14
Walker, M. A., Volpi, S., Sims, K. B., Walter, J. E., and Traggiai, E. (2014). Powering the immune system: mitochondria in immune function and deficiency. J. Immunol. Res. 2014:164309. doi: 10.1155/2014/164309
Wang, J., Jiang, M., Chen, X., and Montaner, L. J. (2020a). Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J. Leukoc. Biol. 108, 17–41. doi: 10.1002/JLB.3COVR0520-272R
Wang, P., Yang, L., Cheng, G., Yang, G., Xu, Z., You, F., et al. (2013). UBXN1 interferes with Rig-I-like receptor-mediated antiviral immune response by targeting MAVS. Cell Rep. 3, 1057–1070. doi: 10.1016/j.celrep.2013.02.027
Wang, R., Zhu, Y., Ren, C., Yang, S., Tian, S., Chen, H., et al. (2020b). Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy 1–16. doi: 10.1080/15548627.2020.1725375 [Epub ahead of print]
Weinberg, S. E., Sena, L. A., and Chandel, N. S. (2015). Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417. doi: 10.1016/j.immuni.2015.02.002
Wu, Y., Liu, Q., Zhou, J., Xie, W., Chen, C., Wang, Z., et al. (2017). Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov. 3, 1–14. doi: 10.1038/celldisc.2017.6
Xu, H., He, X., Zheng, H., Huang, L. J., Hou, F., Yu, Z., et al. (2014). Structural basis for the prion-like MAVS filaments in antiviral innate immunity. Elife 3:e01489. doi: 10.7554/elife.01489
Yabal, M., Calleja, D. J., Simpson, D. S., and Lawlor, K. E. (2019). Stressing out the mitochondria: mechanistic insights into NLRP3 inflammasome activation. J. Leukoc. Biol. 105, 377–399. doi: 10.1002/JLB.MR0318-124R
Yap, J. K. Y., Moriyama, M., and Iwasaki, A. (2020). Inflammasomes and pyroptosis as therapeutic targets for COVID-19. J. Immunol. 205, 307–312. doi: 10.4049/jimmunol.2000513
Yin, Y., and Wunderink, R. G. (2018). MERS, SARS and other coronaviruses as causes of pneumonia. Respirology 23, 130–137. doi: 10.1111/resp.13196
Yu, C., Chen, R., Li, J. J., Li, J. J., Drahansky, M., Paridah, M., et al. (2012). We are IntechOpen, the world’s leading publisher of Open Access books built by scientists, for scientists TOP 1%. Intech, 13.
Yuan, X., Shan, Y., Yao, Z., Li, J., Zhao, Z., Chen, J., et al. (2006). Mitochondrial location of severe acute respiratory syndrome coronavirus 3b protein. Mol. Cells 21, 186–191.
Zamarin, D., García-Sastre, A., Xiao, X., Wang, R., and Palese, P. (2005). Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 1:e4. doi: 10.1371/journal.ppat.0010004
Zhang, L., Qin, Y., and Chen, M. (2018). Viral strategies for triggering and manipulating mitophagy. Autophagy 14, 1665–1673. doi: 10.1080/15548627.2018.1466014
Zhao, Y., Sun, X., Nie, X., Sun, L., Tang, T. S., and Chen, D., et al. (2012). COX5B regulates MAVS-mediated antiviral signaling through interaction with ATG5 and repressing ROS production. PLoS Pathog. 8:e1003086. doi: 10.1371/journal.ppat.1003086
Zhao, Z., Tao, M., Han, W., Fan, Z., Imran, M., Cao, S., et al. (2019). Nuclear localization of Zika virus NS5 contributes to suppression of type I interferon production and response. J. Gen. Virol. doi: 10.1099/jgv.0.001376 [Epub ahead of print]
Zheng, Y., Liu, Q., Wu, Y., Ma, L., Zhang, Z., Liu, T., et al. (2018). Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS 1-caspase-1 axis. EMBO J. 37:e99347. doi: 10.15252/embj.201899347
Keywords: mitochondria, innate immunity, mitochondrial antiviral signaling, retinoic acid inducible gene-I, RNA virus, cytokine storm
Citation: Dutta S, Das N and Mukherjee P (2020) Picking up a Fight: Fine Tuning Mitochondrial Innate Immune Defenses Against RNA Viruses. Front. Microbiol. 11:1990. doi: 10.3389/fmicb.2020.01990
Edited by:
Miguel A. Martín-Acebes, National Institute for Agricultural and Food Research and Technology (INIA), SpainReviewed by:
Maria Teresa Sanchez-Aparicio, Icahn School of Medicine at Mount Sinai, United StatesCopyright © 2020 Dutta, Das and Mukherjee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Piyali Mukherjee, cGl5YWxpLmRic0BwcmVzaXVuaXYuYWMuaW4=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.