 Rajender Kumar
Rajender Kumar Jeanette E. Bröms
Jeanette E. Bröms Anders Sjöstedt
Anders Sjöstedt
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 11 August 2020
Sec. Evolutionary and Genomic Microbiology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.01928
Pan-genome analysis is a powerful method to explore genomic heterogeneity and diversity of bacterial species. Here we present a pan-genome analysis of the genus Francisella, comprising a dataset of 63 genomes and encompassing clinical as well as environmental isolates from distinct geographic locations. To determine the evolutionary relationship within the genus, we performed phylogenetic whole-genome studies utilizing the average nucleotide identity, average amino acid identity, core genes and non-recombinant loci markers. Based on the analyses, the phylogenetic trees obtained identified two distinct clades, A and B and a diverse cluster designated C. The sizes of the pan-, core-, cloud-, and shell-genomes of Francisella were estimated and compared to those of two other facultative intracellular pathogens, Legionella and Piscirickettsia. Francisella had the smallest core-genome, 692 genes, compared to 886 and 1,732 genes for Legionella and Piscirickettsia respectively, while the pan-genome of Legionella was more than twice the size of that of the other two genera. Also, the composition of the Francisella Type VI secretion system (T6SS) was analyzed. Distinct differences in the gene content of the T6SS were identified. In silico approaches performed to identify putative substrates of these systems revealed potential effectors targeting the cell wall, inner membrane, cellular nucleic acids as well as proteins, thus constituting attractive targets for site-directed mutagenesis. The comparative analysis performed here provides a comprehensive basis for the assessment of the phylogenomic relationship of members of the genus Francisella and for the identification of putative T6SS virulence traits.
The genus Francisella belongs to the γ-subclass of Proteobacteria, but shows no close relationship to other human pathogens (Sjöstedt, 2005). The genus is diverse with many species adapted to specific ecological niches and some of the pathogenic species to a very broad range of mammals, as well as fish (Sjöstedt, 2007; Birkbeck et al., 2011; Colquhoun and Duodu, 2011; Sjödin et al., 2012; Pilo, 2018; Yon et al., 2019). A feature of the genus is an unusual fatty acid composition and a high lipid content of the cell wall (Sjöstedt, 2005). The important human pathogen, F. tularensis, has for 50 years been divided into several subspecies (Keim et al., 2007; Kingry et al., 2013), the most important being subsp. holarctica and subsp. tularensis, both harboring isolates that cause human tularemia (Tärnvik and Berglund, 2003). This disease is rather common in many countries of the Northern hemisphere, however, isolates of subsp. tularensis are found in North America only (Kingry et al., 2013). Isolates of subsp. tularensis, in particular the lineage A1b, are the most virulent, both in humans but also in animal models (Kugeler et al., 2009). The designations of type A and type B are often used to designate subsp. tularensis and holarctica, but these have no formal approval. In addition, there is a third subspecies, subsp. mediasiatica, represented by strains from the Central Asian republics of former Soviet Union, but in contrast to the other subspecies, it has low virulence and has not been reported as a human pathogen (Olsufiev et al., 1959). The three subspecies demonstrate distinct genomic differences as demonstrated by multiple whole-genome sequences present in current databases. Some 30 years ago, F. novicida was recognized, a rare human pathogen with many isolates derived from environmental sources (Hollis et al., 1989; Kingry et al., 2013). This is also true for a second species of the genus, F. philomiragia, which possesses distinct biochemical characteristics compared to F. tularensis (Hollis et al., 1989). As for F. novicida, the few cases of human F. philomiragia-infections that have been described are healthy individuals with a history of contact with natural water, e.g., near-drowning, or which are immunocompromised (Robles-Marhuenda et al., 2018).
In contrast to the aforementioned, since long recognized members of the genus Francisella, a large number of new species have been described during the last decade, often identified by genomic characterization of one or a few isolates. The rapidly expanding number of species demonstrate that the genus Francisella is very diverse, likely exists globally, and many species are adapted to highly specialized environmental niches (Hollis et al., 1989; Clarridge et al., 1996; Barns et al., 2005; Kuske et al., 2006; Siddaramappa et al., 2011, 2012; Qu et al., 2013; Challacombe et al., 2017). The best-described example is F. noatunensis, an economically important pathogen that globally causes serious disease in farmed and wild fish in both salt and fresh water (Birkbeck et al., 2011; Colquhoun and Duodu, 2011; McDermott and Palmeiro, 2013). Two subspecies have been recognized, subsp. noatunensis and subsp. orientalis. Recently, however, the latter was proposed to form a novel species; Francisella orientalis sp. nov., and an additional subspecies within the species F. noatunensis was suggested, i.e., subsp. chilensis subsp. nov. (Ramirez-Paredes et al., 2020). In addition, a multitude of potentially new Francisella species has been isolated globally from environmental sources, e.g., cooling water systems, from a wide variety of tick endosymbionts, as well as from human samples, e.g., skin lesions, or from immunocompromised patients near-drowning, with respiratory disease, or with cerebrospinal infection (Hollis et al., 1989; Wenger et al., 1989; Clarridge et al., 1996; Whipp et al., 2003; Barns et al., 2005; Kuske et al., 2006; Kugeler et al., 2008; Petersen et al., 2009; Huber et al., 2010; Siddaramappa et al., 2011, 2012; Kreizinger et al., 2013; Qu et al., 2013; Respicio-Kingry et al., 2013; Rydzewski et al., 2014; Challacombe et al., 2017; Wang Y. et al., 2018; Vallesi et al., 2019).
In view of the rapidly evolving diversity within many bacterial genera and families, the need to obtain additional data to provide a robust platform for species delineation is essential. This is particularly true of the genus Francisella, since for many decades, there has been much ambiguity regarding the taxonomical relationships between many species and subspecies, further emphasized by the discoveries of previously unrecognized bacterial isolates with unclear taxonomic belonging. The rapidly evolving diversity within the genus Francisella many times challenges the traditional taxonomical classification, since several of the aforementioned isolates have only been identified by means of genetic characterization and may be unculturable, or are phenotypically ill-defined. To this end, recent work is attempting to define unambiguous criteria that can be generally applied to delineate bacterial species in Francisella as well as in other genera (Sjödin et al., 2012; Challacombe et al., 2017). In this regard, the utility of the dramatically increasing amount of genomic data has to be incorporated in the species definition alongside other relevant, more traditional taxonomic data.
For the genus Francisella, a large number of completed and draft genome assemblies are available in biological sequence databases, such as the National Center for Biotechnology Information (NCBI) assembly database and the Joint Genome Institute (JGI) Genome Portal. These huge sequence datasets offer not only the possibility to understand the functional and evolutionary repertoire of bacterial genera, but also open up possibilities for developing therapies and engineering applications. The objective of this study was to elucidate the core- and pan-genome features of the Francisella genus to shed light on its diversity and characteristics, as well as to identify putative T6SS substrates in silico. Our analysis identifies conceptual and technical approaches that may be used for studies of pathogenicity, especially related to secretion systems.
All 62 publicly available (January 2018) whole genome sequences of Francisella bacteria were downloaded from the NCBI assembly database1 and used for analysis. Allofrancisella guangzhouensis, a species previously considered to be a member of the genus (Qu et al., 2016), was also included in the analysis, thus making the number of genomes analyzed 63. These complete genome assemblies cover almost the complete genus Francisella, comprising 14 species with various number of subspecies (a total number of 26; Table 1). As a starting point, any plasmid sequences present were removed from the assemblies. In the next step, whole genome comparisons were performed, and the average nucleotide identity (ANI) calculated in the pyani program (Pritchard et al., 2016), using the BlastN alignment tool with 1,020 nt long fragments as input sequences and other parameters used were default. For each pairwise genome comparison, an ANI matrix was generated along with a dendrogram. The same methodology was also applied to the genus Legionella (77 complete genomes) and Piscirickettsia (19 complete genomes), to allow comparisons to be made between the three genera. Only one representative of highly related species (ANI ≥ 99.5%) was used for further analysis of the pan-genome, phylogenomic analysis etc.
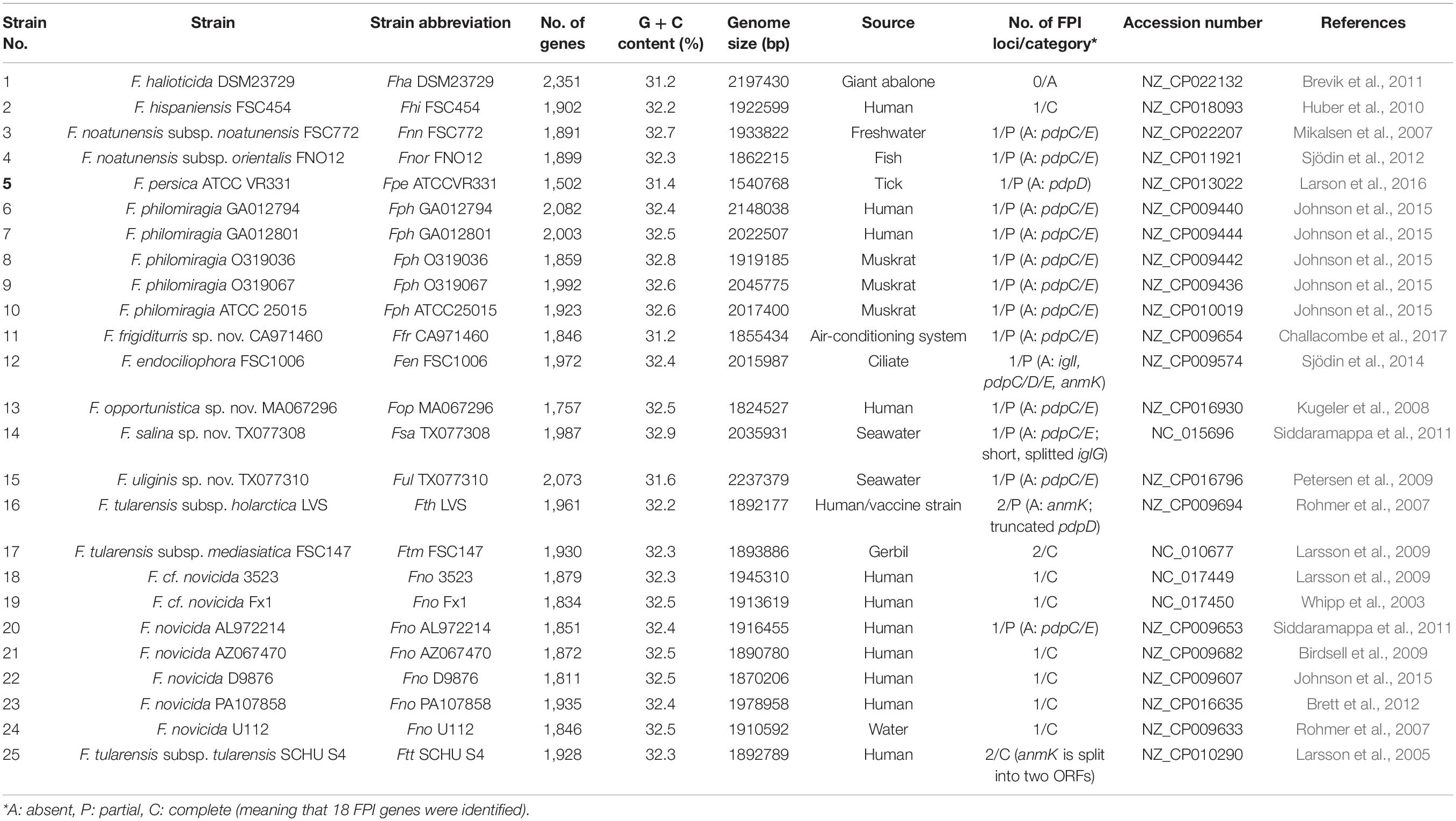
Table 1. The 25 representative and complete Francisella genome assemblies, including their annotation, the bacterial niche as well as a description of their Francisella Pathogenicity Island (FPI).
The Francisella core- and pan-genome size was assessed in a manner similar to that previously reported, using iterative and combinatorial approaches (Tettelin et al., 2005; Meric et al., 2014; Mosquera-Rendon et al., 2016). To estimate the number of orthologous genes within the genus, we used the GET_HOMOLOGUES tool (Contreras-Moreira and Vinuesa, 2013) and the three clustering algorithms (i) bidirectional best-hit (BDBH) (Wolf and Koonin, 2012) COGtriangles (Kristensen et al., 2010) and (iii) OrthoMCL (Ortho Markov Cluster Algorithm) (Li et al., 2003). Orthologous genes were clustered using an E-value of >1e-05 and a query coverage of > 50%. Finally, the core-genome was defined as the set of genes shared by representative species/strains, while the pan-genome was defined as the sum of the core-genome and the set of auxiliary (i.e., available in more than 1 and less than 26 genomes) and exclusive (i.e., available in only one genome) genes. We validated the result of the pan-genome analysis by BPGA (Bacterial Pan Genome Analysis tool) that uses the USEARCH algorithm for fastest clustering (Chaudhari et al., 2016). The core- and pan-genomes, as well as their predicted sizes and trajectories, were obtained using the method proposed by Knight (Knight et al., 2017), the models/regression algorithms given by Tettelin (Tettelin et al., 2005, 2008), and the binomial mixture model of Snipen (Snipen et al., 2009). For each method, the parameters used were default.
Curve fitting of the pan-genome was performed using a power-law regression based on Heaps’ law [y = ApanxBpan + Cpan], as previously described (Tettelin et al., 2005, 2008; Rasko et al., 2008). The same protocol was also applied to estimate the core- and pan-genomes for the genera Legionella and Piscirickettsia. Further, the common core-genome shared by all three genera was estimated, based on individual core sets for each genus as input. In the next step, this “core of core” was functionally characterized using COG (Clusters of Orthologous Groups) and KEGG (Kyoto Encyclopedia of Genes and Genomes) annotations.
For whole-genome phylogenetic analysis of closely related F. tularensis strains, we used multiple approaches. First, we used the UBCG (Up-to-date bacterial core genes) approach, by utilizing its pipeline and default parameters2 (Na et al., 2018). First, all 26 genome assemblies were converted into bcg files using the UBCG.jar extract command. These files contain a label with full information about the strain/genome and strain details. Next, all markers, i.e., a set of 92 bacterial core genes, were identified from an up-to-date genome database using the hmmsearch program and default parameters.3 In the next step, multiple alignments were performed for each gene using the UBCG.jar align command with the MAFFT (Multiple Alignment Fast Fourier Transform) alignment program4 using default parameters. Each of the UBCG genes were aligned separately, before being concatenated into a single alignment. A highly resolved maximum likelihood tree was obtained using FastTree5 and visualized using the iTOL server.6 A bootstrap analysis was performed to determine the reliability of the branches obtained.
We also constructed a marker-based phylogenetic tree, by using the GET_PHYLOMARKERS software package in the default mode (Vinuesa et al., 2018), and with sets of single copy orthologous core-genomes as input. This analysis allows us to identify high-quality markers to estimate robust genome phylogenies from the UBCG, thereby resolving poor tree topologies. During the phylogenetic tree reconstruction, a set of sequential filters was applied to exclude recombinant alignments and horizontal gene transfer. A maximum likelihood (ML) phylogenetic tree was estimated from the concatenated set of top-ranking alignments at the DNA as well as at the protein levels, using the advanced general amino-acid replacement matrix model (LG) (Le and Gascuel, 2008) and MFP feature in the IQ-TREE (IQT) software (Nguyen et al., 2015). The remaining parameters were kept as default. The tree was visualized using the iTOL server. A bootstrap analysis was performed to determine the reliability of the branches obtained.
Comparative analyses of FPI/T6SS clusters were performed using the MultiGeneBlast program with default parameters7 (Medema et al., 2013). This program offers a BLAST-based tool to perform “architecture searches” with operons or gene clusters as basic units, instead of single genes. This allows for the identification of genomic loci containing homologs of specific user-specified gene combinations. As input query, we used sequences corresponding to the FPI cluster of the F. novicida strain U112 (accession number NZ_CP009633) to search a database containing all of the 26 representative Francisella species. To generate blast hits, we set the minimal sequence identify to 25% and the sequence coverage to 30%, while the rest of the parameters were kept as default. Using the same parameters, we also tested the FPI cluster of U112 against the bacterial domain in the NCBI gene bank database to look for the presence of FPI homologous genes in other bacterial genera. To estimate the G + C contents for the FPI cluster and for the whole genome, the following formula was used: (G + C)/(A + T + G + C) ∗ 100%. We also analyzed the amino acid composition of the FPI proteins (encoded by pdpA to anmK) and compared it with the amino acid composition of the rest of the genome. The first was calculated using the concatenated all FPI proteins only, while the second was calculated using the concatenated all protein sequences after excluding the FPI proteins. The genomes included in the analysis, in addition to F. novicida U112, were F. tularensis subsp. tularensis SCHU S4 (NZ_CP010290), F. cf. novicida Fx1 (NC_017450), and F. tularensis subsp. holarctica LVS (NZ_CP009694).
The Bastion6 program8 predicts T6SS effectors using a two-layer SVM-based ensemble model with optimized parameters (Wang J. W. et al., 2018). We employed this program to search for putative T6SS effectors encoded within the Francisella genomes, using the complete genome sequence of F. tularensis subsp. tularensis SCHU S4 as a reference genome. Predicted promiscuous effectors were selected based on an ensemble model result score of ≥0.5, and were functionally described and Gene Ontology (GO)-annotated with respect to their predicted biological process, molecular function or cellular component, using the PANNZER2 (Protein annotation with Z-scoRE) server (Toronen et al., 2018). We also used our hits to search the Pfam database9 for conserved domains of unknown functions, DUFs.
To specifically search for homologs of T6SS-dependent, ion-selective pore-forming effectors (Mariano et al., 2019) within the Francisella genus, we used the sequence for the effector Ssp6 (SMDB11_4673) of Serratia marcescens Db10 as query against the NR database (set as Francisella group) using the PSI-BLASTP program with default parameters. To specifically search for MIX effectors (Salomon et al., 2014) within the Francisella genome, we used the NR database from NCBI using the position specific iterative (PSI)-BLASTP with four iterations and other parameters kept as default. As queries, we used representative sequences for each of the following five classes of MIX effectors: MIX I - Vibrio parahaemolyticus VP1388 (accession: NP_797767), MIX II - Proteus mirabilis IdsD (accession SPY42138), MIX III - Burkholderia thailandensis BTH_I2691 (accession ABC38088), MIX IV - Vibrio cholerae VCA0020 (accession NP_232421) and MIX V – V. parahaemolyticus VPA1263 (accession NP_800773). Each query generated a set of identified hit protein sequences, which we used in a multiple sequence alignment analysis to identify the conserved sequence and predict putative signal peptides. Furthermore, by using the MultiGeneBlast program, the chromosomal location of the identified hits as well as the upstream and downstream ORFs were analyzed for the 26 representative complete genome sequence data set.
Whole-genome comparisons have the power to discriminate between strains and species with high resolution. For this purpose, all completely sequenced available Francisella genomes (a total of 63 when this study was initiated, see Supplementary Table S1) were selected for further analysis, out of which five were excluded since they were found to represent duplicated genomes. For the remaining 57 genomes, whole-genome sequence comparisons were performed in a pairwise fashion, by calculating and comparing the ANI (average nucleotide identity) (Yon et al., 2019), for each genome pair (Supplementary Table S2). ANI is a well-documented and robust method for comparing genomes and assessing species relationships (Konstantinidis et al., 2006). The pair-wise comparisons showed a minimum ANI of ∼74.2% for the most distant strains, while strains of the same subspecies showed an ANI of >97.0%. Only one representative of highly related species (ANI ≥ 99.5%) was used for further pan-genome analysis. This allowed us to down-select the genome set aimed to represent the entire genus Francisella to a total of 26 genomes (Figure 1). For pairwise ANI comparisons of the 26 genomes, see Supplementary Table S3. Noteworthy, we observed that Francisella philomiragia GA012794 and Francisella philomiragia GA012801, while named as belonging to the same species, show only about 93% ANI, according to the comparable algorithms ANIb (93.63%) and OrthoANI (93.9%), thus questioning their species belonging (Supplementary Table S3). The 26 genomes were found to represent two major groups; a large cluster which comprised all the human pathogens and for which the strains showed an ANI of 97.0 - 99.5%, and a second cluster that comprised strains that predominantly are environmental or water-related, and with ANI values of 74.2–90.4% (Figure 1). Importantly, the minor variation (32.3 ± 0.4) in the G + C content of this genome dataset was indicative of a stable boundary delineation within the genus (Table 1).
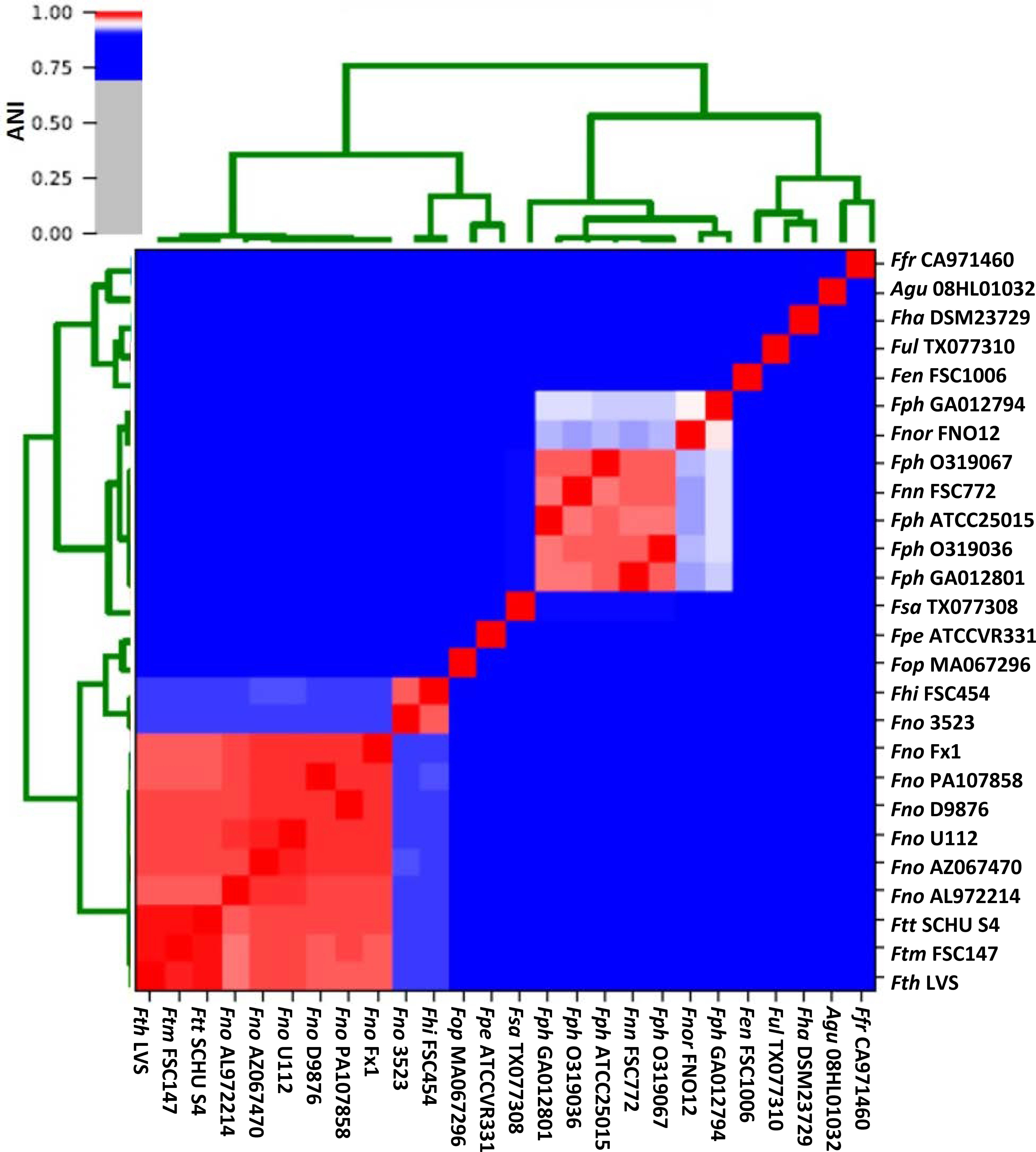
Figure 1. Average Nucleotide Identity (ANI) demonstrating nucleotide-level genomic similarity between the coding regions of indicated Francisella genomes. Pairwise comparisons for all 26 complete genomes were computed by BlastN using the Pyani Program. For strain abbreviations, see Table 1.
In the genus Legionella, a total of 77 complete genome assemblies were used for ANI analysis. Using the same down-selection process as for Francisella, 35 genomes were selected for further pan-genome analysis. The total genome size was larger than that of Francisella, and more diverse in sequence, since the minimum ANI was approximately 71%. The largest cluster within the genus belonged to species L. pneumophila and strains thereof, and showed an ANI of > 96%. All available genomes from the genus Piscirickettsia (19 in total) were derived from only two species, P. salmonis and P. litoralis, and showed ANI values ranging from 95.7–99.9% (data not shown).
Bacterial genomes are dynamic entities that harbor essential genes and accessory elements, which may be unique to each community. The so called ‘core’ genomes constitute conserved genes present in all strains studied, while ‘dispensable’ genomes (also known as flexible or accessory genomes) are composed of genes absent from one or more of the strains (Tettelin et al., 2005). The latter usually pertains to supplementary biochemical pathways and functions that may confer a selective advantage to the microbe, such as ecological adaptation, antibiotic resistance, virulence mechanisms, or colonization of a new host. To estimate the pan- and core-genome sizes of Francisella, we used our down-selected 26 genomes, from 14 Francisella species, and the binomial mixture model of Snipen and collaborators (Snipen et al., 2009) and Tettelin and collaborators (Tettelin et al., 2005). We observed that the more genomes analyzed (i.e., increasing the data set), the bigger the estimated pan-genome size. At the same time, the rate of the increase was going down (Figure 2A). Thus, since the core/pan-genome ratio did not reach a distinct sharp plateau, we conclude that Francisella has an open pan-genome (Figures 2A,B).

Figure 2. Pan-genome analysis of 26 Francisella genomes from 14 species. Estimates of pan-genome size (A) and (B) core-genome size, both with the Tettelin fit.
Based on the 26 Francisella genomes (Table 1), and the use of three different algorithms (for details see section “Materials and Methods”) the pan-genome of the genus was predicted to comprise 4,053 genes. Amongst these, 692 genes (709 including paralogs) constituted the core genome, i.e., genes present in all genomes included in the analysis (Table 2 and Figure 3A). The core genome in turn, constituted approximately 36.1% of the mean number of CDS (692 vs. 1,915) (Table 2). Together with the soft-core genomes, i.e., genes present in 95% of all genomes included in the analysis (Supplementary Figure S1; Kaas et al., 2012), these 977 and highly conserved genes may provide information about the evolutionary history of the members of a genus. The remaining genes of the pan-genome were accessory genes, of which 2,179 constituted the cloud genome, i.e., strain-specific and rare genes present only in a few genomes (Vernikos et al., 2015), which might be rapidly gained or lost (Collins and Higgs, 2012). The remaining 897 genes constituted the shell genome, i.e., moderately conserved and dispensable genes, present in one or several genomes (Supplementary Figure S1). The cloud and shell genome subsets reflect both the evolutionary history of a lineage as well as adaptation of an organism to its particular environment (Nelson and Stegen, 2015).

Table 2. Comparative core- and pan-genome analysis of the genera Francisella, Legionella, and Piscirickettsia.
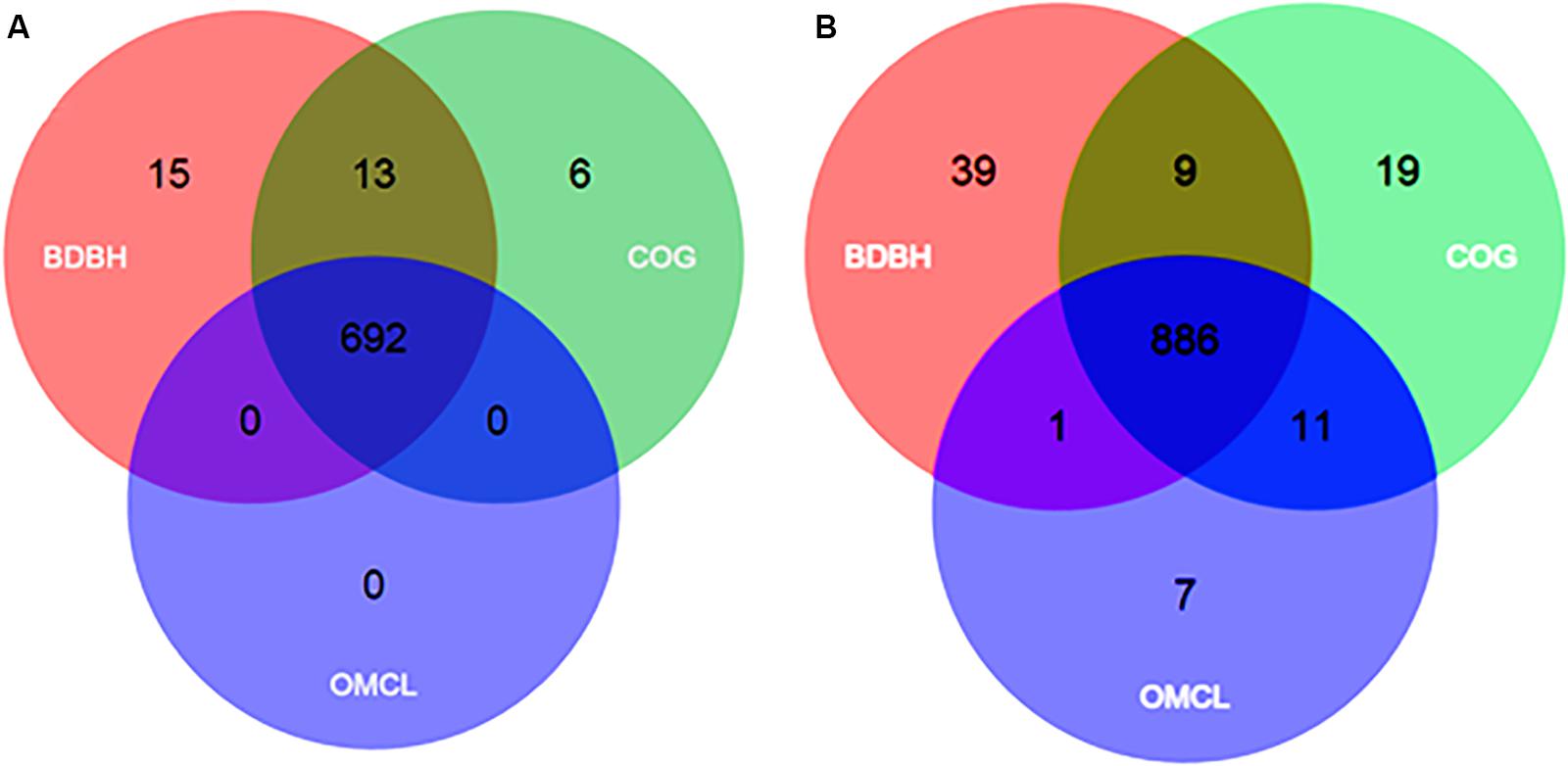
Figure 3. Venn diagrams of core genomes from Francisella (A) and Legionella (B) generated by the BDBH, COG and OMCL strategies, using the GET_HOMOLOGUES tool. Singletons (genes present in only one copy in any genome) from 26 and 77 representative species sequences respectively were used as input.
In Francisella, approximately 71% of the strain-specific genes were predicted to encode hypothetical proteins, while 29% encode functionally characterized proteins. The total number of coding genes and the genome size for each of the 26 representative Francisella genomes are provided in Table 1. From the core genome analyses, AAI (Average Amino-acid identity) was calculated using protein-coding sequences (CDSs) of the 26 selected genomes. A heat-map representing the degree of similarity of the genomes based on the average amino acid identities of their CDSs was constructed (Figure 4), demonstrating the formation of two distinct groups. The observation also illustrates the microbial evolution and displays a functional relationship between different Francisella strains as well as species obtained from variable environments.
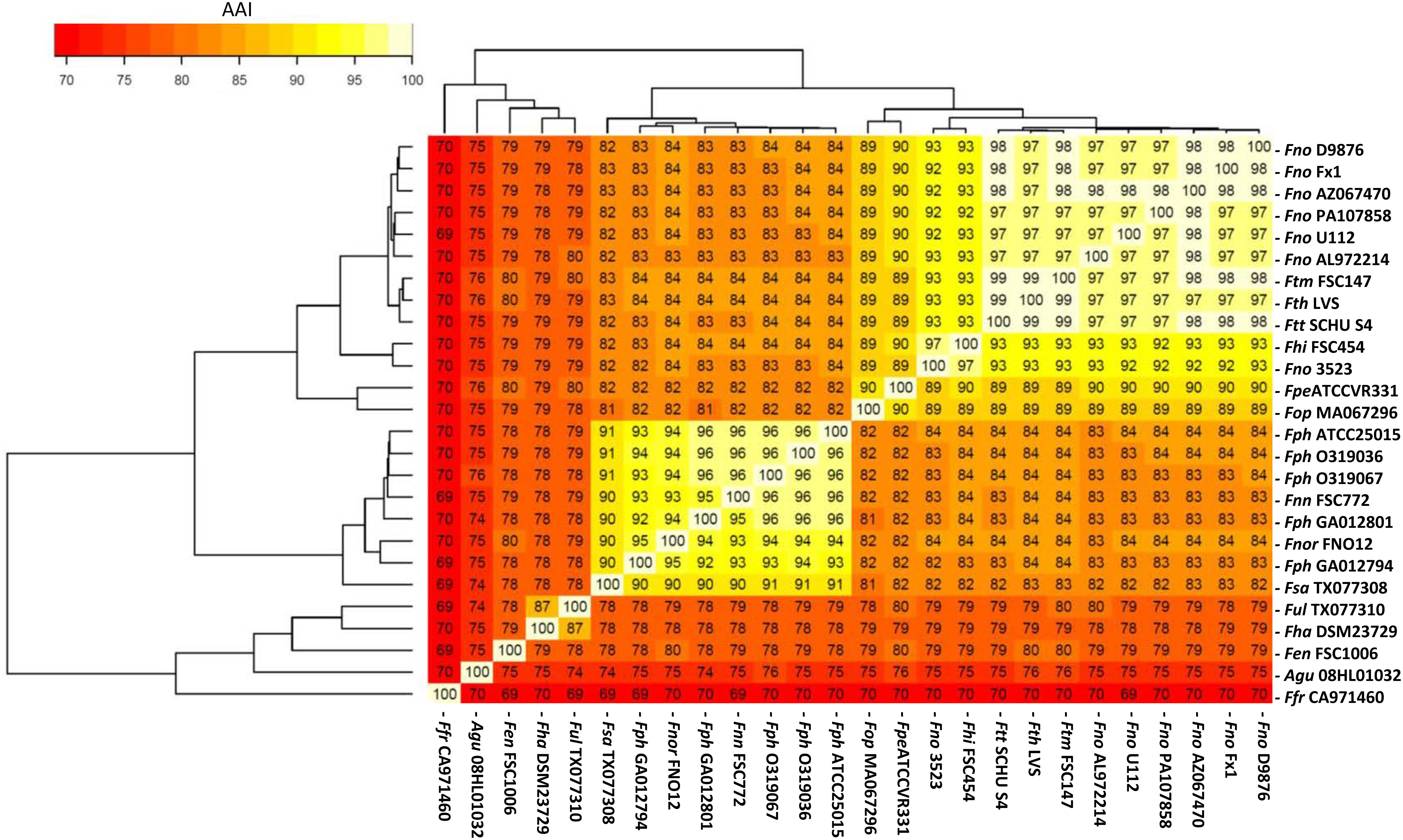
Figure 4. A heatmap representing the degree of similarity of genomes based on the average amino acid identities of their protein coding genes. The heatmap was derived from the high similarity (light yellow) and low similarity (dark orange) of CDSs derived from the 26 Francisella genomes. For strain abbreviations, see Table 1.
By using the same approach as for Francisella, the core- and pan-genomes of the genus Legionella were estimated to be 886 and 8,413 genes, respectively, while the corresponding numbers for Piscirickettsia were 1,324 and 4,178 genes, respectively (Table 2 and Figure 3B). It should be noted that the core-genome size of Piscirickettsia may be affected by the lack of genomes of other species than P. salmonis and P. litoralis, and therefore appear to be larger than those of Francisella and Legionella (Nourdin-Galindo et al., 2017). We also compared the core-genome size to the mean number of CDS per genome. For Legionella this corresponded to 29.2% (886 vs. 3031) and for Piscirickettsia to 44.2% (1323 vs. 2995) (Table 2). Furthermore, the “core of core” within all the three genera comprised 263 genes, while the corresponding numbers within Francisella and Legionella were 383 genes, within Piscirickettsia and Francisella 399 genes, and within Legionella and Piscirickettsia 472 genes (Table 3). To assign biological functions to the genus orthologs (“core of core”), the corresponding amino acid sequences for all 263 shared genes were annotated using COG. This revealed that the majority (25.7%) of the proteins belonged to the COG category Translation, ribosomal structure and biogenesis, 7.5% to Energy production and conversion, 7.6% to Post-translational modification, protein turnover, and chaperones, and 3.3% were poorly categorized, or with unknown function (Supplementary Figure S2). We also mapped the protein cellular functions using KEGG. Genes were divided into five branches according to the biological pathways they are likely to participate in and the percentage of genes belonging to a particular category calculated to be as follows: (A), Cellular Processes (1.7%); (B), Environmental Information and Cellular Processing (6.8%); (C), Genetic Information Processing (26.9%); (D), Metabolism (63.1%); (E), Organismal Systems (0.39%), and (F), Human diseases (2.8%) (Supplementary Figure S3).

Table 3. The size of the common core-genome shared between different genera.
Phylogenetic relationship of bacteria is usually estimated by comparing sequences of homologous genes, typically the 16S rRNA gene. In the case of Francisella, however, the differences within the 16S rRNA sequence are very few (Challacombe et al., 2017), requiring the use of an alternative approach. While single gene-based phylogenetic trees have low inter-species discriminatory power, multi-gene approaches offer the possibility to create more robust phylogenetic trees (Castresana, 2007; Satoh et al., 2013). Thus, we explored the genetic diversity within the genus Francisella by inferring the phylogenomic relationship based on the genomic content. For this purpose, we used the up-to-date bacterial core gene set, UBCG, consisting of 92 core genes from 1,492 bacterial species covering 28 phyla. This robust phylogenomic method is universally applicable to any phyla of the domain Bacteria (Na et al., 2018). The obtained results clearly indicated two major and distinct clades, A and B, and an additional and diverse cluster designated C (Figure 5). Most strains within clade A are pathogenic to mammals, e.g., members of the species F. tularensis and subspecies thereof, while clade B includes strains found in the marine environment, most of which are pathogenic to fish, but also some potentially pathogenic to humans, e.g., F. philomiragia and F. noatunensis and its subspecies. Clade B is more disparate than clade A. Clade C was found to comprise A. guangzhouensis 08HL01032T, the species F. frigiditurris sp. nov. CA971460, F. endociliophora FSC1006, F. uliginis sp. nov. TX077310, and F. halioticida DSM23729, most of which are associated with the marine environment (Table 1). Four species, F. hispaniensis FSC454, F. cf. novicida 3523, F. opportunistica sp. nov. MA067296, and F. persica ATCC VR331, differentiated into two groups and formed a small cluster phylogenetically rather close to clade A (Figure 5). This global phylogenomic-based analysis also supported the ANI and AAI hierarchical cluster-based dendrograms (Figures 1, 4). In addition to the aforementioned phylogenetic approaches, we also assessed the phylogeny based on the non-recombinant loci alignment, as a means to construct a phylogenetic tree of more accurate and precise topology. The 692 core genes (Table 2) were used for evaluating the phylogenies based on encoded proteins as well as DNA content. Top scoring phylogenetic markers were selected based on the criteria recommended by Vinuesa (Vinuesa et al., 2018), i.e., they should (i) be non-recombinant (Kaas et al., 2012), (ii) show a robust phylogenetic signal, and (iii) result in a coherent phylogenetic tree. In total, 43 proteins and 236 DNA-based markers were used for maximum likelihood (ML) phylogenetic estimation, generating two trees of almost identical topology (Figure 6 and data not shown), confirming that our phylogeny is correct and optimal. Like the UBCG as well as ANI-based phylogenetic trees, the marker-based phylogenetic tree also formed three main clades (Figure 6).
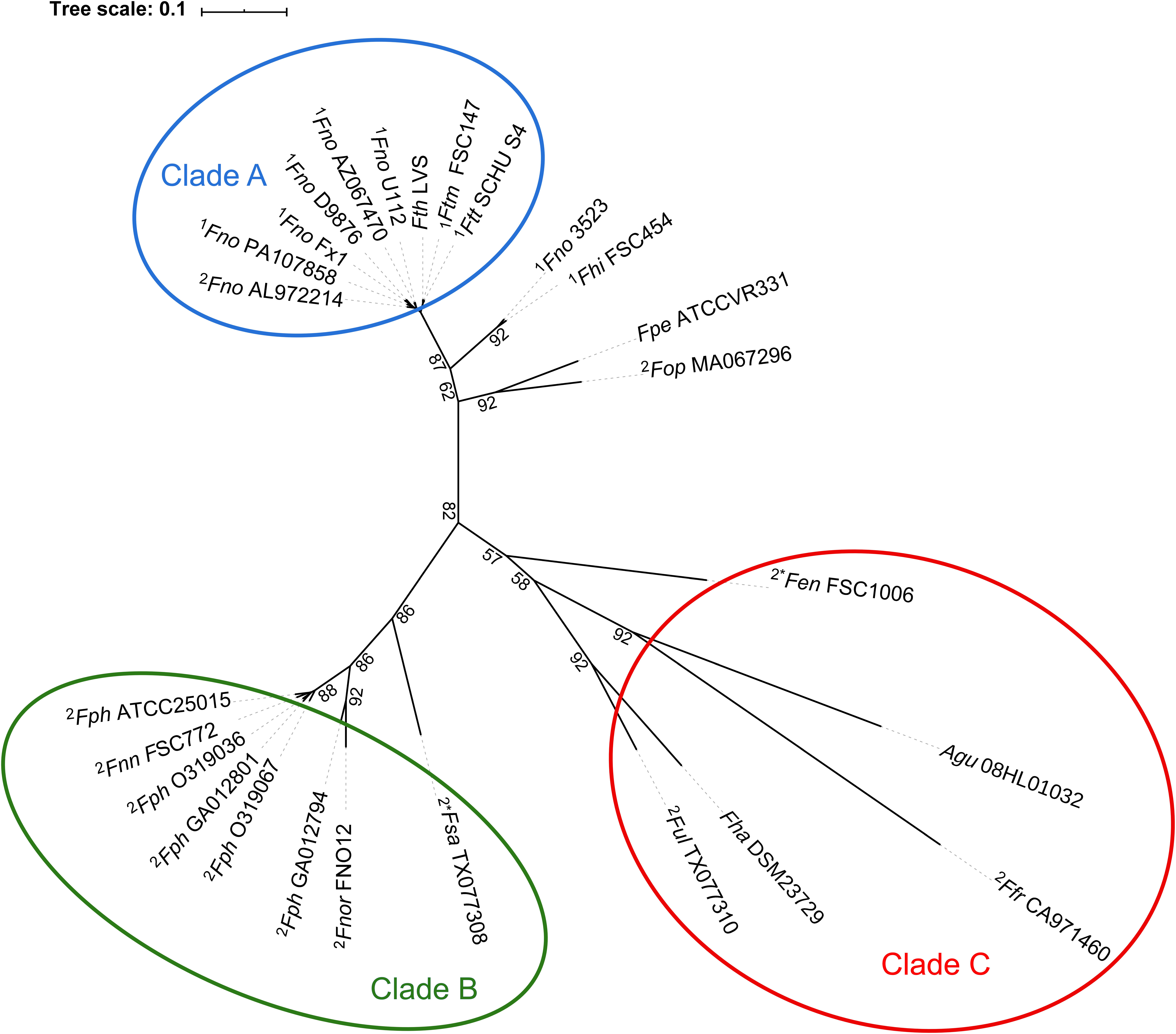
Figure 5. Phylogenomics tree reconstruction by the UBCG software, using standard settings based on 92 up-to-date bacterial core genes, revealing three major clades (A–C). Bootstrap values are presented at the branching points. For strain abbreviations, see Table 1. The numbers 1 or 2 indicate that a given genome belongs to one of the two major groups identified with respect to FPI gene content; group 1 (complete FPI island with 18 genes) or group 2 (incomplete FPI, lacking the pdpC and pdpE genes). An asterisk indicates that additional FPI genes are missing for group 2 members. For genomes without numbers, see Table 1 for a description of their FPI gene content. Scale bar equals 0.1 substitutions per nucleotide position.

Figure 6. A maximum likelihood (ML) phylogenetic tree based on the non-recombinant loci concatenated set of top-ranking phylogenetic markers, revealing three major clades (A–C). Bootstrap values are presented at the branching points. For strain abbreviations, see Table 1. Scale bar equals 0.1 substitutions per nucleotide position.
Sjödin et al. (2012) previously reported a divergence of the Francisella genus into two distinct clades, with clade A comprising F. tularensis, F. novicida, F. hispaniensis, and F. persica, and clade B containing F. philomiragia and F. noatunensis. Our comprehensive phylogenetic analysis also confirmed this bifurcation of Francisella into two clades, with the addition of a third clade, clade C. Notably, in the UBCG analysis, the F. persica ATCC VR331 and F. opportunistica sp. nov. MA067296 showed a common ancestor and were closer to clade A, while in the selected marker-based trees, these species are more disparate (Figure 6). The clade A of the marker-based phylogenetic tree comprised F. tularensis and subspecies thereof, with the addition of F. cf. novicida Fx1 (Figure 6). Overall, our results based on selected markers therefore give additional support to the core-genome-based phylogenomic tree of the genus Francisella.
The Francisella pathogenicity island (FPI) is a cluster of 16–19 genes, present in most of the Francisella genomes that have been sequenced to date. Although 16 FPI genes are highly conserved, 2–3 genes are absent or interrupted by stop codons in some strains (Nano and Schmerk, 2007). Intriguingly, the highly virulent Francisella strains contain two copies of the entire FPI, while the less virulent Francisella strains have a single copy (Spidlova and Stulik, 2017). We found that depending on species analyzed, the overall G + C content of the FPI was 3–5% lower than for the rest of the Francisella genome, ∼32% (Supplementary Table S4). Moreover, significant variations in G + C content within this region were also noted (data not shown; Nano et al., 2004). In support, a comparison of proteins encoded within the FPI and outside of the FPI demonstrated that the most over-represented amino acids within the FPI correspond to lysine, asparagine and serine, all of which are encoded by GC-poor codons (Supplementary Table S5). In contrast, the most under-represented amino acids within the FPI corresponded to alanine, glycine, valine, tryptophan, i.e., GC-rich codons, as well as methionine (Supplementary Table S5). Similar results were obtained for all of the four genomes investigated, i.e., F. tularensis subsp. holarctica LVS, F. noatunensis subsp. noatunensis FSC772, F. cf. novicida Fx1 and F. novicida U112 (Supplementary Table S5). To search for FPI genes within our 26 representative genomes, we used the MultiGeneBlast program and the FPI island of F. novicida U112 as query. Our results show that all of the 26 Francisella genomes had at least one copy of the FPI, except for A. guangzhouensis 08HL01032T (data not shown) and F. halioticida DSM23729, for which only the genes encoding IglA and IglB, i.e., the T6SS sheath proteins, were detected. The F. tularensis subsp. holarctica LVS, subsp. mediasiatica FSC147, and subsp. tularensis SCHU S4 all have two copies of the FPI as shown in Table 1 and Supplementary Figure S4. Two out of the 26 genomes, those from F. endociliophora FSC1006 and F. salina sp. nov. TX077308, have a single FPI copy with three or more of the FPI genes missing or inactivated (Table 1 and Supplementary Figure S4). Interestingly, F. philomiragia GA012794 and F. endociliophora FSC1006 possess two additional T6SS clusters, both of which lack significant homology to the FPI cluster. Instead, our phylogenomic analysis suggested that they show most similarity to the T6SS of Escherichia coli (data not shown).
Based on FPI gene content and organization, two major groups could be distinguished within the Francisella genus. The first is characterized by the presence of an intact FPI cluster and includes, e.g., F. hispaniensis FSC454, F. tularensis subsp. mediasiatica FSC147 and F. tularensis subsp. tularensis SCHU S4, F. novicida U112, F. novicida PA107858, F. novicida D9876, F. novicida AZ067470, F. cf. novicida 3523, and F. cf. novicida Fx1. Most of the species belonging to this group clustered to clade A in the phylogenetic tree analysis. The second group is characterized by the presence of an FPI cluster, which lacks both the pdpC and pdpE genes. This group included, e.g., all strains of F. philomiragia and F. noatunensis, and F. noatunensis subsp. orientalis, F. frigiditurris sp. nov. CA971460, F. opportunistica sp. nov. MA067296, F. uliginis sp. nov. TX077310, F. salina sp. nov. TX077308, F. novicida AL972214, and F. endociliophora FSC1006, the latter being unique in that its FPI also lacks pdpD, anmK, and iglI, and exhibits gene rearrangements (Table 1, Supplementary Figure S4, and Figure 7). With the exception of F. opportunistica sp. nov. MA067296 and F. novicida AL972214, all of this group belong to clade B or clade C according to our analysis. In addition, other variants of the FPI cluster were predicted from the analysis (Figure 7). For example, strain F. persica was found to lack the entire pdpD gene, while the same gene is truncated in both loci of the F. tularensis subsp. holarctica strain LVS. The anmK gene exists as two distinct truncated forms in F. tularensis subsp. tularensis, but is absent in subsp. holarctica (Figure 7 and Supplementary Figure S4). Recently, Brodmann et al., 2017 reported that pdpC, pdpD, pdpE and anmK are dispensable for T6S.
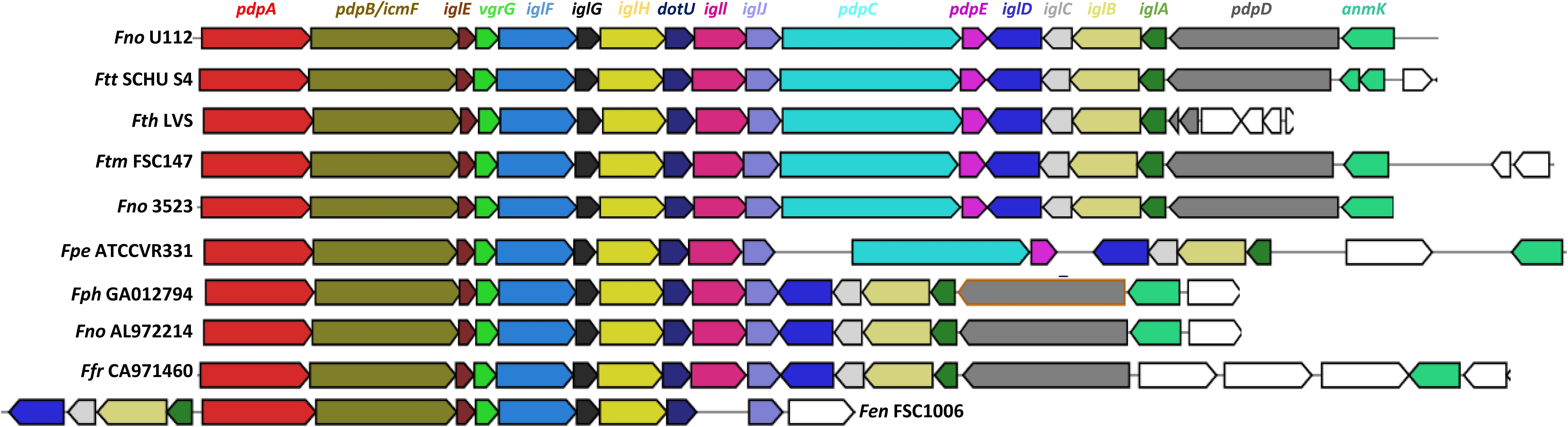
Figure 7. Comparative analysis of T6SS clusters in the genus Francisella. Shown are clusters from some representative species belonging to clades A and B that were identified in Figure 5. For strain abbreviations, see Table 1.
We also searched the NCBI database for FPI homologs present outside of the genus Francisella using the BlastP program. As reported before, a few FPI proteins had homologs in T6SSs belonging to a wide range of species, e.g., IglA, IglB, and DotU, many of which have been demonstrated to be functionally conserved (De Bruin et al., 2007; Bröms et al., 2010, 2012). Interestingly, this category also included IglG, and to some extent IglI, both of which previously were reported to lack homologs in other bacteria (Bröms et al., 2011). Also homologs of AnmK were found in other bacterial species, as well as outside of the FPI cluster within Francisella. Based on homology, anmK is predicted to encode an anhydro-N-acetylmuramic acid kinase. In contrast, we could not find any homolog to PdpC outside of the genus Francisella. For the remaining FPI components, only one or a few homolog(s) outside of the genus exist(s), and then primarily in species closely related to Francisella, such as Piscirickettsia sp., Cysteiniphilum sp., Fangia hongkongensis, and Pseudofrancisella aestuarii. Taken together, our comparative analysis of the FPI gene cluster demonstrates that the FPI genes are highly similar within the genus, but share low similarities with T6SS genes of other bacterial species.
Effector protein identification is critical for the understanding of how the Francisella FPI promotes pathogenesis. So far, a few putative effectors encoded within the FPI have been identified by the use of different reporter assays (Barker et al., 2009; Bröms et al., 2012) and, more recently by a proteome-based approach combined with quantitative mass spectrometry (Eshraghi et al., 2016). Interestingly, the latter study also identified putative effector proteins encoded outside of the FPI for F. novicida, including OpiA. In a follow up study, this protein was shown to possess phosphatidylinositol 3-kinase-activity, alter phagosomal maturation, and, thereby, promote intracellular growth of F. novicida (Ledvina et al., 2018).
To search for putative T6SS effector proteins within the genome of F. tularensis subsp. tularensis SCHU S4, we used the Bastion6 machine learning predictor to identify putative T6SS effectors. A total of 144 promising candidates, all with a predicted ensemble score above 50%, were retrieved using Bastion6. All candidates, except for PdpB and PdpD, were encoded outside of the FPI. For further details about the hits, see supporting information in Supplementary Table S6. PANNZER2 in combination with gene ontology were used to functionally describe and annotate the putative effectors further. This analysis demonstrated that more than 1/3 of the putative effectors are predicted to act on cellular targets including the peptidoglycan cell wall (hydrolases), cellular nucleic acids and proteins (nucleases and proteolytic enzymes respectively), as well as the inner membrane (phospholipases) (see Supplementary Table S7 for more details about the proteins putative function and localization). Among the top-ranked hits, three were predicted to possess hydrolase activity and, according to the Carbohydrate-Active Enzymes (CAZymes: http://www.cazy.org/) analysis, constitute members of the glycosyl hydrolases family 18. Four putative effectors had protein domains of no characterized function, i.e., DUF1338, DUF2147, DUF4124, and DUF4440 (Supplementary Table S7). Further investigation using the Pfam database suggested that these hits may be a putative metal hydrolase, a member of the lipocalin family, to possess an immunoglobulin-like (Ig-like) fold and to be a member of the nuclear transport factor 2 (NTF-2)-family, respectively. Among the putative effectors, we also identified an OmpA family protein (outer membrane lipoprotein; Supplementary Table S7). OmpA is a peptidoglycan-binding protein that is not physically part of the T6SS clusters, but has been suggested to share a functional relationship with some T6SS proteins (Shrivastava and Mande, 2008).
While we failed to identify any homologs to the ion-selective pore-forming T6SS effectors that were recently identified and suggested to be widespread within Enterobacteriaceae (Mariano et al., 2019), we also carried out an analysis searching for effectors with the previously identified N−terminal domains named MIX (Marker for type six effectors). Previously, T6SS effectors of various Proteobacteria were demonstrated to share this conserved motif and to group into five clans named MIX I-V (Salomon et al., 2014). We used known MIX sequences from representative clan members to search for MIX effectors in the genus Francisella. We failed to identify putative MIX effectors belonging to the MIX-II, III and IV clans, however, two Francisella proteins showed low sequence similarity to either MIX-I or MIX-V clan members. Both predicted effectors are mainly found in the marine and fish-pathogenic strains (Clade B), and in some mammalian pathogenic-species of Francisella (Clade A). The first putative effector (MIX-I) is a conserved hypothetical protein (locus tag: “FTT_1768c”), functionally predicted to be a Chitinase/glycoside hydrolase family 18 protein and also identified as a putative effector in the Bastion6 machine learning based predictor for T6SS effectors (Supplementary Table S7). The second (MIX-V) is an uncharacterized protein of the DUF3568 family (locus tag: “FTT_1416c”). Upon further comparative analysis of this protein, we found that members of this family are approximately 120–130 amino acids long and contain a highly conserved cysteine residue within the N-terminus. In agreement with a putative role as lipoproteins, the first 25 amino acids of the N-terminus were predicted to form a signal peptide, suggesting that prelipoproteins belonging to this family would be cleaved directly upstream of the conserved cysteine. Interestingly, some Francisella genomes were found to have duplicate or triplicate copies of the DUF3568-containing gene, including F. endociliophora strain FSC1006, F. halioticida strain DSM23729, and F. tularensis subsp. tularensis strain WY96. A DUF3568 neighborhood analysis did not provide any evidence for an association with the T6SS (data not shown). Remarkably, the DUF3568 domain-containing protein originally reported as F. tularensis Virulence Determinant protein (i.e., Flpp3) has been suggested to share structural homology to Bet v1 allergen proteins (Zook et al., 2015). Taken together, this analysis has revealed the presence of putative T6SS substrates encoded outside of the FPI within the Francisella genome. Functional characterization will be needed to determine whether they are indeed T6S substrates and if they contribute to bacterial virulence.
Bacterial taxonomy based on 16S rRNA sequencing has since long been the most important parameter to explore the phylogenetic relationships of bacteria and to assign genus- and species-belonging. A drawback, however, is that the resolution of the method is normally not sufficient to discriminate subspecies and that it is vulnerable to biases depending on primer sequence-matching in different species (Chan et al., 2012; Rosselli et al., 2016). Also, phenotypical and biochemical characteristics have been used as a basis for phylogenetic determination, however, these are traits that to some extent can be affected by choice of culture medium and other conditions (Tindall et al., 2010). Therefore, objective methods that show high resolution need to be implemented. One promising and rather often used method in this regard is based on determination of the relatedness by calculating the average nucleotide identity, as previously described (Han et al., 2016).
The present study constitutes a comprehensive comparative genomic characterization of the genus Francisella. The characterized divergences and similarities identified here represent an important contribution toward understanding the biology and evolution of Francisella. Importantly, the minor variation (32.3 ± 0.4) in the G + C content of this genome dataset was indicative of a stable boundary delineation within the genus. The distinctly lower G + C content of the FPI suggests that horizontal gene transfer has been a major factor driving the evolution of the FPI of Francisella. Indeed, Nano et al., suggested that the FPI originally had been acquired through horizontal gene transfer from an organism with a lower G + C content (Nano et al., 2004). Our findings support their conclusion and, additionally, we could demonstrate a distinct bias for GC-poor codons within the FPI. Thus, our findings are in agreement with findings in eubacterial and archaeal genomes demonstrating that a biased nucleotide-content causes a divergent amino acid composition of the encoded proteins (Singer and Hickey, 2000). In contrast, Larsson et al., postulated that the ancestor had been an organism with a higher G + C content, but our findings do not support the hypothesis (Larsson et al., 2009).
Our phylogenetic trees were based on analyses including the core genome, ANI, and non-recombinant loci alignment of 26 completely sequenced genomes. Since a multitude of analyses, including the established method UBCG that includes up-to-date core genes in the analysis, were performed and gave congruent results, the findings strongly corroborate previous phylogenetic analyses and further refine the relationships within the genus. This is the first time that UBCG has been implemented for the genus Francisella. Regardless of method used, the analysis provided unequivocal evidence for the existence of two genogroups, Clade A and Clade B, which has also been reported previously (Sjödin et al., 2012). Our phylogenetic trees closely resemble those previously reported by Sjödin et al. and Challacombe et al., but the variety of methods used in our study add much more robustness to the composition of the phylogenetic trees obtained. Clade A comprised mostly human pathogenic strains, predominantly belonging to F. tularensis, whereas clade B was more diverse and encompassed fish pathogens and strains rarely pathogenic to humans, such as F. noatunensis and F. philomiragia. The analysis also identified phylogenetic positions for recently characterized strains such as F. cf. novicida 3523, F. frigiditurris sp. nov. CA971460, F. opportunistica sp. nov. MA067296, F. uliginis sp. nov. TX077310, and F. salina sp. nov. TX077308.
The study by Challacombe et al. characterized four new species of the genus Francisella and demonstrated that the demarcation of new species in bacteria is quite challenging (Challacombe et al., 2017). This is in particular the case for isolates with similar genomic characteristics, but different physiological features, e.g., some being pathogenic, whereas others are opportunistic pathogens, or even non-pathogenic (Challacombe et al., 2017). The analyses by Challacombe, based on ANI, 16S rRNA, or a multilocus sequence typing scheme, gave congruent results and overall also agree with the taxonomic positions we identified. Collectively, the findings support the use of genomic analyses as a basis for species delineation and demonstrate a robustness in the phylogenetic trees of the genus. Thereby, the methods utilized herein are potent tools for a precise delineation of the taxonomical belonging of strains that will be identified in the future. In addition to the aforementioned study, Dietrich et al. reported the identification of three isolates of F. opportunistica sp. nov., from human blood and cerebrospinal fluid, which showed ANI inter-strain similarities of 99.9%, and 88.6% to the closest relative, the tick endosymbiont F. persica (Dietrich et al., 2019). In agreement, our ANI analysis of 26 complete genomes of Francisella demonstrated ANI values > 95% within species, and 74–95% between species. These values also concurred with the conclusions of the study by Appelt et al. (2019) in which F. tularensis isolates from Switzerland were analyzed. In this study, an ANI threshold of 99.5% was postulated to distinguish subspecies from each other.
In our analyses, we also included A. guangzhouensis strain 08HL01032T to determine its phylogenetic relationship with the genus Francisella. Prior to 2016, this strain was considered a member of the genus Francisella, however, based upon 16S RNA- and multilocus sequence typing-based analyses, it was reclassified as a separate genus (Qu et al., 2016). To date, this is the only complete genome available for this genus, but a scaffold assembly exists for A. inopinata. Interestingly, the phylogenetic tree obtained from the core genome comparative analysis clearly indicated that A. guangzhouensis 08HL01032T is an outlier, separate from the two main clusters of Francisella strains. However, it clustered with F. frigiditurris sp. nov. and the same relationship was also confirmed in the protein marker-based phylogenetic tree. Our further in-depth analysis concluded that these two strains of A. guangzhouensis and F. frigiditurris sp. nov. exhibited very similar ANI values vs. the SCHU S4 strain (74.5% vs ∼74%), the latter being the lowest value of all 26 Francisella genomes analyzed. Interestingly, in the recent study by Challacombe et al., F. frigiditurris sp. nov. was suggested to be a new member of the genus Francisella (Challacombe et al., 2017). Thus, A. guangzhouensis 08HL01032T may be closer to the genus Francisella than previously considered (Qu et al., 2016), and the classification of this strain as a member of a separate genus is therefore not clear-cut.
The nucleotide diversity was rather similar for Francisella and Legionella, 74% and 71%, respectively. The pan-genome of the latter was considerably larger, comprising 8,413 genes, whereas that of Francisella encompassed 4,053 genes. Of these, 692 genes, represented the core-genome, whereas the corresponding number for Legionella was 886 genes. The core genes are expected to play a role in the ability of these intracellular pathogens to survive within the specialized environment of phagocytic cells and protozoa, respectively. Still, as evidenced by the differences in the size of their pangenomes, both pathogens demonstrate a distinct genetic composition that likely contributes to unique features for the two genera. In this regard, a drawback in the genetic analysis of Francisella is the plethora of unannotated genes, however, a majority of these could still be assigned a function using COG or KEGG.
The FPI is essential for the virulence of Francisella and encodes a Type VI secretion system (T6SS) (Nano et al., 2004; Bröms et al., 2010). All of the 26 Francisella genomes possess at least one FPI copy, with the exception of F. halioticida DSM23729, for which only the genes encoding the T6SS sheath proteins, IglA and IglB, were detected. A. guangzhouensis 08HL01032T also lacked the island, as reported previously (Challacombe et al., 2017). Both, together with F. frigiditurris sp. nov. CA971460, exhibited among the lowest ANI values overall in our analysis. Since the latter strain possesses a typical FPI, low ANI values does not correlate with the absence of the FPI in the genome. Upon analyzing FPI gene content and organization, several groups could be distinguished, including those that (i) lacked the entire FPI, i.e., F. halioticida DSM23729, (ii) possessed one complete FPI copy or more, e.g., F. hispaniensis FSC454 (1 copy) and F. tularensis subsp. tularensis SCHU S4 (2 copies), (iii) lacked both of pdpC and pdpE, e.g., F. philomiragia, (iv) lacked a functional pdpD gene, i.e., F. persica ATCC VR331 and F. tularensis subsp. holarctica LVS, or (v), lacked all of pdpC, pdpD and pdpE genes (F. endociliophora FSC1006). Similar results were obtained in the previous study by Challacombe et al., which was based on 31 Francisella genomes in total (Challacombe et al., 2017). The advantage of using a larger genome data set is the possibility of finding unique FPI patterns not discovered before, however, all of the additional genomes that we included in our study could be sorted into the previously categorized FPI groups. We did, however, make one interesting observation, since we observed an additional FPI genogroup as represented by F. endociliophora FSC1006. The strain lacks pdpC, pdpD, pdpE as well as anmK, as previously reported (Challacombe et al., 2017), but, in addition, we identified a lack of the iglI gene. Thus, the repertoire of FPI variants is more diverse than previously reported. The additional genomes that have been sequenced upon completion of this study may add to this complexity. The lack of pdpC and pdpD in certain strains was reported previously (Eshraghi et al., 2016). The two genes have previously been suggested to encode effector proteins, in fact, pdpD was identified as an effector also in our computational screen. Thus, the acquisition of pdpC and pdpD genes may have been an important step toward pathogenesis in mammals, possibly facilitating host tropism. The role of pdpE is less clear, since studies indicate that mutant is as virulent as the parental strain (Bröms et al., 2011). Nevertheless, since loss of pdpE always is accompanied by loss of pdpC, our results suggest that these two proteins somehow may interact.
While the repertoire of effector proteins is quite abundant for some T6SS, e.g., V. cholerae, a modest number of substrates has been identified for the Francisella T6SS (Bröms et al., 2012; Eshraghi et al., 2016). Naturally, this could simply be a consequence of low effector abundance, choice of strain and/or method to quantify secretion. Our findings of 144 promising candidates, most of them encoded outside of the FPI, therefore constitute interesting targets for site-directed mutagenesis. Among the top-ranked hits, we identified, e.g., glycosyl hydrolase active enzymes. One of the candidates was FTT_1768c, which shares some homology to MIX-I effector proteins, and was functionally predicted to be a Chitinase/glycoside hydrolase family 18 protein. In fact, the FTT_1768c protein was identified in a high-throughput yeast two-hybrid assay, revealing putative physical interactions to human proteins, including Vps35 (Vacuolar protein sorting-associated protein 35) (Dyer et al., 2010). The latter is a core component of the retromer complex, which controls vesicular transport within eukaryotic cells and consists of a membrane-associated sorting nexin dimer and a vacuolar protein sorting (Vps) trimer. Because of its essential role in vesicle trafficking, this transport pathway has emerged as an important target for intracellular bacterial pathogens to promote their survival and replication. For example, VPS35 and VPS26A, both components of the retromer, were recently shown to be required for the diversion of Brucella-containing vacuoles (BCVs) from the endolysosomal pathway and the establishment of the intracellular replicative niche (Casanova et al., 2019). Moreover, the Dot/Icm effector RidL of L. pneumophila inhibits retromer activity to promote intracellular replication by directly binding to the retromer subunit VPS29 (Finsel et al., 2013), thereby outcompeting essential retromer regulators (Yao et al., 2018). This raises the question of whether our identified hit, the Chitinase/glycoside hydrolase family 18 protein, plays a similar role in vesicle trafficking and intracellular survival of Francisella, and whether this involves a direct physical interaction with the retromer. To our knowledge, this has not been investigated. Interestingly, this putative effector is highly conserved among the different subspecies of F. tularensis, >99% identity, but is less conserved within the species F. philomiragia and F. noatunensis, 37–52%, that only rarely infect humans, possibly reflecting a difference in function. These candidate genes may therefore constitute interesting targets for designing novel strategies to prevent and control infections with species that belong to this highly diverse and environmentally adapted genus.
Collectively, the comparative genomic analysis performed provides a comprehensive basis for the assessment of the phylogenomic relationship of members of the genus Francisella and for the identification of putative T6SS virulence traits.
All datasets presented in this study are included in the article/Supplementary Material.
RK, AS, and JB designed the study, analyzed the data, and wrote the manuscript. RK performed all experiments.
RK acknowledges the MIMS and UCMR for funding at Umeå University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Igor Golovlev and Athar Alam for fruitful discussions. We acknowledge research funding for this work by grants 2013-4581 and 2013-8621 from the Swedish Research Council and a Biotechnology grant from the Medical Faculty, Umeå University, Umeå, Sweden (FS 2.1.6-2291-18), and the JC Kempe Memorial Foundation (JCK-1624).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01928/full#supplementary-material
AAI, Average Amino acid Identity; ANI, Average Nucleotide Identity; DUF, domain of unknown function; F, Francisella; FPI, Francisella Pathogenicity Island; MIX, Marker for type six effectors; T6SS, Type VI secretion system.
Appelt, S., Koppen, K., Radonic, A., Drechsel, O., Jacob, D., Grunow, R., et al. (2019). Genetic diversity and spatial segregation of Francisella tularensis subspecies holarctica in Germany. Front. Cell. Infect. Microbiol. 9:376. doi: 10.3389/Fcimb.2019.00376
Barker, J. R., Chong, A., Wehrly, T. D., Yu, J. J., Rodriguez, S. A., Liu, J. R., et al. (2009). The Francisella tularensis pathogenicity island encodes a secretion system that is required for phagosome escape and virulence. Mol. Microbiol. 74, 1459–1470. doi: 10.1111/j.1365-2958.2009.06947.x
Barns, S. M., Grow, C. C., Okinaka, R. T., Keim, P., and Kuske, C. R. (2005). Detection of diverse new Francisella-like bacteria in environmental samples. Appl. Environ. Microbiol. 71, 5494–5500. doi: 10.1128/AEM.71.9.5494-5500.2005
Birdsell, D. N., Stewart, T., Vogler, A. J., Lawaczeck, E., Diggs, A., Sylvester, T. L., et al. (2009). Francisella tularensis subsp. novicida isolated from a human in Arizona. BMC Res. Notes 2:223. doi: 10.1186/1756-0500-2-223
Birkbeck, T. H., Feist, S. W., and Verner-Jeffreys, D. W. (2011). Francisella infections in fish and shellfish. J. Fish Dis. 34, 173–187. doi: 10.1111/j.1365-2761.2010.01226.x
Brett, M., Doppalapudi, A., Respicio-Kingry, L. B., Myers, D., Husband, B., Pollard, K., et al. (2012). Francisella novicida bacteremia after a near-drowning accident. J. Clin. Microbiol. 50, 2826–2829. doi: 10.1128/JCM.00995-12
Brevik, Ø, Ottem, K., Kamaishi, T., Watanabe, K., and Nylund, A. (2011). Francisella halioticida sp. nov., a pathogen of farmed giant abalone (Haliotis gigantea) in Japan. J. Appl. Microbiol. 111, 1044–1056. doi: 10.1111/j.1365-2672.2011.05133.x
Brodmann, M., Dreier, R. F., Broz, P., and Basler, M. (2017). Francisella requires dynamic type VI secretion system and ClpB to deliver effectors for phagosomal escape. Nat. Commun. 8:15853. doi: 10.1038/Ncomms15853
Bröms, J., Sjöstedt, A., and Lavander, M. (2010). The role of the Francisella tularensis pathogenicity island in type VI secretion, intracellular survival, and modulation of host cell signaling. Front. Microbiol. 1:136. doi: 10.3389/fmicb.2010.00136
Bröms, J. E., Lavander, M., Meyer, L., and Sjöstedt, A. (2011). IglG and IglI of the Francisella pathogenicity island are important virulence determinants of Francisella tularensis LVS. Infect. Immunity 79, 3683–3696. doi: 10.1128/iai.01344-10
Bröms, J. E., Meyer, L., Sun, K., Lavander, M., and Söstedt, A. (2012). Unique substrates secreted by the Type VI secretion system of Francisella tularensis during intramacrophage infection. PLoS One 7:e50473. doi: 10.1371/journal.pone.0050473
Casanova, A., Low, S. H., Québatte, M., Sedzicki, J., Tschon, T., Ketterer, M., et al. (2019). A role for the VPS retromer in Brucella intracellular replication revealed by genomewide siRNA screening. mSphere 4:e00380-19.
Castresana, J. (2007). Topological variation in single-gene phylogenetic trees. Genome Biol. 8:216. doi: 10.1186/Gb-2007-8-6-216
Challacombe, J. F., Petersen, J. M., Gallegos-Graves, V., Hodge, D., Pillai, S., and Kuske, C. R. (2017). Whole-genome relationships among Francisella bacteria of diverse origins define new species and provide specific regions for detection. Appl. Environ. Microbiol. 83:e02589-16. doi: 10.1128/AEM.02589-16
Chan, J. Z. M., Halachev, M. R., Loman, N. J., Constantinidou, C., and Pallen, M. J. (2012). Defining bacterial species in the genomic era: insights from the genus Acinetobacter. BMC Microbiol. 12:302. doi: 10.1186/1471-2180-12-302
Chaudhari, N. M., Gupta, V. K., and Dutta, C. (2016). BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6:24373. doi: 10.1038/Srep24373
Clarridge, J. E.III, Raich, T. J., Sjöstedt, A., Sandstrom, G., Darouiche, R. O., Shawar, R. M., et al. (1996). Characterization of two unusual clinically significant Francisella strains. J. Clin. Microbiol. 34, 1995–2000. doi: 10.1128/jcm.34.8.1995-2000.1996
Collins, R. E., and Higgs, P. G. (2012). Testing the infinitely many genes model for the evolution of the bacterial core genome and pangenome. Mol. Biol. Evol. 29, 3413–3425. doi: 10.1093/molbev/mss163
Colquhoun, D. J., and Duodu, S. (2011). Francisella infections in farmed and wild aquatic organisms. Vet. Res. 42:47. doi: 10.1186/1297-9716-42-47
Contreras-Moreira, B., and Vinuesa, P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701. doi: 10.1128/Aem.02411-13
De Bruin, O. M., Ludu, J. S., and Nano, F. E. (2007). The Francisella pathogenicity island protein IglA localizes to the bacterial cytoplasm and is needed for intracellular growth. BMC Microbiol. 7:1. doi: 10.1186/1471-2180-7-1
Dietrich, E. A., Kingry, L. C., Kugeler, K. J., Levy, C., Yaglom, H., Young, J. W., et al. (2019). Francisella opportunistica sp. nov., isolated from human blood and cerebrospinal fluid. Int. J. Syst. Evol. Microbiol. 70, 1145–1151. doi: 10.1099/ijsem.0.003891
Dyer, M. D., Neff, C., Dufford, M., Rivera, C. G., Shattuck, D., Bassaganya-Riera, J., et al. (2010). The human-bacterial pathogen protein interaction networks of Bacillus anthracis, Francisella tularensis, and Yersinia pestis. PLoS One 5:e12089. doi: 10.1371/journal.pone.0012089
Eshraghi, A., Kim, J., Walls, A. C., Ledvina, H. E., Miller, C. N., Ramsey, K. M., et al. (2016). Secreted effectors encoded within and outside of the Francisella pathogenicity Island promote intramacrophage growth. Cell Host Microbe 20, 573–583. doi: 10.1016/j.chom.2016.10.008
Finsel, I., Ragaz, C., Hoffmann, C., Harrison, C. F., Weber, S., van Rahden, V. A., et al. (2013). The Legionella effector RidL inhibits retrograde trafficking to promote intracellular replication. Cell Host Microbe 14, 38–50. doi: 10.1016/j.chom.2013.06.001
Han, N., Qiang, Y. J., and Zhang, W. (2016). ANItools web: a web tool for fast genome comparison within multiple bacterial strains. Database 2016:baw084. doi: 10.1093/database/baw084
Hollis, D. G., Weaver, R. E., Steigerwalt, A. G., Wenger, J. D., Moss, C. W., and Brenner, D. J. (1989). Francisella philomiragia comb. nov. (formerly Yersinia philomiragia) and Francisella tularensis biogroup novicida (formerly Francisella novicida) associated with human disease. J. Clin. Microbiol. 27, 1601–1608. doi: 10.1128/jcm.27.7.1601-1608.1989
Huber, B., Escudero, R., Busse, H. J., Seibold, E., Scholz, H. C., Anda, P., et al. (2010). Description of Francisella hispaniensis sp. nov., isolated from human blood, reclassification of Francisella novicida (Larson et al. 1955) Olsufiev et al., 1959 as Francisella tularensis subsp. novicida comb. nov. and emended description of the genus Francisella. Int. J. Syst. Evol. Microbiol. 60(Pt 8), 1887–1896. doi: 10.1099/ijs.0.015941-0
Johnson, S. L., Daligault, H. E., Davenport, K. W., Coyne, S. R., Frey, K. G., Koroleva, G. I., et al. (2015). Genome sequencing of 18 Francisella strains to aid in assay development and testing. Microbiol. Resour. Announc. 3:e00147-15. doi: 10.1128/genomeA.00147-15
Kaas, R. S., Friis, C., Ussery, D. W., and Aarestrup, F. M. (2012). Estimating variation within the genes and inferring the phylogeny of 186 sequenced diverse Escherichia coli genomes. BMC Genomics 13:577. doi: 10.1186/1471-2164-13-577
Keim, P., Johansson, A., and Wagner, D. M. (2007). Molecular epidemiology, evolution, and ecology of Francisella. Ann. N. Y. Acad. Sci. 1105, 30–66. doi: 10.1196/annals.1409.011
Kingry, L. C., Cummings, J. E., Brookman, K. W., Bommineni, G. R., Tonge, P. J., and Slayden, R. A. (2013). The Francisella tularensis FabI enoyl-acyl carrier protein reductase gene is essential to bacterial viability and is expressed during infection. J. Bacteriol. 195, 351–358. doi: 10.1128/jb.01957-12
Knight, D. R., Squire, M. M., Collins, D. A., and Riley, T. V. (2017). Genome analysis of Clostridium difficile PCR Ribotype 014 lineage in Australian Pigs and humans reveals a diverse genetic repertoire and signatures of long-Range interspecies transmission. Front. Microbiol. 7:2138. doi: 10.3389/Fmicb.2016.02138
Konstantinidis, K. T., Ramette, A., and Tiedje, J. M. (2006). Toward a more robust assessment of intraspecies diversity, using fewer genetic markers. Appl. Environ. Microbiol. 72, 7286–7293. doi: 10.1128/Aem.01398-06
Kreizinger, Z., Hornok, S., Dan, A., Hresko, S., Makrai, L., Magyar, T., et al. (2013). Prevalence of Francisella tularensis and Francisella-like endosymbionts in the tick population of Hungary and the genetic variability of Francisella-like agents. Vector Borne Zoon. Dis. 13, 160–163. doi: 10.1089/vbz.2012.1065
Kristensen, D. M., Kannan, L., Coleman, M. K., Wolf, Y. I., Sorokin, A., Koonin, E. V., et al. (2010). A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics 26, 1481–1487. doi: 10.1093/bioinformatics/btq229
Kugeler, K. J., Mead, P. S., Janusz, A. M., Staples, J. E., Kubota, K. A., Chalcraft, L. G., et al. (2009). Molecular epidemiology of Francisella tularensis in the United States. Clin. Infect. Dis. 48, 863–870. doi: 10.1086/597261
Kugeler, K. J., Mead, P. S., McGowan, K. L., Burnham, J. M., Hogarty, M. D., Ruchelli, E., et al. (2008). Isolation and characterization of a novel Francisella sp. from human cerebrospinal fluid and blood. J. Clin. Microbiol. 46, 2428–2431. doi: 10.1128/JCM.00698-08
Kuske, C. R., Barns, S. M., Grow, C. C., Merrill, L., and Dunbar, J. (2006). Environmental survey for four pathogenic bacteria and closely related species using phylogenetic and functional genes. J. Forensic. Sci. 51, 548–558. doi: 10.1111/j.1556-4029.2006.00131.x
Larson, M. A., Nalbantoglu, U., Sayood, K., Zentz, E. B., Cer, R. Z., Iwen, P. C., et al. (2016). Reclassification of Wolbachia persica as Francisella persica comb. nov and emended description of the family Francisellaceae. Int. J. Syst. Evol. Microbiol. 66, 1200–1205. doi: 10.1099/ijsem.0.000855
Larsson, P., Elfsmark, D., Svensson, K., Wikstrom, P., Forsman, M., Brettin, T., et al. (2009). Molecular evolutionary consequences of niche restriction in Francisella tularensis, a facultative intracellular pathogen. PLoS Pathog. 5:e1000472. doi: 10.1371/journal.ppat.1000472
Larsson, P., Oyston, P. C., Chain, P., Chu, M. C., Duffield, M., Fuxelius, H. H., et al. (2005). The complete genome sequence of Francisella tularensis, the causative agent of tularemia. Nat. Genet. 37, 153–159. doi: 10.1038/ng1499
Le, S. Q., and Gascuel, O. (2008). An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320. doi: 10.1093/molbev/msn067
Ledvina, H. E., Kelly, K. A., Eshraghi, A., Plemel, R. L., Peterson, S. B., Lee, B., et al. (2018). A phosphatidylinositol 3-kinase effector alters phagosomal maturation to promote intracellular growth of Francisella. Cell Host Microbe 24:285. doi: 10.1016/j.chom.2018.07.003
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Mariano, G., Trunk, K., Williams, D. J., Monlezun, L., Strahl, H., Pitt, S. J., et al. (2019). A family of Type VI secretion system effector proteins that form ion-selective pores. Nat. Commun. 10, 1–15.
McDermott, C., and Palmeiro, B. (2013). Selected emerging infectious diseases of ornamental fish. Vet. Clin. North Am. Exot. Anim. Pract. 16, 261–282. doi: 10.1016/j.cvex.2013.01.006
Medema, M. H., Takano, E., and Breitling, R. (2013). Detecting sequence homology at the gene cluster level with multigeneblast. Mol. Biol. Evol. 30, 1218–1223. doi: 10.1093/molbev/mst025
Meric, G., Yahara, K., Mageiros, L., Pascoe, B., Maiden, M. C. J., Jolley, K. A., et al. (2014). A reference pan-genome approach to comparative bacterial genomics: identification of novel epidemiological markers in pathogenic Campylobacter. PLoS One 9:e92798. doi: 10.1371/journal.pone.0092798
Mikalsen, J., Olsen, A. B., Tengs, T., and Colquhoun, D. J. (2007). Francisella philomiragia subsp. noatunensis subsp. nov., isolated from farmed Atlantic cod (Gadus morhua L.). Int. J. Syst. Evol. Microbiol. 57, 1960–1965. doi: 10.1099/ijs.0.64765-0
Mosquera-Rendon, J., Rada-Bravo, A. M., Cardenas-Brito, S., Corredor, M., Restrepo-Pineda, E., and Benitez-Paez, A. (2016). Pangenome-wide and molecular evolution analyses of the Pseudomonas aeruginosa species. BMC Genomics 17:45. doi: 10.1186/s12864-016-2364-4
Na, S. I., Kim, Y. O., Yoon, S. H., Ha, S. M., Baek, I., and Chun, J. (2018). UBCG: up-to-date bacterial core gene set and pipeline for phylogenomic tree reconstruction. J. Microbiol. 56, 280–285. doi: 10.1007/s12275-018-8014-6
Nano, F. E., and Schmerk, C. (2007). The Francisella pathogenicity island. Ann. N. Y. Acad. Sci. 1105, 122–137. doi: 10.1196/annals.1409.000
Nano, F. E., Zhang, N., Cowley, S. C., Klose, K. E., Cheung, K. K., Roberts, M. J., et al. (2004). A Francisella tularensis pathogenicity island required for intramacrophage growth. J. Bacteriol. 186, 6430–6436. doi: 10.1128/jb.186.19.6430-6436.2004
Nelson, W. C., and Stegen, J. C. (2015). The reduced genomes of Parcubacteria (OD1) contain signatures of a symbiotic lifestyle. Front. Microbiol. 6:713. doi: 10.3389/Fmicb.2015.00713
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nourdin-Galindo, G., Sanchez, P., Molina, C. F., Espinoza-Rojas, D. A., Oliver, C., Ruiz, P., et al. (2017). Comparative pan-genome analysis of Piscirickettsia salmonis reveals genomic divergences within genogroups. Front. Cell. Infect. Microbiol. 7:459. doi: 10.3389/Fcimb.2017.00459
Olsufiev, N. G., Emelyanova, O. S., and Dunayeva, T. N. (1959). Comparative study of strains of B. tularense in the old and new world and their taxonomy. J. Hyg. Epidemiol. Microbiol. Immunol. 3, 138–149.
Petersen, J. M., Carlson, J., Yockey, B., Pillai, S., Kuske, C., Garbalena, G., et al. (2009). Direct isolation of Francisella spp. from environmental samples. Lett. Appl. Microbiol. 48, 663–667. doi: 10.1111/j.1472-765X.2009.02589.x
Pilo, P. (2018). Phylogenetic lineages of Francisella tularensis in animals. Front. Cell. Infect. Microbiol. 8:258. doi: 10.3389/fcimb.2018.00258
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/c5ay02550h
Qu, P. H., Chen, S. Y., Scholz, H. C., Busse, H. J., Gu, Q., Kampfer, P., et al. (2013). Francisella guangzhouensis sp. nov., isolated from air-conditioning systems. Int. J. Syst. Evol. Microbiol. 63(Pt 10), 3628–3635. doi: 10.1099/ijs.0.049916-0
Qu, P. H., Li, Y., Salam, N., Chen, S. Y., Liu, L., Gu, Q., et al. (2016). Allfrancisella inopinata gen. nov., sp. nov. and Allofrancisella frigidaquae sp. nov., isolated from water-cooling systems, and transfer of Francisella guangzhouensis Qu et al., 2013 to the new genus as Allofrancisella guangzhouensis comb. nov. Int. J. Syst. Evol. Microbiol. 66, 4832–4838. doi: 10.1099/ijsem.0.001437
Ramirez-Paredes, J. G., Larsson, P., Thompson, K. D., Penman, D. J., Busse, H. J., Ohrman, C., et al. (2020). Reclassification of Francisella noatunensis subsp. orientalis Ottem et al. 2009 as Francisella orientalis sp. nov., Francisella noatunensis subsp. chilensis subsp. nov. and emended description of Francisella noatunensis. Int. J. Syst. Evol. Microbiol. 70, 2034–2048. doi: 10.1099/ijsem.0.004009
Rasko, D. A., Rosovitz, M. J., Myers, G. S. A., Mongodin, E. F., Fricke, W. F., Gajer, P., et al. (2008). The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J. Bacteriol. 190, 6881–6893. doi: 10.1128/Jb.00619-08
Respicio-Kingry, L. B., Byrd, L., Allison, A., Brett, M., Scott-Waldron, C., Galliher, K., et al. (2013). Cutaneous infection caused by a novel Francisella sp. J. Clin. Microbiol. 51, 3456–3460. doi: 10.1128/JCM.01105-13
Robles-Marhuenda, A., Vaca, M., Romero, P., Ferreira, A., Lopez-Granados, E., and Arnalich, F. (2018). Francisella philomiragia: think of chronic granulomatous disease. J. Clin. Immunol. 38, 257–259. doi: 10.1007/s10875-018-0498-7
Rohmer, L., Fong, C., Abmayr, S., Wasnick, M., Larson Freeman, T. J., Radey, M., et al. (2007). Comparison of Francisella tularensis genomes reveals evolutionary events associated with the emergence of human pathogenic strains. Genome Biol. 8:R102. doi: 10.1186/gb-2007-8-6-r102
Rosselli, R., Romoli, O., Vitulo, N., Vezzi, A., Campanaro, S., de Pascale, F., et al. (2016). Direct 16S rRNA-seq from bacterial communities: a PCR-independent approach to simultaneously assess microbial diversity and functional activity potential of each taxon. Sci. Rep. 6:32165. doi: 10.1038/Srep32165
Rydzewski, K., Schulz, T., Brzuszkiewicz, E., Holland, G., Luck, C., Fleischer, J., et al. (2014). Genome sequence and phenotypic analysis of a first German Francisella sp. isolate (W12-1067) not belonging to the species Francisella tularensis. BMC Microbiol. 14:169. doi: 10.1186/1471-2180-14-169
Salomon, D., Kinch, L. N., Trudgian, D. C., Guo, X. F., Klimko, J. A., Grishin, N. V., et al. (2014). Marker for type VI secretion system effectors. Proc. Natl. Acad. Sci. U.S.A. 111, 9271–9276. doi: 10.1073/pnas.1406110111
Satoh, S., Mimuro, M., and Tanaka, A. (2013). Construction of a phylogenetic tree of photosynthetic prokaryotes based on average similarities of whole genome sequences. PLoS One 8:e70290. doi: 10.1371/journal.pone.0070290
Shrivastava, S., and Mande, S. S. (2008). Identification and functional characterization of gene components of Type VI secretion system in bacterial genomes. PLoS One 3:e2955. doi: 10.1371/journal.pone.0002955
Siddaramappa, S., Challacombe, J. F., Petersen, J. M., Pillai, S., Hogg, G., and Kuske, C. R. (2011). Common ancestry and novel genetic traits of Francisella novicida-like isolates from North America and Australia as revealed by comparative genomic analyses. Appl. Environ. Microbiol. 77, 5110–5122. doi: 10.1128/AEM.00337-11
Siddaramappa, S., Challacombe, J. F., Petersen, J. M., Pillai, S., and Kuske, C. R. (2012). Genetic diversity within the genus Francisella as revealed by comparative analyses of the genomes of two North American isolates from environmental sources. BMC Genomics 13:422. doi: 10.1186/1471-2164-13-422
Singer, G. A., and Hickey, D. A. (2000). Nucleotide bias causes a genomewide bias in the amino acid composition of proteins. Mol. Biol. Ecol. 17, 1581–1588. doi: 10.1093/oxfordjournals.molbev.a026257
Sjödin, A., Svensson, K., Öhrman, C., Ahlinder, J., Lindgren, P., Duodu, S., et al. (2012). Genome characterisation of the genus Francisella reveals insight into similar evolutionary paths in pathogens of mammals and fish. BMC Genomics 13:268. doi: 10.1186/1471-2164-13-268
Sjödin, A., Ohrman, C., Backman, S., Larkeryd, A., Granberg, M., Lundmark, E., et al. (2014). Complete genome sequence of Francisella endociliophora strain FSC1006, isolated from a laboratory culture of the marine ciliate Euplotes raikovi. Genome Announc. 26:e01227-14. doi: 10.1128/genomeA.01227-14
Sjöstedt, A. (2005). “Family XVII. Francisellaceae, genus I. Francisella,” in Bergey’s Manual of Systematic Bacteriology, ed. D. J. Brenner (New York, NY: Springer-Verlag).
Sjöstedt, A. (2007). Tularemia: history, epidemiology, pathogen physiology, and clinical manifestations. Ann. N. Y. Acad. Sci. 1105, 1–29. doi: 10.1196/annals.1409.009
Snipen, L., Almoy, T., and Ussery, D. W. (2009). Microbial comparative pan-genomics using binomial mixture models. BMC Genomics 10:385. doi: 10.1186/1471-2164-10-385
Spidlova, P., and Stulik, J. (2017). Francisella tularensis type VI secretion system comes of age. Virulence 8, 628–631. doi: 10.1080/21505594.2016.1278336
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955.
Tettelin, H., Riley, D., Cattuto, C., and Medini, D. (2008). Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11, 472–477. doi: 10.1016/j.mib.2008.09.006
Tindall, B. J., Rossello-Mora, R., Busse, H. J., Ludwig, W., and Kampfer, P. (2010). Notes on the characterization of prokaryote strains for taxonomic purposes. Int. J. Syst. Evol. Microbiol. 60, 249–266. doi: 10.1099/ijs.0.016949-0
Toronen, P., Medlar, A., and Holm, L. (2018). PANNZER2: a rapid functional annotation web server. Nucleic Acids Res. 46, W84–W88. doi: 10.1093/nar/gky350
Vallesi, A., Sjödin, A., Petrelli, D., Luporini, P., Taddei, A. R., Thelaus, J., et al. (2019). A new species of the gamma-Proteobacterium Francisella, F. adeliensis Sp. Nov., Endocytobiont in an Antarctic marine ciliate and potential evolutionary forerunner of pathogenic species. Microb. Ecol. 77, 587–596. doi: 10.1007/s00248-018-1256-3
Vernikos, G., Medini, D., Riley, D. R., and Tettelin, H. (2015). Ten years of pan-genome analyses. Curr. Opin. Microbiol. 23, 148–154. doi: 10.1016/j.mib.2014.11.016
Vinuesa, P., Ochoa-Sanchez, L. E., and Contreras-Moreira, B. (2018). GET_PHYLOMARKERS, a software package to select optimal orthologous clusters for phylogenomics and inferring pan-genome phylogenies, used for a critical geno-taxonomic revision of the Genus Stenotrophomonas. Front. Microbiol. 9:771. doi: 10.3389/Fmicb.2018.00771
Wang, J. W., Yang, B. J., Leier, A., Marquez-Lago, T. T., Hayashida, M., Rocker, A., et al. (2018). Bastion6: a bioinformatics approach for accurate prediction of type VI secreted effectors. Bioinformatics 34, 2546–2555. doi: 10.1093/bioinformatics/bty155
Wang, Y., Mao, L., Sun, Y., Wang, Z., Zhang, J., Zhang, J., et al. (2018). A Novel Francisella-Like Endosymbiont in Haemaphysalis longicornis and Hyalomma asiaticum, China. Vector Borne Zoonotic Dis. 18, 669–676. doi: 10.1089/vbz.2017.2252
Wenger, J. D., Hollis, D. G., Weaver, R. E., Baker, C. N., Brown, G. R., Brenner, D. J., et al. (1989). Infection caused by Francisella philomiragia (formerly Yersinia philomiragia). A newly recognized human pathogen. Ann. Intern. Med. 110, 888–892. doi: 10.7326/0003-4819-110-11-888
Whipp, M. J., Davis, J. M., Lum, G., de Boer, J., Zhou, Y., Bearden, S. W., et al. (2003). Characterization of a novicida-like subspecies of Francisella tularensis isolated in Australia. J. Med. Microbiol. 52(Pt 9), 839–842. doi: 10.1099/jmm.0.05245-0
Wolf, Y. I., and Koonin, E. V. (2012). A tight link between orthologs and bidirectional best hits in bacterial and archaeal genomes. Genome Biol. Evol. 4, 1286–1294. doi: 10.1093/gbe/evs100
Yao, J., Yang, F., Sun, X., Wang, S., Gan, N., Liu, Q., et al. (2018). Mechanism of inhibition of retromer transport by the bacterial effector RidL. Proc. Natl. Acad. Sci. U.S.A. 115, E1446–E1454.
Yon, L., Duff, J. P., Agren, E. O., Erdelyi, K., Ferroglio, E., Godfroid, J., et al. (2019). Recent changes in infectious diseases in European wildlife. J. Wildl. Dis. 55, 3–43. doi: 10.7589/2017-07-172
Keywords: whole-genome analysis, T6SS, Francisella, ANI, core-genome and pan-genome
Citation: Kumar R, Bröms JE and Sjöstedt A (2020) Exploring the Diversity Within the Genus Francisella – An Integrated Pan-Genome and Genome-Mining Approach. Front. Microbiol. 11:1928. doi: 10.3389/fmicb.2020.01928
Received: 05 May 2020; Accepted: 22 July 2020;
Published: 11 August 2020.
Edited by:
Iain Sutcliffe, Northumbria University, United KingdomReviewed by:
Jean Challacombe, UC San Diego Health, United StatesCopyright © 2020 Kumar, Bröms and Sjöstedt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anders Sjöstedt, YW5kZXJzLnNqb3N0ZWR0QHVtdS5zZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.