 Samson Hailemariam
Samson Hailemariam Shengguo Zhao
Shengguo Zhao Jiaqi Wang
Jiaqi Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 14 August 2020
Sec. Systems Microbiology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.01826
The unclassified Succinivibrionaceae lineages are abundant in high yielding multiparous cows, and their presence is positively correlated with milk yield and fat percentage and reduces methane emissions. However, it is still unclear which species are associated with the most efficient feed nutrient utilization and productivity. Here, we used integrated whole genome sequencing and matrix-assisted laser desorption/ionization mass spectrometry, coupled with phenotypic and chemotaxonomic analysis, to characterize S. dextrinosolvens Z6, a species in Succinivibrionaceae isolated from the rumen. To assess the role of S. dextrinosolvens Z6 in nitrogen metabolism, cells grown in different nitrogen sources were analyzed by RNA sequencing. The whole genome sequence result revealed a genome size of 3.47 Mbp with 38.9% of G + C content. A total of 2993 encoding sequences account for 98%. The genes for regulating carbohydrate (10.6%) and amino acid (9%) transport and metabolism were the most abundant. ANI (Average nucleotide identity) showed that SD-Z6 was most closely related to SD-22B (99.96%). The whole genome alignment of SD-Z6 with SD-22B showed a more than 0.34 Mb nucleotide difference. Growth of SD-Z6 occurred at a temperature 36–42°C with an optimum at 39.7°C, pH 6–8; the optimum pH was 6.9 and with 0–1% (w/v) NaCl. The maximum growth (OD600 0.825 ± 0.12) and microbial crude protein (MCP) (178.2 μg/ml) were observed in cells grown in amino acid. The maximum concentration of ammonia (3.96 ± 1.2) was observed in urea containing media and 1.06 mM (26.7% of the produced) remained after 24 h incubation. Activities of urease and glutamine synthase (P < 0.01) and glutamate dehydrogenase (P < 0.05) were significantly different in nitrogen and growth phase. Glutamate synthetase (P < 0.01) was significantly different only at different growth phases. In total, 1246 differentially expressed genes (DEGs) were identified in all nitrogen. Among DEGs, 33 were related to nitrogen metabolism. Their expression correlated with nitrogen sources and the intensity of enzyme activity. This result enhances our understanding of the roles of Succinivibrionaceae in the efficient nitrogen utilization and on environmental protection.
The rumen is a complex ecosystem in which consumed nutrients are digested and converted to diverse end products by microorganisms. The main end products of fermentation are volatile fatty acids (VFAs) and microbial protein, both of which are used by the host ruminant as sources of energy and protein and are important to the health and productivity of the animals (Bryant, 1959; Russell and Hespell, 1981). However, the efficiency of VFA and microbial protein production by the fermentation in the rumen is highly variable and dependent on genetic and functional aspects of the rumen microbiome (Khafipour et al., 2009; Kruger Ben Shabat et al., 2016; Zeng et al., 2019). The unclassified Succinivibrionaceae is among different ruminal bacteria families, whose presence in the rumen has been associated with feed efficiency and animal productivity for dairy cattle fed with high total mixed ration (TMR) (Hernandez-Sanabria et al., 2012; Indugu et al., 2017; Zeng et al., 2019).
Studies by Indugu et al. (2017) and Xue et al. (2018) revealed that Succinivibrionaceae lineages were abundant in high yielding primiparous and multiparous cows and that their presence was positively correlated with milk yield and milk fat composition. Groups of bacteria in this family have been reported to produce succinate, a precursor of propionate (Blackburn and Hungate, 1963) and acetate, which affect lactation performance and milk fat content, respectively. Moreover, Pope et al. (2011), McCabe et al. (2015), and Danielsson et al. (2017) reported that unclassified Succinivibrionaceae growth in the rumen reduces methane emissions since bacteria in this family utilize hydrogen to produce succinate, which is also an input for methanogenesis. However, these studies did not target specific Succinivibrionaceae species and the main focus was on the contribution of the bacteria in this family to carbohydrate metabolism in the rumen. Notably, their contribution to nitrogen metabolism has not been studied in detail, despite the fact that the balance of carbon and nitrogen utilization is a key factor in the growth and performance of ruminants (Seo et al., 2013).
The Succinivibrionaceae family consists of gammaproteobacteria, which are gram-negative, strictly anaerobic, non-spore-forming species, and comprises mainly ruminant inhabitant members of the Succinivibrio, Succinimonas, and Ruminobacter genera. Succinivibrio dextrinosolvens is a well-studied species of the Succinivibrionaceae family and Succinivibrio genus (Bryant and Small, 1955). Previously isolated S. dextrinosolvens cells were shown to be motile in vibrating movements and to appear singly or in pairs; however, some isolated strains commonly form helical or twisted filaments of two to four cell coils (Stackebrandt and Hespell, 2006). Succinivibrio strains ferment glucose, resulting in the production of large amounts of acetic and succinic acids (Bryant and Small, 1955). Moreover, a few strains have been shown to possess all the enzymes necessary for nitrogen-containing compound degradation and assimilation. For example, S. dextrinosolvens C18 has glutamine synthetase (GS), urease, glutamate dehydrogenase, and several other nitrogen assimilation enzymes, whereas strain 24 lack urease (Patterson and Hespell, 1985). Such differences between S. dextrinosolvens strains could be important for their ability to participate in nitrogen metabolism. However, the absence of complete genome information for this bacterium has been a major impediment to functional gene studies, and the different descriptions of S. dextrinosolvens strains have mainly been based on morphological, biochemical (Bryant and Small, 1955; O’Herrin and Kenealy, 1993), or partial genomic characteristics. In addition, little is known about the transcriptional response of this bacterium to various nitrogen sources. We report the genomic, biochemical, molecular, and transcriptome features of S. dextrinosolvens Z6 and propose that this information will benefit the manipulation of its genetic potential for better feed utilization and the production performance of ruminant animals.
The basal media contained 30 ml of clarified rumen fluid, 10 ml of urea (20% w/v), 0.05 g of glucose, 0.05 g of maltose, 0.05 g of soluble starch, 38 ml of solution 1 (0.3% K2HPO4), 38 ml of solution 2 (0.3% KH2PO4, 0.6% NaC1, 0.06% MgSO2, and 0.06% CaCl2), 0.1 ml of Pfennig trace elements (300 mg of H3BO3, 100 mg of ZnSO4⋅7H2O, 30 mg of MnCl2⋅4H2O, 20 mg of CoCl2⋅6H2O, 30 mg of Na2MoO4⋅2H2O, 10 mg of Na2SeO3, 20 mg of NiCl2, 10 mg of CuCl2⋅2H2O, and 150 mg of FeCl2⋅4H2O per 1000 ml of distilled H2O), 5 ml of hemin (0.05%), 0.31 ml of VFA mix (17 ml of acetic acid, 6 ml of propionic, 4 ml of n-butyric, and 1 ml each of n-valeric, isovaleric, isobutyric, and 2-methylbutyric acids), 0.5 g of NaHCO3, 0.1 ml of resazurin (0.1%), 0.05 g of L-cysteine HCl, and 0.0012 g of phenol red per 100 ml. Media one (M1) consisted of basal media plus 0.05 g of tryptone and 0.05 g of yeast extract, while basal media alone was used as media two (M2), media three (M3) was basal media and 1.5 g agar per 100 ml, and media four (M4) was basal media without urea. After boiling, a gassing probe was used to purge the solution with food-grade CO2 for at least 2 h while the media cooled. Subsequently, 10 ml of media was dispensed anaerobically into a Hungate tube in an anaerobic chamber, and autoclaved for 15 min at 100 kPa, 121°C. After cooling, urea, vitamins, and phenol red were added (as above) and the media was stored at 4°C in the dark.
S. dextrinosolvens Z6 was isolated from rumen fluid obtained from dairy cows fed with basic TMR as previously described (Jin et al., 2016). To prepare the rumen microbe inoculum, the rumen fluid sample was aspirated with a syringe and added to an anaerobically prepared 30% (v/v) glycerol stock solution in equal volumes. The inoculum samples were stored on dry ice and immediately brought back to the laboratory and stored at −80°C.
For the enrichment, 200 μl of rumen microbe inoculum was inoculated by using a syringe inside the anaerobic chamber into 10 ml of M1 media and cultured at 39°C for 24 h as the first generation. The culture was then transferred to new M1 media to produce the second and third generations. The third-generation culture was diluted and spread on M3 media and incubated at 39°C for 2–4 days in an incubator inside an anaerobic controlled atmosphere chamber (Lansing, MI, United States). Pink colonies were picked and cultured in M2 media at 39°C for 16 h for genomic DNA extraction using the TIANamp Bacteria DNA Kit (TIANGEN, China). In parallel, the same amount of bacterial liquid solution was mixed with 30% (v/v) glycerol cryopreservation solution, and the strain was stored at -80°C.
PCR primers [UreC-F (5’-TGGGCCTTAAAATHCAYGA RGAYTGGG-3’) and UreC-R (5’-SGGTGGTGGCACACCAT NANCATRTC-3)] that amplify the urease alpha subunit encoding gene (ureC) were used to determine the number of ureC gene copies (Reed, 2001). The 16S rRNA gene from total bacteria was quantified using the universal primers 27F (5′-AGAGTTTGATCMTGGCTCAG-3′) and 1492R (5′-TACGGYTACCTTGTTACGACTT-3′) (Frank et al., 2008). All PCR reactions were performed with a MyCycler Thermal Cycler (Bio-Rad, United States) in a 25 μl mixture containing 2.5 μl of PCR buffer (Invitrogen), 0.75 μl of MgCl2 (50 mM), 0.5 μl of dNTP (10 mM), 1.5 μl of each forward and reverse primer, 0.3 μl of platinum Taq DNA polymerase (Invitrogen), 1 μl of template DNA, and 16.95 μl of ddH2O. PCR amplification began with a 3 min denaturing step at 94°C, followed by 30 cycles at 94°C for 30 s, 58°C for 30 s, and 72°C for 1.5 min, and the final extension was at 72°C for 7 min and 10°C for 10 min for 16S rRNA amplification. For the urease amplification, cycling conditions were as follows: 5 min 94°C and 30 cycles of 94°C for 30 s, 50°C for 30 s, and 72°C for 30 s, with a final extension at 72°C for 15 min and 10°C 10 min. The PCR products were sequenced by Sanger sequencing (Applied Biosystems 3730XL, Foster City, CA, United States).
For urease activity testing, cell-free extracts were used as crude enzymes in an experiment modified from previous methods (Richmond and Yep, 2019). Briefly, 2 ml of each anaerobic culture was harvested by centrifugation at 12,000 g for 20 min at 4°C, washed once with 50 mM PBS buffer, and resuspended in PBS buffer. The cells were disrupted by sonication at 80 W (30 cycles of a 30 s pulse followed by 1 min of cooling in an ice water bath). After centrifugation (12,000 g, 20 min, 4°C), the supernatant was collected as the enzyme solution for urease activity measurement.
Genomic DNA from S. dextrinosolvens Z6 isolated as above was sequenced using Oxford Nanopore Technology (Oxford, United Kingdom). For ONT sequencing, high-quality genomic DNA was extracted using the QIAamp genomic DNA kit (Qiagen, California, United States) following the manufacturer’s instructions. DNA quality was confirmed using gel electrophoresis and a NanoDrop One (Thermo Fisher Scientific, CA, United States), and the DNA was quantified using a Qubit (Thermo Fisher Scientific, CA, United States). The library was prepared using the ONT 1D ligation sequencing kit (Oxford, United Kingdom), and the final library was loaded onto a flow cell and sequenced on a GridION Sequencer device (Oxford, United Kingdom) performing single-molecule DNA sequencing to obtain raw sequence data.
The following criteria were used as a cutoff in a quality control of the raw data: mean Q score template ≥ 7 and sequence length ≥1000 bp. After quality control, the data were assembled using canu (Koren et al., 2017) and then corrected by Pilon (Walker et al., 2014). After removing redundant sequences, circlator (Hunt et al., 2015) was implemented to move the origin of the sequence to the start site of the genome replication to obtain the final genome sequence. The data were aligned to the assembled genome using minimap2 (Li, 2018), and the sequence depth of each locus was calculated by SAMtools (Li et al., 2009). Plasmid sequence was identified by aligning the sequences to a plasmid database (Jesus et al., 2019) using blastn software (Camacho et al., 2009), and coding genes were predicted by prodigal (Hyatt et al., 2010). The tRNA genes were predicted by tRNAscan-SE (Lowe and Chan, 2016), rRNA genes were predicted by RNAmmer (Lagesen et al., 2007), and other ncRNAs were predicted by infernal (Nawrocki and Eddy, 2013) searching the Rfam database (Kalvari et al., 2018). CRISPR was predicted by minced (Bland et al., 2007), and Islander (Hudson et al., 2015) was used for predicting gene islands. Interproscan (Jones et al., 2014) was used to annotate the predicted genes from the TIGRFAMs (Haft et al., 2013), Pfam (Kalvari et al., 2018), and GO (Ashburner et al., 2000) databases. Blastp was used to align the protein sequences to the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al., 2014) and RefSeq databases (O’Leary et al., 2016). The protein sequences were also aligned to the clusters of orthologous groups (COG) database using rpsblast (Camacho et al., 2009) for COG annotation.
For genome comparison between S. dextrinosolvens Z6 and other related members of the genus, draft genome sequences of four S. dextrinosolvens strains and one Succinimonas amylolytica strain were downloaded from the NCBI database (accession numbers: GCA_900167015.1, GCA_900116345.1, GCA_000702045.1, GCA_900114195.1, and NZ_KB899636.1 for strain DSM 3072, ACV-10, H5, 22B, and S. amylolytica DSM_2873 used as an outgroup). Circular comparison plots between these genomes were prepared using BLAST Ring Image Generator (BRIG) (version 0.95) (Alikhan et al., 2011), and inference of clusters of orthologous groups was performed with OrthoFinder (version 1.1.5) (Emms and Kelly, 2015). A total of 1044 gene families exclusively containing single copy genes were selected as potential phylogenetic markers. For each of these clusters of orthologous groups, protein family alignments were built with muscle (version 3.8) (Edgar, 2004). Phylogenetic trees were constructed with RAxML (version 8.2.9 SSE3) (Stamatakis, 2014). To calculate the average nucleotide identity (ANI) between strains, the OAT (Orthologous ANI Tool) software package was used (Lee et al., 2016). Venn maps were made using the VennDiagram R package (Chen and Boutros, 2011).
Cell morphology was examined using scanning electron microscopy (Hitachi FE-SEM SU-8010). Gram staining was performed using Hucker’s modification (Hucker, 1921). The growth of S. dextrinosolvens Z6 was analyzed at different temperatures (15, 20, 25, 30, 37, 40, and 45°C) under anaerobic conditions at 39°C at different pH values (pH 5, 6, 7, 8, 9, and 10), and in different NaCl concentrations [0, 0.5, 1, 2, 3, 4, and 5% (w/v)] to test salt tolerance. M2 media without carbohydrate sources was used to test carbohydrate (glycerol, D-cellobiose, D-mannose, D-raffinose, D-sorbitol, L-rhamnose, D-trehalose, D-glucose, D-mannitol, D-lactose, D-saccharose, D-maltose, salicin, D-xylose, starch, and L-arabinose) utilization. Each carbohydrate was added to the carbohydrate free medium to a 0.5% final concentration and the media was prepared in triplicate and filter sterilized, before fresh overnight cultures were inoculated and incubated for 72 h at 37°C. The utilization of carbohydrate was determined by OD600 readings as previously described by Bertin et al. (2013), and the activities of 20 enzymes were determined using the API ZYM system (BioMerieux, Marcy, France) according to the manufacturer’s instructions.
S. dextrinosolvens Z6 cells were grown anaerobically at 39°C in M4 media supplemented with different nitrogen sources [ammonia ((NH4)2SO4), Amicase amino acid mix (Sigma-Aldrich, United States), and urea] in a concentration of 9.4 mM NH4-N. Five independent biological replicates were analyzed for each treatment. Growth was measured in five replicates by determining the increase in optical density at 600 nm (OD600) using a Visible Spectrophotometer V-5600PC (Shanghai Metash Instruments Co., Ltd., Shanghai, China) for 72 h. The maximum growth rate of the bacteria was determined as in Hall et al. (2014). Samples for chemical and enzymatic assays were harvested at early, mid, and late log growth phases by centrifugation at 8350 g for 5 min. The cell pellets and supernatants were stored at −80°C until analysis. The transcriptome samples were harvested in the mid-exponential growth phase.
Ammonia concentration was determined in the supernatant using an automated phenol-hypochlorite method (Broderick and Kang, 1980). The concentration of urea was determined using a urea assay kit (Sigma-Aldrich) based on the manufacturer’s protocol. The concentration of total free α-amino acids in the supernatant of the mixed amino acid-grown cultures was analyzed using ninhydrin as previously described (Jones et al., 2002). Microbial crude protein (MCP) was determined by the Folin phenol method (Makkar et al., 1982). Urease and glutamine synthase (GS) activity were determined using urease and glutamine synthase assay kits (Sigma-Aldrich), respectively, and glutamate dehydrogenase (GDH) and glutamate synthetase (GOGAT) activity were measured using activity detection kits (Solarbio, Beijing, China) according to the manufacturer’s instructions.
S. dextrinosolvens Z6 was grown in three different nitrogen sources in five biological replications and then harvested in the mid-exponential growth phase by centrifugation at 10,000 g for 5 min at 4°C to stop further growth of the bacteria. Total RNA was extracted from bacterial cell samples using TRIzol (Invitrogen, CA, United States) and purified using a QIAGEN RNeasy Mini Kit (Qiagen) following the manufacturer’s instructions. The concentration and purity of the extracted RNA were detected on a NanoDrop2000 Spectrophotometer (Thermo Fisher Scientific, MA, United States), and RNA integrity was visualized by agarose gel electrophoresis and on an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, United States). Ribosomal RNA (rRNA) was removed from total RNA by use of a Ribo-off rRNA Depletion Kit (Vazyme Biotech, Nanjing, China), and the libraries were constructed using the TruSeq Stranded Total RNA Library Prep Kit (Vazyme Biotech, Nanjing, China). The libraries were then sequenced using the Illumina HiSeq platform for 2 × 150 bp sequencing. RNA-seq quality control of raw data was performed using the FastQC1 and HTQC tools (Yang et al., 2013). Once the samples had passed the quality assessment, depending on the quality assessment results, the RNA-seq read quality trimming was performed using the sickle software (Version1.33) (Fass, 2011) and SeqPrep2 program. The high-quality sequences were aligned to the complete genome of S. dextrinosolvens Z6 using Burrow-Wheeler Aligner (BWA) and DIAMOND (Buchfink et al., 2014). High-throughput functional annotation was performed using Blast2GO (Version 2.5). The reference genome was analyzed using six major databases (NR, Swiss-Prot, Pfam, COG, GO, and KEGG) to obtain gene function information. The quantitative expression software RSEM (Li and Dewey, 2011) was used to quantitatively analyze the expression levels of genes and transcripts.
Differentially expressed genes (DEGs) between different nitrogen source groups were determined using the R/Bioconductor package, DESeq (Anders and Huber, 2010) with read counts calculated by HTseq (Anders et al., 2015). Read count normalization was performed using the regularized logarithm (rlog) method in DESeq. DEGs were selected if the adjusted p < 0.05 and the fold change ≥ 1.5. The top DEGs related to nitrogen metabolism were selected for this analysis.
To validate the RNA-seq data, quantitative reverse transcription PCR (qRT-PCR) analyses of selected genes were performed. The total RNA was extracted from S. dextrinosolvens Z6 grown in different nitrogen sources and reverse transcribed into cDNA by using first-strand cDNA Synthesis Kit (Genenode Biotech, Beijing). Quantitative PCR was performed using SYBR green (Genenode Biotech, Beijing, China) by using a FS384 real-time PCR system (Changzhou Fusheng Biotechnology, China) in a 10 μl mixture containing DNA template 2 μl, forward and reverse primer 0.25 μl each, 2 × SYBR Green qPCR Mix (Antibody) 5 μl, and DEPC-ddH2O 2.5 μl. The PCR amplification began with a 2 min denaturing step at 94°C, followed by 40 cycles at 94°C for 15 s, and 60°C for 30 s of annealing extension. The primers designed for RT-qPCR in this study are listed in Supplementary Table 10. The relative quantification of a gene is defined as the change in expression of the target gene relative to the reference genes. The relative expression level of each gene was calculated by the formula (2–ΔΔCT).
The enzyme activities (urease, GDH, GS, and GOGAT) and MCP data were analyzed using the mixed model, and real-time PCR data were analyzed by one-way ANOVA in SAS 9.2 (SAS Institute SAS/STAT(R), 2009). The model included fixed effects of time and nitrogen sources (ammonia, amino acid, and urea), and the results are presented as histograms made using GraphPad Prism version 8.0.0 for Windows, GraphPad Software, San Diego, California, United States3. Least square means are reported, and the significance was set to P < 0.05. The correlations between the activity of the analyzed enzymes and the expression of the corresponding DEGs were described by using Pearson’s correlation coefficient.
The complete genome sequence for S. dextrinosolvens strain Z6 has been deposited in GenBank under the accession number CP047056, BioProject number PRJNA545926, and BioSample number SAMN13552832. The raw genome sequence data are available in the Sequence Read Archive (SRA) database under accession number SRR10680263. The raw data for transcriptome sequencing are available in the same Bioproject number, Biosample number SAMN15009434, and in the SRA database under accession number SRR11855358-SRR11855371.
Following enrichment and spreading on plates, a single bacterial colony was isolated based on its morphological growth (red color) on the solid M3 media. PCR amplification using urease gene primers with DNA from the selected colony as a template resulted in a clear band, and extracts derived from cells from the colony had urease activity (85.4 ± 1.77 NH3 nmol/min/mg protein). Furthermore, 16S rRNA sequencing with subsequent comparison to NCBI database sequences using BLAST revealed that the selected colony showed the highest identity to S. dextrinosolvens strain 0554 (98.13% identity). We named the strain isolated here, S. dextrinosolvens Z6.
We determined that the S. dextrinosolvens Z6 genome is 3.47 Mbp, with a G + C content of 38.9% (Figure 1A), a total of 2993 coding sequences, 21 rRNA genes (7 copies of each 5S rRNA, 16S rRNA, and 23S rRNA), 69 tRNAs, 7 ncRNAs, 3 gene islands, and 5 CRISPR repeats (Table 1). Functional annotation of the genome indicated that the percentage of protein-encoding genes was 98%, while 1268 genes (42.4% of the total protein-encoding gene) are of unknown function. The genes with an annotated function included those involved in “regulating carbohydrate transport and metabolism” (182 genes), “translation, ribosomal structure and biogenesis” (181), accounting for ∼10% each, and “regulation of amino acid transport and metabolism” (155). KEGG annotation indicated that most of the annotated genes were involved in carbohydrate metabolism (138 genes), membrane transport (129 genes), and amino acid metabolism (119 genes) (Figures 1B,C).
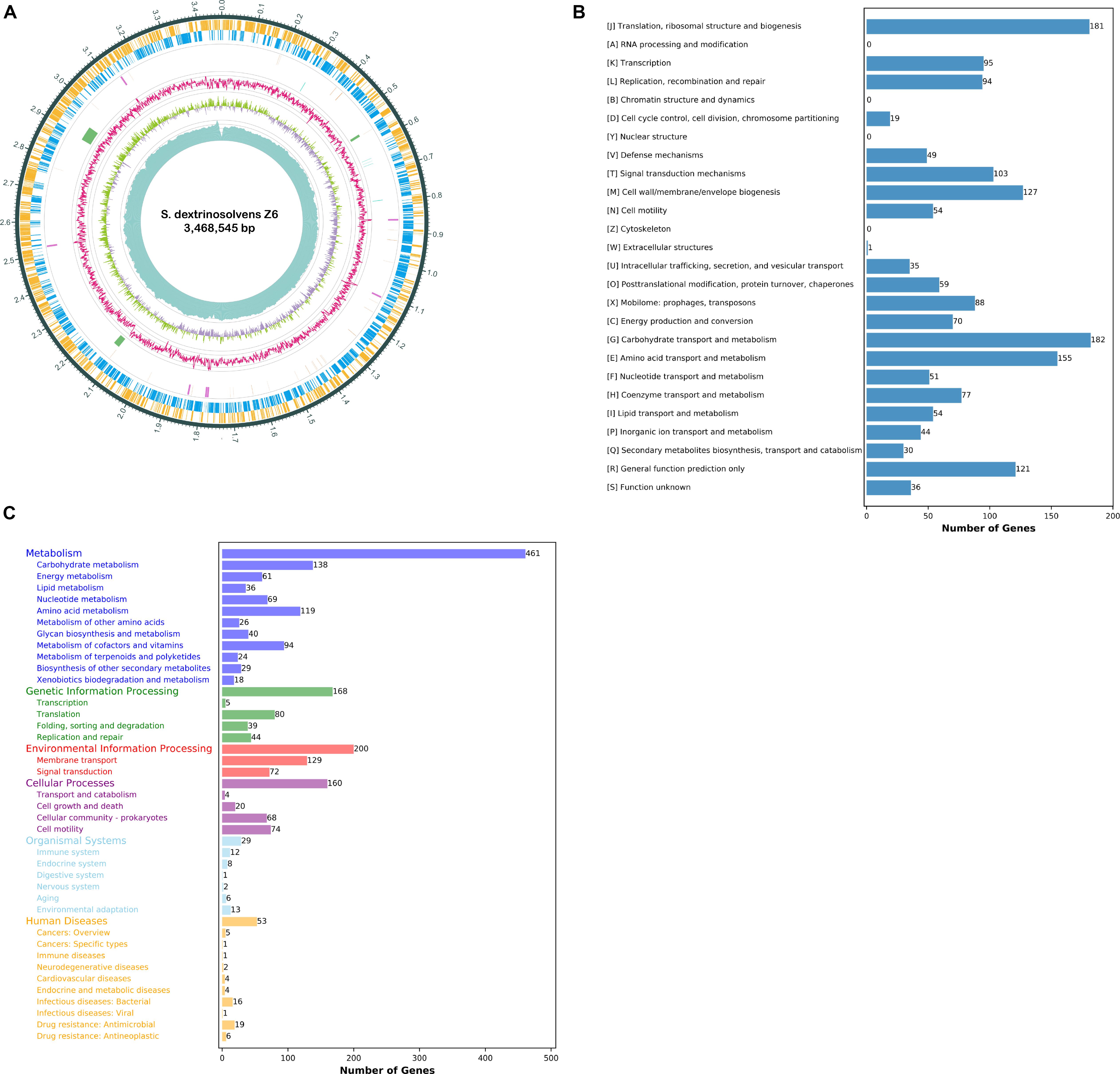
Figure 1. Succinivibrio dextrinosolvens strain Z6 genome information. (A) Circle diagram showing the nuclear genome. From the outside to the inside are the coding genes (sense), coding genes (antisense), tRNAs (orange) and rRNAs (purple), CRISPRs (blue) and gene islands (green), GC ratio, GC-skew, and sequencing depth. (B) Functional classification of genomically encoded proteins in clusters of orthologous groups (COG). (C) Functional classification of genomically encoded proteins using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database.
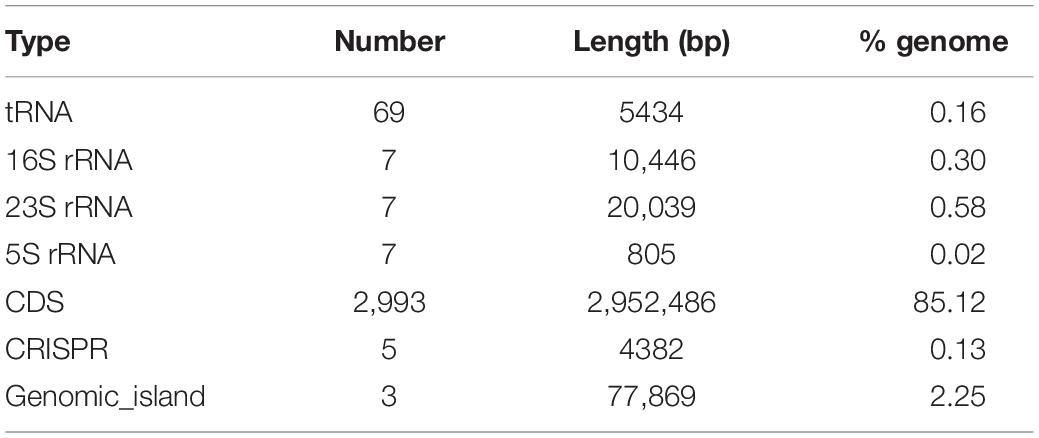
Table 1. Statistics for genomic structure prediction of S. dextrinosolvens Z6.
Circular genome comparison (Figure 2A) of S. dextrinosolvens Z6 with related strains in the genus revealed examples of dissimilarity, such as gaps, indicating the uniqueness of this strain, and a whole genome alignment between S. dextrinosolvens Z6 and the very closely related S. dextrinosolvens 22B showed more than 0.34 Mb of nucleotide polymorphisms (gaps) (Supplementary Table 1). The strain isolated here was fully sequenced, whereas the other strains in the genus are only represented by partial genome sequences (Supplementary Table 1). A phylogenetic tree analysis of S. dextrinosolvens strains showed that strain Z6 was most closely related to S. dextrinosolvens 22B (Figure 2B). ANI values between S. dextrinosolvens strain Z6 and S. dextrinosolvens 22B, S. dextrinosolvens ACV-10, S. dextrinosolvens H5, and S. dextrinosolvens DSM_3072 were 99.96% (64.09% genome coverage), 86.73% (44.24% genome coverage), 83.87% (39.89% genome coverage), and 83.14% (41.14% genome coverage), respectively (Figure 2C and Supplementary Table 2), which is above the recommended cutoff for species delineation (<95–96% ANI) for S. dextrinosolvens 22B.
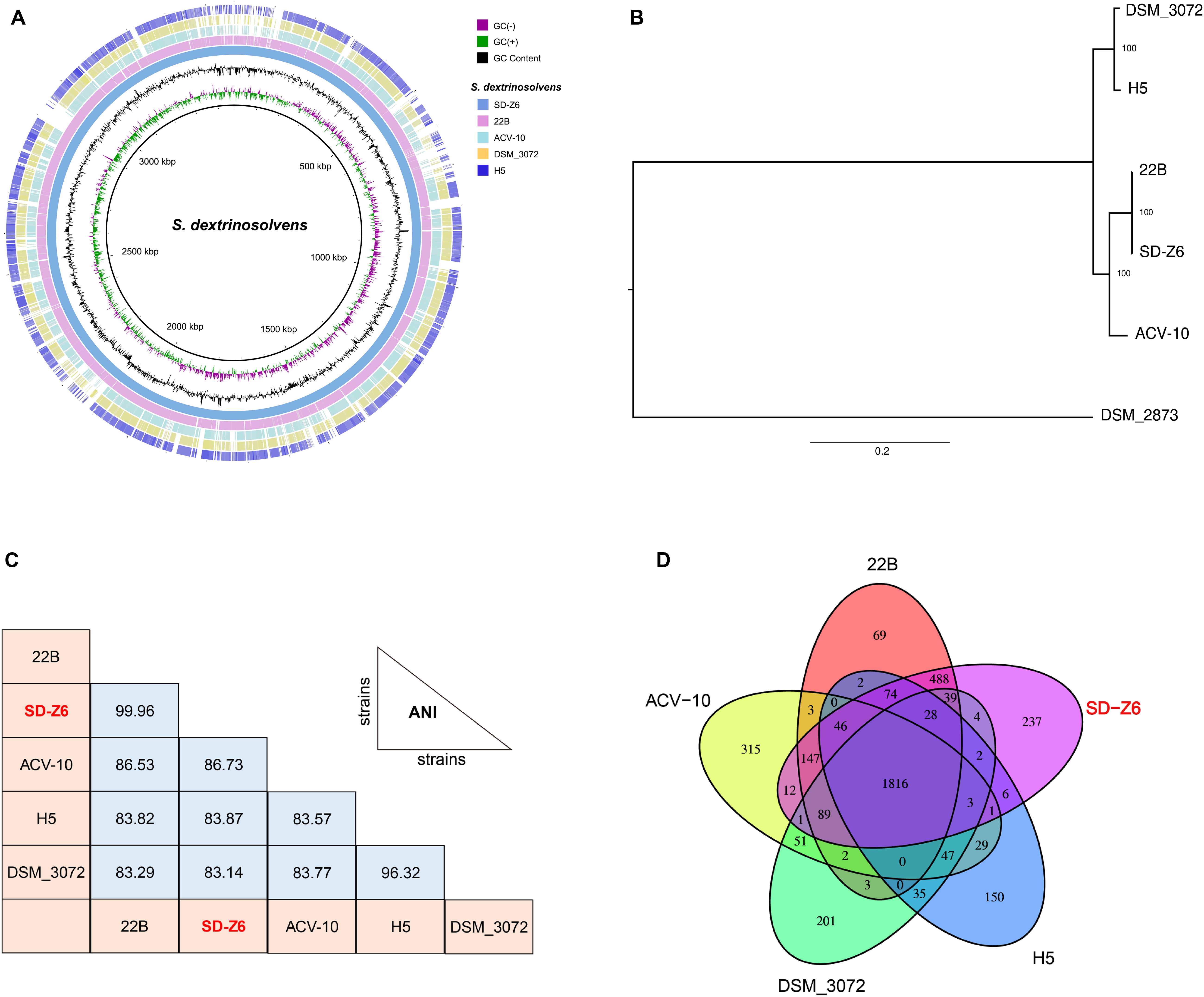
Figure 2. Comparative genome analysis of Succinivibrio dextrinosolvens strain Z6 and closely related strains. (A) BLAST Ring Image Generator (BRIG) analysis of S. dextrinosolvens strain Z6 and the genomes of S. dextrinosolvens 22B, S. dextrinosolvens ACV-10, S. dextrinosolvens H5, and S. dextrinosolvens DSM_3072. The innermost ring represents the skew (- and +), followed by GC content. The third ring shows S. dextrinosolvens strain Z6 (blue), ring 4 S. dextrinosolvens 22B (pink), ring 5 S. dextrinosolvens ACV-10 (light blue), ring 6 S. dextrinosolvens DSM_3072 (yellow), and ring 7 S. dextrinosolvens H5 (dark blue). (B) Phylogenetic tree highlighting the relationship of S. dextrinosolvens strain Z6 with other strains in the Succinivibrio genus. The evolutionary history was inferred using the maximum likelihood method and showed that S. dextrinosolvens Z6 is a member of the Succinivibrio genus. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Succinimonas amylolytica DSM_2873 is an outgroup. (C) Pairwise comparisons of average nucleotide identity (ANI), and (D) global gene conservation in Succinivibrio. Each oval represents the total number of genes in each genome. Overlapping regions depict the number of genes shared between the respective genomes.
S. dextrinosolvens Z6 had the highest gene and protein number of strains reported from the genus (Supplementary Table 1). A comparative gene-by-gene distribution analysis (Figure 2D) between S. dextrinosolvens Z6, S. dextrinosolvens 22B, S. dextrinosolvens ACV-10, S. dextrinosolvens H5, and S. dextrinosolvens DSM_3072 showed that 1816 genes (61%) were conserved among each Succinivibrio genomes. Furthermore, 237 genes were unique to S. dextrinosolvens Z6, compared to 69 for S. dextrinosolvens 22B, 315 for S. dextrinosolvens ACV-10, 150 for S. dextrinosolvens H5, and 201 for S. dextrinosolvens DSM_3072. Thus, the genome sequence of S. dextrinosolvens Z6 adds considerable information about the genetic diversity within the Succinivibrio genus.
Nitrogen metabolism–related genes in the genome of strain Z6 were grouped as genes involved in urea hydrolysis, amino acid- and peptide-degrading genes, and genes involved in ammonia assimilation and amino acid biosynthesis (Figure 3). The open read frames (ORFs) for the urea hydrolysis-related urease (ureA, B, C, E, F and D) and the urea transporter (urtE, D, C, B, and A) genes, which encode a protein of urease enzyme and urea transporter, respectively, clustered together and involved in urea hydrolysis (Table 2). The genes involved in amino acid and peptide hydrolysis, such as pepN, T, P and B, PldB, ilvE, fbl, ArgE, and DadA, were found to be scattered throughout the genome. They encode enzymes that degrade proteins, peptides, and amino acids at different structural sites. Genes involved in ammonia assimilation and the biosynthesis of amino acids (asnB, gltBD, and dapF), such as alanine, aspartate, glutamate, and lysine, were also identified in the genome. However, other genes known to be involved in regulation of nitrogen metabolism and amino acid biosynthesis were spread throughout the genome.
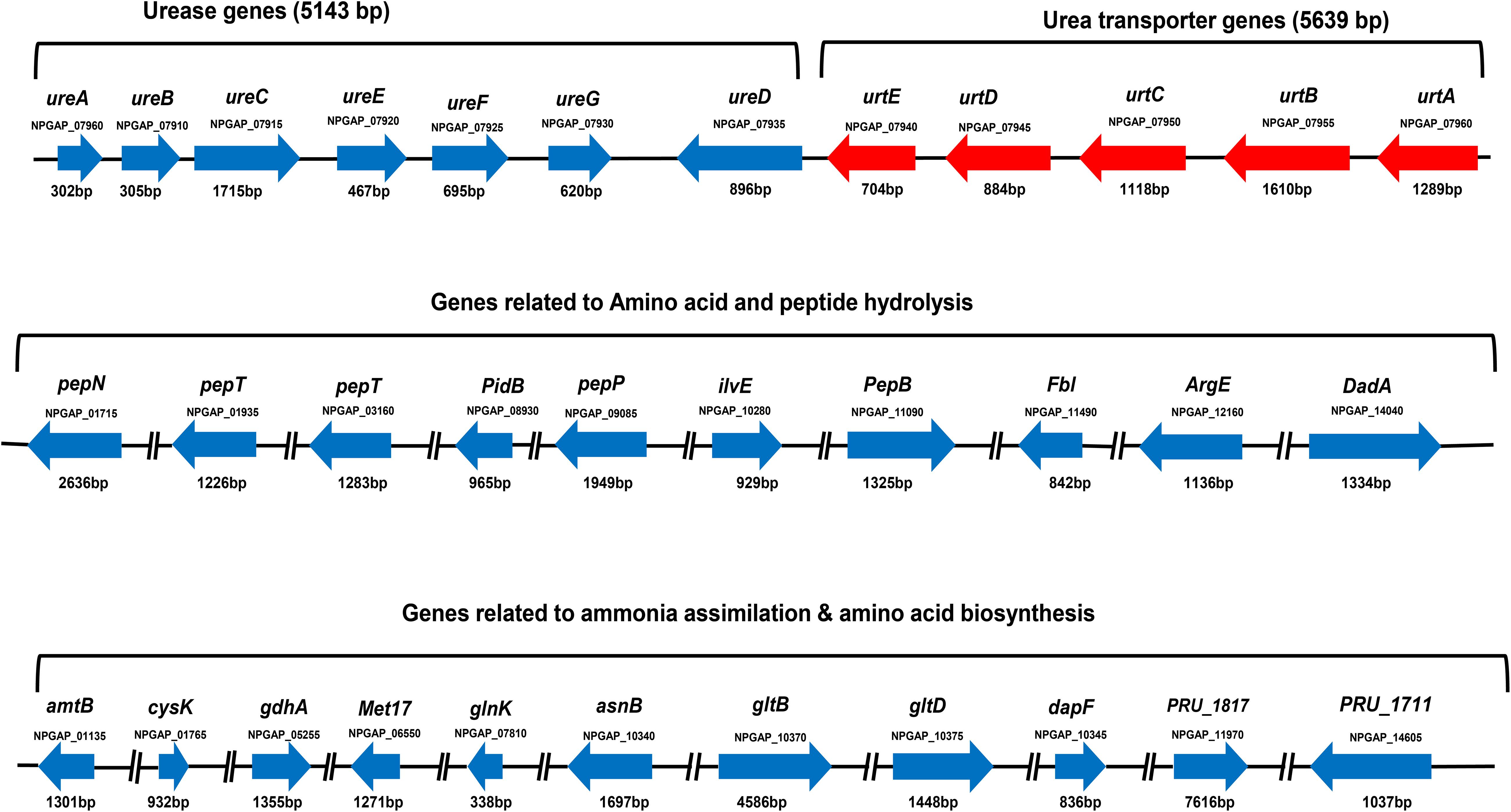
Figure 3. Gene clusters for urea-nitrogen hydrolysis and transportation; gene groups for amino acid and peptide degradation; and ammonia assimilation and amino acid biosynthesis. The code written under the gene represents locus tag. The solid line between the genes indicates the genes on the same cluster and the broken line indicates the genes found in different clusters.
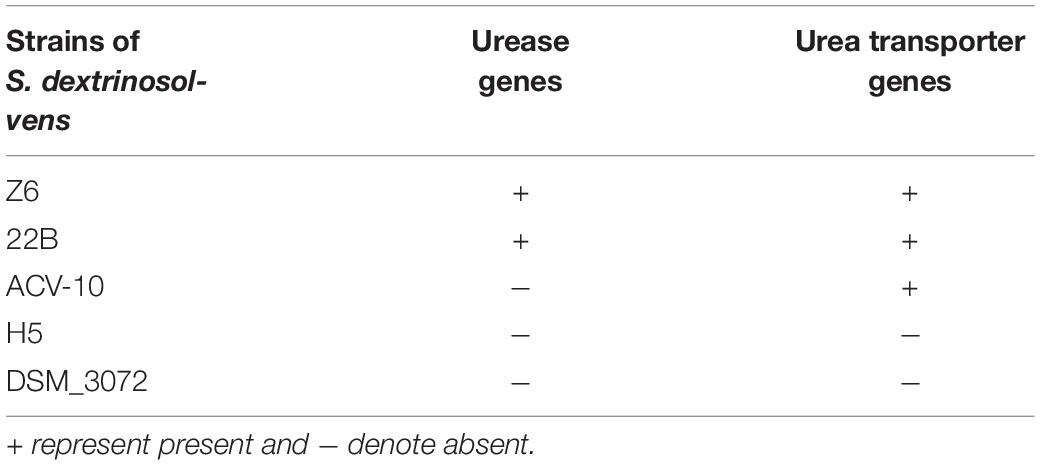
Table 2. The comparison between Succinivibrio dextrinosolvens strains in urease subunit and urea transporter genes.
S. dextrinosolvens Z6 is a strictly anaerobic, gram-negative bacterium with rod-shaped cells 0.12–0.47 μm × 1.01–1.7 μm (width × length). The cells are found singly or in pairs as shown by scanning electron microscopy (SEM) (Figures 4A,B) and have the basic morphological characteristics reported for other members of the Succinivibrio genus. Growth takes place at temperatures from 36 to 42°C with an optimum of 39.7°C (Figure 5A), and a pH range from 6 to 8, with an optimum of 6.9 (Figure 5B). Supplementation with 0–1% (w/v) NaCl supported growth, with maximum growth at 1% (Figure 5C).
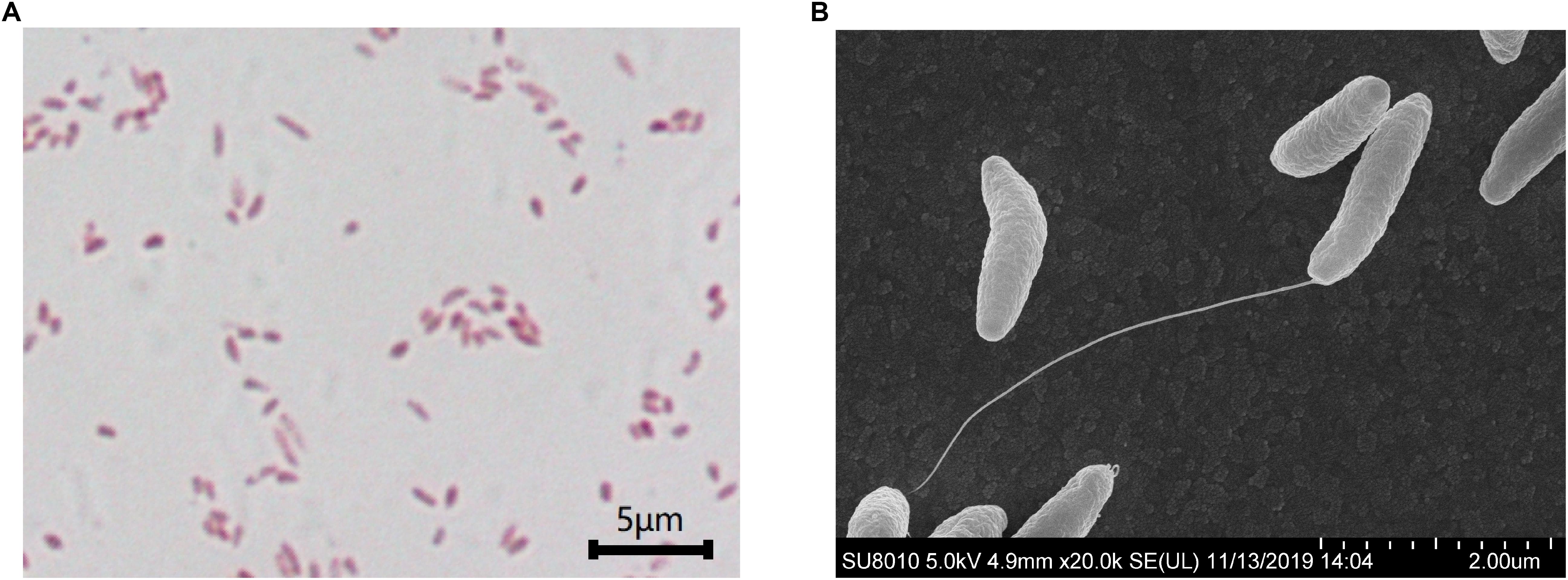
Figure 4. Scanning electron microscopy images of Succinivibrio dextrinosolvens strain Z6. (A) Gram staining of S. dextrinosolvens strain Z6. (B) Scanning electron micrographs of S. dextrinosolvens strain Z6. Scale as indicated on the respective panel.
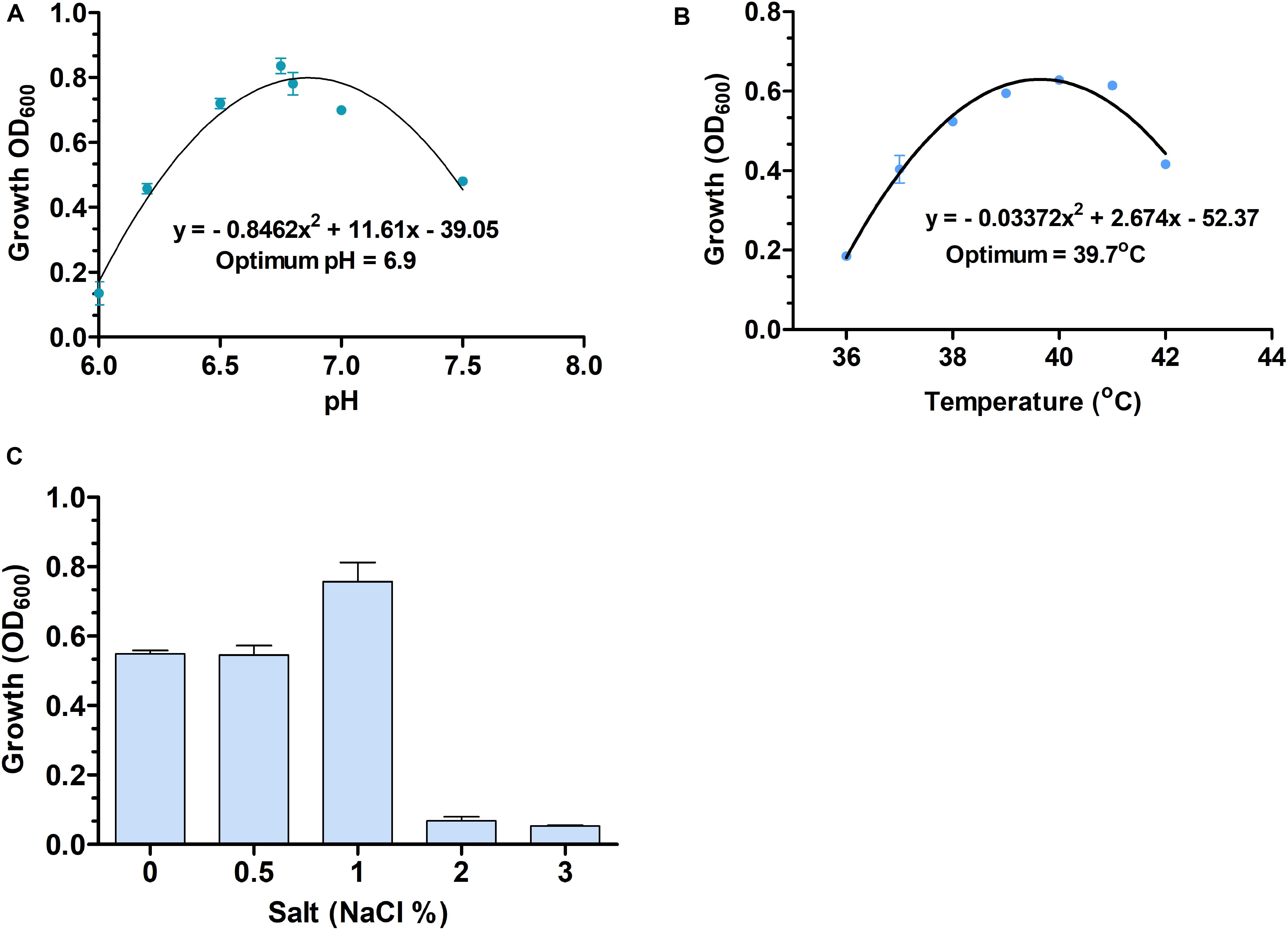
Figure 5. The optimum temperature and pH (A,B), and different salt (% NaCl) concentrations (C) used for Succinivibrio dextrinosolvens strain Z6 growth. Data points represent averages from triplicate experiments. Error bars represent standard deviations.
The predominant detected polar lipids in S. dextrinosolvens Z6 were phosphatidyl glycerol (PG), phosphatidyl ethanolamine (PE), unidentified aminophospholipid (UAPL), and unidentified lipids (UL 1–4), and the cellular fatty acid profile showed that C16:0 FAME (35.34%) and summed feature 10 (C18:1 c11/t9/t6 FAME) (30.93%) were the major components. The major cell wall sugars were ribose and glucose, and the detected DAP (2, 6-diaminopimelic acid) composition in the cell wall was meso-DAP.
A carbohydrate utilization assay revealed that S. dextrinosolvens Z6 fermented D-glucose, D-xylose, maltose, L-arabinose, esculin, D-sorbitol, D-cellobiose, and L-rhamnose, while trehalose, lactose, starch, cellulose, glycerol, inositol, and D-melezitose were not fermented (Supplementary Table 2). API ZYM strips showed that S. dextrinosolvens Z6 was positive for the alkaline phosphatase, esterase lipase, α-chymotrypsin, acid phosphatase, naphthol-AS-BI-phosphohydrolase, leucine arylamidase, valine arylamidase, cysteine arylamidase, and N-acetyl-β-glucosaminidase enzymes, while lipase, trypsin, galactosidase (α and β), glucosidase (α and β), β-glucuronidase, and α-fucosidase tests were negative. A MALDI-TOF spectrum of our strains was compared with spectra in the database and the score values of our strain was less than 1.7 as shown in Supplementary Figure 1.
S. dextrinosolvens Z6 was grown in media containing either urea, ammonia, or amino acids as a nitrogen source, and preferential amino acid utilization was observed (Figure 6A). The maximum growth rate on different nitrogen sources during the exponential growth phase was lower with urea (0.065 ± 0.006 h–1) and amino acids (0.091 ± 0.026 h–1) than with ammonia (0.11 ± 0.016 h–1). The maximum cell density at OD600 was 0.825 ± 0.018, 0.697 ± 0.056, and 0.687 ± 0.024 for growth on amino acid, ammonia, and urea, respectively (Supplementary Table 4). The concentrations of ammonia and urea used by S. dextrinosolvens Z6 were inversely related to the growth phase of the bacteria, whereas the level of amino acid utilization showed a small decline, and after 9 h of incubation, we observed a slight increase and a subsequent stationary profile (Figures 6B–D). The concentration of ammonia in the medium supplemented with ammonia sulfate showed a continuous decrease throughout the exponential growth phase and was almost depleted within 21 h. During growth in urea, concentrations of ammonia increased between 3 and 6 h (Figure 6B), with a continuous decrease in urea concentration (Figure 6D). Finally, the ammonia concentration in the medium supplemented with amino acids was shown to increase between 6 and 12 h, but slower than the rate of ammonia produced by urea.
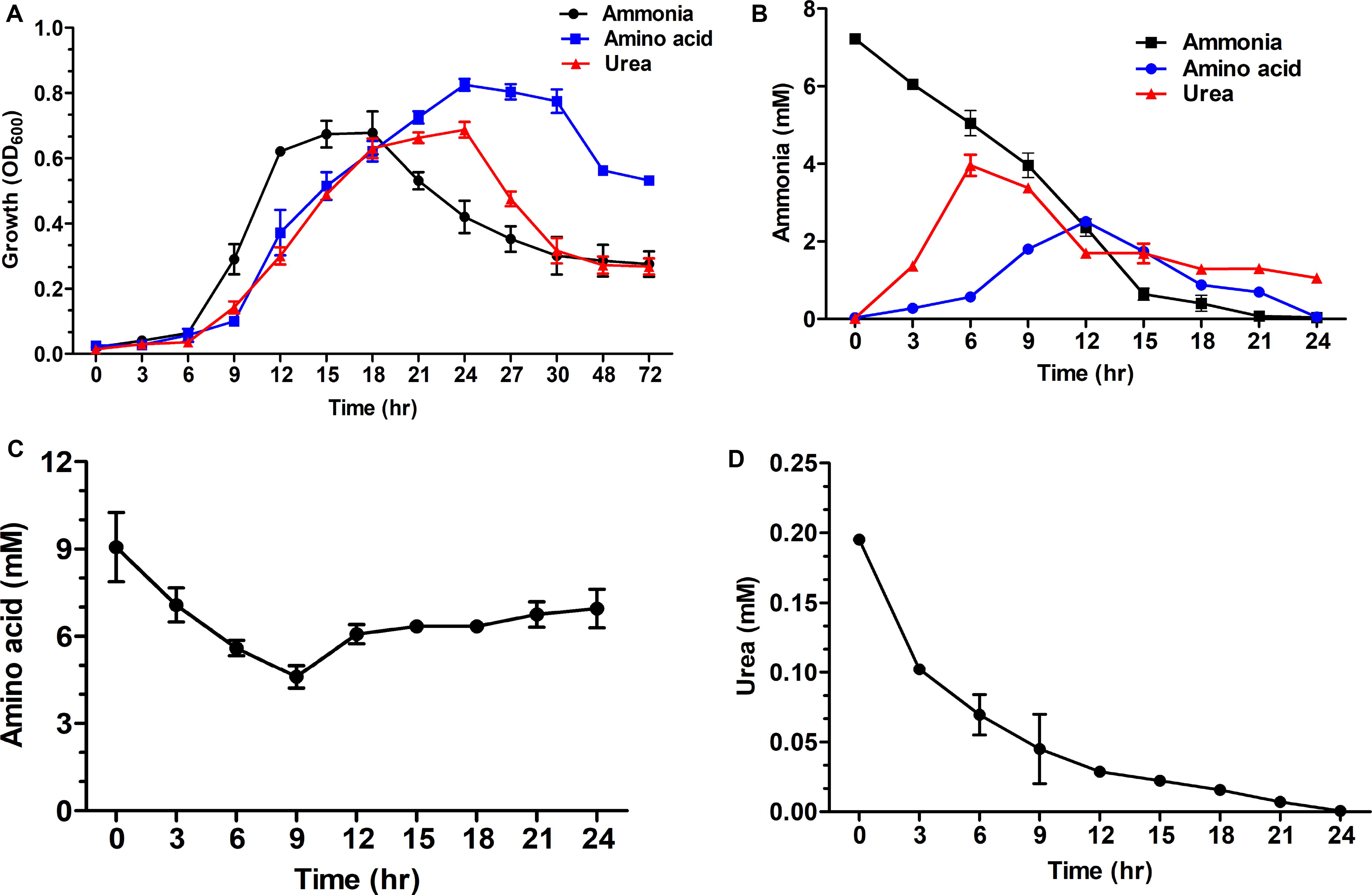
Figure 6. Time course measurements of the effect of nitrogen sources on growth of S. dextrinosolvens Z6. Data points represent averages for triplicate cultures. Error bars represent standard deviations. (A) Growth. (B) Ammonia utilization and production. (C) Amino acid utilization. (D) Urea utilization.
Urease (P < 0.01), GS (P < 0.01), and GDH (P < 0.05) activities were significantly different in response to different nitrogen sources, while GOGAT activity was not significantly different (P = 0.26) for different nitrogen sources. However, all enzyme activities were significantly different (P < 0.01) in the three growth phases. The results for the early and mid-exponential growth phases showed that urease and GS activities were low when cells were grown in a medium with ammonia. These enzyme activities were far higher for the bacteria grown in medium supplemented with urea and amino acids, especially during the earlier growth phase for GS (Figure 7C). In contrast, GDH activity was significantly higher for cells grown in ammonia-containing medium. Notably, urease activity was higher for bacteria grown in urea-containing medium in the late exponential growth phase and a continuous increase was shown throughout the growth phase. The cultures with ammonia and amino acids were shown to have the highest GDH and GOGAT activities in the mid-exponential growth phase (Figures 7B,D). The MCP was significantly different (P < 0.01) for both different nitrogen sources and growth phases.
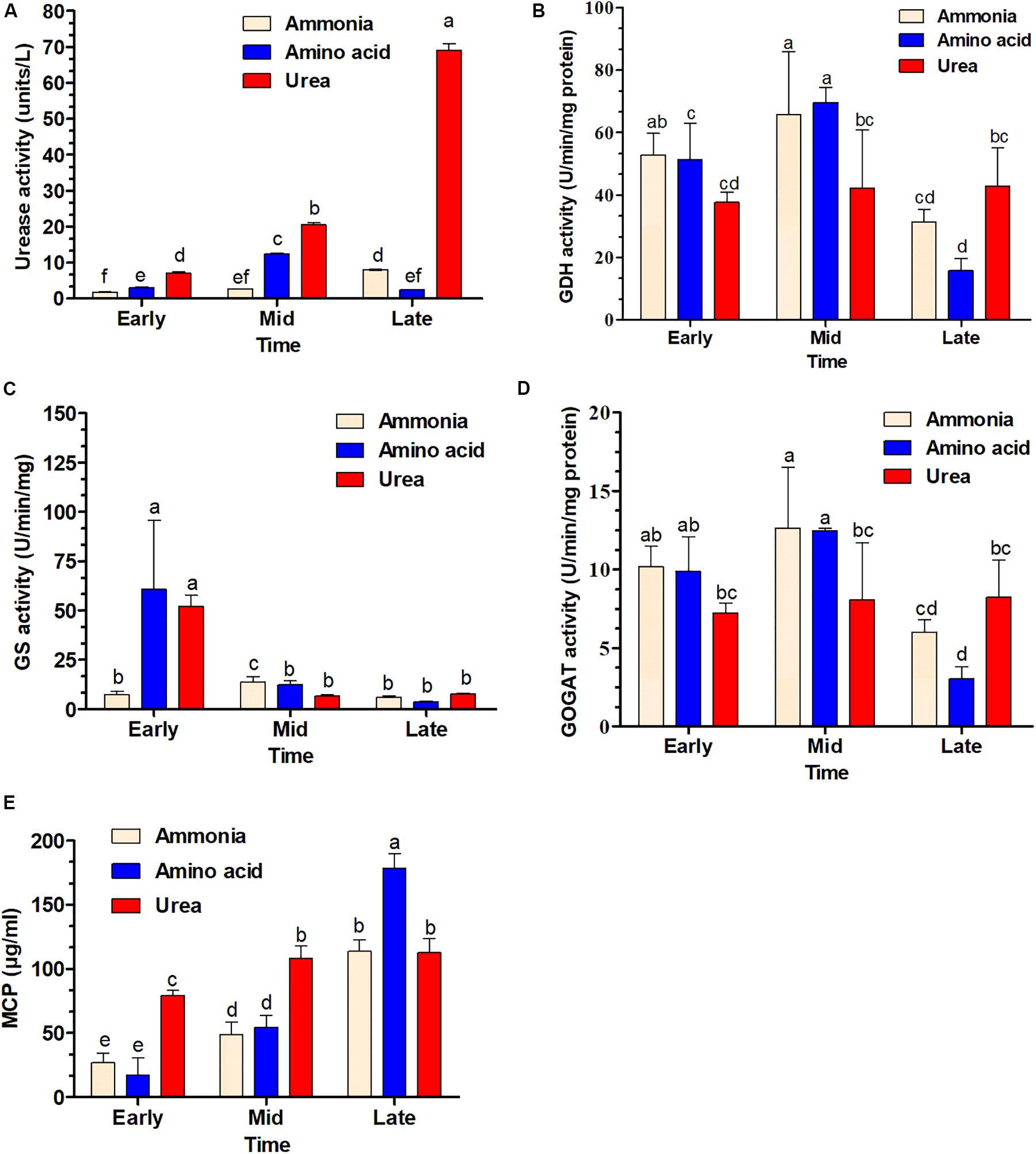
Figure 7. Time course determination of enzyme activity and soluble microbial crude protein production (MCP) in S. dextrinosolvens Z6 grown on different nitrogen sources. The bars represent averages for triplicate cultures used to measure enzyme activity. Error bars represent standard deviations. (A) Urease activity. (B) Glutamate dehydrogenase (GDH) activity. (C) Glutamine synthetase (GS) activity. (D) Glutamate synthase (GOGAT) activity. (E) Soluble microbial crude protein. The data points represent early, mid, and late exponential growth phases. Bars with different letters are significantly different.
Fifteen libraries were established with the following designations: AA1, AA2, AA3, AA4, and AA5 were from amino acid supplement; AMO1, AMO2, AMO3, AMO4, and AMO5 were ammonia supplement, while U1, U2, U3, U4, and U5 were urea-supplemented groups with five biological replicates, respectively. The cDNA libraries were prepared and sequenced by Illumina HiSeq platform, which generated a total of 812,540,682 raw reads (Supplementary Table 5). After removing the adaptors and low-quality reads, 787,498,536 clean reads were obtained with a high quality of Q30_96.23%. We then mapped the trimmed clean reads onto the reference genome and 36.98–85.64% of the clean reads were mapped uniquely to the genome, whereas CDs mapped reads 26.88–86.18% onto the genome (Supplementary Table 6).
S. dextrinosolvens Z6 showed differential gene expression patterns during growth in different nitrogen sources. A total of 1246 genes displayed statistically significant fold changes in their transcript abundances during growth on amino acids, ammonia, or urea. Among the DEGs were 33 related to nitrogen metabolism (Figures 8A–C). Genes (ilvE, CysK, PurF, and FabG) related to amino acid degradation and biosynthesis (mainly branched chain amino acid aminotransferase; enzymes involved in cys/met metabolism, and in phenylalanine, tyrosine, and tryptophan biosynthesis) were significantly differentially expressed in cells grown on amino acids than on urea and ammonia. The highest detected log fold changes in transcript abundances corresponded to the clustered genes encoding the branched chain amino acid aminotransferase (2.55 log-fold changes) followed by 3-deoxy-7-phosphoheptulonate synthase and pyridoxal-phosphate-dependent enzymes involved in amino acid biosynthesis (Supplementary Table 7).
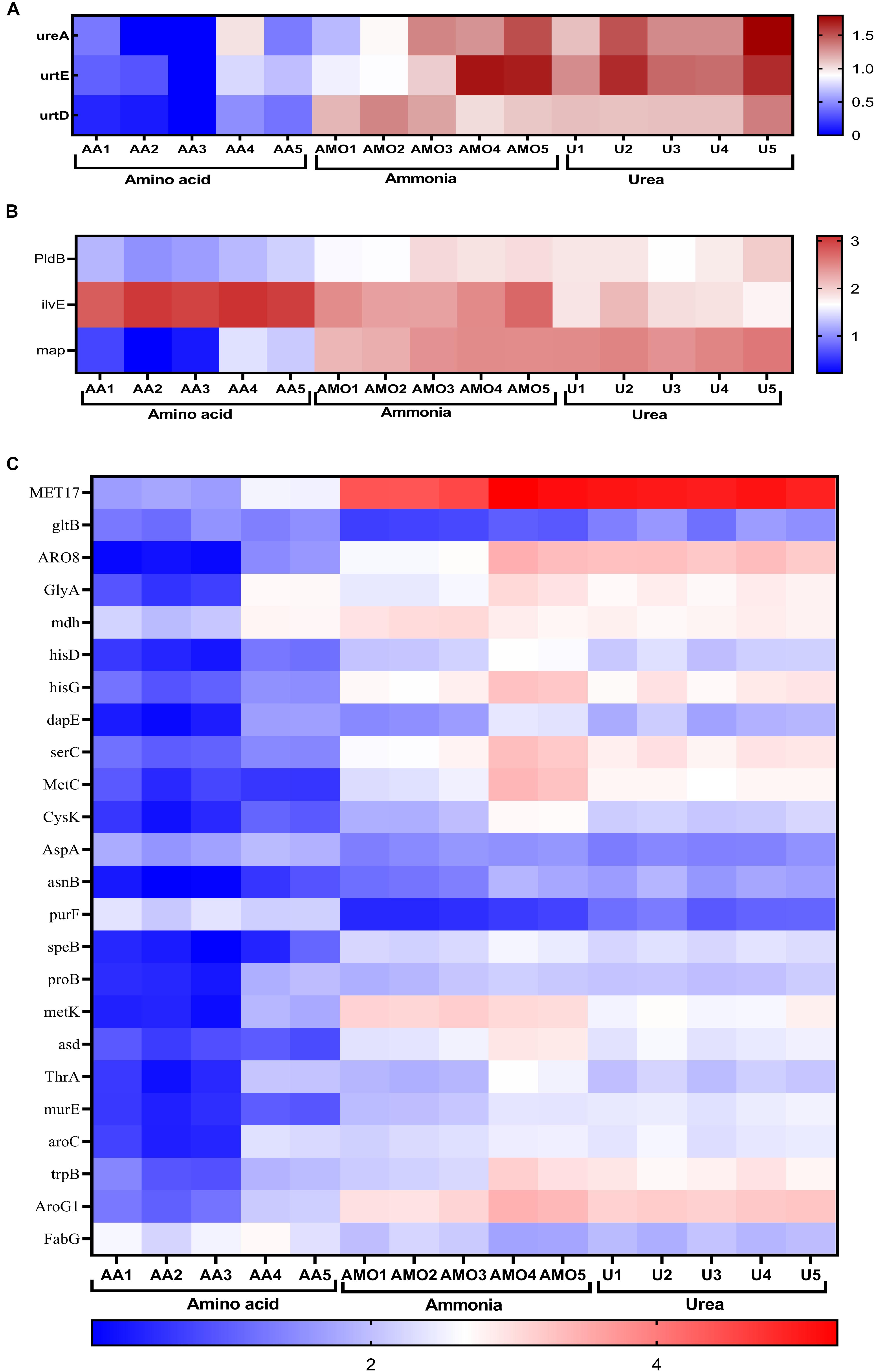
Figure 8. Comparison of differentially expressed genes in cells grown in media containing amino acids, ammonia, or urea. Heat map displaying log-transformed normalized transcript abundances for a subset of genes involved in nitrogen metabolism. (A) Urease and urea transporter genes. (B) Genes involved in amino acid and peptide degradation. (C) Genes involved in ammonia assimilation and amino acid biosynthesis. Abbreviations AA1-5, AMO1-5, and U1-5 represent the five replications of amino acid, ammonia, and urea, respectively.
In this study, a total of eight nitrogen metabolism-related candidate genes found to be differentially expressed in S. dextrinosolvens Z6 grown in a medium containing urea, ammonia, or amino acid were selected for validation to understand their expression level. Quantitative real-time PCR analysis reveals that all the eight assessed candidate genes were differentially expressed in relation to the type of nitrogen sources (Supplementary Figure 2).
Nitrogen is an essential macronutrient for microbial growth and basic metabolic processes and nitrogen sources utilized by different ruminal bacteria include urea, free amino acids, and peptides as non-protein nitrogen-containing compounds (NPN), and true proteins. The degradation and utilization rate of nitrogen sources can vary depending on the species and strain of bacteria. Accordingly, identifying the representative bacteria and understanding their role in the utilization of different nutrients available will provide knowledge that can help manipulate different related microbes for better utilization of feed resources.
In the present study, the ureolytic bacterium S. dextrinosolvens Z6 was isolated from rumen fluid samples collected from dairy cows fed with roughage and concentrate mix. To enrich and analyze its urease-producing capacity, a urea-containing medium was used since, as previously described (Jin et al., 2016); a color change in the medium supplemented with indicators can identify urease-producing organisms (Moore, 1977). Other isolates of this species have previously been obtained from rumen of sheep, cattle, and wallaby guts (Bryant and Small, 1955; Morrison et al., 2011), and have shown significant differences in terms of nitrogen utilization and physiological requirements. The isolated strain was examined morphologically by SEM and was found to be much shorter than the strain isolated by Bryant and Small (1955). Strain 22B and our strain Z6 have clusters of urease and urea transporter genes (Figure 7), which is an indication for the potential of this bacteria in utilizing urea as a nitrogen source as shown by Bryant and Small (1955).
The effect of different nitrogen sources on S. dextrinosolvens Z6 growth was examined by comparing in vitro growth on ammonia, amino acid, and urea as sole nitrogen sources (Figure 6A). S. dextrinosolvens Z6 preferentially utilized amino acids, which is consistent with a previous report (Gomez-Alarcon et al., 1982). Although the maximum growth was lower than for amino acids, strain Z6 was also able to grow on ammonia and urea, and showed a relatively faster growth rate on ammonia. This distinguishes strain Z6 from other reported strains, which prefer ammonia as the nitrogen sources for optimal growth. This was confirmed by the genome sequence and transcriptome analysis, in that strain Z6 contained genes encoding enzymes such as urease, deaminase, aminotransferase and peptidase, which are involved in urea, amino acid, and peptide utilization and metabolism.
In this study to keep RNA integrity/quality, bacteria growth and further enzymatic action have stopped immediately at the time of mid-exponential growth phase; we have used 4°C for harvesting bacteria pellet by centrifuge, but this temperature might cause cold shock response by the bacteria that affect genes’ transcriptional expression (Gao et al., 2006; Liu et al., 2014; Rastrojo et al., 2019). However, we have limited the time to 5 min as it has been applied by Liu et al. (2014) which may reduce this effect on the RNA expression. The enzyme branched-chain amino acid aminotransferase encoded by ilvE catalyzes the conversion of branched-chain amino acids (L-leucine, L-isoleucine, and L-valine) and α-ketoglutarate into branched chain α-keto acids and glutamate. This enzyme was upregulated in cells grown in amino acid-containing media. This suggests that S. dextrinosolvens Z6 can utilize branched chain amino acids for growth and is consistent with studies showing that ruminal bacteria are inhibited and yields of microbial protein decreased in goats fed on a diet from which all branched chain amino acids (BCAA) were removed (Wang et al., 2008; Apajalahti et al., 2019). Furthermore, as has been indicated by different studies, this enzyme is vital for the growth and survival of bacteria (Zhang et al., 2013; Amorim Franco and Blanchard, 2017; Li et al., 2017). However, the level of free amino acids declined for the first 9 h of incubation and subsequently was constant, which might be explained by the production of different nitrogen-containing compounds in the medium. Consistent with this idea, other studies have shown that bacteria can produce some amino acids and release them to the surrounding environment (Lam et al., 2009; Aliashkevich et al., 2018).
We observed an increased transcript abundance of the glutamine phosphoribosylpyrophosphate amidotransferase encoding gene (purF) during growth in amino acid-containing media, unlike with the other two media, which suggests that S. dextrinosolvens Z6 can use L-glutamine to produce purine via the purine biosynthesis pathway (Yee et al., 2015). Purine is a precursor for GTP synthesis and thus plays an important role in energy production and growth regulation (Kriel et al., 2012).
Urease activity was higher in all growth phases in the presence of urea than in the presence of ammonia or amino acids. This is also consistent with the transcript abundance of the urease gene structural components (ureA) and the urea transporter protein-encoding genes, urtE and urtD, which were expressed at higher levels in the presence of urea. A combination of the two gene clusters would support bacterial growth in the urea-containing medium and expand the range of potential nitrogen sources. This result revealed that strain Z6 utilizes an energy-dependent system including the urea transporter, which is encoded by the urtABCDE gene cluster, for taking up urea for hydrolysis. This is consistent with certain other bacterial isolates (Beckers et al., 2004) in which the transporter genes are transcribed in response to nitrogen limitation.
When S. dextrinosolvens Z6 was grown in ammonia, we observed higher glutamate dehydrogenase (NADH-GDH) activity compared to growth on amino acids and urea. However, the transcript of the gene related to GDH was not detectable during growth on any nitrogen source. The activity of GS during the early exponential growth phase was significantly higher for cells grown on amino acids and urea, likely due to limited ammonia levels, since previous studies reported that the activity and expression of GS and its gene were higher when cells were grown at low ammonia concentrations (Hua et al., 2004; Kim et al., 2012).
HisD and hisG are involved in the biosynthesis of histidine in bacteria and other organisms and were expressed at higher levels as a result of growth in urea as compared to growth in ammonia-containing media. These genes encode the protein product of histidinol dehydrogenase and ATP phosphoribosyltransferase, respectively, and were among the DEGs that were more highly expressed. However, other studies have reported that the presence of urea inhibits the activities of these enzymes (Yourno, 1968; Andorn and Aronovitch, 1982; Senior, 1989; Mizukami et al., 1994).
It is evident from the results of this current study that different nitrogen sources affect the enzyme activity and transcript abundance of certain proteins and genes in S. dextrinosolvens Z6, suggesting that there is potential for regulating its nitrogen metabolism. Because of the availability of different nitrogen sources, concentration, and passage rate, it is hard to make a direct comparison between in vitro experiment and real ruminal activities. However, this finding gives a basis to understand the role of the whole ruminal bacteria community.
All datasets presented in this study are included in the article/Supplementary Material.
The animal study was reviewed and approved by the Ethics Committee of Institute of Animal Sciences of CAAS (No. IAS2019-14).
SH and SZ equally contributed to this study’s conceptualization, methodology, software handling, formal analysis, investigation, writing – original draft preparation, and writing – review and editing. JW contributed to validation, supervision, project administration, and funding acquisition. All authors have read and agreed to the published version of the manuscript.
This research was supported by the National Key Research and Development Program (2017YFD0500502), the National Natural Science Foundation of China (31430081), the Scientific Research Project for Major Achievements of the Agricultural Science and Technology Innovation Program (ASTIP) (No. CAAS-ZDXT2019004), and the Modern Agro-Industry Technology Research System of the PR China (CARS-36).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01826/full#supplementary-material
Aliashkevich, A., Alvarez, L., and Cava, F. (2018). New insights into the mechanisms and biological roles of D-amino acids in complex eco-systems. Front. Microbiol. 9:683. doi: 10.3389/fmicb.2018.00683
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Amorim Franco, T. M., and Blanchard, J. S. (2017). Bacterial branched-chain amino acid biosynthesis: structures, mechanisms, and drugability. Biochemistry 56, 5849–5865. doi: 10.1021/acs.biochem.7b00849
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11:R106. doi: 10.1186/gb-2010-11-10-r106
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638
Andorn, N., and Aronovitch, J. (1982). Purification and properties of histidinol dehydrogenase from Escherichia coli B. J. Gen. Microbiol. 128, 579–584. doi: 10.1099/00221287-128-3-579
Apajalahti, J., Vienola, K., Raatikainen, K., Holder, V., and Moran, C. A. (2019). Conversion of branched-chain amino acids to corresponding isoacids - an in vitro tool for estimating ruminal protein degradability. Front. Vet. Sci. 6:311. doi: 10.3389/fvets.2019.00311
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556.Gene
Beckers, G., Bendt, A. K., Krämer, R., and Burkovski, A. (2004). Molecular identification of the urea uptake system and transcriptional analysis of urea transporter- and urease-encoding genes in Corynebacterium glutamicum. J. Bacteriol. 186, 7645–7652. doi: 10.1128/JB.186.22.7645-7652.2004
Bertin, Y., Chaucheyras-Durand, F., Robbe-Masselot, C., Durand, A., de la Foye, A., Harel, J., et al. (2013). Carbohydrate utilization by enterohaemorrhagic Escherichia coli O157: H7 in bovine intestinal content. Environ. Microbiol. 15, 610–622. doi: 10.1111/1462-2920.12019
Blackburn, T. H., and Hungate, R. E. (1963). Succinic acid turnover and propionate production in the bovine rumen. Appl. Microbiol. 11, 132–135. doi: 10.1128/aem.11.2.132-135.1963
Bland, C., Ramsey, T. L., Sabree, F., Lowe, M., Brown, K., Kyrpides, N. C., et al. (2007). CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8:209. doi: 10.1186/1471-2105-8-209
Broderick, G. A., and Kang, J. H. (1980). Automated simultaneous determination of ammonia and total amino acids in ruminal fluid and in vitro media. J. Dairy Sci. 63, 64–75. doi: 10.3168/jds.S0022-0302(80)82888-8
Bryant, M. (1959). Bacterial species of the rumen. Bacteriol. Rev. 23, 125–153. doi: 10.1128/mmbr.23.3.125-153.1959
Bryant, M. P., and Small, N. (1955). Characteristics of two new genera of anaerobic curved rods isolated from the rumen of cattle. J. Dairy Sci. 72, 22–26. doi: 10.1128/jb.72.1.22-26.1956
Buchfink, B., Xie, C., and Huson, D. H. (2014). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Chen, H., and Boutros, P. C. (2011). VennDiagram: a package for the generation of highly-customizable venn and euler diagrams in R. BMC Bioinformatics 12:35. doi: 10.1186/1471-2105-12-35
Danielsson, R., Dicksved, J., Sun, L., Gonda, H., Müller, B., Schnürer, A., et al. (2017). Methane production in dairy cows correlates with rumen methanogenic and bacterial community structure. Front. Microbiol. 8:226. doi: 10.3389/fmicb.2017.00226
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:157. doi: 10.1186/s13059-015-0721-2
Fass, J. J. N. (2011). Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files (Version 1.33).
Frank, J. A., Reich, C. I., Sharma, S., Weisbaum, J. S., Wilson, B. A., and Olsen, G. J. (2008). Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl. Environ. Microbiol. 74, 2461–2470. doi: 10.1128/AEM.02272-07
Gao, H., Yang, Z. K., Wu, L., Thompson, D. K., and Zhou, J. (2006). Global transcriptome analysis of the cold shock response of Shewanella oneidensis MR-1 and mutational analysis of its classical cold shock proteins. J. Bacteriol. 188, 4560–4569. doi: 10.1128/JB.01908-05
Gomez-Alarcon, R. A., O’Dowd, C., Leedle, J. A. Z., and Bryant, M. P. (1982). 1,4-Naphthoquinone and other nutrient requirements of Succinivibrio dextrinosolvens. Appl. Environ. Microbiol. 44, 346–350. doi: 10.1128/aem.44.2.346-350.1982
Haft, D. H., Selengut, J. D., Richter, R. A., Harkins, D., Basu, M. K., and Beck, E. (2013). TIGRFAMs and genome properties in 2013. Nucleic Acids Res. 41, 387–395. doi: 10.1093/nar/gks1234
Hall, B. G., Acar, H., Nandipati, A., and Barlow, M. (2014). Growth rates made easy. Mol. Biol. Evol. 31, 232–238. doi: 10.1093/molbev/mst187
Hernandez-Sanabria, E., Goonewardene, L. A., Wang, Z., Durunna, O. N., Moore, S. S., and Guan, L. L. (2012). Impact of feed efficiency and diet on adaptive variations in the bacterial community in the rumen fluid of cattle. Appl. Environ. Microbiol. 78, 1203–1214. doi: 10.1128/AEM.05114-11
Hua, Q., Yang, C., Oshima, T., Mori, H., and Shimizu, K. (2004). Analysis of gene expression in Escherichia coli in response to changes of growth-limiting nutrient in chemostat cultures. Society 70, 2354–2366. doi: 10.1128/AEM.70.4.2354
Hucker, G. J. (1921). A new modification and application of the gram stain. J. Bacteriol. 6, 395–397. doi: 10.1128/jb.6.4.395-397.1921
Hudson, C. M., Lau, B. Y., and Williams, K. P. (2015). Islander: a database of precisely mapped genomic islands in tRNA and tmRNA genes. Nucleic Acids Res. 43, D48–D53. doi: 10.1093/nar/gku1072
Hunt, M., Silva, N., De Otto, T. D., Parkhill, J., Keane, J. A., and Harris, S. R. (2015). Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. 16, 1–10. doi: 10.1186/s13059-015-0849-0
Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Indugu, N., Vecchiarelli, B., Baker, L. D., Ferguson, J. D., Vanamala, J. K. P., and Pitta, D. W. (2017). Comparison of rumen bacterial communities in dairy herds of different production. BMC Microbiol. 17:190. doi: 10.1186/s12866-017-1098-z
Jesus, T. F., Ribeiro-Gonçalves, B., Silva, D. N., Bortolaia, V., Ramirez, M., and Carriço, J. A. (2019). Plasmid ATLAS: plasmid visual analytics and identification in high-Throughput sequencing data. Nucleic Acids Res. 47, D188–D194. doi: 10.1093/nar/gky1073
Jin, D., Zhao, S., Wang, P., Zheng, N., Bu, D., Beckers, Y., et al. (2016). Insights into abundant rumen ureolytic bacterial community using rumen simulation system. Front. Microbiol. 7:1006. doi: 10.3389/fmicb.2016.01006
Jones, D. L., Owen, A. G., and Farrar, J. F. (2002). Simple method to enable the high resolution determination of total free amino acids in soil solutions and soil extracts. Soil Biol. Biochem. 34, 1893–1902. doi: 10.1016/S0038-0717(02)00203-1
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kalvari, I., Argasinska, J., Quinones-Olvera, N., Nawrocki, E. P., Rivas, E., Eddy, S. R., et al. (2018). Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 46, D335–D342. doi: 10.1093/nar/gkx1038
Kanehisa, M., Goto, S., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, 199–205. doi: 10.1093/nar/gkt1076
Khafipour, E., Li, S., Plaizier, J. C., and Krause, D. O. (2009). Rumen microbiome composition determined using two nutritional models of subacute ruminal acidosis. Appl. Environ. Microbiol. 75, 7115–7124. doi: 10.1128/AEM.00739-09
Kim, J. N., Cann, I. K. O., and Mackie, R. I. (2012). Purification, characterization, and expression of multiple glutamine synthetases from Prevotella ruminicola 23. J. Bacteriol. 194, 176–184. doi: 10.1128/JB.05916-11
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive κ-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kriel, A., Bittner, A. N., Kim, S. H., Liu, K., Tehranchi, A. K., Zou, W. Y., et al. (2012). Direct regulation of GTP homeostasis by (p)ppGpp: a critical component of viability and stress resistance. Mol. Cell 48, 231–241. doi: 10.1016/j.molcel.2012.08.009
Kruger Ben Shabat, S., Sasson, G., Doron-Faigenboim, A., Durman, T., Yaacoby, S., Berg Miller, M. E., et al. (2016). Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 10, 2958–2972. doi: 10.1038/ismej.2016.62
Lagesen, K., Hallin, P., Rødland, E. A., Stærfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lam, H., Oh, D. C., Cava, F., Takacs, C. N., Clardy, J., De Pedro, M. A., et al. (2009). D-amino acids govern stationary phase cell wall remodeling in bacteria. Science 325, 1552–1555. doi: 10.1126/science.1178123
Lee, I., Kim, Y. O., Park, S. C., and Chun, J. (2016). OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 66, 1100–1103. doi: 10.1099/ijsem.0.000760
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, K. H., Yu, Y. H., Dong, H. J., Zhang, W., Ma, J. C., and Wang, H. H. (2017). Biological functions of ilvC in branched-chain fatty acid synthesis and diffusible signal factor family production in Xanthomonas campestris. Front. Microbiol. 8:2486. doi: 10.3389/fmicb.2017.02486
Liu, B., Zhang, Y., and Zhang, W. (2014). RNA-seq-based analysis of cold shock response in Thermoanaerobacter tengcongensis, a bacterium harboring a single cold shock protein encoding gene. PLoS One. 9: e93289. doi: 10.1371/journal.pone.0093289
Lowe, T. M., and Chan, P. P. (2016). tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57. doi: 10.1093/nar/gkw413
Makkar, H. P., Sharma, O. P., Dawra, R. K., and Negi, S. S. (1982). Simple determination of microbial protein in rumen liquor. J. Dairy Sci. 65, 2170–2173. doi: 10.3168/jds.S0022-0302(82)82477-6
McCabe, M. S., Cormican, P., Keogh, K., O’Connor, A., O’Hara, E., Palladino, R. A., et al. (2015). Illumina MiSeq phylogenetic amplicon sequencing shows a large reduction of an uncharacterised succinivibrionaceae and an increase of the Methanobrevibacter gottschalkii clade in feed restricted cattle. PLoS One 10:e0133234. doi: 10.1371/journal.pone.0133234
Mizukami, T., Hamu, A., Ikeda, M., Oka, T., and Katsumata, R. (1994). Cloning of the ATP phosphoribosyl transferase gene of Corynebacterium glutamicum and application of the gene to L-histidine production. Biosci. Biotechnol. Biochem. 58, 635–638. doi: 10.1271/bbb.58.635
Moore, W. E. (1977). Urease assay and urease-producing species of anaerobes in the bovine rumen and human feces. Urease assay and urease-producing species of anaerobes in the bovine rumen and human feces. Appl. Environ. Microbiol. 33, 1097–1104. doi: 10.1128/aem.33.5.1097-1104.1977
Morrison, M., Pope, P. B., Smith, W., Denman, S. E., Tringe, S. G., Barry, K., et al. (2011). Isolation of Succinivibrionaceae implicated in low methane emissions from tammar wallabies. Science 333, 646–648. doi: 10.1126/science.1205760
Nawrocki, E. P., and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
O’Herrin, S. M., and Kenealy, W. R. (1993). Glucose and carbon dioxide metabolism by Succinivibrio dextrinosolvens. Appl. Environ. Microbiol. 59, 748–755. doi: 10.1128/aem.59.3.748-755.1993
O’Leary, N. A., Wright, M. W., Brister, J. R., Ciufo, S., Haddad, D., McVeigh, R., et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. doi: 10.1093/nar/gkv1189
Patterson, J. A., and Hespell, R. B. (1985). Glutamine synthetase activity in the ruminal bacterium Succinivibrio dextrinosolvens. Appl. Environ. Microbiol. 50, 1014–1020. doi: 10.1128/aem.50.4.1014-1020.1985
Pope, P. B., Smith, W., Denman, S. E., Tringe, S. G., Barry, K., Hugenholtz, P., et al. (2011). Isolation of Succinivibrionaceae implicated in low methane emissions from Tammar wallabies. Science 333, 646–648.
Rastrojo, A., Corvo, L., Lombraña, R., Solana, J. C., Aguado, B., and Requena, J. M. (2019). Analysis by RNA-seq of transcriptomic changes elicited by heat shock in Leishmania major. Sci. Rep. 9:6919. doi: 10.1038/s41598-019-43354-9
Reed, K. E. (2001). Restriction enzyme mapping of bacterial urease genes: using degenerate primers to expand experimental outcomes. Biochem. Mol. Biol. Educ. 29, 239–244. doi: 10.1016/S1470-8175(01)00095-9
Richmond, S., and Yep, A. (2019). Quantification of urease activity. Methods Mol. Biol. 2021, 85–96.
Russell, J. B., and Hespell, R. B. (1981). Microbial rumen fermentation. J. Dairy Sci. 64, 1153–1169. doi: 10.3168/jds.S0022-0302(81)82694-X
Seo, J. K., Kim, M. H., Yang, J. Y., Kim, H. J., Lee, C. H., Kim, K. H., et al. (2013). Effects of synchronicity of carbohydrate and protein degradation on rumen fermentation characteristics and microbial protein synthesis. Asian Austral. J. Anim. Sci. 26, 358–365. doi: 10.5713/ajas.2012.12507
Stackebrandt, E., and Hespell, R. B. (2006). “The family Succinivibrionaceae”, in The Prokaryotes, eds. M. Dworking, S. Falkow, E. Rosenberg, K. H. Schleifer, and E. Stackebrandt, (Cham: Springer International Publishing) 419–429.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e0112963. doi: 10.1371/journal.pone.0112963
Wang, M., Wang, H., Cao, H., Li, G., and Zhang, J. (2008). Effects of limiting amino acids on rumen fermentation and microbial community In vitro. Agric. Sci. China 7, 1524–1531. doi: 10.1016/S1671-2927(08)60412-5
Xue, M., Sun, H., Wu, X., Guan, L. L., and Liu, J. (2018). Assessment of rumen microbiota from a large dairy cattle cohort reveals the pan and core bacteriomes contributing to varied phenotypes. Appl. Environ. Microbiol. 84, 1–13. doi: 10.1128/AEM.00970-18
Yang, X., Liu, D., Liu, F., Wu, J., Zou, J., Xiao, X., et al. (2013). HTQC: a fast quality control toolkit for Illumina sequencing data. BMC Bioinformatics 14:33. doi: 10.1186/1471-2105-14-33
Yee, R., Cui, P., Shi, W., Feng, J., and Zhang, Y. (2015). Genetic screen reveals the role of purine metabolism in Staphylococcus aureus persistence to rifampicin. Antibiotics 4, 627–642. doi: 10.3390/antibiotics4040627
Yourno, J. (1968). Composition and subunit structure of histidinol dehydrogenase from Salmonella typhimurium. J. Biol. Chem. 243, 3277–3288.
Zeng, H., Guo, C., Sun, D., Seddik, H., and Mao, S. (2019). The ruminal microbiome and metabolome alterations associated with diet-induced milk fat depression in dairy cows. Metabolites 9:154. doi: 10.3390/metabo9070154
Keywords: Succinivibrio dextrinosolvens, nitrogen metabolism, genome sequence, enzyme activity, rumen
Citation: Hailemariam S, Zhao S and Wang J (2020) Complete Genome Sequencing and Transcriptome Analysis of Nitrogen Metabolism of Succinivibrio dextrinosolvens Strain Z6 Isolated From Dairy Cow Rumen. Front. Microbiol. 11:1826. doi: 10.3389/fmicb.2020.01826
Received: 30 March 2020; Accepted: 10 July 2020;
Published: 14 August 2020.
Edited by:
George Tsiamis, University of Patras, GreeceReviewed by:
Stephan Schmitz-Esser, Iowa State University, United StatesCopyright © 2020 Hailemariam, Zhao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shengguo Zhao, emhhb3NoZW5nZ3VvMTk4NEAxNjMuY29t; Jiaqi Wang, amlhcWl3YW5nQHZpcC4xNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.