 Takashi Abe
Takashi Abe Yu Akazawa1†
Yu Akazawa1† Tomoya Baba
Tomoya Baba- 1Department of Information Engineering, Faculty of Engineering, Niigata University, Niigata, Japan
- 2Comparative Genomics Laboratory, National Institute of Genetics, Mishima, Japan
- 3Advanced Genomics Center, National Institute of Genetics, Mishima, Japan
- 4Microbial Physiology Laboratory, National Institute of Genetics, Mishima, Japan
- 5Joint Support-Center for Data Science Research, Research Organization of Information and Systems, Tokyo, Japan
Horizontal gene transfer (HGT) has been widely suggested to play a critical role in the environmental adaptation of microbes; however, the number and origin of the genes in microbial genomes obtained through HGT remain unknown as the frequency of detected HGT events is generally underestimated, particularly in the absence of information on donor sequences. As an alternative to phylogeny-based methods that rely on sequence alignments, we have developed an alignment-free clustering method on the basis of an unsupervised neural network “Batch-Learning Self-Organizing Map (BLSOM)” in which sequence fragments are clustered based solely on oligonucleotide similarity without taxonomical information, to detect HGT candidates and their origin in entire genomes. By mapping the microbial genomic sequences on large-scale BLSOMs constructed with nearly all prokaryotic genomes, HGT candidates can be identified, and their origin assigned comprehensively, even for microbial genomes that exhibit high novelty. By focusing on two types of Alphaproteobacteria, specifically psychrotolerant Sphingomonas strains from an Antarctic lake, we detected HGT candidates using BLSOM and found higher proportions of HGT candidates from organisms belonging to Betaproteobacteria in the genomes of these two Antarctic strains compared with those of continental strains. Further, an origin difference was noted in the HGT candidates found in the two Antarctic strains. Although their origins were highly diversified, gene functions related to the cell wall or membrane biogenesis were shared among the HGT candidates. Moreover, analyses of amino acid frequency suggested that housekeeping genes and some HGT candidates of the Antarctic strains exhibited different characteristics to other continental strains. Lys, Ser, Thr, and Val were the amino acids found to be increased in the Antarctic strains, whereas Ala, Arg, Glu, and Leu were decreased. Our findings strongly suggest a low-temperature adaptation process for microbes that may have arisen convergently as an independent evolutionary strategy in each Antarctic strain. Hence, BLSOM analysis could serve as a powerful tool in not only detecting HGT candidates and their origins in entire genomes, but also in providing novel perspectives into the environmental adaptations of microbes.
Introduction
Although genetic information is generally inherited vertically, i.e., from an organism’s ancestor to its offspring, it can occasionally be transferred horizontally in a process called horizontal gene transfer (HGT) between organisms that do not share an ancestor-offspring relationship (Andam and Gogarten, 2011). HGT was first experimentally demonstrated in Escherichia coli in the late 1940s (Tatum and Lederberg, 1947; Soucy et al., 2015), however, the potential evolutionary implications of cross-species HGT was first appreciated only in the late 1980s (Syvanen, 1985, 2012). Since this time, HGT studies have focused primarily on the phylogenetic relationships among organisms, as revealed through similarity searches of nucleotide or amino acid sequences, and were related to taxonomy, the last universal common ancestor, and the Tree of Life (Andam and Gogarten, 2011; Syvanen, 2012; Soucy et al., 2015). Recent innovations in DNA sequencing have led to a massive increase in the amount of genomic and metagenomic data leading to not only an expansion of the interpretations of the Tree of Life through the identification of previously unknown genes, genomic information, or organisms using cultivation-independent genomics approaches, but also the prediction of potentially enormous numbers of HGT cases (Hug et al., 2016; Parks et al., 2017; Schulz et al., 2017; Williams et al., 2017; Castelle and Banfield, 2018).
In terms of its biological significance, HGT plays a dominant role in the evolution and environmental adaptation of an organism (Ochman et al., 2000; Gogarten and Townsend, 2005; Keeling and Palmer, 2008; Darmon and Leach, 2014; Booth et al., 2016). For instance, certain HGTs have been suggested to be associated with adaptation to living in various adverse environments, such as cold (Blanc et al., 2012; Raymond and Kim, 2012; Zeng et al., 2016; Mock et al., 2017), hot and acidic (Schonknecht et al., 2013), or high-salt environments (Harding et al., 2017), as well as environments that induce oxidative stress or ultraviolet radiation stress (Fast et al., 2003; Slamovits and Keeling, 2004), and those featuring the combined stresses of desiccation, oxidation, and osmotic pressure (Eyres et al., 2015). Several bioinformatics and genome-wide studies concerning HGT genes have been conducted, including the estimation of HGT gene frequency (Dagan et al., 2008; Zamani-Dahaj et al., 2016), and HGT gene functional analysis (Jain et al., 1999; Nakamura et al., 2004; Beiko et al., 2005; Choi and Kim, 2007). These studies confirmed the global applicability of HGT and their importance in the evolution of species. Furthermore, they employed approaches based on the phylogenetic relationships that rely on sequence alignments and reference datasets. However, currently predicted HGTs remain limited as HGT is typically identified using phylogeny-based methods that rely on sequence alignments. Additionally, the individual evaluation of HGT genes in each genome analysis remains a challenge to be overcome. Using phylogeny-based methods, an HGT is detected once the phylogenetic location of a query sequence in a tree is determined to not match that of a reference phylogeny. Although this approach is standard, the frequency of HGT events might be underestimated, particularly when information on donor sequences is lacking (Tamames and Moya, 2008).
An alternative approach is the composition-based method, which relies on nucleotide compositional features such as G + C percentage or codon usage (Kanaya et al., 1999, 2001; Garcia-Vallve et al., 2000; Nakamura et al., 2004), and thus does not require sequence alignments. Although G + C percentage has been used as a fundamental phylogenetic characteristic of microbial genomes, it is too simple a parameter for the differentiation of a wide variety of genomes. However, oligonucleotide composition can distinguish between species even when they feature the same G + C percentage, as the oligonucleotide composition varies substantially among genomes and thus constitutes a “genome signature” (Karlin et al., 1998; Mrazek, 2009). Moreover, composition-based methods are generally effective at identifying recently acquired HGT events since ancient HGT genes eventually evolve or ameliorate to become similar to the other genome signatures of host organisms (Douglas and Langille, 2019). However, compositional and phylogenetic methods have different sensitivities as they likely target HGT events of different ages, with the compositional method primarily identifying recent transfers, while phylogenetic methods generally detect older events, although sufficient variation is required to alter tree topology of the region imported by HGT (Tamames and Moya, 2008).
To improve existing compositional methods, we have focused on the oligonucleotide composition in genomic sequences and developed an alignment-free clustering method based on an unsupervised neural network: “Batch-Learning Self-Organizing Map (BLSOM).” Neural networks are a typical machine learning method based on the mathematical model of the neuron circuit; meanwhile the Kohonen’s Self-Organizing Map, which is the basis of BLSOM, was proposed based on the mathematical model of self-organization in the cerebral visual cortex (Kohonen, 1990; Kohonen et al., 1996). In this method, sequence fragments are clustered based solely on the similarity of oligonucleotides and in the absence of taxonomical information (Kanaya et al., 2001; Abe et al., 2003). We have previously optimized the BLSOM method for the phylogenetic classification of genomic sequences obtained from the mixed genomes of environmental microbes (Iwasaki et al., 2013; Nakao et al., 2013; Qiu et al., 2019), and analyzed tetranucleotide frequencies (256-dimensional vectorial data) in 5-kb sequence fragments. Furthermore, by focusing on HGT events in relation to the symbiosis between eukaryotes and prokaryotes, we have successfully detected HGT candidates from diverse bacterial origins in the tsetse fly genome despite their low similarity to currently available genomic sequences (Nakao et al., 2016).
In this study, we applied our BLSOM method to detect HGTs in the genomes of two Antarctic bacteria, Sphingomonas sp. HMP6 and HMP9, as well as other Sphingomonas strains isolated from other continents. The two Antarctic bacteria are taxonomically closely related and were isolated from the same environment, i.e., an Antarctic moss pillar (Nakai et al., 2012). The genus Sphingomonas belongs to the family Sphingomonadaceae of the class Alphaproteobacteria (Takeuchi et al., 2001) and has been isolated from various environments, including water (Yabuuchi et al., 2001), plants (Eevers et al., 2015), and soil (Huang et al., 2009; Tabata et al., 2011; Wei et al., 2015), as well as in the form of cosmopolitan bacteria in all continents on the planet (Huang et al., 2017). Antarctica is an extreme environment, especially for terrestrial organisms, due to its freezing temperatures, desiccation, oligotrophic environment, and intense ultraviolet radiation (Chown et al., 2015). As a consequence, organisms living in Antarctica have evolved to adjust to their environment and have thrived in unique biospheres, including “moss pillars” in some ultra-oligotrophic lakes in Skarvsnes, East Antarctica (Imura et al., 1999). Here we have sought to elucidate the development of evolutionary adaptations of Antarctic bacteria using genome sequence analysis; and have identified many coding HGT genes in these genomes using phylogeny-based methods. However, many genes remain to be identified. As part of a growing fundamental enquiry into the mechanisms responsible for microbe adaptation in recovering and developing biospheres, it is imperative to determine the proportion of coding HGT genes in their genomes, the origin of these HGT genes, as well as which types of functional HGT genes have become incorporated into a host organism’s genome and their role in the organism’s adaptation to its living environment. This study is a first step in analyzing the genomes of these organisms to elucidate the strategies used to adapt to their living environment. By focusing on the genomes of two Antarctic Sphingomonas species, we detected HGTs and sought to unravel the process by which the HGT candidate genes were acquired. BLSOM analysis could serve as a powerful tool for facilitating the detection of HGTs and their origins in entire genomes.
Materials and Methods
Isolation and Taxonomic Analysis of Antarctic Sphingomonas Strains
We collected Antarctic Sphingomonas strains from an Antarctic moss pillar on January 19, 2000 from lake Hotoke-Ike at 69°28’ S and 39°34’ E in Skarvsnes, East Antarctica (Nakai et al., 2012). To isolate the bacteria, the frozen Antarctic moss pillar sample was slowly melted at room temperature, after which the resultant lake water was recovered, diluted aseptically with sterilized water, spread onto BG11-agar plates (Stanier et al., 1971) containing 1% glucose, and incubated at 10°C for several weeks. Single colonies growing on these plates were purified by transferring them onto new plates and incubating for several additional weeks at 10°C. The isolated strains were subsequently grown in liquid BG11 medium with 1% glucose at 10°C and stored at –80°C after the addition of glycerol to reach a final concentration of 25%.
For the 16S rRNA gene sequencing and taxonomic analysis, we performed PCR using the bacterium-specific 27F and the universal 1492R primers (DeLong, 1992) for 16S rRNA gene amplification, following the reaction conditions described by Naganuma et al. (2007). The amplified segments were cloned using a TOPO TA cloning kit (Invitrogen, Carlsbad, CA, United States), and were used to transform E. coli TOP10 (Invitrogen). The resultant 16S rRNA gene inserts were sequenced using an ABI 3100 automatic DNA-sequencer (Applied Biosystems, Foster City, CA, United States), and the obtained 16S rRNA nucleotide sequences were analyzed phylogenetically using MEGA 6.0 (Tamura et al., 2013) and the maximum-likelihood method based on the Tamura-Nei model (Tamura and Nei, 1993).
Bacterial growth at different temperatures (4, 10, 15, 20, 25, and 30°C) was investigated in liquid BG11 medium containing 1% glucose for up to 8 weeks. The optimal growth was also evaluated at 15°C in these liquid media: BG11, BG11 including 1% glucose, and R2A (Reasoner and Geldreich, 1985).
Genome Sequencing and Bioinformatic Analysis of Genomic Data
The Antarctic Sphingomonas strains were grown in 5 mL of R2A medium at 15°C with shaking (150 rpm) until the OD600 reached ∼1.0. Cells were harvested by centrifugation at 8000 × g for 5 min, and the genomic DNA was isolated from the pelleted cells by using a Qiagen Genomic-tip 100/G column (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The genome sequences of the Sphingomonas strains were determined using Illumina paired-end sequencing (Illumina Inc., San Diego, CA, United States) and single-molecule real-time DNA sequencing (Pacific Biosciences, Menlo Park, CA, United States). First, whole-genome sequencing data were generated on a PacBio RSII platform and assembled using the HGAP2 assembler (Pacific Biosciences). The assembled genome sequences were corrected using the Quiver software (Pacific Biosciences). Next, Illumina paired-end libraries were constructed and sequenced using an Illumina MiSeq, and the Illumina sequence data were mapped against the genome sequences by BWA v0.7.7 with the option “-M” (Li and Durbin, 2009) for error correction.
The refined genomic data were annotated using various software tools. Genes were identified by MiGAP (Sugawara et al., 2009) and MetaGeneAnnotator (Noguchi et al., 2008), while GLIMMER (Delcher et al., 1999) predicted the coding sequences (CDSs), after which manual curation was performed. For example, in the case of the predicted CDS, wherein the start position of the chromosomal replication initiator protein DnaA was at a “GTG” codon following an “ATG,” the CDS start position was manually curated at the “ATG.” The predicted CDSs were translated and used to search for genes in other organisms in the non-redundant databases provided by the National Center for Biotechnology Information (NCBI_nr) in Feb. 2016, UniProt release 2016_2 (The UniProt, 2018) and STRING v9.1 (Szklarczyk et al., 2019), through BLASTp (Altschul et al., 1990). Top hits with an E-value threshold of 1e-5 or lower were used to describe each of the predicted proteins encoded by the identified genes. Genes encoding tRNAs were manually annotated using the tRNADB-CE database group’s strategy (Abe et al., 2011). This tRNA prediction was performed by running three prediction programs used for tRNA gene search, namely tRNAscan-SE v2.0 (Lowe and Eddy, 1997), ARAGORN v1.2.38 (Laslett and Canback, 2004), and tRNAfinder v1.4 (Kinouchi and Kurokawa, 2006). The tRNA genes obtained were defined as reliable without further checks. Next, the residual discordant cases were checked manually based on tRNA secondary structure. Non-coding genes and miscellaneous features were predicted using RNAmmer (Lagesen et al., 2007).
To assign taxonomic information using sequence similarity searches for the strains HMP6 and HMP9, we performed BLASTp against NCBI_nr in February 2016. We examined the taxonomic information of sequences showing a best-hit with an E-value of 1e-5 or less, with a similarity to both sequence identity and coverage of 70% or more as the threshold value.
For the comparative analysis based on an orthologous gene group, orthologous genes were extracted by surveying bidirectional best-hit (BBH) relationships among Sphingomonas strains using BLASTp with an E-value threshold of 1e-5.
To ascertain the types of function acquired by HGT candidates, we compared their functional classification based on the Clusters of Orthologous Groups of Proteins (COG), a gene ortholog database provided by NCBI (Galperin et al., 2015). The functional categories in COG were determined through BLASTp analysis performed using the amino acid sequences registered in COG released in 2014. The COG functional categories of the genes were then obtained using the top-hit with an E-value of 1e-5 or less as the threshold value.
Batch-Learning Self-Organizing Map
Kohonen’s Self-Organizing Map (SOM), an unsupervised neural network algorithm, is a powerful tool for clustering and visualizing high-dimensional complex data on a two-dimensional map (Kohonen, 1990; Kohonen et al., 1996). We modified the conventional SOM for genome informatics on the basis of batch-learning to develop BLSOM, aiming to make the learning process and the resulting map independent of the order of data input (Kanaya et al., 2001; Abe et al., 2003). The newly developed SOM is suitable for high-performance parallel computing and therefore for big data analysis.
The initial weight vectors were defined using principal component analysis (PCA) based on the variance-covariance matrix, rather than by using random values, as PCA can classify gene sequences into groups of recognized biological categories. The weight vectors (wij) were arranged in a two-dimensional lattice denoted by i (=0, 1,.., I-1) and j (=0, 1,.., J-1) (where I and J represent the size of two-dimensional lattice points in first and second dimensions, respectively) and were set and updated as described previously (Kanaya et al., 2001; Abe et al., 2003). A BLSOM program suitable for PC cluster systems is available on our website1.
Detecting HGT Candidates in Microbial Genomes and Predicting Their Origin
An overview of the proposed workflow is presented in Figure 1. To create sequence datasets for BLSOM analysis, bacterial nucleotide and draft genome sequences were obtained from GenBank2 and the whole-genome shotgun division in DDBJ3, respectively, in January 2016. The registered sequence length of 5 kb or more was extracted and grouped by species. Here, allogeneic strains were grouped as the same species, and plasmid sequences were excluded. When the total number of nucleotides grouped by each species reached 10 kb or more, these sequences were aggregated per genus and concatenated by adding ten undetermined nucleotides (Ns) between the sequences, resulting in a single concatenated sequence per genus. When the number of Ns in a sequence exceeded 10% of the window size, the sequence was omitted from analysis, while when it was less than 10%, the oligonucleotide frequencies were normalized to the length without Ns and included in the analysis. The genus names and the number of 5 kb segments, which were used as sequence datasets, are shown in Supplementary Table S1.
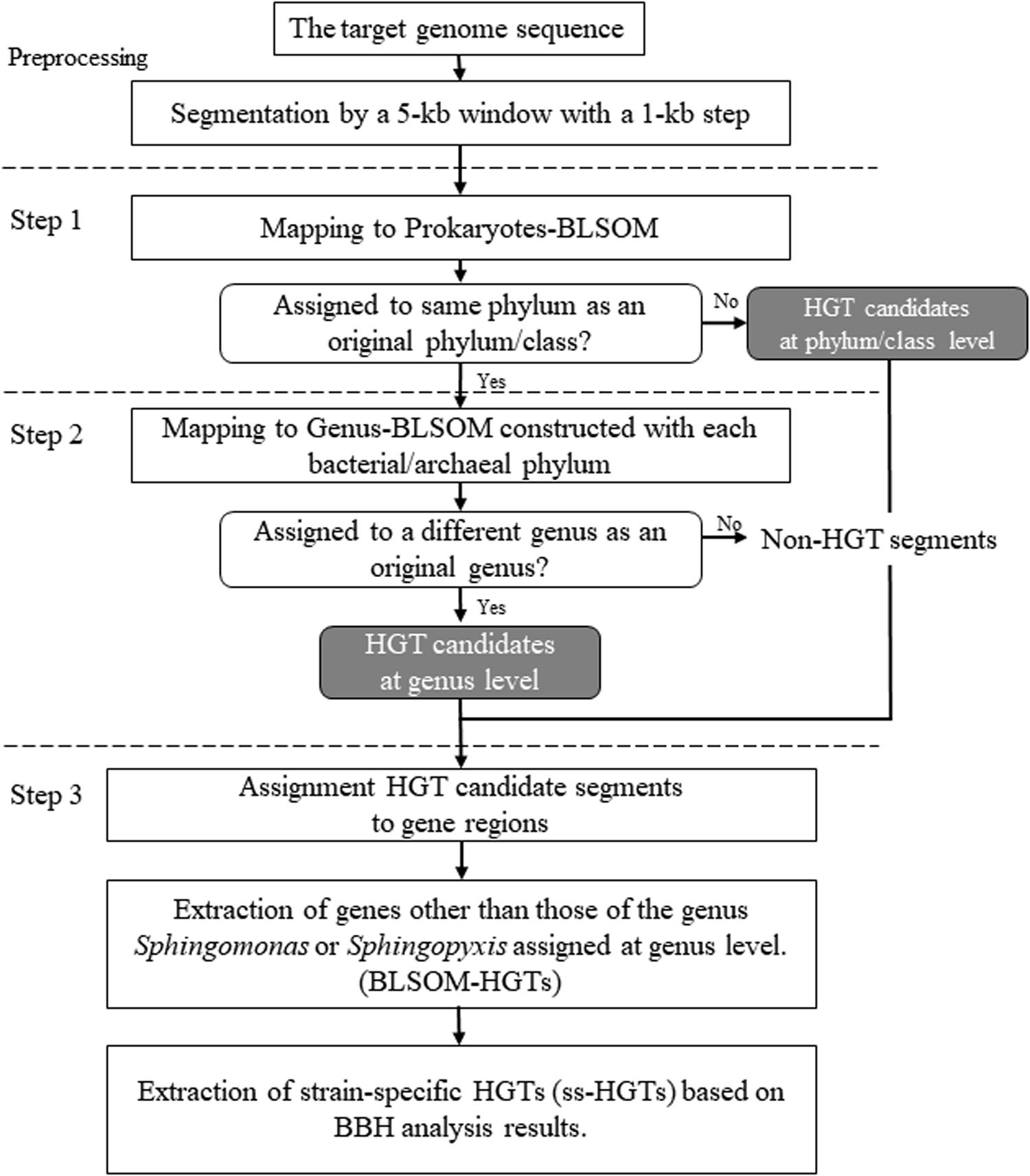
Figure 1. Workflow for detecting HGT candidates in the microbial genomes using BLSOM.
In this workflow, two types of large-scale BLSOMs, namely Prokaryotes-BLSOM and Genus-BLSOM, were constructed to identify HGT candidates in microbial genomes. These BLSOMs were constructed using a degenerate tetranucleotide composition totaling 5,200,000 5-kb sequences from 1,217 genera, of which at least 10 kb of sequence was available from DDBJ/ENA/GenBank. The degenerate tetranucleotide composition refers to the composition of degenerate sets in which a pair of complementary tetranucleotides was added (e.g., ATGC and GCAT). Hence, our previous BLSOM analysis of a wide range of species revealed that sequences from a single genome often form a vertically mirror-symmetrical cluster (Abe et al., 2003), e.g., according to the replicational direction of the genomic fragments.
During preprocessing, a 5-kb window featuring a 1-kb step was used (“Preprocessing” in Figure 1), which was mapped to Prokaryotes-BLSOM by identifying the lattice point with the minimum Euclidian distances of the degenerate tetranucleotide composition between the genomic segment and the lattice points (“Step 1” in Figure 1). For every lattice point at which microbial genomic segments were mapped into phylum/class territories, the most abundant phylum/class was identified, and the mapped microbial genomic segments were assigned to this phylum/class by BLSOM. To identify the taxonomic origin of the microbial genomic segments that were mapped to the prokaryotic territories on Prokaryotes-BLSOM, the segments were mapped on Genus-BLSOM (“Step 2” in Figure 1). The microbial genomic segments were mapped in a similar stepwise manner on Genus-BLSOMs constructed using sequences from more detailed taxonomic categories (e.g., genera).
To assign the microbial genomic segments to specific gene regions, the taxonomic category of a gene was assigned the taxonomic category of the microbial genomic fragment that covered the entire gene region and whose start position was the closest (“Step 3” in Figure 1). Next, the genes other than those of the genus Sphingomonas or Sphingopyxis assigned by Genus-BLSOM were defined as HGT candidates (abbreviated as BLSOM-HGTs). Furthermore, from these BLSOM-HGTs, by performing BBH analysis among the Sphingomonas strains, only those genes that showed similarity only to themselves or whose BBH was not established were defined as strain-specific HGT candidates (abbreviated as ss-HGTs).
The HGT candidate detection of private genomic sequence segments, such as that of new species, against Prokaryotes- and Genus-BLSOM can be performed by applying the phylogenetic estimation software for metagenomic sequences that we have previously developed. The software and BLSOM data are available on our website4.
Amino Acid Frequency Analysis for Investigating Low-Temperature Adaptation in Antarctic Sphingomonas Strains
Cold-adapted enzymes feature a low arginine content and a low Arg/(Arg + Lys) ratio, as these are well-suited for their low-temperature activity and thermal instability (Russell, 2000). We sought to further elucidate the role of the arginine content to comprehensively investigate low-temperature adaptation in the Antarctic environment by comparing the amino acid frequencies in proteins encoded by genes (4,280 and 1,712 genes from continental and Antarctic strains, respectively), for which BBH relationships were established in eight strains, including a Zymomonas strain. By following this selection strategy, a t-test was performed for each amino acid. Among the amino acids that showed significant differences, Lys, Ser, Thr, and Val were selected as the amino acids with frequencies higher in Antarctic strains than in continental strains, whereas Ala, Arg, Glu, and Leu were selected as the amino acids with frequencies lower in Antarctic strains than in continental strains. The low-temperature relation ratio for Antarctic Sphingomonas strains based on the eight selected amino acids is as follows:
This ratio is based on the ratio of amino acids that are decreased in the Antarctic environment, and thus the value is low in proteins encoded by the genes of Antarctic strains.
Statistical Analysis
Considering that the statistical analyzes used differed depending on the normality and equal variance of the two samples compared, Kolmogorov–Smirnov test was first performed to test normality. If the two compared samples were normally distributed, F-test for equality of variance was performed. We also performed the two-sample test for assessing differences between two population means to compare amino acid frequencies. Student’s t-test was used when normal distribution and equality of variance were observed, Welch’s t-test was applied when normal distribution was observed without equality of variance, and Wilcoxon rank-sum test was used if normality was not observed. Finally, the p-value obtained was adjusted by Benjamini-Hochberg procedure. All statistical tests were performed in R package (R Core Team, 2019).
Results and Discussion
Sphingomonas Strains HMP6 and HMP9 Are Closely Related to Psychrotolerant Bacteria
Comparative 16S rRNA sequence analysis of the two Sphingomonas strains HMP6 and HMP9 revealed a 95.5% similarity. Results from our previous study analyzing the microflora of the Antarctic moss pillar, using 16S rRNA clone libraries, suggested the existence of Sphingomonas species as one 16S rRNA clone, “MPB2-63” (Nakai et al., 2012). MPB2-63 showed 94.4% sequence similarity to both HMP6 and HMP9, and the highest similarity to MPB2-63 based on the current 16S rRNA database (accessed on Jan. 2020; O’Leary et al., 2016)5 was found in the case of Sphingomonas antarctica 200 (Huang et al., 2017). A phylogenetic tree obtained using the maximum-likelihood method in MEGA 6.0 (Tamura et al., 2013) software is presented in Figure 2, which shows the phylogenetic relationships based on the 16S rRNA sequence data of 12 Sphingomonas strains and the clone MPB2-63 (Nakai et al., 2012). Zymomonas mobilis ATCC 10988 (Skotnicki et al., 1981), a genome-sequenced type strain (Pappas et al., 2011), was used as the tree outgroup. The multiple sequence alignment in the FASTA format and the phylogenetic tree in the Newick format in Figure 2 are presented as Supplementary Materials S1, S2, respectively. The publicly available isolated strains that represent the closest relatives to the Antarctic Sphingomonas strains HMP6 and HMP9 and the clone MPB2-63, are Sphingomonas psychrolutea MDB1-A (Liu et al., 2015), Sphingomonas aerolata NW12 (Busse et al., 2003), and S. antarctica 200 (Huang et al., 2017), respectively. These Sphingomonas strains are reported as psychrotolerant bacteria isolated from low-temperature environments, such as glacial ice in Tibet (Liu et al., 2015), airborne in United Kingdom (Busse et al., 2003), and tundra soil in Antarctica (Huang et al., 2017). The Sphingomonas strains HMP6 and HMP9 were found to grow at 4–25°C, with an optimal growth at 15°C, suggesting that they exhibit the growth feature of psychrotolerant bacteria. We also analyzed the phylogenetic relationships based on the 16S rRNA sequence data with an additional 24 completely sequenced Sphingomonas strains (Supplementary Figure S1). The multiple sequence alignment in the FASTA format and the phylogenetic tree in the Newick format in this figure are presented as Supplementary Materials S3, S4, respectively. The Antarctic Sphingomonas strains, namely HMP6, HMP9, and S. antarctica 200 (Huang et al., 2017) and the clone MPB2-63 belong to three independent groups.
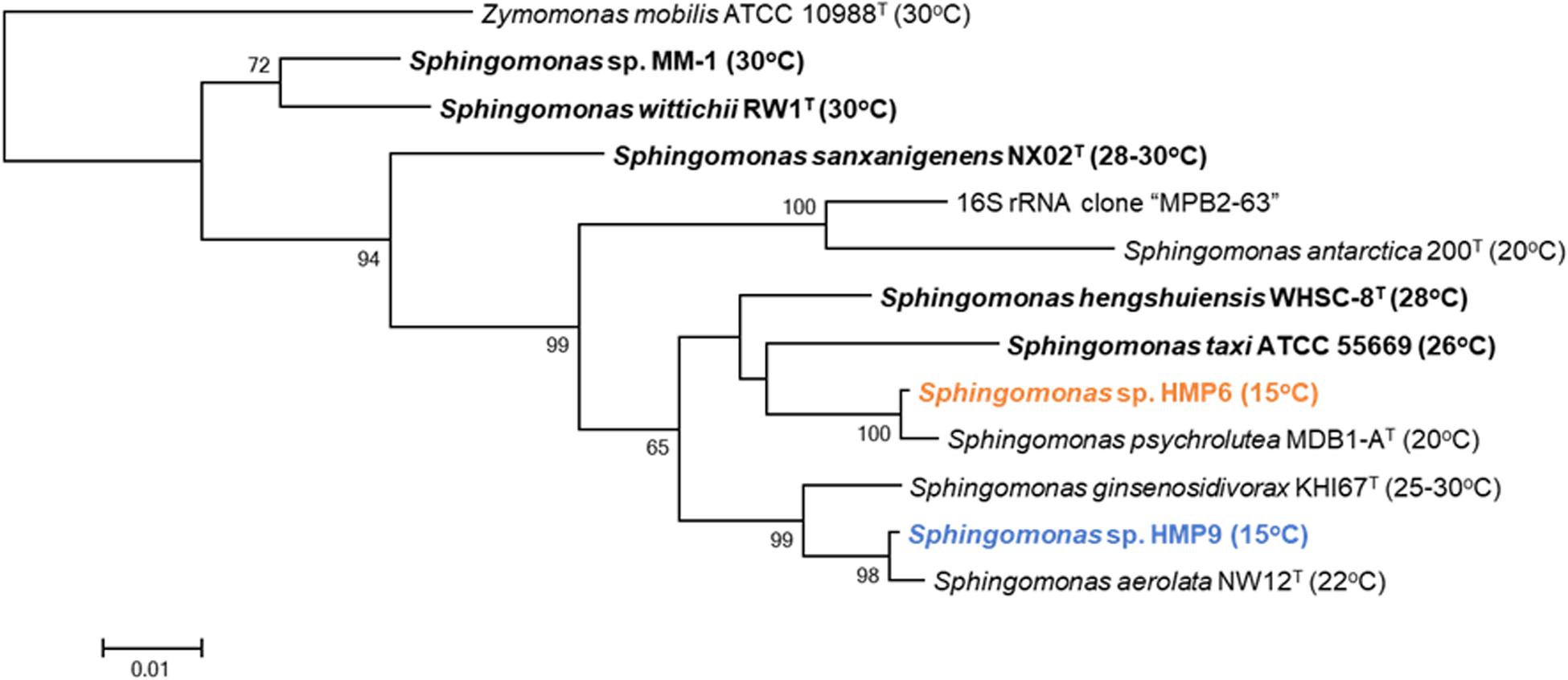
Figure 2. Phylogenetic tree of the 12 Sphingomonas strains based on 16S rRNA sequence data constructed using the maximum-likelihood method under the Tamura-Nei model. Species names in bold are genome-sequenced strains. The numbers before the branching points correspond to bootstrap percentage values for 1000 replicates, whereas the superscript “T” indicates type strains. Values inside parentheses are the temperatures for optimum growth. Orange and blue text represents the Antarctic HMP6 and HMP9 strains, respectively.
The optimal growth temperature of each Sphingomonas species has not yet been comprehensively reported, and limited information is presented in Supplementary Tables S2, S3. Depending on the optimal growth temperature, different culture media are used (Supplementary Table S2). For example, Sphingomonas sp. MM-1, Sphingomonas wittichii RW1, Sphingomonas sanxanigenens NX02, and Sphingomonas hengshuiensis WHSC-8, which showed growth at ∼30°C, used relatively nutrient rich media, such as 1/3 LB medium, Difco nutrient broth, NK medium, and NB medium; meanwhile, psychrotolerant S. aerolata NW12, S. antarctica 200, Sphingomonas sp. HMP6, and Sphingomonas sp. HMP9 used the nutrient poor R2A medium. Moreover, Sphingomonas ginsenosidivorax KHI67, which showed intermediate growth at temperatures between 25 and 30°C, also used the R2A medium. Although comparison of the optimal growth temperatures for all Sphingomonas species using the same culture medium is necessary, here we only evaluated them roughly, while focusing on the Sphingomonas strains with well-reported genomic features and their growing temperatures, as well as those of the newly sequenced strains as listed in Figure 2.
Genome Structures of the Antarctic Strains Are Not Conserved in Continental Strains
The summary genome information of the Antarctic Sphingomonas strains HMP6 and HMP9, as well as the other genome-sequenced Sphingomonas strains, are presented in Table 1. The detailed gene annotation data for these genomes are shown in Supplementary Tables S4–S6. In addition to a circular chromosome, HMP9 carries a plasmid (designated as pHMP9) ∼43 kb in size, whereas HMP6 does not carry a plasmid. The G + C percentage of pHMP9 is 63%, which is lower than that of the HMP9 genome (66%). The plasmid contains 46 predicted genes, one of which, pHMP9_0003, likely encodes the replication protein RepB (RefSeq: WP_010165325), which shows a high level of conservation with MC45_RS18375 on Sphingomonas taxi ATCC 55669 plasmid STP2 (Eevers et al., 2015) and with G432_21330 on Sphingomonas sp. MM-1 plasmid pISP1 and G432_RS21815 on plasmid pISP4 (Wu et al., 2017). A phylogenetic relationship for the RepB genes among these plasmids is shown in Supplementary Figure S2, wherein multiple sequence alignment in the FASTA format and the phylogenetic tree in the Newick format are shown in Supplementary Material S5, S6, respectively. Besides pHMP9_0003, certain other genes encoded on pHMP9 are conserved in S. taxi ATCC 55669 plasmid STP2 (Eevers et al., 2015) and in Sphingomonas sp. MM-1 plasmids pISP0, pISP1, pISP2, pISP3, and pISP4 (Wu et al., 2017), suggesting that pHMP9 represents one of the typical plasmid structures harbored in the genus Sphingomonas.
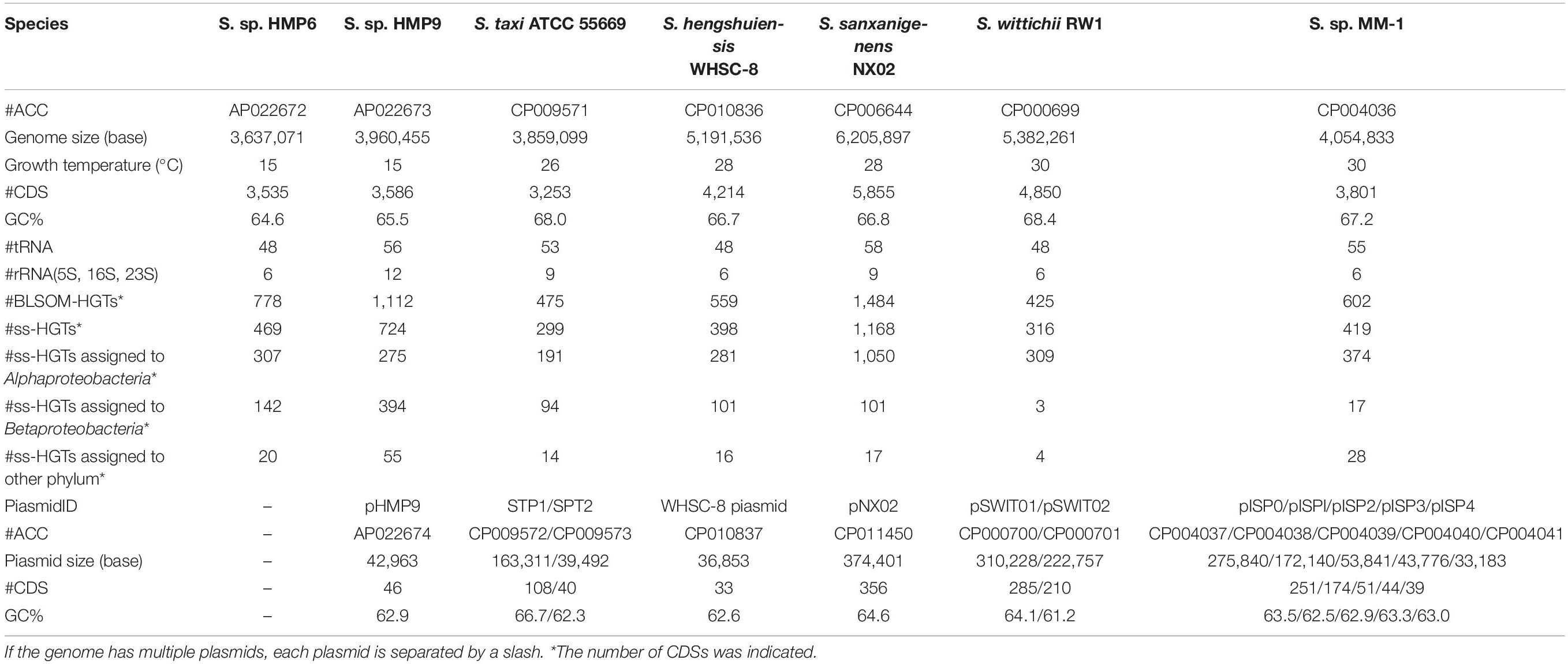
Table 1. Genome information and number of HGT candidate genes.
The Antarctic Sphingomonas strains HMP6 and HMP9 feature small genome sizes (<4 Mb) and lower G + C percentages (<66%) than those of the other Sphingomonas strains and harbor a similar number of protein-coding genes. However, a twofold difference exists in the number of their rRNA genes (Table 1): HMP6 carries two sets of rRNA operons, whereas HMP9 carries four. Previous genomic studies reported that Sphingomonas strains in this genus harbor two or three sets of rRNA operons in their genomes (Miller et al., 2010; Tabata et al., 2013; Eevers et al., 2015; Wu et al., 2017). However, from our complete genome sequence data, the Sphingomonas strains appear to harbor one to five sets of rRNA operons in their genomes, the minimum of which are found in Sphingomonas indica Dd16, Sphingomonas ginsengisoli KACC 16858, Sphingomonas sp. AE3, Sphingomonas sp. YZ-8, Sphingomonas sp. XS-10, and Sphingomonas sp. HKS19, while the maximum is in Sphingomonas sp. IC081 (Supplementary Figure S1 and Supplementary Table S7). The number of rRNA operons in bacterial genomes was suggested to predict the growth rate and growth efficiency in response to resource availability (Roller et al., 2016). Although we did not observe a markedly enhanced growth rate in HMP9 relative to that of HMP6, the growth conditions we examined were only roughly detected and were limited to the liquid media of BG11, BG11 including 1% glucose, and R2A. To better evaluate the relationship between growth and the number of rRNA operons in HMP6 or HMP9, it will be necessary to continuously measure the growth rate and growth efficiency under experimentally controlled conditions in which several nutrients or temperatures are tested. In terms of the numbers of tRNA genes in Sphingomonas genomes, HMP6 harbors 48 genes, equal to that in Sphingomonas hengshuiensis WHSC-8 and S. wittichii RW1, which both carry two sets of rRNA operons. In contrast, HMP9 harbors 56 tRNA genes, similar to S. taxi ATCC 55669 and S. sanxanigenens NX02, which carry three sets of rRNA operons. In the same survey of complete genome sequence data, Sphingomonas strains were found to contain 44—64 tRNA genes in their genomes, of which the minimum 44 is in Sphingomonas sp. YZ-8, whereas the maximum 64 are in Sphingomonas sp. IC081 (Supplementary Figure S1). These Sphingomonas strains present similar “linked rRNA operon structures” (Brewer et al., 2020), consisting of 16S rRNA, Ile-tRNA, Ala-tRNA, 23S rRNA, 5S rRNA, and Met-tRNA. HMP9 includes two additional rRNA operon structures as compared with HMP6, and thus six additional tRNA genes are present in the HMP9 genome.
Although the Sphingomonas strains HMP6 and HMP9 did not form a clade (Figure 2 and Supplementary Figure S1), they did exhibits features of psychrotolerant bacteria. This suggests that the adaptation to Antarctic’s low-temperature environment may be achieved via convergent adaptation (independent evolution of similar traits). Furthermore, the genomes of the strains HMP6 and HMP9 were smaller and had lower GC content than those of the other Sphingomonas strains (Table 1). According to gene annotation for predicting CDSs by using BLASTp against the NCBI_nr database, only ∼65% of the CDSs of both HMP6 and HMP9 strains were found to be similar to the genes in the genus Sphingomonas; ∼10% of the CDSs were similar to the genes in other genera and >25% remained as “no-hits.” These results suggest that the genome structures of HMP6 and HMP9 consist of a large number of genes that do not show conservation with genes in the genus Sphingomonas, thereby implying that these strains carry horizontally transferred genes. To test this hypothesis, we used an alignment-free HGT-detection method.
HGTs of HMP6 and HMP9 Have Different Origins
We used BLSOM to detect HGT candidates and their origins in two Antarctic Sphingomonas strains, HMP6 and HMP9 (hereafter, Antarctic strains), and in five strains isolated from continents other than Antarctica with their entire genomes previously decoded (hereafter, continental strains). Figure 3 shows the proportions of the genomic sequence segments of each strain assigned to each taxonomic rank by BLSOM. Firstly, among the continental strains in the genus Sphingomonas, S. wittichii featured the highest proportion (90.8%), whereas S. sanxanigenens had the lowest (75.5%), with the average proportion being 82.8%. Conversely, between the Antarctic strains, the assigned proportions were 67.8% for HMP6 and 67.2% for HMP9. Thus, the proportions assigned to the genus Sphingomonas tended to be lower in the Antarctic than in the continental strains. Next, at the Sphingomonadaceae family level, the highest proportion was 90.9% for S. wittichii, and the lowest was 76.8% for S. sanxanigenens, with the average at 84.6%, whereas the proportions were 78.5 and 68.2% for HMP6 and HMP9, respectively. Lastly, for the proportion of genomic sequence segments assigned to the class Alphaproteobacteria of the same phylum/class, the highest was 99.8% in S. wittichii and the lowest was 93.5% in S. taxi, with an average of 96.7% among the continental strains; by comparison, HMP6 featured a high proportion (92.2%) similar to the continental strains, whereas HMP9 was 77.2%, which was lower than those for HMP6 and the continental strains. The remaining 22.8% was assigned to other phylum/classes. This suggests that the Antarctic strains have acquired a higher proportion of HGT candidates than the continental strains, which further supports that the Antarctic strain HMP6 has acquired most of the HGTs from the same family or phylum/class. In contrast, HMP9 contains a higher proportion of HGT candidates acquired from another phylum/class than from either S. taxi, a closely related species in the phylogenetic tree, or HMP6, which was isolated from the same Antarctic environment. Thus, HGTs of HMP9 have origins that differ from those of HMP6 and the continental strains.
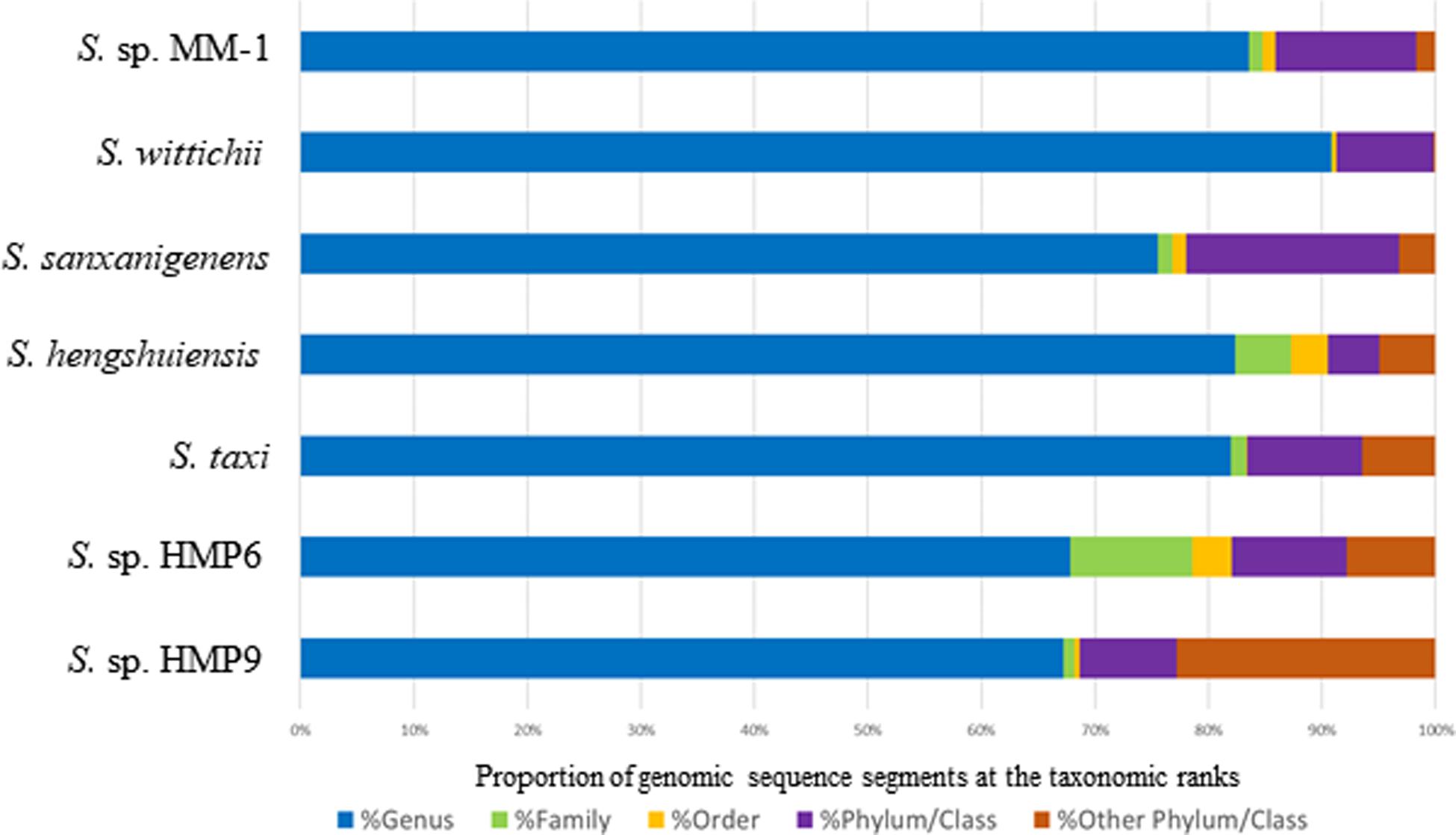
Figure 3. Proportion of genomic sequence segments in the Sphingomonas genome-sequenced strains assigned according to taxonomic rank. The horizontal bars represent proportion of genomic segments assigned to each taxonomic rank by BLSOM with taxonomic ranks color-coded: genus ( ), family (
), family ( ), order (
), order ( ), phylum/class (
), phylum/class ( ), and other phylum/class (
), and other phylum/class ( ).
).
To determine whether our proposed BLSOM analysis is applicable to draft genomes, we applied BLSOM to detect HGT candidates for the genome sequence segments of S. ginsenosidivorax KHI67 and S. aerolata NW12, whose draft genomes were decoded in type strains phylogenetically closely related to the Antarctic strain HMP9 (Supplementary Figure S3). The Antarctic strain HMP9 and its phylogenetically closely related strains may have a higher proportion derived from a phylum that differs from that of the other Sphingomonas strains, suggesting a tendency to acquire genes from another phylum (Figure 3 and Supplementary Figure S3). In addition, taxonomic assignment was performed on the Sphingomonas plasmid genomes (Supplementary Figure S4). pHMP9, as well as the plasmids of other Sphingomonas strains, was predicted to be derived from the class Alphaproteobacteria, suggesting that it is the common ancestral plasmid of other Sphingomonas strains, which is similar to the phylogenetic tree for plasmid RepB gene (Supplementary Figure S2).
Next, we assigned the origin of each gene based on the taxonomic prediction results for genomic sequence segments obtained using BLSOM (Figure 4A). Here, we defined genes other than those assigned to Sphingomonas or Sphingopyxis by BLSOM as HGT candidates (BLSOM-HGTs). Table 1 shows the number of BLSOM-HGTs. Among the continental strains, S. sanxanigenens featured the highest proportion of BLSOM-HGTs (25.4%), whereas S. wittichii was the lowest (8.8%), with the average at 15.6%. In comparison, the proportions for HMP6 and HMP9 were 22.0 and 31.0%, respectively. Similar to the prediction results for genomic sequence segments, the prevalence of HGT candidates was higher in the Antarctic than in the continental strains.
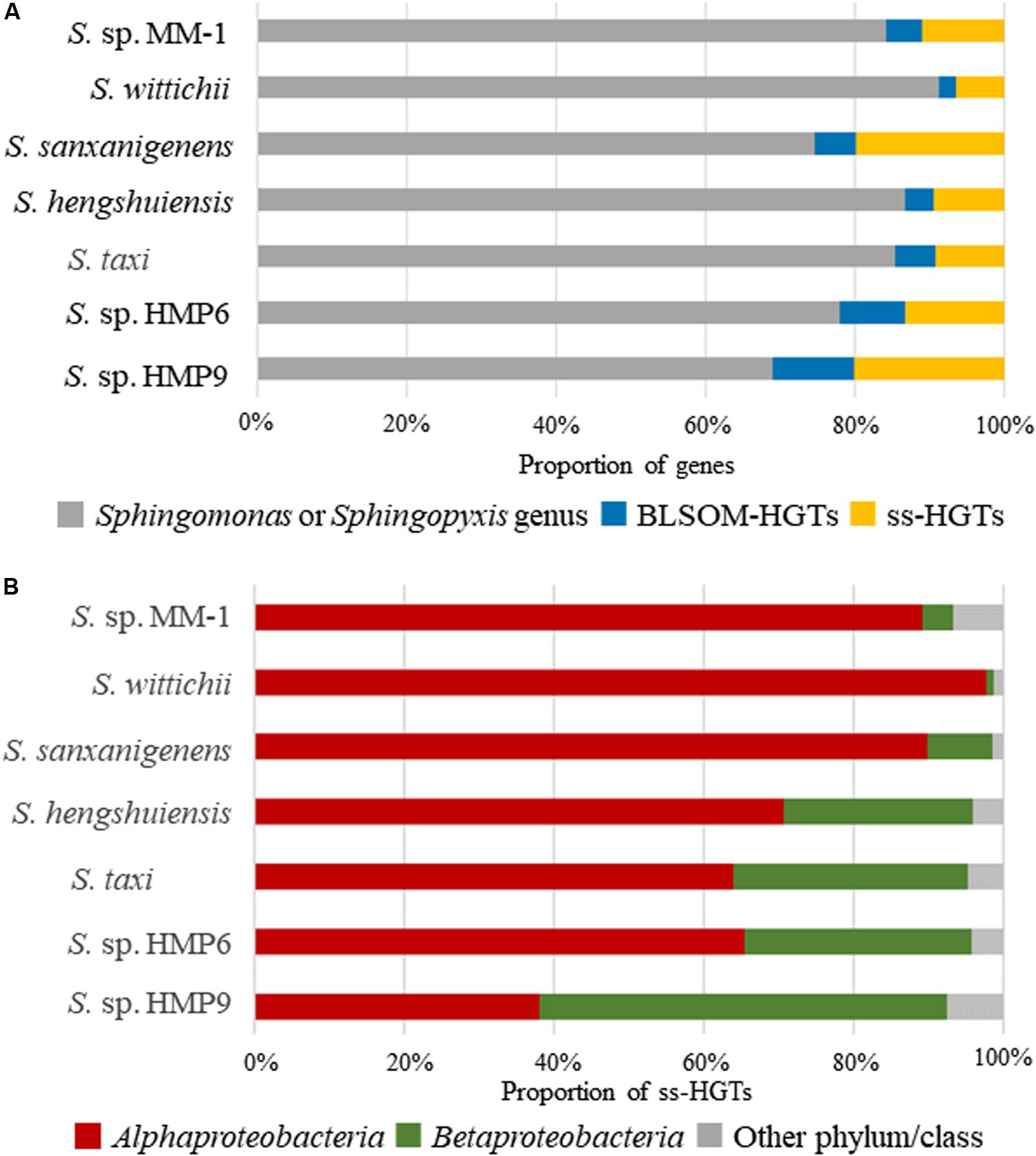
Figure 4. Proportion of HGT candidate genes in the Sphingomonas strains assigned by BLSOM and BBH. (A) Proportion of HGT candidate genes. Gray ( ): proportion of genes assigned to Sphingomonas or Sphingopyxis genus; blue (): BLSOM-HGTs; orange (): strain-specific HGT candidate genes (ss-HGTs). (B) Proportion of ss-HGTs at phylum/class level through BBH analysis from BLSOM-HGTs. Brown (
): proportion of genes assigned to Sphingomonas or Sphingopyxis genus; blue (): BLSOM-HGTs; orange (): strain-specific HGT candidate genes (ss-HGTs). (B) Proportion of ss-HGTs at phylum/class level through BBH analysis from BLSOM-HGTs. Brown ( ): Alphaproteobacteria; dark green (
): Alphaproteobacteria; dark green ( ): Betaproteobacteria; gray (): other phylum/class.
): Betaproteobacteria; gray (): other phylum/class.
To clarify the role of HGT candidates in Antarctic adaptation, we extracted ss-HGT candidates from the BLSOM-HGTs. Here, by using the results of BBH analysis of the Antarctic and continental strains, we defined “ss-HGTs” as genes forming a BBH group only with themselves, or as genes not forming a BBH group. In the Antarctic strains, the proportion of ss-HGTs to BLSOM-HGTs was as high as ∼63%. The ss-HGTs were then comprehensively analyzed as HGT candidates that are highly likely to be involved in Antarctic environmental adaptation. The origin of the ss-HGTs was examined at the phylum/class level as shown in Figure 4B. The continental strains and HMP6 were most frequently assigned to the class Alphaproteobacteria, in which the genus Sphingomonas belongs, and their next most frequently assigned origin was from the class Betaproteobacteria. Conversely, in HMP9, the proportion of ss-HGTs derived from the class Betaproteobacteria was the highest, accounting for more than half of the total ss-HGTs. This suggests that HMP9 obtained more HGT candidates from more distantly related bacteria than S. taxi and HMP6, which are phylogenetically related. We further examined the origin of the ss-HGTs derived from the class Alphaproteobacteria at the family level (Supplementary Figure S5A) and found that the proportion derived from the family Bradyrhizobiaceae differed among the strains, whereas that from the family Erythrobacteraceae was higher in HMP6 (24.1%) than in HMP9 (4.7%). Moreover, in the Antarctic strains, the family Burkholderiaceae, containing Burkholderia spp., accounted for >80% of the ss-HGTs from the class Betaproteobacteria, which suggests similar origins of the acquired ss-HGTs within the class Betaproteobacteria (Supplementary Figure S5B). Many of these families assigned to the class Alphaproteobacteria or Betaproteobacteria have also been reported by previous meta-16S rRNA gene analyses for moss pillars (Nakai et al., 2012), suggesting that they are likely candidate donor organisms.
Overall, HMP9 contained a high proportion of ss-HGTs obtained from more distantly related bacteria, and the two Antarctic strains have different ss-HGT origins.
It is considered that only recently acquired HGT events could be identified effectively using composition-based methods as ancient HGT genes have likely eventually evolved or ameliorated to become similar to the rest of the genome signatures (Douglas and Langille, 2019). However, we are especially interested in recently acquired HGT events in recovering Antarctic terrestrial biospheres since these biospheres in Antarctica, for example in Antarctic lake environments, had been destroyed during the glacial period and were recovered in the subsequent interglacial period (Chown et al., 2015). We would like to evaluate the recently acquired HGT events by recovering and developing the biosphere in an Antarctic lake environment after the last glacial maximum approximately 10,000 years ago (Chown et al., 2015).
BLSOM Has Higher Detection and Classification Capabilities Than BLASTp Searches
To validate the effectiveness of the taxonomic prediction by BLSOM in terms of HGT candidate detection, we compared the taxonomic information obtained by BLSOM with that of amino acid sequence searches from BLASTp, a sequence similarity search program. Since it was difficult to define the complete positive or negative HGT gene sets in the genus Sphingomonas, we compared our BLSOM results with the taxonomic information obtained by BLASTp to validate our proposed method’s taxonomic prediction capacity. Figure 5 shows the genes of the Antarctic strains classified according to the taxonomic rank and obtained using these methods. The detection capability of BLSOM is higher than that of BLASTp searches with all genomic sequence segments assigned to recognized microbial origins when checking for new microbes. In contrast, ∼30% of the genes were not classified by BLASTp searches. Moreover, BLSOM can assign taxonomic information comprehensively even for microbial genomes that exhibit high novelty, which cannot be detected using sequence similarity searches.
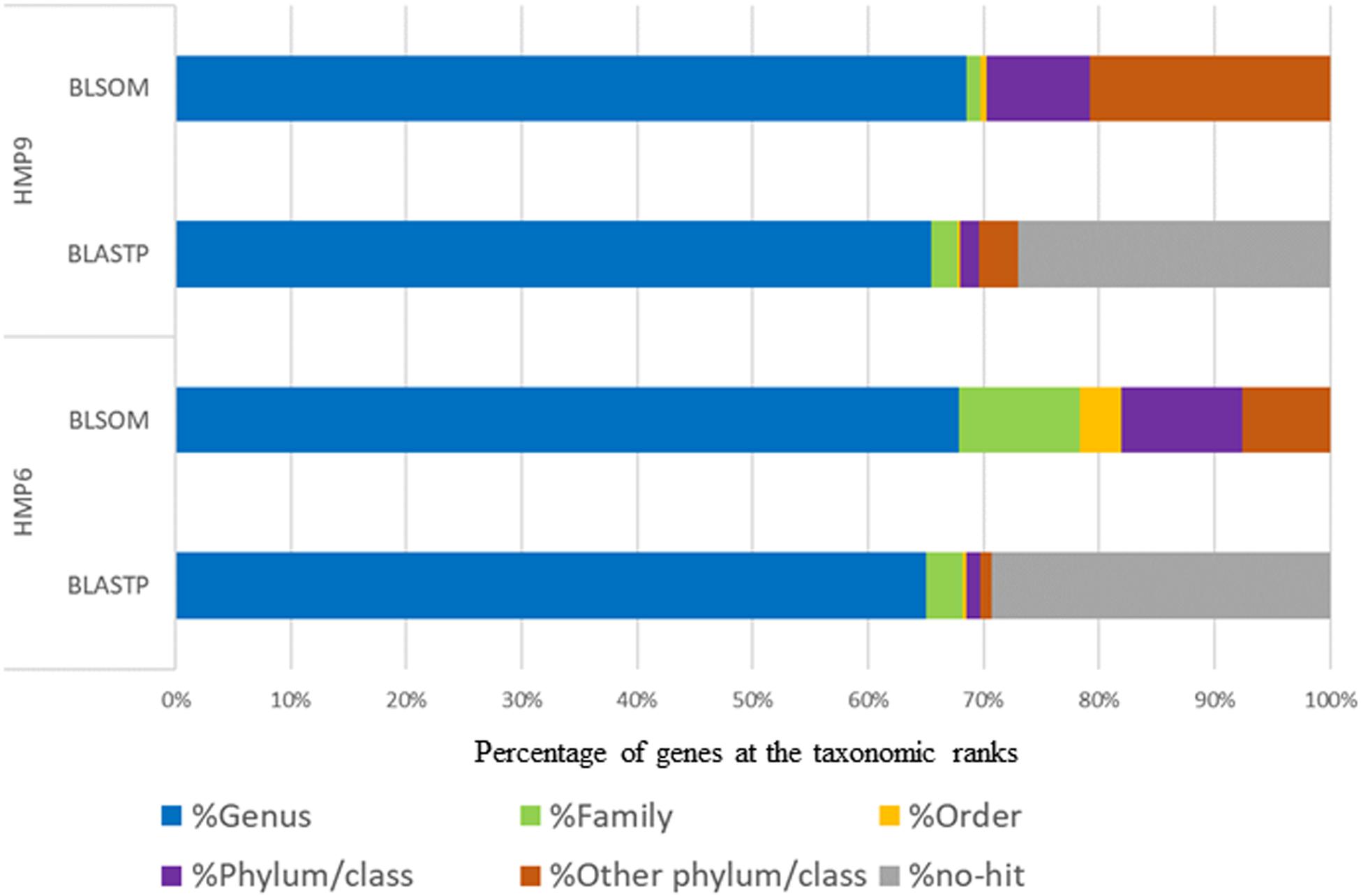
Figure 5. Distribution of the detected genes in Antarctic HMP6 and HMP9 strains by BLSOM and BLASTp assigned according to taxonomic rank. The horizontal bars represent proportions of the detected genes assigned to each taxonomic rank by BLSOM and BLASTp with taxonomic ranks color-coded: genus (), family (), order (), phylum/class (), other phylum/class (), and no-hit ().
To confirm assignment capacity, we examined the percentage of matching in comparison with taxonomic information obtained by BLSOM and BLASTp at each taxonomic rank and found that, at the genus level, the percentage of matching between BLSOM and BLASTp was 66.2% for HMP6 and 70.0% for HMP9. This low percentage of matching obtained using BLASTp was likely due to the associated difficulty in distinguishing Sphingomonas and Sphingopyxis in Genus-BLSOM, as 88.3% of Sphingopyxis genomic segments was mixed with Sphingomonas genomic segments. Therefore, the percentage of matching obtained including the two genera, Sphingomonas and Sphingopyxis, was 77.9% for HMP6 and 71.0% for HMP9. Conversely, at the family level, the percentage of matching was 81.3% for HMP6 and 72.9% for HMP9, and at the phylum/class level, the percentage of matching was 93.4% for HMP6 and 81.1% for HMP9. The percentage of matching for HMP9 was lower than HMP6 since the genes derived from the class Betaproteobacteria were assigned by BLSOM, and these could not be readily detected by BLASTp.
Antarctic Strains Acquired Horizontally Transferred Genes to Adapt to the Antarctic Environment
To ascertain the types of functions acquired by the ss-HGTs of HMP6 and HMP9, we compared functional classification based on COG. The COG functional categories of the genes are shown in Supplementary Tables S4–S6.
Figure 6 shows the total genes and the ss-HGTs in HMP6 and HMP9 according to COG functional categories, shown on the right. Focusing on the ss-HGTs whose functions could be identified, the functional category in which the most ss-HGTs were detected was “Mobilome: prophages, transposons (X),” followed by “Cell wall/membrane/envelope biogenesis (M).” The ss-HGTs in 44 genes for HMP6 and 75 genes for HMP9 acquired through horizontal transfer belonged to “X,” which indicates that the ss-HGTs detected using BLSOM are highly reliable candidates from the viewpoint of gene functions. The functional category could not be established for 700 genes in HMP6 and 579 genes in HMP9; meanwhile, ss-HGTs was observed for 164 and 170 genes, respectively (“No COG” in Figure 6).
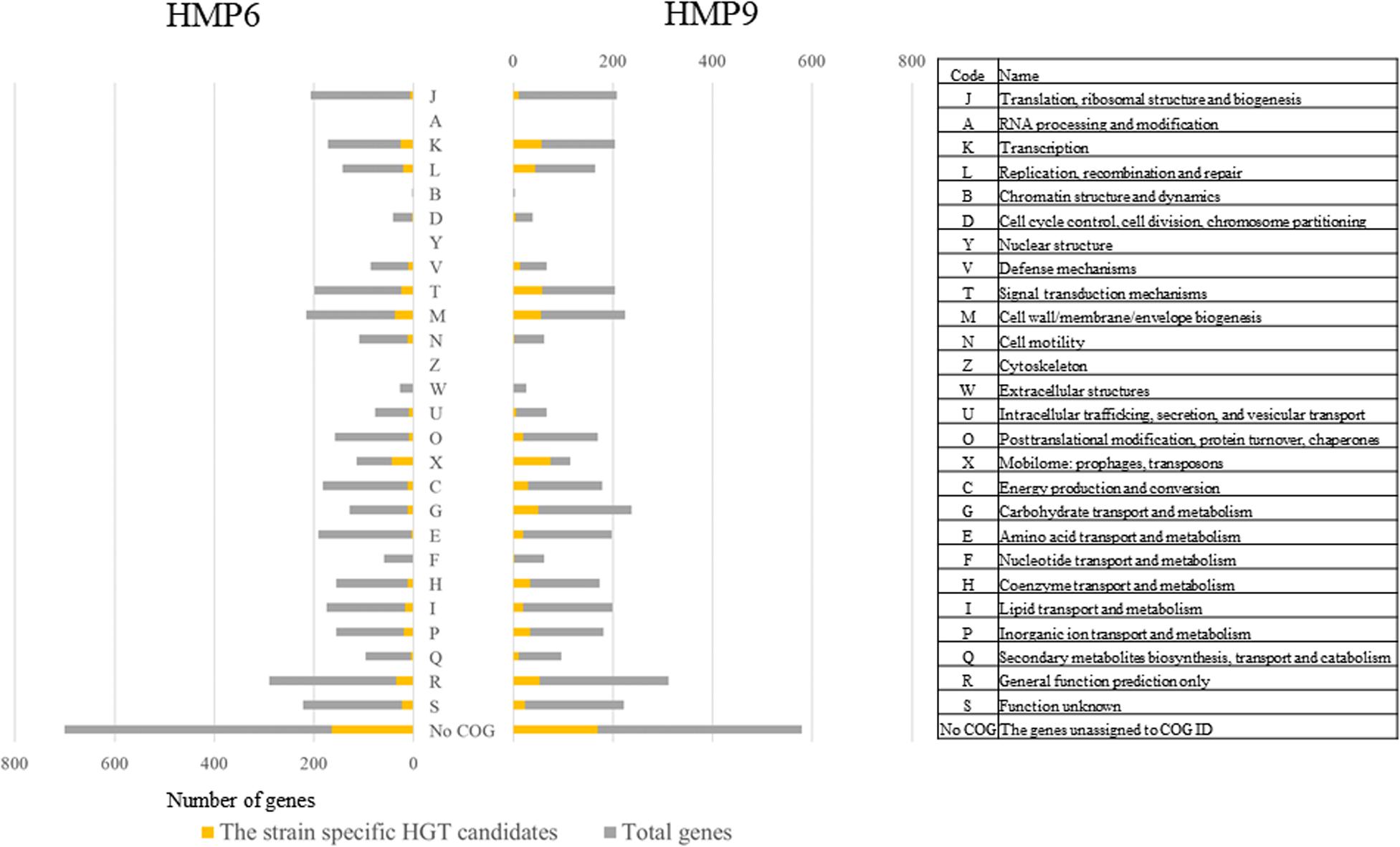
Figure 6. Distribution of the detected genes in Antarctic HMP6 and HMP9 strains grouped according to their COG functional categories. The COG functional codes and detailed description of COG functional categories are presented in the right-side list. Gray (): total number of total genes; orange (): number of strain-specific HGT candidates.
To analyze this comprehensively, we compared the percentages of the ss-HGTs among the genes identified in each functional category in the continental strains and in HMP6 and HMP9 (Figure 7). The proportion of ss-HGTs was significantly higher in M for HMP6 and in ten functional categories for HMP9 (K, L, T, M, O, X, C, G, E, H) compared with the continental strains. According to a gene-function analysis of interdomain HGTs from bacteria to archaea in 134 sequenced archaeal genomes, the function was unknown for 37% of the HGTs; however, the predicted gene-function categories in descending order of HGTs were E, C, G, P, T, H, M, and K (Nelson-Sathi et al., 2015). Here, the pink functional categories indicate those also detected for HMP9. Similarly, in a study conducted using a metagenomic approach on the gene functions of interdomain HGTs from bacteria to archaea, the function was unknown for 44% of the HGTs, and the predictable functions in descending order of HGTs were found to be in C, E, H, J, G, I, L, and M (Brochier-Armanet et al., 2011). Here, the pink functional categories indicate those detected for HMP9. Although the HGT predictions in these studies were based on phylogenic analyses using BLASTp, their results showed similar functional categories predicted using our BLSOM analysis.
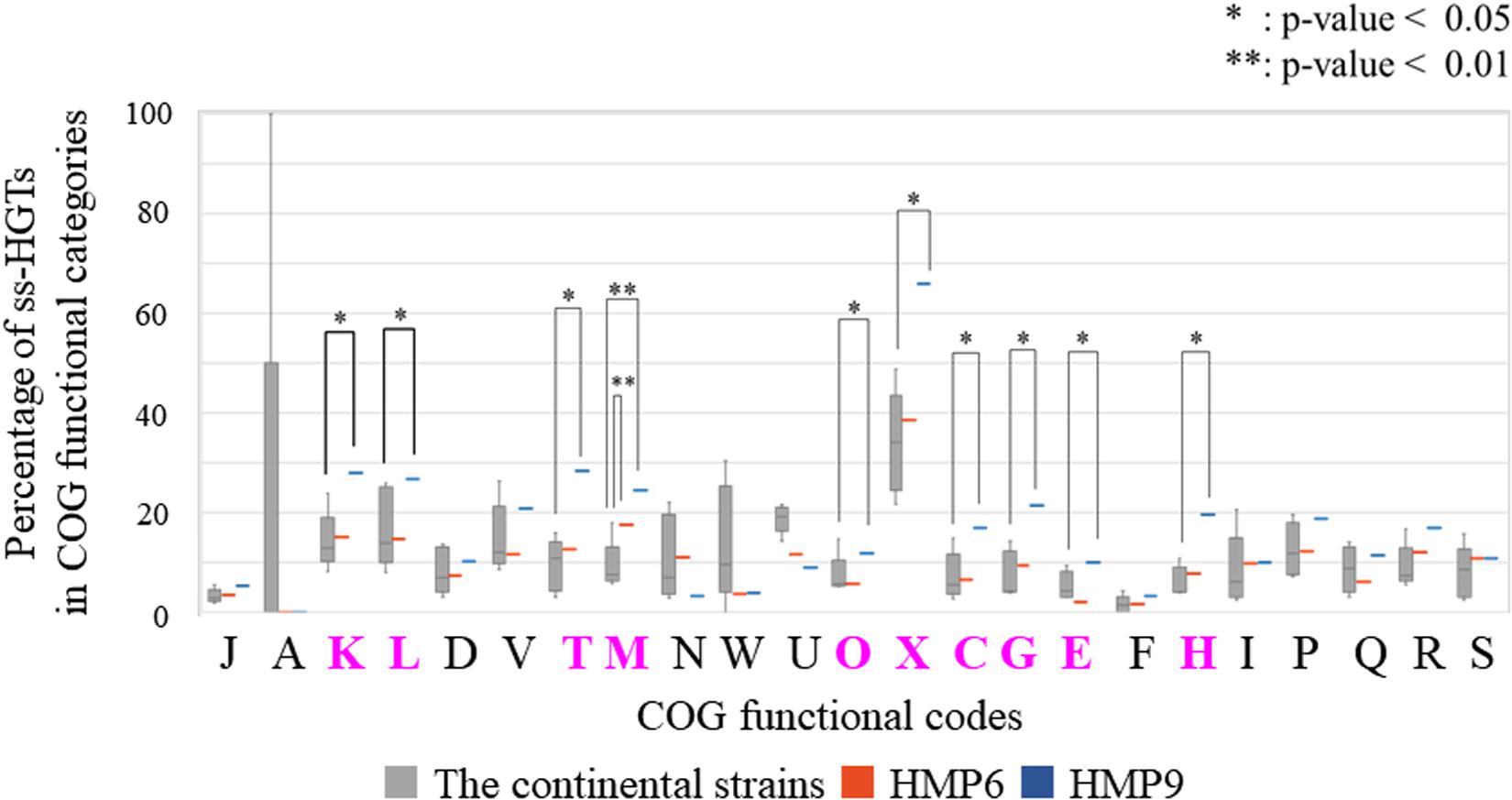
Figure 7. Percentage of the strain-specific HGT candidates (ss-HGTs) per COG functional category in the continental and HMP6 and HMP9 strains. Gray (): continental strains; orange bar (): HMP6 strain; blue bar (): HMP9 strain. Asterisks: significant difference between continental and Antarctic strains. Pink functional categories represent those with significant differences between the continental and Antarctic HMP6 or HMP9 strains.
To investigate the functional differences between the ss-HGTs of the Antarctic strains that were assigned to the classes Alphaproteobacteria and Betaproteobacteria, we compared the number of ss-HGTs between the continental and the Antarctic strains according to each origin among the ten functional categories in which the proportion of the ss-HGTs was predominant in the Antarctic strains (Figure 8). In the ss-HGTs derived from the class Alphaproteobacteria, no significant differences were found between the continental and Antarctic strains in any functional category. Meanwhile, in the ss-HGTs derived from the class Betaproteobacteria, a majority of the ten functional categories contained more Antarctic ss-HGTs than continental-strain ss-HGTs. Further, the number of ss-HGTs in the continental strains differed significantly in “M” for HMP6 and in ten functional categories for HMP9. Specifically, in terms of the number of ss-HGTs in HMP9, the ss-HGTs detected frequently were those belonging to “Carbohydrate transport and metabolism (G)” and “M,” which was also detected for HMP6.
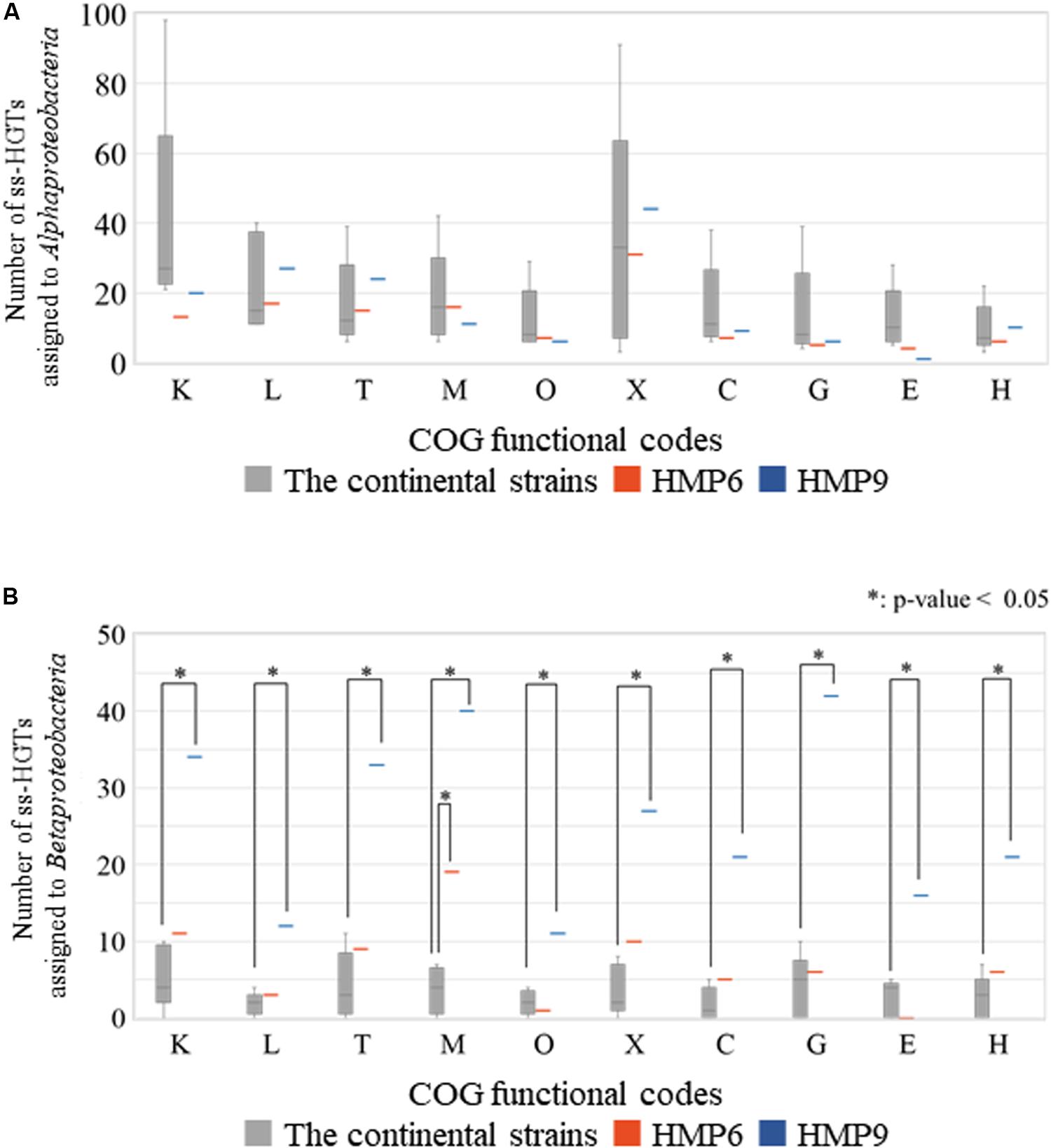
Figure 8. Number of strain-specific HGT candidates (ss-HGTs) assigned to (A) Alphaproteobacteria and (B) Betaproteobacteria according to their COG functional categories showing significant differences between the continental and the Antarctic HMP6 and HMP9 strains. Gray (): continental strains; orange bar (): HMP6 strain; blue bar (): HMP9 strain. Asterisks: significant difference between continental and Antarctic strains.
Furthermore, we also constructed a 2 × 2 contingency table, calculated the odds ratio, and performed Fisher’s exact test (Supplementary Table S8). These additional tests yielded similar results as those described above.
The adaptation strategies used by psychrophiles to survive in low-temperature environments are generally suggested to include the enhancement of membrane fluidity and the biosynthesis or accumulation of compatible solutes, mainly carbohydrates related to the control of cytoplasmic osmotic pressure (De Maayer et al., 2014). For instance, HGTs in the functional categories “M” and “G” were reported in genome analysis studies of psychrophiles in the genus Alteromonas as part of marine bacteria of the class Gammaproteobacteria (Math et al., 2012), as well as of bacteria in the genus Psychroflexus (phylum Bacteroidetes) isolated from sea-ice (Feng et al., 2014). These strategies for adaptation to low-temperature environments are related to the various environmental factors such as desiccation, low nutrient availability, high or low osmotic pressure, ultraviolet radiation, and combined stresses. For example, freezing causes desiccation and results in high osmotic pressure for microbes (De Maayer et al., 2014). In the Antarctic strains HMP6 and HMP9, the functions of their genes obtained through HGTs are associated with the adaptation to environmental changes, not only to the low-temperature, but also to the conditions in Antarctic lakes, i.e., ultra-oligotrophic environment and fluctuations in sunshine and temperature during “white-night” in summer and “polar-night” in winter.
Our findings strongly suggest that these Antarctic strains might have acquired horizontally transferred genes from the class Betaproteobacteria for adaptation to the Antarctic environment. Moreover, HMP6 and HMP9 are suggested to harbor HGT candidates from distinct origins. However, in terms of the gene-function of the ss-HGTs derived from the class Betaproteobacteria, HMP6 and HMP9 had incorporated 19 and 40 genes belonging to the functional category “M,” respectively. In addition, HMP9 increased the number of ss-HGTs in ten functional categories, including “G.” Regarding the individual functions under “G,” in six genes for HMP6 and 42 genes for HMP9, the types of gene functions were nearly the same, however, the number of the ss-HGTs detected in HMP9 was frequently higher. Gene clusters consisting of these ss-HGTs including “M” or “G” were also identified. Although ss-HGTs derived from the class Betaproteobacteria appear to have been obtained through independent HGT events, confirming whether adjacent HGT candidates in the gene cluster are acquired in the same HGT event or via gene duplication after an HGT event, requires further investigation through BBH relationships and phylogenetic tree analysis. It is, however, imperative that these further investigations take into account taxonomic assignment and construction of gene families, when conducting best BLAST hit analysis (Dick et al., 2017). Thus, the Antarctic strains might have acquired some of the gene functions required for adaptation to the Antarctic environment from the class Betaproteobacteria, and multiple HGT acquisition mechanisms might underlie these HGT events.
Amino Acid Frequency Analysis to Examine Relationship Between HGT Candidates and Adaptation to Antarctic Environment
Adaptations to low temperatures and oligotrophic environments are examples of special adaptations found in the extreme environment of the Antarctic (Feng et al., 2014). Low-temperature adaptation is necessary for psychrophilic bacteria to thrive at low temperatures, wherein the chemical reaction rate is low, and for stable mass transport and metabolism reactions to occur. Specifically, low-temperature adaptation is influenced by modifications of coding for lysine and arginine in cold-adapted genes that exhibit this tendency, and it has been reported that the low-temperature adaptation of the affected proteins can be examined by investigating their low arginine content and Arg/(Arg + Lys) ratio (Russell, 2000).
To examine the relationship between HGT candidates and adaptation to the Antarctic environment, we focused on the amino acid frequency of proteins of the Antarctic and continental strains. First, we searched for significant differences between the Antarctic and continental strains, in terms of the amino acid sequences of proteins encoded by 856 housekeeping genes, whose conservation was determined by BBH analysis and reported complete genomic sequences and optimal growth temperatures (Figure 2 and Supplementary Table S2). The ratio of the amino acid frequencies of housekeeping genes between the Antarctic and continental strains are shown in Figure 9A, whereas the actual numbers, means, and significant differences are listed in Supplementary Table S9. In the Antarctic strains, increased frequencies for five amino acids (Asn, Lys, Ser, Thr, Val), and decreased frequencies for five (Ala, Arg, Glu, Leu, Pro), were observed for housekeeping genes. Secondly, we identified significant differences between the Antarctic and continental strains in terms of the amino acid frequencies of the ss-HGTs, the ratio of which are shown in Figure 9B, and the actual numbers, means, and significant differences are listed in Supplementary Table S10. The ss-HGTs showed a similar tendency as those of the housekeeping genes, save for two amino acids (Met and Trp) that indicated opposite tendencies (Figures 9A,B and Supplementary Tables S9, S10). Our findings regarding the amino acid frequencies of housekeeping genes and ss-HGTs between the Antarctic and continental strains are consistent with those of a previous study that reported low arginine content and an Arg/(Arg + Lys) ratio in cold-adapted genes (Russell, 2000).
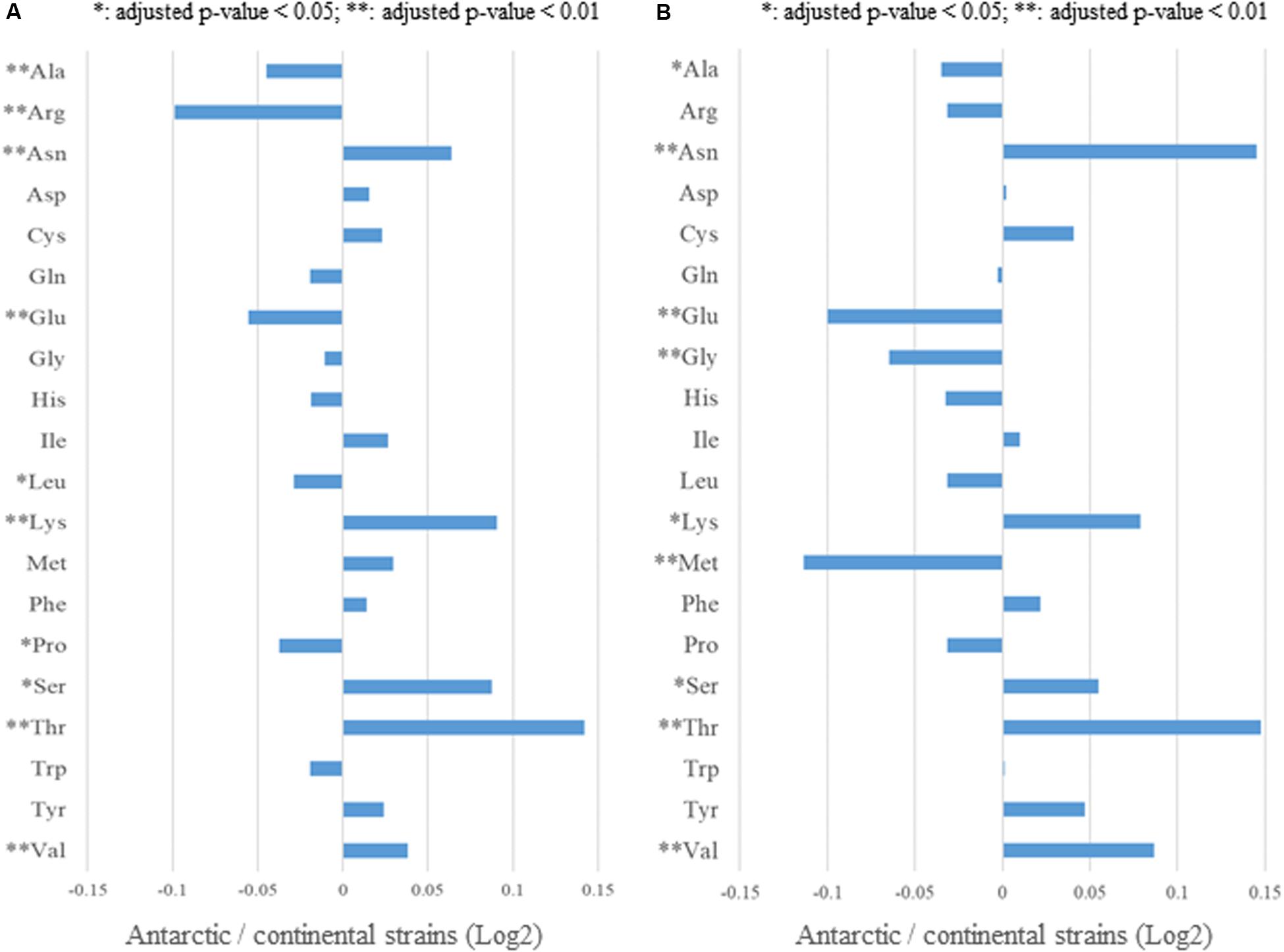
Figure 9. Comparison of amino acid usage between the Antarctic and continental strains. (A) Ratio of amino acid frequencies in the housekeeping genes of the Antarctic and continental strains. Asterisks: significant difference between Antarctic and continental strains. (B) Ratio of amino acid frequencies in the strain-specific HGT candidates of the Antarctic and continental strains.
We also examined the amino acid frequencies of housekeeping genes compared to each clade of Antarctic strain HMP6 and HMP9 and found that HMP6 formed a clade with S. psychrolutea MDB1-A (Figure 2). However, as the genomic sequence data has not been previously reported for MDB1-A we instead analyzed Sphingomonas sp. AAP5 and Sphingomonas panacis DCY99 with HMP6 (Supplementary Figure S2). Although the complete genome sequences have been reported for these Sphingomonas strains, their optimal growth temperatures have not (Supplementary Table S7). Our results show that HMP9 formed a clade with S. aerolata NW12 and S. ginsenosidivorax KHI67 (Figure 2), however, this genome sequencing data was not complete but rather was draft data containing sequence gaps (Supplementary Table S7). The distribution of amino acid frequencies in comparison with the strains in each HMP6 or HMP9 clade and the continental strains are shown in Supplementary Figures S6, S7, respectively. It was suggested that the amino acid frequencies of housekeeping genes in these strains in each clade showed a similar tendency in Arg, Glu, Ser, and Thr with the Antarctic strain HMP6 and HMP9. From these results, it may be suggested that the Antarctic strains HMP6 and HMP9 evolved independently in each clade and that their low-temperature adaptations occurred convergently (Supplementary Figure S2).
Regarding the properties of the amino acids, Ser and Thr are hydrophilic, whereas Ala and Leu are hydrophobic. Moreover, Arg can form more salt bridges and hydrogen bonds with surrounding residues through guanidino groups than Lys, thus helping to stabilize the proteins (Russell, 2000). Psychrophilic strains of the genus Shewanella and Gammaproteobacteria were found to have decreased G + C content and proteome Ala, Pro, and Arg contents (Zhao et al., 2010). Since the Antarctic strains and psychrophilic bacteria exhibit similar characteristics in terms of amino acid frequency, the Antarctic strains may have adapted to low-temperature environments by increasing the flexibility of their protein structures and decreasing the activation energy of a chemical reaction (Russell, 2000; Marx et al., 2007).
To confirm the low-temperature adaptation, we investigated the Arg/(Arg + Lys) ratio, and its relationship with optimal growth temperature, which is displayed in Figure 10A. Based on this relationship (Zeldovich et al., 2007), we selected Lys, Ser, Thr, and Val as amino acids that were increased in the Antarctic strains, whereas Ala, Arg, Glu, and Leu were those decreased in the Antarctic strains. From this we developed a formula (see section “Materials and Methods”) defined as the low-temperature relation ratio for Antarctic Sphingomonas strains. The relationship between the optimal growth temperature and the low-temperature relation ratio is plotted in Figure 10B. In terms of the housekeeping genes (blue dots in Figures 10A,B), the continental strains and the Antarctic strains showed high and low values on the low-temperature relation ratio, respectively, suggesting that changes in amino acid frequency for low-temperature adaptation occurred in the Antarctic strains. Conversely, in terms of the ss-HGTs (orange dots), the ratios for the ss-HGTs of the Antarctic strains were as low as those of the housekeeping genes of the Antarctic strains, suggesting that changes in amino acid frequency for low-temperature adaptation occurred in not only the housekeeping genes but also in the ss-HGTs, which indicates these changes potentially occurred in the entire genome.
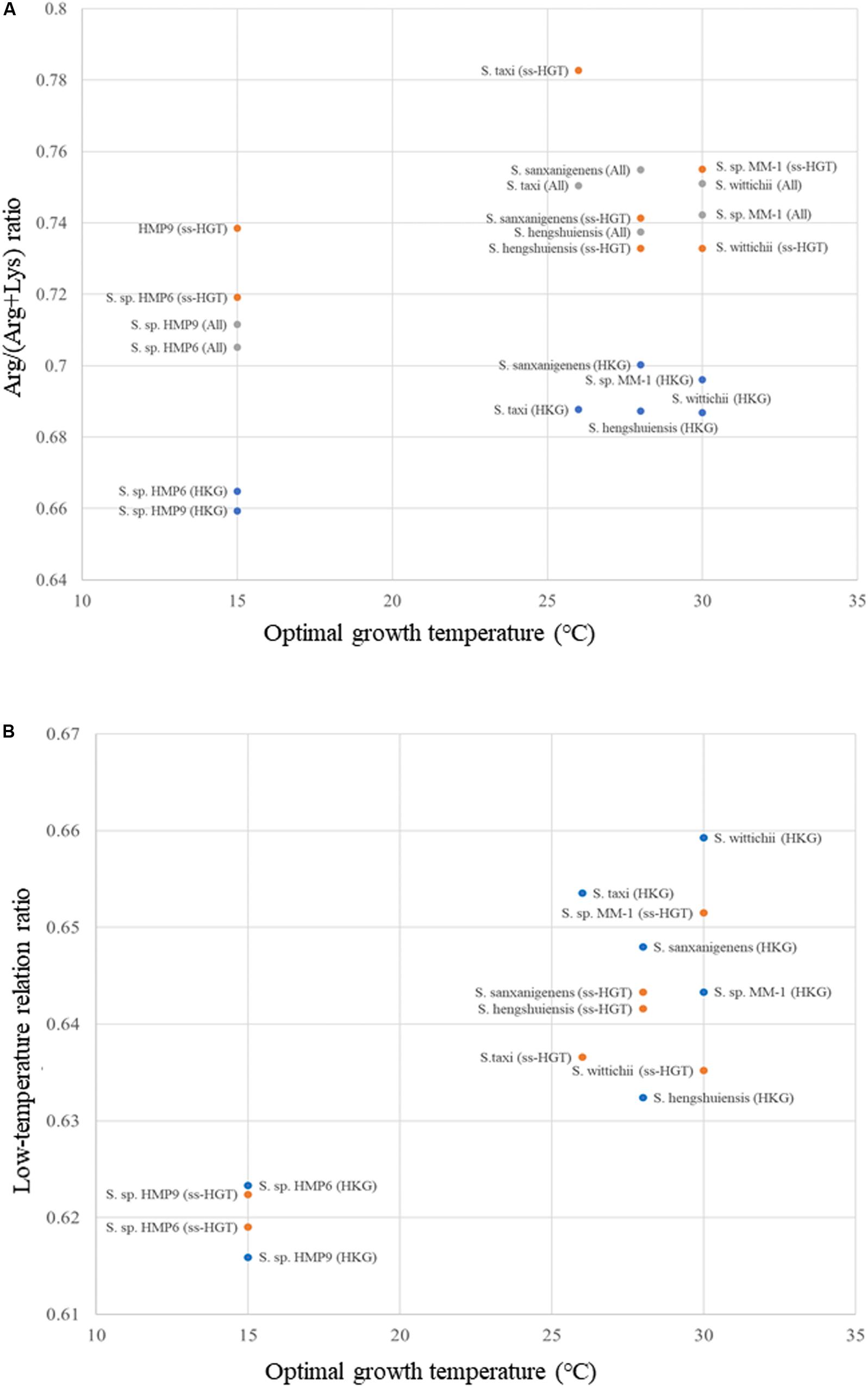
Figure 10. Scatter plot of amino acid ratio versus optimal growth temperature of Sphingomonas strains. (A) Arg/(Arg + Lys) ratio. Blue: housekeeping genes (HKG); orange: strain-specific HGT candidates (ss-HGT); gray: all genes (All). (B) Low-temperature relation ratio. Blue: housekeeping genes (HKG); orange: strain-specific HGT candidates.
We also compared the low-temperature relation ratios for individual genes (Supplementary Figure S8). The low-temperature relation ratios of both the housekeeping genes and ss-HGTs were spread widely, however, the spread ranges were similar among the Sphingomonas strains. The changes in the amino acid frequencies for low-temperature adaptation could potentially occur in the entire genome based on the results of the housekeeping genes. The relationship between the HGT genes and changes in the amino acid frequency after the HGT events is an important issue to be considered for further study as it could serve as a key factor in the elucidation of the low-temperature adaptation process in Antarctic bacteria. Therefore, we conclude that the ss-HGTs may be involved in the adaptation of genes to the Antarctic environment, including that to low temperatures, although the origins of the ss-HGTs of the two Antarctic strains are different.
Conclusion
The method we have developed for detecting HGT candidates employs BLSOM, an algorithm based on oligonucleotide usage that does not depend on sequence similarity. In other words, HGT candidates are identified and their origin is assigned with high efficiency based solely on references to oligonucleotide usage. In our proposed workflow, although BLSOM was constructed using a degenerate tetranucleotide composition and 5-kb window featuring 1-kb step, similar results can also be obtained with different conditions, for example using segment length or oligonucleotides, as shown by our previous studies (Abe et al., 2003; Iwasaki et al., 2013). Our method is applicable to both complete and draft genomes.
From the ss-HGT candidates we detected in two Antarctic strains of Sphingomonas, HMP6 and HMP9, through BLSOM and BBH analysis, we found that HMP6 harbored a higher number of genes acquired through horizontal transfer from the class Alphaproteobacteria, which is closely related to Sphingomonas spp., whereas HMP9 harbored a higher number of genes acquired from the class Betaproteobacteria, which is relatively more distantly related to Sphingomonas spp. This finding clearly shows an origin difference in the horizontally transmitted genes found in the two Antarctic strains, thus suggesting that horizontally transferred genes were obtained through multiple acquisition processes occurring in the same Antarctic environment.
In terms of gene functions, both Antarctic strains have obtained genes related to cell walls or cell membranes mainly from the class Betaproteobacteria. Particularly, HMP9 has acquired ten gene functions, including those involved in carbohydrate metabolism from the class Betaproteobacteria. Moreover, in relation to adaptability to the Antarctic environment, these ss-HGT candidates exhibited changes in amino acid frequency suited for low-temperature adaptation, and these changes have occurred throughout the genome. Similarly, this frequency change was observed in ss-HGTs. Thus, it is highly likely that the ss-HGTs are related to adaptation in the Antarctic environment, including that in low-temperature. The origins and the genetic functions of the horizontally transferred genes suggest that multiple pathways and strategies underlie adaptation to the Antarctic environment.
Our results indicate that our proposed BLSOM analysis could serve as a powerful tool for detecting HGT candidates and their origins in entire genomes. Our findings not only the enhance current understanding of how the two Antarctic Sphingomonas strains have adapted to their living environment, but also provide more general insights into the adaptation process in terms of amino acid composition changes that occur via HGT.
Generally, the compositional method primarily identifies recent transfers, while the phylogenetic method is more suitable for the detections of older events (Tamames and Moya, 2008). We have recently developed a new hierarchical BLSOM according to data class, such as phylotype (Kikuchi et al., 2015), and will aim to develop a new powerful method that makes use of the characteristics of both the compositional and phylogenetic methods to detect recent and older HGTs efficiently. This platform will allow for HGT acquisition and adaptation processes combined with the amino acid frequency analysis in future studies.
The large-scale destruction and reconstruction of ecosystems are presumed to have occurred in the glacial and interglacial cycles in Antarctica (Chown et al., 2015), and it is considered that various adaptation strategies have been tested in each organism or biosphere during this process. Functional genes for cellular process and energy metabolism that were acquired by horizontal transfer are considered to have been affected by changes in the amino acid frequency of the proteins that these genes encode, and Antarctic bacteria have acquired genes from other bacteria already adapted to low-temperature environments; therefore, these genes and the bacteria have shown high environmental adaptability even under conditions in which genetic resources are limited, such as in Antarctica. The adaptability has occurred convergently as independent evolutionary strategies in each Antarctic strain.
Data Availability Statement
The datasets generated for this study can be found in the DNA Data Bank of Japan under accession numbers AP022672 for Sphingomonas sp. HMP6, AP022673 for Sphingomonas sp. HMP9 and AP022674 for its plasmid named pHMP9.
Author Contributions
TA and TB designed the research. TA, YA, and TB performed the research. AT and HN contributed new materials. TA, YA, AT, and TB analyzed the data. All authors contributed to the article and approved the submitted version.
Funding
The work described in this report was funded by a grant from the Systematic Analysis for Global Environmental Change and Life on Earth (SAGE) project promoted by the Transdisciplinary Research Integration Center, under the Research Organization of Information and Systems, Japan, and Grants-in-Aid for Scientific Research (C: no. 17K00401) and Challenging Exploratory Research (no. 15K14595) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The genome sequencing was done by the staff of the Advanced Genomics Center at the National Institute of Genetics. The computation was done with the Earth Simulator of the Japan Agency for Marine-Earth Science and Technology. We would like to thank Editage (www.editage.com) for English language editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01486/full#supplementary-material
Footnotes
- ^ http://bioinfo.ie.niigata-u.ac.jp/?BLSOM
- ^ https://ftp.ncbi.nlm.nih.gov/genbank/
- ^ ftp://ftp.ddbj.nig.ac.jp/ddbj_database/wgs/
- ^ http://bioinfo.ie.niigata-u.ac.jp/?PEMS4HGT
- ^ https://www.ncbi.nlm.nih.gov/refseq/targetedloci/
References
Abe, T., Ikemura, T., Sugahara, J., Kanai, A., Ohara, Y., Uehara, H., et al. (2011). tRNADB-CE 2011: tRNA gene database curated manually by experts. Nucleic Acids Res. 39, D210–D213. doi: 10.1093/nar/gkq1007
Abe, T., Kanaya, S., Kinouchi, M., Ichiba, Y., Kozuki, T., and Ikemura, T. (2003). Informatics for unveiling hidden genome signatures. Genome Res. 13, 693–702. doi: 10.1101/gr.634603
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/s0022-2836(05)80360-80362
Andam, C. P., and Gogarten, J. P. (2011). Biased gene transfer in microbial evolution. Nat. Rev. Microbiol. 9, 543–555. doi: 10.1038/nrmicro2593
Beiko, R. G., Harlow, T. J., and Ragan, M. A. (2005). Highways of gene sharing in prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 14332–14337. doi: 10.1073/pnas.0504068102
Blanc, G., Agarkova, I., Grimwood, J., Kuo, A., Brueggeman, A., Dunigan, D. D., et al. (2012). The genome of the polar eukaryotic microalga Coccomyxa subellipsoidea reveals traits of cold adaptation. Genome Biol. 13:R39. doi: 10.1186/gb-2012-13-5-r39
Booth, A., Mariscal, C., and Doolittle, W. F. (2016). The modern synthesis in the light of microbial genomics. Annu. Rev. Microbiol. 70, 279–297. doi: 10.1146/annurev-micro-102215-195456
Brewer, T. E., Albertsen, M., Edwards, A., Kirkegaard, R. H., Rocha, E. P. C., and Fierer, N. (2020). Unlinked rRNA genes are widespread among bacteria and archaea. ISME J. 14, 597–608. doi: 10.1038/s41396-019-0552-553
Brochier-Armanet, C., Deschamps, P., Lopez-Garcia, P., Zivanovic, Y., Rodriguez-Valera, F., and Moreira, D. (2011). Complete-fosmid and fosmid-end sequences reveal frequent horizontal gene transfers in marine uncultured planktonic archaea. ISME J. 5, 1291–1302. doi: 10.1038/ismej.2011.16
Busse, H. J., Denner, E. B., Buczolits, S., Salkinoja-Salonen, M., Bennasar, A., and Kampfer, P. (2003). Sphingomonas aurantiaca sp. nov., Sphingomonas aerolata sp. nov. and Sphingomonas faeni sp. nov., air- and dustborne and Antarctic, orange-pigmented, psychrotolerant bacteria, and emended description of the genus Sphingomonas. Int. J. Syst. Evol. Microbiol. 53(Pt 5) 1253–1260. doi: 10.1099/ijs.0.02461-2460
Castelle, C. J., and Banfield, J. F. (2018). Major new microbial groups expand diversity and alter our understanding of the tree of life. Cell 172, 1181–1197. doi: 10.1016/j.cell.2018.02.016
Choi, I. G., and Kim, S. H. (2007). Global extent of horizontal gene transfer. Proc. Natl. Acad. Sci. U.S.A. 104, 4489–4494. doi: 10.1073/pnas.0611557104
Chown, S. L., Clarke, A., Fraser, C. I., Cary, S. C., Moon, K. L., and McGeoch, M. A. (2015). The changing form of Antarctic biodiversity. Nature 522, 431–438. doi: 10.1038/nature14505
Dagan, T., Artzy-Randrup, Y., and Martin, W. (2008). Modular networks and cumulative impact of lateral transfer in prokaryote genome evolution. Proc. Natl. Acad. Sci. U.S.A. 105, 10039–10044. doi: 10.1073/pnas.0800679105
Darmon, E., and Leach, D. R. (2014). Bacterial genome instability. Microbiol. Mol. Biol. Rev. 78, 1–39. doi: 10.1128/mmbr.00035-13
De Maayer, P., Anderson, D., Cary, C., and Cowan, D. A. (2014). Some like it cold: understanding the survival strategies of psychrophiles. EMBO Rep. 15, 508–517. doi: 10.1002/embr.201338170
Delcher, A. L., Harmon, D., Kasif, S., White, O., and Salzberg, S. L. (1999). Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27, 4636–4641. doi: 10.1093/nar/27.23.4636
DeLong, E. F. (1992). Archaea in coastal marine environments. Proc. Natl. Acad. Sci. U.S.A. 89, 5685–5689. doi: 10.1073/pnas.89.12.5685
Dick, A. A., Harlow, T. J., and Gogarten, J. P. (2017). Short branches lead to systematic artifacts when BLAST searches are used as surrogate for phylogenetic reconstruction. Mol. Phylogenet. Evol. 107, 338–344. doi: 10.1016/j.ympev.2016.11.016
Douglas, G. M., and Langille, M. G. I. (2019). Current and promising approaches to identify horizontal gene transfer events in metagenomes. Genome Biol. Evol. 11, 2750–2766. doi: 10.1093/gbe/evz184
Eevers, N., Van Hamme, J. D., Bottos, E. M., Weyens, N., and Vangronsveld, J. (2015). Sphingomonas taxi, isolated from Cucurbita pepo, proves to be a DDE-degrading and plant growth-promoting strain. Genome Announc 3:e00489-15. doi: 10.1128/genomeA.00489-415
Eyres, I., Boschetti, C., Crisp, A., Smith, T. P., Fontaneto, D., Tunnacliffe, A., et al. (2015). Horizontal gene transfer in bdelloid rotifers is ancient, ongoing and more frequent in species from desiccating habitats. BMC Biol. 13:90. doi: 10.1186/s12915-015-0202-209
Fast, N. M., Law, J. S., Williams, B. A., and Keeling, P. J. (2003). Bacterial catalase in the microsporidian Nosema locustae: implications for microsporidian metabolism and genome evolution. Eukaryot. Cell 2, 1069–1075. doi: 10.1128/ec.2.5.1069-1075.2003
Feng, S., Powell, S. M., Wilson, R., and Bowman, J. P. (2014). Extensive gene acquisition in the extremely psychrophilic bacterial species Psychroflexus torquis and the link to sea-ice ecosystem specialism. Genome Biol. Evol. 6, 133–148. doi: 10.1093/gbe/evt209
Galperin, M. Y., Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2015). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269. doi: 10.1093/nar/gku1223
Garcia-Vallve, S., Romeu, A., and Palau, J. (2000). Horizontal gene transfer in bacterial and archaeal complete genomes. Genome Res. 10, 1719–1725. doi: 10.1101/gr.130000
Gogarten, J. P., and Townsend, J. P. (2005). Horizontal gene transfer, genome innovation and evolution. Nat. Rev. Microbiol. 3, 679–687. doi: 10.1038/nrmicro1204
Harding, T., Roger, A. J., and Simpson, A. G. B. (2017). Adaptations to high salt in a halophilic protist: differential expression and gene acquisitions through duplications and gene transfers. Front. Microbiol. 8:944. doi: 10.3389/fmicb.2017.00944
Huang, H. D., Wang, W., Ma, T., Li, G. Q., Liang, F. L., and Liu, R. L. (2009). Sphingomonas sanxanigenens sp. nov., isolated from soil. Int. J. Syst. Evol. Microbiol. 59(Pt 4) 719–723. doi: 10.1099/ijs.0.000257-250
Huang, Y., Wei, Z., Danzeng, W., Kim, M. C., Zhu, G., Zhang, Y., et al. (2017). Sphingomonas antarctica sp. nov., isolated from Antarctic tundra soil. Int. J. Syst. Evol. Microbiol. 67, 4064–4068. doi: 10.1099/ijsem.0.002253
Hug, L. A., Baker, B. J., Anantharaman, K., Brown, C. T., Probst, A. J., Castelle, C. J., et al. (2016). A new view of the tree of life. Nat. Microbiol. 1:16048. doi: 10.1038/nmicrobiol.2016.48
Imura, S., Bando, T., Saito, S., Seto, K., and Kanda, H. (1999). Benthic moss pillars in Antarctic lakes. Polar Biol. 22, 137–140. doi: 10.1007/s003000050401
Iwasaki, Y., Abe, T., Wada, K., Wada, Y., and Ikemura, T. (2013). A Novel Bioinformatics Strategy to Analyze Microbial Big Sequence Data for Efficient Knowledge Discovery: Batch-Learning Self-Organizing Map (BLSOM). Microorganisms 1, 137–157. doi: 10.3390/microorganisms1010137
Jain, R., Rivera, M. C., and Lake, J. A. (1999). Horizontal gene transfer among genomes: the complexity hypothesis. Proc. Natl. Acad. Sci. U.S.A. 96, 3801–3806. doi: 10.1073/pnas.96.7.3801
Kanaya, S., Kinouchi, M., Abe, T., Kudo, Y., Yamada, Y., Nishi, T., et al. (2001). Analysis of codon usage diversity of bacterial genes with a self-organizing map (SOM): characterization of horizontally transferred genes with emphasis on the E. coli O157 genome. Gene 276, 89–99. doi: 10.1016/s0378-1119(01)00673-674
Kanaya, S., Yamada, Y., Kudo, Y., and Ikemura, T. (1999). Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene 238, 143–155. doi: 10.1016/s0378-1119(99)00225-225
Karlin, S., Campbell, A. M., and Mrazek, J. (1998). Comparative DNA analysis across diverse genomes. Annu. Rev. Genet. 32, 185–225. doi: 10.1146/annurev.genet.32.1.185
Keeling, P. J., and Palmer, J. D. (2008). Horizontal gene transfer in eukaryotic evolution. Nat. Rev. Genet. 9, 605–618. doi: 10.1038/nrg2386
Kikuchi, A., Ikemura, T., and Abe, T. (2015). Development of self-compressing BLSOM for comprehensive analysis of big sequence data. Biomed. Res. Int. 2015:506052. doi: 10.1155/2015/506052
Kinouchi, M., and Kurokawa, K. (2006). tRNAfinder: a Software System To Find All tRNA genes in the DNA Sequence Based on the Cloverleaf Secondary Structure. J. Comp. Aid Chem. 7, 116–124. doi: 10.2751/jcac.7.116
Kohonen, T., Oja, E., Simula, O., Visa, A., and Kangas, J. (1996). Engineering applications of the self-organizing map. Proc. IEEE 84, 1358–1384. doi: 10.1109/5.537105
Lagesen, K., Hallin, P., Rodland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Laslett, D., and Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16. doi: 10.1093/nar/gkh152
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Liu, Q., Liu, H. C., Zhang, J. L., Zhou, Y. G., and Xin, Y. H. (2015). Sphingomonas psychrolutea sp. nov., a psychrotolerant bacterium isolated from glacier ice. Int. J. Syst. Evol. Microbiol. 65, 2955–2959. doi: 10.1099/ijs.0.000362
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Marx, J. C., Collins, T., D’Amico, S., Feller, G., and Gerday, C. (2007). Cold-adapted enzymes from marine Antarctic microorganisms. Mar. Biotechnol. 9, 293–304. doi: 10.1007/s10126-006-6103-6108
Math, R. K., Jin, H. M., Kim, J. M., Hahn, Y., Park, W., Madsen, E. L., et al. (2012). Comparative genomics reveals adaptation by Alteromonas sp. SN2 to marine tidal-flat conditions: cold tolerance and aromatic hydrocarbon metabolism. PLoS One 7:e35784. doi: 10.1371/journal.pone.0035784
Miller, T. R., Delcher, A. L., Salzberg, S. L., Saunders, E., Detter, J. C., and Halden, R. U. (2010). Genome sequence of the dioxin-mineralizing bacterium Sphingomonas wittichii RW1. J. Bacteriol. 192, 6101–6102. doi: 10.1128/jb.01030-1010
Mock, T., Otillar, R. P., Strauss, J., McMullan, M., Paajanen, P., Schmutz, J., et al. (2017). Evolutionary genomics of the cold-adapted diatom Fragilariopsis cylindrus. Nature 541, 536–540. doi: 10.1038/nature20803
Mrazek, J. (2009). Phylogenetic signals in DNA composition: limitations and prospects. Mol. Biol. Evol. 26, 1163–1169. doi: 10.1093/molbev/msp032
Naganuma, T., Miyoshi, T., and Kimura, H. (2007). Phylotype diversity of deep-sea hydrothermal vent prokaryotes trapped by 0.2- and 0.1-microm-pore-size filters. Extremophiles 11, 637–646. doi: 10.1007/s00792-007-0070-75
Nakai, R., Abe, T., Baba, T., Imura, S., Kagoshima, H., Kanda, H., et al. (2012). Microflorae of aquatic moss pillars in a freshwater lake, East Antarctica, based on fatty acid and 16S rRNA gene analyses. Polar Biol. 35, 425–433. doi: 10.1007/s00300-011-1090-1092
Nakamura, Y., Itoh, T., Matsuda, H., and Gojobori, T. (2004). Biased biological functions of horizontally transferred genes in prokaryotic genomes. Nat. Genet. 36, 760–766. doi: 10.1038/ng1381
Nakao, R., Abe, T., Funayama, S., and Sugimoto, C. (2016). Horizontally transferred genetic elements in the tsetse fly genome: an alignment-free clustering approach using batch learning self-organising map (BLSOM). Biomed. Res. Int. 2016:3164624. doi: 10.1155/2016/3164624
Nakao, R., Abe, T., Nijhof, A. M., Yamamoto, S., Jongejan, F., Ikemura, T., et al. (2013). A novel approach, based on BLSOMs (Batch Learning Self-Organizing Maps), to the microbiome analysis of ticks. ISME J. 7, 1003–1015. doi: 10.1038/ismej.2012.171
Nelson-Sathi, S., Sousa, F. L., Roettger, M., Lozada-Chavez, N., Thiergart, T., Janssen, A., et al. (2015). Origins of major archaeal clades correspond to gene acquisitions from bacteria. Nature 517, 77–80. doi: 10.1038/nature13805
Noguchi, H., Taniguchi, T., and Itoh, T. (2008). MetaGeneAnnotator: detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res. 15, 387–396. doi: 10.1093/dnares/dsn027
Ochman, H., Lawrence, J. G., and Groisman, E. A. (2000). Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304. doi: 10.1038/35012500
O’Leary, N. A., Wright, M. W., Brister, J. R., Ciufo, S., Haddad, D., McVeigh, R., et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. doi: 10.1093/nar/gkv1189
Pappas, K. M., Kouvelis, V. N., Saunders, E., Brettin, T. S., Bruce, D., Detter, C., et al. (2011). Genome sequence of the ethanol-producing Zymomonas mobilis subsp. mobilis lectotype strain ATCC 10988. J. Bacteriol. 193, 5051–5052. doi: 10.1128/jb.05395-5311
Parks, D. H., Rinke, C., Chuvochina, M., Chaumeil, P. A., Woodcroft, B. J., Evans, P. N., et al. (2017). Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2, 1533–1542. doi: 10.1038/s41564-017-0012-17
Qiu, Y., Abe, T., Nakao, R., Satoh, K., and Sugimoto, C. (2019). Viral population analysis of the taiga tick, Ixodes persulcatus, by using Batch Learning Self-Organizing Maps and BLAST search. J. Vet. Med. Sci. 81, 401–410. doi: 10.1292/jvms.18-0483
R Core Team (2019). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Raymond, J. A., and Kim, H. J. (2012). Possible role of horizontal gene transfer in the colonization of sea ice by algae. PLoS One 7:e35968. doi: 10.1371/journal.pone.0035968
Reasoner, D. J., and Geldreich, E. E. (1985). A new medium for the enumeration and subculture of bacteria from potable water. Appl. Environ. Microbiol. 49, 1–7. doi: 10.1128/aem.49.1.1-7.1985
Roller, B. R., Stoddard, S. F., and Schmidt, T. M. (2016). Exploiting rRNA operon copy number to investigate bacterial reproductive strategies. Nat. Microbiol. 1:16160. doi: 10.1038/nmicrobiol.2016.160
Russell, N. J. (2000). Toward a molecular understanding of cold activity of enzymes from psychrophiles. Extremophiles 4, 83–90. doi: 10.1007/s007920050141
Schonknecht, G., Chen, W. H., Ternes, C. M., Barbier, G. G., Shrestha, R. P., Stanke, M., et al. (2013). Gene transfer from bacteria and archaea facilitated evolution of an extremophilic eukaryote. Science 339, 1207–1210. doi: 10.1126/science.1231707
Schulz, F., Eloe-Fadrosh, E. A., Bowers, R. M., Jarett, J., Nielsen, T., Ivanova, N. N., et al. (2017). Towards a balanced view of the bacterial tree of life. Microbiome 5:140. doi: 10.1186/s40168-017-0360-369
Skotnicki, M. L., Lee, K. J., Tribe, D. E., and Rogers, P. L. (1981). Comparison of ethanol production by different zymomonas strains. Appl. Environ. Microbiol. 41, 889–893. doi: 10.1128/aem.41.4.889-893.1981
Slamovits, C. H., and Keeling, P. J. (2004). Class II photolyase in a microsporidian intracellular parasite. J. Mol. Biol. 341, 713–721. doi: 10.1016/j.jmb.2004.06.032
Soucy, S. M., Huang, J., and Gogarten, J. P. (2015). Horizontal gene transfer: building the web of life. Nat. Rev. Genet. 16, 472–482. doi: 10.1038/nrg3962
Stanier, R. Y., Kunisawa, R., Mandel, M., and Cohen-Bazire, G. (1971). Purification and properties of unicellular blue-green algae (order Chroococcales). Bacteriol. Rev. 35, 171–205. doi: 10.1128/mmbr.35.2.171-205.1971
Sugawara, H., Ohyama, A., Mori, H., and Kurokawa, K. (2009). “Microbial Genome Annotation Pipeline (MiGAP) for diverse users,” in Proceedings of the 20th International Conference on Genome Informatics, (Tokyo: Yokohama).
Syvanen, M. (1985). Cross-species gene transfer; implications for a new theory of evolution. J. Theor. Biol. 112, 333–343. doi: 10.1016/s0022-5193(85)80291-80295
Syvanen, M. (2012). Evolutionary implications of horizontal gene transfer. Annu. Rev. Genet. 46, 341–358. doi: 10.1146/annurev-genet-110711-155529
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. doi: 10.1093/nar/gky1131
Tabata, M., Endo, R., Ito, M., Ohtsubo, Y., Kumar, A., Tsuda, M., et al. (2011). The lin genes for gamma-hexachlorocyclohexane degradation in Sphingomonas sp. MM-1 proved to be dispersed across multiple plasmids. Biosci. Biotechnol. Biochem. 75, 466–472. doi: 10.1271/bbb.100652
Tabata, M., Ohtsubo, Y., Ohhata, S., Tsuda, M., and Nagata, Y. (2013). Complete Genome Sequence of the gamma-Hexachlorocyclohexane-Degrading Bacterium Sphingomonas sp. Strain MM-1. Genome Announc 1:e00247-13. doi: 10.1128/genomeA.00247-213
Takeuchi, M., Hamana, K., and Hiraishi, A. (2001). Proposal of the genus Sphingomonas sensu stricto and three new genera, Sphingobium, Novosphingobium and Sphingopyxis, on the basis of phylogenetic and chemotaxonomic analyses. Int. J. Syst. Evol. Microbiol. 51(Pt 4) 1405–1417. doi: 10.1099/00207713-51-4-1405
Tamames, J., and Moya, A. (2008). Estimating the extent of horizontal gene transfer in metagenomic sequences. BMC Genomics 9:136. doi: 10.1186/1471-2164-9-136
Tamura, K., and Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526. doi: 10.1093/oxfordjournals.molbev.a040023
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tatum, E. L., and Lederberg, J. (1947). Gene Recombination in the Bacterium Escherichia coli. J. Bacteriol. 53, 673–684. doi: 10.1128/jb.53.6.673-684.1947
The UniProt, C. (2018). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515. doi: 10.1093/nar/gky1049
Wei, S., Wang, T., Liu, H., Zhang, C., Guo, J., Wang, Q., et al. (2015). Sphingomonas hengshuiensis sp. nov., isolated from lake wetland. Int. J. Syst. Evol. Microbiol. 65, 4644–4649. doi: 10.1099/ijsem.0.000626
Williams, T. A., Szollosi, G. J., Spang, A., Foster, P. G., Heaps, S. E., Boussau, B., et al. (2017). Integrative modeling of gene and genome evolution roots the archaeal tree of life. Proc. Natl. Acad. Sci. U.S.A. 114, E4602–E4611. doi: 10.1073/pnas.1618463114
Wu, M., Huang, H., Li, G., Ren, Y., Shi, Z., Li, X., et al. (2017). The evolutionary life cycle of the polysaccharide biosynthetic gene cluster based on the Sphingomonadaceae. Sci. Rep. 7:46484. doi: 10.1038/srep46484
Yabuuchi, E., Yamamoto, H., Terakubo, S., Okamura, N., Naka, T., Fujiwara, N., et al. (2001). Proposal of Sphingomonas wittichii sp. nov. for strain RW1T, known as a dibenzo-p-dioxin metabolizer. Int. J. Syst. Evol. Microbiol. 51(Pt 2) 281–292. doi: 10.1099/00207713-51-2-281
Zamani-Dahaj, S. A., Okasha, M., Kosakowski, J., and Higgs, P. G. (2016). Estimating the frequency of horizontal gene transfer using phylogenetic models of gene gain and loss. Mol. Biol. Evol. 33, 1843–1857. doi: 10.1093/molbev/msw062
Zeldovich, K. B., Berezovsky, I. N., and Shakhnovich, E. I. (2007). Protein and DNA sequence determinants of thermophilic adaptation. PLoS Comput. Biol. 3:e5. doi: 10.1371/journal.pcbi.0030005
Zeng, Z., Liu, X., Yao, J., Guo, Y., Li, B., Li, Y., et al. (2016). Cold adaptation regulated by cryptic prophage excision in Shewanella oneidensis. ISME J. 10, 2787–2800. doi: 10.1038/ismej.2016.85
Keywords: horizontal gene transfer, Antarctic environment, Sphingomonas genome, Batch-Learning Self-Organizing Map, oligonucleotide, low-temperature adaptation, amino acid frequency, convergent evolution
Citation: Abe T, Akazawa Y, Toyoda A, Niki H and Baba T (2020) Batch-Learning Self-Organizing Map Identifies Horizontal Gene Transfer Candidates and Their Origins in Entire Genomes. Front. Microbiol. 11:1486. doi: 10.3389/fmicb.2020.01486
Received: 05 February 2020; Accepted: 08 June 2020;
Published: 03 July 2020.
Edited by:
Koji Yahara, National Institute of Infectious Diseases (NIID), JapanReviewed by:
Haruo Suzuki, Keio University, JapanFrancisco (Paco) Barona-Gomez, Center for Research and Advanced Studies of the National Polytechnic Institute, Mexico
Copyright © 2020 Abe, Akazawa, Toyoda, Niki and Baba. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takashi Abe, dGFrYWFiZUBpZS5uaWlnYXRhLXUuYWMuanA=; Tomoya Baba, dG9iYWJhQG5pZy5hYy5qcA==
†These authors have contributed equally to this work