 Danielle L. Peters
Danielle L. Peters Jaclyn G. McCutcheon
Jaclyn G. McCutcheon Jonathan J. Dennis
Jonathan J. Dennis
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 24 June 2020
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.01358
This article is part of the Research TopicBacteriophages Isolation From The Environment And Their Antimicrobial Therapeutic PotentialView all 21 articles
A novel Siphoviridae phage specific to the bacterial species Stenotrophomonas maltophilia was isolated from a pristine soil sample and characterized as a second member of the newly established Delepquintavirus genus. Phage DLP3 possesses one of the broadest host ranges of any S. maltophilia phage yet characterized, infecting 22 of 29 S. maltophilia strains. DLP3 has a genome size of 96,852 bp and a G+C content of 58.4%, which is significantly lower than S. maltophilia host strain D1571 (G+C content of 66.9%). The DLP3 genome encodes 153 coding domain sequences covering 95% of the genome, including five tRNA genes with different specificities. The DLP3 lysogen exhibits a growth rate increase during the exponential phase of growth as compared to the wild type strain. DLP3 also encodes a functional erythromycin resistance protein, causing lysogenic conversion of the host D1571 strain. Although a temperate phage, DLP3 demonstrates excellent therapeutic potential because it exhibits a broad host range, infects host cells through the S. maltophilia type IV pilus, and exhibits lytic activity in vivo. Undesirable traits, such as its temperate lifecycle, can be eliminated using genetic techniques to produce a modified phage useful in the treatment of S. maltophilia bacterial infections.
Stenotrophomonas maltophilia is a robust, non-sporulating, obligate aerobe Gram-negative bacillus (Brooke, 2012). The genus name Stenotrophomonas was originally chosen due to the perceived limited nutritional range of the bacterium (Palleroni and Bradbury, 1993), though many studies have since shown an impressive metabolic versatility (Chatelut et al., 1995; Berg et al., 1999; Hauben et al., 1999; Minkwitz and Berg, 2001; Svensson-Stadler et al., 2012). S. maltophilia is often found in close association with plant rhizospheres where they actively promote plant growth through the secretion of growth-promoting compounds (Ryan et al., 2009). This plant growth promotion is now being used commercially; S. maltophilia strains are used as biofertilizers due to their ability to fix nitrogen, produce growth-promoting plant hormones, and protect plant roots from phytopathogens (Rathi and Nandabalan, 2017). Additionally, the vast metabolic diversity observed in S. maltophilia enables these bacteria to be used in bioremediation, from heavy metal detoxification of soils and waterways to the degradation of insecticides and volatile organic compounds such as benzene. However, their widespread use in biotechnology and agriculture is problematic due to their ability to cause disease in humans (Berg and Martinez, 2015).
S. maltophilia is an important multidrug resistant, opportunistic pathogen that is most commonly associated with pneumonia and bacteremia in immunocompromised patients. S. maltophilia has also been identified as the cause of soft tissue infections, osteomyelitis, meningitis, endocarditis, otitis and scleritis (Brooke, 2012). Several risk factors are associated with S. maltophilia infections in the general population, including malignancy, human immunodeficiency virus, cystic fibrosis (CF), intravenous drug use, surgical and accidental trauma, prolonged hospitalization, mechanical ventilation, indwelling catheters, corticosteroids, immunosuppressive therapy, and treatment with broad-spectrum antibiotics (Al-Anazi and Al-Jasser, 2014). One study estimated the nosocomial mortality rates attributed to S. maltophilia bacteremia at 16.7%, and the overall mortality rate of patients infected with S. maltophilia was 25% (Naidu and Smith, 2012). A recent study on the clinical outcomes of cancer patients with bloodstream infections (BSI) and pneumonia caused by S. maltophilia infections in Mexico City indicated that 31.6% died within the first month; 22.1% of the deaths were due to pneumonia and 9.5% were due to BSI (Velazquez-Acosta et al., 2018). A similar study on BSI mortality rates associated with 937 German intensive care units found S. maltophilia infections had the highest mortality rates (28.4%), followed next by non-albicans-Candida spp. (27.1%) and Pseudomonas aeruginosa (25.8%) (Schwab et al., 2018).
Due to a wide array of pathogenicity factors, S. maltophilia can be difficult to clear once an infection has been established. S. maltophilia pathogenicity mechanisms include swimming and twitching motility (from flagella and type IV pili, respectively), a DNA hypermutator mechanism, iron uptake transporters, biofilm formation, lipopolysaccharide (LPS), many extracellular enzymes encoded such as fibrolysin, lipases, esterase, DNase, RNase, proteases and lecithinase, type II protein secretion systems, host cell invasion, and quorum sensing signaling systems (Brooke, 2012). In addition, clinical S. maltophilia isolate culture supernatants applied to Hep-2, HeLa, or Vero cells resulted in cytotoxic effects such as intensive rounding, loss of intercellular junctions, and membrane blebbing followed by cell death within 24 h (Figueiredo et al., 2006). In some cases, endocytosis, hemolytic activity, and cell aggregation were observed, and the addition of protease inhibitors did not prevent cell cytotoxicity. LPS production was shown to be required for attachment to surfaces, and colonization and virulence in a rat lung model (McKay et al., 2003). As well, it was shown that an O-polysaccharide synthesis mutant was susceptible to complement-mediated cell killing (Brooke et al., 2008). S. maltophilia can form biofilms on many surfaces such as glass, plastics, implanted medical devices, and lung epithelial cells (Huang et al., 2006; Pompilio et al., 2008, 2010; Brooke, 2014). A study focusing on the biofilm formation of CF-derived bronchial epithelial cell monolayers revealed that all S. maltophilia isolates were able to form biofilms on bronchial epithelial cells (Pompilio et al., 2010). Type-1 fimbriae genes have only been identified in clinical S. maltophilia isolates, which suggests they may play a direct role in the colonization of infected CF individuals (Nicoletti et al., 2011). In vitro tissue culture assays have indicated the S. maltophilia fimbriae 1 (SMF-1) protein is important for adherence to eukaryotic cells and glass, and anti-SMF-1 antibodies inhibit adherence to eukaryotic cells and glass if the antibodies were applied during early stages of infection (de Oliveira-Garcia et al., 2003). The ability of S. maltophilia isolates to form biofilms and invade host cells contributes to the prolonged infections noted in hematology and oncology patients, despite aggressive antibiotic treatments (Lai et al., 2006).
Innate antibiotic resistance in S. maltophilia infections is a major contributing factor to treatment failure. Patients who receive the wrong antibiotic at initial infection diagnosis have an increased risk of mortality compared to patients who received appropriate S. maltophilia antibiotic treatment initially (Velazquez-Acosta et al., 2018). S. maltophilia infections can be further complicated by the emergence of mutants with pleiotropic antibiotic resistance (Denton and Kerr, 1998). Intrinsic resistance is attributed to mechanisms such as reduced membrane permeability (Mett et al., 1988), chromosomally encoded multidrug efflux pumps, and chromosomally encoded Qnr pentapeptide repeat proteins (Sanchez et al., 2008; Sanchez and Martinez, 2010; Chang et al., 2011). Additionally, antibiotic inactivating enzymes such as inducible L1 and L2 β-lactamases (Walsh et al., 1997; Crowder et al., 1998; Okazaki and Avison, 2008), a TEM-2 β-lactamase from a Tn1-like transposon (Avison et al., 2000), and aminoglycoside- inactivating enzymes (Okazaki and Avison, 2007; Vetting et al., 2008) have been identified in many S. maltophilia isolates. The L1 protein is a molecular class B zinc-dependent metallo-β-lactamase which hydrolyzes virtually all classes of β-lactams, including penicillins, cephalosporins and carbapenems (Avison et al., 2001), whereas the L2 protein is a molecular class A clavulanic acid-sensitive cephalosporinase (Chang et al., 2015). The drug of choice to treat S. maltophilia infections continues to be trimethoprim/sulfamethoxazole (TMP/SMX) (Chung et al., 2013; Wang et al., 2016), although resistance to TMP/SMX across clinical isolates is common (30.5%) (Ochoa-Sanchez and Vinuesa, 2017). However, higher rates of TMP/SMX resistance, ranging from 16% to nearly 80%, have been noted in patients with cancer, CF, and in several specific countries (Chang et al., 2015; Rhee et al., 2016). The majority of TMP/SMX resistance is due to the presence of a sul gene encoding a sulfonamide resistance protein located on a class 1 integron (sul1) (Barbolla et al., 2004; Chung et al., 2015), and also on insertion sequence common region (ISCR) elements (sul2) (Toleman et al., 2007) rather than through the overexpression of efflux pumps. A dhfr gene encoding a dihydrofolate reductase associated with a class 1 integron has also been shown to cause some level of resistance against TMP/SMX (Hu et al., 2011), along with the activity from several RND family efflux pumps, although not to the same degree as the resident sul genes (Huang et al., 2013; Lin et al., 2015; Sanchez, 2015). Thus, research into alternative treatment options is critical due to increased resistance to TMP/SMX noted with specific comorbidities and geographic locations.
Related to this high-level antibiotic resistance, S. maltophilia is also capable of tolerating biocides and harsh environments, which makes this bacterium difficult to eradicate from hospital settings. Nosocomial isolation sources for S. maltophilia include ultra-pure water, hemodialysis water, nebulizers, hand-washing soap, hospital antiseptic solution, chlorhexidine-cetrimide topical antiseptic solution, hypochlorite cleaners, triclosan, sodium dodecyl sulfate, and antiseptics containing quaternary ammonium compounds (Brooke, 2012; Kampmeier et al., 2017). Non-clinical multidrug resistant isolates of S. maltophilia have also been recovered from environments such as soil, water, plants and food sources (Berg et al., 2005; Qureshi et al., 2005; Brooke, 2012; Lin et al., 2017; Furlan and Stehling, 2018; Kim et al., 2018), and have the potential to cause community-acquired infections (Falagas et al., 2009; Chang et al., 2014).
High levels of innate antibiotic resistance in S. maltophilia isolates highlights the need for alternative treatment options to combat these infections. The use of bacteriophages, or phages, as viruses specifically lytic toward bacteria, is being investigated as a viable option. Phage therapy requires the isolation and characterization of phages prior to their use as prophylactic or therapeutic agents. Phage therapy holds several advantages over traditional antibiotics (Sulakvelidze et al., 2001; Burrowes et al., 2011; Golkar et al., 2014). First, phages possess a narrow host range, only infecting a few strains within a species, enabling precise targeting of the treatment, and preventing disruption of the patient’s normal microbiota. Second, phages are self-replicating, such that phage replication can significantly increase phage abundance at the site of the bacterial infection. Third, phages use a different mechanism of action to kill bacteria than chemical antibiotics, and so are virulent against antibiotic resistant bacteria. Fourth, few side effects have been reported during or after phage application, including immune responses, suggesting that higher organisms are adapted to the presence of phages within tissues or on body surfaces. In addition, the high abundance of phages in the environment make them relatively simple to isolate. As well, phages are relatively easy to manipulate through molecular approaches to create therapeutically enhanced phages which exhibit improved performance in vivo, and phage biology is becoming better understood such that the development of new therapeutic phages will progress more rapidly than it has in the past. Ideally, a therapeutic cocktail of multiple phages targeting several bacterial strains with different phage-receptors will be developed to increase the host range of the cocktail and protect against receptor-mutation mediated resistance (Burrowes et al., 2011; Golkar et al., 2014).
Twenty phages specific to S. maltophilia have been isolated and characterized to date. Eleven phages (Sm1, IME13, IME15, S3, Sm14, ΦSMA5, DLP1, DLP2, DLP4, DLP5, DLP6) have been isolated and characterized specifically for their therapeutic potential. For example, the Myoviridae phage ΦSMA5 has an extensive host range successfully infecting 61 out of 87 strains tested (Chang et al., 2005), while Sm1 is the first S. maltophilia phage used in a murine model where it was found to provide 100% protection against S. maltophilia infection. The broad host range of ΦSMA5 and significant in vivo protection Sm1 provides in a murine model are promising examples of therapeutic phages that can be used for the treatment of antibiotic resistant S. maltophilia infections.
Initial phage isolation was accomplished with five clinical S. maltophilia strains (D1585, D1571, D1614, D1576, D1568) from the Canadian Burkholderia cepacia complex Research and Referral Repository (CBCCRRR; Vancouver, BC, Canada). An additional 22 S. maltophilia clinical isolates were obtained from the Provincial Laboratory for Public Health - North (Microbiology), Alberta Health Services for host range analysis. Strains SMDP92 and ATCC 13637 were gifted by Dr. Jorge Giron. Strains were grown aerobically at 30°C on Luria-Bertani solid media until single colonies were visible (16–36 h) or in LB broth with shaking at 225 RPM.
The phage DLP3 (vB_SmaS_DLP_3) was isolated from soil collected in Edmonton, Alberta, Canada using host strain S. maltophilia D1571. No plants were associated with the soil sample. Approximately 10 ml of soil was mixed with 10 ml of LB broth, 1 ml of modified suspension media (SM) (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 10 mM MgSO4) and 100 μl of a D1571 overnight culture. The slurry was incubated overnight at 30°C with shaking at 225 RPM. The supernatant was filter-sterilized with a Millex-HA 0.45 μm syringe-driven filter (Millipore, Billerica, MA, United States) and stored at 4°C, as performed previously (Peters et al., 2015, 2017, 2019). A single plaque was picked to propagate a working-stock solution for analysis using top-agar overlays. Briefly, 100 μl of overnight D1571 culture and 100 μl DLP3 stock (∼109 PFU/ml) were mixed and incubated for 5 min at room temperature then added to 3 ml of 0.7% LB top agar. The mixture was poured onto an LB plate and incubated for 18 h at 30°C. The top agar of plates showing confluent lysis was scraped into a 50 ml Falcon tube. A 3 ml aliquot of SM was added for each plate scraped and the slurry was shaken for 1 min followed by centrifugation (5 min at 10,000 × g) and filter sterilization. Host range analysis was performed on 29 S. maltophilia and 19 Pseudomonas aeruginosa strains using serially diluted DLP3 lysate into SM. A 10 μl aliquot of each concentration was spotted in triplicate onto a plate containing bacterial culture in a top-agar overlay and incubated overnight at 30°C.
Phage lysate for electron microscopy was prepared using LB plates and top agar made with agarose and filter sterilized using a 0.22 μm filter. A carbon-coated copper grid was overlaid with 10 μl of phage lysate for 2 min then stained with 4% uranyl acetate for 30 s. A Philips/FEI (Morgagni) transmission electron microscope (TEM) with charge-coupled device camera at 80 kV (University of Alberta Department of Biological Sciences Advanced Microscopy Facility) was used to obtain TEM images. The capsid diameter, tail length and tail width of ten virions were measured using ImageJ and averages calculated using Microsoft Excel.
Genomic DNA was isolated from a high-titer DLP3 stock (109 PFU/ml). Lysate was clarified by spinning 10,000 × g for 10 min and the supernatant was subsequently treated with 100 μl 100x DNase I buffer (1 M Tris–HCl, 0.25 M MgCl2, 10 mM CaCl2), 10 μl DNase I (Thermo Scientific, Waltham, MA, United States), 6 μl RNase (Thermo Scientific) and incubated 1 h at 37°C. A 400 μl aliquot of 0.5 M EDTA (pH 8.0), SDS (final concentration of 2%) and Proteinase K (final concentration of 400 μg/ml) was added followed by incubation at 55°C overnight. A 1/2 volume of 6 M NaCl was added and the solution was vortexed at high speed for 30 s followed by centrifugation at 17,900 × g for 30 min. The supernatant was transferred to a fresh tube with an equal volume of 100% isopropanol and stored at −20°C for at least 1 h to overnight. The DNA was pelleted with centrifugation at 17,900 × g for 20 min at 4°C followed by three 70% ethanol washes. The pellet was dried at room temperature and resuspended in nuclease-free water. Purity and concentrations of eluted DNA were checked with a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Waltham, MA, United States). DLP3 genomic (gDNA) DNA was sequenced using both Illumina and Pacific Biosciences technologies. A Nextera XT library was generated for paired-end sequencing on MiSeq (Illumina) platform using MiSeq v2 reagent kit. Restriction fragment length polymorphism (RFLP) analysis was used with 15 FastDigest (Thermo Scientific) restriction enzymes: EcoRI, XbaI, BamHI, HindIII, KpnI, SmaI, SphI, PstI, SacI, SaII, ApaI, ClaI, NdeI, SpeI, Xhol. Restriction reactions were set up using 1 μl FastDigest enzyme, 2 μl FastDigest restriction buffer, 1 μg of phage DNA and nuclease-free water to bring the final volume to 20 μl. Reactions were separated on a 0.8% (wt/vol) agarose gel in 1x TAE (pH 8.0).
A 96,852 bp contig assembled from the Illumina reads with SPAdes 3.8.0 was identified for further analysis. No gaps or ambiguous sites were found in the assembly, which has a mean coverage of 114 reads and Q40 of 93.8%. Prediction of open reading frames (ORFs) was accomplished with the GLIMMER plugin (Delcher et al., 2007) for Geneious (Kearse et al., 2012) using the Bacteria and Archaea setting, as well as GeneMarkS for phage (Besemer et al., 2001). Conserved domain searches were performed using CD-Search with the CDD v3.16 – 50369 PSSMs database (Marchler-Bauer et al., 2011). Phyre (Kelley et al., 2015), HHblits (Remmert et al., 2011; Zimmermann et al., 2018) and ITASSER (Yang and Zhang, 2015) were used to gain insights into possible functions of hypothetical proteins or to provide more support for putative functions. BLASTn and BLASTp were used to gain information on relatives based on genomic data and individual proteins, respectively (Altschul et al., 1997). The NCBI non-redundant protein sequence and nucleotide collection databases (update dates for both: 2018/08/26) were used for the BLASTp and BLASTn searches, respectively. BLASTp results above 1.00E-03 were annotated as hypothetical proteins. tRNAs were identified using the general tRNA model with tRNAscan-SE software (Schattner et al., 2005).
A pilT clean deletion mutant of S. maltophilia D1585 was constructed using overlap-extension PCR and allele exchange as previously described (McCutcheon et al., 2018). Briefly, regions upstream and downstream of the pilT gene in D1585 were PCR amplified using Phusion High-Fidelity DNA Polymerase (New England Biolabs) with primer pairs UpF-HindIII (GGGCAAGCTTCAGTACCTGCGGCTTCACTG) and UpR-OE (CTCGAACAGGCGCTTGGACGCTTTGTTCTTTACGG) for the upstream region, and DnF-OE (AAGAACAAAGCGTC CAAGCGCCTGTTCGAGTAAGG) and DnR-XbaI (GGGC TCTAGACTTCAGCTTGTGGATCTCGC) for the downstream region. Overlap regions are italicized and restriction enzyme recognition sites are bolded. Following overlap-extension PCR, the deletion cassette was ligated into pEX18Tc and this plasmid, pD1585ΔpilT, was transformed into E. coli S17-1 for bacterial mating with D1585. Single crossover transconjugants were selected on LB agar containing 100 μg/mL tetracycline and merodiploid status was confirmed by colony PCR with pilT specific primers pilTF (GTTCCGTTGAATCAGGAGGC) and pilTR (GAGGGCATGTACCAGGAAAC). Positive merodiploids were grown in LB broth and plated on LB with 10% sucrose to select for double crossover ΔpilT mutants that were confirmed by colony PCR as above. For complementation, the D1585 pilT gene was cloned into pBBR1MCS using pilTF and pilTR primers with tails for HindIII and XbaI restriction enzymes (Thermo Fisher) to create pD1585pilT.
DLP3 plaquing ability on wildtype and mutant strains of D1585 and 280 was determined by spot assay on bacterial lawns as previously described (McCutcheon et al., 2018). Briefly, bacterial strains were grown in 1/2 LB supplemented with 35 μg/mL chloramphenicol at 30°C with aeration at 225 RPM for 18 h. 100 μL of overnight culture was used in a top agar overlay containing 35 μg/mL chloramphenicol and allowed to solidify. DLP3 lysate standardized to 1010 PFU/mL on S. maltophilia D1571 was ten-fold serially diluted in SM to 103 PFU/mL and 5 μL of each dilution was spotted on the prepared plates in triplicate. Plates were incubated upright at 30°C and imaged after 16 h. Each experiment was repeated in biological and technical triplicate.
Isolation of DLP3 protein for SDS-PAGE analysis was accomplished following a protocol for the formation of ghost particles (Boulanger, 2009). Briefly, sterile DLP3 lysate (∼1 × 109) was clarified twice with 10,000 × g centrifugations and treated with nucleases following the DNA isolation protocol described above. After the incubation, an equal volume of 10 M LiCl was added and the solution was incubated at 46°C for 10 min, followed by 10-fold dilutions into sterile Milli-Q water. The released DLP3 gDNA was digested with an addition of 10 mM MgCl2 and 50 U of RNase-free DNaseI per 1 × 1012 PFU. This solution was incubated overnight at 37°C, followed by ultracentrifugation at 28,700 × g for 1.2 h. The supernatant was discarded and pellets were resuspended with 100 μl SM. An aliquot of the sample was diluted in half with 2x Laemmli sample buffer (10% [v/v] beta-mercaptoethanol [BME], 6% [w/v] SDS, 20% [v/v] glycerol, 0.2 mg/ml bromophenol blue) and incubated 10 min at 99°C.
An SDS-PAGE gel with a 4% stack and a 7.5% resolving portion was made with 40% 37.5:1 acrylamide/bis-acrylamide solution (Bio-Rad) and fresh 10% ammonium persulfate. The gel was loaded into a Mini-PROTEAN electrophoresis chamber (Bio-Rad) using 1x running buffer. A 6 μl aliquot of PageRuler Plus Prestained Protein Ladder (Thermo Scientific) was used as a molecular weight standard and 6–12 μl of DLP3 ghost particles in 1x sample buffer was loaded into the remaining wells. The gel was run at 180 kV for 75 min and placed in Coomassie R-250 stain for 1 h with gentle rocking. The gel was destained over 2 h, with the destaining solution replaced every 30 min. The gel was placed in a 50 ml Falcon tube with Milli-Q to transport the gel for mass spectrometry analysis at the Alberta Proteomics and Mass Spectrometry (APM) facility located at the University of Alberta.
In-gel trypsin digestion was performed on the samples. The lane was cut into seven equal gel sections, destained twice in 100 mM ammonium bicarbonate/acetonitrile (ACN) (50:50), reduced (10 mM BME–100 mM bicarbonate) and then alkylated (55 mM iodoacetamide–100 mM bicarbonate). After dehydration, trypsin digestion (6 ng/μl) was allowed to proceed overnight at room temperature. Tryptic peptides were extracted from the gel using 97% water–2% acetonitrile–1% formic acid followed by a second extraction using 50% of the initial extraction buffer and 50% acetonitrile. Fractions containing tryptic peptides were resolved and ionized using nanoflow high-performance liquid chromatography (HPLC) (Easy-nLC 1000; Thermo Scientific) coupled to a Q Exactive Orbitrap mass spectrometer (MS) (Thermo Scientific). Nanoflow chromatography and electrospray ionization were accomplished by using a Pico- Frit fused silica capillary column (ProteoPepII; C18) with a 100-μm inner diameter (New Objective) (300 Å, 5 μm pore size). Peptide mixtures were injected onto the column at a flow rate of 3,000 nl/min and resolved at 500 nl/min using 75-min linear gradients of 4% to 40% (vol/vol) aqueous ACN with 0.2% (vol/vol) formic acid. The mass spectrometer was operated in data-dependent acquisition mode, recording high-accuracy and high-resolution Orbitrap survey spectra using external mass calibration, with a resolution of 35,000 and m/z range of 400 to 2,000. The 15 most intensely multiply charged ions were sequentially fragmented by HCD fragmentation. After two fragmentations, all precursors selected for dissociation were dynamically excluded for 60 s. Data were processed using Proteome Discoverer 1.4 (Thermo Scientific). The UniProt Stenotrophomonas database and all DLP3 proteins were searched using SEQUEST (Thermo Scientific). Search parameters included a precursor mass tolerance of 10 ppm and a fragment mass tolerance of 0.8 Da. Peptides were searched with carbamidomethyl cysteine as a static modification and oxidized methionine and deamidated glutamine and asparagine as dynamic modifications.
Top agar overlay plates showing confluent lysis of D1571 by DLP3 were used to obtain resistant colonies. Briefly, 3 ml of SM was added to the plates and a sterile glass rod was used to gently skim the agar. The SM was collected and placed into microcentrifuge tubes, then centrifuged at 5,000 × g for 5 min. The supernatant was discarded and 1 ml of fresh SM was added to resuspend the pellet, followed by centrifugation at 5,000 × g for 5 min. This wash step was repeated three times in total. Following the final wash centrifugation, the supernatant was removed and the pellet was resuspended in 500 μl LB broth. Cells were serially diluted with LB and plated on LB plates, then incubated at 30°C for 16 h. Single colony isolates were selected for further study and tested for superinfection resistance using overnight cultures of every isolate in a top agar overlay assay with DLP3. After an 18 h incubation at 30°C, the plates were observed for plaque development. Single colony isolates without plaque development were retained for analysis.
Triplicate minimal inhibitory concentration (MIC) experiments on S. maltophilia strains D1571 and D1571:DLP3 were conducted using established protocols (Wiegand et al., 2008) to study the functionality of the DLP3-encoded Erm. Overnight cultures were grown at 30°C in 5 mL LB. A 1:100 subculture was grown at 30°C to an OD600 of 0.1 in Mueller-Hinton broth (MH) and used in 96 well plates containing an erythromycin dilution series (MP Biomedicals). Following a 16 h incubation, OD600 absorbance was obtained using a Wallac 1420 VICTOR2 multilabel counter (PerkinElmer, Waltham, MA, United States) and values were averaged using Excel. Statistical analysis was conducted using GraphPad Prism 7 (GraphPad Software Inc., San Diego, CA, United States) to perform a two-way analysis of variance (ANOVA) with Sidak’s multiple comparisons.
Single colony triplicate overnight cultures of wild type D1571 and the lysogen D1571:DLP3 were grown in LB broth at 30°C with shaking. Subcultures (1:100) for each sample were performed using LB broth and each subculture was grown to an OD600 of ∼0.32 at 30°C with 225 RPM shaking. Subcultures were distributed in triplicate aliquots of 200 μl in 96 well plates; an LB broth control was included for each plate. The OD600 was then obtained for each plate using a Wallac 1420 VICTOR2 multilabel counter (PerkinElmer, Waltham, MA) at the following time points: 0, 2, 4, 6, 8 h. The OD600 data were used to determine the growth rate (μ) with the established formula: log10 N − log10 N0 = (μ/2.303) (t − t0), whereby N0 is the time zero (t0) OD600 reading and N is the final OD600 reading obtained at a specific time (t) in the experiment. Resulting data were analyzed with GraphPad Prism 7 (GraphPad Software Inc., San Diego, CA, United States) to graph the growth curve and growth rate. Statistical analysis of the growth curve and growth rate data were performed in GraphPad Prism 8 using two-way repeated measurement ANOVA with multiple comparisons (Sidak’s).
G. mellonella infections were performed as previously described, with modifications (Seed and Dennis, 2008; Kamal et al., 2019). Single colony triplicate overnight cultures of wildtype strain D1571 and the DLP3 lysogen, D1571:DLP3, were grown aerobically at 37°C in LB for 19 h corresponding to approximately 2 × 1010CFU/mL. Cultures were standardized by OD600, washed once in 1× phosphate-buffered saline (PBS, pH 7.4) and serially diluted tenfold in PBS. G. mellonella larvae were bred in-house at 30°C using artificial food (wheat germ: 264 g, brewer’s yeast: 132 g, beeswax: 210 g, glycerol: 132 g, honey: 132 g, water: 66 g) and larvae weighing approximately 250 mg were selected for experiments. Each experiment consisted of ten larvae per group and 5 μL aliquots of bacterial culture were injected into the rear left proleg of each larva using a 250 μL Hamilton syringe fitted with a repeating dispenser. Sterile PBS injected larvae were used as negative controls and showed 100% survival for the duration of the experiment. Colony counts on LB agar were used to determine the CFUs injected. Following injection, larvae were placed in a static incubator in the dark at 37°C and scored for death every 24 h until 120 h post-infection (hpi). Larvae were considered dead when they did not respond to touch with movement.
For DLP3 phage rescue trials, wildtype D1571 culture was prepared as described above and an inoculum of approximately 8 × 106CFU/larvae was chosen. DLP3 lysate was used at 8.9 × 1010 PFU/mL. Larvae were injected with 10 μL of DLP3 lysate dilutions to give MOIs of approximately 100 and 50 into the rear right proleg 1.5 hpi with D1571. Aliquots of 5 μL PBS and 10 μL SM were used in place of bacteria and phage, respectively, for negative controls. Each worm was therefore injected with 15 μL total volume. Larvae were incubated and scored for survival as above. Results from three separate trials were combined and survival at each timepoint was plotted using the Kaplan–Meier method with error bars for standard error using GraphPad Prism 8. Statistical analysis of survival differences was completed using the Log-rank (Mantel–Cox) test.
Bacteriophage DLP3 (vB_SmaS-DLP_3) was isolated from soil using clinical S. maltophilia strain D1571. Transmission electron microscopy (TEM) enabled the classification of DLP3 as a Siphoviridae of the B1 morphotype (Ackermann and Eisenstark, 1974) due to the long, non-contractile tail averaging 202.2 ± 5.7 nm and isometric capsid with a length and width of 92.8 ± 4.1 and 84.0 ± 2.8 nm, respectively (Figure 1). No tail fibers were observed in the TEM images. The host range of DLP3 against all 29 clinical S. maltophilia isolates reveals a broad tropism through the successful infection of 22 strains, although DLP3 is not capable of infecting the P. aeruginosa strains tested (Table 1). The restriction fragment length polymorphism analysis revealed DLP3 genomic DNA is resistant to the 15 restriction enzymes screened. These results suggest DLP3 contains modified DNA, although the exact types of modifications are currently undetermined.
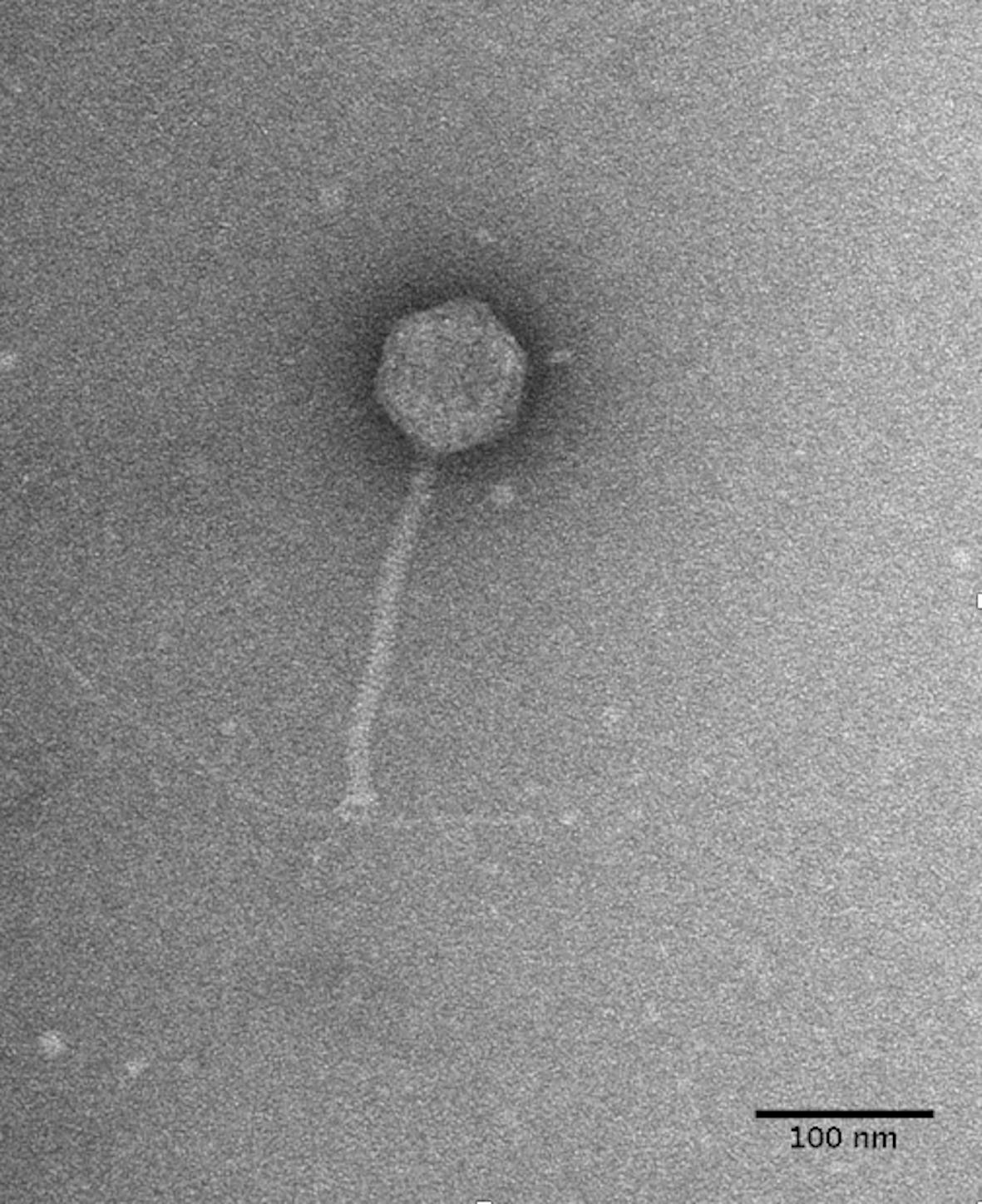
Figure 1. DLP3 Siphoviridae morphology. Phage lysate was applied to a carbon-coated copper grid and stained with 4% uranyl acetate. Transmission electron micrographs were obtained at 180,000× magnification. A S. maltophilia D1571 pili is shown to be interacting with the baseplate portion of the DLP3 tail. The averaged measurements for tail length, capsid length and width from ten virions is 202, 92, and 84 nm, respectively.

Table 1. Host range analysis for S. maltophilia bacteriophage DLP3 on S. maltophilia and P. aeruginosa strains.
The DLP3 genome is 96,852 bp long with a 58.3% global GC content. No low coverage or ambiguous regions were identified with the assembled contig, which has a mean coverage of 114 and a Q40 of 99.6%. NCBI non-redundant protein sequence database searches indicate that DLP3 shares a high identity to the Siphoviridae phage vB_SmaS_DLP_5 (DLP5) (Peters and Dennis, 2018). DLP5 is the type species of the new genus Delepquintavirus and based on the genomic similarities between DLP3 and DLP5, DLP3 is also a member of this new genus. Open reading frame calling with Glimmer and GeneMarkS identified a total of 148 protein coding domain sequences (CDS) covering 95% of the genome (Figure 2 and Table 2). DLP3 also encodes five tRNA genes with different specificities: Tyr (GTA), Sup (CTA), Ser (GCT), Ile (GAT), Glu (TTC). A total of 97 proteins could not be assigned functions due to lack of significant results from both BLASTp and CD-Search. The DLP3 genome with putative gene annotations has been deposited in GenBank under accession number MT110073.
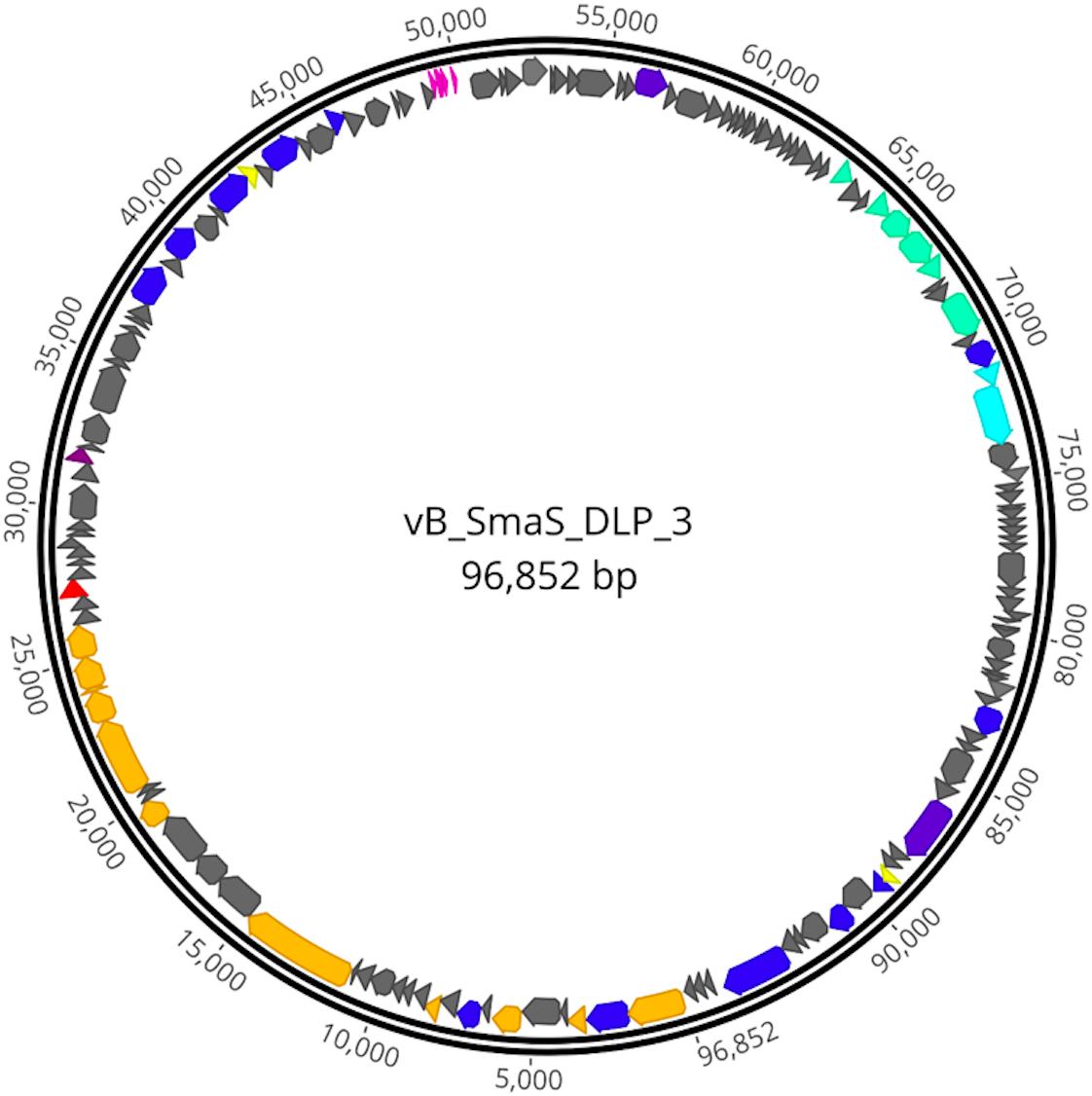
Figure 2. Genome map of DLP3. Scale in bp is shown on the outer periphery. Predicted functions are grouped by color: teal; moron, gray; hypothetical, light blue; DNA packaging, pink; tRNA, red; lysis, green; virion morphogenesis, dark blue; DNA replication and repair, purple; auxiliary metabolism, and yellow; regulatory.
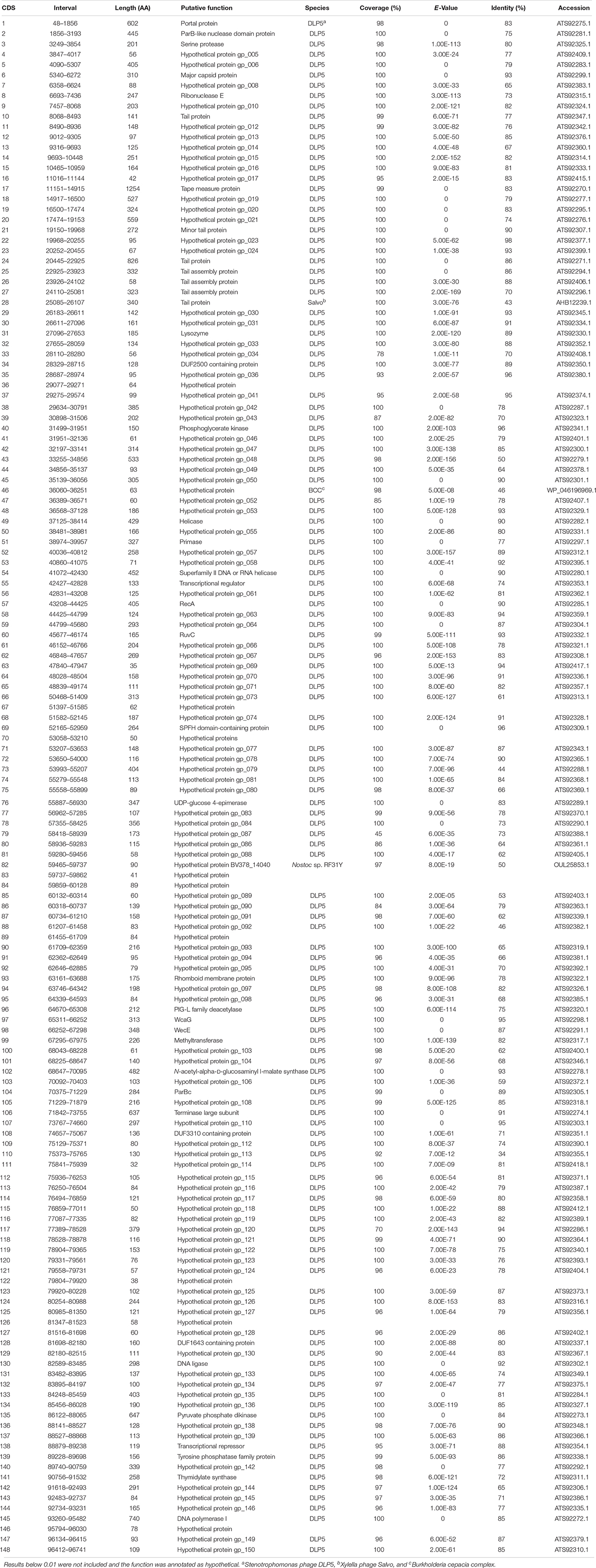
Table 2. Genome annotations for DLP3 obtained from BLASTp and CD-Search data.
The CD-Search did yield 37 DLP3 proteins with conserved domains predicted (Table 3). Three of the domains identified are domains of unknown function (DUF) which tended to be distributed throughout Gram-negative bacteria and to a lesser extent in Gram-positive bacteria, according to the species distribution for each DUF in pfam (Finn et al., 2014). There are six conserved domains (CD) identified which are involved in virion morphogenesis: phage portal protein superfamily, gp1; phage capsid family, gp6; phage tail proteins, gp10 and gp24; laminin G, gp28; and tape measure protein domain, gp17. Nine proteins with domains involved in DNA replication and repair identified with the CD-Search include two ParB domains, gp2 and 104; two helicases, gp49 and 54; Holliday junction resolvase, gp60; RecA recombinase, gp57; topoisomerase primase, gp51; DNA ligase, gp130; and DNA polymerase A, gp145. The remaining CD results appear to be quite diverse in their functions such as the SpoVK family domain involved in sporulation (gp38) (Fan et al., 1992), protein-tyrosine phosphatases (gp139 and gp141) typically involved in signal transduction (Whitmore and Lamont, 2012), and a membrane-associated serine protease of the rhomboid family (gp93). One CD identified that is of particular interest is the glycosyltransferase domain of gp102. T-even bacteriophages have been shown to use glycosyltransferases for DNA modification by linking a glycosyl group to hydroxymethyl-cytosine, thus protecting the DNA against digestion by bacterial restriction systems (Bair and Black, 2007). Another role for glycosyltransferases within bacteriophages is highlighted by some Shigella phages which have been shown to seroconvert their host by modifying the O-antigen polysaccharides to prevent infection of the bacteria by other O-antigen receptor phages (Allison and Verma, 2000). The specific glycosyltransferase family is RfaB, a protein involved in the assembly of the LPS core of Escherichia coli K-12 (Pradel et al., 1992). This result suggests DLP3 may use the glycosyltransferase to modify the host LPS, similar to the seroconverting Shigella phages, though this has yet to be confirmed experimentally.
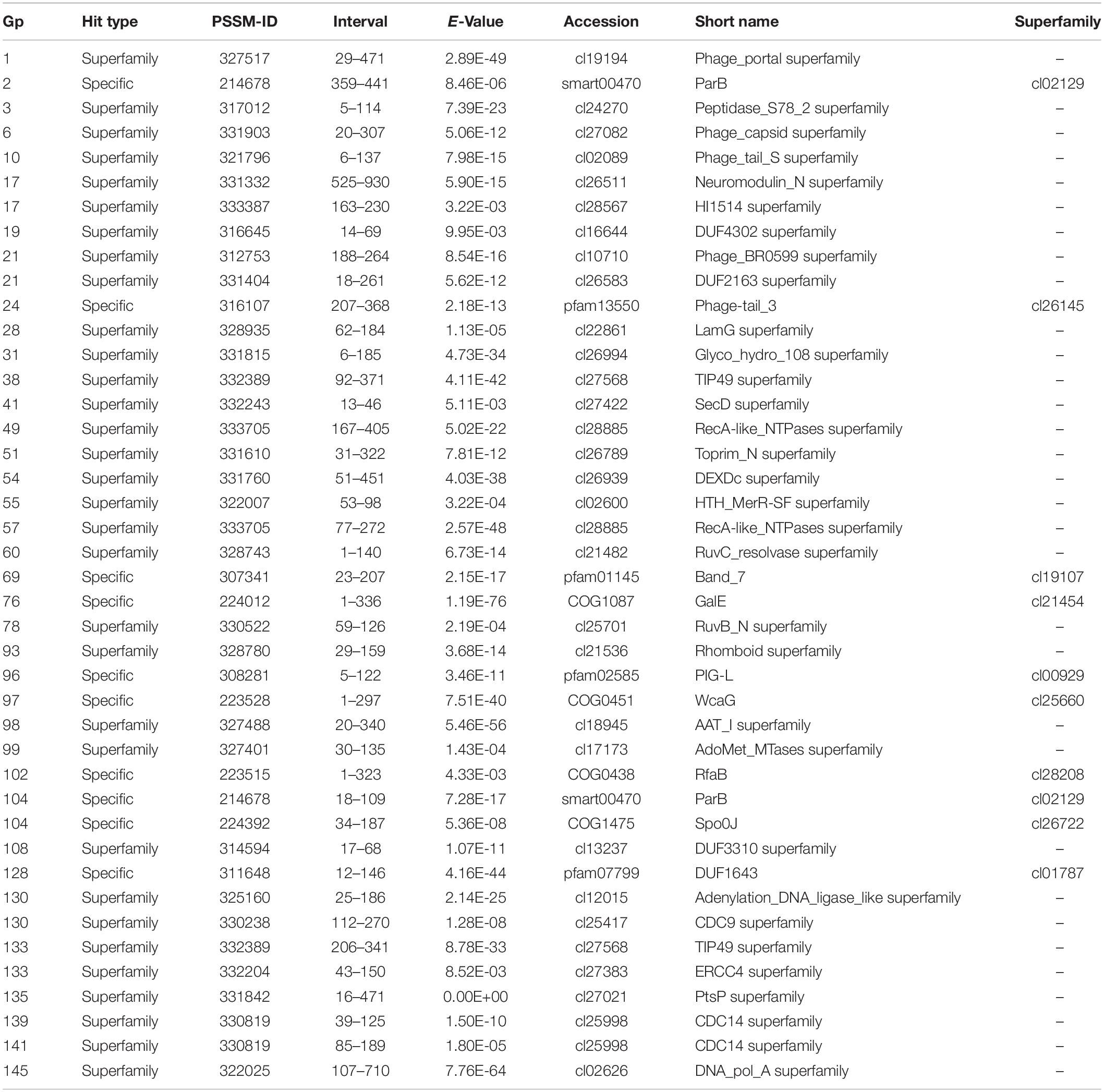
Table 3. The conserved domains found in the 148 DLP3 gene products.
While characterizing the Siphoviridae phage DLP3 genome, it was evident through BLASTn and BLASTp searches that phages DLP3 and DLP5 are closely related. The genome size of DLP3 and DLP5 are similar (96,852 versus 96,542 bp), as is their GC content (58.3 versus 58.4%). BLASTn analysis of DLP3 against DLP5 revealed 81% identity over 90% of the DLP5 genome (0.0 E-value). A LASTZ alignment (Harris, 2007) of the phages using DLP5 as the reference sequence shows sequence identity >30% between their genomes, represented as a mustard yellow color in the consensus (Figure 3). BLASTn alignment between DLP3 to DLP5 gives a query coverage of 89% and 81% identity. Two small stretches with a breakdown in identity are observed between the DLP5 and DLP3 genomes, which is viewed as a breakdown in the alignment blocks of the dot plot around 30,000 and 60,000 bp (Figure 3). These stretches correspond to four genes (DLP05_037 to DLP05_039, plus DLP05_087) encoding hypothetical proteins with no significant matches in the NCBI database, no conserved domains, and no significant results when using structural prediction software such as HHpred and Phyre. Two additional genes, DLP05_045 and DLP05_068, which encode hypothetical proteins, are not present in the DLP3 genome. The DLP05_045 gene product shares 93% coverage with 36% identity (1.0E-08) to a Polaromonas naphthalenivorans hypothetical protein which contains no conserved domains, whereas the DLP05_068 gene product did not have similar sequences in the NCBI database or conserved domains predicted. Both phages encode five tRNAs, with four of the five tRNAs sharing the same specificity: Sup-CTA, Glu-TTC, Ser-GCT, Tyr-GTA. Phage DLP3 differs from DLP5 with respect to the fifth tRNA, which is Ile-GAT in DLP3 but Gly-TCC in DLP5. The amino acid usage of each phage does not explain the differences, as they each have the same usage rates for isoleucine (4.7%) and glycine (7.7%), but there are several nucleotide changes observed in this region when comparing DLP3 to DLP5.
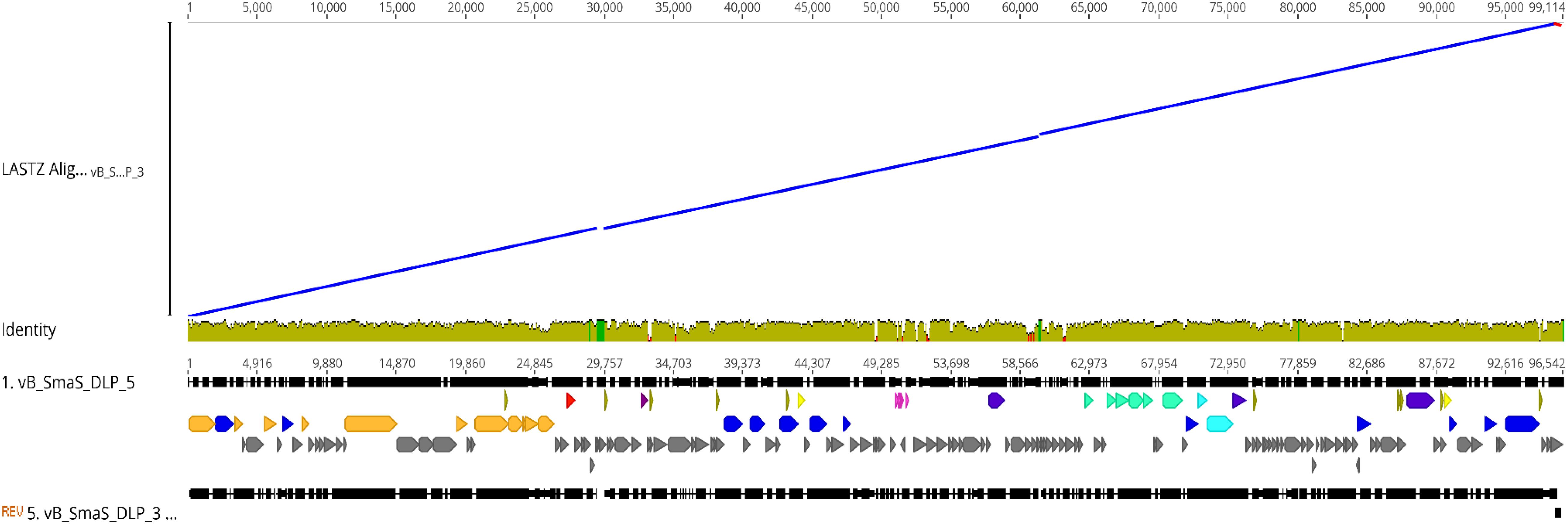
Figure 3. Genomic alignment of DLP3 to DLP5 using the Large-Scale Genome Alignment Tool. Identity is indicated by color: mustard yellow; >30%, green; deletion in DLP3, red; deletion in DLP5.
Besides the genomic similarities observed between DLP3 and DLP5, they also share morphological similarities. The DLP3 and DLP5 phages have the same measurement averages for their head width (84 nm) and length (92 nm), as well as tail length (202 nm). The host range of DLP3 versus DLP5 is significantly different, with DLP3 exhibiting a broad host range infecting 22 S. maltophilia strains, whereas DLP5 only infects six (Table 1). Of the six strains DLP5 is capable of infecting (102, 249, D1571, D1614, D1576, SMDP92), DLP3 is only unable to infect the DLP5 host strain D1614. The virion morphogenesis proteins of DLP3 range in sequence identity to the DLP5 equivalents from 43.8% (gp28) to 93.2% (gp6), with all but gp28 having sequence identity greater than 70%. The gp28 proteins of DLP3 and DLP5 are related to the Xylella phage Sano tail fiber protein. The annotation of tail fiber for the Sano protein was based solely on synteny to the Burkholderia phage BcepNazgul255. There are no shared conserved domains identified between these two proteins, and the Bcep Nazgul protein is 1,270 amino acids long whereas the DLP3 and DLP5 gp28 proteins and the Sano tail fiber protein are only approximately 340 amino acids long; thus, we consider the assignment of tail fiber function to the Sano phage protein to be incorrect.
The gp28 proteins of DLP3 and DLP5 may play a role in host range due to the variability observed between these proteins. The DLP5 gp28 protein shares 62.5% identity with the Sano tail fiber, while DLP3 only has 39.6% identity to the Sano protein and 41.8% identity to the DLP5 protein. The first 242 N-terminal amino acids of the DLP3 and DLP5 gp28 consensus show a high degree of variability at 24.5% pairwise identity. The remaining 104 amino acids from 243 to the C-terminal have high pairwise identity at 88.5%. Variability in the N-terminal region was also observed with a MUSCLE alignment using all three proteins from DLP3, DLP5 and Sano (Supplementary Figure S1). The first 258 N-terminal amino acids of the protein consensus share only 35.4% pairwise identity, which increases to 77.8% over the remaining 104 amino acids. The alignment shows at least nine insertion/deletion events have occurred within the DLP3 gene resulting in gaps and insertions in the translated DLP3 protein compared to the Sano and DLP5 proteins. Further investigation into gp28 of DLP3 and DLP5 as a host recognition protein is currently underway to help elucidate whether this protein plays a role in the host range of the Delepquintavirus phages.
Three Siphoviridae phages, DLP1, DLP2 and DLP4, previously isolated on S. maltophilia strain D1585, were found to bind the type IV pilus as their cell surface receptor across their host ranges (McCutcheon et al., 2018; Peters et al., 2019). Despite isolating DLP3 on strain D1571, we assessed DLP3 plaquing ability on the previously constructed D1585 and 280 ΔpilA mutants lacking the major pilin subunit. Similar to the three previously characterized phages, S. maltophilia strains lacking type IV pili are also resistant to infection by DLP3, shown by an absence of cell lysis at high titer (Figure 4). Complementation with the endogenous genes restores phage infection to wildtype levels. Unfortunately, the pilA gene is undetectable in the incompletely assembled D1571 genome, which prevented construction of pili mutants in this background. In contrast to host range data collected when DLP3 was initially isolated from the environment (Table 1), DLP3 efficiency of plating on D1585 and 280 is much lower than initially documented, showing phage activity at 109 PFU/mL compared to plaque formation at 105 PFU/mL (Figure 4). We attribute this reduced plaquing ability on some hosts to the repeated propagation of DLP3 on strain D1571. It is hypothesized that propagating DLP3 on a single host has selected for phages optimized to that host over time, resulting in the differences observed.
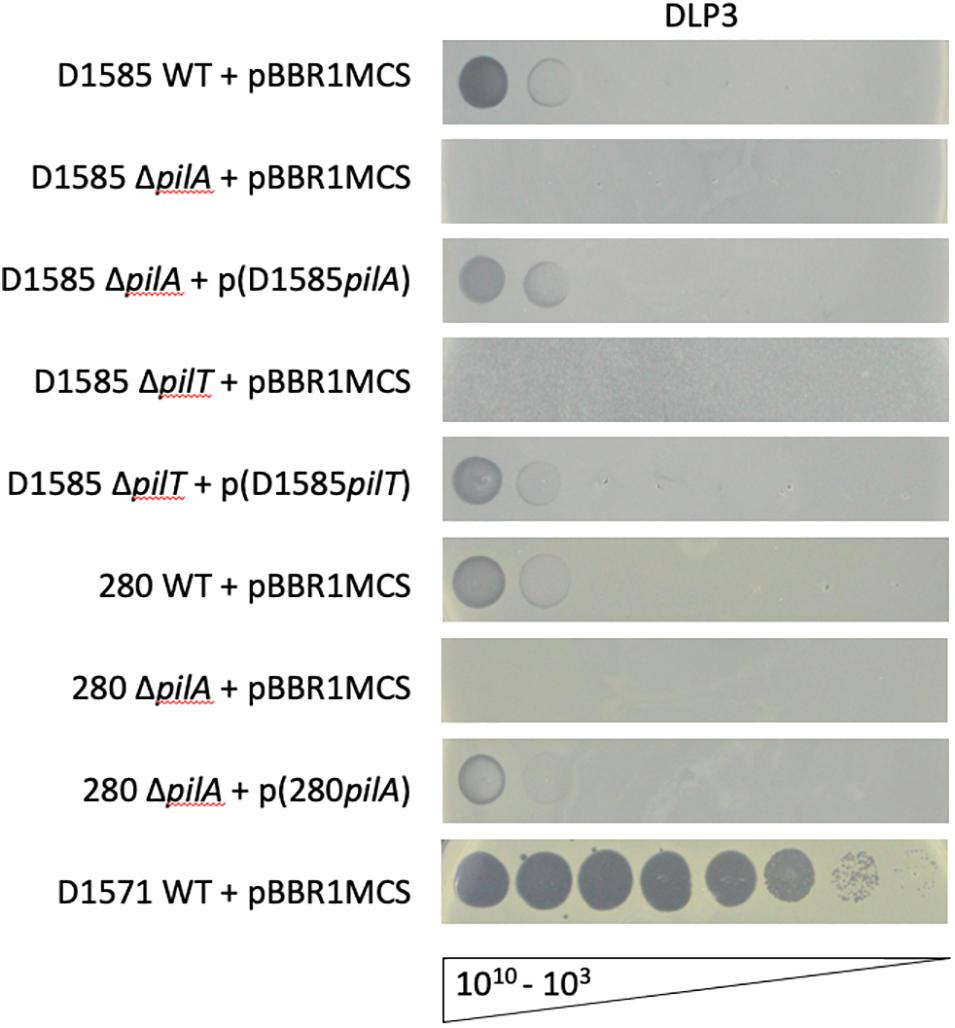
Figure 4. DLP3 requires a functional type IV pilus to infect host strains D1585 and 280. Phage DLP3 is capable of infecting wildtype S. maltophilia strains D1585 and 280, infecting both at 109 PFU/mL, whereas phage infection is blocked in the ΔpilA and ΔpilT mutants. Complementation with the endogenous genes restores phage infection to wildtype levels. DLP3 plaquing ability on strain D1571 is shown for titer calculation, however, no mutants were constructed in this background. Images are representative of three biological replicates, each with three technical replicates.
An additional mutant was constructed in strain D1585, ΔpilT, lacking the retraction ATPase required for depolymerization of the pilus (Burrows, 2005). This mutation results in hyperpiliated, non-motile cells having numerous non-functional pili projecting from the cell surface. Similar to the ΔpilA mutants, D1585 ΔpilT is resistant to DLP3 phage infection. This strain grows poorly in liquid, as observed in the speckled overlay lawn, however, this phenotype, as well as susceptibility to phage, is restored by complementation with the pilT gene (Figure 4). These results indicate that DLP3 uses the type IV pilus as a cell surface receptor and requires a functional pilus capable of retraction to reach the cell surface for successful host infection. This is the fourth documented S. maltophilia phage to use the type IV pilus as its receptor, however, we have yet to identify the receptor for the highly similar phage DLP5. Minor differences in the genomes of these two phages likely explains the significant differences observed in phage host range due to the binding of different receptors.
To further investigate DLP3 morphology, phage structural proteins were analyzed by HPLC-MS and screened against DLP3 proteins and the Stenotrophomonas database in UniProt. The SEQUEST results from searching all DLP3 proteins identified 21 (Table 4), though only 11 proteins were classified as virion morphogenesis using BLASTp and CD-Search data (Supplementary Figure S2). The most abundant protein isolated was the major capsid protein (gp6), which is the main structural component of a bacteriophage capsid (Hendrix and Johnson, 2012). The second most abundant protein found was the portal protein (gp1), which forms the entry site for phage DNA to be packaged into the capsid by the large terminase. The portal protein also functions like a DNA sensor, measuring the amount of DNA packaged into the capsid and signaling the large terminase to end genome-packaging once full (Lokareddy et al., 2017).
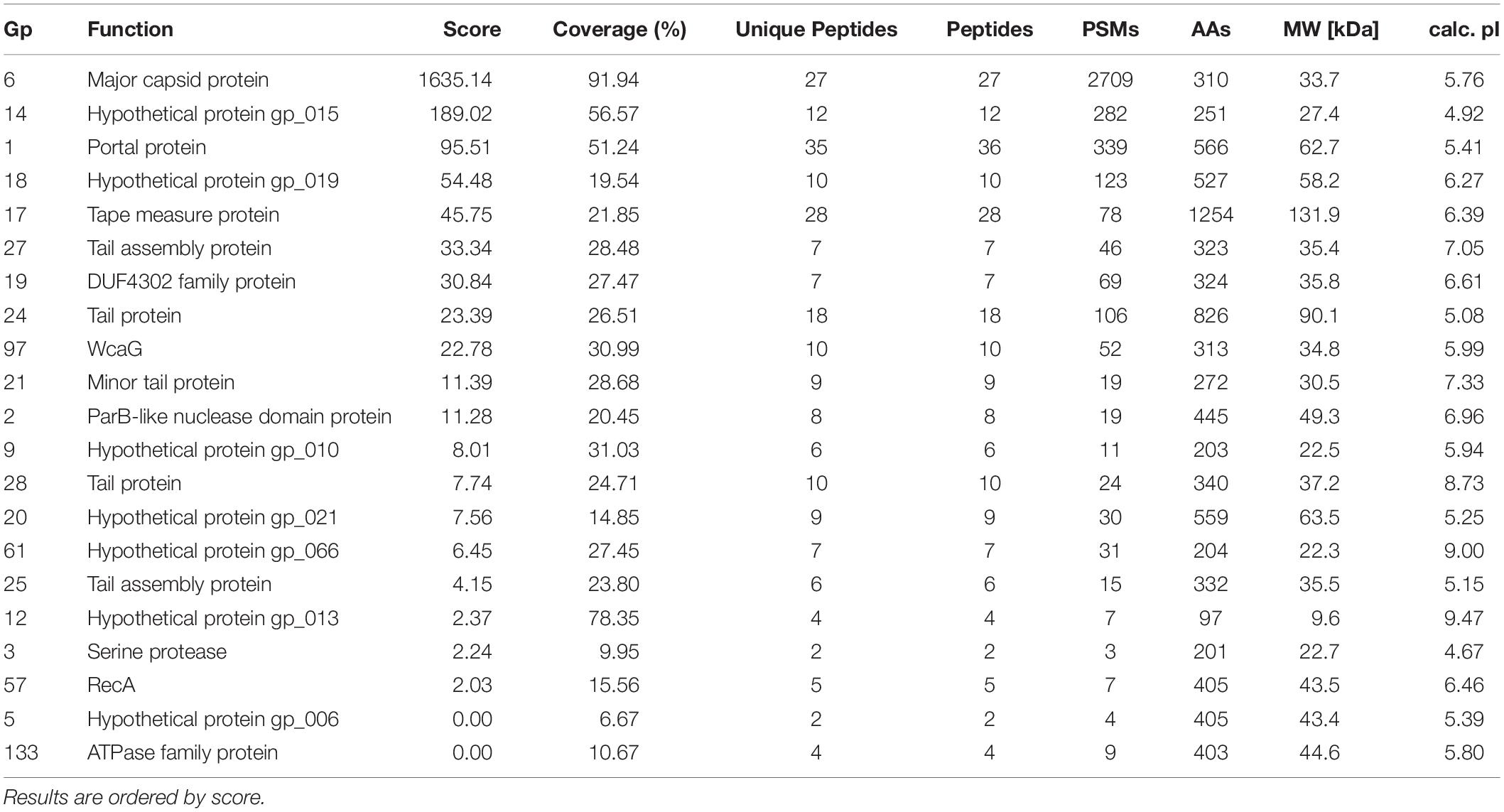
Table 4. Mass spectrometry protein results with the DLP3 protein database.
Gene products gp14 and gp18 were also identified. Both gene products are hypothetical proteins without conserved domains. Also, they do not have significant results using HHpred; therefore, their structural function is unknown (Table 4). Phyre2 analysis of these proteins only suggested a putative function for gp14, which showed structural identity to the major tropism determinant P1 (MTD-P1) of Bordetella phage BPP-1 (38.7% confidence, 25% identity). The MTD-P1 protein in BPP-1 is responsible for phage receptor binding (Miller et al., 2008) and uses specific variable residues for target recognition similar to an antibody (Miller, 2006). Another abundant protein identified is gp24, which is predicted to be a structural component of the DLP3 tail with a phage-tail_3 family (PF13550) conserved domain. The tape measure protein (gp17), which is responsible for setting the tail length (Belcaid et al., 2011), is the largest protein identified and the sixth most abundant, though the ∼130 kDa band was faint after fully destaining (Supplementary Figure S2). The remaining DLP3 proteins identified and ordered by relative abundance are gene products gp19, gp97, gp27, gp61, gp20, gp28, gp2, gp21, gp25, gp9, gp133, gp12, gp57, gp5, and gp3. Some of the proteins identified are surprising, such as gp2 (Par-B like nuclease domain protein) and gp5 (RecA), but based on the results obtained by screening the Stenotrophomonas protein database, it is evident that there were proteins present from the bacterial cell; thus, there may also be DLP3 proteins present that are not structural in function (Table 4 and Supplementary Table S2).
Stable DLP3 lysogens of S. maltophilia strain D1571 have been isolated and due to the presence of ParB domains in two of the DLP3 proteins, DLP3 may lysogenize its host as a phagemid. The closely related phage DLP5 was also found to encode two proteins with ParB domains and was successfully isolated as a phagemid from the lysogenized strain D1614 (Peters and Dennis, 2018). Blastn and PHAST analysis (Altschul et al., 1997; Zhou et al., 2011) of GenBank-deposited Stenotrophomonas genomes indicates that phage DLP3 (or phages closely related to DLP3) has also lysogenized S. maltophilia strains ABB550, FDAARGOS_325 and ICU331. Phage DLP5 has been previously shown to cause lysogenic conversion of S. maltophilia strain D1571 (Peters and Dennis, 2018). Therefore, we sought to further investigate the characteristics of the D1571:DLP3 lysogen relative to wildtype S. maltophilia strain D1571.
Annotation of the DLP3 genome revealed the presence of a methyltransferase domain in the gene product encoded by DLP3_099. A specific domain identified is the AdoMet_MTases superfamily comprised of class I S-adenosylmethionine-dependent methyltransferases. The class I family is the largest and most diverse, with members targeting a range of substrate specificities such as small molecules, lipids, nucleic acids, and different target atoms for methylation (Struck et al., 2012). Members of this family are known to maintain structural similarity even when the amino acid sequence diverges to as little as 10% (Schubert et al., 2003). Further investigation into gp69 with HHblits revealed high sequence identity to Erm(45) of Staphylococcus fleurettii (HHblits: 100% probability, 2.4e-104 E-value). Researchers identified Erm(45) as the enzyme responsible for increased erythromycin resistance in some strains of S. fleuretti (Wipf et al., 2015). The erm gene (erythromycin ribosome methylase) encodes a methylase enzyme that provides macrolide resistance by methylating the erythromycin target-site on the ribosome (Prunier et al., 2002). Together, these findings suggested that the DLP3 encoded methyltransferase is an erythromycin resistance protein similar to Erm(45) and could cause an increase in erythromycin resistance of the host cell during lysogeny.
To test the functionality of the DLP3 Erm protein, minimum inhibitory concentration (MIC) assays of the wild type D1571 and lysogen D1571:DLP3 were completed. The results show a statistically significant difference in erythromycin resistance with DLP3 lysogeny over a wide range of concentrations tested (Figure 5). There are differences noted between the wild type and lysogenized strains at lower concentrations of erythromycin, with wild type having a statistically significant increase in optical density at lower concentrations (6 and 34 μg/ml; P < 0.05, 12 and 23 μg/ml; P < 0.01). The cause of the decreased resistance in the lysogen noted at lower concentrations of erythromycin is currently unknown. This trend is reversed at higher concentrations of erythromycin, with the lysogen having significantly higher OD600 readings at 144 and 188 μg/ml erythromycin concentrations compared to wild type control (P < 0.0001). The increased resistance noted at higher erythromycin concentrations indicates lysogenic conversion of D1571 by lysogenized DLP3 and confirms the functionality of the predicted Erm protein. Fortuitously, S. maltophilia strains are already highly resistant toward macrolides, and macrolides such as erythromycin are not considered to be a frontline treatment option.
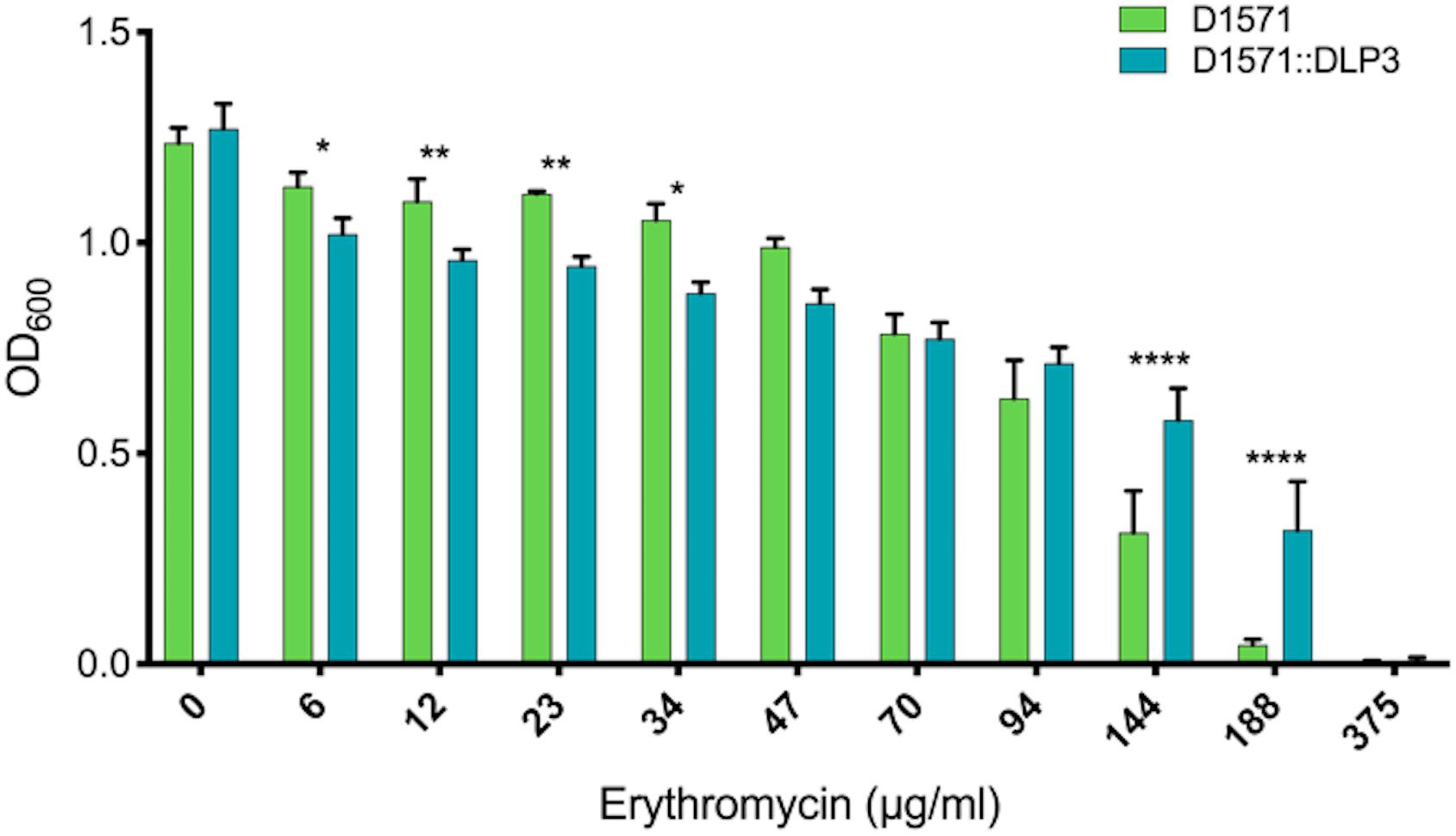
Figure 5. Erythromycin resistance of D1571:DLP3 lysogen increases compared to wild type control D1571. Minimum inhibitory concentration assay was completed in biological and mechanical triplicates. Two-way ANOVA with Sidak’s multiple comparisons test was performed on the MIC data. Statistical significance is represented as: ****P < 0.0001; **P < 0.01; and *P < 0.05.
While working with the D1571:DLP3 lysogen and wild type D1571 for the erythromycin resistance assay, it was evident that the lysogen exhibited a faster growth rate as compared to the wild type strain. This difference in growth rate was also observed when growing single colony isolates of the lysogen and wild type on an LB agar plate, with the lysogen having larger single colonies after a 16 h incubation at 30°C as compared to the wild type D1571 (data not shown). To investigate this phenotypic difference further, a growth curve was conducted in mechanical and biological triplicates for the lysogen and wild type over 8 h. The plotted growth curve clearly shows the lysogenized strain exhibiting a faster growth rate than the wild type strain, with significance found following a two-way repeated measures ANOVA with multiple comparisons at 2 h, P < 0.05; 4 h, P < 0.0001; and 6 h, P < 0.01 (Figure 6A). This observation was confirmed by converting the OD600 data into growth rate (μ) and plotting the resulting data (Figure 6B). A two-way repeated measures ANOVA with multiple comparisons revealed significant (P < 0.0001) differences between 0- to 2-h and 2- to 4-h time intervals for the lysogen and wild type D1571. The increased growth rate is only observed during the lag and early exponential phase of growth and disappears in the 4- to 6-h and 6- to 8-h growth rate intervals. The cause of the growth rate differences observed with DLP3 lysogenization is currently unknown, though DLP3 does encode many hypothetical and moron genes which may be expressed to produce this phenotype.
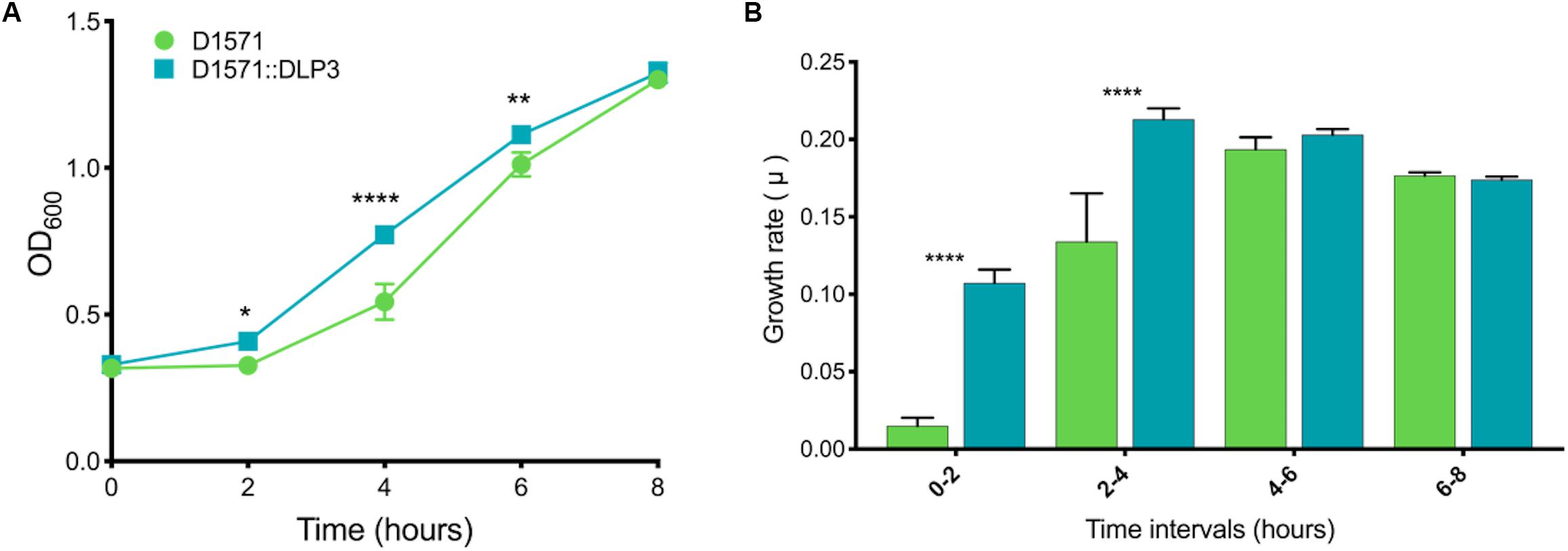
Figure 6. In vitro growth analysis of D1571 wild type and D1571:DLP3 lysogen. (A) Growth curve of wildtype D1571 compared to D1571:DLP3 strains grown in LB broth over 8 h. Results from biological and mechanical triplicate experiments were averaged and the mean plotted with the standard deviation represented as error bars. (B) Growth curve OD600 data was converted to the growth rate for wildtype and DLP3 lysogen with the mean plotted and error bars representing standard deviation. Two-way repeated measures ANOVA with multiple comparisons indicates statistical significance (*P < 0.05; **P < 0.01; ****P < 0.0001).
Based on the differing growth characteristics of D1571:DLP3 lysogen and the wildtype strain, we examined if the increased growth rate of the lysogen observed in vitro affected the virulence of strain D1571 in vivo using the G. mellonella larvae infection model. G. mellonella have been used as a non-mammalian eukaryotic model for assessing the virulence of many bacterial pathogens, including S. maltophilia (Betts et al., 2014; Tsai et al., 2016; Melloul et al., 2018), as well as a model to study the efficacy of novel antimicrobial compounds and phages (Seed and Dennis, 2009; Kamal and Dennis, 2015; Cutuli et al., 2019). Injection of G. mellonella larvae with S. maltophilia D1571 results in dose-dependent killing, with the lethal dose for this strain greater than 107 CFU per larva (Figure 7A). Coinciding with the faster growth rate observed in vitro, the D1571:DLP3 lysogen was more virulent than the wildtype D1571 strain, resulting in significantly lower survival for G. mellonella larvae injected with 107 (P < 0.01) or 106 (P < 0.001) CFU over 120 h (Figure 7A). This increased virulence may be due to the faster growth rate of D1571:DLP3, however, CFU were not recovered from the larvae following infection.
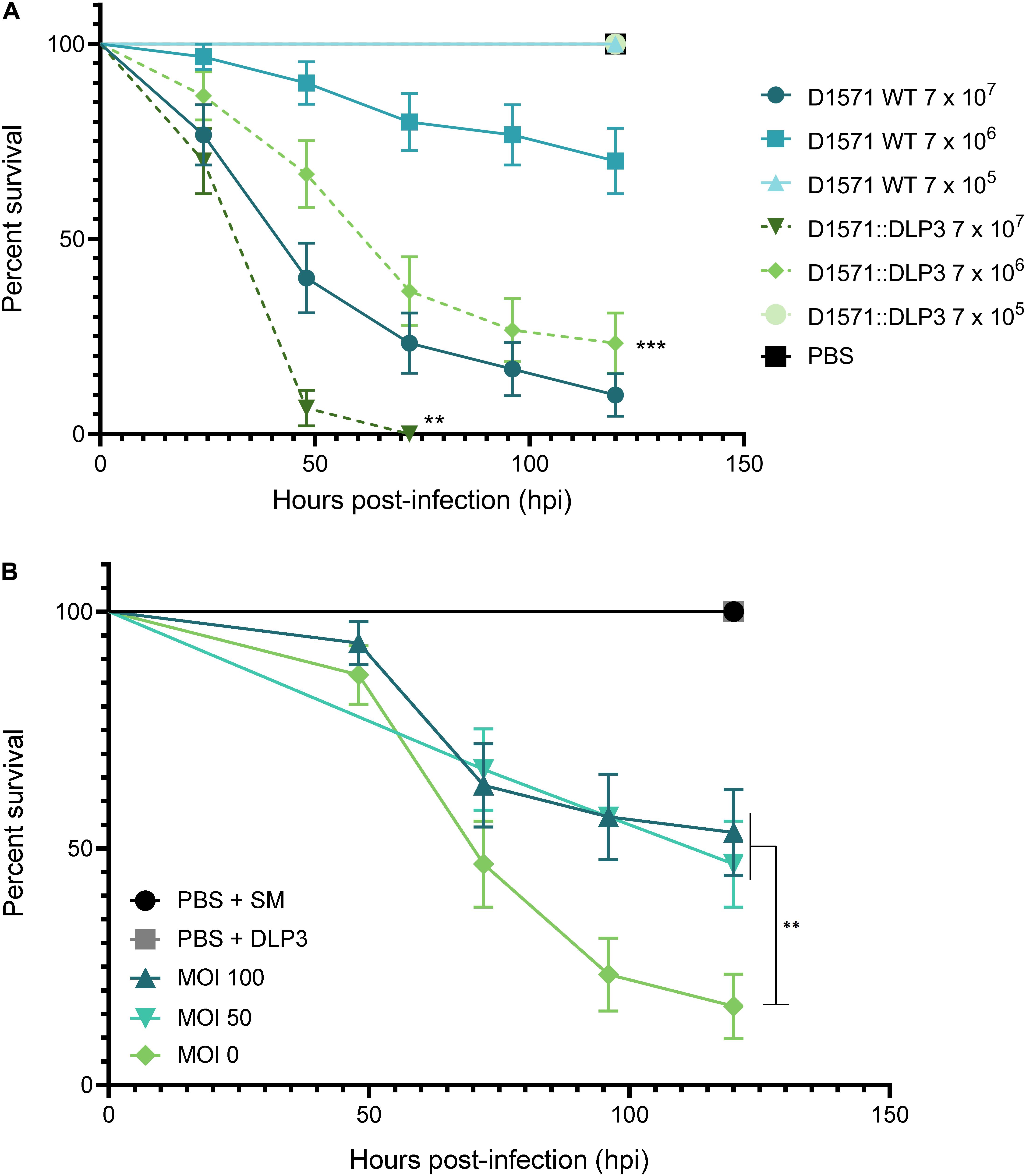
Figure 7. Effect of DLP3 on the virulence of D1571 in G. mellonella. (A) Survival of G. mellonella larvae over 120 h following infection with S. maltophilia D1571 wildtype or D1571:DLP3 lysogen at varying CFU. Larvae infected with D1571:DLP3 showed significantly lower survival at 107 (**P < 0.01) and 106 (***P < 0.001) CFU than larvae injected with the same inoculum of D1571 (log-rank test). (B) Survival of G. mellonella larvae injected with 8 × 108CFU of D1571 over 120 h treated with DLP3 at an MOI of 100 (8.9 × 108 PFU), 50 (4.5 × 108 PFU) or 0 at 1.5 h post-infection. For controls, sterile PBS and SM were used in place of bacteria and phage injections, respectively. A significant difference in survival was observed between untreated larvae (MOI 0) and either phage treatment (**P < 0.01; log-rank test). Ten larvae were injected per group and results were obtained from three separate trials and plotted using the Kaplan–Meier method with standard error bars.
Despite the DLP3 lysogen showing increased virulence in vivo, we sought to determine if DLP3 could rescue G. mellonella from wildtype D1571 infection. An inoculum of approximately 106 CFU per larva was chosen to allow for phage rescue at a multiplicity of infection of at least 100, as DLP3 does not propagate higher than 1010 PFU/mL. Larvae were injected with DLP3 lysate at 1.5 h post-infection with D1571 and survival monitored over 120 h. Compared to treatment with SM buffer, significantly more larvae survived when given phage at a MOI of 50 or 100 (Figure 7B, P < 0.01), with an average survival at 120 h of 53% or 47% for worms treated at an MOI of 100 or 50, respectively, compared to 17% survival of untreated larvae. Attempts to concentrate DLP3 to a titer greater than 1012 PFU/mL without causing melanization of larvae following phage injection were unsuccessful, however, we expect that treatment at a higher MOI would increase the survival of infected larvae. Increased survival may also occur with repeated phage injections over the course of the experiment, however, this was not tested. Overall, this preliminary investigation using G. mellonella indicates the potential of DLP3 as a therapeutic for the treatment of S. maltophilia infections.
The novel temperate phage DLP3 isolated from a soil sample enabled the identification of a second member of the newly established Delepquintavirus genus. DLP3 has a genome size of 96,852 bp and a GC content of 58.4%, which is significantly lower than the host strain D1571 which has a GC content of 66.9%. DLP3 encodes two proteins with ParB conserved domains enabling the stable lysogenization of the host strain D1571. Lysogenization by DLP3 leads to a growth rate increase during the lag and early exponential phase of growth for the host when compared to the wild type strain. DLP3 also encodes a functional Erm protein, allowing for the lysogenic conversion of the host D1571 strain which is observed though increased resistance to erythromycin at 144 and 188 μg/ml concentrations. Despite the temperate lifestyle of this phage, DLP3 is capable of lytic activity in vivo.
Like DLP3, many of the S. maltophilia temperate phages characterized to date feature moron genes that encode virulence factors or antibiotic resistance proteins that could cause lysogenic conversion of their host (Hagemann et al., 2006; Garcia et al., 2008; Peters and Dennis, 2018; Peters et al., 2019). Genetically modifying these phages by removing adverse genes, such as those encoding virulence factors or lysogeny proteins, will enable the creation of safe and highly targeted therapeutic agents (Lynch et al., 2010; Dedrick et al., 2019). Modified phages have been used in a case study against Mycobacterium abscessus (Dedrick et al., 2019) and a clinical trial (NCT04191148) sponsored by Locus Bioscience treating urinary tract infections with their modified crPhage cocktail. The genetically modified phage therapy examples set the precedence for other phages, such as DLP3, to be genetically modified and included in therapeutic phage cocktails. To this end, further study of DLP3 receptor binding proteins will possibly allow for the modification of other S. maltophilia phages showing therapeutic potential to target a wider host range.
The datasets generated for this study can be found in GenBank under accession number MT110073.
DP isolated phage DLP3, performed experiments to characterize the genome and viral particle, wrote the original manuscript draft and edited the manuscript. JM performed experiments to characterize the virus, wrote parts of the manuscript, and edited the manuscript. JD conceived of the research, directed the experiments, funded the research, and edited the manuscript. All authors contributed to the article and approved the submitted version.
This research was supported by operating funds to JD from the Natural Sciences and Engineering Research Council of Canada (NSERC-238414-2018). DP gratefully acknowledges Alberta Innovates AITF, and NSERC PGS-D graduate scholarships. JM was kindly supported by a CGS-D award from NSERC and an AIGSS award from Alberta Innovates.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Arlene Oatway from the University of Alberta Department of Biological Sciences Advanced Microscopy Facility for assistance with electron microscopy. The authors thank the Canadian Burkholderia cepacia complex Research and Referral Repository (CBCCRRR, Vancouver, BC, Canada) and The Provincial Laboratory for Public Health – North (Microbiology), Alberta Health Services, for gifts of the S. maltophilia strains used in this study. The authors thank Dr. Andrew Keddie from the University of Alberta for supplying the G. mellonella larvae used to establish their lab colony, and thank Dr. Keddie and Dr. Brent Weber for providing information and advice on breeding G. mellonella.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01358/full#supplementary-material
FIGURE S1 | MUSCLE multiple sequence alignment of gp28 proteins from phages DLP3 and DLP5, and tail fiber protein of Xylella phage Sano. Note the increased amino acid conservation toward the C-termini of the proteins versus the N-termini as observed through protein alignment.
FIGURE S2 | SDS-PAGE gel of DLP3 ghost particles (L) compared to a PageRuler Plus Pre-stained Protein Ladder (Thermo Scientific). Approximate masses are labeled on the right of the gel in kDa. Protein bands labeled using mass spectrometry results from SEQUEST scan of DLP3 and Stenotrophomonas protein databases. Stacking and resolving portions of the gel were 4 and 7.5%, respectively. The gel was stained with Coomassie R-250.
TABLE S1 | Bacterial strains and plasmids used in this study.
TABLE S2 | Mass spectrometry protein results using the Stenotrophomonas protein database in UniProt. The results are organized by score.
Ackermann, H. W., and Eisenstark, A. (1974). The present state of phage taxonomy. Intervirology 3, 201–219. doi: 10.1159/000149758
Al-Anazi, K. A., and Al-Jasser, A. M. (2014). Infections caused by Stenotrophomonas maltophilia in recipients of hematopoietic stem cell transplantation. Front. Oncol. 4:232. doi: 10.3389/fonc.2014.00232
Allison, G. E., and Verma, N. K. (2000). Serotype-converting bacteriophages and O-antigen modification in Shigella flexneri. Trends Microbiol. 8, 17–23. doi: 10.1016/s0966-842x(99)01646-7
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Avison, M. B., Higgins, C. S., von Heldreich, C. J., Bennett, P. M., and Walsh, T. R. (2001). Plasmid location and molecular heterogeneity of the L1 and L2 beta-lactamase genes of Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 45, 413–419. doi: 10.1128/AAC.45.2.413-419.2001
Avison, M. B., von Heldreich, C. J., Higgins, C. S., Bennett, P. M., and Walsh, T. R. (2000). A TEM-2 beta-lactamase encoded on an active Tn1-like transposon in the genome of a clinical isolate of Stenotrophomonas maltophilia. J. Antimicrob. Chemother. 46, 879–884. doi: 10.1093/jac/46.6.879
Bair, C., and Black, L. W. (2007). Exclusion of glucosyl-hydroxymethylcytosine DNA containing bacteriophages. J. Mol. Biol. 366, 779–789. doi: 10.1016/j.jmb.2006.11.049
Barbolla, R., Catalano, M., Orman, B. E., Famiglietti, A., Vay, C., Smayevsky, J., et al. (2004). Class 1 integrons increase trimethoprim-sulfamethoxazole MICs against epidemiologically unrelated Stenotrophomonas maltophilia isolates. Antimicrob. Agents Chemother. 48, 666–669. doi: 10.1128/AAC.48.2.666-669.2004
Belcaid, M., Bergeron, A., and Poisson, G. (2011). The evolution of the tape measure protein: units, duplications and losses. BMC Bioinformatics 12(Suppl. 9):S10. doi: 10.1186/1471-2105-12-S9-S10
Berg, G., Eberl, L., and Hartmann, A. (2005). The rhizosphere as a reservoir for opportunistic human pathogenic bacteria. Environ. Microbiol. 7, 1673–1685. doi: 10.1111/j.1462-2920.2005.00891.x
Berg, G., and Martinez, J. L. (2015). Friends or foes: can we make a distinction between beneficial and harmful strains of the Stenotrophomonas maltophilia complex? Front. Microbiol. 6:241. doi: 10.3389/fmicb.2015.00241
Berg, G., Roskot, N., and Smalla, K. (1999). Genotypic and phenotypic relationships between clinical and environmental isolates of Stenotrophomonas maltophilia. J. Clin. Microbiol. 37, 3594–3600. doi: 10.1128/jcm.37.11.3594-3600.1999
Besemer, J., Lomsadze, A., and Borodovsky, M. (2001). GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29, 2607–2618. doi: 10.1093/nar/29.12.2607
Betts, J. W., Phee, L. M., Woodford, N., and Wareham, D. W. (2014). Activity of colistin in combination with tigecycline or rifampicin against multidrug-resistant Stenotrophomonas maltophilia. Eur. J. Clin. Microbiol. Infect. Dis. 33, 1565–1572. doi: 10.1007/s10096-014-2101-3
Boulanger, P. (2009). Bacteriophages: Methods and Protocols, Molecular and Applied Aspects, Vol. 2, eds M. R. J. Clokie and A. M. Kropinski (Totowa, NJ: Humana Press), 227–238.
Brooke, J. S. (2012). Stenotrophomonas maltophilia: an emerging global opportunistic pathogen. Clin. Microbiol. Rev. 25, 2–41. doi: 10.1128/cmr.00019-11
Brooke, J. S. (2014). New strategies against Stenotrophomonas maltophilia: a serious worldwide intrinsically drug-resistant opportunistic pathogen. Exp. Rev. Anti Infect. Ther. 12, 1–4. doi: 10.1586/14787210.2014.864553
Brooke, J. S., Vo, A., Watts, P., and Davis, N. A. (2008). Mutation of a lipopolysaccharide synthesis gene results in increased biofilm of Stenotrophomonas maltophilia on plastic and glass surfaces. Ann. Microbiol. 58, 35–40. doi: 10.1007/bf03179442
Burrowes, B., Harper, D. R., Anderson, J., McConville, M., and Enright, M. C. (2011). Bacteriophage therapy: potential uses in the control of antibiotic-resistant pathogens. Expert Rev. Anti Infect. Ther. 9, 775–785. doi: 10.1586/eri.11.90
Burrows, L. (2005). Weapons of mass retraction. Mol. Microbiol. 57, 878–888. doi: 10.1111/j.1365-2958.2005.04703.x
Chang, H. C., Chen, C. R., Lin, J. W., Shen, G. H., Chang, K. M., Tseng, Y. H., et al. (2005). Isolation and characterization of novel giant Stenotrophomonas maltophilia phage phi SMA5. Appl. Environ. Microbiol. 71, 1387–1393. doi: 10.1128/aem.71.3.1387-1393.2005
Chang, Y. C., Tsai, M. J., Huang, Y. W., Chung, T. C., and Yang, T. C. (2011). SmQnrR, a DeoRtype transcriptional regulator, negatively regulates the expression of Smqnr and SmtcrA in Stenotrophomonas maltophilia. J. Antimicrob. Chemother. 66, 1024–1028. doi: 10.1093/jac/dkr049
Chang, Y. T., Lin, C. Y., Chen, Y. H., and Hsueh, P. R. (2015). Update on infections caused by Stenotrophomonas maltophilia with particular attention to resistance mechanisms and therapeutic options. Front. Microbiol. 6:893. doi: 10.3389/fmicb.2015.00893
Chang, Y. T., Lin, C. Y., Lu, P. L., Lai, C. C., Chen, T. C., Chen, C. Y., et al. (2014). Stenotrophomonas maltophilia bloodstream infection: comparison between community-onset and hospital-acquired infections. J. Microbiol. Immunol. Infect. 47, 28–35. doi: 10.1016/j.jmii.2012.08.014
Chatelut, M., Dournes, J. L., Chabanon, G., and Marty, N. (1995). Epidemiological typing of Stenotrophomonas (Xanthomonas) maltophilia by PCR. J. Clin. Microbiol. 33, 912–914. doi: 10.1128/jcm.33.4.912-914.1995
Chung, H. S., Hong, S. G., Kim, Y. R., Shin, K. S., Whang, D. H., Ahn, J. Y., et al. (2013). Antimicrobial susceptibility of Stenotrophomonas maltophilia isolates from Korea, and the activity of antimicrobial combinations against the isolates. J. Korean Med. Sci. 28, 62–66. doi: 10.3346/jkms.2013.28.1.62
Chung, H. S., Kim, K., Hong, S. S., Hong, S. G., Lee, K., and Chong, Y. (2015). The sul1 gene in Stenotrophomonas maltophilia with high-level resistance to trimethoprim/sulfamethoxazole. Ann. Lab. Med. 35, 246–249. doi: 10.3343/alm.2015.35.2.246
Crowder, M. W., Walsh, T. R., Banovic, L., Pettit, M., and Spencer, J. (1998). Overexpression, purification, and characterization of the cloned metallo-β-lactamase L1 from Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 42, 921–926. doi: 10.1128/aac.42.4.921
Cutuli, M. A., Petronio, G. P., Vergalito, F., Magnifico, I., Pietrangelo, L., Venditti, N., et al. (2019). Galleria mellonella as a consolidated in vivo model hosts: new developments in antibacterial strategies and novel drug testing. Virulence 10, 527–541. doi: 10.1080/21505594.2019.1621649
de Oliveira-Garcia, D., Dall’Agnol, M., Rosales, M., Azzuz, A. C., Alcántara, N., Martinez, M. B., et al. (2003). Fimbriae and adherence of Stenotrophomonas maltophilia to epithelial cells and to abiotic surfaces. Cell. Microbiol. 5, 625–636. doi: 10.1046/j.1462-5822.2003.00306.x
Dedrick, R. M., Guerrero-Bustamante, C. A., Garlena, R. A., Daniel, A., Ford, K., Harris, K., et al. (2019). Engineered bacteriophages for treatment of a patient with a disseminated drug resistant Mycobacterium abscessus. Nat. Med. 25, 730–733. doi: 10.1038/s41591-019-0437-z
Delcher, A. L., Bratke, K. A., Powers, E. C., and Salzberg, S. L. (2007). Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679. doi: 10.1093/bioinformatics/btm009
Denton, M., and Kerr, K. G. (1998). Microbiological and clinical aspects of infection associated with Stenotrophomonas maltophilia. Clin. Microbiol. Rev. 11, 57–80. doi: 10.1128/cmr.11.1.57
Falagas, M. E., Kastoris, A. C., Vouloumanou, E. K., and Dimopoulos, G. (2009). Community-acquired Stenotrophomonas maltophilia infections: a systematic review. Eur. J. Clin. Microbiol. Infect. Dis. 28, 719–730. doi: 10.1007/s10096-009-0709-5
Fan, N., Cutting, S., and Losick, R. (1992). Characterization of the Bacillus subtilis sporulation gene spoVK. J. Bacteriol. 174, 1053–1054. doi: 10.1128/jb.174.3.1053-1054.1992
Figueiredo, P. M. S., Furumura, M. T., Santos, A. M., Sousa, A. C., Kota, D. J., Levy, C. E., et al. (2006). Cytotoxic activity of clinical Stenotrophomonas maltophilia. Lett. Appl. Microbiol. 43, 443–449. doi: 10.1111/j.1472-765X.2006.01965.x
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Furlan, J. P. R., and Stehling, E. G. (2018). Detection of blaPER on an IncA/C plasmid in Stenotrophomonas maltophilia isolated from Brazilian soil. Water Air Soil Pollut. 229:142.
Garcia, P., Monjardín, C., Martín, R., Madera, C., Soberón, N., Garcia, E., et al. (2008). Isolation of new Stenotrophomonas bacteriophages and genomic characterization of temperate phage S1. Appl. Environ. Microbiol. 74, 7552–7560. doi: 10.1128/aem.01709-08
Golkar, Z., Bagasra, O., and Pace, D. G. (2014). Bacteriophage therapy: a potential solution for the antibiotic resistance crisis. J. Infect. Dev. Ctries. 8, 129–136. doi: 10.3855/jidc.3573
Hagemann, M., Hasse, D., and Berg, G. (2006). Detection of a phage genome carrying a zonula occludens like toxin gene (zot) in clinical isolates of Stenotrophomonas maltophilia. Arch. Microbiol. 185, 449–458. doi: 10.1007/s00203-006-0115-7
Hanahan, D., Jessee, J., and Bloom, F. R. (1991). Plasmid transformation of Escherichia coli and other bacteria. Methods Enzymol. 204, 63–113. doi: 10.1016/0076-6879(91)04006-a
Harris, R. S. (2007). Improved Pairwise Alignment of Genomic DNA. Ph.D. thesis, The Pennsylvania State University, State College, PA.
Hauben, L., Vauterin, L., Moore, E. R. B., Hoste, B., and Swings, J. (1999). Genomic diversity of the genus Stenotrophomonas. Int. J. Syst. Bacteriol. 49, 1749–1760. doi: 10.1099/00207713-49-4-1749
Hendrix, R. W., and Johnson, J. E. (2012). Bacteriophage HK97 capsid assembly and maturation. Adv. Exp. Med. Biol. 726, 351–363. doi: 10.1007/978-1-4614-0980-9_15
Hoang, T. T., Karkhoff-Schweizer, R. R., Kutchma, A. J., and Schweizer, H. P. (1998). A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212, 77–86. doi: 10.1016/s0378-1119(98)00130-9
Hu, L. F., Chang, X., Ye, Y., Wang, Z. X., Shao, Y. B., Shi, W., et al. (2011). Stenotrophomonas maltophilia resistance to trimethoprim/sulfamethoxazole mediated by acquisition of sul and dfrA genes in a plasmid-mediated class 1 integron. Int. J. Antimicrob. Agents 37, 230–234. doi: 10.1016/j.ijantimicag.2010.10.025
Huang, T. P., Somers, E. B., and Wong, A. C. L. (2006). Differential biofilm formation and motility associated with lipopolysaccharide/exopolysaccharide-coupled biosynthetic genes in Stenotrophomonas maltophilia. J. Bacteriol. 188, 3116–3120. doi: 10.1128/JB.188.8.3116-3120.2006
Huang, Y. W., Hu, R. M., and Yang, T. C. (2013). Role of the pcm-tolCsm operon in the multidrug resistance of Stenotrophomonas maltophilia. J. Antimicrob. Chemother. 68, 1987–1993. doi: 10.1093/jac/dkt148
Kamal, F., and Dennis, J. J. (2015). Burkholderia cepacia complex phage-antibiotic synergy (PAS): antibiotics stimulate lytic phage activity. Appl. Environ. Microbiol. 81, 1132–1138. doi: 10.1128/aem.02850-14
Kamal, F., Peters, D. L., McCutcheon, J. G., Dunphy, G. B., and Dennis, J. J. (2019). Use of greater wax moth larvae (Galleria mellonella) as an alternative animal infection model for analysis of bacterial pathogenesis. Methods Mol. Biol. 1898, 163–171. doi: 10.1007/978-1-4939-8940-9_13
Kampmeier, S., Pillukat, M. H., Pettke, A., Kossow, A., Idelevich, E. A., and Mellmann, A. (2017). Evaluation of a Stenotrophomonas maltophilia bacteremia cluster in hematopoietic stem cell transplantation recipients using whole genome sequencing. Antimicrob. Resist. Infect. Control 6:115. doi: 10.1186/s13756-017-0276-y
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Kim, Y. J., Park, J. H., and Seo, K. H. (2018). Presence of Stenotrophomonas maltophilia exhibiting high genetic similarity to clinical isolates in final effluents of pig farm wastewater treatment plants. Int. J. Hyg. Environ. Health 221, 300–307. doi: 10.1016/j.ijheh.2017.12.002
Kovach, M. E., Phillips, R. W., Elzer, P. H., Roop, R. M., and Peterson, K. M. (1994). pBBR1MCS: a broad-host-range cloning vector. Biotechniques 16, 800–802.
Lai, C. H., Wong, W. W., Chin, C., Huang, C. K., Lin, H. H., Chen, W. F., et al. (2006). Central venous catheter-related Stenotrophomonas maltophilia bacteraemia and associated relapsing bacteraemia in haematology and oncology patients. Clin. Microbiol. Infect. 12, 986–991. doi: 10.1111/j.1469-0691.2006.01511.x
Lin, L., Wang, S. F., Yang, T. Y., Hung, W. C., Chan, M. Y., and Tseng, S. P. (2017). Antimicrobial resistance and genetic diversity in ceftazidime nonsusceptible bacterial pathogens from ready-to-eat street foods in three Taiwanese cities. Sci. Rep. 7:15515.
Lin, Y. T., Huang, Y. W., Chen, S. J., Chang, C. W., and Yang, T. C. (2015). The SmeYZ efflux pump of Stenotrophomonas maltophilia contributes to drug resistance, virulence-related characteristics, and virulence in mice. Antimicrob. Agents Chemother. 59, 4067–4073. doi: 10.1128/aac.00372-15
Lokareddy, R. K., Sankhala, R. S., Roy, A., Afonine, P. V., Motwani, T., Teschke, C. M., et al. (2017). Portal protein functions akin to a DNA-sensor that couples genome-packaging to icosahedral capsid maturation. Nat. Commun. 8:14310. doi: 10.1038/ncomms14310
Lynch, K. H., Seed, K. D., Stothard, P., and Dennis, J. J. (2010). Inactivation of Burkholderia cepacia complex phage KS9 gp41 identifies the phage repressor and generates lytic virions. J. Virol. 84, 1276–1288. doi: 10.1128/JVI.01843-09
Marchler-Bauer, A., Lu, S., Anderson, J. B., Chitsaz, F., Derbyshire, M. K., DeWeese-Scott, C., et al. (2011). CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–D229. doi: 10.1093/nar/gkq1189
McCutcheon, J. G., Peters, D. L., and Dennis, J. J. (2018). Identification and characterization of type IV pili as the cellular receptor of broad host range Stenotrophomonas maltophilia bacteriophages DLP1 and DLP2. Viruses 10:E338. doi: 10.3390/v10060338
McKay, G. A., Woods, D. E., MacDonald, K. L., and Poole, K. (2003). Role of phosphoglucomutase of Stenotrophomonas maltophilia in lipopolysaccharide biosynthesis, virulence, and antibiotic resistance. Infect. Immun. 71, 3068–3075. doi: 10.1128/iai.71.6.3068-3075.2003
Melloul, E., Roisin, L., Durieux, M., Woerther, P., Jenot, D., Risco, V., et al. (2018). Interactions of Aspergillus fumigatus and Stenotrophomonas maltophilia in an in vitro mixed biofilm model: does the strain matter? Front. Microbiol. 9:2850. doi: 10.3389/fmicb.2018.02850
Mett, H., Rosta, S., Schacher, B., and Frei, R. (1988). Outer membrane permeability and beta-lactamase content in Pseudomonas maltophilia clinical isolates and laboratory mutants. Rev. Infect. Dis. 10, 765–769. doi: 10.1093/clinids/10.4.765
Miller, J. L. (2006). Understanding Receptor Specificity though the Massively Variable Major Tropism Determinant of Bordetella Bacteriophage. Ph.D. thesis, University of California, Oakland, CA.
Miller, J. L., Le Coq, J., Hodes, A., Barbalat, R., Miller, J. F., and Ghosh, P. (2008). Selective ligand recognition by a diversity-generating retroelement variable protein. PLoS Biol. 6:e131. doi: 10.1371/journal.pbio.0060131
Minkwitz, A., and Berg, G. (2001). Comparison of antifungal activities and 16S ribosomal DNA sequences of clinical and environmental isolates of Stenotrophomonas maltophilia. J. Clin. Microbiol. 39, 139–145. doi: 10.1128/JCM.39.1.139-145.2001
Naidu, P., and Smith, S. (2012). A review of 11 years of Stenotrophomonas maltophilia blood isolates at a tertiary care institute in Canada. Can. J. Infect. Dis. Med. Microbiol. 23, 165–169. doi: 10.1155/2012/762571
Nicoletti, M., Iacobino, A., Prosseda, G., Fiscarelli, E., Zarrilli, R., De Carolis, E., et al. (2011). Stenotrophomonas maltophilia strains from cystic fibrosis patients: genomic variability and molecular characterization of some virulence determinants. Int. J. Med. Microbiol. 301, 34–43. doi: 10.1016/j.ijmm.2010.07.003
Ochoa-Sanchez, L. E., and Vinuesa, P. (2017). Evolutionary genetic analysis uncovers multiple species with distinct habitat preferences and antibiotic resistance phenotypes in the Stenotrophomonas maltophilia complex. Front. Microbiol. 8:1548. doi: 10.3389/fmicb.2017.01548
Okazaki, A., and Avison, M. B. (2007). Aph(3′)-IIc, an aminoglycoside resistance determinant from Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 51, 359–360. doi: 10.1128/aac.00795-06
Okazaki, A., and Avison, M. B. (2008). Induction of L1 and L2 β-lactamase production in Stenotrophomonas maltophilia is dependent on an AmpR-type regulator. Antimicrob. Agents Chemother. 52, 1525–1528. doi: 10.1128/aac.01485-07
Palleroni, N. J., and Bradbury, J. F. (1993). Stenotrophomonas, a new bacterial genus for Xanthomonas maltophilia (Hugh 1980) Swings et al. 1983. Int. J. Syst. Bacteriol. 43, 606–609. doi: 10.1099/00207713-43-3-606
Peters, D. L., and Dennis, J. J. (2018). Complete genome sequence of temperate Stenotrophomonas maltophilia bacteriophage DLP5. Genome Announc. 6:e00073-18.
Peters, D. L., Lynch, K. H., Stothard, P., and Dennis, J. J. (2015). The isolation and characterization of two Stenotrophomonas maltophilia bacteriophages capable of cross-taxonomic order infectivity. BMC Genomics 16:664. doi: 10.1186/s12864-015-1848-y
Peters, D. L., McCutcheon, J. G., Stothard, P., and Dennis, J. J. (2019). Novel Stenotrophomonas maltophilia temperate phage DLP4 is capable of lysogenic conversion. BMC Genomics 20:300. doi: 10.1186/s12864-019-5674-5
Peters, D. L., Stothard, P., and Dennis, J. J. (2017). The isolation and characterization of Stenotrophomonas maltophilia T4-like bacteriophage DLP6. PLoS One 12:e0173341. doi: 10.1371/journal.pone.0173341
Pompilio, A., Crocetta, V., Confalone, P., Nicoletti, M., Petrucca, A., Guarnieri, S., et al. (2010). Adhesion to and biofilm formation on IB3-1 bronchial cells by Stenotrophomonas maltophilia isolates from cystic fibrosis patients. BMC Microbiol. 10:102. doi: 10.1186/1471-2180-10-102
Pompilio, A., Piccolomini, R., Picciani, C., D’Antonio, D., Savini, V., and Di Bonaventura, G. (2008). Factors associated with adherence to and biofilm formation on polystyrene by Stenotrophomonas maltophilia: the role of cell surface hydrophobicity and motility. FEMS Microbiol. Lett. 287, 41–47. doi: 10.1111/j.1574-6968.2008.01292.x
Pradel, E., Parker, C. T., and Schnaitman, C. A. (1992). Structures of the rfaB, rfaI, rfaJ, and rfaS genes of Escherichia coli K-12 and their roles in assembly of the lipopolysaccharide core. J. Bacteriol. 174, 4736–4745.
Prunier, A. L., Malbruny, B., Tande, D., Picard, B., and Leclercq, R. (2002). Clinical isolates of Staphylococcus aureus with ribosomal mutations conferring resistance to macrolides. Antimicrob. Agents Chemother. 46, 3054–3056. doi: 10.1128/aac.46.9.3054-3056.2002
Qureshi, A., Mooney, L., Denton, M., and Kerr, K. G. (2005). Stenotrophomonas maltophilia in salad. Emerg. Infect. Dis. 11, 1157–1158. doi: 10.3201/eid1107.040130
Rathi, M., and Nandabalan, Y. K. (2017). Copper-tolerant rhizosphere bacteria-characterization and assessment of plant growth promoting factors. Environ. Sci. Pollut. Res. 24, 9723–9733. doi: 10.1007/s11356-017-8624-2
Remmert, M., Biegert, A., Hauser, A., and Soding, J. (2011). HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 9, 173–175. doi: 10.1038/nmeth.1818
Rhee, J. Y., Song, J. H., and Ko, K. S. (2016). Current situation of antimicrobial resistance and genetic differences in Stenotrophomonas maltophilia complex isolates by multilocus variable number of tandem repeat analysis. Infect. Chemother. 48, 285–293. doi: 10.3947/ic.2016.48.4.285
Ryan, R. P., Monchy, S., Cardinale, M., Taghavi, S., Crossman, L., Avison, M. B., et al. (2009). The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat. Rev. Microbiol. 7, 514–525. doi: 10.1038/nrmicro2163
Sanchez, M. B. (2015). Antibiotic resistance in the opportunistic pathogen Stenotrophomonas maltophilia. Front. Microbiol. 6:658. doi: 10.3389/fmicb.2015.00658
Sanchez, M. B., Hernandez, A., Rodriguez-Martinez, J. M., Martinez-Martinez, L., and Martinez, J. L. (2008). Predictive analysis of transmissible quinolone resistance indicates Stenotrophomonas maltophilia as a potential source of a novel family of Qnr determinants. BMC Microbiol. 8:148. doi: 10.1186/1471-2180-8-148
Sanchez, M. B., and Martinez, J. L. (2010). SmQnr contributes to intrinsic resistance to quinolones in Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 54, 580–581. doi: 10.1128/aac.00496-09
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Schubert, H. L., Blumenthal, R. M., and Cheng, X. (2003). Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci. 28, 329–335. doi: 10.1016/s0968-0004(03)00090-2
Schwab, F., Geffers, C., Behnke, M., and Gastmeier, P. (2018). ICU mortality following ICU acquired primary bloodstream infections according to the type of pathogen: a prospective cohort study in 937 Germany ICUs (2006-2015). PLoS One 13:e0194210. doi: 10.1371/journal.pone.0194210
Seed, K. D., and Dennis, J. J. (2008). Development of Galleria mellonella as an alternative infection model for the Burkholderia cepacia complex. Infect. Immun. 76, 1267–1275. doi: 10.1128/iai.01249-07
Seed, K. D., and Dennis, J. J. (2009). Experimental bacteriophage therapy increases survival of Galleria mellonella larvae infected with clinically relevant strains of the Burkholderia cepacia Complex. Antimicrob. Agents Chemother. 53, 2205–2208. doi: 10.1128/aac.01166-08
Simon, R., Priefer, U., and Puhler, A. (1983). A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat. Biotechnol. 1, 784–791. doi: 10.1038/nbt1183-784
Struck, A. W., Thompson, M. L., Wong, L. S., and Micklefield, J. (2012). S-adenosyl-methionine dependent methyltransferases: highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. Chem. Biochem. 13, 2642–2655. doi: 10.1002/cbic.201200556
Sulakvelidze, A., Alavidze, Z., and Morris, J. G. Jr. (2001). Bacteriophage therapy. Antimicrob. Agents Chemother. 45, 649–659. doi: 10.1128/AAC.45.3.649-659.2001
Svensson-Stadler, L. A., Mihaylova, S. A., and Moore, E. R. (2012). Stenotrophomonas interspecies differentiation and identification by gyrB sequence analysis. FEMS Microbiol. Lett. 327, 15–24. doi: 10.1111/j.1574-6968.2011.02452.x
Toleman, M. A., Bennett, P. M., Bennett, D. M., Jones, R. N., and Walsh, T. R. (2007). Global emergence of trimethoprim/sulfamethoxazole resistance in Stenotrophomonas maltophilia mediated by acquisition of sul genes. Emerg. Infect. Dis. 13, 559–565. doi: 10.3201/eid1304.061378
Tsai, C. J., Loh, J. M. S., and Proft, T. (2016). Galleria mellonella infection models for the study of bacterial diseases and for antimicrobial drug testing. Virulence 7, 214–229. doi: 10.1080/21505594.2015.1135289
Velazquez-Acosta, C., Zarco-Marquez, S., Jiminez-Andrade, M. C., Volkow-Fernandez, P., and Cornejo-Juarez, P. (2018). Stenotrophomonas maltophilia bacteremia and pneumonia at a tertiary-care oncology center: a review of 16 years. Support. Care Cancer 26, 1953–1960. doi: 10.1007/s00520-017-4032-x
Vetting, M. W., Park, C. H., Hegde, S. S., Jacoby, G. A., Hooper, D. C., and Blanchard, J. S. (2008). Mechanistic and structural analysis of aminoglycoside N-acetyltransferase AAC(6′)-Ib and its bifunctional, fluoroquinolone-active AAC(6′)-Ib-cr variant. Biochemistry 47, 9825–9835. doi: 10.1021/bi800664x
Walsh, T. R., MacGowan, A. P., and Bennett, P. M. (1997). Sequence analysis and enzyme kinetics of the L2 serine β-lactamase from Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 41, 1460–1464. doi: 10.1128/aac.41.7.1460
Wang, C. H., Lin, J. C., Lin, H. A., Chang, F. Y., Wang, N. C., Chiu, S. K., et al. (2016). Comparisons between patients with trimethoprim-sulfamethoxazole-susceptible and trimethoprim-sulfamethoxazole-resistant Stenotrophomonas maltophilia monomicrobial bacteremia: a 10-year retrospective study. J. Microbiol. Immunol. Infect. 49, 378–386. doi: 10.1016/j.jmii.2014.06.005
Whitmore, S. E., and Lamont, R. J. (2012). Tyrosine phosphorylation and bacterial virulence. Int. J. Oral Sci. 4, 1–6. doi: 10.1038/ijos.2012.6
Wiegand, I., Hilpert, K., and Hancock, R. E. W. (2008). Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3, 163–175. doi: 10.1038/nprot.2007.521
Wipf, J. R. K., Schwendener, S., Nielsen, J. B., Westh, H., and Perreten, V. (2015). The new macrolide-lincosamide-streptogramin b resistance gene erm(45) is located within a genomic island in Staphylococcus fleurettii. Antimicrob. Agents Chemother. 59, 3578–3581. doi: 10.1128/aac.00369-15
Yang, J., and Zhang, Y. (2015). Protein structure and function prediction using I-TASSER. Curr. Protoc. Bioinformatics 52, 5.8.1–5.8.15. doi: 10.1002/0471250953.bi0508s52
Zhou, Y., Liang, Y., Lynch, K. H., Dennis, J. J., and Wishart, D. S. (2011). PHAST: a fast phage search tool. Nucleic Acids Res. 39, W347–W352. doi: 10.1093/nar/gkr485
Keywords: Stenotrophomonas, bacteriophage, phage, phage therapy, antimicrobial resistance
Citation: Peters DL, McCutcheon JG and Dennis JJ (2020) Characterization of Novel Broad-Host-Range Bacteriophage DLP3 Specific to Stenotrophomonas maltophilia as a Potential Therapeutic Agent. Front. Microbiol. 11:1358. doi: 10.3389/fmicb.2020.01358
Received: 25 February 2020; Accepted: 27 May 2020;
Published: 24 June 2020.
Edited by:
Sutthirat Sitthisak, Naresuan University, ThailandReviewed by:
Kitiya Vongkamjan, Prince of Songkla University, ThailandCopyright © 2020 Peters, McCutcheon and Dennis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan J. Dennis, am9uLmRlbm5pc0B1YWxiZXJ0YS5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.