
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 05 June 2020
Sec. Virology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.01316
This article is part of the Research TopicCoronavirus Disease (COVID-19): Pathophysiology, Epidemiology, Clinical Management and Public Health ResponseView all 400 articles
 Tsuyoshi Sekizuka1
Tsuyoshi Sekizuka1 Sanae Kuramoto2Eri Nariai2Masakatsu Taira3Yushi Hachisu3Akihiko Tokaji4Michiyo Shinohara5Tsuyoshi Kishimoto5Kentaro Itokawa1Yusuke Kobayashi6Keisuke Kadokura6Hajime Kamiya6Tamano Matsui6Motoi Suzuki6
Sanae Kuramoto2Eri Nariai2Masakatsu Taira3Yushi Hachisu3Akihiko Tokaji4Michiyo Shinohara5Tsuyoshi Kishimoto5Kentaro Itokawa1Yusuke Kobayashi6Keisuke Kadokura6Hajime Kamiya6Tamano Matsui6Motoi Suzuki6 Makoto Kuroda1*
Makoto Kuroda1*Japan has reported 26 cases of coronavirus disease 2019 (COVID-19) linked to cruise tours on the River Nile in Egypt between March 5 and 15, 2020. Here, we characterized the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genome of isolates from 10 travelers who returned from Egypt and from patients possibly associated with these travelers. We performed haplotype network analysis of SARS-CoV-2 isolates using genome-wide single-nucleotide variations. Our analysis identified two potential Egypt-related clusters from these imported cases, and these clusters were related to globally detected viruses in different countries.
The current pandemic of coronavirus disease 20191 (COVID-19) is caused by a positive-sense RNA virus, named the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Coronaviridae Study Group of the International Committee on Taxonomy of V., 2020). As of April 6, 2020, Japan has confirmed 3,985 cases in total, excluding the cases in Diamond Princess cruise ship. This makes Japan one of the developed countries least affected by SARS-CoV-2. The Japanese government has focused on the identification and mitigation of emerging COVID-19 clusters before further expansion, a strategy considered optimal in a low infection rate situation. Japan has sustained moderate spread by focusing on COVID-19 outbreak clusters; however, an ever-increasing number of COVID-19 cases has made it difficult to identify all infection routes.
From the beginning of March 2020, 46 travelers who returned to Japan from abroad were suspected to have imported COVID-19; these cases account for roughly 10% of all new cases recorded in Japan. Among these imported cases, as many as 26 have been linked to cruise tours on the River Nile in Egypt between March 5 and 15, 2020. Most of the travelers visited Egypt from late February to early March and embarked on Nile River cruise ship tours between Cairo and Luxor for 3–4 days. Soon after they returned to Japan, they experienced the onset of fever and sore throat. They visited their respective local consultation centers for recent arrivals from abroad and underwent PCR testing, which confirmed them as being positive for SARS-CoV-2. A field epidemiological study was conducted on the people closely associated with them, such as family members, who might have been exposed to the virus. In this study, we have evaluated viral genome sequences from SARS-CoV-2-positive travelers who returned from Egypt, and characterized the haplotype networks to demonstrate possible routes of the spread.
We evaluated viral genome sequences from 10 SARS-CoV-2-positive travelers who returned from Egypt, as well as their close contacts, to identify possible routes of spread. The travel histories, clinical courses, and PCR testing results are summarized in Figure 1. To characterize the potential origins and routes of the suspected imported cases, we determined the whole-genome sequence of SARS-CoV-2 using a multiplex PCR-based RNA-Seq by ARTIC Network protocol2 based on PrimalSeq (Quick et al., 2017; Grubaugh et al., 2019) with modified primers and protocol (Itokawa et al., 2020). For the obtained genome sequences, haplotype network analysis using genome-wide single-nucleotide variations (SNVs) on the core regions from positions 99 to 29,796 nt in the Wuhan-Hu-1 reference genome sequence (GISAID ID, EPI_ISL_402125; GenBank ID, MN908947.3) was performed (Figure 2). SARS-CoV-2 genome sequences with nearly full-length information (≥ 29 kb) were retrieved from the GISAID EpiCoV database on March 30, 2020, and we generated haplotype networks by median-joining network analysis using PopART software3 to highlight and trace a potential infectious route among COVID-19 patient populations.
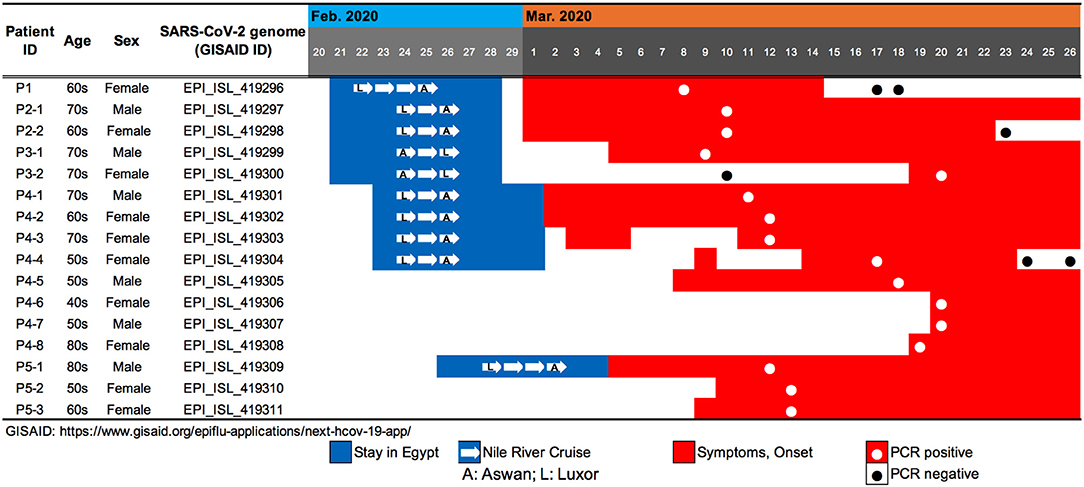
Figure 1. Summary of travel history, clinical course, and PCR testing for 10 SARS-CoV-2-positive travelers who returned to Japan from Egypt, as well as the associated patients who were their close contacts.
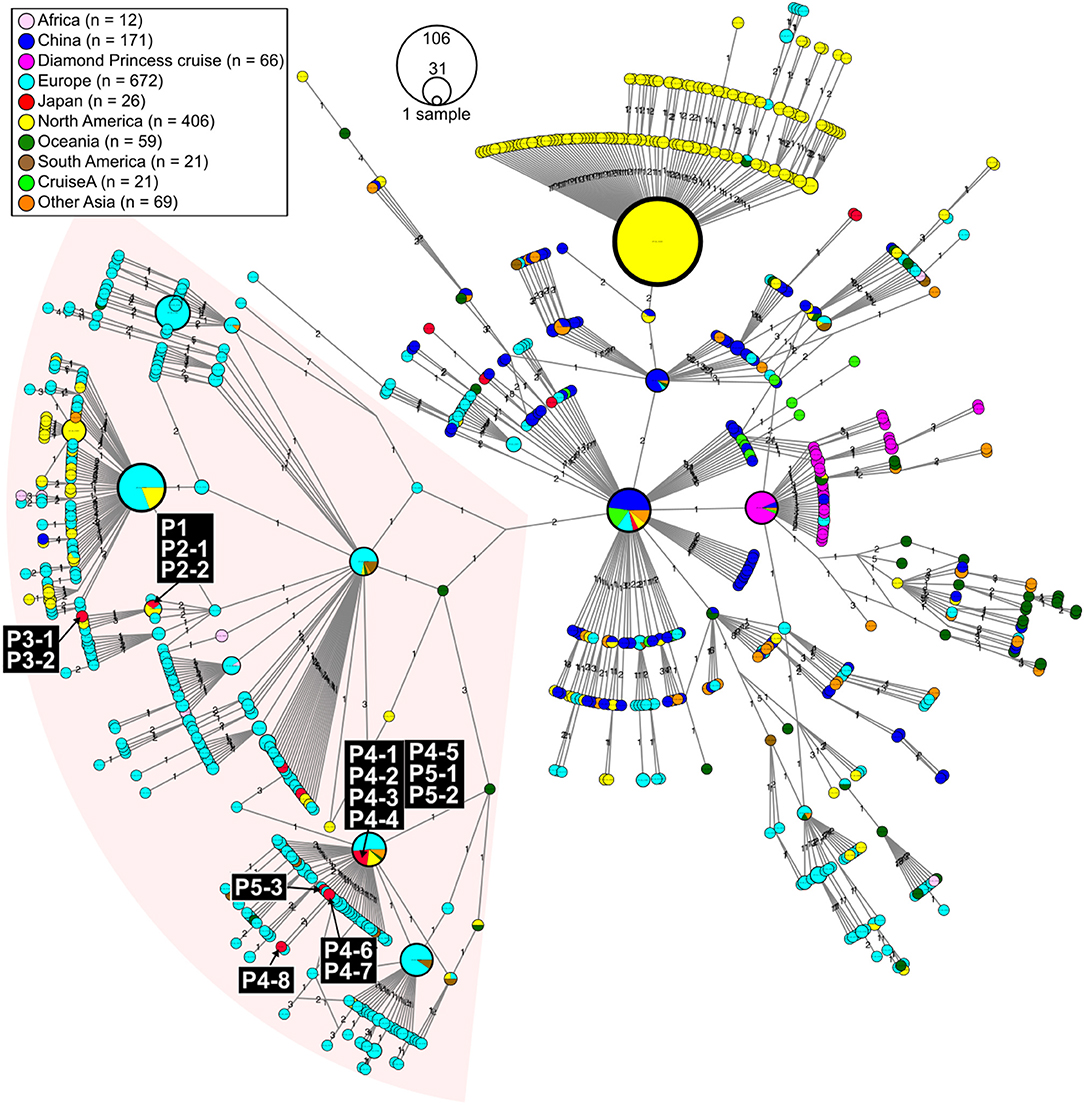
Figure 2. Haplotype network using genome-wide single-nucleotide variations (HN-GSNVs) of SARS-CoV-2 isolates in the world. Whole-genome sequences of SARS-CoV-2 isolates from 10 travelers who returned from Egypt and possible patients linked to them (Figure 1) were compared with all GISAID-available SARS-CoV-2 genomes (n = 1,507, updated on March 30, 2020. See Table S1) by median-joining SNV network analysis. The numbers on the edges indicate differential SNVs between pair-wise nodes (isolates). SARS-CoV-2 disseminated from the end of December, 2019, from Wuhan City in China, one of the potential origins of Wuhan-Hu-1, isolated on December 26, 2020 (GenBank ID: MN908947). Wuhan-Hu-1 is plotted at the center of the haplotype network. Currently, at least three clades have disseminated globally in a region-specific manner.
Patient P1 (P1; hereafter, patients are designated in this manner) arrived at Cairo airport from Tokyo on February 22 and embarked on a Nile River cruise ship for 4 days (Figure 1). The SARS-CoV-2 genome sequence of the P1 isolate shows a close lineage with European isolates, with several SNVs (Figure 3). P2-1 and P2-2 had visited Egypt together and traveled aboard the same Nile River cruise ship, and SARS-CoV-2 genome sequences isolated from them are identical with that of P1 (Figure 3).
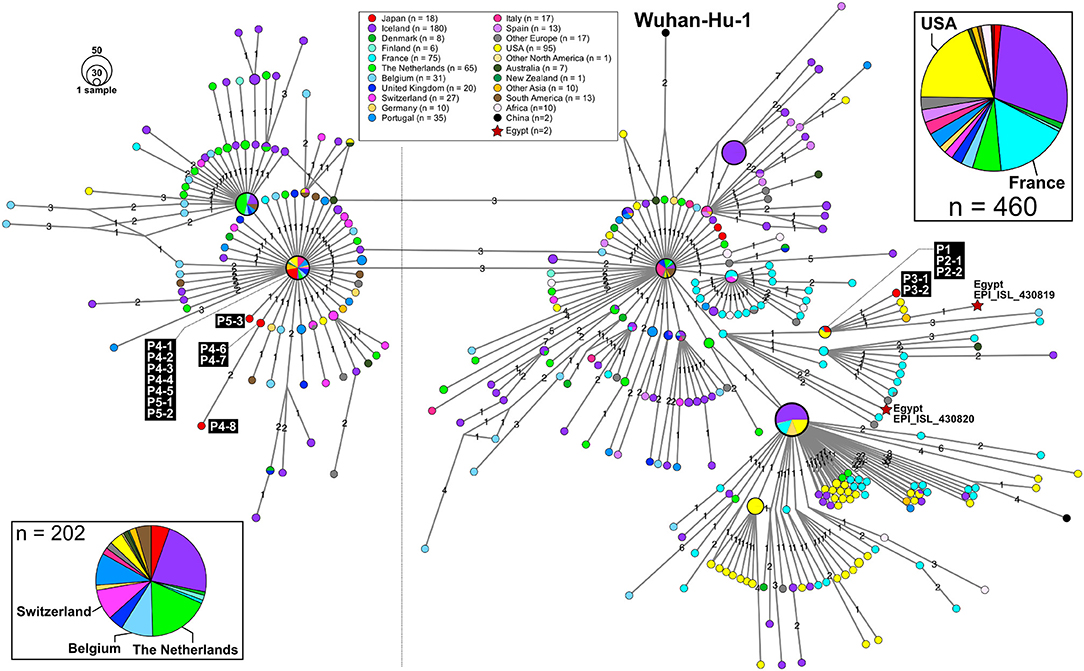
Figure 3. An excerpt of HN-GSNVs from Japanese travelers returning from Egypt and patients associated with them. The haplotype of SARS-CoV-2 genome sequences for 16 patients (Figure 1) was found in the two marked clusters, which comprised the most European isolates (Figure 2). See the legends in Figure 2 for details.
The couple P3-1 and P3-2 visited Egypt together, on the same Tokyo to Cairo flight as the above P1 patient but boarded a different Nile River cruise ship. The husband, P3-1, showed flu-like symptoms on March 5 and was confirmed with COVID-19 on March 9, while the wife, P3-2, was asymptomatic and PCR negative on March 10 despite their close contact during the trip. 10 days later, however, on March 20, P3-2 exhibited symptoms and was confirmed with COVID-19. These two SARS-CoV-2 isolates show identical genome sequence (Figure 3) and are distinct from the genome sequences (P1, P2-1, and P2-2) by only one SNV (Figure 3).
Meanwhile, compared to the genome sequences of the above five patients, the SARS-CoV-2 genome sequence obtained from following P4 and P5 patients showed clearly different haplotype lineage, with at least five differential SNVs (Figure 3). Four patients (P4-1 to P4-4) had visited Egypt at the end of February, a few days after the above-mentioned patients, and exhibited symptoms after returning back to Japan. Intriguingly, the genome sequences of four additional patients (P4-5–P4-8), having no history of recent overseas travel or contact with the above patients, were markedly close to the P4-related isolates. P4-6 and P4-7 are coworkers with P4-5, and P4-8 is mother of P4-5, indicating that three patients (P4-6–P4-8) were close contacts to P4-5 as original source. This finding demonstrated the identification of a potential hidden link to the import of infection from Egypt.
P5-1 had visited Egypt and embarked on a Nile River cruise ship different from the other travelers mentioned above. He showed symptoms and tested positive by PCR after returning home. His relatives, P5-2 (daughter) and P5-3 (sister), who had close contact with him, subsequently showed symptoms and tested positive for SARS-CoV-2 by PCR (Figure 1). The SARS-CoV-2 genome sequence of P5-1 was identical to that of P5-2, distinct only by one SNV from that of P5-3, indicating direct infections from P5-1 to P5-2 and P5-3, (Figure 3).
Thus far, two genome sequences of SARS-CoV-2 isolates in Egypt (isolation date: 2020/03/18; GISAID ID: EPI_ISL_430819 and EPI_ISL430820) are available in GISAID, and the haplotype network exhibits that P1 and P3 patients are closely related to those Egypt isolates with 2 or 3 SNVs (Figure 3).
In this study, we found two SARS-CoV-2 genome lineages from Egypt-related imported cases. These virus lineages belonged to a single clade rooted to the major indexed isolates, which diverged from Wuhan-Hu-1 by several clades. The members in this clade are considered to be circulating in multiple countries, mainly in the Europe and South America. The Egypt-related isolates described in this study are divided into two distinct SARS-CoV-2 haplotype lineages, with two or three additional SNVs from the major indexed isolates; one lineage, including P1–P3, included most haplotypes isolated from France and Egypt (Figure 3), whereas the other lineage, including P4 and P5, included the Netherlands/Belgium/Switzerland isolates.
On 6 March, the Egyptian Health Ministry confirmed 12 COVID-19 cases among the Egyptian crew staff aboard a Nile River cruise ship. On 7 March, the Egyptian health authorities announced that 45 people on board that ship had tested positive, and that the ship had been subjected to quarantine at a dock in Luxor. It is also speculated that Egypt probably has a large burden of COVID-19 cases that are unreported, and Egypt might be a source of COVID-19 export that is not yet accounted for by many public health initiatives (Tuite et al., 2020). Since early March this year, the number of reported COVID-19 cases has been rapidly increasing in Europe countries. This study suggested that patients with a history of travel to Egypt and embarking on Nile River cruises between mid-February and early March could be one of the potential sources of COVID-19 cases imported into Japan.
Pharyngeal specimens were collected from patients, and a quantitative reverse transcription PCR (RT-qPCR) testing for SARS-CoV-2 (Jung et al., 2020; Shirato et al., 2020) was performed.
Basically, whole genome sequences of SARS-CoV-2 was obtained by PrimalSeq protocol to enrich cDNA of SARS-CoV-2 genome by multiplex RT-PCR amplicons using a multiplexed PCR primer set which was proposed by Wellcome Trust ARTIC Network. We found particular two amplicons regularly showed low to zero coverage due to primer dimerization as described in Itokawa et al. (2020), we used the modified primer for the multiplex PCR amplifications (Itokawa et al., 2020). The PCR products from same clinical sample was pooled, purified and subjected for Illumina library construction using QIAseq FX DNA Library Kit (QIAGEN, Hilden Germany). NextSeq 500 platform (Illumina, San Diego, USA) was used for sequencing the indexed libraries. The NGS reads were mapped to the SARS-CoV-2 Wuhan-Hu-1 reference genome sequence (29.9 kb ss-RNA; GenBank ID: MN908947), resulting to the specimen-specific SARS-CoV-2 genome sequence by fully mapping on the reference. These mapped reads of SARS-CoV-2 sequences were assembled using A5-miseq v.20140604 (Coil et al., 2015) to determine the full genome sequence (see the details in Table S1). The SNV sites and marked heterogeneity were extracted by the read-mapping at ≥10 × depth and from 99 to 29,796 nt region of Wuhan-Hu-1 genome sequence (see the details in Table S2).
The nearly full-length genome sequence (≥ 29 kb) of SARS-CoV-2 were retrieved from GISAID EpiCoV database in March 10, 2020, followed by multiple alignment using MAFFT v7.222 (Katoh and Standley, 2013). The poorly aligned regions in 5′ and 3′ end were trimmed; we determined that the core regions were from 99 to 29,796 nt position against Wuhan-Hu-1 genome sequences (GISAID ID, EPI_ISL_402125; GenBank ID, MN908947.3). Gap-containing sequences in the core region were excluded; sequences of 1,507 isolates in GISAID database were eventually used in subsequent analyses (updated on March 30, 2020. See Table S1). The genome sequences were aligned using MAFFT program together with sequences retrieved from database, followed by extraction of SNV and deletion sites. The SNV median-joining network analysis was performed by PopART software3.
The new sequences have been deposited in GISAID with accession IDs EPI_ISL_419296 – EPI_ISL_419311, in addition, in DDBJ/NCBI/EBI with accession IDs LC547518 – LC547533.
The studies involving human participants were reviewed and approved by the National Institute of Infectious Diseases in Japan (Approval No. 1091). It was conducted according to the principles of the Declaration of Helsinki, in compliance with the Law Concerning the Prevention of Infections and Medical Care for Patients of Infections of Japan. The ethical committee waived the need for written consent regarding the research into the viral genome sequence. The personal data related to the clinical information were anonymized, and our procedure is not to request written consent for all patients suffering from COVID-19. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
TS, KI, and MK designed and organized the genome study. SK, EN, MT, YH, AT, MSh, and TK performed the laboratory detection. TS and KI performed the genome analysis. YK, KK, HK, TM, and MSu contributed to the field epidemiological study. MK wrote the manuscript.
This study was supported by a Grant-in Aid from the Japan Agency for Medical Research and Development (AMED) under Grant number JP19fk0108103 and JP19fk0108104. The funding agencies had no role in the study design, data collection or analysis, decision to publish, or manuscript preparation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are deeply thankful to the staff of the local public health centers in the prefectures of Kochi, Ishikawa, Chiba, and Saitama for the field epidemiological study and for preserving and providing the clinical specimens. We thank the Field Epidemiology Training Program (FETP) team; the Ministry of Health, Labor and Welfare; and local governments for their assistance with administrative matters, field investigation, data collection, and laboratory testing. We would like to thank all the authors who have kindly deposited and shared genome data on GISAID. A table with genome sequence acknowledgments can be found in Table S3.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01316/full#supplementary-material
1. ^Coronavirus Disease 2019 (COVID-19) Pandemic: Increased Transmission in the EU/EEA and the UK–Seventh. Available online at: https://www.ecdc.europa.eu/sites/default/files/documents/RRA-seventh-update-Outbreak-of-coronavirus-disease-COVID-19.pdf (accessed March 25, 2020).
2. ^ARTIC Network protocol. Available online at: https://artic.network/ncov-2019.
3. ^PopART_software: http://popart.otago.ac.nz.
Coil, D., Jospin, G., and Darling, A. E. (2015). A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 31, 587–589. doi: 10.1093/bioinformatics/btu661
Coronaviridae Study Group of the International Committee on Taxonomy of V. (2020). The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 5, 536–544. doi: 10.1038/s41564-020-0695-z
Grubaugh, N. D., Gangavarapu, K., Quick, J., Matteson, N. L., De Jesus, J. G., Main, B. J., et al. (2019). An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol 20:8. doi: 10.1186/s13059-018-1618-7
Itokawa, K., Sekizuka, T., Hashino, M., Tanaka, R., and Kuroda, M. (2020). A proposal of an alternative primer for the ARTIC Network's multiplex PCR to improve coverage of SARS-CoV-2 genome sequencing. bioRxiv. doi: 10.1101/2020.03.10.985150. [Epub ahead of print].
Jung, Y. J., Park, G-S., Moon, J. H., Ku, K., Beak, S-H., Kim, S., et al. (2020). Comparative analysis of primer-probe sets for the laboratory confirmation of SARS-CoV-2. bioRxiv. doi: 10.1101/2020.02.25.964775. [Epub ahead of print].
Katoh, K., and Standley, D.M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Quick, J., Grubaugh, N. D., Pullan, S. T., Claro, I. M., Smith, A. D., Gangavarapu, K., et al. (2017). Multiplex PCR method for MinION and illumina sequencing of zika and other virus genomes directly from clinical samples. Nat. Protoc. 12, 1261–1276. doi: 10.1038/nprot.2017.066
Shirato, K., Nao, N., Katano, H., Takayama, I., Saito, S., Kato, F., et al. (2020). Development of genetic diagnostic methods for novel coronavirus 2019 (nCoV-2019) in Japan. Jpn J. Infect Dis. doi: 10.7883/yoken.JJID.2020.061. [Epub ahead of print].
Keywords: cruise ship, imported case, genome epidemiology, single-nucleotide variations, haplotype network
Citation: Sekizuka T, Kuramoto S, Nariai E, Taira M, Hachisu Y, Tokaji A, Shinohara M, Kishimoto T, Itokawa K, Kobayashi Y, Kadokura K, Kamiya H, Matsui T, Suzuki M and Kuroda M (2020) SARS-CoV-2 Genome Analysis of Japanese Travelers in Nile River Cruise. Front. Microbiol. 11:1316. doi: 10.3389/fmicb.2020.01316
Received: 06 April 2020; Accepted: 25 May 2020;
Published: 05 June 2020.
Edited by:
Tatsuo Shioda, Osaka University, JapanReviewed by:
Ahmed Mohamed Kandeil, National Research Centre, EgyptCopyright © 2020 Sekizuka, Kuramoto, Nariai, Taira, Hachisu, Tokaji, Shinohara, Kishimoto, Itokawa, Kobayashi, Kadokura, Kamiya, Matsui, Suzuki and Kuroda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Makoto Kuroda, bWFrb2t1cm9AbmlpZC5nby5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.