 V. Kavi Sidharthan
V. Kavi Sidharthan Amitha Mithra Sevanthi
Amitha Mithra Sevanthi Sarika Jaiswal
Sarika Jaiswal V. K. Baranwal
V. K. Baranwal
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 05 June 2020
Sec. Virology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.01232
This article is part of the Research Topic Next-Generation Sequencing and CRISPR-Cas Editing in Plant Virology View all 17 articles
Next-generation sequencing (NGS) based virome analyses of mRNA and sRNA have recently become a routine approach for reliable detection of plant viruses and viroids. In the present study we identified the viral/viroidal spectrum of several Indian grapevine cultivars and reconstructed their whole genomes using the publically available mRNAome and sRNAome datasets. Twenty three viruses and viroids (including two variants of grapevine leafroll associated virus 4) were identified from two tissues (fruit peels and young leaves) of three cultivars among which nine unique grapevine viruses and viroids were identified for the first time in India. Irrespective of the assemblers and tissues used, the mRNA based approach identified more acellular pathogens than the sRNA based approach across cultivars. Further, the mRNAome was on par with the whole transcriptome in viral identification. Through de novo assembly of transcriptomes followed by mapping against reference genome, we reconstructed 19 complete/near complete genomes of identified viruses and viroids. The reconstructed viral genomes included four larger RNA genomes (>13 kb), a DNA genome (RG grapevine geminivirus A), a divergent genome (RG grapevine virus B) and a genome for which no reference is available (RG grapevine virus L). A large number of SNPs detected in this study ascertained the quasispecies nature of viruses. Detection of three recombination events and phylogenetic analyses using reconstructed genomes suggested the possible introduction of viruses and viroids into India from several continents through the planting material. The whole genome sequences generated in this study can serve as a resource for reliable indexing of grapevine viruses and viroids in quarantine stations and certification programs.
Grapevine (Vitis vinifera L.) is an important cash crop grown worldwide (McGovern, 2003). Being a clonally propagated crop, grapevine is amenable for coinfection by different viruses and viroids (Jo et al., 2018). It is reported to be susceptible to the largest number of acellular pathogens compared to other crop species (Beuve et al., 2018; Hily et al., 2018). Till date, more than 70 viruses and 7 viroids have been reported to infect grapevine (Singhal et al., 2019). Many a time, grapevine viruses deviate from the classical ‘one pathogen – one disease’ concept, i.e., interaction among more than one viral agent leads to disease development (Byrd and Segre, 2016).
Planting pathogen free crop propagules is of paramount importance in grapevine for increasing the productive life of vineyards (Kumar et al., 2015). Traditional detection techniques like ELISA, PCR and their variants are employed for indexing of a few selected viruses of grapevine while certifying the planting material for commercial planting. But these methods can only answer whether the pathogen(s) under investigation is present or not, leaving the status of all other untested viruses in the planting material unknown (Czotter et al., 2018). To assure the health of the planting material of grapevine that remains productive in the field for an average of 15 years in India, it would be necessary to subject it to rigorous indexing for all possible grapevine infecting viruses/viroids. For this, it would be essential to study the virome (total viral population) of the mother stock, the results of which can then be used for developing appropriate detection assays of all pathogens for screening the clonal propagules. Next-generation sequencing (NGS) approaches can provide us with a snapshot of the virome present in the propagule as they are effective not only in detecting the known viral pathogens and their variants, if any, but also in unravelling unknown one(s) (Jo et al., 2018). Among the various NGS based approaches, sRNA (sRNAome) and mRNA (mRNAome) sequencing are commonly used to reveal the virome of a given sample (Pantaleo et al., 2010; Pirovano et al., 2015; Jones et al., 2017; Maliogka et al., 2018; Pooggin, 2018; Massart et al., 2019). Recently, a few studies attempted to reveal the virome of different crops like grapevine, apple, and pepper from publically available mRNAome data (Jo et al., 2015, 2016, 2017).
Both sRNA and mRNA pools can effectively capture single as well as double strand RNA viruses and some DNA viruses (Seguin et al., 2014; Roossinck et al., 2015; Jo et al., 2017). However, the relatively lower representation of viral RNA in the background of total plant RNA limits the use of mRNAome compared to sRNAome for viral detection (Beuve et al., 2018; Maliogka et al., 2018). As mRNA based methods can give longer contigs, they are more useful for variant detection, especially when significant genetic diversity exists as found in some of the grapevine viruses such as grapevine leafroll associated virus 3 (GLRaV3) (Xiao et al., 2019). Thus, it would be worthwhile to study the virome using both these methods for robust identification of entire virome of a plant species.
Though India grows grapevine on 137,000 hectares and exports 185,172 tonnes of grapes annually (FAOSTAT, 2017), only a few studies have been attempted to detect grapevine viruses in India. All these studies targeted only one/few virus(es)/ viroid(s) at a time using traditional detection methods (Kumar et al., 2012, 2013; Sahana et al., 2013; Adkar-Purushothama et al., 2014; Rai et al., 2017; Marwal et al., 2019; Singhal et al., 2019). The current study is the first virome report of grapevines from India using sRNA and mRNA datasets of three Indian grapevine cultivars available in the public domain (Tirumalai et al., 2019) identifying a large number of viruses and viroids.
Detailed information on plant materials and library construction is available in Tirumalai et al. (2019). In brief, total RNA was isolated from fruit peels (FP) and young leaves (YL) of three grapevine cultivars- Bangalore Blue (BB), Dilkush (DK), and Red Globe (RG). mRNA-seq and sRNA-seq libraries with two biological replicates, 24 in total, were constructed from isolated total RNA according to the NEXT flex Rapid directional mRNA-seq bundle library protocol (Trapnell et al., 2012) and the TruSeq Small RNA Sample Preparation Guide (Illumina, San Diego, CA, United States) respectively. Sequencing was performed on the Illumina NextSeq500 platform which yielded 75 bp single end reads. Thus, a total, of 12 mRNA and 12 sRNA libraries obtained from two tissues (FP, YL) of three grapevine cultivars (BB, DK, RG) in two biological replicates were analyzed in the current study. The details of the materials used and the complete processing pipeline are indicated in Figure 1.
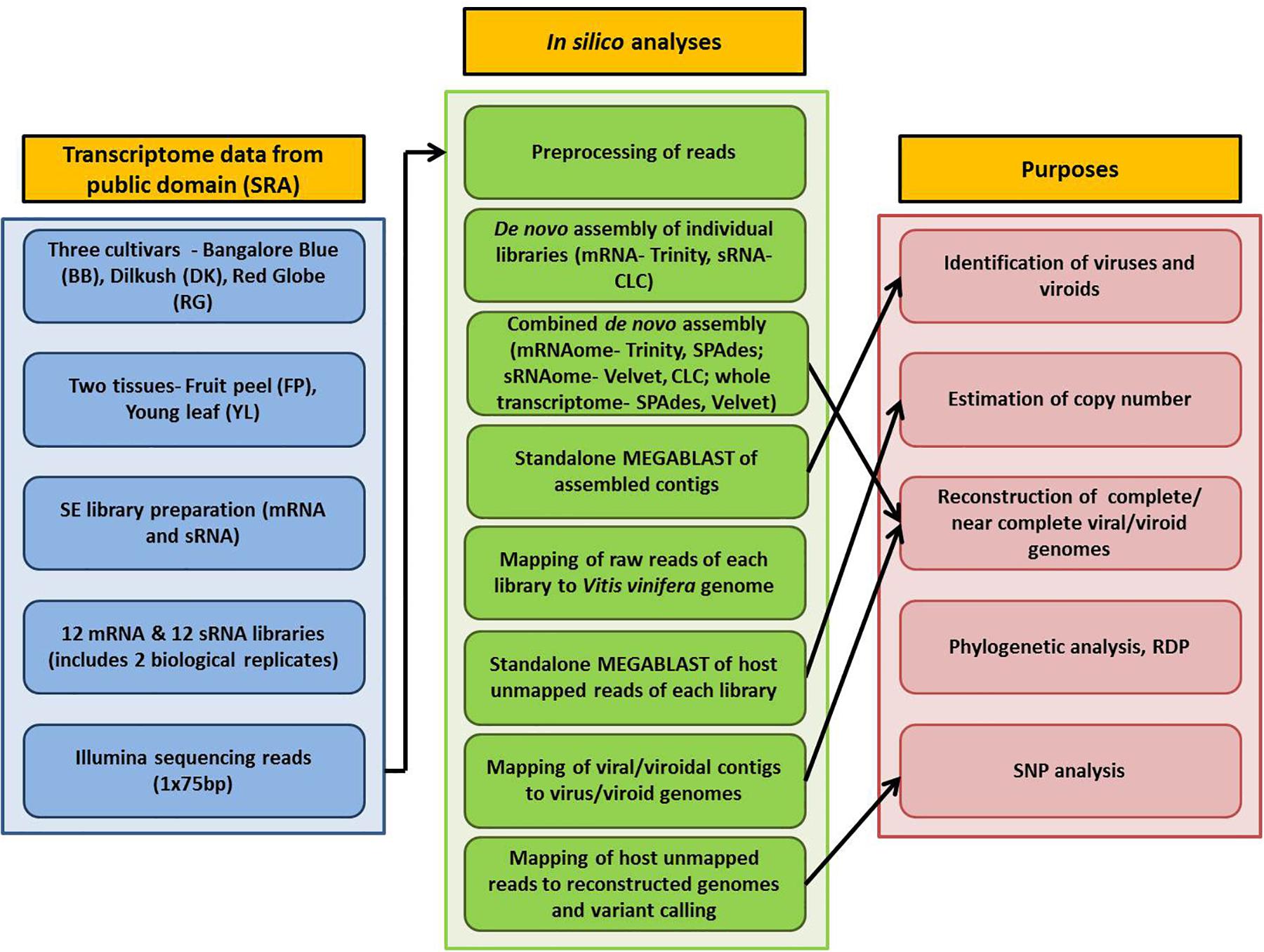
Figure 1. Schematic representation of the viromic study of three Indian grapevine cultivars.
The bioinformatics analyses were performed using Advanced Super Computing Hub for Omics Knowledge in Agriculture (ASHOKA) facility at ICAR-IASRI, New Delhi, India. Raw data of 24 libraries were downloaded from SRA database and converted to FASTQ files using the SRA toolkit version 2.9.6 (Leinonen et al., 2010). Three approaches were followed for de novo assembly. In the first approach, mRNA and sRNA libraries, 12 each, were individually assembled using Trinity version 2.5.1 (Grabherr et al., 2011) and CLC genomics workbench 12 de novo assembly tool, respectively with default parameters. For the second approach, combined mRNAome or sRNAome for each cultivar was obtained by aggregating corresponding mRNA or sRNA reads, respectively from four libraries (including two tissues and two replicates) of each individual cultivar. Similarly, whole transcriptome of each cultivar was obtained by aggregating both mRNA and sRNA reads from eight libraries of individual cultivar in the third approach. Trinity (k = 25), SPAdes (k = 21,23,25) version 3.13.1 (Bushmanova et al., 2019) and CLC (automatic word size = 20), Velvet (k = 17) version 1.2.10 (Zerbino and Birney, 2008) were used to assemble combined mRNAomes and sRNAomes, respectively while whole transcriptomes were assembled using SPAdes (k = 17,19,21) and Velvet (k = 21) assemblers.
All the assembled contigs were subjected to standalone MEGABLAST analysis (e-value cut off: 1e-5; query coverage: ≥ 80%) against the complete reference sequences of viruses and viroids1 using NCBI blast+ version 2.9.0. Only contigs of greater than 50 (for sRNAome) and 200 nucleotides (mRNAome and whole transcriptome) were considered for analyses. To validate the viruses/viroids identified through assembly, the reads of each mRNA/sRNA library were first mapped to the Vitis vinifera genome (GCF_000003745.3) using CLC workbench mapping tool with default parameters (match score-1, mismatch cost-2, length fraction-0.5, similarity fraction-0.8). The unmapped reads were then analyzed using MEGABLAST algorithm (e-value cut off: 1e-5; query coverage: ≥80%) against the reference genomes of viruses and viroids. Only those viruses/vioids that were detected through assembly (from sRNAome/mRNAome/whole transcriptome) and BLAST analysis of reads from at least two libraries (derived from the particular nucleic acid pool from which the contigs were obtained) of the corresponding cultivar were considered. To arrive at the copy number for a virus/viroid, the number of reads associated with either RdRp ORF (in case of viruses that use sub-genomic RNA (sgRNA) for translation) or the entire polyprotein [in grapevine fleck virus (GFkV), grapevine rupestris vein feathering virus (GRVFV)] or the entire genome (in viroids) was multiplied with 75 (for mRNA)/ 24 (for sRNA) followed by division with the size (bp) of the corresponding genomic region of the virus/viroid. Intact mRNA reads were used for copy number estimation while the pre-processed reads were used in case of sRNA. The average length of pre-processed sRNA reads in all libraries was near to 24. Hence the factors 75 and 24 were used for mRNA and sRNA libraries, respectively. As a reference genome for grapevine virus L (GVL) was not available in NCBI, we included the de novo assembled GVL genome of the present study (that was identified by performing BLASTn analysis of larger contigs against “non-redundant” (nr) (NCBI) database) for MEGABLAST analysis.
Virus/viroid associated contigs were filtered from the total contigs using SAM tools version 1.9 (Li et al., 2009). The detailed procedure followed for genome reconstruction is given in Supplementary Figure S1. In brief, the Trinity assembled longer contigs from combined mRNAomes were examined for the presence of intact viral/viroidal genome. Further, the SPAdes assembled longer contigs from combined mRNAomes and whole transcriptomes were examined followed by inspection of Trinity assembled larger contigs in individual mRNA libraries. Next, the Trinity assembled viral/viroidal contigs from combined mRNAomes were mapped against the NCBI designated reference genomes of identified viruses and viroids (CLC workbench mapping tool). In cases where the Trinity assembled contigs were insufficient to reconstruct the entire genome, SPAdes assembled contigs from combined mRNAomes and whole transcriptomes were supplemented during mapping. Still, if the genome could not be obtained, the most closely related genome was used as reference during mapping. The full length consensus sequence, if obtained, after mapping/directly by de novo assembly was considered as the complete/near complete genome for a particular virus/viroid. To find ORFs in assembled viral genomes, we used NCBI ORF finder2.
The complete genomes retrieved from NCBI along with the viral/viroid genomes reconstructed in this study were aligned using CLUSTALW tool in MEGA7 software version 7.0.26 (Kumar et al., 2016). Aligned sequences were subjected to pairwise distance analysis and phylogenetic tree construction using neighborhood joining (NJ) method and Kimura 2-parameter (K2P) model with 1000 bootstrap replicates. For grapevine geminivirus A (GGVA), grapevine latent viroid (GLVd), grapevine leafroll associated virus 4 (GLRaV4), grapevine virus B (GVB), GVL, grapevine rootstock stem lesion associated virus (GRSLaV) and GRVFV, all the respective complete genomes available in NCBI were used for analysis. Owing to the availability of a large number of genome sequences for GLRaV3, only those sequences showing 100% query coverage in BLASTn analysis against nr (NCBI) database were taken for analysis. Similarly, in cases of Australian grapevine viroid (AGVd), grapevine yellow speckle viroid-1, -2 (GYSVd1, GYSVd2), and hop stunt viroid (HSVd), only 10 non-redundant genomes that were highly similar to each isolate of a viroid were used. In all the cases, an outgroup (except for pairwise distance analysis) and the NCBI designated reference genome (except GVL, for which there is no designated reference sequence) were included.
The host unmapped reads of individual cultivars were mapped against the complete/near complete viral/viroid genomes assembled from the corresponding cultivar using the mapping tool available in CLC workbench using default parameters (match score-1, mismatch cost-2, length fraction-0.5, similarity fraction-0.8). For SNP detection, the mapped files were subjected to fixed ploidy variant detection using CLC workbench. As viral genomes are haploid, ploidy value was considered as one throughout the analyses.
Using CLUSTALW aligned MEGA file as input, recombination analysis was performed using RDP4 package version 4.39 (Martin et al., 2015) employing nine different algorithms. Only recombination events detected by at least five algorithms in the reconstructed viral genomes were considered. Only viral sequences used for phylogenetic analyses were used for detection of recombinants.
The number of raw reads ranged from 10.5 to 40.2 million with an average of 23.3 million for mRNA and 2.9 to 8.3 million with an average of 4.3 million for sRNA libraries (Table 1). As mRNA reads were of acceptable quality (without adapter sequences; phred-score > 20), we proceeded directly for de novo assembly while sRNA reads were filtered to remove adapter sequences and poor quality reads (quality scores < 0.05).
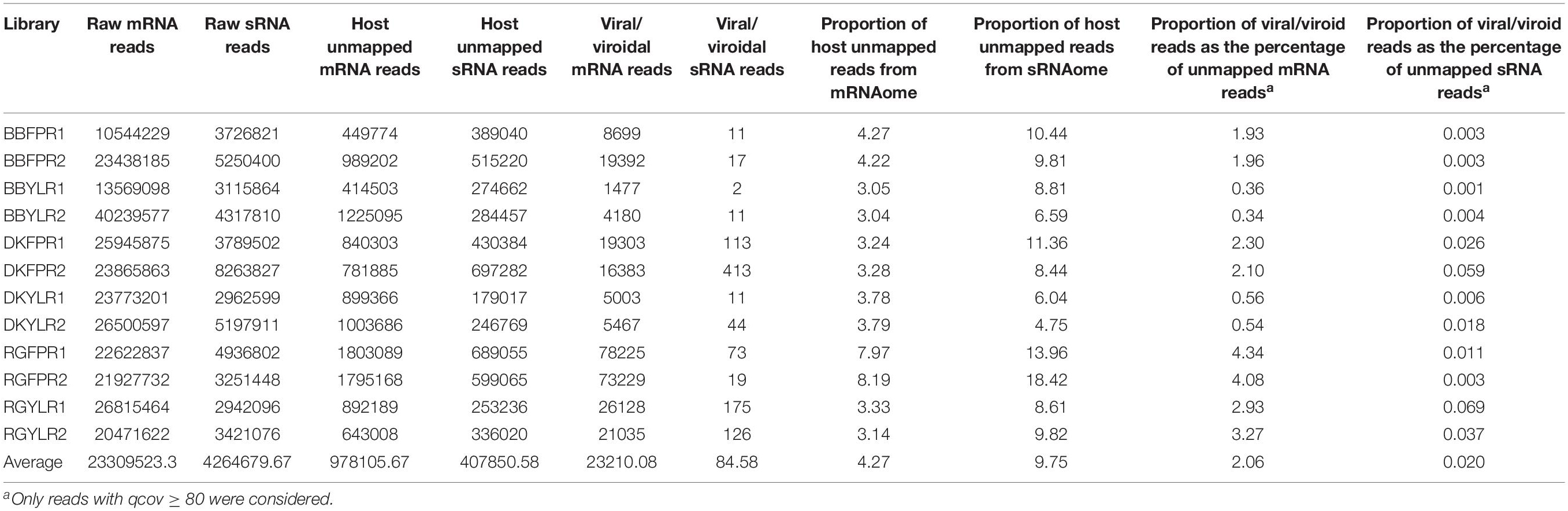
Table 1. Raw reads, host unmapped reads, and viral/viroid associated reads of each library.
We identified more viruses and viroids from mRNAome (23) than sRNAome (7) across cultivars and tissues (Supplementary Table S1). The only exception for this was the FP-specific sRNA datasets of cv. DK which identified six viruses and viroids while the corresponding mRNAome could identify only five. Among the two tissues, relatively more viruses/viroids were identified in FP than YL in all cultivars except DK from mRNA libraries. However, nearly similar number of viruses/viroids was identified from sRNA libraries across tissues and cultivars (Supplementary Figure S2 and Supplementary Table S1). Combined mRNAome assembly using Trinity identified the same number of viruses and viroids (23) across cultivars as compared to the individual mRNA libraries (23). However, on a closer look, we found that combining the reads of the two tissues of each cultivar did offer some advantage in case of mRNAome, since additional virus(es)/viroid(s) were identified in BB (1), DK (2) and RG (1). The only exception to this is GVE that was detected in individual mRNA libraries but not in combined mRNAome (Figures 2A,B, Supplementary Figures S3A,B, and Supplementary Table S2). Similarly, combined sRNAome assembly using CLC was more effective as it could identify two unique viruses (GVF in DK and GRSLaV in RG) in addition to the seven viruses and viroids identified by the individual library approach across cultivars (Figures 2D,E, Supplementary Figures S4A,B, and Supplementary Table S3). Between the combined sRNAome and combined mRNAome, the former could identify only a fraction of viruses and viroids (12) identified by the latter even after accounting for the viruses and viroids identified by all the assemblers. Interestingly, from combined mRNAomes and whole transcriptomes exactly the same number of viruses/viroids was identified in BB, DK and RG cultivars (6, 10, and 21), representing a total of 23 viruses/viroids though the identities of a few differed in cvs. DK and RG. The identified acellular pathogens included 14 grapevine viruses (including two GLRaV4 variants), four mitoviruses and five viroids – Alternaria alternata chrysovirus 1 (AaCV1), Alternaria arborescens mitovirus 1 (AaMV1), AGVd, Erysiphe necator mitovirus 1 (EnMV1), Erysiphe necator mitovirus 3 (EnMV3), GFkV, GGVA, GLVd, GLRaV3, grapevine leafroll associated virus -4, -5, -6 (GLRaV4, GLRaV5, GLRaV6), GVA, GVB, GVL, GVE, GVF, GRSLaV, GRVFV, GYSVd1, GYSVd2, HSVd and tobacco streak virus (TSV). It is worthy of mention that while TSV was detected from cvs. BB, RG in the mRNAome, none of the sRNA libraries could detect it (Supplementary Tables S2, S3). Interestingly, GLRaV3, GVA, GVB, GYSVd1, and HSVd were identified in all the cultivars (Figures 3A–D and Supplementary Table S4).
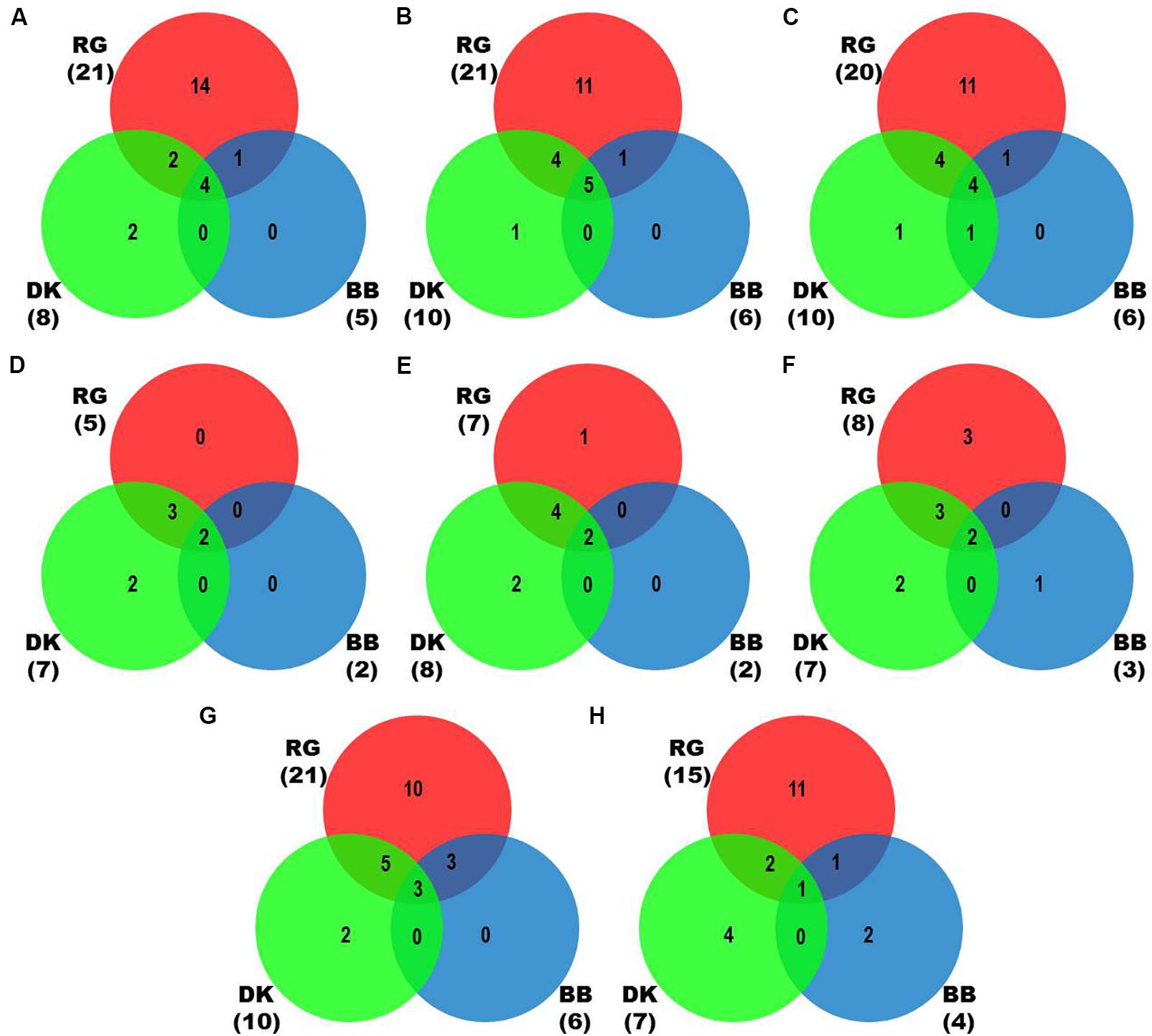
Figure 2. Identification of viruses and viroids in three Indian grapevine cultivars – BB, DK, RG. Venn diagrams display the number of identified viruses and viroids based on Trinity assembled contigs from individual mRNA libraries of each cultivar (A), Trinity (B), SPAdes (k = 21,23,25) assembled contigs from combined mRNAome of each cultivar (C), CLC assembled contigs from individual sRNA libraries of each cultivar (D), CLC (E), Velvet (k = 13,15,17) assembled contigs from combined sRNAome of each cultivar (F), SPAdes (k = 17,19,21) (G) and Velvet (k = 17,19,21) (H) assembled contigs from whole transcriptome of each cultivar. Blue, green and red circles in the Venn diagrams represent the cvs. BB, DK, and RG, respectively.
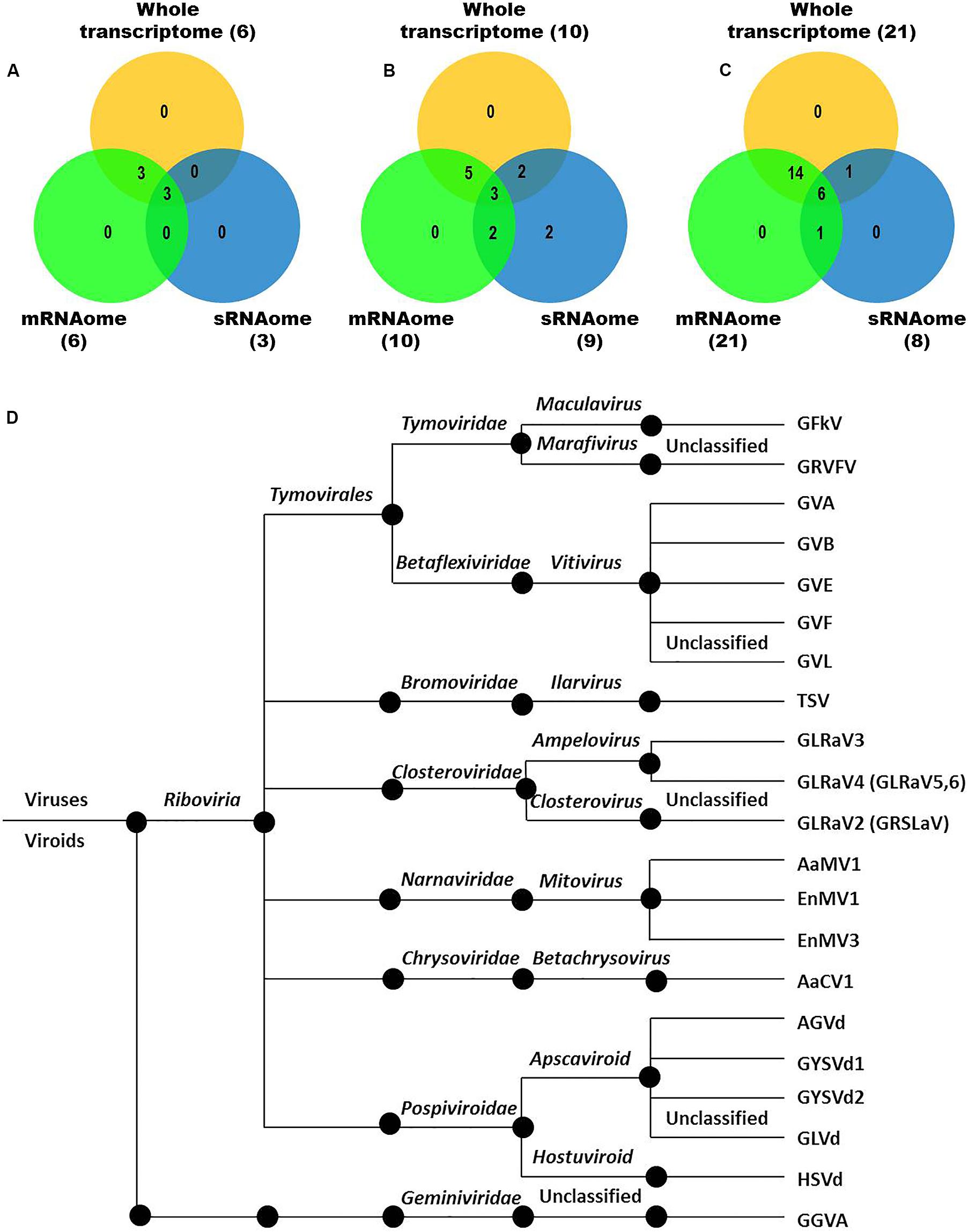
Figure 3. Viruses and viroids identified in three Indian grapevine cultivars – BB, DK, RG. Venn diagram displaying viruses and viroids identified by any of the assemblers from combined mRNAomes, sRNAomes and whole transcriptomes in cultivars BB (A), DK (B), and RG (C). Blue, green, and yellow circles in the Venn diagrams represent the sRNAome, mRNAome, and whole transcriptome, respectively. Classification of the identified viruses and viroids is on the basis of taxonomy (D).
In case of combined mRNAomes, the number of viruses and viroids identified by both the assemblers were similar except in cv. RG where Trinity identified one additional viroid (GYSVd1) compared to SPAdes (Figures 2B,C and Supplementary Table S2). However, both the assemblers identified almost similar number of viral contigs for all but one cultivar (Supplementary Figures S3B,C). In case of combined sRNAomes, Velvet identified one additional virus in BB (GVA) and RG (GVE) than CLC but CLC detected two Velvet undetected virus/viroid in DK (GVF, GYSVd1). Interestingly, GVB was identified in cv. DK by Velvet but not by CLC (Figures 2E,F and Supplementary Table S3). Considering the number of viral and viroid contigs, Velvet identified 67.3, 14.7, and 52.3% more contigs than CLC in BB, DK, and RG (Supplementary Figures S4B,C). In case of whole transcriptome assembly, SPAdes identified more viruses (2, 3, and 6 additional viruses/viroids in cvs. BB, DK, and RG, respectively) and viral contigs as compared to Velvet in all cultivars. Notably Velvet based assembly failed to identify HSVd from any whole transcriptome, or GYSVd2 from DK or GYSVd1,2 from RG, whereas SPAdes identified HSVd in BB and RG, and GYSVd2 in DK and GYSVd1,2 in RG from the corresponding whole transcriptomes. However, Velvet did detect HSVd in each of the combined sRNAomes and GYSVd2 from DK and GYSVd1,2 from RG (Figures 2G,H, Supplementary Figures S5A,B, and Supplementary Tables S3, S5).
The number of host unmapped mRNA and sRNA reads ranged from 0.41 to 1.80M and 0.18 to 0.70M, respectively across libraries. Though the number of host unmapped reads was higher (0.98M) in case of mRNA compared to sRNA (0.41M), the proportion of unmapped reads to total reads was higher in the latter (9.75%) than the former (4.27%). On average, 2.06 and 0.02% of host-unmapped reads from mRNA and sRNA libraries mapped to viral/viroidal genomes (Supplementary Figures S6A,B and Table 1). In general, the proportion of virus/viroid associated reads was relatively higher in mRNA libraries constructed from FP than YL while no such trend was observed in case of sRNA libraries. Based on copy number estimates, HSVd (94–100%) predominated in cv. BB in both mRNA and sRNA libraries. In case of mRNA libraries of cvs. DK and RG, HSVd and GYSVd2 were predominant in FP and YL, respectively. In sRNA libraries of cv. DK and in all but one sRNA libraries of cv. RG, GYSVd2 was predominant irrespective of tissue type (Supplementary Figures S7A,B). Further, both the replicates in each tissue of a cultivar were highly similar not only in detecting the viromes but also in estimating their copy number.
By mapping, the viral/viroid associated contigs from combined mRNAome and whole transcriptome of each cultivar against the NCBI designated reference genomes of identified viruses and viroids we obtained complete or near complete (>99%) genomes of 15 viruses and viroids from three cultivars (Table 2). Some other viral contigs could not be assembled into full genomes using the reference genomes as scaffolds. For the assembly of GLRaV3 and GLRaV4 genomes from cv. RG, the longest Trinity assembled contig of each virus was first blasted against the nr (NCBI) database. The complete genome of the most highly similar isolate was then used as a reference during mapping in each case. Trinity assembly of library RGFPR2 directly yielded the whole genome of GVB. Similarly, we obtained GVL genome from one of the Trinity assembled longest contigs from combined mRNAome of cv. RG through BLAST against nr (NCBI) database. In total, we obtained 19 complete/near complete viral/viroid genomes from three cultivars (Table 2). Trinity yielded relatively longer contigs for most viruses and viroids as compared to SPAdes in all cultivars with mRNA reads (Supplementary Figures S8, S9). On the contrary, SPAdes yielded relatively longer viral/viroid contigs as compared to Velvet in most instances when whole transcriptomes were assembled (Supplementary Figures S10, S11). Though Velvet assembled more viral/viroid contigs from combined sRNAomes, CLC yielded longer contigs for most viruses and viroids as compared to Velvet (Supplementary Figures S12, S13). However, we could not reconstruct any viral genome using contigs assembled from combined sRNAomes. From the reconstructed complete/near complete viral genomes, we could identify all of the anticipated ORFs for all recovered viruses using NCBI ORF finder (Supplementary Table S6). Failure to identify intact ORFs in nearly complete genomes that could be assembled to the tune of >95% (Supplementary Table S7) were still deemed incomplete.
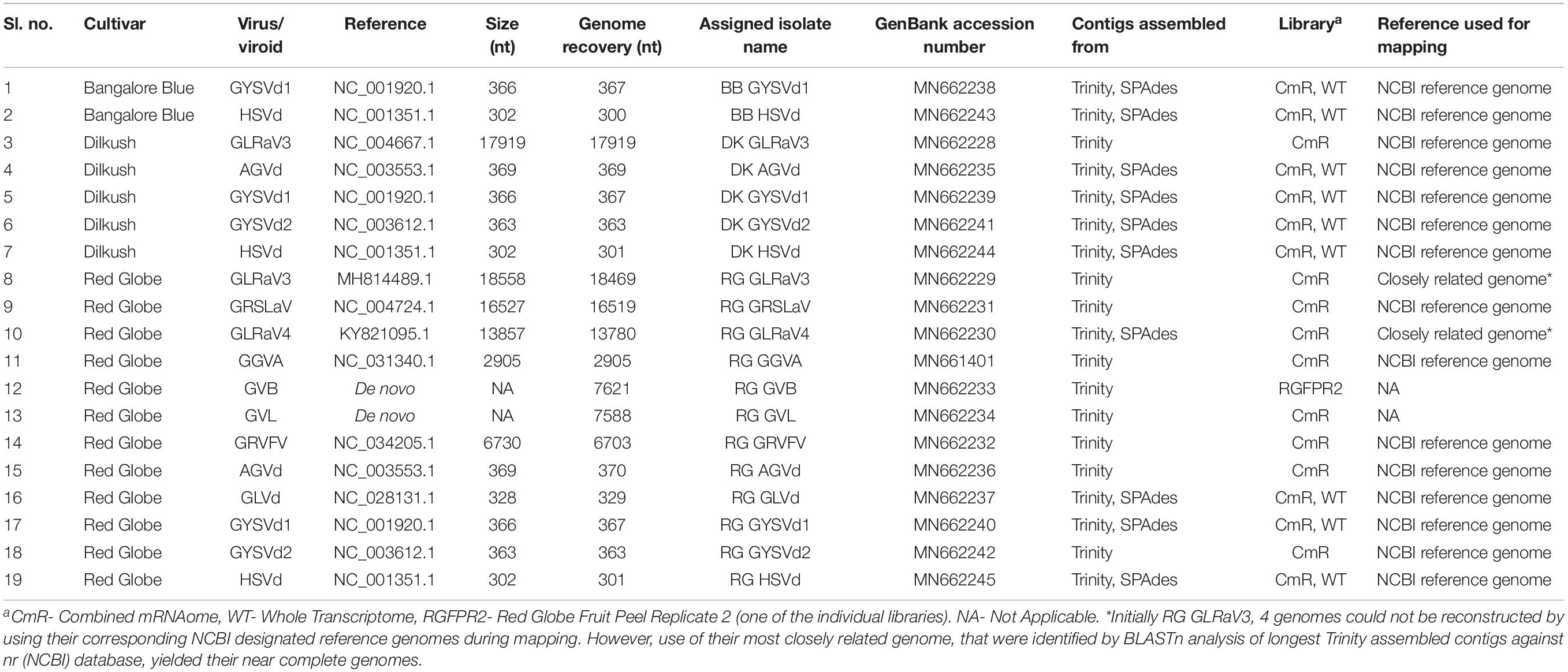
Table 2. Summary of complete/near complete viral/viroidal genomes reconstructed from three Indian grapevine cultivars.
Each of the complete/near complete genomes obtained were subjected to pairwise distance (Supplementary Tables S8–S19) and phylogenetic analyses (Figures 4A–L) along with related complete genomes retrieved from NCBI, and the most closely related genomes are indicated here, including their country of origin.
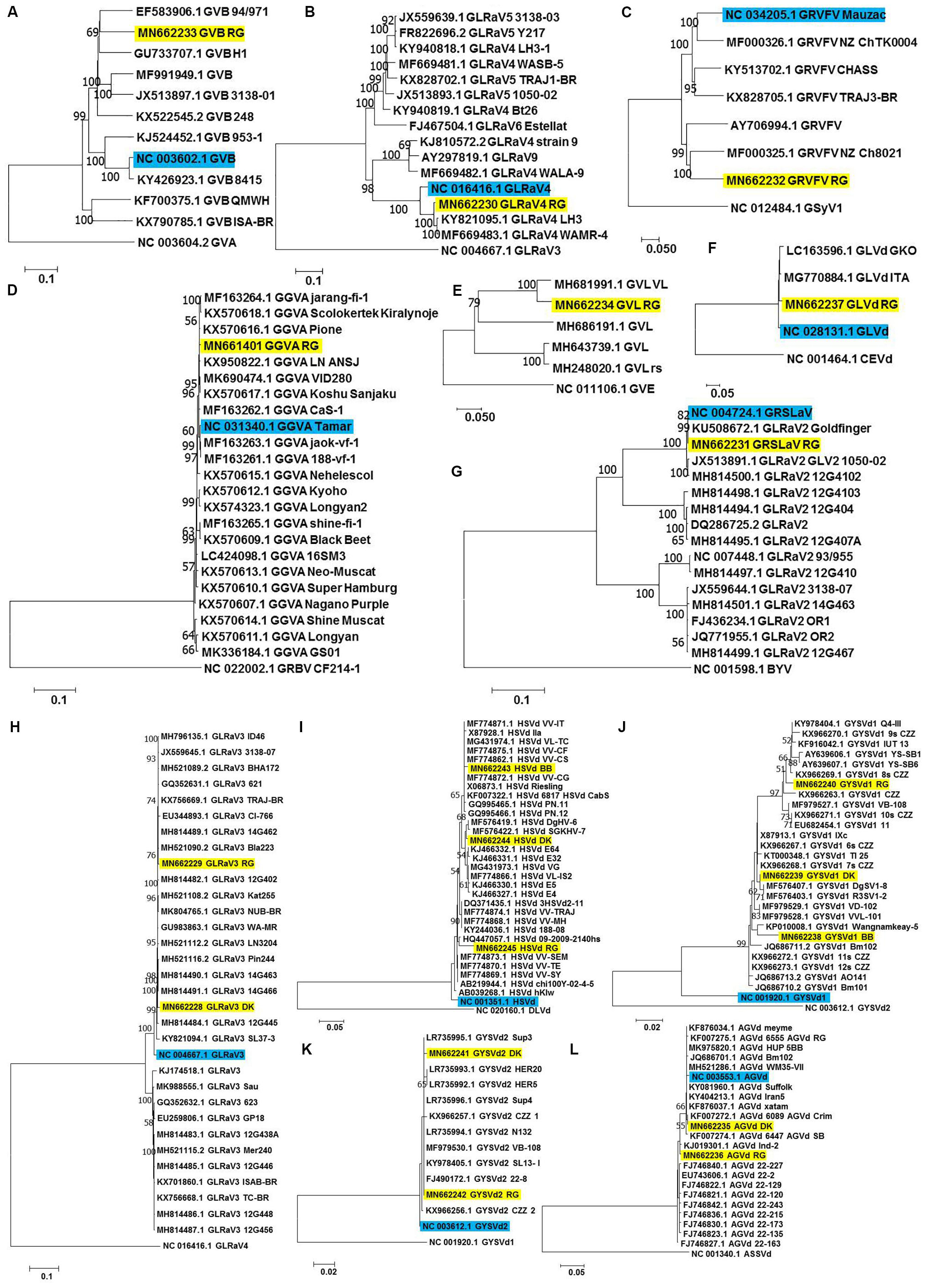
Figure 4. Phylogenetic analyses for genome reconstructed viruses and viroids. Phylogenetic relationships of GVB (A), GLRaV4 (B), GRVFV (C), GGVA (D), GVL (E) GLVd (F), GRSLaV (G), GLRaV3 (H), HSVd (I), GYSVd1 (J), GYSVd2 (K), and AGVd (L) isolates obtained in this study with global isolates. Phylogenetic tree was constructed using Neighborhood joining (NJ) method and Kimura 2-parameter model (K2P) with 1000 bootstrap replicates. Yellow colored boxes indicate the isolates obtained in this study. Blue colored boxes indicate the corresponding NCBI designated reference genome. Only bootstrap values more than 50 are shown. An outgroup was used in the analysis of each virus/viroid.
a-RG GVB (MN662233) was related to a South African isolate GVB-H1 [79.7% nucleotide (nt) identity; GU733707.1] (Figure 4A and Supplementary Table S8).
b-RG GLRaV4 (MN662230) was closely related to a Pakistani isolate LH3 (97.9% nt identity; KY821095.1) (Figure 4B and Supplementary Table S9).
c-RG GRVFV (MN662232) was related to a New Zealand isolate NZ Ch8021 (80.6% nt identity; MF000325.1) (Figure 4C and Supplementary Table S10).
d-RG GGVA (MN661401) was most closely related to a Japanese isolate Pione (99.6% nt identity; KX570616.1) (Figure 4D and Supplementary Table S11).
e-RG GVL (MN662234) was closely related to a Croatian isolate VL (94.0% nt identity; MH681991.1) (Figure 4E and Supplementary Table S12).
f-RG GLVd (MN662237) was closely related to an Italian isolate ITA (98.4% nt identity; MG770884.1) (Figure 4F and Supplementary Table S13).
g-RG GRSLaV (MN662231) was most closely related to a Californian isolate obtained from the cv. RG (99.8% nt identity; NC_004724.1) (Figure 4G and Supplementary Table S14).
h-DK GLRaV3 (MN662228) shared 99.0% nt identity with Canadian isolates- 14G463 (MH814490.1), 14G466 (MH814491.1), 14G462 (MH814489.1), 3138-07 (JX559645.1), a Brazilian isolate TRAJ-BR (KX756669.1) and an US isolate WA-MR (GU983863.1).
i-RG GLRaV3 (MN662229) shared 99.4% nt identity with US isolates- Bla223 (MH521090.2), WA-MR 314 (GU983863.1), Canadian isolates- 14G463 (MH814490.1), 14G462 (MH814489.1) and a Brazilian isolate TRAJ-BR (KX756669.1).
j-GLRaV3 isolates from cvs. DK and RG showed only 1.3% divergence (Figure 4H and Supplementary Table S15).
k-BB HSVd (MN662243) shared 99.3% nt identity with Brazilian isolates- VL-TC (MG431974.1), VV-CF (MF774875.1), VV-CG (MF774872.1), VV-IT (MF774871.1), VV-CS (MF774862.1) and a German isolate obtained from cv. Riesling (X06873.1).
l-DK HSVd (MN662244) was closely related to a Nigerian isolate DgHV-6 (99.3% nt identity; MF576419.1) while RG HSVd (MN662245) was closely related to a New Zealand isolate 09-2009-2140hs (96.8% nt identity; HQ447057.1). Interestingly, RG HSVd diverged largely from DK HSVd (4.7%) and BB HSVd (5.5%) while DK HSVd showed 2.9% divergence from BB HSVd (Figure 4I and Supplementary Table S16).
m-Similarly, while BB GYSVd1 (MN662238) was closely related to a Thailand isolate Wangnamkeay-5 (98.0% nt identity; KP010008.1), DK GYSVd1 (MN662239) shared 99.7% nt identity with a German isolate IXc (X87913.1), two Nigerian isolates DgSV1-8 (MF576407.1), R3SV1-2 (MF576403.1) and two Chinese isolates clone 6s CZZ (KX966267.1), clone 7s CZZ (KX966268.1) and RG GYSVd1 (MN662240) was most closely related to yet another Pakistani isolate Q4-III (99.1% nt identity; KY978404.1).
n-Like HSVd, RG GYSVd1 diverged from DK (3.2%) and BB GYSVd1 (5.0%) while DK GYSVd1 showed 2.9% divergence from BB GYSVd1 (Figure 4J and Supplementary Table S17).
o-RG GYSVd2 (MN662242) was identical (100% nt identity) to Greek isolates- Sup4 (LR735996.1), Sup3 (LR735995.1), N132 (LR735994.1), a Croatian isolate VB-108 (MF979530.1), a Pakistani isolate SL13- I (KY978405.1) and a Chinese isolate clone 22-8 (FJ490172.1) while the DK GYSVd2 (MN662241) isolate shared 99.7% nt identity with all the six closest relatives of RG GYSVd2 and also with the RG GYSVd2 isolate (Figure 4K and Supplementary Table S18).
p-DK AGVd (MN662235) was identical to a Chilean isolate 6089_AGVd_Crim (100% nt identity; KF007272.1) while RG AGVd (MN662236) shared 99.4% nt identity with an Indian isolate Ind-2 (KJ019301.1) and two Chinese isolates- clone 22-2 (EU743606.1), clone 22-129 (FJ746822.1). It is noteworthy that DK and RG AGVd isolates fell within two separate clades (1.9% divergence) (Figure 4L and Supplementary Table S19).
A large number of SNPs was detected for RG GRVFV (168) followed by RG GLRaV3 (117), that were equally distributed throughout the genome, while no SNP was detected in case of RG GGVA, RG GLVd, RG GYSVd2, DK AGVd, and BB GYSVd1. Other viruses that had a good number of SNPs included GLRaV3 from cv. DK (102) and GLRaV4 (100), GVL (64), and GVB (40) from cv. RG (Figure 5A).
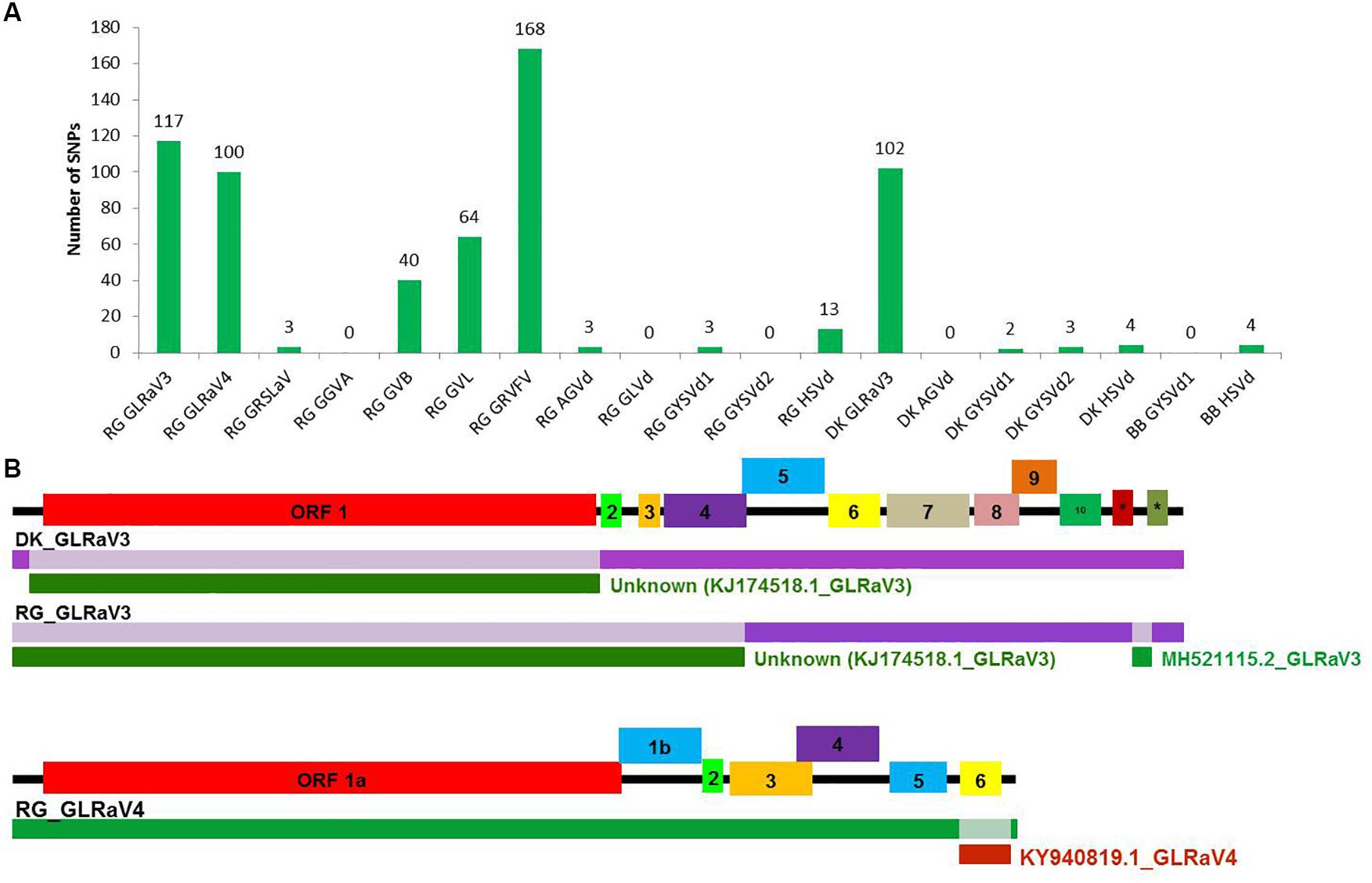
Figure 5. SNP detection and recombination analyses in whole genome reconstructed viruses and viroids. The number of identified SNPs for each virus and viroid (A). Identification of recombination events in reconstructed viral genomes using RDP4 program. (B) The positions of identified recombination events are indicated in a different color in the genome. The corresponding minor parental sequence is indicated by a differently colored bar below each event along with the name. #-ORF 11, *-ORF 12 in GLRaV3.
The reconstructed viral genomes, after alignment, were subjected to detection of recombination events. Among the eight reconstructed viral genomes, recombination events supported by at least five algorithms were detected in only three genomes. In GLRaV3 genomes of DK and RG, a similar recombination event was detected in 5′ region of the genome. An additional recombination event was detected in 3′ region of RG GLRaV3. For RG GLRaV4, we found only one recombinant sequence at 3′ region (Figure 5B and Supplementary Table S20).
In this study, viromes of three Indian grapevine cultivars were determined and some of their whole genomes were reconstructed from publically available mRNAome and sRNAome datasets (Tirumalai et al., 2019). Since the materials used in the present study were obtained from Indian Institute of Horticultural Research (in Bangalore, India) one of the leading grapevine breeding centers in the tropical region (Tirumalai et al., 2019), it is the most appropriate one for performing virome analysis as all the vegetative propagules derived from the breeding stock would be expected to be infected with the same viruses. Interestingly the cv. RG, an introduction from California had the maximum viral load in our study compared to the native cvs., BB and DK.
Uneven distribution of viruses and viroids across tissues of a perennial plant like grapevine (Kominek et al., 2009), suggested that sampling different tissues will reveal a more accurate sanitary status of a plant. We also found pooling samples from different tissues was more reliable than relying on individual tissue for virome analysis. Earlier, Jo et al. (2015), also reported the superiority of tissues-combined assemblies over the individual ones. We further observed that the combined mRNAome and whole transcriptome identified nearly similar acellular pathogens and both these approaches were more sensitive than individual or combined sRNAomes. This might be because of the smaller size and number of reads generated from sRNA libraries. Contrary to the observation of Maliogka et al. (2018), the proportion of viral and viroidal reads in mRNA libraries was higher than sRNA libraries in our study. This might be due to the fact that viral sRNAs are produced only upon activation of host’s antiviral defense while mRNAomes can even detect viruses and viroids that are unrecognized by the host (Hily et al., 2018). Further, similar number of viruses and viroids were identified by Trinity and SPAdes assemblers from mRNAomes and CLC and Velvet assemblers from sRNAomes. However, SPAdes outperformed Velvet in case of whole transcriptomes. So, when more than one assembler was used, one or more viruses that escaped detection by one assembler could be detected by the other (Massart et al., 2019). Thus, use of multiple tissues and assemblers enabled better unraveling of grapevine virome.
In the present study, we identified 19 grapevine viruses and viroids (including two variants of GLRaV4) and four mycoviruses associated with the grapevine fungal pathogens- Erysiphe necator and Alternaria spp. (Kakalikova et al., 2009; Feng et al., 2018). Included among these is GRSLaV, which was earlier reported as a novel virus from California in cv. RG. This indicates the possible introduction of GRSLaV from California along with the RG propagule. However, GRSLaV is now regarded as a strain of grapevine leafroll associated virus 2 (GLRaV2) (as GLRaV-2RG) (Alkowni et al., 2011). Nonetheless, this is the first study that could successfully detect GLRaV-2 or any of its variants in India. Though Kumar et al. (2013) did attempt to detect this virus in India they could not succeed rather they detected GLRaV1 and GLRaV3. Further, GLRaV5 and GLRaV6 are presently regarded as the strains of GLRaV4 (Rai et al., 2017). On this basis, nine grapevine viruses and viroids (GGVA, GLRaV2, GRVFV, GVA, GVE, GVF, GVL, TSV, and GLVd) were detected for the first time in grapevine cultivars grown in Indian soil. Interestingly, we could identify GVL, the reference for which is not yet available in the NCBI, using the GVL genome obtained in this study.
Of the 19 complete/near complete genomes (>99% completion but <100%) obtained in this study, seven viral (including four genomes with > 13 kb) and 1 viroidal genome were recovered for the first time from any Indian grapevine cultivar. None of the viral whole genomes could be recovered from combined sRNAome assembled contigs as reported by Baranwal et al. (2015) and Jo et al. (2016). However, this might be due to the use of lower number of sRNA reads (approximately one-fifth) as compared to the mRNA reads in the current study. Identification of DNA viruses in mRNAome is rare and construction of their whole genome is still scarce (Jo et al., 2017), but we could not only identify GGVA in mRNA of cv. RG but could also reconstruct its genome in entirety with 2905 nucleotides. Initially, the RG GVB and GVL genomes could not be recognized as the former diverged significantly (23%) from the reference genome while there was no reference genome for the latter. However, inspection of Trinity assembled longer contigs of individual and combined mRNA libraries through BLAST analysis against nr (NCBI) database coupled with ORF prediction, identified the whole genomes of these isolates. Complete genomes could not be reconstructed for RG GLRaV3, 4 isolates using reference-based mapping because of their divergence (2.7 and 9.6% divergence of RG GLRaV3, 4 genomes, respectively) from the corresponding reference genome, though their near complete genomes could be reconstructed using the closely related genomes. Thus, examination of larger contigs assembled by various de novo assemblers coupled with usage of increased number of reference genomes of a virus during mapping could increase the chances of whole genome recovery. Identification of large number of viral SNPs in this study ascertains the quasispecies nature of plant viruses (Jo et al., 2018). Hence, the complete/near complete genomes reconstructed in this study were the consensus of viral variants present in a given cultivar.
We followed Jo et al. (2015) for copy number estimation except that we considered reads from only the non sgRNA region to reflect the true abundance of viruses that use sgRNA strategy for translation. Phylogenetic and distance matrix analyses revealed the divergence of AGVd, HSVd and GYSVd1 isolates obtained from different cultivars while the GLRaV3 and GYSVd2 isolates from cvs. DK and RG were related. Recombination analyses revealed that the RG GLRaV3, DK GLRaV3, and RG GLRaV4 isolates were recombinants of global isolates. Owing to the vegetative propagation of grapevine and free trade of planting materials, viruses and viroids can easily spread globally. In addition, coinfection of a single plant with numerous isolates of same/different viruses offers scope for recombination among different isolates (Jo et al., 2016).
Plants do not always express symptoms associated with every virus/viroid present, hence symptomology and individual virus/viroid based detection assays are not sufficient to determine the full spectrum of viruses/viroids present in a plant. Rather, use of available or newer transcriptome datasets is a better choice for profiling of viromes that can serve as a reliable base for indexing of planting materials in plant quarantine stations and during certification.
The datasets analyzed in this study are available in the NCBI repository under the Bioprojects PRJNA421907 (sRNA-seq) and PRJNA421908 (mRNA-seq). The whole genomes of 19 viruses and viroids reconstructed in this study have been submitted to GenBank (MN662228 to MN662245 and MN661401).
VS, AS, SJ, and VB conceptualized and formulated the study and read and approved the final manuscript. VS performed the bioinformatics analyses. VS and AS have drafted the manuscript. VB and SJ edited the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are thankful to ICAR for the ASHOKA facility at ICAR-IASRI, New Delhi. They also acknowledge the support from Head, Division of Plant Pathology, ICAR-IARI, Dean, ICAR-IARI and Director, ICAR-IARI, New Delhi. VS is grateful to the DST for INSPIRE fellowship. The authors are also thankful to Mr. S. Saravana Kumar (SRF at IISER, Trivandrum), Mr. V. Sureshkumar (Young Professional at ICAR-NIPB, New Delhi) and Dr. Susheel Kumar Sharma (Scientist at ICAR-RC NEH, Manipur centre, Imphal) for providing technical guidance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01232/full#supplementary-material
Adkar-Purushothama, C. R., Kanchepalli, P. R., Yanjarappa, S. M., Zhang, Z., and Sano, T. (2014). Detection, distribution, and genetic diversity of Australian grapevine viroid in grapevines in India. Virus Genes 49, 304–311. doi: 10.1007/s11262-014-1085-5
Alkowni, R., Zhang, Y. P., Rowhani, A., Uyemoto, J. K., and Minafra, A. (2011). Biological, molecular, and serological studies of a novel strain of Grapevine leafroll-associated virus 2. Virus Genes 43, 102–110. doi: 10.1007/s11262-011-0607-7
Baranwal, V. K., Jain, P., Saritha, R. K., Jain, R. K., and Gautam, N. K. (2015). Detection and partial characterization of Cowpea mild mottle virus in mungbean and urdbean by deep sequencing and RT-PCR. Crop. Prot. 75, 77–79. doi: 10.1016/j.cropro.2015.05.015
Beuve, M., Hily, J. M., Alliaume, A., Reinbold, C., Le Maguet, J., Candresse, T., et al. (2018). A complex virome unveiled by deep sequencing analysis of RNAs from a french pinot noir grapevine exhibiting strong leafroll symptoms. Arch. Virol. 163, 2937–2946. doi: 10.1007/s00705-018-3949-9
Bushmanova, E., Antipov, D., Lapidus, A., and Prjibelski, A. D. (2019). rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data. Gigascience 8:giz100. doi: 10.1093/gigascience/giz100
Byrd, A. L., and Segre, J. A. (2016). Adapting Koch’s postulates. Science 351, 224–226. doi: 10.1126/science.aad6753
Czotter, N., Molnar, J., Szabó, E., Demian, E., Kontra, L., Baksa, I., et al. (2018). NGS of virus-derived small RNAs as a diagnostic method used to determine viromes of Hungarian vineyards. Front. Microbiol. 9:122. doi: 10.3389/fmicb.2018.00122
FAOSTAT (2017). Statistical Databases. Available online at: http://faostat.fao.org.
Feng, X., Nita, M., and Baudoin, A. B. (2018). Evaluation of quinoxyfen resistance of Erysiphe necator (Grape Powdery Mildew) in a single virginia vineyard. Plant Dis. 102, 2586–2591. doi: 10.1094/PDIS-11-17-1822-RE
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29:644. doi: 10.1038/nbt.1883
Hily, J. M., Candresse, T., Garcia, S., Vigne, E., Tannière, M., Komar, V., et al. (2018). High-throughput sequencing and the viromic study of grapevine leaves: From the detection of grapevine-infecting viruses to the description of a new environmental Tymovirales member. Front. Microbiol. 9:1782. doi: 10.3389/fmicb.2018.01782
Jo, Y., Choi, H., Cho, J. K., Yoon, J. Y., Choi, S. K., and Cho, W. K. (2015). In silico approach to reveal viral populations in grapevine cultivar Tannat using transcriptome data. Sci. Rep. 5:15841. doi: 10.1038/srep15841
Jo, Y., Choi, H., Kim, S. M., Kim, S. L., Lee, B. C., and Cho, W. K. (2016). Integrated analyses using RNA-Seq data reveal viral genomes, single nucleotide variations, the phylogenetic relationship, and recombination for Apple stem grooving virus. BMC Genomics 17:579. doi: 10.1186/s12864-016-2994-6
Jo, Y., Choi, H., Kim, S. M., Kim, S. L., Lee, B. C., and Cho, W. K. (2017). The pepper virome: natural co-infection of diverse viruses and their quasispecies. BMC Genomics 18:453. doi: 10.1186/s12864-017-3838-8
Jo, Y., Lian, S., Chu, H., Cho, J. K., Yoo, S. H., Choi, H., et al. (2018). Peach RNA viromes in six different peach cultivars. Sci. Rep. 8:1844. doi: 10.1038/s41598-018-20256-w
Jones, S., Baizan-Edge, A., MacFarlane, S., and Torrance, L. (2017). Viral diagnostics in plants using next generation sequencing: computational analysis in practice. Front. Plant Sci. 8:1770. doi: 10.3389/fpls.2017.01770
Kakalikova, L’, Jankura, E., and Šrobárová, A. (2009). First report of Alternaria bunch rot of grapevines in Slovakia. Australas. Plant Dis. Notes 4, 68–69. doi: 10.1071/DN09029
Kominek, P., Glasa, M., and Komínková, M. (2009). Analysis of multiple virus-infected grapevine plant reveals persistence but uneven virus distribution. Acta Virol. 53:281. doi: 10.4149/av_2009_04_281.
Kumar, S., Rai, R., and Baranwal, V. K. (2015). Development of an immunocapture–reverse transcription–polymerase chain reaction (IC-RT-PCR) using modified viral RNA release protocol for the detection of Grapevine leafroll-associated virus 3 (GLRaV-3). Phytoparasitica 43, 311–316. doi: 10.1007/s12600-014-0445-y
Kumar, S., Sawant, S. D., Sawant, I. S., Prabha, K., Jain, R. K., and Baranwal, V. K. (2012). First report of Grapevine leafroll-associated virus 1 infecting grapevines in India. Plant Dis. 96, 1828–1828. doi: 10.1094/PDIS-07-12-0647-PDN
Kumar, S., Singh, L., Ferretti, L., Barba, M., Zaidi, A. A., and Hallan, V. (2013). Evidence of Grapevine leafroll associated virus-1–3, Grapevine fleck virus and Grapevine virus B occurring in Himachal Pradesh, India. Indian J. Virol. 24, 66–69. doi: 10.1007/s13337-013-0129-0
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Leinonen, R., Sugawara, H., and Shumway, M., International Nucleotide Sequence Database Collaboration. (2010). The sequence read archive. Nucleic Acids Res. 39, D19–D21. doi: 10.1093/nar/gkq1019
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Maliogka, V. I., Minafra, A., Saldarelli, P., Ruiz-García, A. B., Glasa, M., Katis, N., et al. (2018). Recent advances on detection and characterization of fruit tree viruses using high-throughput sequencing technologies. Viruses 10:436. doi: 10.3390/v10080436
Martin, D. P., Murrell, B., Golden, M., Khoosal, A., and Muhire, B. (2015). RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 1:vev003. doi: 10.1093/ve/vev003
Marwal, A., Kumar, R., Khurana, S. P., and Gaur, R. K. (2019). Complete nucleotide sequence of a new geminivirus isolated from Vitis vinifera in India: a symptomless host of Grapevine red blotch virus. Virusdisease 30, 106–111. doi: 10.1007/s13337-018-0477-x
Massart, S., Chiumenti, M., De Jonghe, K., Glover, R., Haegeman, A., Koloniuk, I., et al. (2019). Virus detection by high-throughput sequencing of small RNAs: Large-scale performance testing of sequence analysis strategies. Phytopathology 109, 488–497. doi: 10.1094/PHYTO-02-18-0067-R
McGovern, P. E. (2003). Ancient Wine: The Search for the Origins of Viniculture. Princeton, NJ: Princeton University Press.
Pantaleo, V., Saldarelli, P., Miozzi, L., Giampetruzzi, A., Gisel, A., Moxon, S., et al. (2010). Deep sequencing analysis of viral short RNAs from an infected Pinot Noir grapevine. Virology 408, 49–56. doi: 10.1016/j.virol.2010.09.001
Pirovano, W., Miozzi, L., Boetzer, M., and Pantaleo, V. (2015). Bioinformatics approaches for viral metagenomics in plants using short RNAs: model case of study and application to a Cicer arietinum population. Front. Microbiol. 5:790. doi: 10.3389/fmicb.2014.00790
Pooggin, M. M. (2018). Small RNA-omics for plant virus identification, virome reconstruction and antiviral defense characterization. Front. Microbiol. 9:2779. doi: 10.3389/fmicb.2018.02779
Rai, R., Khurana, S. P., Kumar, S., Sharma, S. K., Watpade, S., and Baranwal, V. K. (2017). Characterization of Grapevine leafroll-associated virus 4 from Indian vineyards. J. Plant Pathol. 99, 255–259. doi: 10.4454/jpp.v99i1.3826
Roossinck, M. J., Martin, D. P., and Roumagnac, P. (2015). Plant virus metagenomics: advances in virus discovery. Phytopathology 105, 716–727. doi: 10.1094/PHYTO-12-14-0356-RVW
Sahana, A. B., Adkar-Purushothama, C. R., Chennappa, G., Zhang, Z. X., Sreenivasa, M. Y., and Sano, T. (2013). First report of Grapevine yellow speckle viroid-1 and Hop stunt viroid infecting grapevines (Vitis vinifera) in India. Plant Dis. 97, 1517–1517. doi: 10.1094/PDIS-05-13-0494-PDN
Seguin, J., Rajeswaran, R., Malpica-Lopez, N., Martin, R. R., Kasschau, K., Dolja, V. V., et al. (2014). De novo reconstruction of consensus master genomes of plant RNA and DNA viruses from siRNAs. PLoS One 9:e88513. doi: 10.1371/journal.pone.0088513
Singhal, P., Kumar, S., Rai, R., Singh, S. K., and Baranwal, V. K. (2019). Characterization of viroids infecting grapevine in India. Indian Phytopathol. 72, 333–341. doi: 10.1007/s42360-019-00134-9
Tirumalai, V., Swetha, C., Nair, A., Pandit, A., and Shivaprasad, P. V. (2019). miR828 and miR858 regulate VvMYB114 to promote anthocyanin and flavonol accumulation in grapes. J. Exp. Bot. 70, 4775–4792. doi: 10.1093/jxb/erz264
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. ıNat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Xiao, H., Li, C., Al Rwahnih, M., Dolja, V., and Meng, B. (2019). Metagenomic analysis of riesling grapevine reveals a complex virome including two new and divergent variants of Grapevine leafroll-associated virus 3. Plant Dis. 103, 1275–1285. doi: 10.1094/PDIS-09-18-1503-RE
Keywords: virome, mRNAome, sRNAome, grapevine, India
Citation: Sidharthan VK, Sevanthi AM, Jaiswal S and Baranwal VK (2020) Robust Virome Profiling and Whole Genome Reconstruction of Viruses and Viroids Enabled by Use of Available mRNA and sRNA-Seq Datasets in Grapevine (Vitis vinifera L.). Front. Microbiol. 11:1232. doi: 10.3389/fmicb.2020.01232
Received: 04 March 2020; Accepted: 14 May 2020;
Published: 05 June 2020.
Edited by:
Ahmed Hadidi, Agricultural Research Service, United States Department of Agriculture, United StatesReviewed by:
John Hammond, Agricultural Research Service, United States Department of Agriculture, United StatesCopyright © 2020 Sidharthan, Sevanthi, Jaiswal and Baranwal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: V. K. Baranwal, dmJhcmFud2FsMjAwMUB5YWhvby5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.