 Maria Hoffmann
Maria Hoffmann John Miller
John Miller David Melka
David Melka Marc W. Allard1
Marc W. Allard1
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 23 March 2020
Sec. Evolutionary and Genomic Microbiology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.00478
This article is part of the Research Topic What can long-read technology and comparative microbial genomics reveal about evolution and spreading of pathogens under the ONE Health perspective? View all 10 articles
The largest outbreak of Salmonella Agona in the United States occurred in 1998. It affected more than 400 patients and was linked to toasted oat cereal. Ten years later, a similar outbreak occurred with the same outbreak strain linked to the same production facility. In this study, whole-genome sequence (WGS) data from a set of 46 Salmonella Agona including five isolates associated with the 1998 outbreak and 25 isolates associated with the 2008 outbreak were analyzed. From each outbreak one isolate was sequenced on the Pacific Biosciences RS II Sequencer to determine the complete genome sequence. We reconstructed a phylogenetic hypothesis of the samples using a reference-based method for identifying variable sites. Using Single Nucleotide Polymorphism (SNP) analyses, we were able to distinguish and separate Salmonella Agona isolates from both outbreaks with only a mean of eight SNP differences between them. The phylogeny illustrates that the 2008 outbreak involves direct descendants from the 1998 outbreak rather than a second independent contamination event. Based on these results, there is evidence supporting the persistence of Salmonella over time in food processing facilities and highlights the need for consistent monitoring and control of organisms in the supply chain to minimize the risk of successive outbreaks.
Salmonella enterica subsp. enterica is comprised of more than 1,500 serovars, including Salmonella enterica subsp. enterica serovar Agona (Salmonella Agona). Salmonella Agona was first isolated from cattle in Ghana in 1952 (Guinee et al., 1964). Within the last several years, Salmonella Agona has been one of the top 20 most commonly reported serotypes causing human infections (CDC, 2014). It has been responsible for numerous human outbreaks associated with various dry food products such as dried milk, machacado, dried snacks, and cereal (Clark et al., 1973; Killalea et al., 1996; Taylor et al., 1998). In the United States, the largest known outbreak of Salmonella Agona infections occurred between April and May in 1998 (Russo et al., 2013). During that time, the Centers for Disease Control and Prevention (CDC) investigated a multistate (11 states) outbreak of Salmonella Agona infections comprised of 209 confirmed cases (Centers for Disease Control and Prevention, 1998). Among the 162 case patients for which there was available information, 46 (28%) had been hospitalized (Centers for Disease Control and Prevention, 1998). Collaborative investigative efforts by state and federal officials implicated toasted oat cereal as the source of this Agona outbreak. The pulsed-field gel electrophoresis (PFGE) Xbal pattern (JABX01.0001) of the Agona isolate from the cereal production facility was indistinguishable from the predominant PFGE pattern among outbreak-associated clinical isolates. After the investigation, the implicated production lines from the company’s plant were sealed off, all equipment was removed, all surfaces were stripped to bare concrete, decontaminated, refinished and new production lines were installed (Russo et al., 2013).
A decade later, in April 2008, the CDC announced a 15-state outbreak associated with Salmonella Agona resulting in 28 identified cases, with eight individuals being hospitalized. The federal investigation suggested that unsweetened puffed-rice cereals and unsweetened puffed-wheat cereals from the same company that was associated with the 1998 outbreak were the likely sources of contamination (CDC, 2008). The PFGE pattern of the 2008 outbreak strain had the same Xbal pattern (JABX01.0001) as of Salmonella Agona identified during the 1998 outbreak associated with toasted oat cereal by company X. On April 5, 2008 the same company X voluntarily recalled its unsweetened puffed-rice cereals and unsweetened puffed-wheat cereals after finding Salmonella Agona contamination during routine testing from the same plant (Russo et al., 2013).
In this study, whole genome sequence (WGS) data from a group of closely related Salmonella Agona isolates were analyzed. The majority of the isolates were investigated as a part of the two outbreaks of salmonellosis associated with cereals that were separated by 10 years and our objective was to determine the evolutionary relationships among these outbreak isolates as a means to differentiate among two competing hypotheses: (1) persistence of Salmonella within the facility and (2) reintroduction of the same strain. We also demonstrate how the use of WGS can identify stable SNP targets that can be utilized for differentiating closely related Salmonella Agona isolates and clustering closely related isolates together, such as those of the 1998 and 2008 outbreak. Further, we show that the 2008 outbreak involves direct descendants from the 1998 outbreak rather than a second independent event.
We analyzed 46 Salmonella Agona whole genome sequences, 31 of which are first reported here while the remainder were publicly available from GenBank at NCBI (Table 1). Of these 46 isolates, 30 were linked with the original investigation into the outbreak of salmonellosis associated with breakfast cereal including one clinical isolate and 4 food isolates obtained in 1998 and 5 clinical isolates, 8 food isolates, and 12 environmental isolates acquired in 2008. All 30 outbreak associated isolates exhibited indistinguishable Xbal PFGE patterns (JABX01.0001). We also included one clinical Salmonella Agona isolate SL483 containing the same Xbal PFGE pattern from a different event isolated in Wisconsin 2003. The remaining Salmonella Agona isolates were chosen to provide a measure of the genetic diversity that is found among other isolates within the serovar.
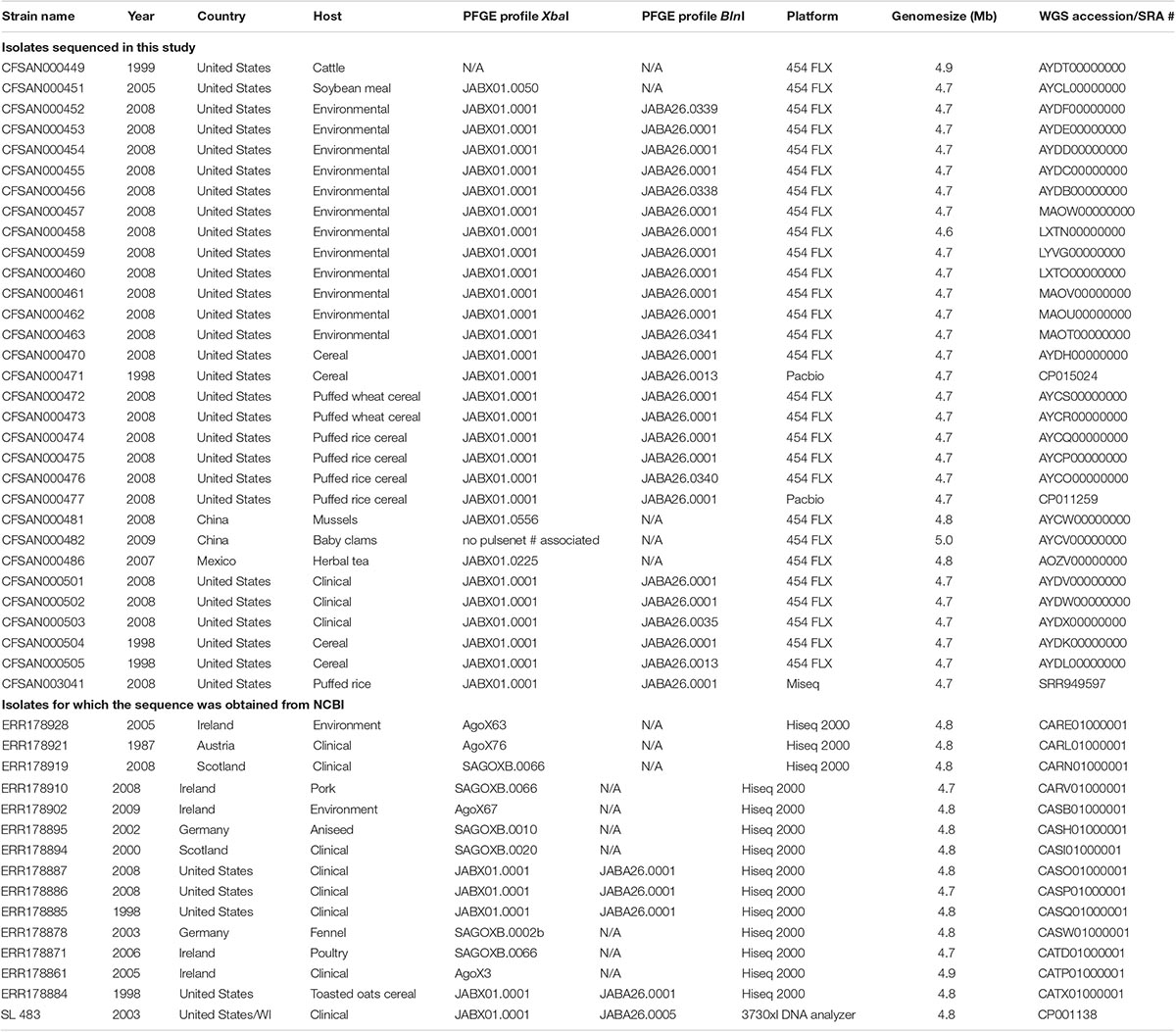
Table 1. Isolates information for the Salmonella Agona included in this study.
Pulsed-field gel electrophoresis was performed according to the PulseNet protocol of the Center for Disease Control and Prevention1 (CDC). Genomic DNA of each strain was isolated from overnight cultures using DNeasy Blood and Tissue kit (Qiagen, CA, United States). Twenty-eight isolates were shotgun sequenced with the Roche Genome Sequencer FLX (454 Life Sciences, Roche, Branford, CT, United States) using the GS FLX Titanium Sequencing Kit XLR70 according to the manufacturer’s protocol. One isolate was sequenced using Illumina’s MiSeq platform (Illumina, Inc., CA, United States). Sample preparation and the sequencing library was prepared using the Nextera XT Sample Preparation Kit and then sequenced for 2 × 151 cycles. Two outbreak associated isolates (1998 and 2008) were sequenced on the Pacific Biosciences (PacBio) RS II Sequencer and assembled as previously described (Hoffmann et al., 2015). Specifically, a single SMRTbell 10-kb library was prepared according to the 10-kb PacBio sample preparation protocol and sequenced using C2 chemistry on two single-molecule real-time (SMRT) cells with a 180-min collection protocol. The 10-kb continuous long read (CLR) data were de novo assembled using the PacBio hierarchical genome assembly process (HGAP3)/Quiver software package, followed by Minimus2, and polished by Quiver (Chin et al., 2013). The assembled sequences were annotated using the NCBI Prokaryotic Genomes Annotation Pipeline (PGAP) and have been deposited at DDBJ/EMBL/GenBank.
We used the CFSAN SNP Pipeline2 (Davis et al., 2015) to construct a SNP matrix among the 46 individuals. This pipeline is a reference-based approach within which Bowtie2 (Langmead and Salzberg, 2012) is used to align reads to the reference and VarScan (Koboldt et al., 2012) is used to detect variants. Within the pipeline, we used Salmonella Agona SL483 (CP001138) as the reference; using NCBI’s methodology (Ciufo et al., 2018) CP001138 has a type strain ANI of 98.6994 confirming taxonomic accuracy. We applied a SNP density filter (i.e., three or more SNPs in a 1000 bp window were removed) to remove variants that are likely the result of recombination or mobile elements (e.g., Petkau et al., 2017).
With the resulting SNP matrix from the CFSAN SNP Pipeline, we constructed a phylogenetic hypothesis of the evolutionary relationships among the 46 isolates investigated using the Genetic Algorithm for Likelihood Inference (GARLI; v2.01.1067; Zwickl, 2006). We used the default settings within GARLI and the best tree was chosen among 10 replicate runs using the observed SNP matrix; topological support was estimated by conducting 1,000 bootstrap replicates. Trees were rooted based on preliminary analyses including the serovar Soerenga as an outgroup. We also applied the ACCTRAN method within the R package phangorn (Schliep, 2011) to provide the number of changes along branches of the best tree from GARLI. We used PopART to construct a minimum spanning network (Bandelt et al., 1999) using the SNP matrix.
A custom python script was written to determine the position of each SNP in the reference genome, the gene within which it was found, and the type of mutation (synonymous or non-synonymous). A custom script was also used to identify fixed SNPs between the outbreak clade and all other isolates. Both scripts are available at https://github.com/CFSAN-Biostatistics/snps_in_genes. Using the COG information in the feature table for the reference found in the Genome database at NCBI, we then assigned each gene to one of the higher functional categories. We compared the abundance of each category between the isolates found in the outbreak and the complete dataset, which indicates what functions might be more pronounced in the former.
To further explore genomic differences among the isolates, the complete chromosomes were aligned with MAUVE aligner version 20150226 using progressive algorithm with default settings (Darling et al., 2004). We constructed a circle plot of genomic differences between the publicly available Agona strain SL483 and the two completely closed outbreak associated isolates CFSAN000477 and CFSAN000471using BRIG (Alikhan et al., 2011).
Public health authorities were informed in 1998 and 2008 of outbreaks involving Salmonella Agona that resulted from consumption of contaminated cereal from the same company X. The PFGE pattern of these highly clonal Agona isolates from 1998 and 2008 were indistinguishable. At that time PFGE was the method used for traceback during an outbreak investigation. However, PFGE does not have the resolution to distinguish a transient strain from a resident strain (Pightling et al., 2018) and, therefore, at that time using this method officials from the state health department were able to correctly identify the source of an outbreak but were not able to determine if the 2008 Agona isolate is the same strain as the 1998 Agona isolate.
Since 2012, several studies have demonstrated the higher resolution and increased discriminatory power of WGS relative to that of PFGE where the former can differentiate highly clonal isolates. This increased resolution of WGS is critical in a successful investigation of foodborne outbreaks to determine the source(s) of contamination (Hoffmann et al., 2014, 2016; Allard et al., 2018). Thus, WGS was invoked retrospectively to the cereal outbreaks of 1998 and 2008 in an attempt to resolve and ameliorate the shortcomings of the other more traditional molecular strategies used for such purposes, as well as to characterize the dynamics of the two outbreak scenarios.
First, using long read sequencing the complete genome sequences of Salmonella Agona isolate CFSAN000471 obtained from cereal during the outbreak in 1998 and of Salmonella Agona isolate CFSAN000477 obtained from unsweetened puffed-rice cereal during the outbreak in 2008 (Hoffmann et al., 2015) were sequenced with the Single Molecule, Real-Time (SMRT) sequencing technology. Isolate CFSAN000471 was sequenced with 110× coverage; its genome size was 4,798,531 bp, and the G + C content was 52.1% and Isolate CFSAN000477 was sequenced with 130× coverage; its genome size was 4,797,172 bp, and the G + C content was also 52.1%. At the time Salmonella Agona SL483 was the only complete genome from Salmonella Agona available in Genbank; it had a genome size of 4,798,660 bp, and a G + C content of 52.1%. The strain was isolated from a clinical sample and is associated with a sporadic case in 2003. Figure 1 shows the pronounced similarity between these three closed Salmonella Agona isolates suggesting that they are highly related with each other. To further characterize them we used the MAUVE aligner to align them and checked manually for differences. The different genome sizes can be explained by the insertion/deletion of transposases. CFSAN000477 and CFSAN000471 carry the same transposase but at different locations within the chromosome (CFSAN000471 transposase insertion: 710,112–711,499; CFSAN000477 (transposase insertion: 987,888–989,229) while SL483 is missing the specific transposase but instead carries an insertion sequence element, IS10, at three different loci. Further, very few SNPS and deletions and insertions were found among the three isolates listed in Supplementary Table 1. Explicitly, CFSAN000471 isolated in 1998 showed only one unique informative SNP in gene int (locus_tag = SEEA8692_015865) not found in CFSAN000477 and SL483. SL483 has one unique informative SNP in gene hslO (locus_tag = SeAg_B3697), two unique SNPs, two unique deletions and two unique insertions in non-coding region, and is missing a hypothetical protein. CFSAN000477 has six unique including five informative SNPs [fusA (locus_tag = SEEA0421_002795), GDP-mannose pyrophosphatase (locus_tag = SEEA0421_021175), transporter gene (locus_tag = SEEA0421_019215), protease (locus_tag = SEEA0421_011315), hypothetical protein (locus_tag = SEEA0421_010320), and fructose-6-phospharte aldolase A (locus_tag = SEEA0421_006170)], four SNPs, one insertion and one deletion in non-coding region.
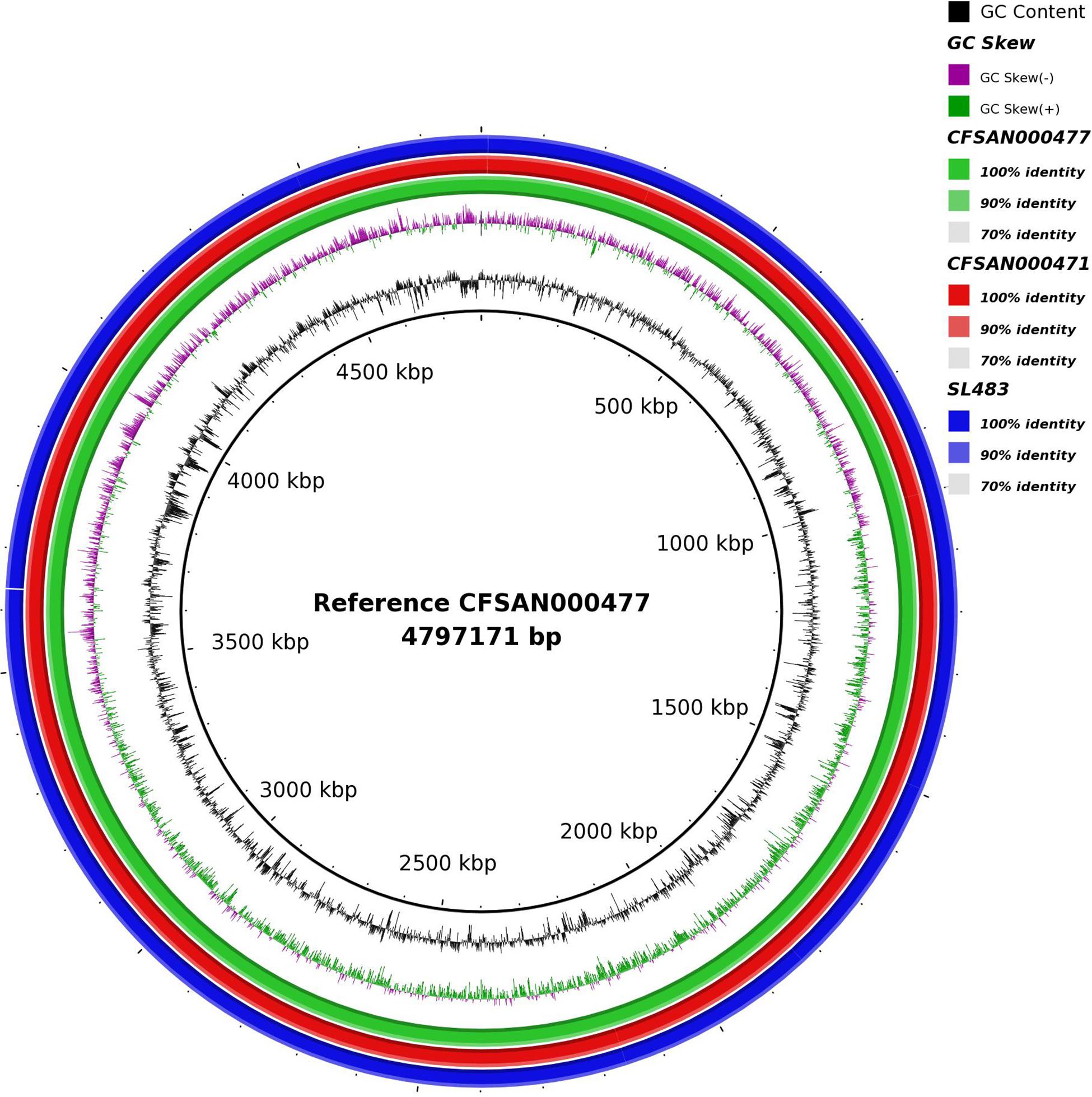
Figure 1. Circle plot showing the similarity between three closed Salmonella Agona isolates associated with the cereal outbreaks. The reference Salmonella Agona CFSAN000477 is associated with the 2008 outbreak, Salmonella Agona CFSAN000471 is associated with the 1998 outbreak and Salmonella Agona SL483 is associated with a sporadic case in 2003.
The genomic size (including extrab. -chromosomal DNA) of the 46 Salmonella Agona isolates varied from 4.6 to 5.1 Mb. Primarily, differences in genomic size was due to the presence or absence of mobile genetic elements, such as phages and genetic islands. Using the complete genome of Salmonella Agona (SL483), we were able to more accurately map the raw reads to the reference genome to build a SNP matrix and subsequently construct a phylogenetic tree (Figure 2). Overall, we identified 441 variable SNPs with 112 not located on a gene. Supplementary Table 2 lists all 441 SNPs including SNP position in the genome, description of the gene where the SNP is located, SNP position in the gene, amino acid change and the substitution type; Supplementary Figure S1 shows the number of changes along branches of the phylogenetic tree inferred with GARLI.
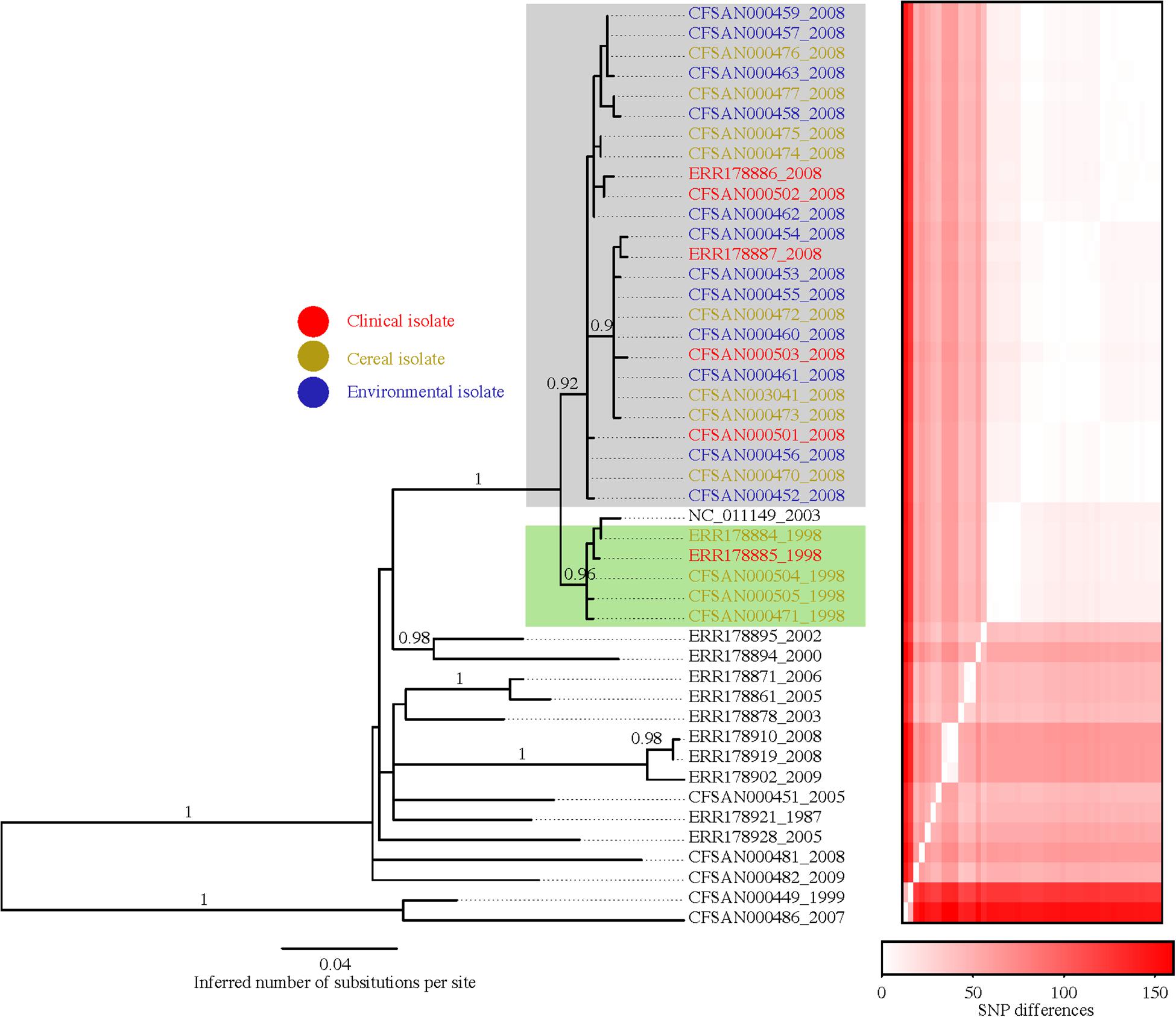
Figure 2. Phylogenetic reconstruction of the 46 Salmonella Agona isolates investigated herein. The tree was constructed from a SNP matrix obtained with the CFSAN SNP Pipeline within which the isolate Salmonella Agona SL483 was used as the reference. The program GARLI v2.01.1067 was used to infer the tree. The heatmap to the right is ordered according to the tips of the tree and shows the pairwise SNP distances among isolates (e.g., the first two columns and last two rows represent the two most basal isolates in the tree that are very different from all other isolates but similar to one another).
The data derived from the phylogenetic analysis of 46 Salmonella Agona isolates (Table 1), collected from various sources between 1987 and 2009 including 5 isolates associated with the 1998 outbreak, and 25 isolates associated with the 2008 outbreak provided critically relevant information. First and most importantly, the 1998 and 2008 outbreak associated isolates clustered together to the exclusion of other isolates with strong support (i.e., 19 fixed SNPs and 100% bootstrap support). Second, the tree showed that all of the isolates associated with the 2008 outbreak, including 6 clinical, 7 cereal, and 12 environmental isolates from the implicated food production facility clustered together with 92% bootstrap support (Figure 2) and three fixed SNPs. Third, the two clinical and three food isolates associated with the 1998 outbreak clustered together with 96% bootstrap support and four fixed SNPs. Interesting, the reference isolate SL483 grouped together with the 1998 outbreak isolates. SL483 is a clinical isolate (sporadic case) obtained in 2003. The analyses suggest that most likely the patient was infected with the same Salmonella Agona strain. Interestingly, there are also two strains from 1998 and two strains from 2008 that were part of a previous publication Zhou et al., 2013 that also cluster with the sample associated with the particular cereal outbreak. The metadata for those samples indicates that they indeed are part of the outbreak event [“A food isolate, isolated at the time of the (1998| 2008) outbreak in the United States”]. Fourth, there is a tendency for isolates to cluster based on geographical location such as United States, Europe and China and proximity. The most distinct isolate in this study was collected from herbal tea in Mexico in 2007.
Given the importance of the tree topology to the accurate interpretation of this study results, we constructed using the same SNP matrix a minimum spanning tree (Figure 3) to further illustrate the relationships among the isolates and to provide additional insight into whether the observed phylogenetic relationships may be influenced by recombination and/or mobile elements. With the phylogenetic analyses linking the 1998 and 2008 strains, the network analysis illustrates that the outbreak isolates form a star-like clade (Figure 3). Star-like clusters are often attributed to rapid diversification and/or a clonal swarm. In this situation, it reveals additional evidence that indeed the 2008 outbreak are direct descendants from the 1998 outbreak rather than a second independent contamination event. Therefore, our analyses suggest that company X had a resident strain surviving inside their facility for a minimum of a decade. Given that the facility was thought to have taken appropriate measures to eliminate the presence of Salmonella, the question arose as to how a strain found 10 years earlier could have persisted in the plant and caused the subsequent outbreak in 2008. One possible explanation is that the bacteria persisted in the cement walls and there is evidence for Salmonella forming dense biofilms on such a surface (Joseph et al., 2001). Based on our SNP analyses where variable positions were mapped to an annotated reference genome, there does appear to be fewer number of SNP differences within genes linked to cell mobility, intracellular transport and transcription within the cereal clade (Figure 4). Such evolutionary changes may be driven by selection pressures from bacterial controls instituted by the food industry or by production processes, resulting in enhanced survival, persistence and even growth within food matrices and in the production environment. These increase the likelihood of foodborne outbreaks with morbidity and mortality that threaten public health. To prevent such outbreaks, we must identify the genetic factors responsible for pathogen persistence across the farm to fork supply chain.
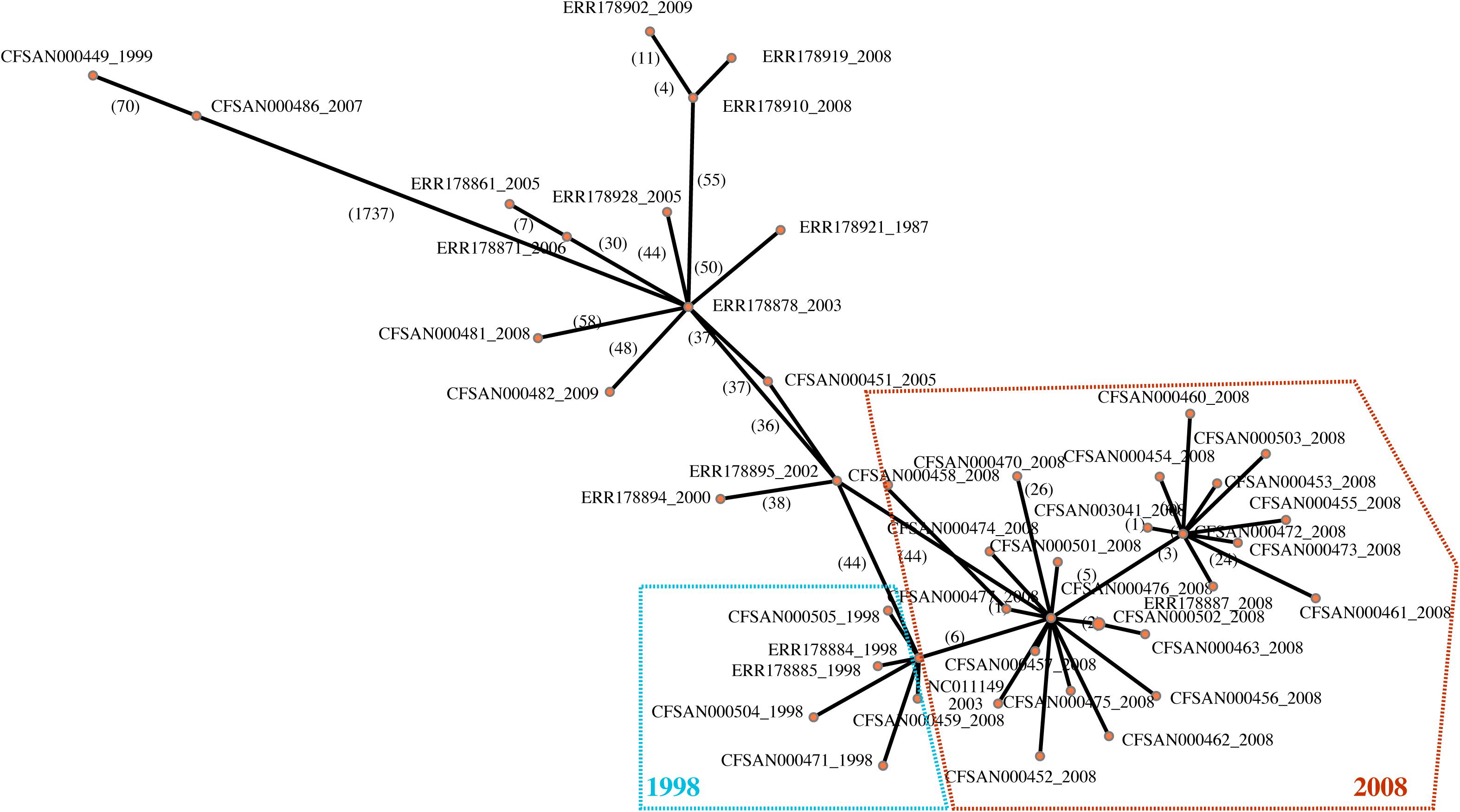
Figure 3. A minimum spanning network constructed with popArt (http://popart.otago.ac.nz/) showing the genetic similarity among 46 Salmonella Agona isolates. Input for the network was the SNP matrix constructed with the CFSAN SNP Pipeline. Edge labels represent the number of SNP differences between nodes.
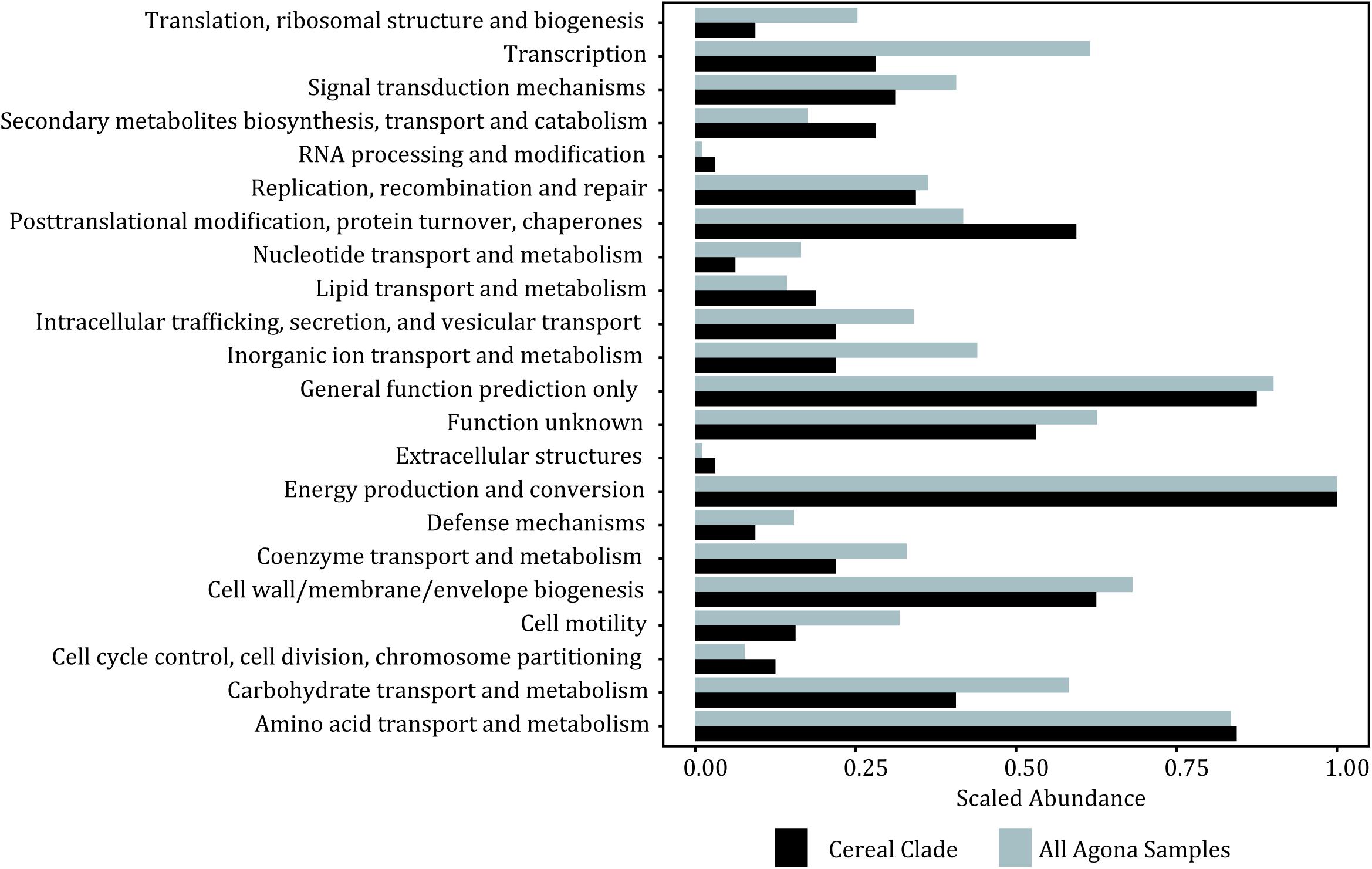
Figure 4. Histogram showing the counts of mutations found within each protein type (COG category) between the outbreak clade and all samples.
The subtyping of isolates associated with a foodborne outbreak event is essential for successful investigation and eventual traceback to a specific food or environmental source (Allard et al., 2012). In this regard, PFGE has been thought of as the “gold standard” by augmenting public health investigations with information regarding genetic similarity of isolation for nearly two decades (Bergholz et al., 2014). However, numerous clonal strains, a particularly common phenomenon within Salmonella, confound epidemiological investigations because PFGE and other traditional molecular typing tools cannot separate these clonally related strains (CDC, 2010). The population and evolutionary dynamics associated with clonal bacterial populations can now be investigated with much greater resolution afforded by next generation sequencing (NGS) data. We are now at a stage that WGS can identify if a company’s product is contaminated with a new strain of a pathogen from the environment or if they have a resident strain within their facility that has persisted over a period of time (e.g., multiple years).
In this study, WGS data from a set of Salmonella Agona isolates were analyzed to provide insight into the evolutionary relationships among strains linked to two outbreaks of salmonellosis separated by ten years. All of the Salmonella Agona strains associated with the 1998 and 2008 outbreaks could be traced to the same cereal facility in the United States and the analyses of genomic data suggests that the same strain from 1998 was responsible for the 2008 outbreak and it was able to survive in the facility. This highlights the persistence of Salmonella and that it can survive in dry food production environments for years.
GenBank accession numbers for all new sequences are listed in Table 1.
All authors played an integral part of project conception. Each author has read and approved the final version of the manuscript. Specifically, MH, MA, EB, and JP conceived and designed the experiments. MH performed the experiments. JP, MH, JM, and DM analyzed the data. MH and JP wrote the manuscript.
This project was supported by the internal FDA/CFSAN research funding.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Some of the results of this study were previously presented at two conferences: Hoffmann, M., Allard, M. W., Brown, E. W., and Pettengill, J. B. 2017. Whole-Genome Sequencing to Determine Origin of a Recurrent Multistate Outbreak of Salmonella Agona. Advances in Genome Biology and Technology (AGBT), Hollywood, FL, United States (poster). Hoffmann, M., Allard, M. W., Brown, E. W., and Pettengill, J. B. 2016. Temporal and Population Dynamics of Salmonella enterica ssp. enterica Serovar Agona Isolates from a Recurrent Multistate Outbreak. International Association for Food Protection Annual Meeting (IAFP), St. Louis, MO, United States (seminar).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00478/full#supplementary-material
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genom. 12:402. doi: 10.1186/1471-2164-12-402
Allard, M. W., Bell, R., Ferreira, C. M., Gonzalez-Escalona, N., Hoffmann, M., Muruvanda, T., et al. (2018). Genomics of foodborne pathogens for microbial food safety. Curr. Opin. Biotechnol. 49, 224–229. doi: 10.1016/j.copbio.2017.11.002
Allard, M. W., Luo, Y., Strain, E., Li, C., Keys, C. E., Son, I., et al. (2012). High resolution clustering of Salmonella enterica serovar Montevideo strains using a next-generation sequencing approach. BMC Genom. 13:32. doi: 10.1186/1471-2164-13-32
Bandelt, H. J., Forster, P., and Rohl, A. (1999). Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48. doi: 10.1093/oxfordjournals.molbev.a026036
Bergholz, T. M., Moreno Switt, A. I., and Wiedmann, M. (2014). Omics approaches in food safety: fulfilling the promise? Trends Microbiol. 22, 275–281. doi: 10.1016/j.tim.2014.01.006
CDC (2008). Multistate Outbreak of Salmonella Agona Infections Linked to Rice & Wheat Puff Cereal (Final Update). Atlanta, GA: Centers for Disease Control and Prevention.
CDC (2010). Multistate Outbreak of Human Salmonella Montevideo infections (final ubdate). Atlanta, GA: Centers for Disease Control and Prevention.
CDC (2014). National Enteric Disease Surveillance: Salmonella Annual Report and Appendices, 2013. Atlanta, GA.: Centers for Disease Control and Prevention.
Centers for Disease Control and Prevention (1998). Multistate outbreak of Salmonella serotype Agona infections linked to toasted oats cereal–United States, April-May, 1998. MMWR Morb. Mortal. Wkly Rep. 47, 462–464.
Chin, C. S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Ciufo, S., Kannan, S., Sharma, S., Badretdin, A., Clark, K., Turner, S., et al. (2018). Using average nucleotide identity to improve taxonomic assignments in prokaryotic genomes at the NCBI. Int. J. Syst. Evol. Microbiol. 68, 2386–2392. doi: 10.1099/ijsem.0.002809
Clark, G. M., Kaufmann, A. F., Gangarosa, E. J., and Thompson, M. A. (1973). Epidemiology of an international outbreak of Salmonella agona. Lancet 2, 490–493. doi: 10.1016/s0140-6736(73)92082-5
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Davis, S., Pettengill, J. B., Luo, Y., Payne, J., Shpuntoff, A., Rand, H., et al. (2015). CFSAN SNP Pipeline: an automated method for constructing SNP matrices from next-generation sequence data. PeerJ Comput. Sci. 1:e20. doi: 10.7717/peerj-cs.20
Guinee, P. A., Kampelmacher, E. H., Willems, H. M., and Spithout, H. (1964). Twelve New Salmonella Types: S. Heerlen, S. Sloterdijk, S. Maartensdijk, S. Maastricht, S. Parera, S. Putten, S. Hoograven, S. Schalkwijk, S. Hilversum, S. Harmelen, S. Breukelen and S. Maarssen. Antonie Van Leeuwenhoek 30, 168–175.
Hoffmann, M., Luo, Y., Monday, S. R., Gonzalez-Escalona, N., Ottesen, A. R., Muruvanda, T., et al. (2016). Tracing origins of the Salmonella bareilly strain causing a food-borne outbreak in the United States. J. Infect. Dis. 213, 502–508. doi: 10.1093/infdis/jiv297
Hoffmann, M., Zhao, S., Pettengill, J., Luo, Y., Monday, S. R., Abbott, J., et al. (2014). Comparative genomic analysis and virulence differences in closely related salmonella enterica serotype heidelberg isolates from humans, retail meats, and animals. Genome Biol. Evol. 6, 1046–1068. doi: 10.1093/gbe/evu079
Hoffmann, M., Payne, J., Roberts, R. J., Allard, M. W., Brown, E. W., and Pettengill, J. B. (2015). Complete Genome Sequence of Salmonella enterica subsp. enterica Serovar Agona 460004 2-1, Associated with a Multistate Outbreak in the United States. Genome Announc 3:e00690-15. doi: 10.1128/genomeA.00690-15
Joseph, B., Otta, S. K., Karunasagar, I., and Karunasagar, I. (2001). Biofilm formation by Salmonella spp. on food contact surfaces and their sensitivity to sanitizers. Int. J. Food Microbiol. 64, 367–372. doi: 10.1016/s0168-1605(00)00466-9
Killalea, D., Ward, L. R., Roberts, D., de Louvois, J., Sufi, F., Stuart, J. M., et al. (1996). International epidemiological and microbiological study of outbreak of Salmonella agona infection from a ready to eat savoury snack–I: England and Wales and the United States. BMJ 313, 1105–1107. doi: 10.1136/bmj.313.7065.1105
Koboldt, D. C., Zhang, Q., Larson, D. E., Shen, D., McLellan, M. D., Lin, L., et al. (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. doi: 10.1101/gr.129684.111
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Petkau, A., Mabon, P., Sieffert, C., Knox, N. C., Cabral, J., Iskander, M., et al. (2017). SNVPhyl: a single nucleotide variant phylogenomics pipeline for microbial genomic epidemiology. Microb. Genom. 3:e000116. doi: 10.1099/mgen.0.000116
Pightling, A. W., Pettengill, J. B., Luo, Y., Baugher, J. D., Rand, H., and Strain, E. (2018). Interpreting whole-genome sequence analyses of foodborne bacteria for regulatory applications and outbreak investigations. Front. Microbiol. 9:1482. doi: 10.3389/fmicb.2018.01482
Russo, E. T., Biggerstaff, G., Hoekstra, R. M., Meyer, S., Patel, N., Miller, B., et al. (2013). A recurrent, multistate outbreak of salmonella serotype agona infections associated with dry, unsweetened cereal consumption. United States, 2008. J. Food Prot. 76, 227–230. doi: 10.4315/0362-028X.JFP-12-209
Schliep, K. P. (2011). phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593. doi: 10.1093/bioinformatics/btq706
Taylor, J. P., Barnett, B. J., del Rosario, L., Williams, K., and Barth, S. S. (1998). Prospective investigation of cryptic outbreaks of Salmonella agona salmonellosis. J. Clin. Microbiol. 36, 2861–2864. doi: 10.1128/jcm.36.10.2861-2864.1998
Zhou, Z., McCann, A., Litrup, E., Murphy, R., Cormican, M., Fanning, S., et al. (2013) Neutral genomic microevolution of a recently emerged pathogen, Salmonella enterica serovar Agona. PLoS Genet. 9:e1003471. doi: 10.1371/journal.pgen.1003471
Keywords: dormancy, outbreak, Salmonella, whole genome sequencing, PacBio, SNP analyses
Citation: Hoffmann M, Miller J, Melka D, Allard MW, Brown EW and Pettengill JB (2020) Temporal Dynamics of Salmonella enterica subsp. enterica Serovar Agona Isolates From a Recurrent Multistate Outbreak. Front. Microbiol. 11:478. doi: 10.3389/fmicb.2020.00478
Received: 04 September 2019; Accepted: 05 March 2020;
Published: 23 March 2020.
Edited by:
Feng Gao, Tianjin University, ChinaReviewed by:
Jason Sahl, Northern Arizona University, United StatesCopyright © 2020 Hoffmann, Miller, Melka, Allard, Brown and Pettengill. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Hoffmann, bWFyaWEuaG9mZm1hbm5AZmRhLmhocy5nb3Y=; James B. Pettengill, SmFtZXMuUGV0dGVuZ2lsbEBmZGEuaGhzLmdvdg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.