 Yongchuang Liu
Yongchuang Liu Xiangrong He
Xiangrong He Pingping Zhu
Pingping Zhu Minggen Cheng
Minggen Cheng Qing Hong
Qing Hong Xin Yan
Xin Yan- 1Key Laboratory of Agricultural Environmental Microbiology, Ministry of Agriculture, College of Life Sciences, Nanjing Agricultural University, Nanjing, China
- 2Jiangsu Provincial Key Lab for Organic Solid Waste Utilization, Nanjing Agricultural University, Nanjing, China
Due to their fast growth rate and robustness, some haloalkalitolerant methanotrophs from the genus Methylotuvimicrobium have recently become not only promising biocatalysts for methane conversion but also favorable materials for obtaining fundamental knowledge on methanotrophs. Here, to realize unmarked genome modification in Methylotuvimicrobium bacteria, a counterselectable marker (CSM) was developed based on pheS, which encodes the α-subunit of phenylalanyl-tRNA synthetase. Two-point mutations (T252A and A306G) were introduced into PheS in Methylotuvimicrobium buryatense 5GB1C, generating PheSAG, which can recognize p-chloro-phenylalanine (p-Cl-Phe) as a substrate. Theoretically, the expression of PheSAG in a cell will result in the incorporation of p-Cl-Phe into proteins, leading to cell death. The Ptac promoter and the ribosome-binding site region of mmoX were employed to control pheSAG, producing the pheSAG-3 CSM. M. buryatense 5GB1C harboring pheSAG-3 was extremely sensitive to 0.5 mM p-Cl-Phe. Then, a positive and counterselection cassette, PZ (only 1.5 kb in length), was constructed by combining pheSAG-3 and the zeocin resistance gene. A PZ- and PCR-based strategy was used to create the unmarked deletion of glgA1 or the whole smmo operon in M. buryatense 5GB1C and Methylotuvimicrobium alcaliphilum 20Z. The positive rates were over 92%, and the process could be accomplished in as few as eight days.
Introduction
Methane, the principal component of natural gas and biogas, is a major candidate source of carbon for (bio)chemical synthesis (Hanson and Hanson, 1996), and the conversion of methane into valuable products has been pursued off and on for almost half a century (Strong et al., 2015). Methane-oxidizing bacteria (methanotrophs) are able to use methane as their sole source of carbon and energy and thus are promising systems for methane-based bioconversion (Hanson and Hanson, 1996; Fei et al., 2014). Since the 1970s, most studies and biotechnological efforts have focused on well-characterized species, such as Methylococcus capsulatus Bath, Methylosinus trichosporium OB3b, and Methylocystis parvus OBBP (Hou et al., 1979; Kalyuzhnaya et al., 2015). In recent years, due to their fast growth rate and robustness, some haloalkaliphilic Methylotuvimicrobium bacteria, such as Methylotuvimicrobium buryatense 5GB1C and Methylotuvimicrobium alcaliphilum 20Z, have been considered particularly promising methanotrophs for industrial use (Trotsenko et al., 2005; Andrea et al., 2015; Nguyen et al., 2018). For strains 5GB1C and 20Z, the genome, transcriptome, and metabolic pathway have been well characterized (Kalyuzhnaya et al., 2013; Strong et al., 2016), the genetic tools have been established (Kalyuzhnaya et al., 2015; Puri et al., 2015; Yan et al., 2016; Nguyen et al., 2018), and metabolic engineering to generate value-added products from methane has been attempted (Andrea et al., 2015; Demidenko et al., 2016; Henard et al., 2016; Nguyen et al., 2018). Promising strains and synthetic biology give new optimism for a realized methane-based bio-industry.
Markerless chromosomal modification is considered an ideal genetic manipulation with no polar effects and guaranteed safety against gene flow by evicting the resistance marker gene. The Flp/FRT site-specific recombination system and a counterselectable marker (CSM), sacB, have been employed to generate a markerless chromosomal modification in Methylotuvimicrobium (Puri et al., 2015; Yan et al., 2016; Nguyen et al., 2018). However, the Flp/FRT system leaves an FRT site at the replacement locus (Hoang et al., 1998; Kühn and Torres, 2002), which interferes with subsequent rounds of manipulations in the same host; counterselection based on sacB and sucrose does not leave a scar on the chromosome, but the positive rate is typically 5–50% in Methylotuvimicrobium according to previous reports (Yan et al., 2016) and our experience. Therefore, an efficient CSM is still needed in Methylotuvimicrobium.
The pheS gene encodes the α-subunit of phenylalanyl-tRNA synthetase, which is highly conserved among bacteria. PheS with T251A and A294G substitutions (PheSAG) has the ability to aminoacylate the phenylalanine analog p-chloro-phenylalanine (p-Cl-Phe) in Escherichia coli (Kast and Hennecke, 1991; Kentaro, 2015). Incorporation of p-Cl-Phe into proteins lead to cell death (Kast and Hennecke, 1991; Kast, 1994). Therefore, pheSAG has been used as a CSM in various bacteria for marker-free genome modification (Kristich et al., 2007; Barrett et al., 2008; Zhoujie et al., 2011; Carr et al., 2015; Xin et al., 2017; Ishikawa et al., 2018). This work aimed to develop pheS as an efficient CSM for Methylotuvimicrobium and to establish a fast marker-free genome modification method for these industrially promising methanotrophs.
Materials and Methods
Bacterial Strains and Culture Conditions
All strains were cultured in an atmosphere of 25% methane in air at 30°C. M. buryatense 5GB1C and its derived strains were grown in NMS2 medium (Puri et al., 2015). M. alcaliphilum 20Z and its derived strains were grown in NMS3 medium (Khmelenina et al., 1997). Plates were incubated in sealed jars (Oxoid Limited, Hampshire, United Kingdom), while liquid cultures were grown in 100 ml glass serum bottles sealed with rubber stoppers and aluminum seals. The total of 30 μg/ml zeocin (Zeo) was added if required.
DNA Manipulation Techniques
Oligonucleotide synthesis (listed in Supplementary Table S1) and DNA sequencing were performed by Sangon Biotech Co., Ltd. (Shanghai, China). The isolation and manipulation of DNA were carried out using standard techniques. All enzymes were commercial preparations and were used as specified by the supplier (NEB, Shanghai, China).
Fusion of Multiple DNA Fragments by Overlap PCR
Fusion of multiple DNA fragments by overlap PCR was carried out as described by Shevchuk et al. (2004). In brief, overlaps of approximately 30–40 nucleotides were introduced between each of 2 fragments through primers. The reaction mixture of step A contained 11.5 μl of water, 4 μl of Phusion buffer (×5), 2 μl of deoxynucleoside triphosphate (dNTP) mix (2.5 mM each), 2 μl of gel-purified fragments (approximately 100 ng each), and 0.5 μl of Phusion DNA polymerase. The cycling parameters were an initial denaturation at 98°C for 3 min and subsequent steps of 98°C for 15 s, annealing at 55°C for 10 s and extension at 72°C for 3 min for 15 cycles total. The reaction mixture of step B contained 34.5 μl of water, 10 μl of Phusion buffer, 4 μl of dNTPmix, 2 μl of forward and reverse primers (10 mM) specific for the expected fragment, and 1 μl of the unpurified PCR product from step A and 0.5 μl of Phusion DNA polymerase. The cycling parameters were an initial denaturation at 98°C for 2 min and subsequent steps of 98°C for 10 s, annealing at 58°C for 10 s, and extension at 72°C for 3 min for 30 cycles total.
Construction of pheSAG
The site-directed mutations in pheSAG (ACC252GCA) and (GCA306GGT) were generated by overlap extension with phes1F/phes1R, phes2F/phes2R, and phes3F/phes3R primer pairs using the genomic DNA of M. buryatense 5GB1C as a template. The ACC252GCA mutation was introduced by the primer phes1R and the GCA306GGT mutation was introduced by the primer phes3F. The pheSAG mutant was named pheSAG-1. The expression cassette pheSAG-2 was constructed based on pheSAG-1. The promoter region of pheSAG-1 was replaced by Ptac with the ZPtac1F/zeoR primer pair, and Ptac was introduced by ZPtac1F. The ribosome-binding site (RBS) region of pheSAG-2 was replaced by the RBS region of mmoX using the ZPtac2F/zeoR primer pair, thereby generating pheSAG-3.
p-Cl-Phe Sensitivity Assessment
Expression cassettes pheSAG-1, pheSAG-2, and pheSAG-3 were individually inserted at chromosome loci between the genes METBUDRAFT_2794 and METBUDRAFT_2795 in strain 5GB1C. The insertion construct containing each cassette and the flanking regions were assembled using PCR with the primers indicated in Supplementary Table S1. The assembled products were transformed into M. buryatense 5GB1C by the electroporation method (Yan et al., 2016). To assess p-Cl-Phe sensitivity, the bacterial strain was grown in NMS2 or NMS3 medium with the corresponding antibiotics with OD600 = 1.0, then the cell cultures were serially diluted 1:10; a sample from each serial dilution was spotted onto agar plates containing p-Cl-Phe at different concentrations, and the plates were incubated at 30°C with methane vapor for 5 days.
Construction of the PZ Cassette
The PZ cassette was constructed in two steps using overlap PCR. First, the zeo gene, together with its RBS sequence, from 5GB1C-Ppmo-xylE was amplified with zeoF/zeoR primer pair. Then, the zeo gene was fused to the 3′ end of pheSAG-3 with phes1F and zeoR primers. Thus, pheSAG-3 and zeo were arranged into an operon under the control of Ptac and RBSmmoX.
Construction of Mutants With glgA1 Deleted in M. buryatense 5GB1C
To generate the glgA1 deletion mutation in strain 5GB1C, the left flanking (LF) region (∼800 bp), direct repeat (DR) sequence (∼450 bp), and right flanking (RF) region (∼800 bp) from strain 5GB1C were amplified using the gA1LF-F/gA1LF-R, gA1DR-F/gA1DR-R, and gA1RF-F/gA1RF-R primer pairs, respectively. The PZ cassette was amplified with the gA1PZ-F/gA1PZ-R primer pair using pheSAG-3 as a template. These four fragments were fused using overlap PCR in the order of the LF, DR, PZ cassette, and RF. The resulting ∼3.5-kb glgA1 deletion amplicon (PCR product) was directly transformed into strain 5GB1C by the electroporation method (Yan et al., 2016). The DR and PZ cassette were inserted immediately upstream of the target region via a double-crossover recombination event without any deletions, which was selected by Zeor (for approximately 3 days). The transformants were transferred to NMS2 plates and maintained overnight at 30°C. Then, the cells were transferred to 5 ml of NMS2 medium and cultivated for approximately 10 h at 30°C to an OD600 = 1.0. Then, 100 μl of cells were spread onto selective plates (for approximately 3–4 days). Mutants growing on selective plates were further confirmed by PCR and DNA sequencing.
Deletion of the 10-kb smmo Operon in M. buryatense 5GB1C
Deletion of the smmo operon was carried out using the same strategy as described for glgA1 deletion. The LF, DR, PZ cassette, and RF fragments were amplified using the MOLF-F/MOLF-R, MODR-F/MODR-R, MOPZ-F/MOPZ-R, and MORF-F/MORF-R primer pairs, respectively. The four fragments were fused to generate the ∼3.5-kb smmo deletion amplicon, which was subsequently transformed into strain 5GB1C.
Construction of the PZ∗ Cassette
The PZ∗ cassette was constructed based on pheSAG-3. We engineered a series of silent mutations in pheSAG-3 to reduce its similarity to wild-type pheS. The artificially synthesized pheS, including a tac promoter, RBS of mmoX (RBSmmoX), and mutated pheS (pheSAG), was fused to the zeo gene to obtain the PZ∗ cassette using the PZ∗LF-F/PZ∗LF-R and PZ∗RF-F/PZ∗RF-R primer pairs.
Deletion of glgA1 in M. alcaliphilum 20Z
The LF region (∼800 bp), DR sequence (∼450 bp), and RF region (∼800 bp) from strain 20Z were amplified using the gALF-F/gALF-R, gADR-F/gADR-R, and gARF-F/gARF-R primer pairs, respectively. The PZ∗ cassette was amplified with the gAPZ-F/gAPZ-R primer pair. These four fragments were fused to generate the ∼3.5-kb glgA1-deletion amplicon and were directly transformed into strain 20Z by electroporation.
Naphthalene Assay
To detect the activity of soluble methane monooxygenase (sMMO), M. buryatense strains were grown in an NMS2 medium without copper at 30°C. A naphthalene oxidation assay was routinely used for the qualitative detection of sMMO activity (Graham et al., 1992). Approximately 50 mg of crushed naphthalene crystals were added to a 3 ml batch culture of M. buryatense and the mixture was shaken at 30°C for 2 h. The cell suspension was then centrifuged for 2 min at 12,000 g. A total of 50 μl of 5 mg ml–1 freshly prepared tetrazotized o-dianisidine was added to the supernatant. The deep purple color of the mixture indicated sMMO activity.
Results
p-Cl-Phe Sensitivity of M. buryatense 5GB1C and M. alcaliphilum 20Z
To determine whether p-Cl-Phe could be used for counterselection with its marker pheSAG in M. buryatense 5GB1C and M. alcaliphilum 20Z, the resistance of both strains to different concentrations of p-Cl-Phe was tested. As shown in Figure 1, strains 5GB1C and 20Z exhibited similar sensitivity to p-Cl-Phe. The growth of both strains was slightly inhibited in the presence of 0.5 mM p-Cl-Phe (with survival rates of 96 and 95%), moderately inhibited at 0.8 or 1.0 mM p-Cl-Phe (with survival rates of 81 to 75%), and greatly inhibited with the addition of 2 mM p-Cl-Phe (with survival rates less than 1%). These results indicate that the concentration of p-Cl-Phe to be added during counterselection should be less than 1 mM.
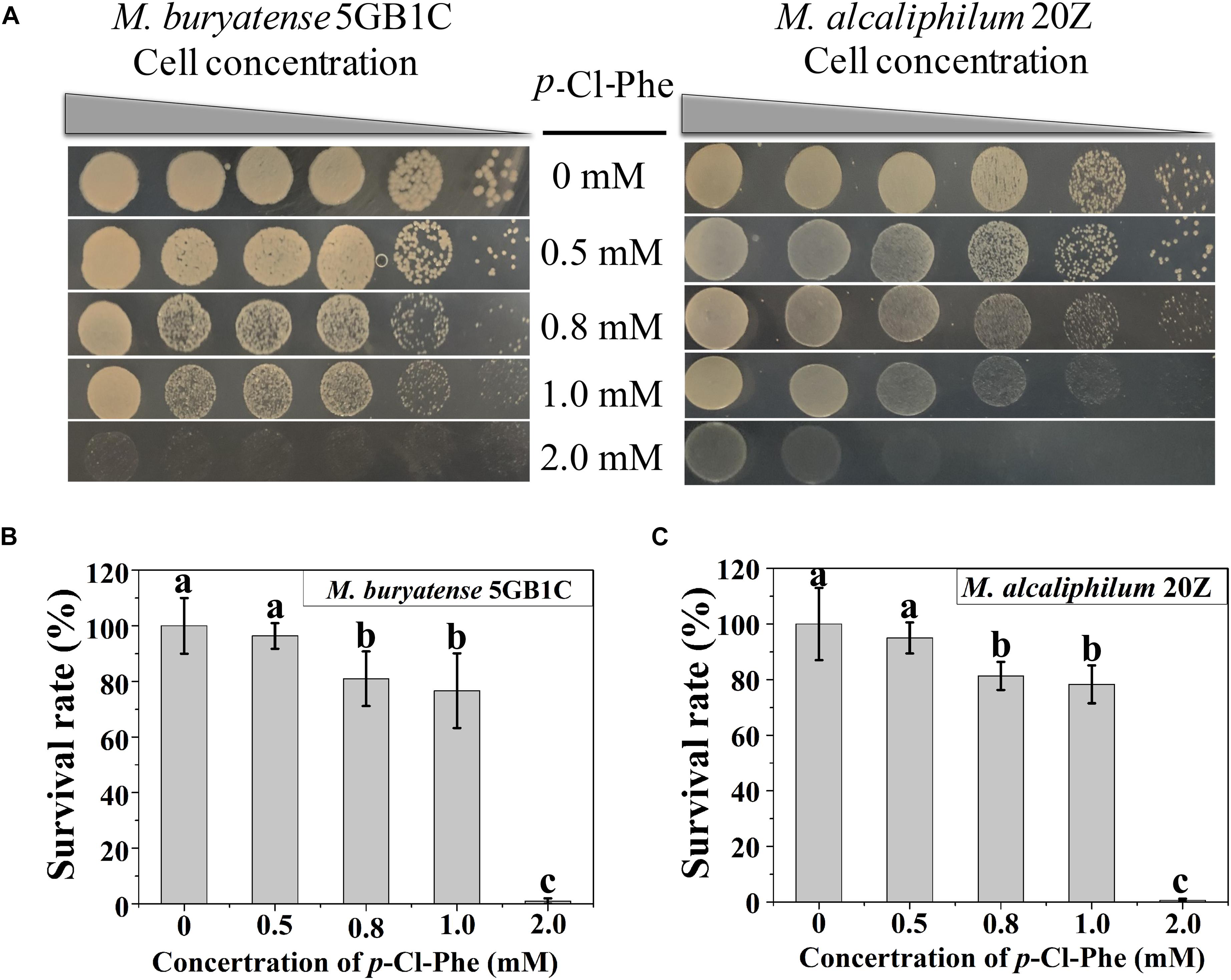
Figure 1. p-Cl-Phe sensitivity of M. buryatense 5GB1C and M. alcaliphilum 20Z. (A) Cell cultures were serially diluted 1:10. Each serial dilution was spotted onto agar plates containing p-Cl-Phe at different concentration. Plates were incubated in sealed jars at 30°C in an atmosphere of 25% methane in air for 5 days. (B,C) Cells were grown to an OD600 of 1.0, diluted 104-fold and plated on NMS2 (M. buryatense 5GB1C) or NMS3 (M. alcaliphilum 20Z) medium containing different concentrations of p-Cl-Phe. All data were expressed as means ± standard deviations (SD) (n = 3). Letters above bars indicate significant differences (P < 0.05). The survival rate of each strain on NMS2 or NMS3 plate was defined as 100%.
Optimizing the Expression Level of pheSAG
To identify the amino acid residues for mutagenesis, multiple sequence alignment of the PheS proteins from various species was carried out using ClustalW2 (Larkin et al., 2007). As shown in Figure 2, the two residues of T252 and A306 in PheSMb or PheSMa corresponded to T251 and A294 in PheSEc, respectively. Therefore, two substitutions of T252A and A306G were introduced into PheSMb through point mutation, generating PheSAG, which would theoretically enhance the p-Cl-Phe sensitivity of M. buryatense 5GB1C. In addition, PheSMb had 91.5% sequence identity with PheSMa, suggesting that they can likely replace each other.

Figure 2. Sequence alignment of PheS. The PheS C-terminal region of strains Escherichia coli K12, Bacillus subtilis 168, M. capsulatus (Bath), M. trichosporium OB3b, M. buryatense 5GB1C, M. alcaliphilum 20Z, and Methylomonas sp. LW13 were aligned using ClustalW2. The residues boxed with a red line indicates the conserved alanine residue that can be subjected to mutagenesis to create p-Cl-Phe sensitivity.
To test whether the expression of PheSAG enhanced the sensitivity of M. buryatense 5GB1C toward p-Cl-Phe, pheSAG-1 (pheSAG with its native transcription and translation signals) (Figure 3A) was inserted between the METBUDRAFT_2794 and METBUDRAFT_2795 genes in the chromosome of strain 5GB1C (Yan et al., 2016). However, strain 5GB1C containing pheSAG-1 displayed subtly enhanced sensitivity toward p-Cl-Phe (Figure 3B), indicating that the expression of PheSAG was insufficient to induce p-Cl-Phe sensitivity. Since Ptac was demonstrated to be a strong promoter in M. buryatense 5GB1C (Puri et al., 2015; Demidenko et al., 2016), the native promoter of pheSAG was replaced by Ptac, thus generating the expression cassette pheSAG-2 (Figure 3A). Unfortunately, cassette pheSAG-2 also failed to induce high sensitivity against 0.5–1 mM p-Cl-Phe in strain 5GB1C (Figure 3B).
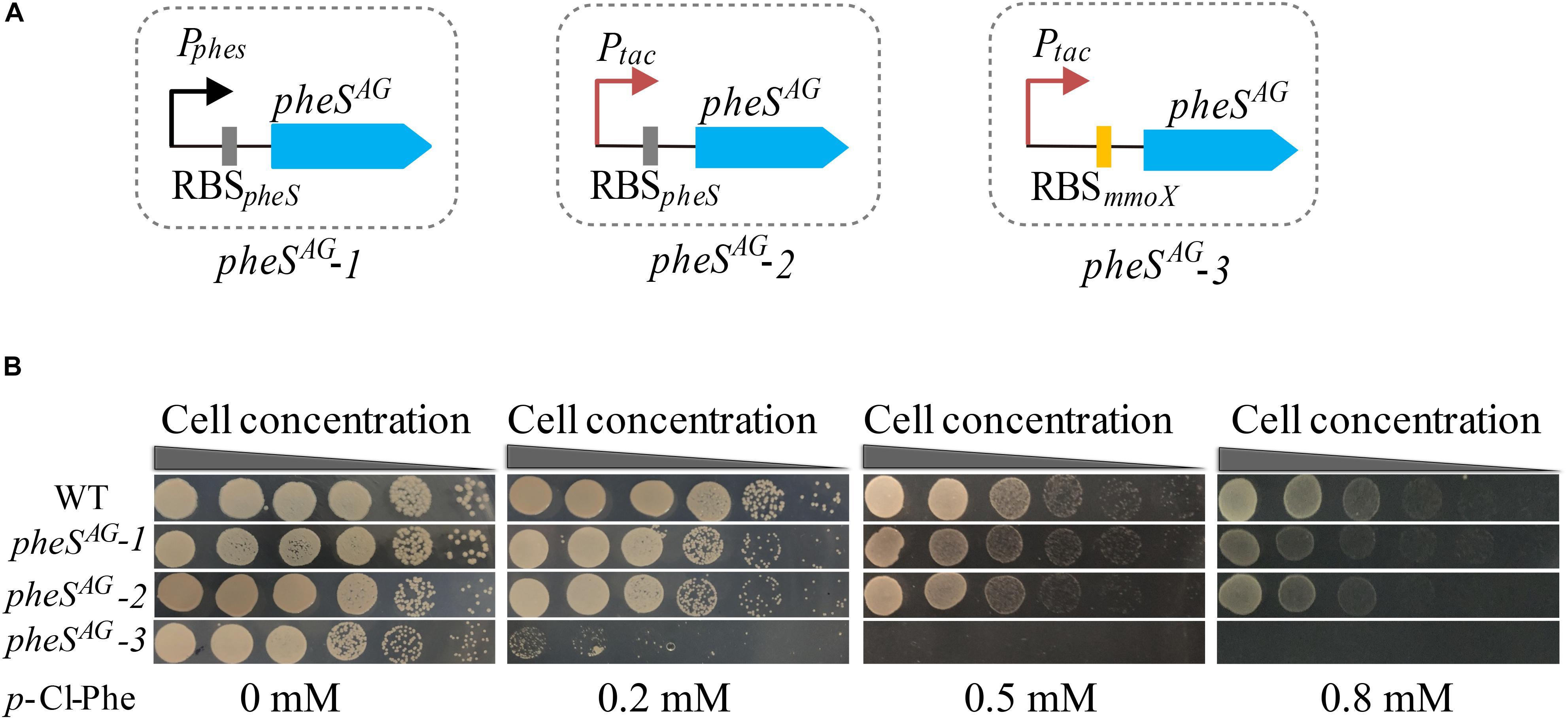
Figure 3. Optimizing the expression level of pheSAG in M. buryatense 5GB1C. (A) Different promoters and RBS were used to control the expression of pheSAG. PpheS and RBSpheS, the promoter and ribosome-binding site (RBS) region of wild type pheS; Ptac, tac promoter; RBSmmoX, the RBS region of mmoX. (B) p-Cl-Phe sensitivity of mutants containing different pheSAG. The sensitivity of 5G (pheSAG-1) and 5G (pheSAG-2) to p-Cl-Phe was not significantly different from that of wild type while the 5G (pheSAG-3) mutant was extremely sensitive to p-Cl-Phe.
The RBS region is crucial to the translation rate, which is usually used to tune protein expression (Barrick et al., 1994; Teramoto et al., 2011; Ravasi et al., 2012; Shi et al., 2018; Opgenorth et al., 2019). Considering that MmoX is an abundantly expressed protein in the absence of copper (Semrau et al., 2010), its RBS region was expected to trigger efficient translation initiation. Therefore, Ptac and RBSmmoX were combined to control the expression of PheSAG, generating the expression cassette pheSAG-3 (Figure 3A). As shown in Figure 3B, M. buryatense 5GB1C containing pheSAG-3 was extremely sensitive to p-Cl-Phe at concentrations over 0.5 mM, suggesting that pheSAG-3 could be used as an efficient CSM in strain 5GB1C.
pheSAG-3, Zeocin Resistance Marker and PCR Based Markerless Deletion Strategies
To verify the feasibility of pheSAG-3 as a CSM, a positive and counterselection cassette named PZ was constructed by assembling the SD sequence of lacZ and the Sh ble gene (zeo) behind pheSAG-3 (Figure 4A). PZ is only approximately 1.5 kb long. The PZ- and PCR-based marker-free deletion was performed according to previous reports (Yan et al., 2016). Briefly, a 450-bp fragment immediately downstream of the target region to be deleted was used as the DR sequence, then, the LF and RF regions (∼800 bp on each side), DR and PZ were fused by overlapping PCR in the order of LF, DR, PZ, and RF, and the product was transferred into strain 5GB1C by electroporation. The DR and PZ were inserted immediately upstream of the target region without any deletion, which was selected by zeocin; finally, the target region together with PZ was excised via recombination between DRs and selected by 0.5 mM p-Cl-Phe (Figure 4B).
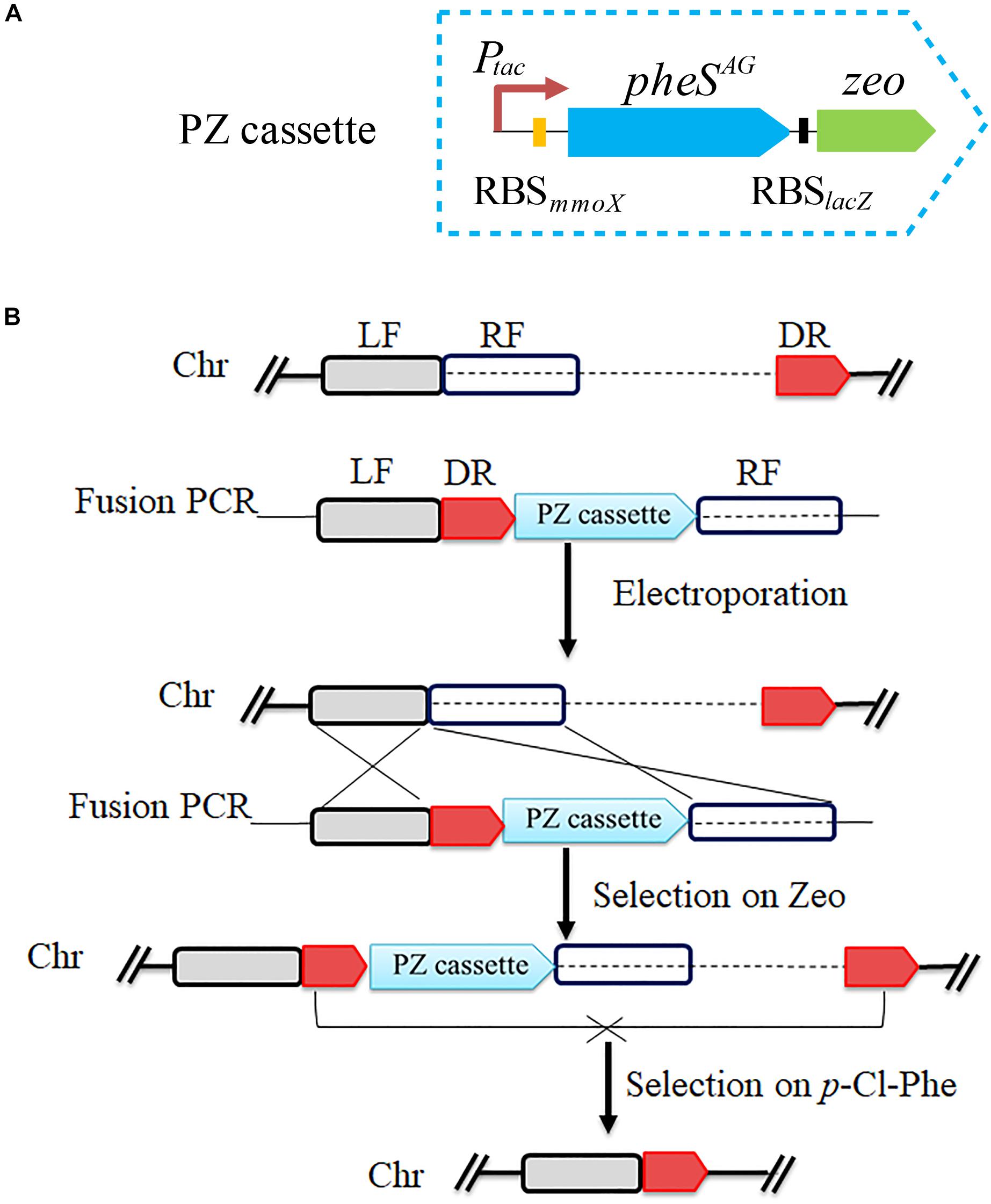
Figure 4. Scheme for pheSAG-3 and PCR-based marker-free DNA fragment deletion. (A) Construction of PZ cassette. The pheSAG-3 was fused with zeocin resistance gene to construct a PZ cassette. (B) PZ cassette and PCR based markerless deletion strategy. To delete a target fragment (dotted line), a ∼450-bp region just downstream of target fragment is used as a direct repeat sequence (DR) and is put ahead of the PZ cassette. Transformants with an insertion of the fragment containing the DR and PZ cassette just upstream of the target region are selected on zeocin. Recombination between two DR sequences excises both the PZ cassette and the target fragment, and the resulting mutant is selected on 0.8 mM p-Cl-Phe. Ptac, tac promoter; zeo, zeocin resistance gene; LF, left flanking region; RF, right flanking region; Chr, chromosome.
Both glgA1 (1.5 kb) and the smmo operon (∼10 kb) were successfully deleted using this strategy in M. buryatense 5GB1C (Figure 5). For each deletion, fifty colonies on the counterselection plate were verified by PCR, and the positive rates were 94 and 92% for glgA1 and the smmo operon, respectively. Moreover, the whole deletion process could be completed within 8 days. Then this strategy was employed to knock out glgA1 in M. alcaliphilum 20Z. Similar to M. buryatense 5GB1C, M. alcaliphilum 20Z harboring the PZ cassette was also extremely sensitive to p-Cl-Phe at concentrations over 0.5 mM (Figure 6A), indicating that PheSAG of M. buryatense could be compiled with PheTM.a. After counterselection, fifty colonies were tested by PCR (Figure 6B), and 46 were positive, with a positive rate of 94%.
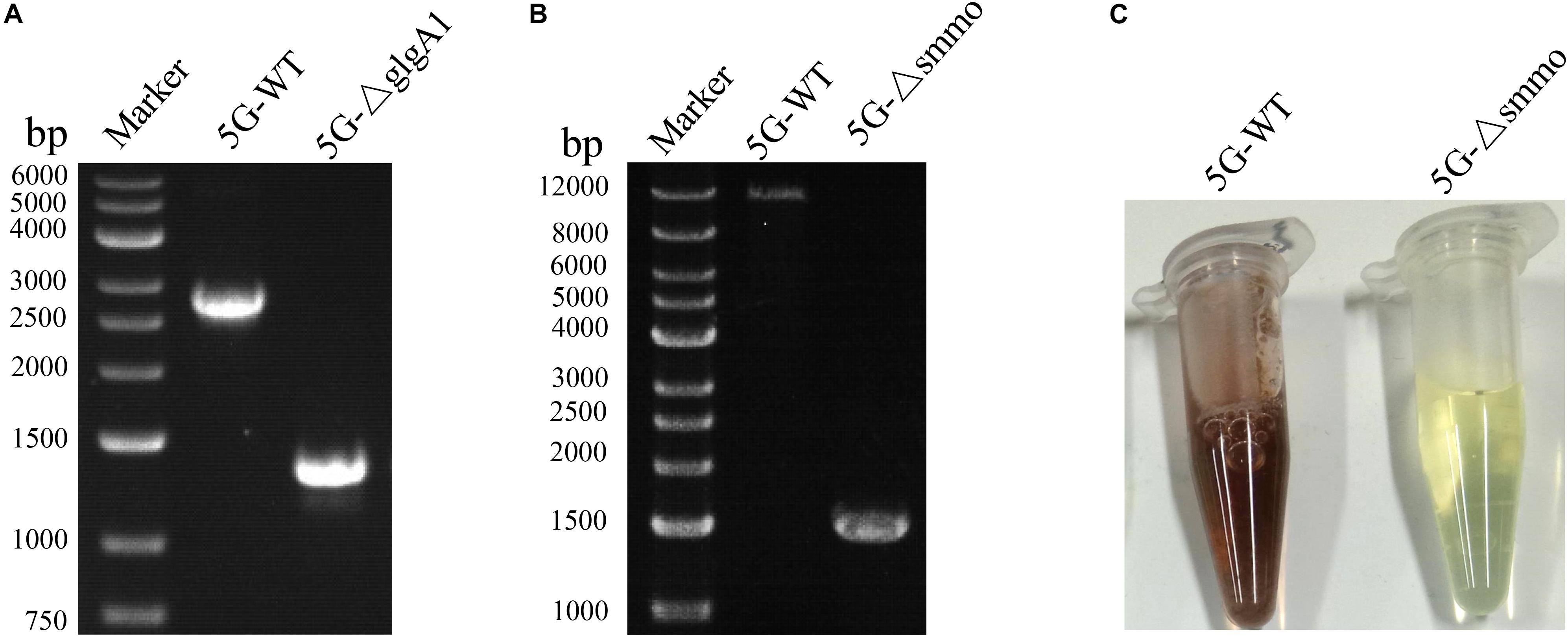
Figure 5. The deletion of glgA1 and smmo operon in M. buryatense 5GB1C. (A) PCR confirmation of glgA1 deletion. PCR was performed using the primers YZ1-F and YZ1-R. The nucleotide sequences of these primers are shown in Supplementary Table S1. From the genome sequence information for M. buryatense 5GB1C, the lengths of PCR amplicons from the wild type and the ΔglgA1 mutant are expected to be 2715 and 1284 bp, respectively. (B) PCR confirmation of smmo deletion. PCR was performed using the primers YZ2-F and YZ2-R. The nucleotide sequences of these primers are shown in Supplementary Table S1. From the genome sequence information for M. buryatense 5GB1C, the lengths of PCR amplicons from the wild type and the ΔsMMO mutant are expected to be 12,199 and 1530 bp, respectively. (C) The sMMO activity of strains 5GB1C and 5G-Δsmmo. Cells grown in NMS2 medium without copper were subjected to naphthalene oxidation assay developed by Graham et al. (1992). The cells expressing sMMO turn deep purple.

Figure 6. PZ cassette-based marker-free DNA fragment deletion. (A) p-Cl-Phe sensitivity of 5G-PZ and 20Z-PZ. Cell cultures were serially diluted 1:10. Each serial dilution was spotted onto agar plates containing p-Cl-Phe at different concentration. Plates were incubated in sealed jars at 30°C in an atmosphere of 25% methane in air for 5 days. (B) The deletion of glgA1 in M. alcaliphilum 20Z. PCR was performed using the primers YZ3-F and YZ3-R. The nucleotide sequences of these primers are shown in Supplementary Table S1. From the genome sequence information for M. alcaliphilum 20Z, the lengths of PCR amplicons from the wild type and the ΔglgA1 mutant are expected to be 2454 and 1023 bp, respectively.
Recoding pheSAG to Avoid Homologous Recombination Between pheSAG and pheS
Since pheSAG was almost identical to pheS, undesired homologous recombination between them may occur during genome modification, leading to false positive results. To avoid this undesired homologous recombination, we sought to decrease the similarity between them by recoding pheSAG. pheSAG was recoded according to the codon usage biases of several highly expressed proteins, namely, particulate methane monooxygenase, sMMO, and methanol dehydrogenase. The new gene pheSAG* shared 68% similarity to pheS (Supplementary Figure S1). Then pheSAG in PZ was replaced by pheSAG^*, generating a new positive and counterselection cassette PZ∗ (Figure 7A). Similar to PZ, PZ∗ also conferred M. buryatense 5GB1C and M. alcaliphilum 20Z with high sensitivity to p-Cl-Phe at concentrations over 0.5 mM (Figure 7B). The marker-free deletion of glgA1 was accomplished using PZ∗ in both strains, with positive rates over 92% (data not shown).
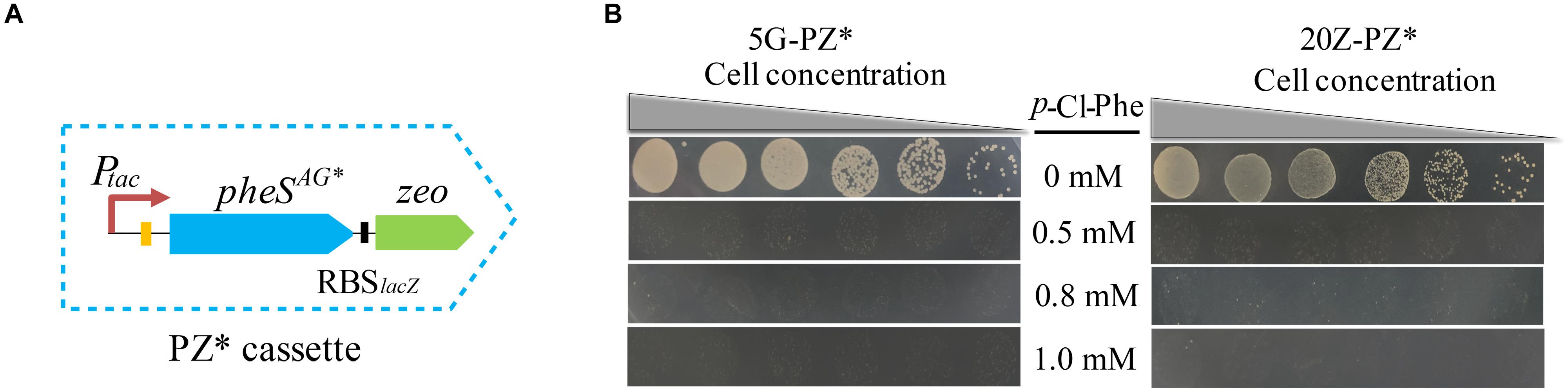
Figure 7. PZ* cassette-based marker-free DNA fragment deletion. (A) Construction of PZ* cassette. (B) p-Cl-Phe sensitivity of 5G-PZ* and 20Z-PZ*. Cell cultures were serially diluted 1:10. Each serial dilution was spotted onto agar plates containing p-Cl-Phe at different concentration. Plates were incubated in sealed jars at 30°C in an atmosphere of 25% methane in air for 5 days.
Discussion
In this study, two mutations, T252A and A306G, were introduced into the PheS of M. buryatense 5GB1C, and the resulting PheSAG mutant could recognize p-Cl-Phe. Then, strong transcription and translation signals (promoter Ptac and RBSmmoX) were used to enhance the expression level of PheSAG to the extent that the host cell failed to grow in the presence of 0.5 mM p-Cl-Phe, demonstrating that the pheSAG-3 expression cassette was an effective CSM. A positive and counterselection cassette PZ∗ was constructed based on pheSAG-3 and the zeocin resistance gene. A PZ∗- and PCR-based method enabled fast and efficient markerless genome deletion in M. buryatense 5GB1C and M. alcaliphilum 20Z, two promising methanotrophs for methane-based bioconversion. Furthermore, point mutations and foreign DNA insertions can also be easily realized using this method.
This method has several advantages over existing methods. First, no scar was left after modification (the Flp/FRT system will leave the FRT site) (Hoang et al., 1998). Second, counterselection was very efficient, with a positive rate greater than 92%. This is much higher than that typically achieved in sacB counterselection, which typically employs a single crossover event to integrate mutant cassettes prior to counterselection (and, in turn, often leads to <50% efficiency) (Puri et al., 2015). Third, the PZ∗ cassette was only 1.5 kb in length, and thus, the deletion construct could easily be assembled by PCR. The CRISPR/Cas9 system may be used with Methylotuvimicrobium in the future, but this system depends on vector construction and usually delivers low efficiency and off-target effects (Slaymaker et al., 2016; Chen et al., 2017; Tapscott et al., 2019). Therefore, the method developed here is a desired unmarked genetic manipulation tool for Methylotuvimicrobium.
This work also offers an alternative strategy to increase the expression level of PheSAG. Since the mutant PheSAG must compete with the endogenous wild-type PheS to form complexes with PheT (Mermershtain et al., 2011), only when the mutant PheSAG is abundantly expressed can the bacteria acquire greater sensitivity sensitive to p-Cl-Phe. According to a previous strategy of using strong promoters (Puri et al., 2015), the promoter Ptac was used to increase the expression of PheSAG, but this effort failed to enhance severe sensitivity toward p-Cl-Phe. Considering the importance of the RBS region in translation (Barrick et al., 1994; Teramoto et al., 2011; Ravasi et al., 2012; Shi et al., 2018; Opgenorth et al., 2019), RBSmmoX was employed to mediate the translation initiation of PheSAG, which resulted in profound sensitivity toward p-Cl-Phe. Therefore, when developing pheS as a CSM in a bacterium, optimizing the RBS region should be considered, and can be easily accomplished using the RBS design software that is now available (Na and Lee, 2010).
Data Availability Statement
The datasets generated for this study can be found in the GenBank accession nos. WP_047601123.1, WP_041851070.1, WP_010960035.1, ATQ70420.1, WP_017840103.1, CCE22593.1, and WP_033158015.1.
Author Contributions
XY developed the project idea and revised the manuscript. YL performed most of the experiments, analyzed the data, and prepared the manuscript. XH and PZ did some data analysis and performed some experiments. MC and QH provided consultation for the work and contributed significantly to the preparation of the manuscript. All authors reviewed the manuscript and agreed with the content.
Funding
This work was supported by the National Natural Science Foundation of China (31770125, 31970099, and 31800095) and Natural Science Foundation for Young Scientists of Jiangsu Province (BK20180541).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Mary Lidstrom from the University of Washington for providing the strain M. buryatense 5GB1C.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00441/full#supplementary-material
References
Andrea, D. L. T., Aisha, M., Frances, C., Laurens, L. M. L., Beck, D. A. C., Pienkos, P. T., et al. (2015). Genome-scale metabolic reconstructions and theoretical investigation of methane conversion in Methylomicrobium buryatense strain 5G(B1). Microb. Cell. Fact. 14:188. doi: 10.1186/s12934-015-0377-3
Barrett, A. R., Yun, K., Inamasu, K. S., Son, M. S., Vukovich, J. M., and Hoang, T. T. (2008). Genetic tools for allelic replacement in Burkholderia species. Appl. Environ. Microbiol. 74:4498. doi: 10.1128/AEM.00531-08
Barrick, D., Villanueba, K., Childs, J., Kalil, R., Schneider, T. D., Lawrence, C. E., et al. (1994). Quantitative analysis of ribosome binding sites in E.coli. Nucleic. Acids. Res. 22, 1287–1295. doi: 10.1093/nar/22.7.1287
Carr, J. F., Danziger, M. E., Huang, A. L., Dahlberg, A. E., and Gregory, S. T. (2015). Engineering the genome of Thermus thermophilus using a counterselectable marker. J. Bacteriol. 197, 1135–1144. doi: 10.1128/JB.02384-14
Chen, J. S., Dagdas, Y. S., Kleinstiver, B. P., Welch, M. M., Sousa, A. A., Harrington, L. B., et al. (2017). Enhanced proofreading governs CRISPR–Cas9 targeting accuracy. Nature 550, 407–410. doi: 10.1038/nature24268
Demidenko, A., Akberdin, I. R., Allemann, M., Allen, E. E., and Kalyuzhanaya, M. G. (2016). Fatty acid biosynthesis pathways in Methylomicrobium buryatense 5G(B1). Front. Microbiol. 7:2167. doi: 10.3389/fmicb.2016.02167
Fei, Q., Guarnieri, M. T., Tao, L., Laurens, L. M., Dowe, N., and Pienkos, P. T. (2014). Bioconversion of natural gas to liquid fuel: opportunities and challenges. Biotechnol. Adv. 32, 596–614. doi: 10.1016/j.biotechadv.2014.03.011
Graham, D. W., Korich, D. G., Leblanc, R. P., Sinclair, N. A., and Arnold, R. G. (1992). Applications of a colorimetric plate assay for soluble methane monooxygenase activity. Appl. Environ. Microbiol. 58, 2231–2236. doi: 10.1128/aem.58.7.2231-2236.1992
Hanson, R. S., and Hanson, T. E. (1996). Methanotrophic bacteria. Microbiol. Rev. 60, 439–471. doi: 10.1006/mpat.1996.0035
Henard, C. A., Smith, H., Dowe, N., Kalyuzhnaya, M. G., Pienkos, P. T., and Guarnieri, M. T. (2016). Bioconversion of methane to lactate by an obligate methanotrophic bacterium. Sci. Rep. 6:21585. doi: 10.1038/srep21585
Hoang, T. T., Karkhoff-Schweizer, R. R., Kutchma, A. J., and Schweizer, H. P. (1998). A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212, 77–86. doi: 10.1016/s0378-1119(98)00130-9
Hou, C. T., Laskin, A. I., and Patel, R. N. (1979). Growth and polysaccharide production by Methylocystis parvus OBBP on methanol. Appl. Environ. Microbiol. 37, 800–804. doi: 10.1128/aem.37.5.800-804.1979
Ishikawa, M., Yokoe, S., Kato, S., and Hori, K. (2018). Efficient counterselection for Methylococcus capsulatus (Bath) by using a mutated pheS gene. App.l Environ. Microbiol. 84:e01875-18. doi: 10.1128/AEM.01875-18
Kalyuzhnaya, M. G., Puri, A. W., and Lidstrom, M. E. (2015). Metabolic engineering in methanotrophic bacteria. Metab. Eng. 29, 142–152. doi: 10.1016/j.ymben.2015.03.010
Kalyuzhnaya, M. G., Yang, S., Rozova, O. N., Smalley, N. E., Clubb, J., Lamb, A., et al. (2013). Highly efficient methane biocatalysis revealed in a methanotrophic bacterium. Nat. Commun. 4:2785. doi: 10.1038/ncomms3785
Kast, P. (1994). pKSS — A second-generation general purpose cloning vector for efficient positive selection of recombinant clones. Gene 138, 109–114. doi: 10.1016/0378-1119(94)90790-0
Kast, P., and Hennecke, H. (1991). Amino acid substrate specificity of Escherichia coli phenylalanyl-tRNA synthetase altered by distinct mutations. J. Mol. Biol. 222, 99–124. doi: 10.1016/0022-2836(91)90740-w
Kentaro, M. (2015). Molecular engineering of a PheS counterselection marker for improved operating efficiency in Escherichia coli. Biotechniques 58, 86–88. doi: 10.2144/000114257
Khmelenina, V. N., Kalyuzhnaya, M. G., Starostina, N. G., Suzina, N. E., and Trotsenko, Y. A. (1997). Isolation and characterization of halotolerant alkaliphilic methanotrophic bacteria from Tuva soda lakes. Curr. Microbiol. 35, 257–261. doi: 10.1007/s002849900249
Kristich, C., Chandler, J., and Gm, D. (2007). Development of a host-genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis. Plasmid 57, 131–144. doi: 10.1016/j.plasmid.2006.08.003
Kühn, R., and Torres, R. M. (2002). Cre/loxP recombination system and gene targeting. Methods Mol. Biol. 180, 175–204. doi: 10.1385/1-59259-178-7:175
Larkin, M. A., Blackshields, G., Brown, N., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Mermershtain, I., Finarov, I., Klipcan, L., Kessler, N., Rozenberg, H., and Safro, M. G. (2011). Idiosyncrasy and identity in the prokaryotic Phe-system: crystal structure of E. coli phenylalanyl-tRNA synthetase complexed with phenylalanine and AMP. Protein. Sci. 20, 160–167. doi: 10.1002/pro.549
Na, D., and Lee, D. (2010). RBS Designer: software for designing synthetic ribosome binding sites that yields a desired level of protein expression. Bioinformatics 26, 2633–2634. doi: 10.1093/bioinformatics/btq458
Nguyen, A. D., Hwang, I. Y., Lee, O. K., Kim, D., Kalyuzhnaya, M. G., Mariyana, R., et al. (2018). Systematic metabolic engineering of Methylomicrobium alcaliphilum 20Z for 2,3-butanediol production from methane. Metab. Eng. 47, 323–333. doi: 10.1016/j.ymben.2018.04.010
Opgenorth, P., Costello, Z., Okada, T., Goyal, G., Chen, Y., Gin, J., et al. (2019). Lessons from two Design-Build-Test-Learn cycles of dodecanol production in Escherichia coli aided by machine learning. ACS Synth. Biol. 6, 1337–1351. doi: 10.1021/acssynbio.9b00020
Puri, A. W., Sarah, O., Frances, C., Ted, C., Beck, D. A. C., Kalyuzhnaya, M. G., et al. (2015). Genetic tools for the industrially promising methanotroph Methylomicrobium buryatense. Appl. Environ. Microbiol. 81, 1775–1781. doi: 10.1128/AEM.03795-14
Ravasi, P., Peiru, S., Gramajo, H., and Menzella, H. G. (2012). Design and testing of a synthetic biology framework for genetic engineering of Corynebacterium glutamicum. Microb. Cell. Fact. 11:147. doi: 10.1186/1475-2859-11-147
Semrau, J. D., Dispirito, A. A., and Yoon, S. (2010). Methanotrophs and copper. FEMS Microbiol. Rev. 34, 496–531. doi: 10.1111/j.1574-6976.2010.00212.x
Shevchuk, N. A., Bryksin, A. V., Nusinovich, Y. A., Cabello, F. C., Sutherland, M., and Ladisch, S. (2004). Construction of long DNA molecules using long PCR-based fusion of several fragments simultaneously. Nucleic Acids Res. 32:e19. doi: 10.1093/nar/gnh014
Shi, F., Luan, M., and Li, Y. (2018). Ribosomal binding site sequences and promoters for expressing glutamate decarboxylase and producing γ-aminobutyrate in Corynebacterium glutamicum. AMB Express 8:61. doi: 10.1186/s13568-018-0595-2
Slaymaker, I. M., Gao, L., Zetsche, B., Scott, D. A., Yan, W. X., and Zhang, F. (2016). Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88. doi: 10.1126/science.aad5227
Strong, P. J., Kalyuzhnaya, M., Silverman, J., and Clarke, W. P. (2016). A methanotroph-based biorefinery: potential scenarios for generating multiple products from a single fermentation. Bioresour. Technol. 215, 314–323. doi: 10.1016/j.biortech.2016.04.099
Strong, P. J., Xie, S., and Clarke, W. P. (2015). Methane as a resource: can the methanotrophs add value? Environ. Sci. Technol. 49, 4001–4018. doi: 10.1021/es504242n
Tapscott, T., Guarnieri, M. T., and Henard, C. A. (2019). Development of a CRISPR/Cas9 system for Methylococcus capsulatus in vivo gene editing. Appl. Environ. Microbiol. 85:e00340-19. doi: 10.1128/AEM.00340-19
Teramoto, H., Watanabe, K., Suzuki, N., Inui, M., and Yukawa, H. (2011). High yield secretion of heterologous proteins in Corynebacterium glutamicum using its own Tat-type signal sequence. Appl. Microbiol. Biotechnol. 91, 677–687. doi: 10.1007/s00253-011-3281-8
Trotsenko, Y. A., Doronina, N. V., and Khmelenina, V. N. (2005). Biotechnological potential of aerobic methylotrophic bacteria: a review of current state and future prospects. Appl. Biochem. Microbiol. 41, 433–441. doi: 10.1007/s10438-005-0078-5
Xin, Y., Guo, T., Mu, Y., and Kong, J. (2017). Development of a counterselectable seamless mutagenesis system in lactic acid bacteria. Microb. Cell. Fact. 16:116. doi: 10.1186/s12934-017-0731-8
Yan, X., Chu, F., Puri, A. W., Fu, Y., and Lidstrom, M. E. (2016). Electroporation-based genetic manipulation in type I methanotrophs. Appl. Environ. Microbiol. 82, 2062–2069. doi: 10.1128/aem.03724-15
Keywords: counterselection, pheS, Methylotuvimicrobium, electroporation-based, markerless mutagenesis
Citation: Liu Y, He X, Zhu P, Cheng M, Hong Q and Yan X (2020) pheSAG Based Rapid and Efficient Markerless Mutagenesis in Methylotuvimicrobium. Front. Microbiol. 11:441. doi: 10.3389/fmicb.2020.00441
Received: 19 November 2019; Accepted: 02 March 2020;
Published: 31 March 2020.
Edited by:
Marina G. Kalyuzhanaya, San Diego State University, United StatesReviewed by:
Michael Thomas Guarnieri, National Renewable Energy Lab (NREL), United StatesMichael Benedik, Texas A&M University, United States
Copyright © 2020 Liu, He, Zhu, Cheng, Hong and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Yan, eWFueGluQG5qYXUuZWR1LmNu