
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 12 March 2020
Sec. Evolutionary and Genomic Microbiology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.00376
 Michael L. Chen1,2
Michael L. Chen1,2 Eric D. Becraft1,3
Eric D. Becraft1,3 Maria Pachiadaki1,4
Maria Pachiadaki1,4 Julia M. Brown1
Julia M. Brown1 Jessica K. Jarett5†
Jessica K. Jarett5† Josep M. Gasol6,7
Josep M. Gasol6,7 Nikolai V. Ravin8
Nikolai V. Ravin8 Duane P. Moser9
Duane P. Moser9 Takuro Nunoura10
Takuro Nunoura10 Gerhard J. Herndl11,12
Gerhard J. Herndl11,12 Tanja Woyke5
Tanja Woyke5 Ramunas Stepanauskas1*
Ramunas Stepanauskas1*
Bacterial candidate phylum PAUC34f was originally discovered in marine sponges and is widely considered to be composed of sponge symbionts. Here, we report 21 single amplified genomes (SAGs) of PAUC34f from a variety of environments, including the dark ocean, lake sediments, and a terrestrial aquifer. The diverse origins of the SAGs and the results of metagenome fragment recruitment suggest that some PAUC34f lineages represent relatively abundant, free-living cells in environments other than sponge microbiomes, including the deep ocean. Both phylogenetic and biogeographic patterns, as well as genome content analyses suggest that PAUC34f associations with hosts evolved independently multiple times, while free-living lineages of PAUC34f are distinct and relatively abundant in a wide range of environments.
Recent advances in cultivation-independent techniques such as metagenomics and single-cell genomics have enabled the study of the coding potential of many “candidate phyla” – uncultivated microbial lineages that are distinct from well-classified bacterial and archaeal phyla (Rinke et al., 2013; Brown et al., 2015; Castelle et al., 2015; Anantharaman et al., 2016; Hug et al., 2016; Parks et al., 2017). In the absence of cultivation, single-cell genomics enables functional characterization of these candidate phyla by providing genetic blueprints for individual cells (Stepanauskas et al., 2017). Candidate phylum “PAUC34f,” also known as “SAUL” (sponge-associated unclassified lineage), is a deeply branching lineage of bacteria that was first discovered in 16S rRNA sequence surveys of prokaryotic communities associated with marine sponges (Hentschel et al., 2002; Schmitt et al., 2012; Thomas et al., 2016; Pita et al., 2018). Early attempts to classify these 16S rRNA sequences placed them within Deltaproteobacteria (Hentschel et al., 2002), Deferribacteres, Acidobacteria (Webster et al., 2011), or left them unclassified (Montalvo and Hill, 2011). Subsequent phylogenetic analyses of the 16S rRNA sequence (Kamke et al., 2014; Astudillo-García et al., 2017) identified PAUC34f as a sister-clade to the candidate phylum Latescibacteria, a widely distributed, putatively saprophytic group (Youssef et al., 2015; Farag et al., 2017), and distantly related to the candidate phylum Poribacteria, a group that represents a substantial fraction of sponge microbiomes (Taylor et al., 2007a; Schmitt et al., 2012; Kamke et al., 2014; Steinert et al., 2017).
Since the initial discovery of PAUC34f, representatives have repeatedly been found in molecular surveys of marine sponge (Porifera) microbiomes from various species, comprising 0.001–20.7% of the identified microbiota (Montalvo and Hill, 2011; Cuvelier et al., 2014; Hardoim and Costa, 2014; Astudillo-García et al., 2017; Jensen et al., 2017; Weigel and Erwin, 2017). Using metagenomic assembly and binning of sponge-associated DNA, two composite genomes (metagenome-assembled genomes, or “MAGs”) of PAUC34f were analyzed recently, significantly expanding our understanding of this group’s phylogeny and metabolic potential. These analyses revealed the potential for degradation of sponge- and algae-derived carbohydrates, phosphate transport and storage, and signatures of host-association, including eukaryotic-like proteins (Astudillo-García et al., 2017).
To date, most of the discussion around PAUC34f has focused on its role in the sponge microbiome, where bacteria are thought to mediate critical biochemical processes (Taylor et al., 2007b; Fan et al., 2013; Webster and Thomas, 2016; Pita et al., 2018). These sponge-associated microbial communities are distinct from those in surrounding seawater and sediment (Thomas et al., 2010, 2016; Reveillaud et al., 2014; Karimi et al., 2017) and differ among sponge species (Webster et al., 2010; Hardoim et al., 2012; Schmitt et al., 2012; Reveillaud et al., 2014; Thomas et al., 2016; Steinert et al., 2017). However, PAUC34f 16S rRNA sequences have also been detected at low abundance in non-sponge environments, including associations with ascidians (Erwin et al., 2014) and corals (Sunagawa et al., 2010; Kimes et al., 2013), tropical stream sediments (Costa et al., 2015), seawater, and marine sediments (Taylor et al., 2013; Thomas et al., 2016; Karimi et al., 2017; Thrash et al., 2017). Recent analyses of two PAUC34f MAGs recovered from the Gulf of Mexico dead zone, coupled with metatranscriptomics, revealed putatively active PAUC34f representatives with evidence of aerobic and facultative aerobic metabolisms, and capacities for complex carbohydrate degradation (Thrash et al., 2017), a similar metabolic profile to PAUC34f MAGs derived from sponges (Astudillo-García et al., 2017).
Here we use single-cell genomics, metagenome fragment recruitment, and comparative genomics to investigate the coding potential and global distribution of PAUC34f. We report 21 PAUC34f single amplified genomes (SAGs) originating from the mesopelagic and bathypelagic ocean, lake sediment, terrestrial groundwater, and a tunicate microbiome (Figure 1). We also utilize previously reported MAGs of PAUC34f from various environments: the Red Sea (Delmont et al., 2017; Tully et al., 2018), the northern Gulf of Mexico dead zone (Thrash et al., 2017), the Mediterranean sponges Aplysina aerophoba and Petrosia ficiformis (Astudillo-García et al., 2017), the East Africa Coast, and the South Atlantic (Tully et al., 2018) (Figure 1). We (i) report the general characteristics of these newly described PAUC34f SAGs and compare them to those of previously analyzed draft genomes; (ii) present an updated 16S rRNA sequence phylogeny, with a focus on the evolution of host-associated, free-living, and other lifestyles; (iii) use a metagenome fragment recruitment approach to assess global environmental abundance, diversity, and specificity of various PAUC34f lineages; and (iv) re-assess putative genomic signatures of host-association, such as eukaryotic-like domains (ELDs) present in PAUC34f lineages from diverse environments.
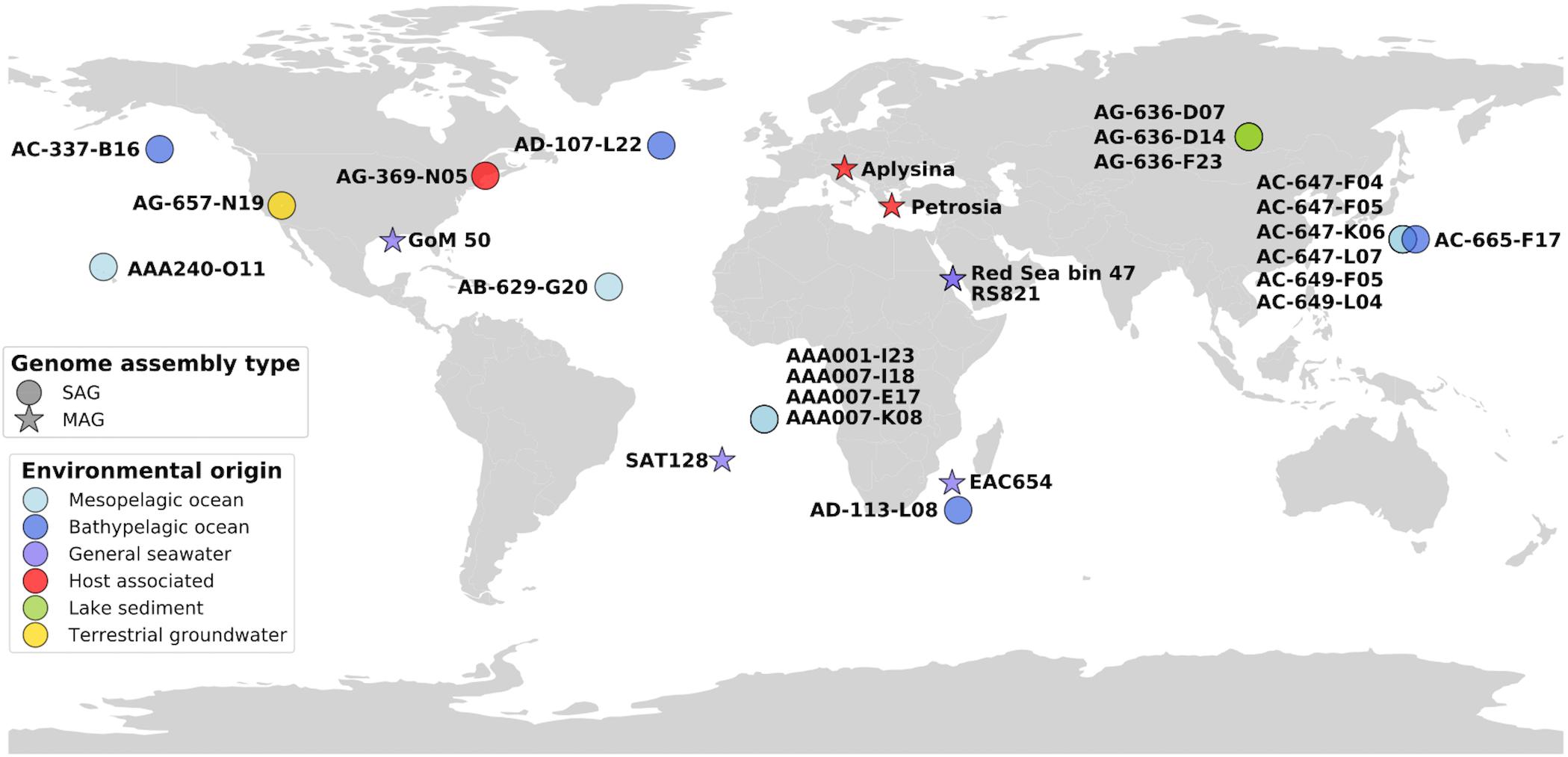
Figure 1. Map of genome origins, colored by sampling environment. SAGs and MAGs are plotted in their approximate geographic areas.
Details of sample collection (dates, locations, and depths) are reported in Table 1. For the marine SAGs, ocean water samples were collected using Niskin bottles. One milliliter aliquots were amended with 5% glycerol and 1× TE buffer (all final concentrations), and stored at −80°C until further analysis. The tunicate sample was immediately placed on ice until processed in the lab. Approximately 100 g of tissue was rinsed, placed in a Ninja blender and homogenized for 10–20 s. The homogenate was passed through a 100 μm filter, amended with 5% glycerol and 1× TE buffer and stored at −80°C.
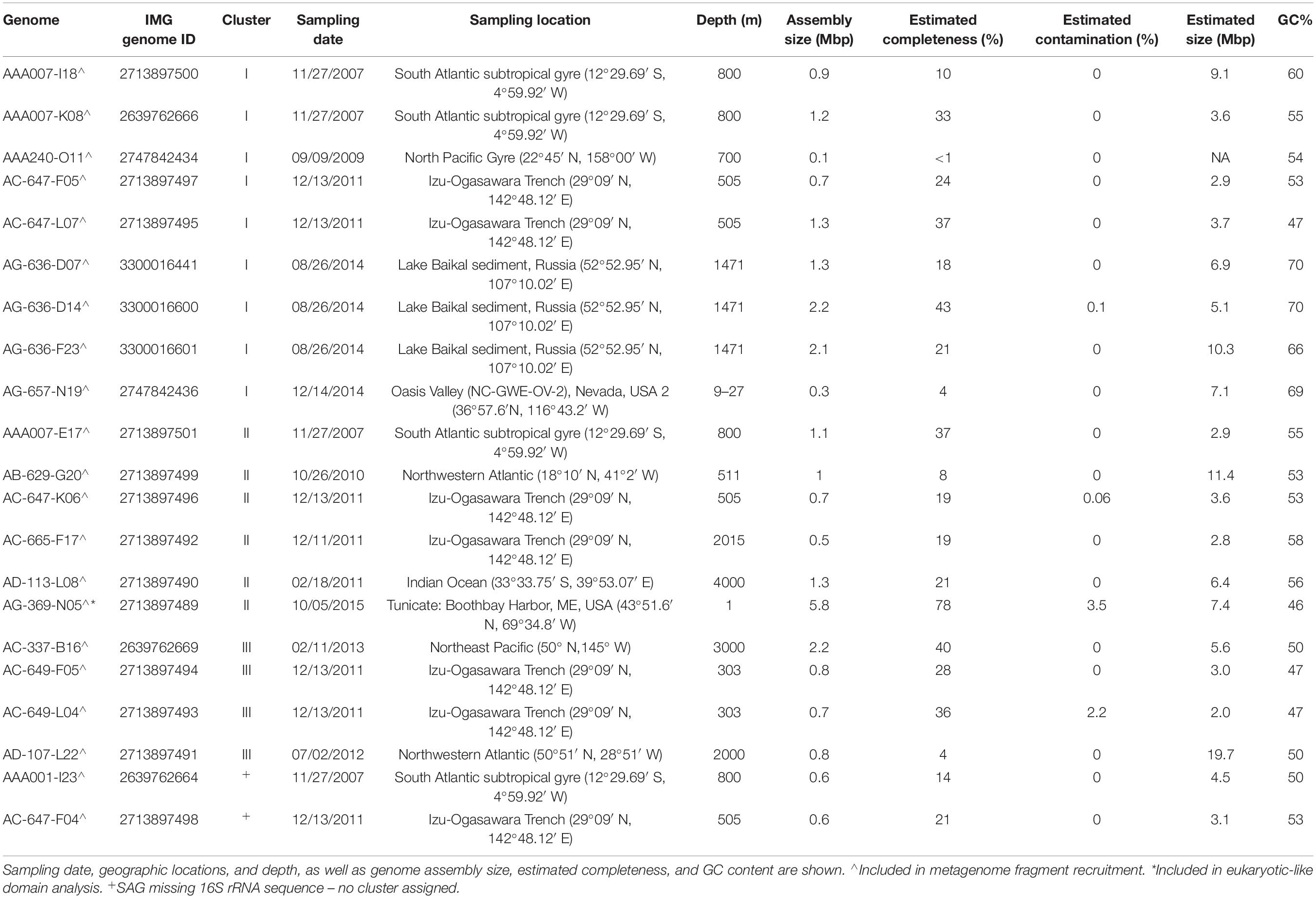
Table 1. Assembly statistics and sampling metadata for the 21 PAUC34f SAGs reported in this study.
Terrestrial groundwater samples (25°C, and pH 7.5) were obtained from a 4-inch nominal PVC-cased monitoring well in Oasis Valley, Nye County, NV, United States (36°57.6′N, 116°43.2′ W, 1,084 m elevation) on 14 December, 2014 (Becraft et al., 2017). The well, NC-GWE-OV-2, was drilled in 2011 and samples an alluvial aquifer (silty sand to gravel) located in the discharge zone of the volcanic Pahute Mesa flow system from a screened interval at 9.1–27.4 m below land surface. Samples were fixed in the same manner as the ocean water samples in the field after discharge of 5,962 L (31 hole volumes) with a Grunfos 30SQ130 submersible pump operated at 25 L.min–1.
Surface sediment samples were collected from Lake Baikal, Russia, using a gravity corer on 26 August, 2014, at water depth of 1471 m, coordinates 52°52.95′ N, 107°10.02′ E. The core’s wet sediment sample had a temperature of 3.8°C and a pH of 7.32, measured immediately after bringing the sample onboard the ship. Five grams of the upper sediment sample (0–1 cm below the lake floor) was mixed with 20 mL of sterile 1× PBS buffer in a 50 ml tube. The sample was centrifuged for 30 s at 2,500 g to remove large particles. One milliliter of supernatant was placed into a sterile cryovial and mixed with 100 μl GlyTE stock (20 ml 100× TE, pH 8.0, 60 ml sterile deionized water, and 100 ml molecular-grade glycerol) and stored at −80°C.
The generation, identification, sequencing, and de novo assembly of SAGs was performed at the Bigelow Laboratory Single Cell Genomics Center.1 Cells were separated with fluorescence-activated cell sorting and lysed with KOH as previously described (Stepanauskas and Sieracki, 2007; Rinke et al., 2014; Stepanauskas et al., 2017). In most cases, genomic DNA amplification was done with multiple displacement amplification (MDA) (Dean et al., 2002) while WGA-X (Stepanauskas et al., 2017) was employed on SAGs with identifiers AG-369 and AG-636. The small subunit (SSU) rRNA gene sequences of all SAGs were obtained by PCR amplification and Sanger sequencing (Stepanauskas and Sieracki, 2007; Stepanauskas et al., 2017). Sequences were then classified using SILVA Mod v128 (Quast et al., 2013) implemented in CREST (Lanzén et al., 2012). SAGs identified as PAUC34f were selected for whole genome sequencing, which was performed as previously described (Stepanauskas et al., 2017). Assembly contamination, completeness, and size were estimated using CheckM (Parks et al., 2015).
Genomes were annotated using BlastKOALA and GhostKOALA (Kanehisa et al., 2016) as well as using the Integrated Microbial Genomes database (Chen et al., 2019). ELDs were predicted from amino acid sequences using the EffectiveDB server’s EffectiveELD tool in Protein Mode, with a minimum Z-score of 4 for enrichment in pathogenic bacteria (Jehl et al., 2011; Eichinger et al., 2016). Numbers of specific domain annotations were counted by keyword searching. Signal peptides were predicted from amino acid sequences using SignalP 4.1 (Petersen et al., 2011). The input was specified as Gram-negative bacteria that may contain TM regions, using default D-cutoff values.
16S rRNA sequences were aligned using the SINA alignment software (Pruesse et al., 2012). MEGA7 (Kumar et al., 2016) was used to manually trim alignments and construct maximum likelihood phylogenetic trees using the Kimura 2-parameter Model (Kimura, 1980), a Gamma distribution with invariable sites, and 95% partial deletion for 200 replicate bootstraps. The resulting Newick tree was visualized using iTOL v3 (Letunic and Bork, 2016).
Recruitment of metagenomic reads was performed using the Burrows-Wheel Aligner (BWA) (Li and Durbin, 2010), as described in Pachiadaki et al. (2017). Ribosomal (rRNA) genes were masked and alignment threshold was set to ≥100 nucleotide overlap and ≥95% nucleotide identity (operational definition of microbial species) (Konstantinidis and Tiedje, 2005; Konstantinidis et al., 2006; Jain et al., 2018). A total of 184 metagenomic datasets (Supplementary Table S3) were used from a variety of environments including the euphotic, mesopelagic and bathypelagic ocean realms, sponges, freshwater lakes, and subsurface aquifer groundwater. The relative abundance of each SAG or MAG’s relatives was determined as the fraction of metagenomic reads mapped per megabase of the reference genome. The heatmap plot was created in R (R Core Team, 2017) using the ggplot2 package (Wickham, 2009).
The 21 PAUC34f SAG assemblies ranged 0.3–5.8 Mbp in size, with estimated completeness of 1–78% (average 25%) and no detectable contamination (Table 1). Full genome sizes, estimated from the most complete SAG assemblies, ranged 5–8 Mbps (average of 6.3 Mbp; Table 1). These results are in line with published PAUC34f MAGs, which were estimated to be 2–7 Mbp (Astudillo-García et al., 2017; Delmont et al., 2017; Thrash et al., 2017; Tully et al., 2018). Previous comparative studies have suggested that, on average, sponge-associated bacteria have larger genomes than free-living bacterioplankton, possibly because of adaptations to a variable, nutrient-rich environment and/or enrichment in mobile genetic elements (Giovannoni et al., 2014; Horn et al., 2016). This is in contrast to the streamlining observed in many other bacterial symbionts (Giovannoni et al., 2014). The GC content of PAUC34f SAGs ranged 47–70%, indicating high genomic variability, with SAGs from Lake Baikal sediment and Oasis Valley groundwater on the high end of the reported GC spectrum for known organisms (66–70%).
Using our expanded PAUC34f dataset, we revisited phylogenetic analyses of this group. We identified 208 16S rRNA sequences in the NCBI database (as of June 2018) with >85% nucleotide similarity to sequences from PAUC34f SAGs (Supplementary Table S1), an increase from the 93 sequences reported in a previous analysis (Astudillo-García et al., 2017). Phylogenetic analysis of the 16S rRNA sequence resulted in three strongly bootstrap-supported clades (labeled Clusters I, II, and III in Figure 2; cluster labels match those reported in Astudillo-García et al., 2017). The phylogenetic topology was similar to that reported by Kamke et al. (2014) and Astudillo-García et al. (2017).
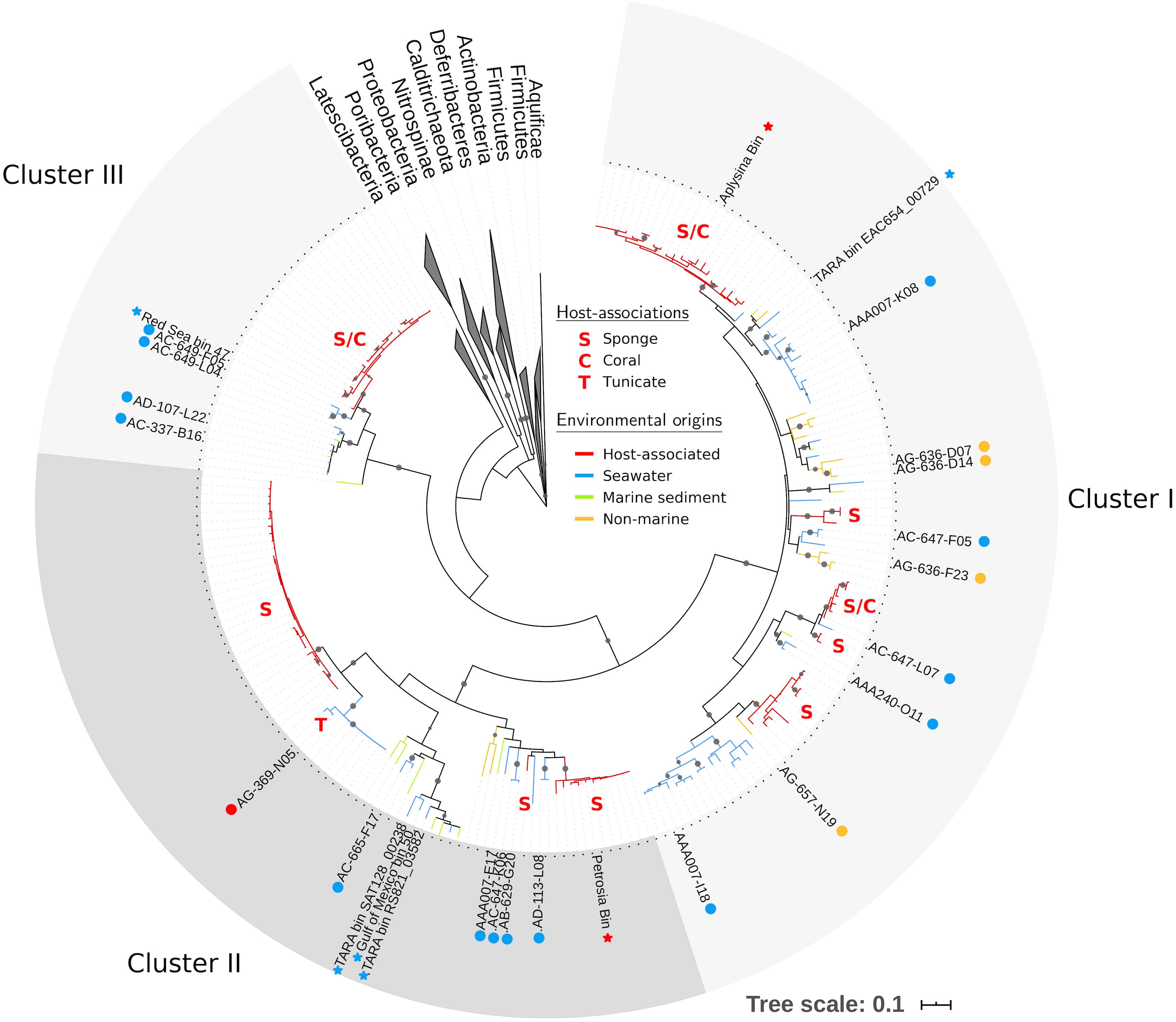
Figure 2. Maximum likelihood tree of PAUC34f using partial 16S rRNA sequences (∼800 bp). Tree includes sequences from PAUC34f genomes (labeled tips) and NCBI sequences with ≥85% similarity to 16S rRNA sequences from PAUC34f genomes (unlabeled tips). Gray circles indicate clades with bootstrap support ≥90%. Branch color indicates the environmental origins of sequences; host-associated clades are additionally annotated with letters indicating the host organism(s) (see Supplementary Table S1 for sequence identifiers and detailed environmental origins). Circles next to labeled tips indicate SAG sequences newly reported here, while stars indicate MAG sequences from other literature – both are colored by environmental origin. Scale bar represents 0.1 nucleotide substitutions per site.
Of all identified PAUC34f 16S rRNA sequences, 53% were derived from associations with a host: 105 from sponges, 4 from corals, and 1 from a tunicate (Table 2). Notably, 96% of these host-derived sequences clustered together, forming strongly supported (>90% bootstrap), monophyletic clades of at least three host-derived lineages (Figure 2 and Supplementary Table S1). This is in line with the sponge- and coral-specific evolutionary clusters that have been reported across multiple phyla (Simister et al., 2012). The clustering of these sequences suggests the evolutionary distinctness of host-associated and non-host-associated PAUC34f representatives, as reported for two lineages within the candidate phylum Poribacteria (Podell et al., 2018). Notably, lineages from the same host species do not always cluster together, and some host species are distributed across all three major PAUC34f clusters. This suggests multiple, historical environmental acquisitions by hosts throughout the diversification of PAUC34f, supporting the occasional horizontal transfer of organisms proposed for some sponge symbionts (Webster et al., 2010; Schmitt et al., 2012; Reveillaud et al., 2014). However, these are likely historical acquisitions, as most sponge symbionts are host-specific; indeed, some host-associated PAUC34f lineages have been observed to have host-specific vertical transmission (Hardoim et al., 2012; Schmitt et al., 2012; Gloeckner et al., 2013; Reveillaud et al., 2014; Thomas et al., 2016). Interestingly, the tunicate-derived SAG AG-369-N05 did not branch closely with any sponge- or coral-derived sequences. This is in agreement with a recent study demonstrating that ascidian microbiomes share some overlap with those of sponges and corals, but also have distinctive features and high levels of host-specificity (Erwin et al., 2014).
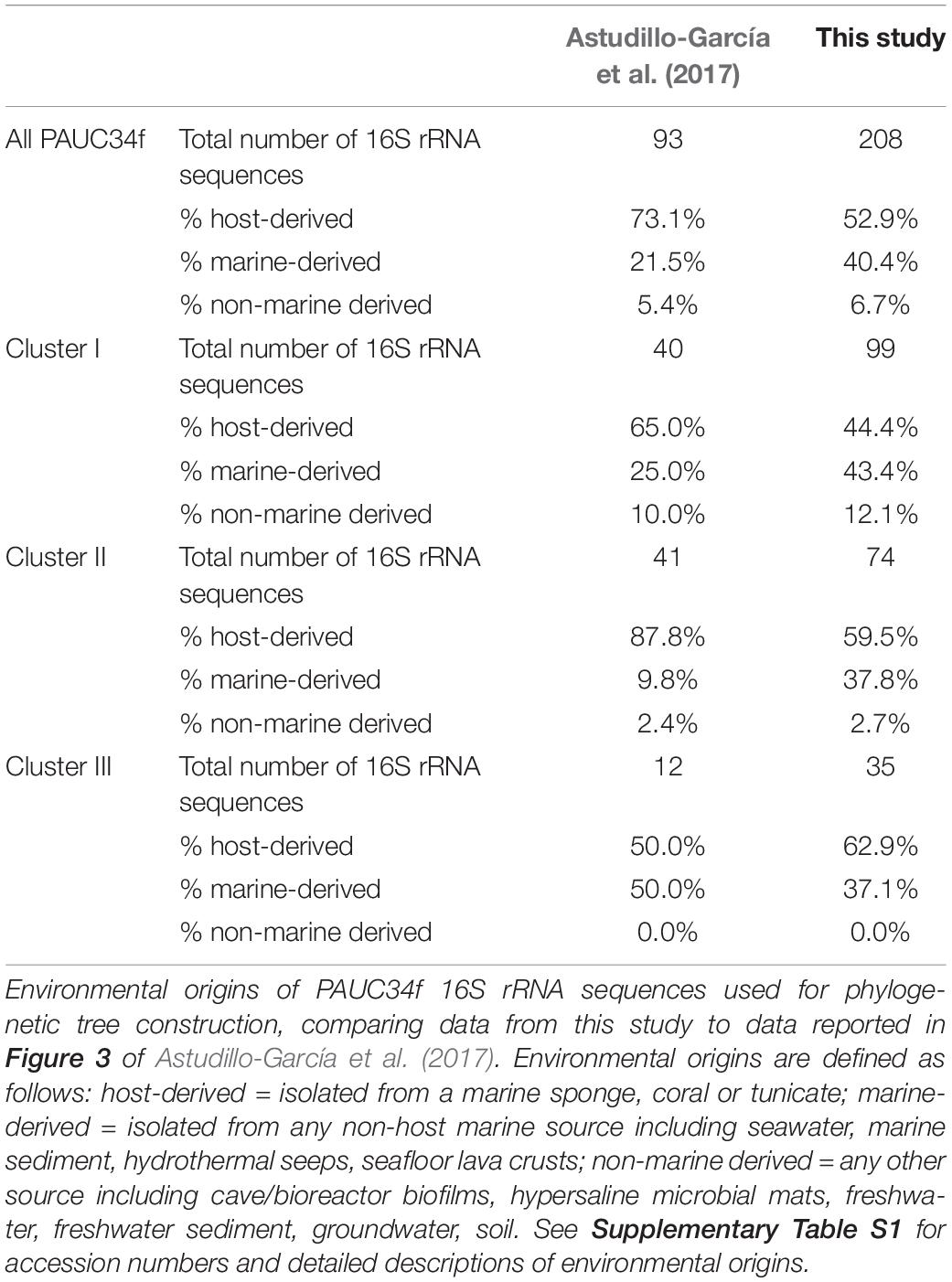
Table 2. Distribution of 16S rRNA sequences.
Surprisingly, about half of the 16S rRNA sequences derived from this study and public databases originate from non-sponge environments, shifting our largely sponge-focused understanding of PAUC34f (Table 2). Sequences from pelagic environments formed several distinct, bootstrap-supported clusters (Figure 2 and Supplementary Table S1). Environment-specific clades from lake sediments and subsurface aquifers were also found, though less abundant. Notably, Cluster III contained only marine (including marine host) sequences, while Clusters I and II contained 12 and 3% non-marine sequences.
In order to further investigate this environmental diversity suggested by the phylogenetic analyses, we used metagenomic fragment recruitment against PAUC34f SAGs and MAGs to assess the distribution and abundance of PAUC34f in various environments. Our main focus was to examine the abundance of PAUC34f in the open ocean, as we recovered multiple SAGs from this environment. Recruitment against Poribacteria and Latescibacteria SAGs and MAGs was also performed for comparative analyses (Table 1). Of the 184 metagenomes used (Supplementary Table S3), 78 were from the Tara Oceans Expedition (Pesant et al., 2015), and additional marine metagenomes were from the Malaspina (Duarte, 2015; Acinas et al., 2019) and other oceanographic expeditions, covering all oceanic realms (from the euphotic to the abyssopelagic zone), including oxygen minimum zones (Tsementzi et al., 2016). Additionally, we included metagenomes from environments where PAUC34f SAGs and MAGs have been identified, including the NC-GWE-OV-2 aquifer, Lake Baikal, and sponges.
We also explored the potential role of particles as a niche for the oceanic PAUC34f. The metagenomic datasets from Tara and Malaspina were generated from filters of different pore sizes, thus enriched in the “free-living” microbial fraction (0.2–0.8 μm for Malaspina; 0.2–0.8 μm or 0.2–3 μm for Tara) or enriched in the “particle-associated” fraction (>0.8 μm for Malaspina; >3 μm for Tara) (Pesant et al., 2015; Acinas et al., 2019). However, only Malaspina data included size-fractionated dark ocean metagenomes (Supplementary Table S4); in these data, PAUC34f’s relative recruitment from the free-living fraction was on average seven times higher compared to the particle-associated fraction (Supplementary Figure S1). This difference was statistically significant (p = 3.318e-06), implying that the oceanic PAUC34f recovered in our study likely have a predominantly free-living lifestyle. Notably, a published dataset from global Malaspina sites – using 16S rRNA amplicon sequencing – showed PAUC34f to be particularly abundant in the small-particle size fraction (0.8–3.0 μm) in the mesopelagic realm, while being more abundant in the planktonic size fraction (0.2–0.8 μm) in the bathypelagic realm (Supplementary Figure S2; Mestre et al., 2018).
The majority of the PAUC34f SAGs of pelagic origin showed high recruitment of metagenomes from the dark ocean (Figure 3). For these SAGs, the percentage of metagenome bases recruited reached up to 0.14% (Figure 3: see bar-graph on x-axis). The same SAGs produced little to no recruitment of metagenomes from the euphotic zone or other environments. The published Malaspina 16S rRNA dataset also reports PAUC34f to be most abundant in the mesopelagic and bathypelagic realms (Supplementary Figure S2; Mestre et al., 2018). In our fragment recruitment, the exceptions are SAGs AC-649-F05 and AAA007-E17, which appear to have cosmopolitan distribution. Although recruitment patterns do not appear to be clade-specific, SAGs with close phylogenetic proximity but different geographical origins have similar distribution patterns (AB-629-G20 from the mesopelagic North Atlantic and AC-647-K06 from the mesopelagic North Pacific recruit similarly, as do AD-107-L22 from the bathypelagic North Atlantic and AC-337-B16 from the bathypelagic North Pacific) (Figure 3). Our observations of recruitment by depth support previous analyses of Tara datasets that identified depth (and its associated physicochemical parameters) as a primary driver of community composition (Sunagawa et al., 2015). Within the dark ocean, we observe no apparent dispersal limitation of SAGs between ocean basins, as observed for other dark ocean groups (Pachiadaki et al., 2017). Similarly there aren’t clear distinctions by latitude, as some low-latitude SAGs (AAA007-I18 and AAA001-I23) recruit similarly to high-latitude ones (AC-337-B16 and AD-107-L22) (Figure 3). SAGs from Lake Baikal (PAUC34f: AG-636-D14, AG-636-D07, and AG-636-F23; Latescibacteria: AG-636-N02, AG-636-I09, and AG-636-I15) and from the Oasis Valley subsurface aquifer (PAUC34f: AG-657-N19) recruited highly and specifically from their respective environmental metagenomes.
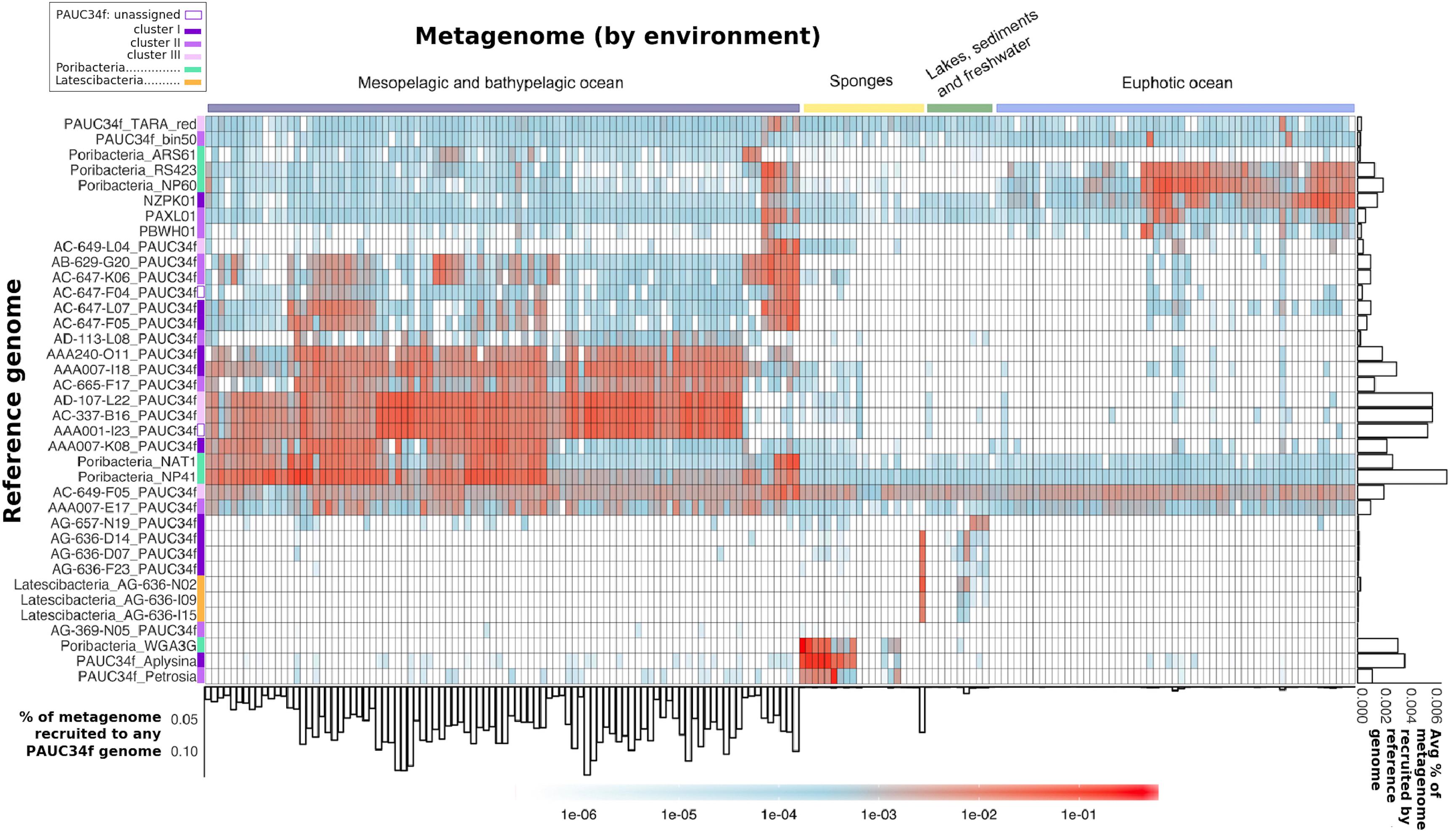
Figure 3. Global distribution of PAUC34f, Poribacteria, and Latescibacteria SAGs and MAGs, as determined by metagenomic fragment recruitment. Reference genomes (SAGs and MAGs) are listed along the y-axis, colored by phylogeny. Metagenomes are listed along the x-axis and colored by environment (see order and sources of metagenomes in Supplementary Table S3). The heatmap color legend indicates the fraction of metagenome bases recruited per megabase of each reference genome. The bar plot below the x-axis indicates the fraction (%) of metagenome bases recruited to at least one of the PAUC34f reference genomes. The bar plot to the right of the y-axis shows the average fraction (%) of metagenome bases recruited by each reference genome across metagenomes.
The sponge-derived MAGs of PAUC34f and Poribacteria recruited almost exclusively sponge metagenomes. Recruitment results corroborate our 16S rRNA sequence results as well as earlier reports indicating that sponge-specific genomes form distinct lineages (Simister et al., 2012). Poribacteria MAGs derived from oceanic metagenomes (“ARS61,” “RS423,” “NP60,” “NAT1,” and “NP41”) recruited largely from open ocean metagenomes and not from sponge metagenomes, supporting their placement within the open ocean-specific Pelagiporibacteria lineage (Podell et al., 2018). The tunicate-derived SAG AG-369-N05 did not recruit reads from any metagenomes analyzed in this study; given that no tunicate-derived metagenomes were included in the analysis, this result is unsurprising and supports the organismal specificity observed in ascidian microbiomes (Erwin et al., 2014).
The observed phylogenetic and biogeographic patterns suggest that many PAUC34f lineages are native to the open ocean. Accordingly, a previous metagenomic and metatranscriptomic study suggested that PAUC34f representatives were active in the Gulf of Mexico dead zone (Thrash et al., 2017). Further support for PAUC34f’s role as an important constituent of the dark ocean bacterioplankton comes from a dataset of 2618 SAGs collected from the dark ocean (FASTA included in Supplementary Data Sheet S2). In this dataset, PAUC34f appeared as the ninth most annotated bacterial phylum (out of 18) by 16S rRNA sequences (Supplementary Table S5), again suggesting this group’s non-trivial presence in the dark ocean. Given these results, it is surprising that this candidate phylum has not been reported as a substantial constituent of marine bacterioplankton until now. This may be due to the frequent misclassifications of molecular data (Hentschel et al., 2002; Montalvo and Hill, 2011; Webster et al., 2011). PAUC34f exemplifies significant gaps that may still exist in our understanding of the still-underexplored microbiome of the dark ocean.
Having observed PAUC34f’s presence and likely specialization in various non-host environments, we sought to investigate genomic and metabolic features that may facilitate these diverse lifestyles. Previous studies have reconstructed metabolic capabilities in MAGs from the Gulf of Mexico hypoxic zone and from sponges, finding aerobic/facultative aerobic metabolisms, complex carbohydrate degradation, and phosphate storage and transport among other features (Astudillo-García et al., 2017; Thrash et al., 2017). We found little to add to these comprehensive reconstructions, so we focused on assessing potential mechanisms of habitat specialization. Genome-wide annotation profiles revealed no clear partitioning of SAGs and MAGs by phylogeny or by environment (data not shown). Thus, we focused on reassessing genomic features linked to host-association, using the eight most complete PAUC34f genome assemblies (SAGs and MAGs).
Eukaryotic-like domains have been shown to be more abundant in healthy sponge microbiomes compared to the surrounding seawater (Thomas et al., 2010; Fan et al., 2013; Reynolds and Thomas, 2016; Karimi et al., 2017). We sought to assess whether such domains are present in non-sponge-associated representatives as well, and whether they are comparatively enriched within the sponge-associated representatives as potentially adaptive features. Recently, an investigation of two sponge-derived PAUC34f MAGs reported the presence of several of these domains (Astudillo-García et al., 2017). Interestingly, ELDs have also been found in both sponge-associated and free-living Poribacteria (Podell et al., 2018). We found predictions in PAUC34f genomes of several domains that have been reported in sponge microbiome literature (Supplementary Table S6). Numbers of each domain were highly variable among genomes and showed no environmental specificity (Supplementary Table S7).
We also compared PAUC34f to two closely related phyla by investigating ELD abundance in SAGs and MAGs of Latescibacteria and Poribacteria (Supplementary Table S2). This comparison is especially interesting because both PAUC34f and Poribacteria consist of many host-associated representatives, while Latescibacteria does not have any known host-association (Farag et al., 2017). Several ELDs showed significant differences in abundance between the three phyla (Figure 4). Of them, Ankyrin and NipSnap were enriched in PAUC34f and Poribacteria, as compared to Latescibacteria, while caspase domains were enriched only in Poribacteria (Supplementary Table S7). Only one domain, FNT3, was enriched in Latescibacteria relative to PAUC34f and Poribacteria.
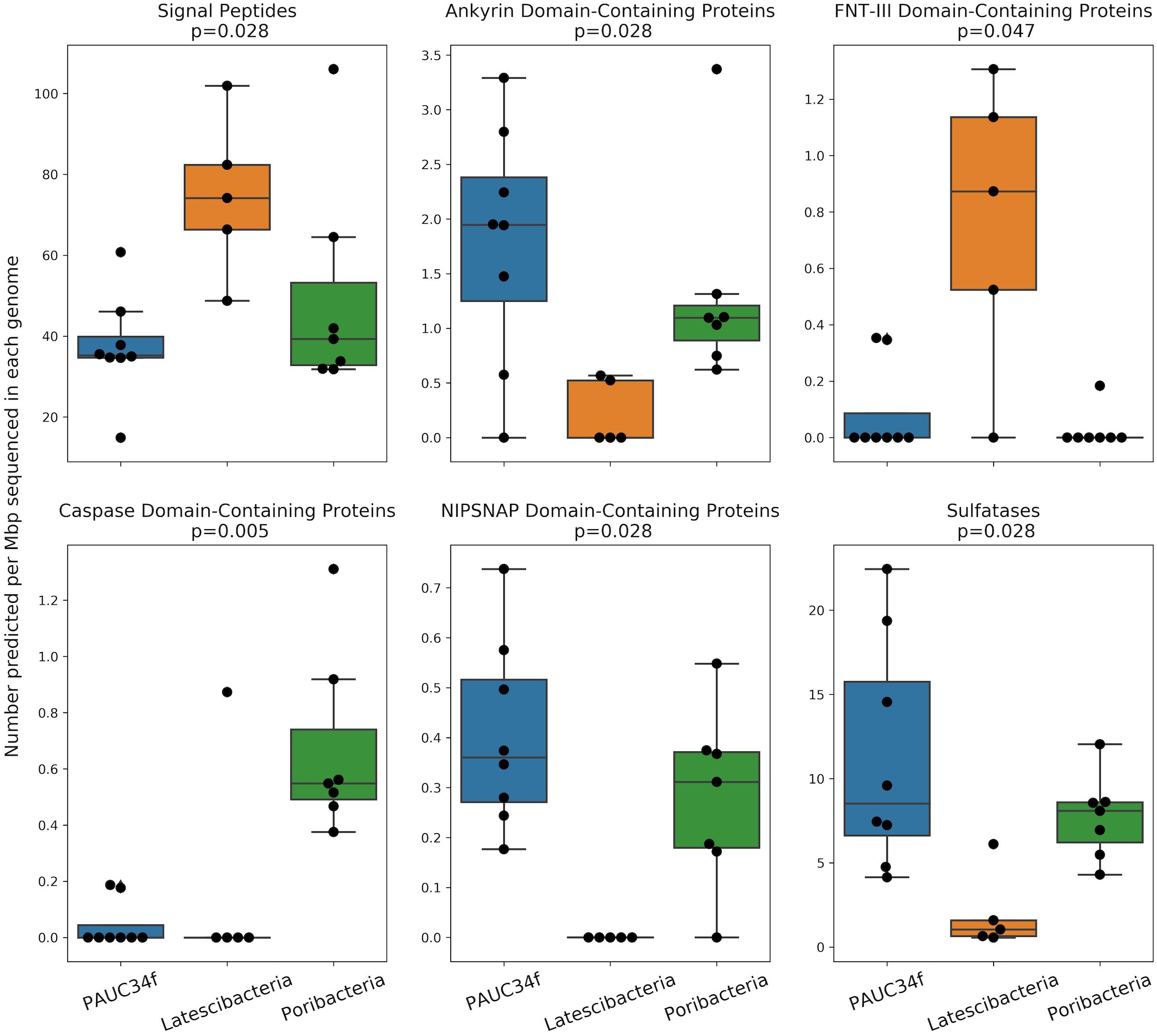
Figure 4. Number of ELDs, ELD-containing proteins, and other sponge-associated features across genomes (SAGs and MAGs) from PAUC34f, Latescibacteria, and Poribacteria. Feature counts are normalized to assembly size (Mbp). Statistics are the results of a Mood’s Median Test (test statistic was Pearson’s chi-squared statistic; values equal to the grand median were counted as below). Only statistically significant comparisons are shown. Remaining comparisons are included in Supplementary Figure S3.
The in situ functions of most ELDs remain elusive, although some domains have been characterized in model systems. Putative functions of these domains include the disruption of phagocytosis (Lee et al., 2002; Nguyen et al., 2014; Reynolds and Thomas, 2016), regulation of gene expression (Viboud and Bliska, 2005), host cell entry (Bierne and Cossart, 2002), cell death, differentiation, signaling, and immune response (Wall and Mccormick, 2014; Huh et al., 2016; Miller et al., 2016), or roles as antioxidants, nutrients, or cofactors (McIntire, 1998; Choi et al., 2008). The FNT3 domain – enriched in Latescibacteria – may facilitate extracellular cellulose degradation (Kataeva et al., 2002), consistent with metabolic analyses of Latescibacteria (Farag et al., 2017). We also found evidence of signal peptides in some of the ELD-containing proteins (Supplementary Table S7), suggesting that these proteins may be active outside the bacterial cell. Notably, the Latescibacteria assemblies were significantly enriched in signal peptides per Mbp (Figure 4). This may reflect the extracellular, cellulosome-affiliated enzymes identified in these bacteria (Farag et al., 2017).
In addition to ELDs, we also examined abundances of several other genome features that have been reported to be enriched in sponge microbiomes, as compared to surrounding seawater and/or sediment, including ABC transporters and sulfatases (Karimi et al., 2017). Neither of these features were more abundant in host-associated vs. free-living PAUC34f genomes (Supplementary Table S5), although PAUC34f and Poribacteria were relatively enriched in sulfatases, as compared to Latescibacteria (Figure 4). Sulfatases are known to facilitate the utilization of sulfated marine polysaccharides (Kamke et al., 2014; Helbert, 2017; Slaby et al., 2017) and are abundant in PAUC34f MAGs from the organic matter-rich Gulf of Mexico hypoxic zone (Thrash et al., 2017). Thus, their presence is not specific to host-associated lifestyle.
These results show the presence of many putatively sponge-related ELDs and other genome features in PAUC34f genomes from various environments. The broad presence of ELDs and sponge-associated features in planktonic lineages of both PAUC34f and Poribacteria (Podell et al., 2018) suggests that these domains may perform different functions in the water column than inside sponge hosts. Seawater contains diverse organic matter-rich microenvironments, such as planktonic cells, detrital particles, and organic gels (Troussellier et al., 2017). Much of the sinking particulate matter in the open ocean is of eukaryotic origin, and ELDs have been suggested as a possible bacterial mechanism to adapt to these microenvironments (Pelve et al., 2017; Podell et al., 2018). The presence of such genome features in ancestral PAUC34f may have facilitated the formation of associations with eukaryotic hosts in some branches of this candidate phylum.
Our findings suggest a substantial, global presence of PAUC34f in non-host-associated environments, such as freshwater sediments, the terrestrial subsurface, and especially the dark ocean bacterioplankton. Meanwhile, the phylogenetic intermix of host-associated and free-living lineages within this candidate phylum indicates that multiple host acquisition events may have taken place throughout the evolutionary history of PAUC34f, leading to symbiotic associations with sponges, corals, and ascidians. Additionally, genomes from diverse environments contain features often linked with host-association; these features may play adaptive roles in these non-host environments or may have facilitated historical host-acquisition events. Together, our results substantially reframe the environmental context of this enigmatic lineage, indicating that these bacteria could be significant players in diverse environments worldwide.
All genome data is available in the Joint Genome Institute Integrated Microbial Genome database.
MC, EB, and MP contributed to data analysis. RS contributed to data creation and served as the PI of the project. JB, JJ, and TW were scientific correspondents. JG, NR, DM, TN, and GH contributed to sample collection and scientific correspondence.
This work was funded by the United States National Science Foundation grants 1460861 (REU site at Bigelow Laboratory for Ocean Sciences), 1441717, 1335810, and 1232982 to RS, and the Simons Foundation (Life Sciences Project Award ID 510023) to RS. NR was supported by the Ministry of Science and Higher Education of Russia. GH was supported by the Austrian Science Fund (FWF) project ARTEMIS (P28781-B21) and the European Research Council under the European Community’s Seventh Framework Program (FP7/2007-2013)/ERC (Grant Agreement No. 268595). JG was supported by Spanish project RTI2018-101025-B-I00. TW and JJ were funded by the U.S. Department of Energy, Joint Genome Institute, a DOE Office of Science User Facility supported under Contract No. DE-AC02-05CH11231.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a past co-authorship with TW and RS.
We thank the staff of the Bigelow Laboratory for Ocean Sciences’ Single Cell Genomics Center for the generation of single-cell genomic data. We also thank Steven Hallam, Silvia Acinas, and Tamara I. Zemskaya for their help obtaining field samples, Darrell Lacy, Levi Kryder, John Klenke, and Jamie Walker of the Nevada Nuclear Waste Repository Program Office (NWRPO) for drilling and pumping of NC-GWE-OV-2, the Nature Conservancy for access to Parker Ranch, and Brittany Kruger, Josh Sackett, and Scott Hamilton-Brehm for NC-GWE-OV-2 sampling assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00376/full#supplementary-material
Acinas, S. G., Sánchez, P., Salazar, G., Cornejo-Castillo, F. M., Sebastián, M., Logares, R., et al. (2019). Metabolic architecture of the deep ocean microbiome. bioRxiv [Preprint] doi: 10.1101/635680
Anantharaman, K., Brown, C. T., Hug, L. A., Sharon, I., Castelle, C. J., Probst, A. J., et al. (2016). Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat. Commun. 7:13219. doi: 10.1038/ncomms13219
Astudillo-García, C., Slaby, B. M., Waite, D. W., Bayer, K., Hentschel, U., and Taylor, M. W. (2017). Phylogeny and genomics of SAUL, an enigmatic bacterial lineage frequently associated with marine sponges. Environ. Microbiol. 20, 561–576. doi: 10.1111/1462-2920.13965
Becraft, E. D., Woyke, T., Jarett, J., Ivanova, N., Godoy-Vitorino, F., Poulton, N., et al. (2017). Rokubacteria: genomic giants among the uncultured bacterial phyla. Front. Microbiol. 8:2264. doi: 10.3389/fmicb.2017.02264
Bierne, H., and Cossart, P. (2002). InlB, a surface protein of Listeria monocytogenes that behaves as an invasin and a growth factor. J. Cell Sci. 115(Pt 17), 3357–3367.
Brown, C. T., Hug, L. A., Thomas, B. C., Sharon, I., Castelle, C. J., Singh, A., et al. (2015). Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 523, 208–211. doi: 10.1038/nature14486
Castelle, C. J., Wrighton, K. C., Thomas, B. C., Hug, L. A., Brown, C. T., Wilkins, M. J., et al. (2015). Genomic expansion of domain archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr. Biol. 25, 690–701. doi: 10.1016/j.cub.2015.01.014
Chen, I. M. A., Chu, K., Palaniappan, K., Pillay, M., Ratner, A., Huang, J., et al. (2019). IMG/M v.5.0: an integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res. 47, D666–D677. doi: 10.1093/nar/gky901
Choi, O., Kim, J., Kim, J., Jeong, Y., Moon, J. S., Park, C. S., et al. (2008). Pyrroloquinoline quinone is a plant growth promotion factor produced by Pseudomonas fluorescens B16. Plant Physiol. 146, 657–668. doi: 10.1104/pp.107.112748
Costa, P. S., Reis, M. P., Ávila, M. P., Leite, L. R., De Araújo, F. M. G., Salim, A. C. M., et al. (2015). Metagenome of a microbial community inhabiting a metal-rich tropical stream sediment. PLoS One 10:e0119465. doi: 10.1371/journal.pone.0119465
Cuvelier, M. L., Blake, E., Mulheron, R., McCarthy, P. J., Blackwelder, P., Thurber, R. L. V., et al. (2014). Two distinct microbial communities revealed in the sponge cinachyrella. Front. Microbiol. 5:581. doi: 10.3389/fmicb.2014.00581
Dean, F. B., Hosono, S., Fang, L., Wu, X., Faruqi, A. F., Bray-Ward, P., et al. (2002). Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. U.S.A. 99, 5261–5266. doi: 10.1073/pnas.082089499
Delmont, T. O., Quince, C., Shaiber, A., Esen, O. C., Lee, S. T. M., Lucker, S., et al. (2017). Nitrogen-fixing populations of planctomycetes and Proteobacteria are abundant in the surface ocean. bioRxiv [Preprint] doi: 10.1101/129791
Duarte, C. M. (2015). Seafaring in the 21st century: the Malaspina 2010 circumnavigation expedition. Limnol. Oceanogr. Bull. 24:11. doi: 10.1002/lob.10008
Eichinger, V., Nussbaumer, T., Platzer, A., Jehl, M. A., Arnold, R., and Rattei, T. (2016). EffectiveDB - updates and novel features for a better annotation of bacterial secreted proteins and type III, IV, VI secretion systems. Nucleic Acids Res. 44, D669–D674. doi: 10.1093/nar/gkv1269
Erwin, P. M., Pineda, M. C., Webster, N., Turon, X., and López-Legentil, S. (2014). Down under the tunic: bacterial biodiversity hotspots and widespread ammonia-oxidizing archaea in coral reef ascidians. ISME J. 8, 575–588. doi: 10.1038/ismej.2013.188
Fan, L., Liu, M., Simister, R., Webster, N. S., and Thomas, T. (2013). Marine microbial symbiosis heats up: the phylogenetic and functional response of a sponge holobiont to thermal stress. ISME J. 7, 991–1002. doi: 10.1038/ismej.2012.165
Farag, I. F., Youssef, N. H., and Elshahed, M. S. (2017). Global distribution patterns and pangenomic diversity of the candidate phylum “Latescibacteria” (WS3). Appl. Environ. Microbiol. 83:e00521-17. doi: 10.1128/AEM.00521-17
Giovannoni, S. J., Cameron Thrash, J., and Temperton, B. (2014). Implications of streamlining theory for microbial ecology. ISME J. 8, 1553–1565. doi: 10.1038/ismej.2014.60
Gloeckner, V., Lindquist, N., Schmitt, S., and Hentschel, U. (2013). Ectyoplasia ferox, an experimentally tractable model for vertical microbial transmission in marine sponges. Microb. Ecol. 65, 462–474. doi: 10.1007/s00248-012-0142-7
Hardoim, C. C. P., and Costa, R. (2014). Temporal dynamics of prokaryotic communities in the marine sponge Sarcotragus spinosulus. Mol. Ecol. 23, 3097–3112. doi: 10.1111/mec.12789
Hardoim, C. C. P., Esteves, A. I. S., Pires, F. R., Gonçalves, J. M. S., Cox, C. J., Xavier, J. R., et al. (2012). Phylogenetically and spatially close marine sponges harbour divergent bacterial communities. PLoS One 7:e53029. doi: 10.1371/journal.pone.0053029
Helbert, W. (2017). Marine polysaccharide sulfatases. Front. Mar. Sci. 4:6. doi: 10.3389/fmars.2017.00006
Hentschel, U., Hopke, J., Horn, M., Anja, B., Wagner, M., Hacker, J., et al. (2002). Molecular evidence for a uniform microbial community in sponges from different oceans molecular evidence for a uniform microbial community in sponges from different oceans. Appl. Environ. Microbiol. 68, 4431–4440. doi: 10.1128/AEM.68.9.4431
Horn, H., Slaby, B. M., Jahn, M. T., Bayer, K., Moitinho-Silva, L., Förster, F., et al. (2016). An enrichment of CRISPR and other defense-related features in marine sponge-associated microbial metagenomes. Front. Microbiol. 7:1751. doi: 10.3389/fmicb.2016.01751
Hug, L. A., Baker, B. J., Anantharaman, K., Brown, C. T., Probst, A. J., Castelle, C. J., et al. (2016). A new view of the tree of life. Nat. Microbiol. 1:16048. doi: 10.1038/nmicrobiol.2016.48
Huh, H. D., Ra, E. A., Lee, T. A., Kang, S., Park, A., Lee, E., et al. (2016). STRAP acts as a scaffolding protein in controlling the TLR2/4 signaling pathway. Sci. Rep. 6:38849. doi: 10.1038/srep38849
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Jehl, M. A., Arnold, R., and Rattei, T. (2011). Effective-A database of predicted secreted bacterial proteins. Nucleic Acids Res. 39, D591–D595. doi: 10.1093/nar/gkq1154
Jensen, S., Fortunato, S. A. V., Hoffmann, F., Hoem, S., Rapp, H. T., Øvreås, L., et al. (2017). The relative abundance and transcriptional activity of marine sponge-associated microorganisms emphasizing groups involved in sulfur cycle. Microb. Ecol. 73, 668–676. doi: 10.1007/s00248-016-0836-3
Kamke, J., Rinke, C., Schwientek, P., Mavromatis, K., Ivanova, N., Sczyrba, A., et al. (2014). The candidate phylum poribacteria by single-cell genomics: new insights into phylogeny, cell-compartmentation, eukaryote-like repeat proteins, and other genomic features. PLoS One 9:e87353. doi: 10.1371/journal.pone.0087353
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Karimi, E., Ramos, M., Gonçalves, J. M. S., Xavier, J. R., Reis, M. P., and Costa, R. (2017). Comparative metagenomics reveals the distinctive adaptive features of the Spongia officinalis endosymbiotic consortium. Front. Microbiol. 8:2499. doi: 10.3389/fmicb.2017.02499
Kataeva, I. A., Seidel, R. D., Shah, A., West, L. T., Li, X. L., and Ljungdahl, L. G. (2002). The fibronectin type 3-like repeat from the Clostridium thermocellum cellobiohydrolase CbHA promotes hydrolysis of cellulose by modifying its surface. Appl. Environ. Microbiol. 68, 4292–4300. doi: 10.1128/AEM.68.9.4292-4300.2002
Kimes, N. E., Johnson, W. R., Torralba, M., Nelson, K. E., Weil, E., and Morris, P. J. (2013). The Montastraea faveolata microbiome: ecological and temporal influences on a Caribbean reef-building coral in decline. Environ. Microbiol. 15, 2082–2094. doi: 10.1111/1462-2920.12130
Kimura, M. (1980). A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. doi: 10.1007/bf01731581
Konstantinidis, K. T., Ramette, A., and Tiedje, J. M. (2006). The bacterial species definition in the genomic era. Philos. Trans. R. Soc. B Biol. Sci. 361, 1929–1940. doi: 10.1098/rstb.2006.1920
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 2567–2572. doi: 10.1073/pnas.0409727102
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lanzén, A., Jørgensen, S. L., Huson, D. H., Gorfer, M., Grindhaug, S. H., Jonassen, I., et al. (2012). CREST–classification resources for environmental sequence tags. PLoS One 7:e49334. doi: 10.1371/journal.pone.0049334
Lee, A. H., Zareei, M. P., and Daefler, S. (2002). Identification of a NIPSNAP homologue as host cell target for Salmonella virulence protein SpiC. Cell. Microbiol. 4, 739–750. doi: 10.1046/j.1462-5822.2002.00225.x
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
McIntire, W. S. (1998). Newly discovered redox cofactors: possible nutritional, medical, and pharmacological relevance to higher animals. Annu. Rev. Nutr. 18, 145–177. doi: 10.1146/annurev.nutr.18.1.145
Mestre, M., Ruiz-González, C., Logares, R., Duarte, C. M., Gasol, J. M., and Sala, M. M. (2018). Sinking particles promote vertical connectivity in the ocean microbiome. Proc. Natl. Acad. Sci. U.S.A. 115, 6799–6807. doi: 10.1073/pnas.1802470115
Miller, J. C., Chezem, W. R., and Clay, N. K. (2016). Ternary WD40 repeat-containing protein complexes: evolution, composition and roles in plant immunity. Front. Plant Sci. 6:1108. doi: 10.3389/fpls.2015.01108
Montalvo, N. F., and Hill, R. T. (2011). Sponge-associated bacteria are strictly maintained in two closely related but geographically distant sponge hosts. Appl. Environ. Microbiol. 77, 7207–7216. doi: 10.1128/AEM.05285-11
Nguyen, M. T. H. D., Liu, M., and Thomas, T. (2014). Ankyrin-repeat proteins from sponge symbionts modulate amoebal phagocytosis. Mol. Ecol. 23, 1635–1645. doi: 10.1111/mec.12384
Pachiadaki, M. G., Sintes, E., Bergauer, K., Brown, J. M., Record, N. R., Swan, B. K., et al. (2017). Major role of nitrite-oxidizing bacteria in dark ocean carbon fixation. Science 358, 1046–1051. doi: 10.1126/science.aan8260
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Parks, D. H., Rinke, C., Chuvochina, M., Chaumeil, P. A., Woodcroft, B. J., Evans, P. N., et al. (2017). Author correction: recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 3:253. doi: 10.1038/s41564-017-0083-5
Pelve, E. A., Fontanez, K. M., and DeLong, E. F. (2017). Bacterial succession on sinking particles in the ocean’s interior. Front. Microbiol. 8:2269. doi: 10.3389/fmicb.2017.02269
Pesant, S., Not, F., Picheral, M., Kandels-Lewis, S., Le Bescot, N., Gorsky, G., et al. (2015). Open science resources for the discovery and analysis of Tara oceans data. Sci. Data 2:150023. doi: 10.1038/sdata.2015.23
Petersen, T. N., Brunak, S., Von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Pita, L., Rix, L., Slaby, B. M., Franke, A., and Hentschel, U. (2018). The sponge holobiont in a changing ocean: from microbes to ecosystems. Microbiome 6:46. doi: 10.1186/s40168-018-0428-1
Podell, S., Blanton, J. M., Neu, A., Agarwal, V., Biggs, J. S., Moore, B. S., et al. (2018). Pangenomic comparison of globally distributed poribacteria associated with sponge hosts and marine particles. ISME J. 13, 468–481. doi: 10.1038/s41396-018-0292-9
Pruesse, E., Peplies, J., and Glöckner, F. O. (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829. doi: 10.1093/bioinformatics/bts252
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
R Core Team, (2017). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for statistical computing.
Reveillaud, J., Maignien, L., Eren, M. A., Huber, J. A., Apprill, A., Sogin, M. L., et al. (2014). Host-specificity among abundant and rare taxa in the sponge microbiome. ISME J. 8, 1198–1209. doi: 10.1038/ismej.2013.227
Reynolds, D., and Thomas, T. (2016). Evolution and function of eukaryotic-like proteins from sponge symbionts. Mol. Ecol. 25, 5242–5253. doi: 10.1111/mec.13812
Rinke, C., Lee, J., Nath, N., Goudeau, D., Thompson, B., Poulton, N., et al. (2014). Obtaining genomes from uncultivated environmental microorganisms using FACS-based single-cell genomics. Nat. Protoc. 9, 1038–1048. doi: 10.1038/nprot.2014.067
Rinke, C., Schwientek, P., Sczyrba, A., Ivanova, N. N., Anderson, I. J., Cheng, J.-F., et al. (2013). Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437. doi: 10.1038/nature12352
Schmitt, S., Tsai, P., Bell, J., Fromont, J., Ilan, M., Lindquist, N., et al. (2012). Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. ISME J. 6, 564–576. doi: 10.1038/ismej.2011.116
Simister, R. L., Deines, P., Botté, E. S., Webster, N. S., and Taylor, M. W. (2012). Sponge-specific clusters revisited: a comprehensive phylogeny of sponge-associated microorganisms. Environ. Microbiol. 14, 517–524. doi: 10.1111/j.1462-2920.2011.02664.x
Slaby, B. M., Hackl, T., Horn, H., Bayer, K., and Hentschel, U. (2017). Metagenomic binning of a marine sponge microbiome reveals unity in defense but metabolic specialization. ISME J. 11, 2465–2478. doi: 10.1038/ismej.2017.101
Steinert, G., Rohde, S., Janussen, D., Blaurock, C., and Schupp, P. J. (2017). Host-specific assembly of sponge-associated prokaryotes at high taxonomic ranks. Sci. Rep. 7:2542. doi: 10.1038/s41598-017-02656-6
Stepanauskas, R., Fergusson, E. A., Brown, J., Poulton, N. J., Tupper, B., Labonté, J. M., et al. (2017). Improved genome recovery and integrated cell-size analyses of individual uncultured microbial cells and viral particles. Nat. Commun. 8:84. doi: 10.1038/s41467-017-00128-z
Stepanauskas, R., and Sieracki, M. E. (2007). Matching phylogeny and metabolism in the uncultured marine bacteria, one cell at a time. Proc. Natl. Acad. Sci. U.S.A. 104, 9052–9057. doi: 10.1073/pnas.0700496104
Sunagawa, S., Coelho, L. P., Chaffron, S., Kultima, J. R., Labadie, K., Salazar, G., et al. (2015). Structure and function of the global ocean microbiome. Science 348:1261359. doi: 10.1126/science.1261359
Sunagawa, S., Woodley, C. M., and Medina, M. (2010). Threatened corals provide underexplored microbial habitats. PLoS One 5:e9554. doi: 10.1371/journal.pone.0009554
Taylor, M. W., Radax, R., Steger, D., and Wagner, M. (2007a). Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 71, 295–347. doi: 10.1128/mmbr.00040-06
Taylor, M. W., Thacker, R. W., and Hentschel, U. (2007b). Evolutionary insights from sponges. Science 316, 1854–1855. doi: 10.1126/science.1144387
Taylor, M. W., Tsai, P., Simister, R. L., Deines, P., Botte, E., Ericson, G., et al. (2013). Sponge-specific bacteria are widespread (but rare) in diverse marine environments. ISME J. 7, 438–443. doi: 10.1038/ismej.2012.111
Thomas, T., Moitinho-Silva, L., Lurgi, M., Björk, J. R., Easson, C., Astudillo-García, C., et al. (2016). Diversity, structure and convergent evolution of the global sponge microbiome. Nat. Commun. 7:11870. doi: 10.1038/ncomms11870
Thomas, T., Rusch, D., DeMaere, M. Z., Yung, P. Y., Lewis, M., Halpern, A., et al. (2010). Functional genomic signatures of sponge bacteria reveal unique and shared features of symbiosis. ISME J. 4, 1557–1567. doi: 10.1038/ismej.2010.74
Thrash, J. C., Seitz, K. W., Baker, B. J., Temperton, B., Gillies, L. E., Rabalais, N. N., et al. (2017). Metabolic roles of uncultivated bacterioplankton lineages in the northern gulf of Mexico “dead zone.” mBio 8:e01017-17. doi: 10.1128/mBio.01017-17
Troussellier, M., Escalas, A., Bouvier, T., and Mouillot, D. (2017). Sustaining rare marine microorganisms: macroorganisms as repositories and dispersal agents of microbial diversity. Front. Microbiol. 8:947. doi: 10.3389/fmicb.2017.00947
Tsementzi, D., Wu, J., Deutsch, S., Nath, S., Rodriguez-R, L. M., Burns, A. S., et al. (2016). SAR11 bacteria linked to ocean anoxia and nitrogen loss. Nature 536, 179–183. doi: 10.1038/nature19068
Tully, B. J., Graham, E. D., and Heidelberg, J. F. (2018). The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci. Data 5:170203. doi: 10.1038/sdata.2017.203
Viboud, G. I., and Bliska, J. B. (2005). Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu. Rev. Microbiol. 59, 69–89. doi: 10.1146/annurev.micro.59.030804.121320
Wall, D. M., and Mccormick, B. A. (2014). Bacterial secreted effectors and caspase-3 interactions. Cell. Microbiol. 16, 1746–1756. doi: 10.1111/cmi.12368
Webster, N. S., Botté, E. S., Soo, R. M., and Whalan, S. (2011). The larval sponge holobiont exhibits high thermal tolerance. Environ. Microbiol. Rep. 3, 756–762. doi: 10.1111/j.1758-2229.2011.00296.x
Webster, N. S., Taylor, M. W., Behnam, F., Lücker, S., Rattei, T., Whalan, S., et al. (2010). Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ. Microbiol. 12, 2070–2082. doi: 10.1111/j.1462-2920.2009.02065.x
Webster, N. S., and Thomas, T. (2016). The sponge hologenome. mBio 7:e00135-16. doi: 10.1128/mBio.00135-16
Weigel, B. L., and Erwin, P. M. (2017). Effects of reciprocal transplantation on the microbiome and putative nitrogen cycling functions of the intertidal sponge, Hymeniacidon heliophila. Sci. Rep. 7:43247. doi: 10.1038/srep43247
Keywords: microbial ecology, uncultivated bacteria, microbial genomics, dark ocean, host-association
Citation: Chen ML, Becraft ED, Pachiadaki M, Brown JM, Jarett JK, Gasol JM, Ravin NV, Moser DP, Nunoura T, Herndl GJ, Woyke T and Stepanauskas R (2020) Hiding in Plain Sight: The Globally Distributed Bacterial Candidate Phylum PAUC34f. Front. Microbiol. 11:376. doi: 10.3389/fmicb.2020.00376
Received: 19 November 2019; Accepted: 20 February 2020;
Published: 12 March 2020.
Edited by:
Haiwei Luo, The Chinese University of Hong Kong, ChinaReviewed by:
Benjamin Tully, University of Southern California, United StatesCopyright © 2020 Chen, Becraft, Pachiadaki, Brown, Jarett, Gasol, Ravin, Moser, Nunoura, Herndl, Woyke and Stepanauskas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ramunas Stepanauskas, cnN0ZXBhbmF1c2thc0BiaWdlbG93Lm9yZw==
†Present address: Jessica K. Jarett, AnimalBiome, Oakland, CA, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.