 Elle M. Barnes
Elle M. Barnes Erin L. Carter
Erin L. Carter J. D. Lewis
J. D. Lewis
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 07 February 2020
Sec. Microbial Symbioses
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.00101
Variation in the microbiome among individual organisms may play a critical role in the relative susceptibility of those organisms to infection, disease, and death. However, predicting microbiome function is difficult because of spatial and temporal variation in microbial diversity, and taxonomic diversity is not predictive of microbiome functional diversity. Addressing this issue may be particularly important when addressing pandemic diseases, such as the global amphibian die-off associated with Bd. Some of the most important factors in probiotic development for disease treatment are whether bacteria with desired function can be found on native amphibians in the local environment. To address this issue, we isolated, sequenced, and assayed the cutaneous bacterial communities of Plethodon cinereus along a gradient of land use change. Our results suggest that cutaneous community composition, but not overall diversity, change with changes in land use, but this does not correspond to significant change in Bd-inhibitory function. We found that Bd-inhibition is a functionally redundant trait, but that level of inhibition varies over phylogenetic, spatial, and temporal scales. This research provides further evidence for the importance of continued examination of amphibian microbial communities across environmental gradients, including biotic and abiotic interactions, when considering disease dynamics.
The diversity of microbes on an individual has been implicated in the health and resilience of organisms in many taxa, from plants (Mendes et al., 2013) to animals (Yildirim et al., 2010; Sanchez et al., 2012; Bahrndorff et al., 2016), including humans (Cho and Blaser, 2012; The Human Microbiome Project Consortium et al., 2012b). For instance, the presence of infection or disease is driven by the diversity of, and interactions between, pathogenic and non-pathogenic microbiota and the host’s immune system (Blaser, 2006; Benson et al., 2009, 2010; Blaser and Falkow, 2009; Maldonado-Contreras et al., 2011; Feng et al., 2017). Yet, the taxonomic diversity of microbes on an organism is an imperfect predictor of functional diversity (Gilbert et al., 2010; Delgado-Baquerizo et al., 2016; Widder et al., 2016). Predicting community function is further confounded because microbiomes vary extensively over space and time. Hence, predicting the role of microbiota in regulating infection and disease requires a better understanding of spatial and temporal variation in both taxonomic and functional diversity (The Human Microbiome Project Consortium et al., 2012a; Lloyd-Price et al., 2017; Kolde et al., 2018).
Bacterial communities may exhibit substantial biogeographic variation at both local (Costello et al., 2009; Bakker et al., 2014; Zhang et al., 2014) and continental scales (Fierer and Jackson, 2006; Meyer et al., 2018). Further, genetic divergence among populations of bacteria is common, due to the limited ability of bacteria to cross geographic barriers, their short generation times, and high population densities (Martiny et al., 2006; Taylor et al., 2006; Kuehne et al., 2007; Krause and Whitaker, 2015). So, while some genera may appear to be cosmopolitan (Fenchel et al., 1997), morphological and functional differences can arise from geographic divergence, creating distinct ecotypes or strains (Cohan, 2002; Bissett et al., 2010; Booth et al., 2016).
An emerging approach to study how microbial communities respond to environmental differences at both local and regional spatial scales is analyzing the effects of land use change. Habitat destruction, intensification of agriculture, and urbanization all alter edaphic properties, with correlated effects on microbial community composition (Zhao and Guo, 2010; Suleiman et al., 2013; Hawkes and Keitt, 2015; Plassart et al., 2019). In particular, urbanization may markedly alter the physicochemical properties of soil, leading to changes in the abundance and taxa of soil microbes (Zhao and Guo, 2010; Xu et al., 2014; Reese et al., 2016). Studies of the effects of land use change, and spatial variation in microbiota more generally, have rapidly increased in prevalence due to improvements in sequencing technologies (Caporaso et al., 2012; Lal and Seshasayee, 2014; Thompson et al., 2017; Averill et al., 2019); of critical importance now is understanding how these differences relate to community function (Robinson et al., 2010; Belzer and de Vos, 2012; Durrant and Bhatt, 2019).
Studies of genes linked to key environmental processes have related disturbances to changes in both microbial taxonomic and functional diversity (Bissett et al., 2011; Paula et al., 2014; Plassart et al., 2019), yet microbiomes usually are classified solely using the 16S rRNA gene. As others note, this gene alone is not a reliable indicator of the functional ability of a microbiome (Mollet et al., 1997; Fukushima et al., 2002; Hilario et al., 2004; Janda and Abbott, 2007; Becker et al., 2015). This weakness partly is because genomic diversity is high within microbial species, and adaptation to the local habitat can change the gene pool, creating sequence-discrete populations (Philippot et al., 2010; Smillie et al., 2011; Mell and Redfield, 2014; Shapiro and Polz, 2014; Becker et al., 2015; Gómez et al., 2016; Garcia et al., 2018; Ellegaard and Engel, 2019). Thus, 16S data may underestimate functional ability of microbiomes, due to strain-level adaptations to local conditions, or overestimate functional ability, due to functional redundancy across taxa (Delgado-Baquerizo et al., 2016; Mori et al., 2016; Vieira-Silva et al., 2016). As a result, understanding the complex interactions that shape microbial community assembly and structure requires complementing taxonomic analyses with assessments of functional differences within species and across environments (Burke et al., 2011; Jiang et al., 2019).
The current global amphibian decline is a critical system for assessing associations between microbial diversity and disease within a conservation context (Wake and Vredenburg, 2008; Ceballos et al., 2015; Bower et al., 2017). The decline is driven by the combined effects of habitat degradation and the fungal disease, Batrachochytrium dendrobatidis (Bd) (Collins and Storfer, 2003; Stuart et al., 2004). Amphibian infection by Bd, however, is blocked by some species of cutaneous bacteria, such as Janthinobacterium lividum, which produce antifungal metabolites and compete for space and resources with each other and the fungus (Longcore et al., 1999; Becker and Harris, 2010; Familiar López et al., 2017; Bates et al., 2018). However, it is still unknown in a variety of systems if these functional defense mechanisms, which many times are assumed to remain consistent with taxonomic identity, actually differ over space and time (Daskin et al., 2014; Jia and Whalen, 2019).
To address this issue, we examined bacterial diversity and function of the salamander microbiome across a 64 km gradient of land use change in the New York metropolitan area (NY, USA). We hypothesized that both composition and diversity of the cutaneous community would differ with urbanization: urban communities comprised of mainly cosmopolitan species, exurban communities of locally unique species, and suburban a mix of both making it the most diverse. In addition, we hypothesized that Bd-inhibitory ability would be a widespread and redundant trait but would increase with urbanization. This is because bacteria regularly come in contact with and compete with other fungi, not only Bd. While Bd prevalence has been suggested to be lower in urban areas (Becker and Zamudio, 2011), large urban areas such as New York are known centers for urban invaders and major points of entry for amphibians and Bd in the wildlife pet trade. Additionally, New York has the optimal climate for Bd persistence even if hosts are unavailable during certain seasons (Schloegel et al., 2009). Given that urban bacteria exist in areas with adverse environmental conditions and an increase in invaders, higher Bd-resistance could be an indirect benefit of their general response to these stressors. Lastly, we also predicted inhibition to vary among isolates of the same species based on location and season due to limitations in dispersal and potential local adaptations. The results of this study further uncover inhibitory bacteria in novel environments, such as urban areas, and aid in the discovery of potential native probiotics for other at-risk amphibian species in this species-rich region.
A terrestrial species of salamander, the eastern redback salamander (Plethodon cinereus), was sampled at each of the nine study sites (n = 65 across all sites). We chose eastern redback salamanders for this study because they are locally abundant and are one of the most commonly used amphibians for studies on Bd-resistance in the United States. Chytridiomycosis has not been demonstrated to affect P. cinereus in nature, but chytridiomycosis symptoms can be induced in artificially infected salamanders by removing or augmenting their microbiome (Harris et al., 2009a,b; Becker and Harris, 2010). Thus, because this salamander can be infected by Bd but is not susceptible to it, studying its microbiome may provide the key to finding anti-fungal bacteria to use on vulnerable individuals. Permission to sample P. cinereus was obtained through the New York State Department of Environmental Conservation (permit no. 1159), the New York City Department of Parks and Recreation (permits 2016-2018), and the New York State Office of Parks, Recreation, and Historic Preservation (permit no. 2016-MP-008) and the Fordham University IACUC #JL-17-01.
This research took place in parks and preserves in New York City and the two counties north of the city (Westchester and Putnam, NY, USA). We sampled from a total of nine study sites (Figure 1), three sites in each of the following categories: urban (Van Cortlandt Park, the New York Botanical Garden, and Pelham Bay Park), suburban (Rockefeller State Park Preserve, Fordham University’s Louis Calder Center, and Westmoreland Sanctuary), and exurban (two sites within Clarence Fahnestock State Park and one within Hudson Highlands State Park Preserve). Study site classification was based upon each site’s distance from Central Park in New York City, human population density, and GIS landcover analysis of percent-developed land within 5 miles of each study site (urban: >80%, suburban: 20–80%, exurban: <20%). All sites were located in mixed deciduous forests in New York State east of the Hudson River.
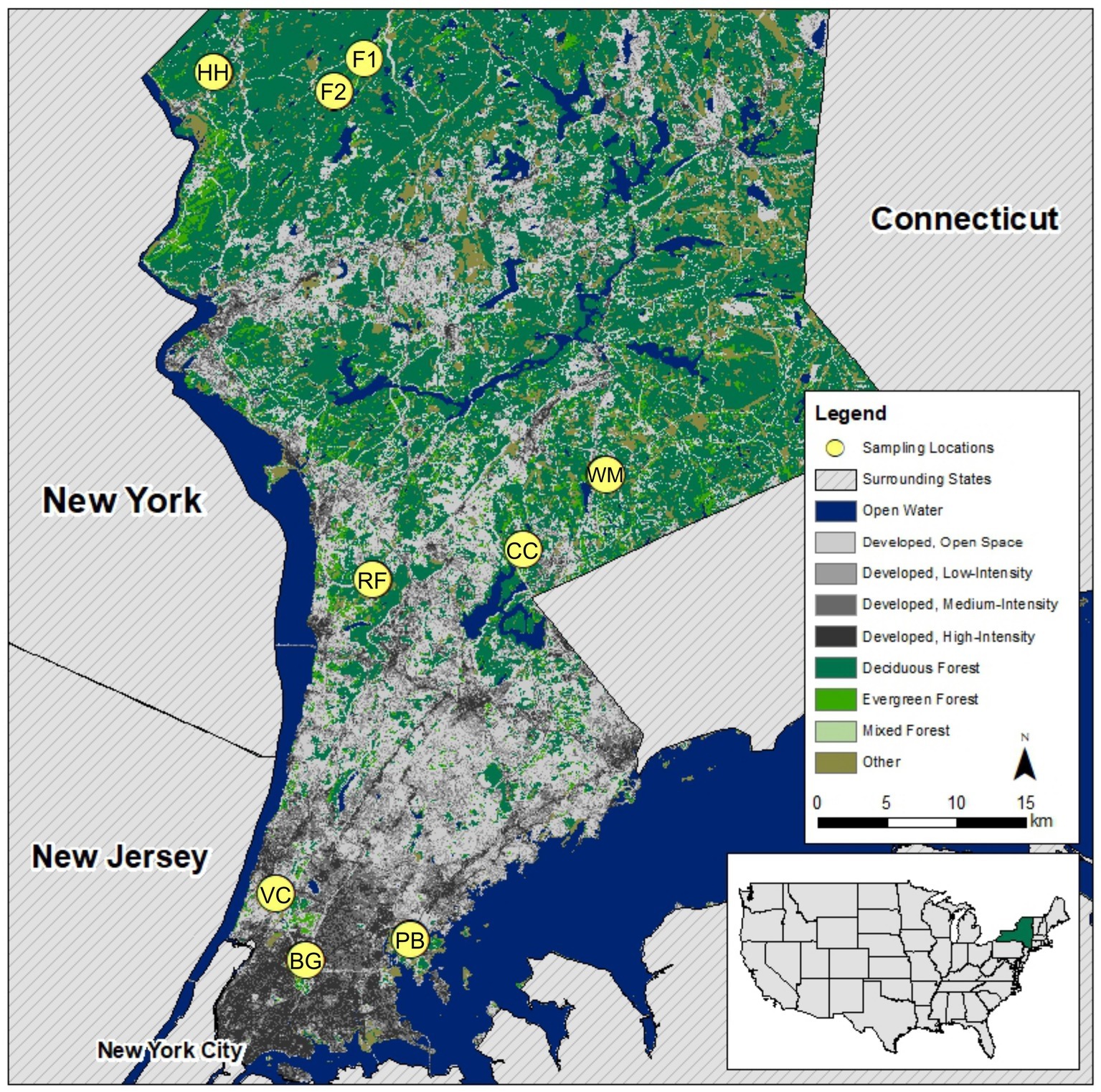
Figure 1. Map of sampling locations along urban-to-exurban gradient showing developed land cover (from low to high intensity) in gray and forest in green adapted from the National Land Cover Database (NLCD) (2011) United States land cover dataset. Site code used throughout the manuscript is shown within the marker to identify each sampling location. Map in bottom-right corner locates New York State within the USA. Sites were classified as urban, suburban, or exurban based upon each site’s distance from Central Park in New York City, human population density, and GIS landcover analysis of percent-developed land within 5 miles of each study site.
Samples were collected in Fall 2016 (September through October) and Spring 2017 (April through May), except for 10 samples taken from salamanders at Fahnestock and Hudson Highlands State Parks in Spring 2018. Salamanders were captured and handled using sterile gloves, which were changed between each individual. A non-invasive sampling method of bacterial swabbing was used based on Boyle et al. (2004). In brief, each salamander was rinsed twice with 25 ml of sterile water and swabbed using a sterile cotton swab on their ventral, right, and left sides (Harris et al., 2006). Swabs were immediately streaked onto LB agar (5 g NaCl, 5 g tryptone, 2.5 g yeast extract, 7.5 g agar, 200 μl 2.5 N NaOH, and 500 ml H2O) and were incubated at ambient temperature for 72 h. We chose to use LB agar to provide a high nutrient alternative to R2A, which is commonly used in amphibian microbiome studies as the nutrient content of salamander skin is still undetermined. Instead, we focused on swabbing a higher number of individuals across our sites to maximize culture diversity regardless of media (Medina et al., 2017b).
For each individual, we isolated bacteria based on morphotype (colony shape, elevation, margin, surface, and color) to pure cultures on LB agar. DNA from these cultures was extracted using the Qiagen DNeasy Blood and Tissue kit (Germantown, MD, USA) following manufacturer’s protocols, with the addition prior to purification of lysozyme incubation for Gram-positive bacteria. DNA was amplified with the 515F (5′-GTGYCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACNVGGGTWTCTAAT-3′) primer pair, which targets the V4 region of the 16S rRNA gene, in 25 μl reactions: 12.5 μl GoTaq Green PCR Master Mix (2X), 1 μl forward primer, 1 μl reverse primer, 2 μl DNA, and 8.5 μl nuclease-free water. PCR protocols included: 95°C for 5 min, followed by 95°C for 1 min, 55°C for 1 min, and 72°C for 1 min for 30 cycles, and 72°C for 5 min. This primer pair was chosen as it targets a small region commonly sequenced by high-throughput 16S rRNA sequencing, allowing our results to be easily compared to results from other studies. Amplicon lengths were assessed using gel electrophoresis (1% agarose), and samples yielding amplicons of ~250 bp were then sequenced using Sanger sequencing at Macrogen (New York, NY, USA).
Forward and reverse amplicon sequences were aligned to obtain a consensus sequence for each isolate in Geneious R11.1 (Biomatters Inc., Auckland 1010, New Zealand). Fourteen sequences were discarded due to poor read quality. The remaining 204 sequences were then run in the EZBioCloud database, a curated and integrated database of all 16S rRNA bacterial sequences from NCBI, to assign taxonomy (Yoon et al., 2017). OTU identification was assigned based on the closest reference sequence match with a taxonomic classification if sequence similarity was >97%. In instances where sequence similarity was >97% match to two or more OTUs, samples were classified based on highest percent query cover and completion of read. Additionally, 16S rRNA gene trees for isolates within each level of urbanization were built with MrBayes v. 3.2.6 (Huelsenbeck and Ronquist, 2001). Our runs consisted of four simultaneous Markov chains, each with 1,000,000 generations, a subsampling frequency of 200, and a burn-in fraction of 0.15. Trees were then visualized and adapted in FigTree v. 1.4.3 (Rambaut, 2012).
All bacterial isolates (including multiple isolates of the same OTU) were challenged with Bd using the 96-well cell-free supernatant (CFS) method developed by Bell et al. (2013). Samples of Bd strain JEL423, a hypervirulent strain from the global panzootic lineage BdGPL (Farrer et al., 2011; DiRenzo et al., 2014), were obtained from Dr. Joyce Longcore (University of Maine). Each isolate was diluted to 1:104 cfu and plated on LB agar plates until colonies formed, after which 1 cm2 of agar with colonies was excised and placed in 7 ml of 1% tryptone broth and allowed to grow at 23°C for 48 h. To obtain Bd used in each microcosm, 1 ml of Bd culture was plated on 1% tryptone plates and grown at 23°C for 4 days, after which 25 ml of 1% tryptone broth was inoculated with decanted zoospores. This Bd liquid culture was then grown at 23°C for 48 h.
Each isolate was co-cultured with 2 × 106 zoospores Bd (100 μl of each) in 800 μl of 1% tryptone, a minimal-nutrient media in which Bd often is grown (Longcore et al., 1999; Piotrowski et al., 2004), for 72 h at 23°C. Cultures were checked for turbidity at OD600 to confirm that they were in late log to early stationary phase and then centrifuged for 5 min at 10,000g. Cultures that did not reach these phases (<5% of isolates) were incubated for an additional 24 h. The supernatant was then filtered through a sterile, 0.22-μm cellulose acetate syringe filter (VWR, Radnor, PA, USA) to obtain CFS. CFS was then assayed with Bd (50 μl CFS: 50 μl Bd-zoospore suspension) in triplicate in a 96-well plate for 10 days at 23°C. The Bd-zoospore suspension consisted of growing Bd on a 1% tryptone agar plate for 4 days, flooding the plate with 3 ml 1% tryptone, and then filtering this suspension through a 20-μm filter. Zoospore concentration was estimated at 2 × 106 zoospores per ml using an INCYTO C-Chip™ hemocytometer (SKC Inc., Covington, GA, USA). Both positive (50 μl Bd CFS: 50 μl Bd-zoospore suspension) and negative controls (50 μl Bd CFS: 50 μl heat-killed Bd-zoospore suspension) were included to control for potential variation in Bd zoospore concentrations, Bd-produced metabolites, and Bd growth when alone. Optical density was measured at 0, 4, 7, and 10 days using an Infinite 200 Pro microplate reader (Tecan Trading AG, Männedorf, Switzerland).
To calculate Bd inhibition, absorbance readings were transformed using the equation: ln[OD/(1-OD)] following Becker et al. (2015). A linear regression was run to determine the growth rate of Bd in the presence of CFS from each isolate. The average slope of each triplicate was then divided by the average growth rate of the positive control, and this value was subtracted from one. This calculated value represented percent Bd inhibition, and bacterial OTUs were then classified by taking the mean percentage inhibition of multiple isolates of the same OTU.
Alpha diversity was calculated using the Shannon index due to its reduced sensitivity to sampling depth differences, as compared to Chao-1 (Haegeman et al., 2013; Preheim et al., 2013). Chi-square test of independence was used to account for the relationship between year and site in our sampling. Beta diversity between sites was measured by generating a Bray-Curtis distance matrix on OTU-level presence-absence data (Legendre and Legendre, 1998) and conducting a permutational multivariate analysis of variance (PERMANOVA) with each site nested by level of urbanization. These data were then used for ordination by nonmetric multidimensional scaling (NMDS) with 1,000 randomized runs. Pairwise analyses were conducted using the package pairwiseAdonis (Martinez, 2019). All analyses were conducted in vegan v. 2.5-2 in R 3.6.0 (Oksanen et al., 2013).
We examined the distribution of isolate inhibition using Hartigan’s dip test (Hartigan and Hartigan, 1985) with the diptest package (Maechler and Ringach, 2013) to test for multimodality. The distribution was then visualized using the EM algorithm in the mixtools package (Benaglia et al., 2009), and the mean and standard deviation of each mode was calculated. We compared each isolate’s percent inhibition with each plate’s Bd control using a Mann-Whitney U-test to classify Bd-inhibitory ability. Differences among bacterial OTUs, genera, phyla, site, and level of urbanization were tested with a Kruskal-Wallis test followed by Dunn post-hoc test. Differences in level of inhibition between the Spring and Fall seasons were tested with a Mann-Whitney U-test.
One of our primary objectives was to characterize the diversity of bacteria on salamanders from a large urban area in the northeastern USA. From 69 salamanders, a total of 218 isolates were cultured, 204 of which could be identified as OTUs (Table 1). Four phyla were identified: Proteobacteria (69%), Firmicutes (21%), Actinobacteria (6%), and Bacteroidetes (4%) (Figure 2). Out of 42 total genera, the three most common genera were Pseudomonas (43 isolates), Bacillus (28 isolates), and Stenotrophomonas (26 isolates). The majority of other genera were represented by less than 10 isolates each. While no one bacterial OTU was found at all nine sites, Stenotrophomonas rhizophila was found across all three levels of urbanization and at seven out of nine sites.
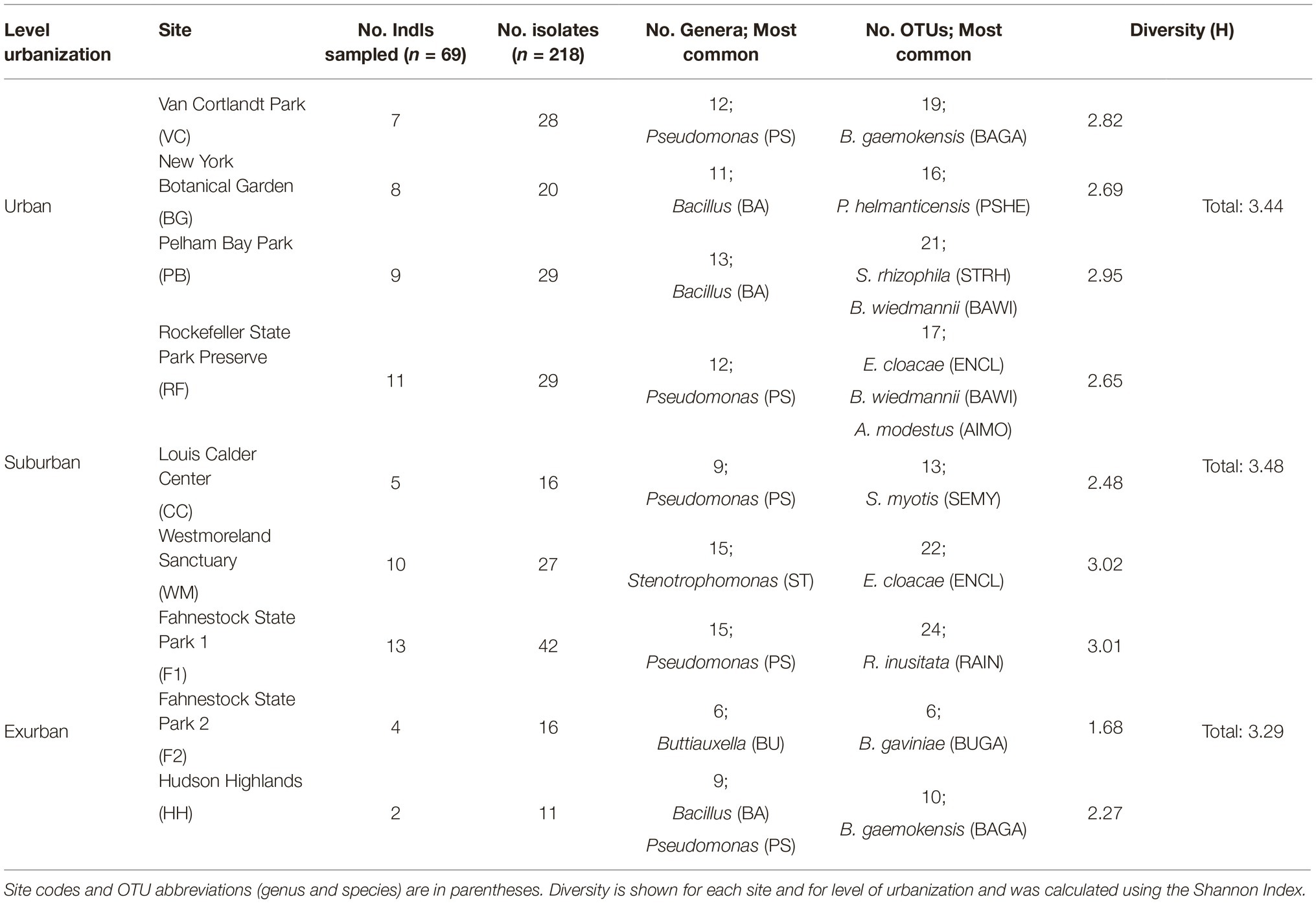
Table 1. Summary of salamanders sampled across nine sites in three levels of urbanization.
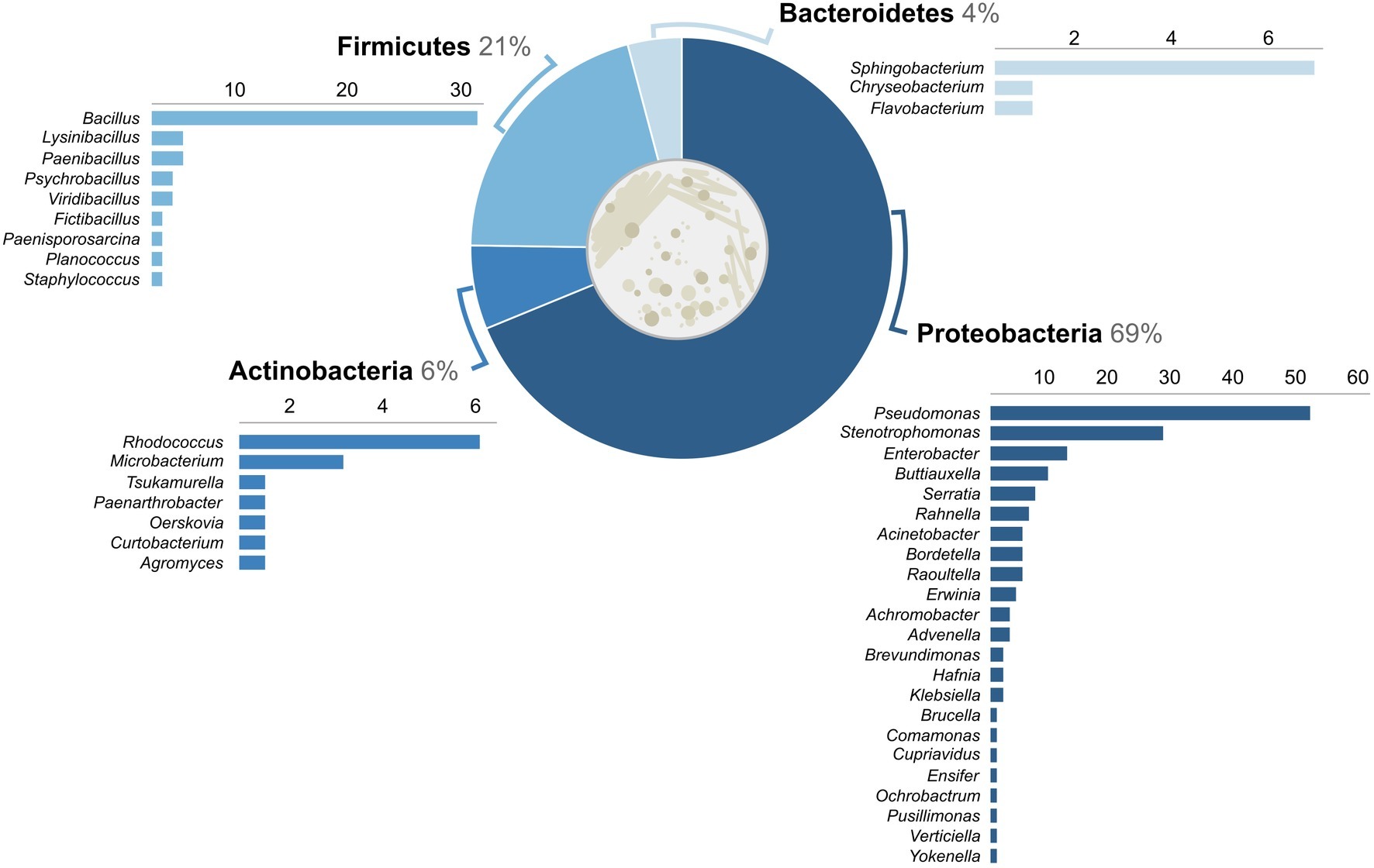
Figure 2. Percent of bacterial isolates (n = 204) across the gradient by phylum. Number of isolates identified within genera is displayed below each phylum.
OTU richness and abundance varied by site (Table 1) and a chi-square test of independence showed that both year and site were correlated with regard to OTU richness (X2 = 137.4, p < 0.05). While there was no trend of richness with land use type (p = 0.73), only 11 genera were found at all three levels of urbanization. These include: Pseudomonas, Bacillus, Stenotrophomonas, Sphingobacterium, Serratia, Rhodococcus, Microbacterium, Lysinibacillus, Enterobacter, Buttiauxella, and Bordetella. The remaining 31 genera were unique to certain levels of urbanization, with 24 genera (42% of total genera) unique to a single site. Interestingly, the most common OTU at each site often belonged to a different genus than the genus found to be most common at that site (Table 1).
While urban sites had the greatest number of individual isolates compared to suburban and exurban sites, suburban sites had the largest overall Shannon’s diversity score, 3.48 (p = 0.56, Table 1). However, high OTU richness did not correlate to phylogenetic diversity (Figure 3), as many of the isolates were closely related and came from a small number of genera. An NMDS of beta diversity indicated that urban, suburban, and exurban bacterial communities grouped by level of urbanization (R2 = 0.36, p = 0.005; Figure 4). Urban and suburban communities overlapped to the left of the plot, but urban communities distributed along the first axis while suburban communities distributed along the second axis. Exurban sites separated to the right of the ordination plot, with some overlap with urban communities and a pairwise PERMANOVA revealed a significant difference with suburban communities (R2 = 0.15, p = 0.003).
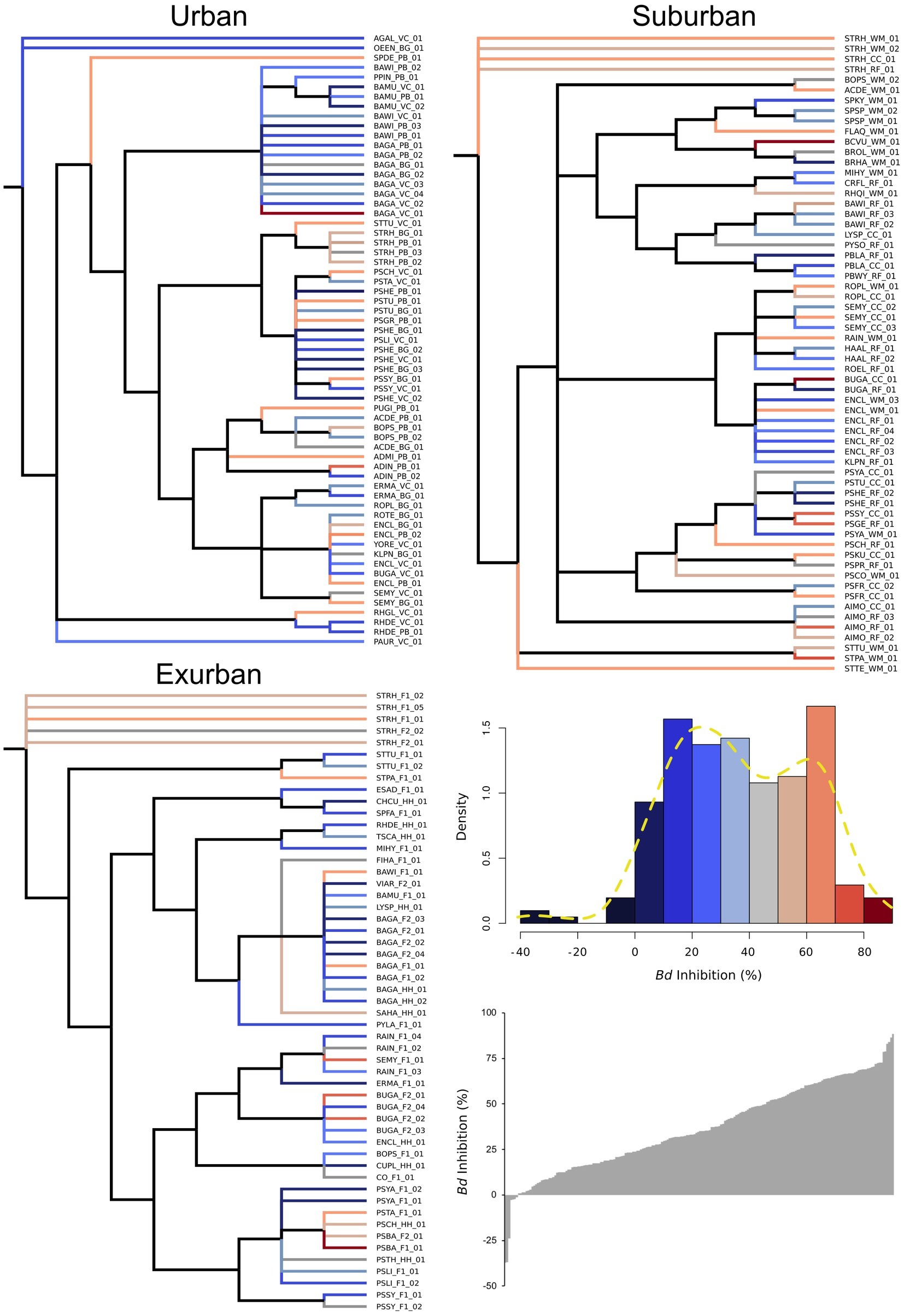
Figure 3. Salamander skin bacterial OTUs (n = 204) at each level of urbanization displayed as phylogenies. OTUs are identified by the first two letters of the genera and species names (from the 16S rRNA sequence) followed by the site code and isolate number. Color of each isolate’s branch on phylogenetic tree corresponds to Bd-inhibitory ability. Bd-inhibitory ability is divided into intervals of 10% and color is denoted as displayed in the density histogram. Cooler colors indicate low Bd-inhibition, whereas warmer colors indicate high Bd-inhibition. Dashed yellow line in the density histogram shows a bimodal distribution of Bd-inhibition (p = 0.030).
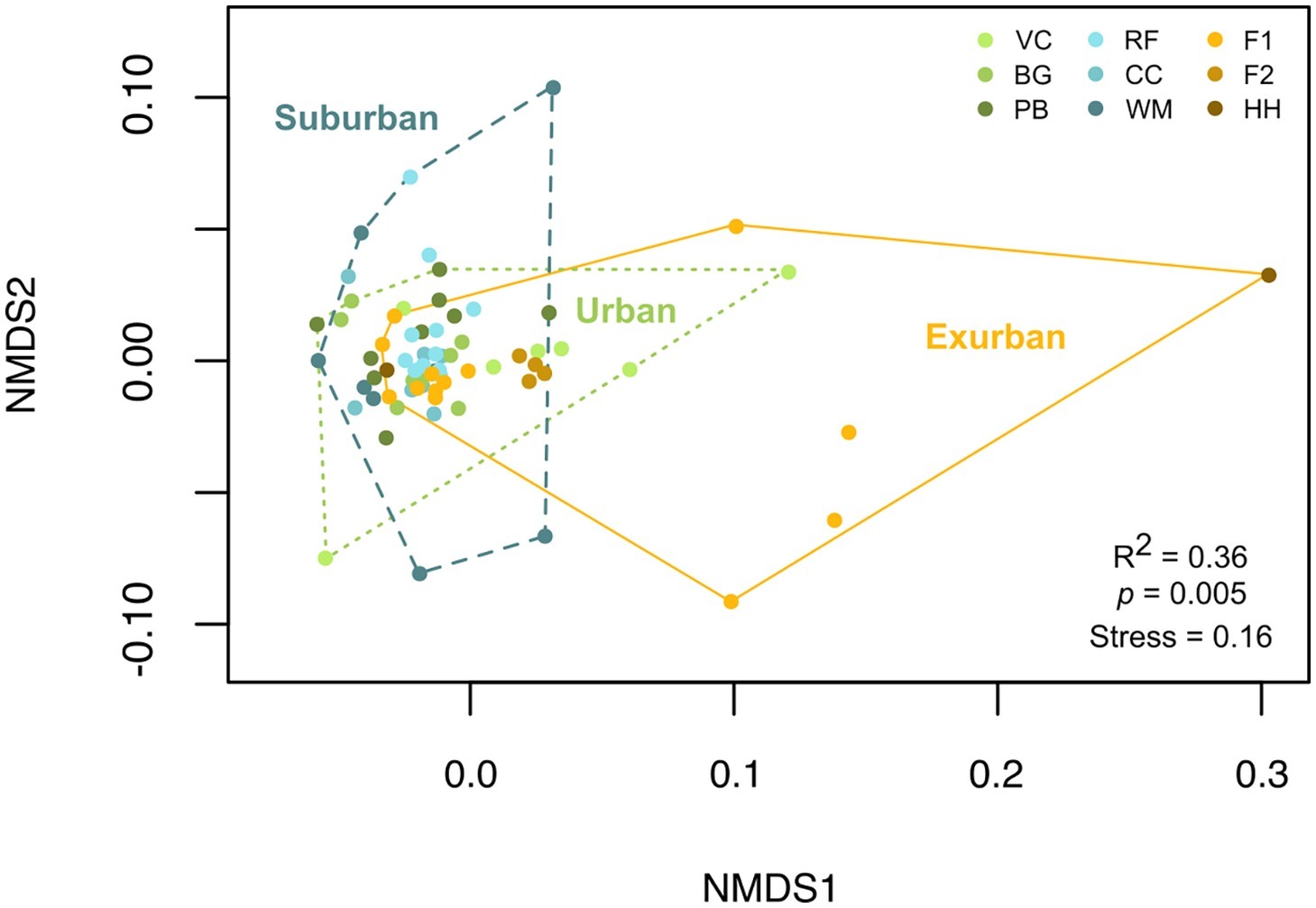
Figure 4. NMDS of beta diversity by individual salamander (n = 69) blocked by level of urbanization. Beta diversity was measured by calculating a Bray-Curtis dissimilarity matrix of OTU-level presence-absence culture data. Color denotes sampling location. PERMANOVA of beta diversity by level of urbanization indicates a significant difference between suburban and exurban skin bacterial communities (R2 = 0.375, p = 0.005).
To assess whether Bd-inhibitory ability was multimodally distributed in our sample of salamanders, we used Hartigan’s dip test. We observed that Bd-inhibitory ability was bimodal (p = 0.030), exhibiting at least two groups of isolates: the first with a mean of 25.9% (SD = 18.5%), and the second with a mean of 63.9% Bd-inhibition (SD = 9.5%) relative to the control (Figure 3). Isolates within 1.5 SD of the second mean (all isolates above 49.6% Bd inhibition) were considered inhibitory—32% of all isolates. Isolates above the mean of the first mode but below 1.5 SD of the second mean (i.e., isolates between 25.9 and 49.6% Bd inhibition) were considered mildly inhibitory—30% of isolates, while isolates below this group but within 1.5 SD of the mean of the first mode were considered to have no effect on Bd-growth—34% of isolates. Lastly, the seven isolates (3%) that fell below this threshold were considered facilitative: increased Bd-growth relative to the control.
To assess whether Bd-inhibitory function was phylogenetically unique vs. redundant across those groups, we analyzed isolate Bd inhibition by phylum, genus, and OTU. When analyzed by phylum, Proteobacteria were on average 68% more inhibitory than Actinobacteria (p = 0.004) and Firmicutes (p < 0.001) and 26% more inhibitory than Bacteroidetes (X2 = 22.1, Figure 5A). At the genus and OTU level, isolates exhibited a large range of inhibitory ability, with few distinct phylogenetic patterns (Figure 3). In some extreme cases, isolates within the same genus, such as Bacillus and Pseudomonas (identified as “BA” and “PS” on the branch tips of the phylogenetic trees), were identified as inhibitory, while others were facilitative to Bd growth (p < 0.05). When we compared a subset of the most common bacterial genera (n ≥ 5 isolates), we found differences between genera (p = 0.012), driven by two genera: Bacillus (non-inhibitory, mean = 25%) and Stenotrophomonas (inhibitory, mean = 54%) (Table 2).
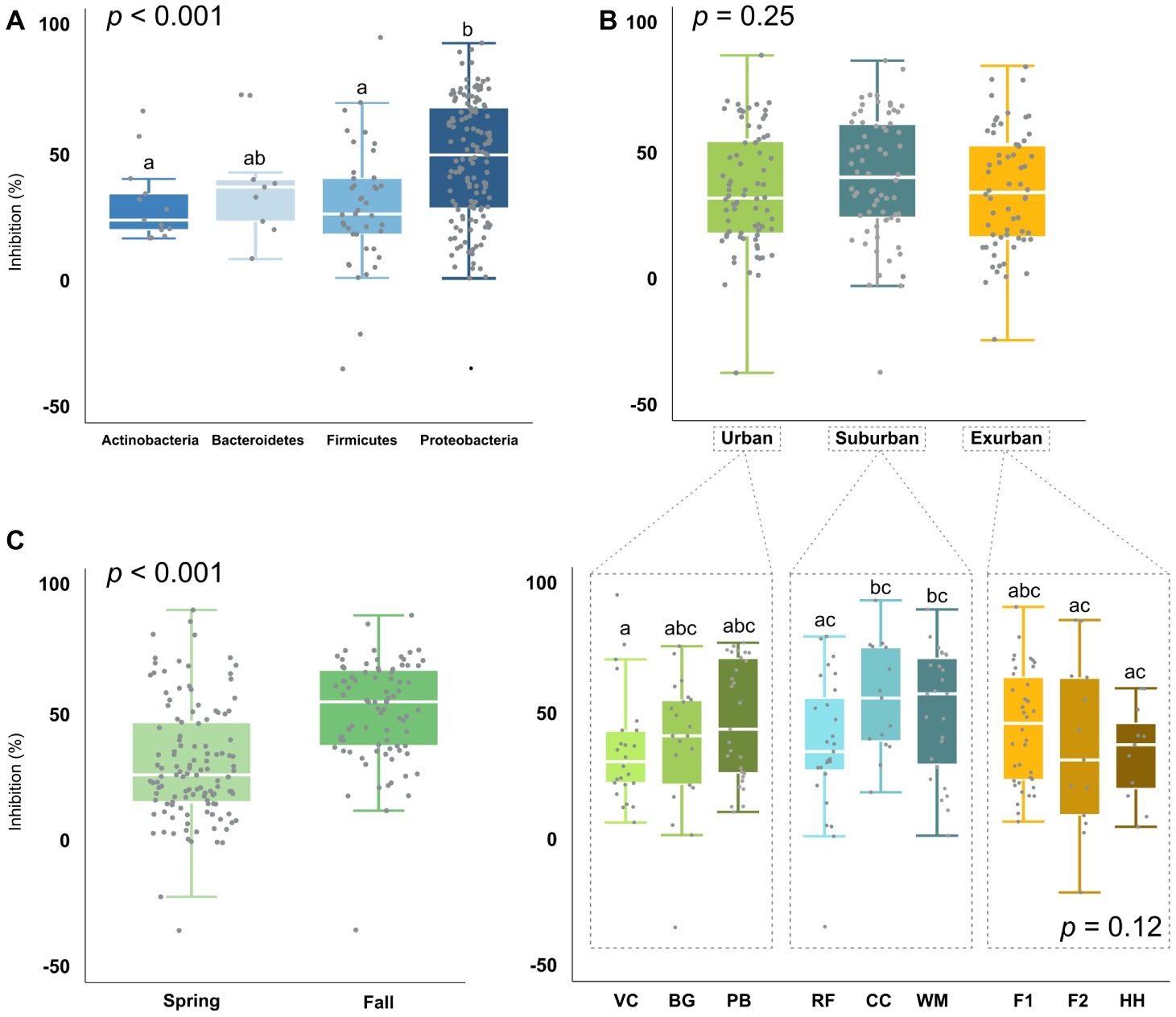
Figure 5. Bd-inhibition of isolates (n = 204) by (A) phylum (p < 0.001), (B) level of urbanization (p = 0.252) and site [p = 0.117, but significant (p < 0.05) pairwise site effects], and (C) season (p < 0.001).
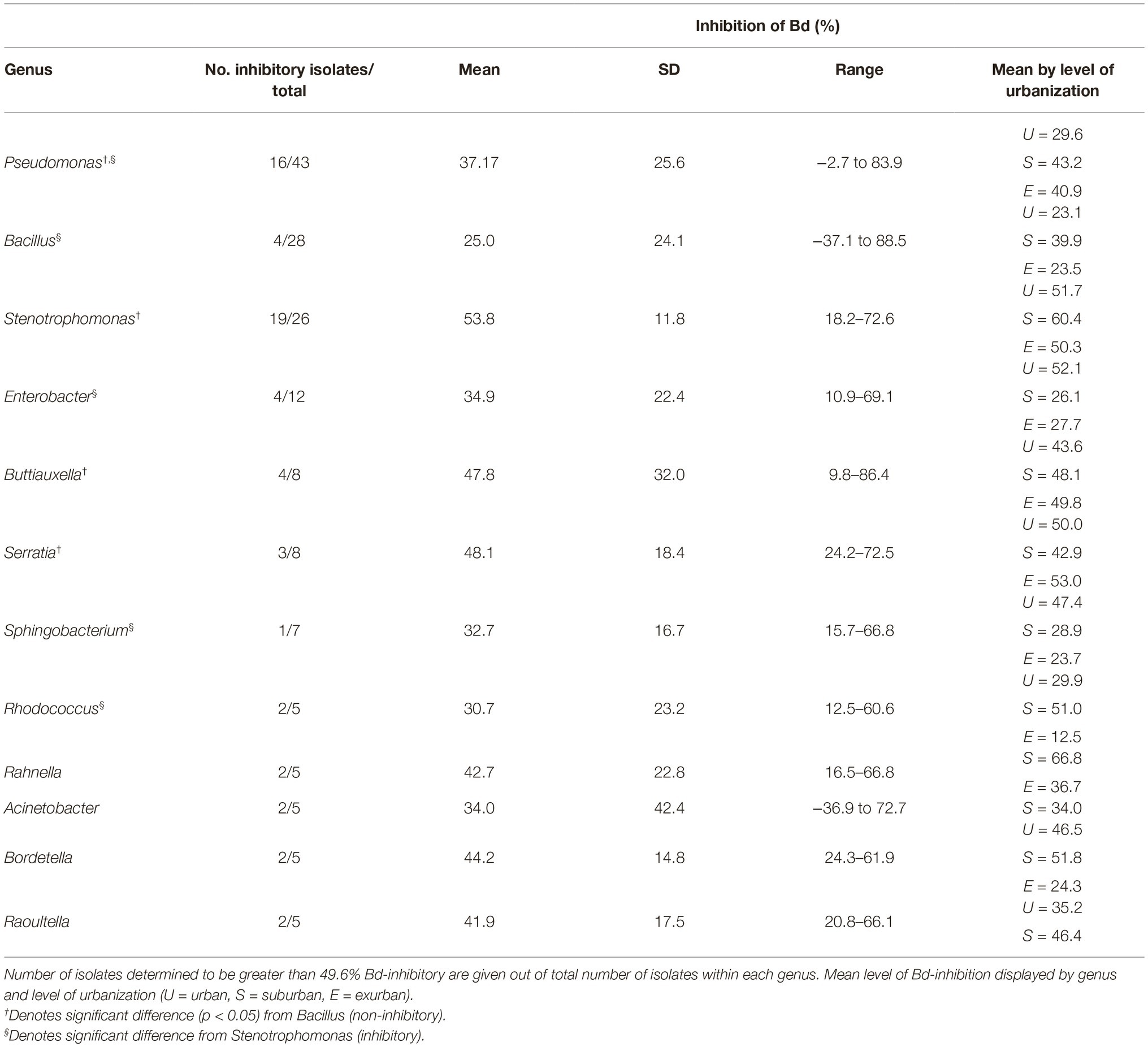
Table 2. Anti-Bd activity of bacterial genera with greater than five isolates.
We further analyzed Bd-inhibitory function by level of urbanization, site, and season. Overall, suburban sites appeared to have the largest proportion of inhibitory isolates with 41%, followed by exurban (34%) and urban (30%). Similarly, individual salamanders also varied in the number of Bd inhibitory isolates cultured from their skin. Depending on the site, 17–50% of isolates per individual were >49.5% inhibitory, with salamanders from suburban sites having on average 15% more inhibitory isolates than individuals from urban or exurban sites. However, the effect of urbanization was not statistically significant (X2 = 2.8, p = 0.25, Figure 5B)—so, while there appeared to be fairly large relative differences in the proportion of inhibitory isolates among levels of urbanization, with suburban communities 10–60% more inhibitory than urban and exurban communities (X2 = 2.5, p = 0.28), there was a lot of variation within genera even at the site level.
In addition, Pseudomonas and Bacillus, which make up a large proportion (35% of all isolates) of the cultured bacterial community, exhibited substantial variation in Bd inhibition, with differences by both site (p < 0.05) and season (Pseudomonas: p < 0.001, Bacillus: p = 0.022). In Bacillus, differences were even observed among isolates at the individual site level, such as B. gaemokensis between VC and F2 (p = 0.021), and B. wiedmannii between PB and F2 (p = 0.021). In contrast, Stenotrophomonas was consistently inhibitory, exhibiting no clear variation in inhibition by level of urbanization, site, season, or OTU. When analyzed by season, bacteria isolated in the fall season were 72% more inhibitory than bacteria in the spring (W = 2291.5, p < 0.001; Figure 5C). This same trend still held when a subset of the bacteria, which were found at the same site in both seasons, were compared (W = 437, p = 0.002). When we compared Bd inhibition by year, we found no significant difference between both the 2017 and 2018 spring seasons (X2 = 43.5, p = 0.40).
Studies on the distribution of bacteria and the variability of their function are limited (Kueneman et al., 2014; Becker et al., 2015; Bletz et al., 2017; Jiménez and Sommer, 2017; Kruger, 2019) but are increasingly relevant to the amphibian microbiome as changes in microbial community structure have been hypothesized to result in changes in anti-fungal function. In line with these studies, our results suggest that predicting microbiome function across space and time is difficult due to strain-level differences and functional redundancy across taxa. This knowledge is foundational to understanding the dynamic nature of host-associated microbiomes and their function against pathogens, especially given the urgent need to conserve and protect vulnerable taxa in environments around the globe.
A key constraint to understand the distribution and variability of the amphibian microbiome is the limited number of studies in certain geographic areas, as indicated by the studies compiled in the Antifungal Isolates Database (Woodhams et al., 2015). These areas notably include the Northeastern United States, which has comparatively high salamander diversity and urban land use (~11%; Nickerson et al., 2011). Thus, we chose to sample Plethodon cinereus along a gradient of land use in the New York metropolitan area. We acknowledge that our sampling design and site selection may result in overlapping effects of land use and site microenvironment. However, this study design ultimately enabled us to better explore how environmental changes over even small geographic distances in these regions can influence variability of host microbial community diversity, composition, and function.
Contrary to our hypothesis, Shannon’s diversity was similar regardless of site or land use. This pattern is in contrast to other studies on the effects of urbanization on plant and animal biodiversity, which suggest that native species richness tends to be reduced in areas with extreme urbanization (Shochat et al., 2006; Faeth et al., 2011; Laforest-Lapointe et al., 2017; Yan et al., 2019). Additionally, the presence of three cosmopolitan genera (Pseudomonas, Bacillus, and Stenotrophomonas) across the entire gradient, representing nearly half (48%) of all isolates, and our observation that urban sites had the highest OTU richness, contrasts with the idea that urbanization generally drives homogenization of taxa in these communities at least for bacteria (McKinney, 2006). However, we did find that site and year were correlated with regard to richness likely due to poor sampling conditions at Fahnestock and Hudson Highlands in 2017 leading us to also sample only those sites in Spring 2018. Despite this, we found no significant difference in Bd inhibition with year regardless of site.
While our use of culturing to examine overall taxonomic diversity is known to miss a proportion of taxa, it allowed us to assess diversity of many actives, and likely functionally relevant, bacteria on salamanders at these locations. However, we do acknowledge that the results of culturing alone are sensitive to the growth media chosen and does not enable us to distinguish bacterial relative abundances as they occur on salamander skin. Our results suggest that while diversity does not differ with urbanization, taxonomic composition does. This pattern has been seen repeatedly in other studies on microbial communities, which suggest that urbanization likely impacts relative abundance of taxa in local communities rather than overall diversity (Cousins et al., 2003; Hall et al., 2009; Xu et al., 2014; Reese et al., 2016; Epp Schmidt et al., 2017; Lehtimäki et al., 2017).
In our comparisons of beta diversity, composition of bacterial communities grouped by level of urbanization. Composition differed at the site level as well. This is likely because 31 genera were unique to certain levels of urbanization with 24 genera (42% of total genera) unique to a single site. Even though three cosmopolitan genera dominated most communities, sites differed in which OTU within these genera were present. This variation we see in OTU-specific composition could partially be explained by dispersal limitation or neutral processes, at least across small geographic distances.
Our finding that composition varies with land use is in line with our hypotheses: we saw the most variation between individual salamander bacterial communities at exurban sites (Figure 4), and more similarity at urban and suburban sites. Further, when we compare the abundance of the 11 genera found at all three levels of urbanization, urban sites had on average 5–12% more cosmopolitan isolates (a total of 78% of urban isolates) than suburban and exurban sites. In these cases, regional variation is likely driven by local environmental conditions associated with urbanization (i.e., changes in nutrient concentrations, habitat fragmentation, heavy metal deposition, etc.) that overwhelm stochastic factors to select for particularly resilient specialist OTUs as well as cosmopolitan generalists.
This larger role of microenvironment versus stochastic factors has repeatedly been seen in soil communities, where soils with similar environmental characteristics have similar microbial communities regardless of geographic distance (Fierer and Jackson, 2006; Lauber et al., 2009; Xu et al., 2014; Lladó et al., 2018; Plassart et al., 2019). Interestingly, we see this same trend in the bacterial communities of the soil-dwelling salamanders sampled in this study. If skin conditions were the primary selective mechanism, we would see no difference in bacterial composition across our gradient. As others have demonstrated, the soil and water environments in which amphibians live in likely function as species pools providing colonists to amphibian hosts (Loudon et al., 2016), and all of the cosmopolitan genera we found on our salamanders are common soil bacteria known to associate with plants or animals. Thus, our results indicate some combination of contemporary environmental variation and lingering historical effects driving distinct microbial biogeography on these salamanders (Martiny et al., 2006; Kraemer and Kassen, 2015). This study serves as an initial investigation into the overall effects of land use associated environmental changes on microbial communities, but further investigation is needed to determine if and how direct land-use associated effects on the soil bacterial reservoir indirectly alter amphibian cutaneous microbiomes.
It is worth noting that we did not find Janthinobacterium lividum, a well-known Bd-inhibitory bacteria (Brucker et al., 2008), on any of the Plethodon cinereus salamanders sampled despite knowledge that strains have been cultured from aquatic and soil environments in the Hudson Valley of New York (O’Brien et al., 2018). Instead, Stenotrophomonas, an antifungal plant mutualist (Wolf et al., 2002; Ryan et al., 2009), was found on salamanders at nearly all sites across our sampling gradient. This genus has a number of favorable characteristics for use as a potential native probiotic in degraded habitats: resistance to heavy metals (including lead), high hydrolytic potential, high bacterial growth rate, and the ability to grow in low-nutrient environments (Ryan et al., 2009). All of these have allowed Stenotrophomonas sp. to colonize and compete in many different biotypes adding to its value as potential anti-Bd isolate for amphibians in our region.
Despite the compositional differences we saw in bacterial taxonomy across sites, overall Bd-inhibitory ability was similar among urban, suburban, and exurban bacterial communities but varied among individuals. Each site possessed at least one bacterial OTU capable of high Bd-inhibition. Given this, Bd-inhibition seems to be a functionally redundant characteristic across the gradient, despite differences in the environment and OTU composition. This pattern is not surprising given that naturally occurring bacteria regularly interact and compete with other microbial taxa, including fungi, for space and resources. Whether inhibitory metabolites are Bd-specific, fungal-specific, or generally anti-microbial is still undetermined for the majority of bacterial strains isolated from amphibian skin in this and other studies and we encourage continued efforts to characterize the amphibian metabolome to identify the mechanisms responsible for anti-Bd function (Woodhams et al., 2014; Belden et al., 2015; Walke et al., 2015).
Despite functional redundancy of Bd-inhibition across the gradient, it does not appear to be a trait associated with phylogenetic relatedness among bacteria (Figure 3). We found variation in inhibition across isolates at the phylum, genus, and OTU levels. For example, in comparing isolates from two different individuals with bacterial communities composed of the same genera, we found Bacillus, Pseudomonas, and Sphingobacterium to vary from 8, 9, and 67% Bd-inhibition at PB (urban) to 61, 44, and 16% at F1 (exurban). Given this, it might be better for researchers to focus on maintaining functional diversity rather than taxonomic diversity in relation to the conservation of amphibian microbiomes. Overall function of the community considers distribution, abundance, and the products of fluctuating interactions between species in the community (Holling, 2001; Alberti, 2005). As shown in this study, taxonomic differences are poor predictors of community functional differences and presence of particular isolates on amphibians in separate locations does not equate to similarities in function.
Contrary to our hypothesis, suburban sites associated with medium levels of environmental change appeared to have the largest proportion of inhibitory isolates with 41%, followed by exurban (34%) and urban (30%) sites. In addition, only 32% of all isolates were inhibitory, which suggests that the majority of the salamander microbiomes cultured in this study show little to no effect on Bd growth in vitro. While inhibition did not significantly differ by level of urbanization, there were site-level differences (Figure 5B). Significant differences among our sites were driven by one suburban site, CC, which consisted of isolates that were on average more inhibitory than all other sites. Differences in Bd inhibition among sites could be due to environmental characteristics, absence of particular isolates, changes in overall community composition, or isolate-level trait differences. Often times, these local factors are not reported or considered in other studies using the Anti-fungal Isolates Database to determine community function from high throughput 16S rRNA sequencing. As mentioned before and reiterated by our results, the 16S rRNA gene alone is not a reliable indicator of Bd-inhibitory function. Thus, averaging the results of globally distributed functional assays likely blurs the fine-scale differences in microbiome function in different regions and can lead researchers to over- or underestimate microbiome function when not complemented by functional assays from local isolates (Mollet et al., 1997; Fukushima et al., 2002; Hilario et al., 2004; Janda and Abbott, 2007; Becker et al., 2015).
We found a high level of anti-Bd functional variation observed across exurban salamander bacterial communities, which was not in line with our initial hypotheses. Our exurban sampling locations (F1, F2, and HH) consist of well-connected hardwood forests and streamside habitats where salamander dispersal is far less limited than in both our urban and suburban sites. However, habitat connectedness or salamander dispersal may not matter at the microbial level. The variation in exurban isolate’s Bd-inhibitory function despite high habitat connectivity is not unexpected given that bacteria can engage in horizontal gene transfer and clonal evolution. Studies have shown that multiple clone-types can exist and swap in dominance based on abiotic and biotic community factors (Fenchel, 2003; Devevey et al., 2015; Wright and Vetsigian, 2016). We also observed isolate-level trait differences in comparisons of isolates across sites from the two most dominant genera, Pseudomonas and Bacillus (Figure 3; Table 2). Less than half of the number of isolates from these genera was inhibitory in urban sites as compared to exurban and suburban sites. These results agree with the findings of Becker et al. (2015), in that there can be substantial functional variation within phylogenetically similar bacterial isolates and provide further evidence for the use of native bacteria in developing potential probiotics.
Differences in an individual bacterial isolate’s interactions with the environment and other microbes are also known to alter bacterial metabolite production (Daskin et al., 2014; Wolz et al., 2017; Medina et al., 2017a; Bird et al., 2018). Thus, another consideration is whether Bd is present and when is it most active, as changes in microbial community composition have been linked with the onset of Bd infection (Pullen et al., 2010; Longo et al., 2015; Clare et al., 2016; Varela et al., 2018). Specifically, Bd is more prevalent during seasons of moderate temperatures (between 17 and 25°C) and high precipitation. In our study, we observed significantly larger Bd-inhibition in the fall as opposed to the spring, which is different than that found in previous studies (Figure 5C; Pullen et al., 2010, Varela et al., 2018).
Despite marginal difference in precipitation, Spring in the New York region is generally cooler than Fall (April/May mean = 15.2°C, September/October mean = 18.2°C according to the National Weather Service’s New York City records over the last 10 years). Based on these temperatures, Spring may be too cold for Bd growth, whereas Fall is within Bd’s optimal temperature range increasing bacterial anti-Bd metabolite response (~17–23°C; Piotrowski et al., 2004). Additionally, amphibians have been shown to lower intensity of Bd infections through over-wintering (Savage et al., 2011; Longo et al., 2015) or raising their own body temperature (Woodhams et al., 2003), which could have confounding effects on their entire microbiome. These temperature differences both in the environment and on the host could also impact which bacteria are present, their physiologies, and their function. Thus, we believe these results warrant further research into seasonal patterns of Bd-inhibition at a range of sites.
Our results indicate omitting functional assays on locally isolated bacteria can lead researchers to over- or underestimate microbiome function with potentially drastic implications for probiotic development or bioaugmentation. While our study of community variation along a relatively small (<65 km) urban land use gradient complements other regional high-throughput taxonomic analyses, further investigation is needed to determine if and how direct land-use associated effects on the soil bacterial reservoir indirectly alter amphibian cutaneous microbiomes. Additionally, our results highlight the need for research into metabolite composition and the genetic mechanisms responsible for their production. These analyses should continue to be performed in both pristine and altered environments to account for the continued habitat degradation occurring alongside the amphibian disease epidemic. In sum, our results provide further evidence for examining the whole microbiome, including micro-differences in species-species interactions and environmental variability, for understanding how it provides disease protection.
The sequencing data generated for this study can be found in GenBank using accession numbers MN582766-MN582943.
The animal study was reviewed and approved by Fordham University, Institutional Animal Care and Use Committee Protocol #JL-17-01.
EB and JL conceived and designed the experiments and wrote the paper. EB and EC performed the experiments and analyzed the data. EB, EC, and JL contributed materials and reagents and reviewed drafts of the paper.
This research was done with the support of the Louis Calder Center graduate student research fund and the Henry Luce Foundation which supported EB through a Clare Boothe Luce fellowship and research fund.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the following organizations for access to their sites for sampling: New York City Department of Parks & Recreation, the Office of Parks and Historic Preservation including the managers of Rockefeller, Fahnestock and Hudson Highlands State Parks, the New York Botanical Gardens, the Westmoreland Sanctuary, and the Louis Calder Center. We thank Steve Kutos, Kiara Taveras, Julia Piccirillo-Stosser, and Sabrina Piccirillo-Stosser for field support.
Alberti, M. (2005). The effects of urban patterns on ecosystem function. Int. Reg. Sci. Rev. 28, 168–192. doi: 10.1177/0160017605275160
Averill, C., Cates, L. L., Dietze, M. C., and Bhatnagar, J. M. (2019). Spatial vs. temporal controls over soil fungal community similarity at continental and global scales. ISME J. 13, 2082–2093. doi: 10.1038/s41396-019-0420-1
Bahrndorff, S., Alemu, T., Alemneh, T., and Lund Nielsen, J. (2016). The microbiome of animals: implications for conservation biology. Research article. Available at: https://www.hindawi.com/journals/ijg/2016/5304028/abs/
Bakker, M. G., Schlatter, D. C., Otto-Hanson, L., and Kinkel, L. L. (2014). Diffuse symbioses: roles of plant–plant, plant–microbe and microbe–microbe interactions in structuring the soil microbiome. Mol. Ecol. 23, 1571–1583. doi: 10.1111/mec.12571
Bates, K. A., Clare, F. C., O’Hanlon, S., Bosch, J., Brookes, L., Hopkins, K., et al. (2018). Amphibian chytridiomycosis outbreak dynamics are linked with host skin bacterial community structure. Nat. Commun. 9:693. doi: 10.1038/s41467-018-02967-w
Becker, M. H., and Harris, R. N. (2010). Cutaneous bacteria of the Redback salamander prevent morbidity associated with a lethal disease. PLoS One 5, 1–6. doi: 10.1371/journal.pone.0010957
Becker, M. H., Walke, J. B., Murrill, L., Woodhams, D. C., Reinert, L. K., Rollins-Smith, L. A., et al. (2015). Phylogenetic distribution of symbiotic bacteria from Panamanian amphibians that inhibit growth of the lethal fungal pathogen Batrachochytrium dendrobatidis. Mol. Ecol. 24, 1628–1641. doi: 10.1111/mec.13135
Becker, C. G., and Zamudio, K. R. (2011). Tropical amphibian populations experience higher disease risk in natural habitats. Proc. Natl. Acad. Sci. USA 108, 9893–9898. doi: 10.1073/pnas.1014497108
Belden, L. K., Hughey, M. C., Rebollar, E. A., Umile, T. P., Loftus, S. C., Burzynski, E. A., et al. (2015). Panamanian frog species host unique skin bacterial communities. Front. Microbiol. 6:1171. doi: 10.3389/fmicb.2015.01171
Bell, S. C., Alford, R. A., Garland, S., Padilla, G., and Thomas, A. D. (2013). Screening bacterial metabolites for inhibitory effects against Batrachochytrium dendrobatidis using a spectrophotometric assay. Dis. Aquat. Org. 103, 77–85. doi: 10.3354/dao02560
Belzer, C., and de Vos, W. M. (2012). Microbes inside—from diversity to function: the case of Akkermansia. ISME J. 6, 1449–1458. doi: 10.1038/ismej.2012.6
Benaglia, T., Chauveau, D., Hunter, D., and Young, D. (2009). Mixtools: an R package for analyzing finite mixture models. J. Stat. Softw. 32, 1–29. Available at: https://hal.archives-ouvertes.fr/hal-00384896
Benson, A. K., Kelly, S. A., Legge, R., Ma, F., Low, S. J., Kim, J., et al. (2010). Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA 107, 18933–18938. doi: 10.1073/pnas.1007028107
Benson, A., Pifer, R., Behrendt, C. L., Hooper, L. V., and Yarovinsky, F. (2009). Gut commensal bacteria direct a protective immune response against toxoplasma gondii. Cell Host Microbe 6, 187–196. doi: 10.1016/j.chom.2009.06.005
Bird, A. K., Prado-Irwin, S. R., Vredenburg, V. T., and Zink, A. G. (2018). Skin microbiomes of California terrestrial salamanders are influenced by habitat more than host phylogeny. Front. Microbiol. 9:442. doi: 10.3389/fmicb.2018.00442
Bissett, A., Richardson, A. E., Baker, G., and Thrall, P. H. (2011). Long-term land use effects on soil microbial community structure and function. Appl. Soil Ecol. 51, 66–78. doi: 10.1016/j.apsoil.2011.08.010
Bissett, A., Richardson, A. E., Baker, G., Wakelin, S., and Thrall, P. H. (2010). Life history determines biogeographical patterns of soil bacterial communities over multiple spatial scales. Mol. Ecol. 19, 4315–4327. doi: 10.1111/j.1365-294X.2010.04804.x
Blaser, M. J. (2006). Who are we? Indigenous microbes and the ecology of human diseases. EMBO Rep. 7, 956–960. doi: 10.1038/sj.embor.7400812
Blaser, M. J., and Falkow, S. (2009). What are the consequences of the disappearing human microbiota? Nat. Rev. Microbiol. 7, 887–894. doi: 10.1038/nrmicro2245
Bletz, M. C., Perl, R. G. B., Bobowski, B. T., Japke, L. M., Tebbe, C. C., Dohrmann, A. B., et al. (2017). Amphibian skin microbiota exhibits temporal variation in community structure but stability of predicted Bd-inhibitory function. ISME J. 11, 1521–1534. doi: 10.1038/ismej.2017.41
Booth, A., Mariscal, C., and Doolittle, W. F. (2016). The modern synthesis in the light of microbial genomics. Annu. Rev. Microbiol. 70, 279–297. doi: 10.1146/annurev-micro-102215-095456
Bower, D. S., Lips, K. R., Schwarzkopf, L., Georges, A., and Clulow, S. (2017). Amphibians on the brink. Science 357, 454–455. doi: 10.1126/science.aao0500
Boyle, D., Boyle, D., Olsen, V., Morgan, J., and Hyatt, A. (2004). Rapid quantitative detection of chytridiomycosis (Batrachochytrium dendrobatidis) in amphibian samples using real-time Taqman PCR assay. Dis. Aquat. Org. 60, 141–148. doi: 10.3354/dao060141
Brucker, R. M., Harris, R. N., Schwantes, C. R., Gallaher, T. N., Flaherty, D. C., Lam, B. A., et al. (2008). Amphibian chemical defense: antifungal metabolites of the Microsymbiont Janthinobacterium lividum on the salamander Plethodon cinereus. J. Chem. Ecol. 34, 1422–1429. doi: 10.1007/s10886-008-9555-7
Burke, C., Steinberg, P., Rusch, D., Kjelleberg, S., and Thomas, T. (2011). Bacterial community assembly based on functional genes rather than species. Proc. Natl. Acad. Sci. USA 108, 14288–14293. doi: 10.1073/pnas.1101591108
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Ceballos, G., Ehrlich, P. R., Barnosky, A. D., García, A., Pringle, R. M., and Palmer, T. M. (2015). Accelerated modern human-induced species losses: entering the sixth mass extinction. Sci. Adv. 1:e1400253. doi: 10.1126/sciadv.1400253
Cho, I., and Blaser, M. J. (2012). The human microbiome: at the interface of health and disease. Nat. Rev. Genet. 13, 260–270. doi: 10.1038/nrg3182
Clare, F. C., Halder, J. B., Daniel, O., Bielby, J., Semenov, M. A., Jombart, T., et al. (2016). Climate forcing of an emerging pathogenic fungus across a montane multi-host community. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371, pii: 20150454. doi: 10.1098/rstb.2015.0454
Cohan, F. M. (2002). What are bacterial species? Annu. Rev. Microbiol. 56, 457–487. doi: 10.1146/annurev.micro.56.012302.160634
Collins, J. P., and Storfer, A. (2003). Global amphibian declines: sorting the hypotheses. Divers. Distrib. 9, 89–98. doi: 10.1046/j.1472-4642.2003.00012.x
Costello, E. K., Lauber, C. L., Hamady, M., Fierer, N., Gordon, J. I., and Knight, R. (2009). Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697. doi: 10.1126/science.1177486
Cousins, J. R., Hope, D., Gries, C., and Stutz, J. C. (2003). Preliminary assessment of arbuscular mycorrhizal fungal diversity and community structure in an urban ecosystem. Mycorrhiza 13, 319–326. doi: 10.1007/s00572-003-0239-4
Daskin, J. H., Bell, S. C., Schwarzkopf, L., and Alford, R. A. (2014). Cool temperatures reduce antifungal activity of symbiotic bacteria of threatened amphibians – implications for disease management and patterns of decline. PLoS One 9:e100378. doi: 10.1371/journal.pone.0100378
Delgado-Baquerizo, M., Giaramida, L., Reich, P. B., Khachane, A. N., Hamonts, K., Edwards, C., et al. (2016). Lack of functional redundancy in the relationship between microbial diversity and ecosystem functioning. J. Ecol. 104, 936–946. doi: 10.1111/1365-2745.12585
Devevey, G., Dang, T., Graves, C. J., Murray, S., and Brisson, D. (2015). First arrived takes all: inhibitory priority effects dominate competition between co-infecting Borrelia burgdorferi strains. BMC Microbiol. 15:1. doi: 10.1186/s12866-015-0381-0
DiRenzo, G. V., Langhammer, P. F., Zamudio, K. R., and Lips, K. R. (2014). Fungal infection intensity and zoospore output of Atelopus zeteki, a potential acute Chytrid Supershedder. PLoS One 9:e93356. doi: 10.1371/journal.pone.0093356
Durrant, M. G., and Bhatt, A. S. (2019). Microbiome genome structure drives function. Nat. Microbiol. 4, 912–913. doi: 10.1038/s41564-019-0473-y
Ellegaard, K. M., and Engel, P. (2019). Genomic diversity landscape of the honey bee gut microbiota. Nat. Commun. 10:446. doi: 10.1038/s41467-019-08303-0
Epp Schmidt, D. J., Pouyat, R., Szlavecz, K., Setälä, H., Kotze, D. J., Yesilonis, I., et al. (2017). Urbanization erodes ectomycorrhizal fungal diversity and may cause microbial communities to converge. Nat. Ecol. Evol. 1:0123. doi: 10.1038/s41559-017-0123
Faeth, S. H., Bang, C., and Saari, S. (2011). Urban biodiversity: patterns and mechanisms. Ann. N. Y. Acad. Sci. 1223, 69–81. doi: 10.1111/j.1749-6632.2010.05925.x
Familiar López, M., Rebollar, E. A., Harris, R. N., Vredenburg, V. T., and Hero, J.-M. (2017). Temporal variation of the skin bacterial community and Batrachochytrium dendrobatidis infection in the terrestrial cryptic frog Philoria loveridgei. Front. Microbiol. 8:2535. doi: 10.3389/fmicb.2017.02535
Farrer, R. A., Weinert, L. A., Bielby, J., Garner, T. W. J., Balloux, F., Clare, F., et al. (2011). Multiple emergences of genetically diverse amphibian-infecting chytrids include a globalized hypervirulent recombinant lineage. Proc. Natl. Acad. Sci. USA 108, 18732–18736. doi: 10.1073/pnas.1111915108
Fenchel, T., Esteban, G. F., and Finlay, B. J. (1997). Local versus global diversity of microorganisms: cryptic diversity of ciliated protozoa. Oikos 80, 220–225. doi: 10.2307/3546589
Feng, K., Zhang, Z., Cai, W., Liu, W., Xu, M., Yin, H., et al. (2017). Biodiversity and species competition regulate the resilience of microbial biofilm community. Mol. Ecol. 26, 6170–6182. doi: 10.1111/mec.14356
Fierer, N., and Jackson, R. B. (2006). The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 103, 626–631. doi: 10.1073/pnas.0507535103
Fukushima, M., Kakinuma, K., and Kawaguchi, R. (2002). Phylogenetic analysis of salmonella, Shigella, and Escherichia coli strains on the basis of the gyrB gene sequence. J. Clin. Microbiol. 40, 2779–2785. doi: 10.1128/JCM.40.8.2779-2785.2002
Garcia, S. L., Stevens, S. L. R., Crary, B., Martinez-Garcia, M., Stepanauskas, R., Woyke, T., et al. (2018). Contrasting patterns of genome-level diversity across distinct co-occurring bacterial populations. ISME J. 12, 742–755. doi: 10.1038/s41396-017-0001-0
Gilbert, J. A., Field, D., Swift, P., Thomas, S., Cummings, D., Temperton, B., et al. (2010). The taxonomic and functional diversity of microbes at a temperate coastal site: a ‘multi-Omic’ study of seasonal and Diel temporal variation. PLoS One 5:e15545. doi: 10.1371/journal.pone.0015545
Gómez, P., Paterson, S., Meester, L. D., Liu, X., Lenzi, L., Sharma, M. D., et al. (2016). Local adaptation of a bacterium is as important as its presence in structuring a natural microbial community. Nat. Commun. 7:12453. doi: 10.1038/ncomms12453
Haegeman, B., Hamelin, J., Moriarty, J., Neal, P., Dushoff, J., and Weitz, J. S. (2013). Robust estimation of microbial diversity in theory and in practice. ISME J. 7, 1092–1101. doi: 10.1038/ismej.2013.10
Hall, S. J., Ahmed, B., Ortiz, P., Davies, R., Sponseller, R. A., and Grimm, N. B. (2009). Urbanization alters soil microbial functioning in the Sonoran Desert. Ecosystems 12, 654–671. doi: 10.1007/s10021-009-9249-1
Harris, R. N., Brucker, R. M., Walke, J. B., Becker, M. H., Schwantes, C. R., Flaherty, D. C., et al. (2009a). Skin microbes on frogs prevent morbidity and mortality caused by a lethal skin fungus. ISME J. 3, 818–824. doi: 10.1038/ismej.2009.27
Harris, R., James, T., Lauer, A., Simon, M., and Patel, A. (2006). Amphibian pathogen Batrachochytrium dendrobatidis is inhibited by the cutaneous bacteria of amphibian species. EcoHealth 3, 53–56. doi: 10.1007/s10393-005-0009-1
Harris, R. N., Lauer, A., Simon, M. A., Banning, J. L., and Alford, R. A. (2009b). Addition of antifungal skin bacteria to salamanders ameliorates the effects of chytridiomycosis. Dis. Aquat. Org. 83, 11–16. doi: 10.3354/dao02004
Hartigan, J. A., and Hartigan, P. M. (1985). The dip test of unimodality. Ann. Stat. 13, 70–84. doi: 10.1214/aos/1176346577
Hawkes, C. V., and Keitt, T. H. (2015). Resilience vs. historical contingency in microbial responses to environmental change. Ecol. Lett. 18, 612–625. doi: 10.1111/ele.12451
Hilario, E., Buckley, T. R., and Young, J. M. (2004). Improved resolution on the phylogenetic relationships among pseudomonas by the combined analysis of atpD, carA, recA and 16S rDNA. Antonie Van Leeuwenhoek 86, 51–64. doi: 10.1023/B:ANTO.0000024910.57117.16
Holling, C. S. (2001). Understanding the complexity of economic, ecological, and social systems. Ecosystems 4, 390–405. doi: 10.1007/s10021-001-0101-5
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Janda, J. M., and Abbott, S. L. (2007). 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J. Clin. Microbiol. 45, 2761–2764. doi: 10.1128/JCM.01228-07
Jia, Y., and Whalen, J. K. (2019). A new perspective on functional redundancy and phylogenetic niche conservatism in soil microbial communities. Pedosphere 30, 18–24. doi: 10.1016/s1002-0160(19)60826-x
Jiang, X., Li, X., Yang, L., Liu, C., Wang, Q., Chi, W., et al. (2019). How microbes shape their communities? A microbial community model based on functional genes. Genomics Proteomics Bioinformatics 17, 91–105. doi: 10.1016/j.gpb.2018.09.003
Jiménez, R. R., and Sommer, S. (2017). The amphibian microbiome: natural range of variation, pathogenic dysbiosis, and role in conservation. Biodivers. Conserv. 26, 763–786. doi: 10.1007/s10531-016-1272-x
Kolde, R., Franzosa, E. A., Rahnavard, G., Hall, A. B., Vlamakis, H., Stevens, C., et al. (2018). Host genetic variation and its microbiome interactions within the human microbiome project. Genome Med. 10:6. doi: 10.1186/s13073-018-0515-8
Kraemer, S. A., and Kassen, R. (2015). Patterns of local adaptation in space and time among soil bacteria. Am. Nat. 185, 317–331. doi: 10.1086/679585
Krause, D. J., and Whitaker, R. J. (2015). Inferring speciation processes from patterns of natural variation in microbial genomes. Syst. Biol. 64, 926–935. doi: 10.1093/sysbio/syv050
Kruger, A. (2019). Functional redundancy of Batrachochytrium dendrobatidis inhibition in bacterial communities isolated from Lithobates clamitans skin. Microb. Ecol. 79, 231–240. doi: 10.1007/s00248-019-01387-7
Kuehne, H. A., Murphy, H. A., Francis, C. A., and Sniegowski, P. D. (2007). Allopatric divergence, secondary contact, and genetic isolation in wild yeast populations. Curr. Biol. 17, 407–411. doi: 10.1016/j.cub.2006.12.047
Kueneman, J. G., Parfrey, L. W., Woodhams, D. C., Archer, H. M., Knight, R., and McKenzie, V. J. (2014). The amphibian skin-associated microbiome across species, space and life history stages. Mol. Ecol. 23, 1238–1250. doi: 10.1111/mec.12510
Laforest-Lapointe, I., Messier, C., and Kembel, S. W. (2017). Tree leaf bacterial community structure and diversity differ along a gradient of urban intensity. mSystems 2:e00087-17. doi: 10.1128/msystems.00087-17
Lal, A., and Seshasayee, A. S. N. (2014). “The impact of next-generation sequencing technology on bacterial genomics” in A systems theoretic approach to systems and synthetic biology II: Analysis and design of cellular systems. eds. V. V. Kulkarni, G-B. Stan, and K. Raman (Dordrecht, Netherlands: Springer), 31–58.
Lauber, C. L., Hamady, M., Knight, R., and Fierer, N. (2009). Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 75, 5111–5120. doi: 10.1128/AEM.00335-09
Legendre, P., and Legendre, L. (1998). Numerical ecology, volume 24, (developments in environmental modelling). Oxford, UK: Elsevier.
Lehtimäki, J., Karkman, A., Laatikainen, T., Paalanen, L., von Hertzen, L., Haahtela, T., et al. (2017). Patterns in the skin microbiota differ in children and teenagers between rural and urban environments. Sci. Rep. 7:45651. doi: 10.1038/srep45651
Lladó, S., López-Mondéjar, R., and Baldrian, P. (2018). Drivers of microbial community structure in forest soils. Appl. Microbiol. Biotechnol. 102, 4331–4338. doi: 10.1007/s00253-018-8950-4
Lloyd-Price, J., Mahurkar, A., Rahnavard, G., Crabtree, J., Orvis, J., Hall, A. B., et al. (2017). Strains, functions and dynamics in the expanded human microbiome project. Nature 550, 61–66. doi: 10.1038/nature23889
Longcore, J. E., Pessier, A. P., and Nichols, D. K. (1999). Batrachochytrium dendrobatidis gen. et sp. nov., a chytrid pathogenic to amphibians. Mycologia 91, 219–227.
Longo, A. V., Savage, A. E., Hewson, I., and Zamudio, K. R. (2015). Seasonal and ontogenetic variation of skin microbial communities and relationships to natural disease dynamics in declining amphibians. R. Soc. Open Sci. 2:140377. doi: 10.1098/rsos.140377
Loudon, A. H., Venkataraman, A., VanTreuren, W., Woodhams, D., Parfrey, L. W., McKenzie, V., et al. (2016). Vertebrate hosts as islands: dynamics of selection, immigration, loss, persistence and potential function of bacteria on salamander skin. Front. Microbiol. 7:333. doi: 10.3389/fmicb.2016.00333
Maechler, M., and Ringach, D. (2013). Diptest: Hartigan’s dip test statistic for Unimodality. R package, Version 0.75-7. Available at: https://CRAN.R-project.org/package=diptest
Maldonado-Contreras, A., Goldfarb, K. C., Godoy-Vitorino, F., Karaoz, U., Contreras, M., Blaser, M. J., et al. (2011). Structure of the human gastric bacterial community in relation to Helicobacter pylori status. ISME J. 5, 574–579. doi: 10.1038/ismej.2010.149
Martinez, P. (2019). pairwiseAdonis: Pairwise multilevel comparison using adonis. R package version 0.3.
Martiny, J. B. H., Bohannan, B. J. M., Brown, J. H., Colwell, R. K., Fuhrman, J. A., Green, J. L., et al. (2006). Microbial biogeography: putting microorganisms on the map. Nat. Rev. Microbiol. 4, 102–112. doi: 10.1038/nrmicro1341
McKinney, M. L. (2006). Urbanization as a major cause of biotic homogenization. Urbanization 127, 247–260. doi: 10.1016/j.biocon.2005.09.005
Medina, D., Hughey, M. C., Becker, M. H., Walke, J. B., Umile, T. P., Burzynski, E. A., et al. (2017a). c. Microb. Ecol. 74, 227–238. doi: 10.1007/s00248-017-0933-y
Medina, D., Walke, J. B., Gajewski, Z., Becker, M. H., Swartwout, M. C., and Belden, L. K. (2017b). Culture media and individual hosts affect the recovery of culturable bacterial diversity from amphibian skin. Front. Microbiol. 8:1574. doi: 10.3389/fmicb.2017.01574
Mell, J. C., and Redfield, R. J. (2014). Natural competence and the evolution of DNA uptake specificity. J. Bacteriol. 196, 1471–1483. doi: 10.1128/JB.01293-13
Mendes, R., Garbeva, P., and Raaijmakers, J. M. (2013). The rhizosphere microbiome: significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 37, 634–663. doi: 10.1111/1574-6976.12028
Meyer, K. M., Memiaghe, H., Korte, L., Kenfack, D., Alonso, A., and Bohannan, B. J. M. (2018). Why do microbes exhibit weak biogeographic patterns? ISME J. 12, 1404–1413. doi: 10.1038/s41396-018-0103-3
Mollet, C., Drancourt, M., and Raoult, D. (1997). rpoB sequence analysis as a novel basis for bacterial identification. Mol. Microbiol. 26, 1005–1011.
Mori, A. S., Isbell, F., Fujii, S., Makoto, K., Matsuoka, S., and Osono, T. (2016). Low multifunctional redundancy of soil fungal diversity at multiple scales. Ecol. Lett. 19, 249–259. doi: 10.1111/ele.12560
National Land Cover Database (NLCD) (2011). Multi-Resolution Land Characteristics Consortium (MRLC). Available at: https://data.nal.usda.gov/dataset/national-land-cover-database-2011-nlcd-2011 (Accessed February 07, 2019).
Nickerson, C., Ebel, R., Borchers, A., and Carriazo, F. (2011). Major uses of land in the United States, 2007, EIB-89. Washington, DC: United States Department of Agriculture, Economic Research Service.
O’Brien, K., Perron, G. G., and Jude, B. A. (2018). Draft genome sequence of a red-pigmented Janthinobacterium sp. native to the Hudson Valley watershed. Genome Announc. 6:e01429-17. doi: 10.1128/genomea.01429-17
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O’hara, R., et al. (2013). Package ‘vegan.’ Community ecology package, version 2
Paula, F. S., Rodrigues, J. L. M., Zhou, J., Wu, L., Mueller, R. C., Mirza, B. S., et al. (2014). Land use change alters functional gene diversity, composition and abundance in Amazon forest soil microbial communities. Mol. Ecol. 23, 2988–2999. doi: 10.1111/mec.12786
Philippot, L., Andersson, S. G. E., Battin, T. J., Prosser, J. I., Schimel, J. P., Whitman, W. B., et al. (2010). The ecological coherence of high bacterial taxonomic ranks. Nat. Rev. Microbiol. 8, 523–529. doi: 10.1038/nrmicro2367
Piotrowski, J. S., Annis, S. L., and Longcore, J. E. (2004). Physiology of Batrachochytrium dendrobatidis, a chytrid pathogen of amphibians. Mycologia 96, 9–15. doi: 10.2307/3761981
Plassart, P., Prévost-Bouré, N. C., Uroz, S., Dequiedt, S., Stone, D., Creamer, R., et al. (2019). Soil parameters, land use, and geographical distance drive soil bacterial communities along a European transect. Sci. Rep. 9:605. doi: 10.1038/s41598-018-36867-2
Preheim, S. P., Perrotta, A. R., Friedman, J., Smilie, C., Brito, I., Smith, M. B., et al. (2013). “Chapter eighteen - computational methods for high-throughput comparative analyses of natural microbial communities” in Methods in enzymology. ed. E. F. DeLong (Cambridge, MA: Academic Press), 353–370.
Pullen, K. D., Best, A. M., and Ware, J. L. (2010). Amphibian pathogen Batrachochytrium dendrobatidis prevalence is correlated with season and not urbanization in Central Virginia. Dis. Aquat. Org. 91, 9–16. doi: 10.3354/dao02249
Rambaut, A. (2012). FigTree v.1.4.3. http://tree.bio.ed.ac.uk/software/figtree/ (Accessed October 12, 2018).
Reese, A. T., Savage, A., Youngsteadt, E., McGuire, K. L., Koling, A., Watkins, O., et al. (2016). Urban stress is associated with variation in microbial species composition[mdash]but not richness[mdash]in Manhattan. ISME J. 10, 751–760. doi: 10.1038/ismej.2015.152
Robinson, C. J., Bohannan, B. J. M., and Young, V. B. (2010). From structure to function: the ecology of host-associated microbial communities. Microbiol. Mol. Biol. Rev. 74, 453–476. doi: 10.1128/MMBR.00014-10
Ryan, R. P., Monchy, S., Cardinale, M., Taghavi, S., Crossman, L., Avison, M. B., et al. (2009). The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat. Rev. Microbiol. 7, 514–525. doi: 10.1038/nrmicro2163
Sanchez, L. M., Wong, W. R., Riener, R. M., Schulze, C. J., and Linington, R. G. (2012). Examining the fish microbiome: vertebrate-derived bacteria as an environmental niche for the discovery of unique marine natural products. PLoS One 7:e35398. doi: 10.1371/journal.pone.0035398
Savage, A. E., Sredl, M. J., and Zamudio, K. R. (2011). Disease dynamics vary spatially and temporally in a North American amphibian. Biol. Conserv. 144, 1910–1915. doi: 10.1016/j.biocon.2011.03.018
Schloegel, L. M., Picco, A. M., Kilpatrick, A. M., Davies, A. J., Hyatt, A. D., and Daszak, P. (2009). Magnitude of the US trade in amphibians and presence of Batrachochytrium dendrobatidis and ranavirus infection in imported North American bullfrogs (Rana catesbeiana). Biol. Conserv. 142, 1420–1426. doi: 10.1016/j.biocon.2009.02.007
Shapiro, B. J., and Polz, M. F. (2014). Ordering microbial diversity into ecologically and genetically cohesive units. Trends Microbiol. 22, 235–247. doi: 10.1016/j.tim.2014.02.006
Shochat, E., Warren, P. S., Faeth, S. H., McIntyre, N. E., and Hope, D. (2006). From patterns to emerging processes in mechanistic urban ecology. Trends Ecol. Evol. 21, 186–191. doi: 10.1016/j.tree.2005.11.019
Smillie, C. S., Smith, M. B., Friedman, J., Cordero, O. X., David, L. A., and Alm, E. J. (2011). Ecology drives a global network of gene exchange connecting the human microbiome. Nature 480, 241–244. doi: 10.1038/nature10571
Stuart, S. N., Chanson, J. S., Cox, N. A., Young, B. E., Rodrigues, A. S. L., Fischman, D. L., et al. (2004). Status and trends of amphibian declines and extinctions worldwide. Science 306, 1783–1786. doi: 10.1126/science.1103538
Suleiman, A. K. A., Manoeli, L., Boldo, J. T., Pereira, M. G., and Roesch, L. F. W. (2013). Shifts in soil bacterial community after eight years of land-use change. Syst. Appl. Microbiol. 36, 137–144. doi: 10.1016/j.syapm.2012.10.007
Taylor, J. W., Turner, E., Townsend, J. P., Dettman, J. R., and Jacobson, D. (2006). Eukaryotic microbes, species recognition and the geographic limits of species: examples from the kingdom fungi. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1947–1963. doi: 10.1098/rstb.2006.1923
The Human Microbiome Project ConsortiumHuttenhower, C., Gevers, D., Knight, R., Abubucker, S., Badger, J. H., et al. (2012a). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
The Human Microbiome Project ConsortiumMethé, B. A., Nelson, K. E., Pop, M., Creasy, H. H., Giglio, M. G., et al. (2012b). A framework for human microbiome research. Nature 486, 215–221. doi: 10.1038/nature11209
Thompson, L. R., Sanders, J. G., McDonald, D., Amir, A., Ladau, J., Locey, K. J., et al. (2017). A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551, 457–463. doi: 10.1038/nature24621
Varela, B. J., Lesbarrères, D., Ibáñez, R., and Green, D. M. (2018). Environmental and host effects on skin bacterial community composition in Panamanian frogs. Front. Microbiol. 9:298. doi: 10.3389/fmicb.2018.00298
Vieira-Silva, S., Falony, G., Darzi, Y., Lima-Mendez, G., Garcia Yunta, R., Okuda, S., et al. (2016). Species–function relationships shape ecological properties of the human gut microbiome. Nat. Microbiol. 1:16088. doi: 10.1038/nmicrobiol.2016.88
Wake, D. B., and Vredenburg, V. T. (2008). Are we in the midst of the sixth mass extinction? A view from the world of amphibians. Proc. Natl. Acad. Sci. USA 105, 11466–11473. doi: 10.1073/pnas.0801921105
Walke, J. B., Becker, M. H., Loftus, S. C., House, L. L., Teotonio, T. L., Minbiole, K. P. C., et al. (2015). Community structure and function of amphibian skin microbes: an experiment with bullfrogs exposed to a Chytrid fungus. PLoS One 10:e0139848. doi: 10.1371/journal.pone.0139848
Widder, S., Allen, R. J., Pfeiffer, T., Curtis, T. P., Wiuf, C., Sloan, W. T., et al. (2016). Challenges in microbial ecology: building predictive understanding of community function and dynamics. ISME J. 10, 2557–2568. doi: 10.1038/ismej.2016.45
Wolf, A., Fritze, A., Hagemann, M., and Berg, G. (2002). Stenotrophomonas rhizophila sp. nov., a novel plant-associated bacterium with antifungal properties. Int. J. Syst. Evol. Microbiol. 52, 1937–1944. doi: 10.1099/00207713-52-6-1937
Wolz, M., Carly, R., Yarwood, S. A., Grant, E. H. C., Fleischer, R. C., and Lips, K. R. (2017). Effects of host species and environment on the skin microbiome of Plethodontid salamanders. J. Anim. Ecol. 87, 341–353. doi: 10.1111/1365-2656.12726
Woodhams, D. C., Alford, R. A., Antwis, R. E., Archer, H., Becker, M. H., Belden, L. K., et al. (2015). Antifungal isolates database of amphibian skin-associated bacteria and function against emerging fungal pathogens. Ecology 96:595. doi: 10.1890/14-1837.1
Woodhams, D., Alford, R., and Marantelli, G. (2003). Emerging disease of amphibians cured by elevated body temperature. Dis. Aquat. Org. 55, 65–67. doi: 10.3354/dao055065
Woodhams, D. C., Brandt, H., Baumgartner, S., Kielgast, J., Küpfer, E., Tobler, U., et al. (2014). Interacting symbionts and immunity in the amphibian skin mucosome predict disease risk and probiotic effectiveness. PLoS One 9:e96375. doi: 10.1371/journal.pone.0096375
Wright, E. S., and Vetsigian, K. H. (2016). Inhibitory interactions promote frequent bistability among competing bacteria. Nat. Commun. 7:11274. doi: 10.1038/ncomms11274
Xu, H.-J., Li, S., Su, J.-Q., Gibson, V., Li, H., and Zhu, Y.-G. (2014). Does urbanization shape bacterial community composition in urban park soils? A case study in 16 representative Chinese cities based on the pyrosequencing method. FEMS Microbiol. Ecol. 87, 182–192. doi: 10.1111/1574-6941.12215
Yan, Y., Klinkhamer, P. G. L., van Veen, J. A., and Kuramae, E. E. (2019). Environmental filtering: a case of bacterial community assembly in soil. Soil Biol. Biochem. 136:107531. doi: 10.1016/j.soilbio.2019.107531
Yildirim, S., Yeoman, C. J., Sipos, M., Torralba, M., Wilson, B. A., Goldberg, T. L., et al. (2010). Characterization of the fecal microbiome from non-human wild primates reveals species specific microbial communities. PLoS One 5:e13963. doi: 10.1371/journal.pone.0013963
Yoon, S.-H., Ha, S.-M., Kwon, S., Lim, J., Kim, Y., Seo, H., et al. (2017). Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 67, 1613–1617. doi: 10.1099/ijsem.0.001755
Zhang, Z., Geng, J., Tang, X., Fan, H., Xu, J., Wen, X., et al. (2014). Spatial heterogeneity and co-occurrence patterns of human mucosal-associated intestinal microbiota. ISME J. 8, 881–893. doi: 10.1038/ismej.2013.185
Keywords: Batrachochytrium dendrobatidis, microbiome, disease, urbanization, functional redundancy, spatial variation
Citation: Barnes EM, Carter EL and Lewis JD (2020) Predicting Microbiome Function Across Space Is Confounded by Strain-Level Differences and Functional Redundancy Across Taxa. Front. Microbiol. 11:101. doi: 10.3389/fmicb.2020.00101
Edited by:
Eria Alaide Rebollar, National Autonomous University of Mexico, MexicoReviewed by:
Reid Harris, James Madison University, United StatesCopyright © 2020 Barnes, Carter and Lewis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elle M. Barnes, ZWJhcm5lczdAZm9yZGhhbS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.