
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 21 January 2020
Sec. Food Microbiology
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.03133
 Nicoletta Guaragnella1,2*†
Nicoletta Guaragnella1,2*† Matteo Chiara3†
Matteo Chiara3† Angela Capece4
Angela Capece4 Patrizia Romano4
Patrizia Romano4 Rocchina Pietrafesa4
Rocchina Pietrafesa4 Gabriella Siesto4
Gabriella Siesto4 Caterina Manzari2
Caterina Manzari2 Graziano Pesole1,2
Graziano Pesole1,2A current trend in winemaking has highlighted the beneficial contribution of non-Saccharomyces yeasts to wine quality. Hanseniaspora uvarum is one of the more represented non-Saccharomyces species onto grape berries and plays a critical role in influencing the wine sensory profile, in terms of complexity and organoleptic richness. In this work, we analyzed a group of H. uvarum indigenous wine strains as for genetic as for technological traits, such as resistance to SO2 and β-glucosidase activity. Three strains were selected for genome sequencing, assembly and comparative genomic analyses at species and genus level. Hanseniaspora genomes appeared compact and contained a moderate number of genes, while rarefaction analyses suggested an open accessory genome, reflecting a rather incomplete representation of the Hanseniaspora gene pool in the currently available genomes. The analyses of patterns of functional annotation in the three indigenous H. uvarum strains showed distinct enrichment for several PFAM protein domains. In particular, for certain traits, such as flocculation related protein domains, the genetic prediction correlated well with relative flocculation phenotypes at lab-scale. This feature, together with the enrichment for oligo-peptide transport and lipid and amino acid metabolism domains, reveals a promising potential of these indigenous strains to be applied in fermentation processes and modulation of wine flavor and aroma. This study also contributes to increasing the catalog of publicly available genomes from H. uvarum strains isolated from natural grape samples and provides a good roadmap for unraveling the biodiversity and the biotechnological potential of these non-Saccharomyces yeasts.
Species of the genus Hanseniaspora, widely known as “apiculate yeasts” due to their lemon-shaped cell morphology, are largely distributed in different environments and includes many species. Expressly, the species Hanseniaspora uvarum is frequently found on mature fruits and particularly on grapes (Zott et al., 2008; Wang et al., 2015a). H. uvarum is also frequently isolated from other fermented beverages, such as cider, palm wine and cashew juice, tequila, sugar-cane aguardente (Owuama and Saunders, 1990; Lachance, 1995; Morais et al., 1997; Valles et al., 2007). This yeast was isolated also from exotic substrates, such as African coffee and in chocolate production (Masoud et al., 2004; Illeghems et al., 2012; Batista et al., 2016).
Hanseniaspora uvarum shows antagonistic properties against the development of molds responsible for fruit spoilage and it was proposed as biocontrol agent against plant pathogens, such as Botrytis cinerea on grapes and strawberries and Penicillium spp. on citrus (Long et al., 2005; Cai et al., 2015). On the other hand, H. uvarum is considered a spoilage yeast in some processes, such as yogurt, orange juice, beer and honey production (Wiles, 1950; Kosse et al., 1997; Renard et al., 2008; Pulvirenti et al., 2009).
In nature, H. uvarum was isolated from different sources, such as soils, plants, insects, birds, mollusk and shrimps, moreover it was occasionally found as clinical isolate in humans where it is considered as opportunistic (Albertin et al., 2016). The widespread diffusion and economic importance of this yeast species demonstrate the high potentiality for application of H. uvarum in food biotechnology and especially in the wine sector in terms of product and process innovation (Tristezza et al., 2016; Capozzi et al., 2019; Berbegal et al., 2017).
In winemaking, these yeasts constitute more than 50% of the total yeast population (Fleet and Heard, 1993), but due to their sensitivity to increasing ethanol concentration, they are gradually replaced by Saccharomyces cerevisiae, the principal wine yeast (Fleet, 2003; Capece et al., 2005). Further studies demonstrated also the existence of interactions mechanisms between H. uvarum and S. cerevisiae during alcoholic fermentation. S. cerevisiae can produce killer toxin (Schmitt and Neuhausen, 1994), and release yet unidentified antimicrobial peptides, which play an important role in reducing non-Saccharomyces yeast population (Albergaria et al., 2010; Wang et al., 2015b).
In the past, this yeast species was traditionally considered as undesirable in winemaking and the addition of sulfites was the traditional way to prevent the risk of its growth at the beginning of the vinification process besides other approaches to limit its proliferation during fermentation (Comitini and Ciani, 2010). The main limit of this species in winemaking is the production of high levels of acetic acid and ethyl acetate, although some authors showed that not all the strains formed high levels of volatile acidity and many of them produced similar levels to those of S. cerevisiae (Bezerra-Bussoli et al., 2013).
However, considering that in the first days of fermentation H. uvarum reaches a very high cell density, it is expected that this yeast contributes significantly to fermentation affecting wine characteristics also if S. cerevisiae is added as starter culture (Capece and Romano, 2019). The actual trend in winemaking has re-evaluated the role of non-Saccharomyces yeasts due to their potential beneficial properties that contribute to increasing the sensory complexity of wines (Berbegal et al., 2017). Indigenous strains of H. uvarum are associated to specific terroir, produce fruity esters and possess a high enzymatic activities (esterases, β-glucosidases, lipases, and proteases), which might contribute to increase the sensory wine complexity (Belda et al., 2016; Tofalo et al., 2016a). In particular, strains of this species have been reported to exhibit β-glucosidase activity 6.6-fold higher than that of indigenous S. cerevisiae strains. This characteristic can be correlated with the increase of volatile compounds contents, such as free terpene, volatile phenols and C13-norisoprenoid, possessing a sensory impact (Martin et al., 2018). Selected H. uvarum strains were used as mixed starter cultures, both as co-inoculation and sequential inoculation with S. cerevisiae, to increase the wine organoleptic quality during industrial production, although until now no H. uvarum strain is commercialized as oenological starter culture (Tristezza et al., 2016; Petruzzi et al., 2017; Roudil et al., 2019). Moreover, H. uvarum has been shown to be compatible with S. cerevisiae and O. oeni in a simultaneous inoculation for the industrial production of regional typical wines, further supporting the use of mixed starter formulation as a promising approach in industrial application (Capozzi et al., 2019). In addition, the presence of H. uvarum has been detected during organic must fermentation in selected wine grape growing regions, supporting the significance to preserve biodiversity of these non-Saccharomyces native yeasts (Suzzi et al., 2012).
Despite this obvious importance of H. uvarum in wine fermentation, data on its genetic makeup is quite scarce. Karyotyping approaches suggested the presence of 7 to 9 chromosomes, with high variability between different isolates (Esteve-Zarzoso et al., 2001; Cadez et al., 2002), whereas the mitochondrial genome of H. uvarum has an exceptional structure among fungi, as it is represented by a short, linear DNA molecule (Pramateftaki et al., 2006). Recently, some brief reports on whole-genome approaches have appeared for Hanseniaspora strains, underlining the growing interest in this yeast, but with limited information regarding genome annotations (Giorello et al., 2014; Sternes et al., 2016; Seixas et al., 2017).
In this study, 26 H. uvarum strains isolated from spontaneous fermentation of grapes from different origin or source were subjected to a preliminary screening for genetic and phenotypic variability. Three strains, possessing different genetic and phenotypic traits, were selected and submitted to genome sequencing and assembly. Comparative genomics analysis at the genus and species levels was performed to identify candidate genes of potential biotechnological relevance.
Hanseniaspora uvarum strains used in this study are listed in Table 1. All strains have been isolated during spontaneous grape must fermentations, performed at lab-scale from grapes of different varieties and directly collected in the vineyard. All the strains were grown on YPD medium (1% yeast extract; 2% peptone; 2% glucose; 2% agar) and maintained at 4°C.
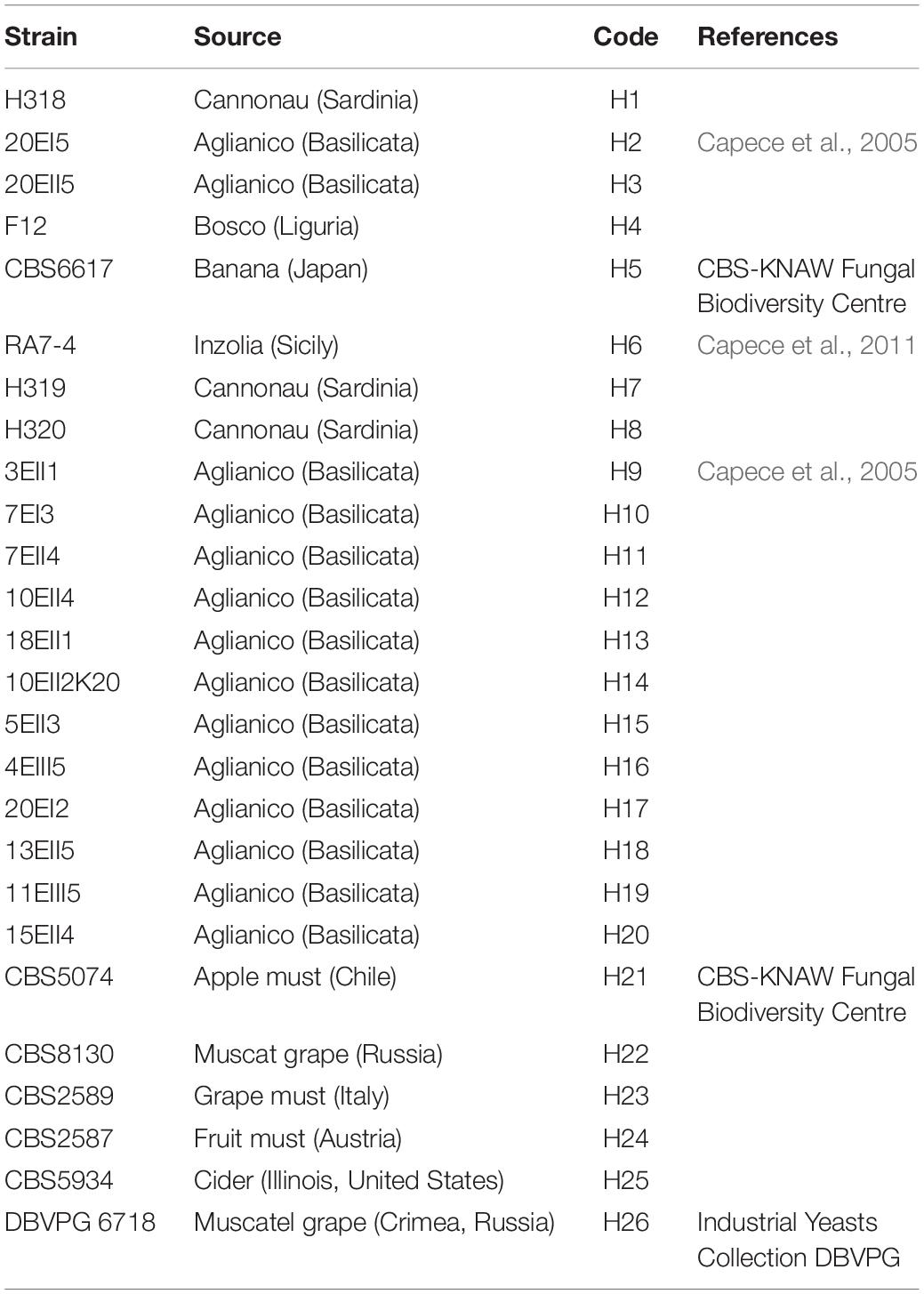
Table 1. List of Hanseniaspora uvarum strains used in this work.
Genomic DNA was isolated by using a synthetic resin (Instagene Bio-Rad Matrix) as previously described in Capece et al. (2011). Genotypic characterization was performed, as previously described for the characterization of non-Saccharomyces strains (Cadez et al., 2002; Capece et al., 2005; Andrade et al., 2006). The M13 (Cadez et al., 2002), P80 (Capece et al., 2005) and microsatellite-primed PCR (MSP-PCR) by using the synthetic oligonucleotide (GTG)5 and (GACA)4, were used for RAPD-PCR and MSP-PCR, respectively. The repeatability of these techniques was assessed in two independent amplification reactions with three repetitions, using H. uvarum reference strain DBVPG 6718. Amplification reactions were performed in a final volume of 50 μL containing 10 μL of Taq Polymerase 5X Buffer (Promega), 4.0 μL of 25 mM MgCl2 (Promega, Milan, Italy), 1 μL of 10 mM dNTP (Promega), 5 μL of 5 μM primer, 0.25 μL (5 U/μL) of Taq DNA polymerase (Promega) and 5 μL of template, with sterile water, added up to final volume. The thermal cycler was programed as follows: initial denaturation at 95°C for 5 min, 35 cycles at 94°C for 1 min for denaturing, 1 min at the primer-specific annealing temperature [54°C for M13 and P80, 52°C for (GTG)5 and 43°C for (GACA)4], 2 min at 72°C for extension and a final step at 72°C for 5 min. PCR products were analyzed by electrophoresis in 1.2% agarose gel, prepared in 1X TAE buffer (40 mM Tris–Acetate, 1 mM EDTA, pH 8.0). The gels were run at 100 V for 90 min, stained with SYBR® Safe (Invitrogen, United States) and captured by the Gel DocTM XR + system (Bio-Rad). The profiles were analyzed with FQWest software v.4.5 (Bio-Rad), using Pearson correlation and the dendrogram was constructed using UPGMA (tolerance 1%, optimization 0.5%). The cophenetic correlation was used to ascertain the consistency of the obtained cluster.
For technological characterization, both extracellular β-glucosidase activity and resistance to sulfur dioxide (SO2) have been determined. Quantitative screening of the extracellular β-glucosidase activity was performed following the protocol described by Mendes Ferreira et al. (2001). Strains were inoculated in 20 mL of YPD liquid medium and, after 48 h of incubation at 26°C, 1 × 106 cells/mL were inoculated in a liquid medium composed by yeast nitrogen base without aminoacids (0.67%), glucose (2%) and 0.4 mL of ferric ammonium citrate solution (1% w/v). Flasks were incubated in an orbital shaker at 150 rpm and 26°C for 24 h. Extracellular β-glucosidase activity was determined in 1 ml of supernatant, recovered by centrifugation at 3000 rpm for 10 min. Enzymatic activity was evaluated by determining the amount of p-nitrophenol (pNP) released from the p-nitrophenyl-β-D-glycoside (pNPG) by adding 0.2 mL of pNPG solution (5 mmol/L) in citrate-phosphate buffer (citric acid 0.1 M, Na2HPO4 0.2 M, pH 5) to 0.2 mL of each supernatant fluid and incubating at 30°C for 1 h. The reaction was stopped by adding 1.2 mL of Na2CO3 solution (0.2M). The amount of pNP released was determined spectrophotometrically at 400 nm. The enzymatic activity was quantified using a standard curve of pNP ranging between 10 and 150 nmol/mL. Results were expressed as nmol of pNP released for mL for hours.
Resistance to SO2 was tested by evaluating strain fermentative performance in natural red grape must (pH 3.4; sugars 225 g L–1; yeast assimilable nitrogen (YAN) 234 mg L–1) pasteurized at 100°C for 20 min, supplemented with 50 mg/L of total SO2 (added as potassium metabisulfite), as reported in Capece et al. (2011). Pasteurized grape must without SO2 addition was used as a control. The fermentations were performed at 26°C. Each sample was inoculated with 107 cell/mL from pre-cultures grown for 24 h in 5 mL of YPD liquid medium. The samples with SO2 were inoculated after 30 min of SO2 addition.
The SO2-resistance was expressed as the ratio between fermentative vigor (amount of CO2 produced at the third day of fermentation) of strains in the presence of SO2 and without SO2.
DNA library preparation was performed by the means of TruSeq DNA Nano Sample Prep kit (Illumina, San Diego, CA, United States), according to manufacturer’s instruction. Inserts size ranges were approximately between 200 and 500 bp. The library obtained from the H2 strain was sequenced on the Illumina NextSeq500 platform. A total of 30 M of 100 bp paired-end reads were produced. While libraries obtained from the H4 and H20 strains were sequenced on an Illumina MiSeq platform in order to obtain 2 × 200 bp paired-end reads. Reads were subjected to quality trimming using the Trimmomatic program, with default parameters (Bolger et al., 2014). Quality trimmed reads were subsequently subjected to assembly Spades (Prjibelski et al., 2014) using the default parameters and the following range of kmers (33,55,77,99) for the 100 bp reads and (33,55,77,99,121) for the 200 bp reads. Scaffolding was performed, using SSPACE (Boetzer and Pirovano, 2014). Gene annotation was performed using the Augustus program (Stanke et al., 2006), with gene models derived from S. cerevisiae and using the default cut-off value of 0.4 for the posterior probability.
All against all BlastP (Altschul et al., 1990) were performed using the BLOSUM80 matrix and accepting only best reciprocal hits with an e-value ≤ 1e-5, which covered at least 40% of the protein length, and where “second-best” hits produce bit scores < 90% of that associated with the best match. Putative COGs were established as groups of best reciprocal blast hits, using a custom utility available at https://github.com/cvulpispaper/compute_aai_and_cogs.
For each number of organisms considered (H. uvarum strains and Hanseniaspora species, respectively) the inferred sizes of core and accessory genomes were recorded for all the possible combinations of genomes. To avoid possible ascertainment biases in the comparison at genus level only one representative genome assembly was considered for species where more than one assembly was available. For H. uvarum and for H. vineae the genome assembly of the AWRI3580 and T02/19AF strain were considered, respectively. Plots were prepared showing mean and standard deviation of these statistics.
Where available (see Supplementary Table S1), heterozygous sites were inferred directly from the reference genomic assemblies based on IUPAC ambiguity codes. Alternatively, raw sequencing reads were obtained from public sequence repositories and aligned to their respective reference genome assembly in order to identify heterozygous sites (see Supplementary Table S1). Alignments were performed using the Bowtie2 (Langmead and Salzberg, 2012) software with default parameters, variant calling was performed by the means of the Freebayes program, again using default parameters (Garrison and Marth, 2012).
All heterozygous sites were masked using a custom Perl script. Complete genome assemblies were aligned using the Minimap2 program (Li, 2018) using the asm20 preset. Genomic identity levels were estimated directly from the Minimap2 output files by the means of a custom Perl script, available at https://github.com/matteo14c/minimap2_to_genome_identity.
Unfortunately due to lack of data (sequencing reads not deposited in any publicly available database) masking of heterozygous sites was not possible for the H. uvarum strain 34-9, H. uvarum strain CBA6001, H. vineae strain T02/19AF and H. vineae strain T02/05AF as indicated in Supplementary Table S1. Considerations regarding the relatively low levels of heterozygosity (from 0.18 to 0.81% depending on the species. Supplementary Table S1), if compared to the average level of identity between species (average 76.45%), would, however, suggest that this is not likely to have a considerable impact on the final clustering.
Hierarchical clustering was performed using the R (version 3.4.4 2018-03-15) implementation of the Neighbor-Joining algorithm from the cluster package (Maechler et al., 2019).
PFAM protein domains were annotated to predict protein-coding genes with the pfam_scan.pl program, using both the Pfam-A and Pfam-B domain models from the Pfam32.0 release of the Pfam database, with default parameters (Finn et al., 2016). The number of occurrences of each Pfam domain in each genome was counted using a Perl custom script. A simple R (version 3.4.4) script based on the hypergeometric distribution and implementing a Bonferroni correction was used to compute the p-value for the over-representation of the domains in each group.
Statistical analyses were performed using the Stats package as provided by the R programing language (version 3.4.4) R Core Team (2018). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria URL https://www.r-project.org/.
Flocculation test was performed according to the method described by Suzzi and Romano (1991), with small changes. Flocculation capacity was tested in liquid YNB (yeast nitrogen base), inoculating 24-h cells. Flocculation capacity was assessed by eye after 2, 15, and 20 days of incubation at 26°C. The flocculation levels, with small changes. Flocculation capacity was tested in liquid YNB (yeast nitrogen base), inoculating 24-h cells. Flocculation capacity was assessed by eye after 2, 15, and 20 days of incubation at 26°C. Flocculation levels were classified based on a rating scale from 0 (not flocculent) to 5 (very flocculent).
All the H. uvarum indigenous strains used in this work and their relative geographical origin are reported in Table 1.
All the 26 strains were subjected to genotypic and phenotypic characterization. H. uvarum strain typing was performed by using four different PCR methods based on RAPD analysis and MSP-PCR as described in Materials and Methods. According to our results, the (GTG)5 primer showed the best discrimination between the H. uvarum strains considered in this work, whereas other primers generated profiles that were too similar and/or had low levels of reproducibility (data not shown). Molecular profiles obtained by PCR fingerprinting with the (GTG)5 primer were clustered using the UPGMA algorithm with Pearson correlation-based distance measures, three major groups (A, B, C) were identified based on a similarity cut-off. Six isolates that did not show similarity with any other isolate were considered singletons (Figure 1). No particular correlation between the source of isolation of the strains and genotypic clustering was observed, with the notable exception of group B, which is composed mainly (7/8) by strains isolated from the Aglianico grape variety.
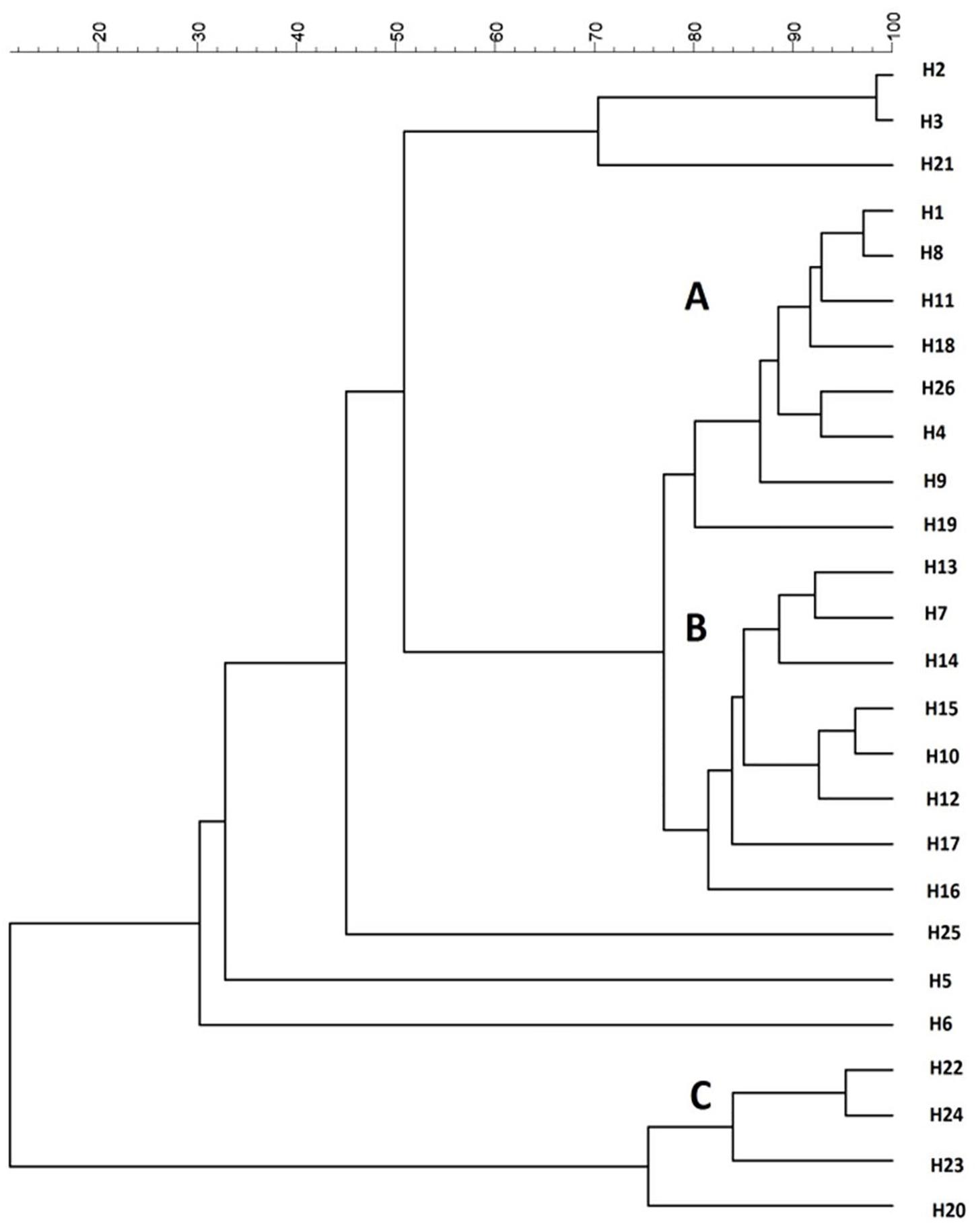
Figure 1. Cluster analysis with UPGMA method and Pearson distance of 26 H. uvarum profiles based on MSP-PCR with (GTG)5 primer.
Phenotypic characterization was performed by measuring two fermentative parameters, such as the levels of β-glucosidase activity and the resistance to SO2 during the early stages of fermentation (Table 2).
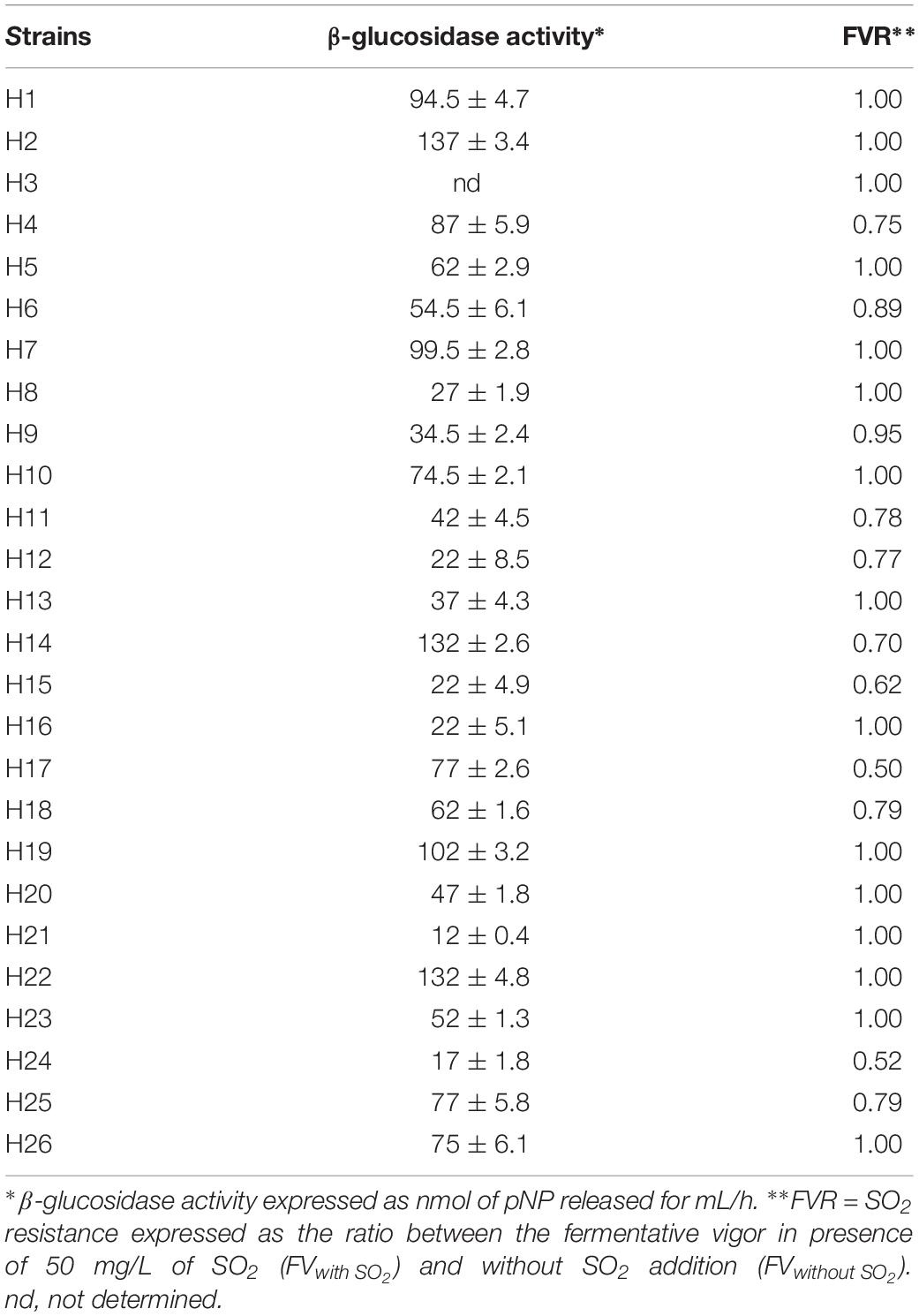
Table 2. Technological characterization of H. uvarum strains.
Quantitative screenings for extracellular β-glucosidase activity revealed high levels of variability among the strains. Of note, only the H3 strain did not exhibit detectable levels of β-glucosidase activity. In two strains (H21 and H24), a weak enzymatic activity was observed (12 and 17 nmol pNP ml–1 h–1), whereas four strains (H2, H14, H19 and H22) exhibited a high level of activity (higher than 100 nmol pNP ml–1 h–1). From a biotechnological point of view, high levels of β-glucosidase activity have been previously associated with increased hydrolysis of bound monoterpenes, which can enhance the fruity character of the wines (Rodriguez et al., 2004; Jolly et al., 2014).
The strains were also tested for the influence of SO2 on the fermentative activity as this compound is normally added to crushed grapes and H. uvarum species is known to be highly sensitive. The resistance of the strains to SO2 was assessed by measuring the fermentative vigor in the presence of 50 mg/L of SO2 (FVwith SO2). For each strain, the ratio (FVR) between FV with SO2 and the fermentative vigor without SO2 addition (FV without SO2) was used to express the SO2 resistance level. Values of this ratio similar to 1 show no differences between strain fermentative vigor with or without sulfur dioxide addition, whereas values lower than 1 indicate that the SO2 addition determines a reduction of fermentative vigor of the strains, indicating strains very sensitive to this compound. As reported in Table 2, fifteen strains were not affected by SO2 (FVR values equals to 1), whereas the remaining strains showed varied levels of sensitivity. The H24 strain, in particular, displayed a marked reduction of about 50% of the fermentative vigor (FVR values of 0.52). On the basis of these results, three wild H. uvarum strains, H2, H4 and H20, which were assigned to different groups according to (GTG)5 profiles (Figure 1) and exhibited different levels of resistance to SO2 and β-glucosidase activity (i.e., high, intermediate and low activity), were selected for whole genome sequencing and subjected to further characterization.
Summary statistics of the genome assemblies of the H2, H4, and H20 strains are presented in Table 3. The complete genome sequences of H2, H4, and H20 H. uvarum strains have been deposited in NCBI and are available from Genbank server under the following accessions numbers: uvarum H2 SAMN12284349; uvarum H4 SAMN12284353; uvarum H20 SAMN12284351.
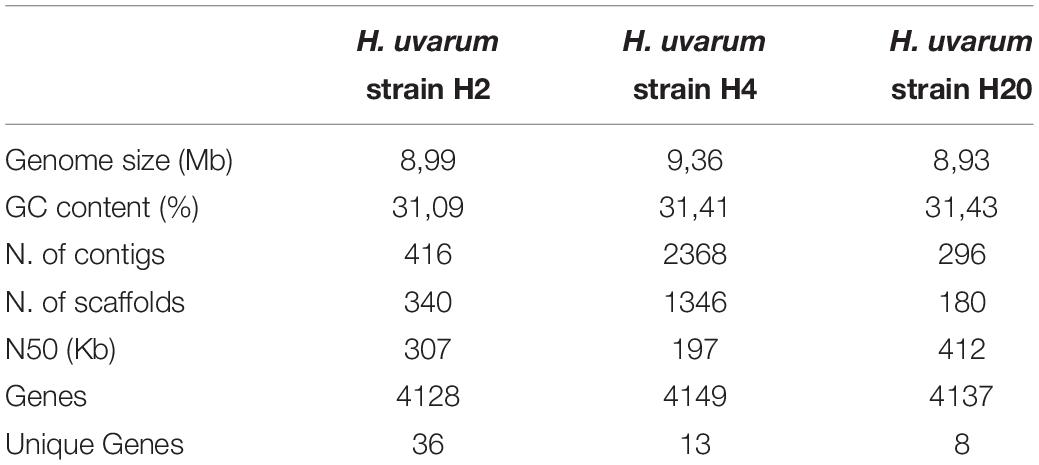
Table 3. Summary of genome assembly for H2, H4, and H20 H. uvarum strains.
All the assemblies are similar in size -ranging from 8,93 Mb in H20 to 9,36 Mb in H4- and composition, with a GC content of approximately 31%. While the H2 and H20 assemblies show a significantly reduced number of contigs (416 and 296 for H2 and H20 respectively, versus 2368 for H4) and an increased overall contiguity (N50 307 and 412 Kb for H2 and H20, 197 Kb for H4) if compared with the assembly obtained for the H4 isolate. This observation is likely a reflection of an overall increase in the number and extent of repetitive sequences in the genome of H4.
Notwithstanding the difference in the contiguity of the assemblies equivalent numbers of genes (about 4000) were predicted for all the assemblies by the Augustus program.
Genome assemblies of H. uvarum H2, H4, and H20 were compared with a selection of the currently publicly available Hanseniaspora genomes, including five strains of H. uvarum and other 10 species of Hanseniaspora (Table 4).
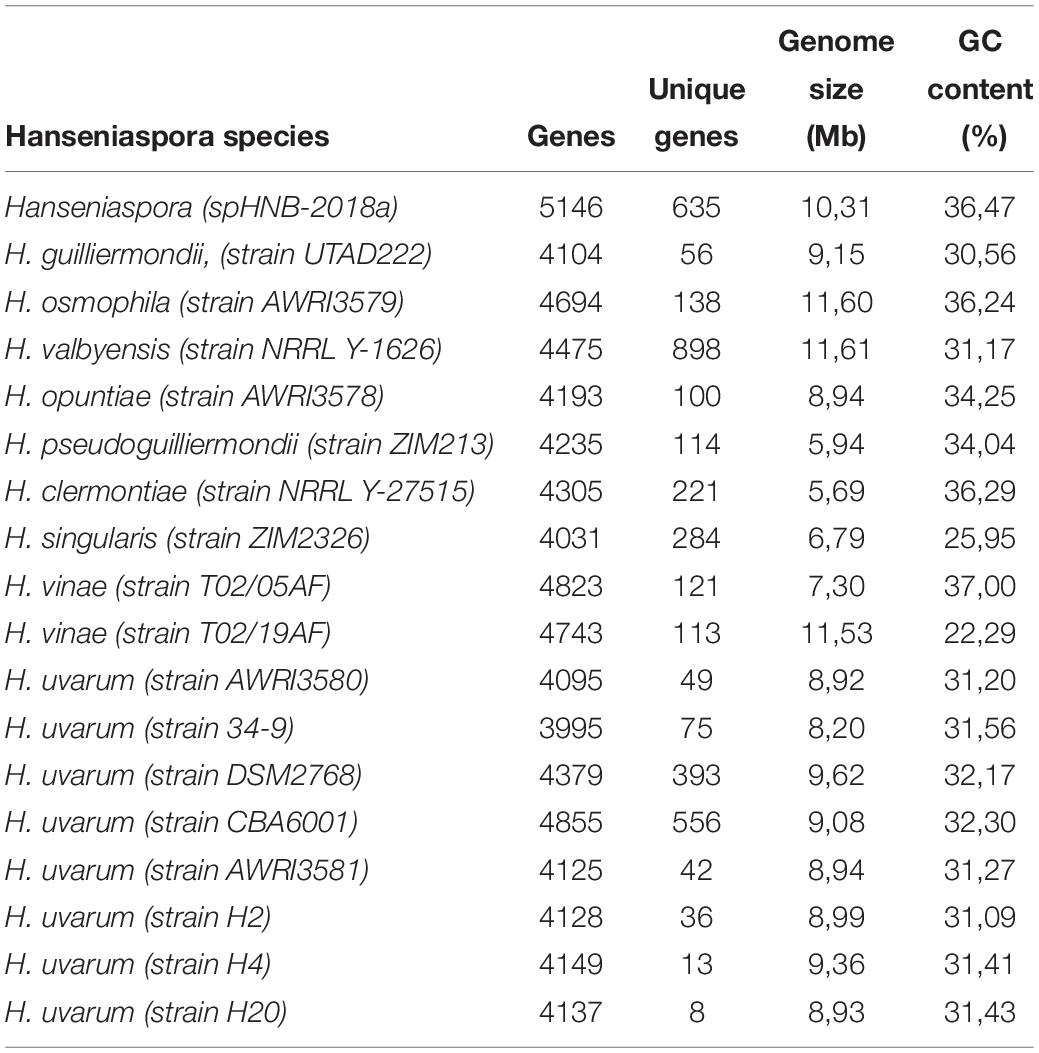
Table 4. Summary of comparative genomic analysis of Hanseniaspora and H. uvarum strains.
Phenetic clustering of Hanseniaspora based on genome identity levels (Figure 2A, see section Materials and Methods) suggest that, notwithstanding slightly different levels of heterozygosity (Supplementary Table S1), all the Hanseniaspora uvarum strains included in our analyses form a well supported monophyletic clade, with an average level of genomic identity of 96.75% or higher. Importantly, while the clusters identified by our analyses are broadly consistent with the phylogeny of Hanseniaspora reported by Steenwyk et al. (2019) and correctly separate fast and slow evolving lineages, we notice some inconsistencies with respect to the position of H. clermontiae, which in our clustering seems to be closely related to the H. singularis and H. valbiensis. Considerations regarding cross-species average genomic identity levels of Hanseniaspora which are remarkably similar (all between 74 and 77% with an average of 76.45%) suggest that this observation is likely the reflection of the reduced resolution of simple methods based on average genomic identity, if compared with sophisticated methods based on complex evolutionary models, in the precise reconstruction of phylogenetic relationships between distantly related species.
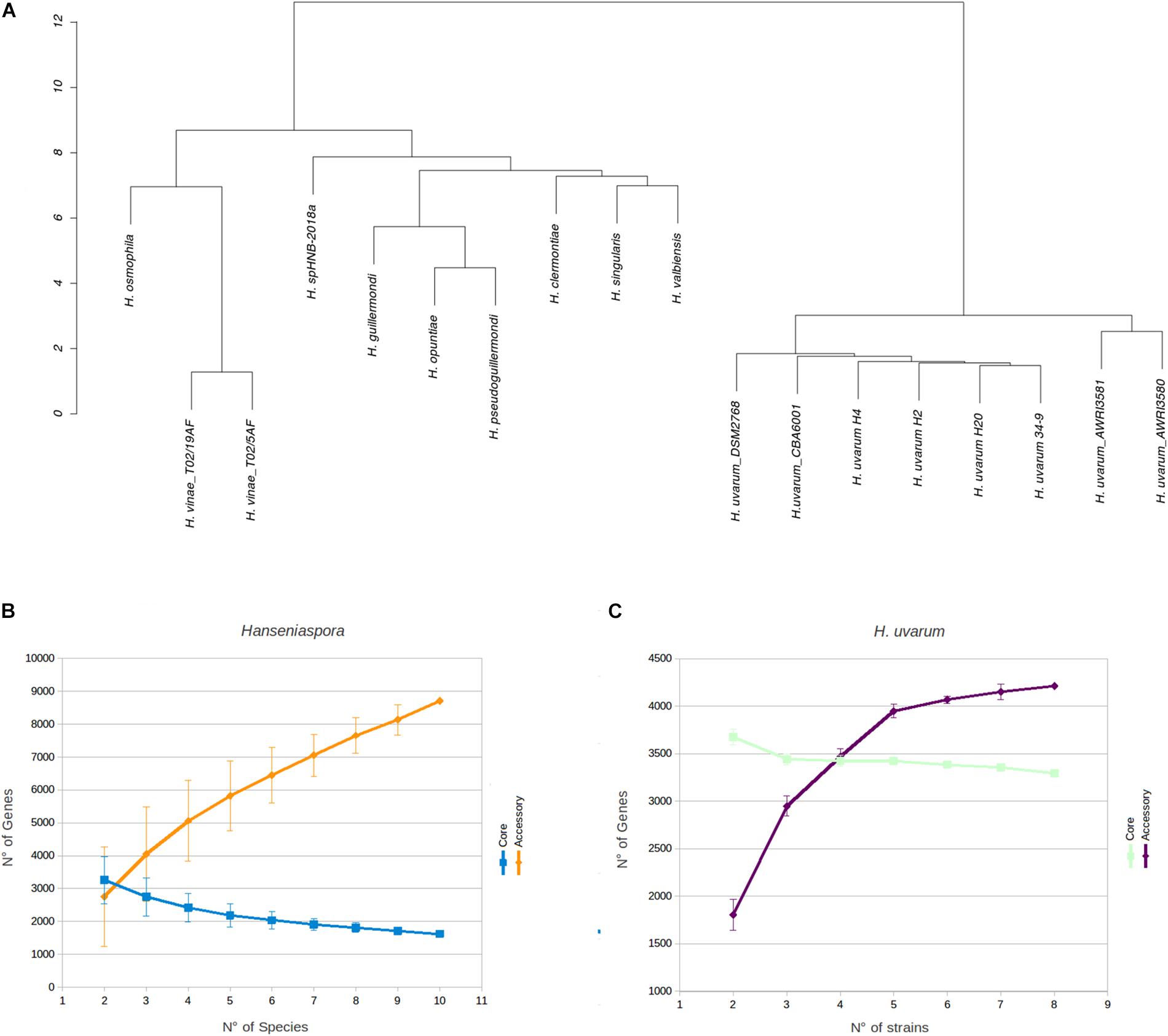
Figure 2. Phenetic clustering of Hanseniaspora and rarefaction analyses. (A) Hierarchical clustering of Hanseniaspora based on genomic identity. Pairwise levels of genomic identity were established by pairwise genome alignment, after masking dimorphic sites in each genome. Clustering was performed based on Euclidean distances of identity profiles. (B) Estimation of completeness of core and accessory genomes by rarefaction analyses at the genus level. Core and accessory genome sizes were calculated for all possible combinations of Hanseniaspora genomes. One representative species was selected for each genus included in the analyses (see section Materials and Methods). (C) Estimation of completeness of core and accessory genomes by rarefaction analyses for H. uvarum. Core and accessory genome sizes were calculated on each possible randomly resampled combinations of H. uvarum genomes.
To avoid possible ascertainment biases all the genomes were subjected to re-annotation using the Augustus program. Predicted genes were subjected to functional annotation of protein domains by the means of the pfam_scan software (see Section Materials and Methods).
Pairwise comparisons of sequence similarity of the predicted proteomes were performed using the blastP program, with the BLOSUM80 similarity matrix. Putative Clusters of Orthologous Genes (COGs) were subsequently established as groups of best reciprocal Blast hits, using a custom Perl script (see Section Materials and Methods). For each assembly, genes that were not assigned to COGs (i.e., did not recover a possible ortholog in any of the genomes included in the analysis) were considered “unique,” that is specific to a particular Hanseniaspora strain or species. Our approach identified a total of 7503 clusters (more than one gene) of putative orthologs as well as 3852 unique genes.
Summary statistics of gene content and numbers of “unique” genes are reported in Table 4.
The complete annotation of PFAM protein domains identified in all the “species-specific” genes is available at the following publicly accessible repository: https://github.com/matteo14c/supplementary_dataset_Guaragnella_et_al. Some of them have been selected for their biotechnological potential and reported on a heatmap (Supplementary Table S5 and Supplementary Figure S1).
Unsurprisingly -in the light of the relatively high levels of similarity between the genomes – we notice that all the H. uvarum strains herein considered show a very limited number of “unique genes” (between 126 for CBA6001 and 5 AWRI3580 and AWRI3581). While the number of unique genes associated with different species of Hanseniaspora is substantially larger (between 100 H. opuntiae and 898 H. valbyensis). This observation is also confirmed by rarefaction analyses. Indeed while estimates of the size of the core genome (Figure 2B) remain fairly stable when additional strains are sampled, suggesting that the currently available genomes provide an almost complete representation of the catalog of core genes Hanseniaspora, the accessory genome remains rather open when additional species are included (Figure 2B), indicating that our sampling of the Hanseniaspora species complex is probably incomplete.
On the other hand (Figures 2C, 3) profiles of gene absence/presence between the 8 H. uvarum strains included in this study (Figure 3) are completely consistent with a compact and relatively closed pan genome, as outlined by the fact that being composed by more than 3200 genes, the core genome accounts for more than 75% of the average (4223 genes) gene content of an isolate. Consistent with this observation, our analyses suggest that the 8 genomes included in this study provide a quite complete representation of the pan genome of H. uvarum, which seems to be composed by less than 4500 genes (Figure 2C).
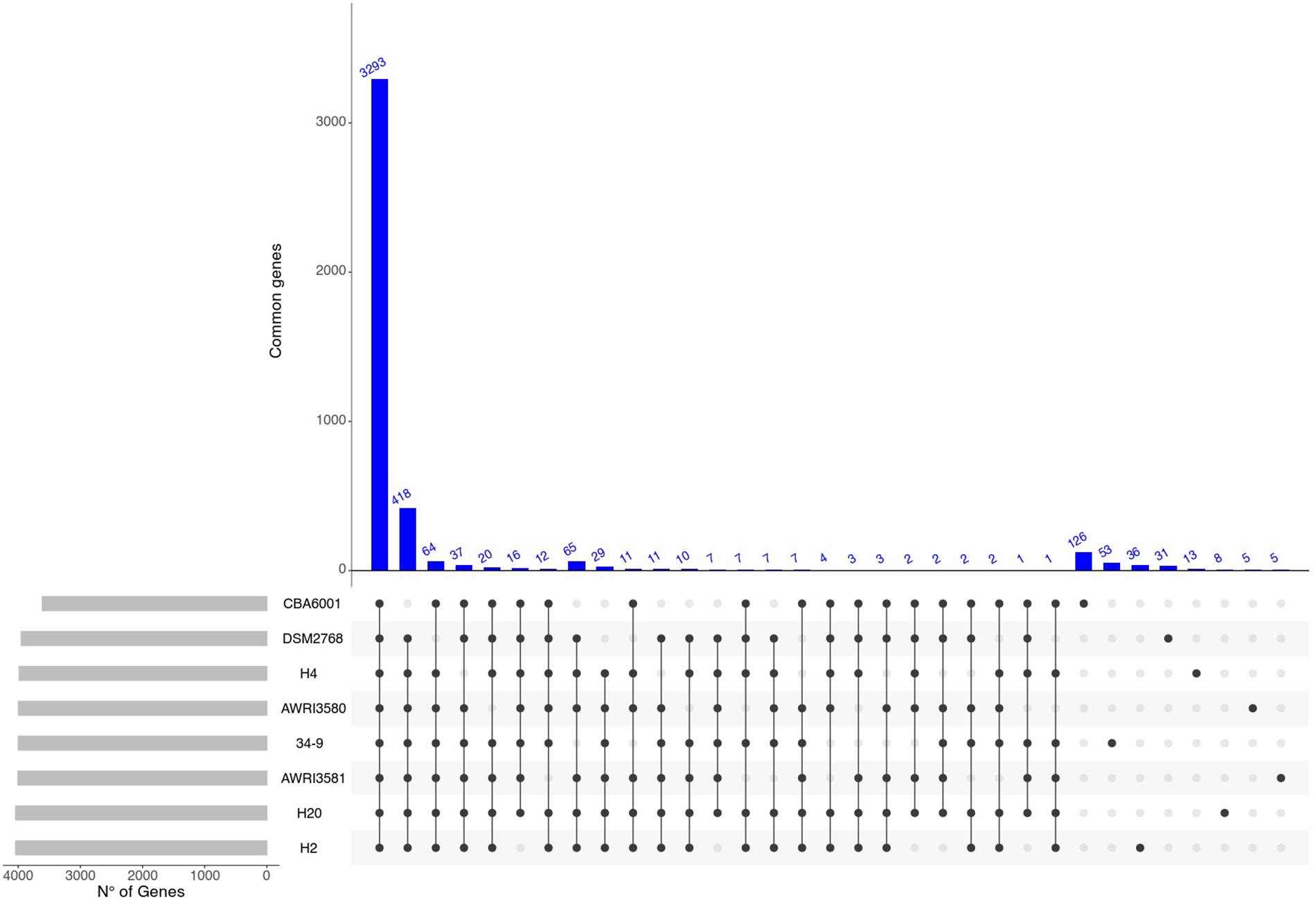
Figure 3. Graphical overview of shared genes. Number of genes shared between different combinations of H. uvarum strains. Blue bars indicate the number of shared genes. Gray bars are used to show the total number of genes for each H. uvarum strains.
In order to identify possible metabolic pathways or phenotypic traits specific for the H2, H20, and H4 H. uvarum strains, functional enrichment analyses of PFAM domains were performed and occurrences of protein domains in each isolate were compared with the equivalent figure in the H. uvarum pan genome by the means of a statistical test based on the hypergeometric distribution (see Section Materials and Methods).
The complete results of this analysis are reported in Supplementary Tables S2–S4. The 10 most over and under-represented domains for each strain are reported in Tables 5–7 for H2, H4, and H20 respectively. Interestingly, contrasting patterns of enrichment for selected domains were observed between the H2, H4 and H20 strains. In this regards, while H2 shows a highly significant enrichment of terms related to flocculin repeat (PF00624) and flocculation (Flo11 (PF10182) (Table 5), flocculation related protein domains such as PA14_2 (PF10528) are consistently under-represented in H4 (p-value 0.024) (Table 6). While we cannot conclusively exclude the possibility that under-representation of Flocculin domains in the H4 genome could be associated with the reduced contiguity of underlying genome assembly resulting in a reduced representation of repetitive of genes associated with repetitive sequence domains in the assembly, considerations regarding the average levels of aminoacidic identity of Flocculin genes (82.5%) and Flocculin domains (92.04%) advocates against this possibility. Indeed, such high levels of diversity are not likely to have a major impact on the assembly. Moreover, analysis based on coverage levels of the contigs (data not shown) suggests that all the Flocculin genes identified in the present study are present in single copy. No significantly over or under-representation of flocculation related terms (PA14_2, p-value 0.254) are observed for H20 (Table 7). A significant enrichment in the OPT domain (PF03169, p-value < 0.05) found in oligopeptide transporters was observed in both H2 and H4 strains. H4 strain showed also the presence of a PTR2 (PF00854) domain encoding for an oligopeptide transporter (p-value < 0.062). In order to verify whether this functional prediction was confirmed at lab-scale, flocculation tests were performed on the three strains. Results obtained confirm at least in part the observations based on genetic data: indeed while a strong flocculation phenotype (value 5) is observed in H2, on the other hand, H4 shows reduced levels of flocculation (value 2) and in H20 no flocculation could be observed (value 0).
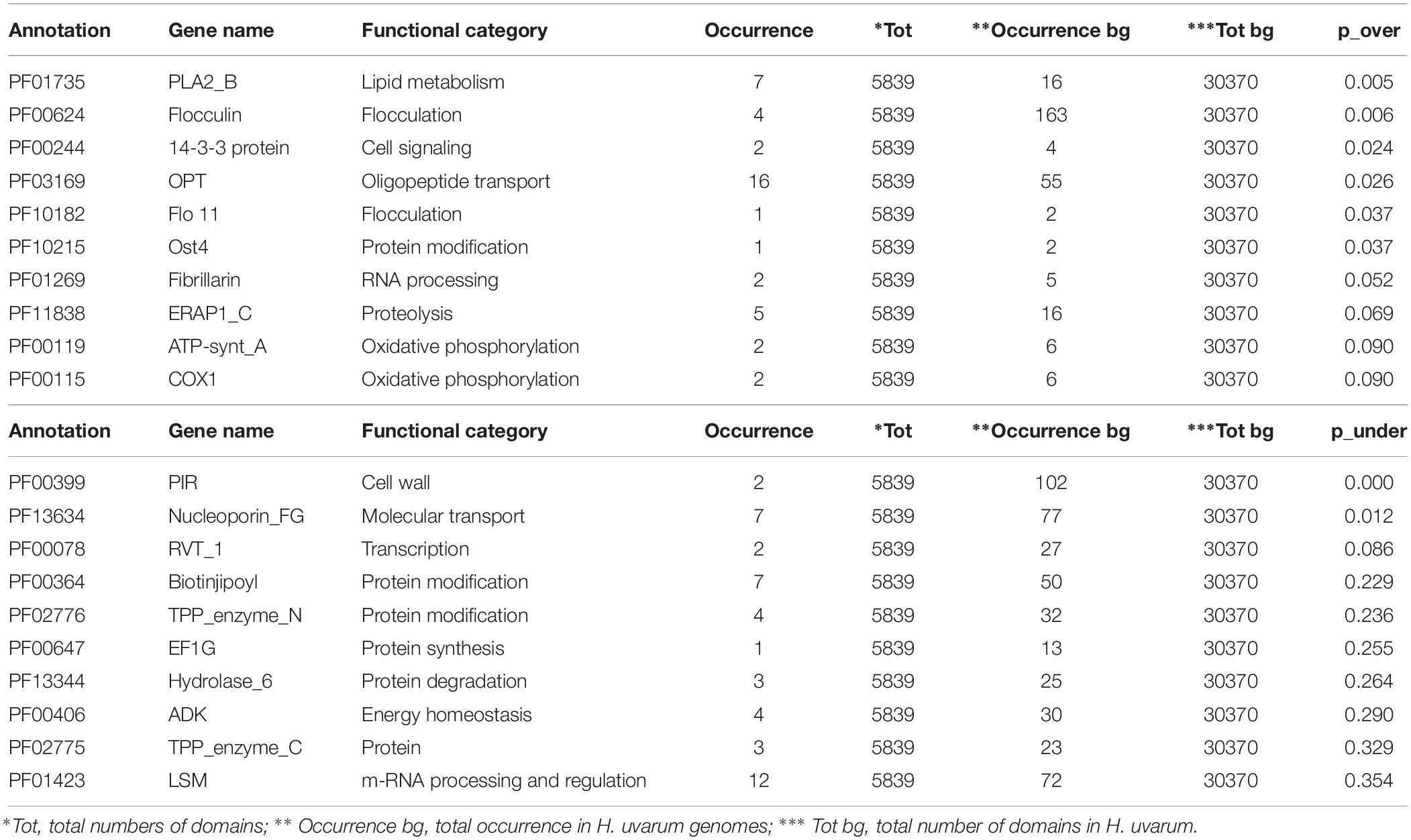
Table 5. The top 10 enriched and under-represented domains of H2 strain.
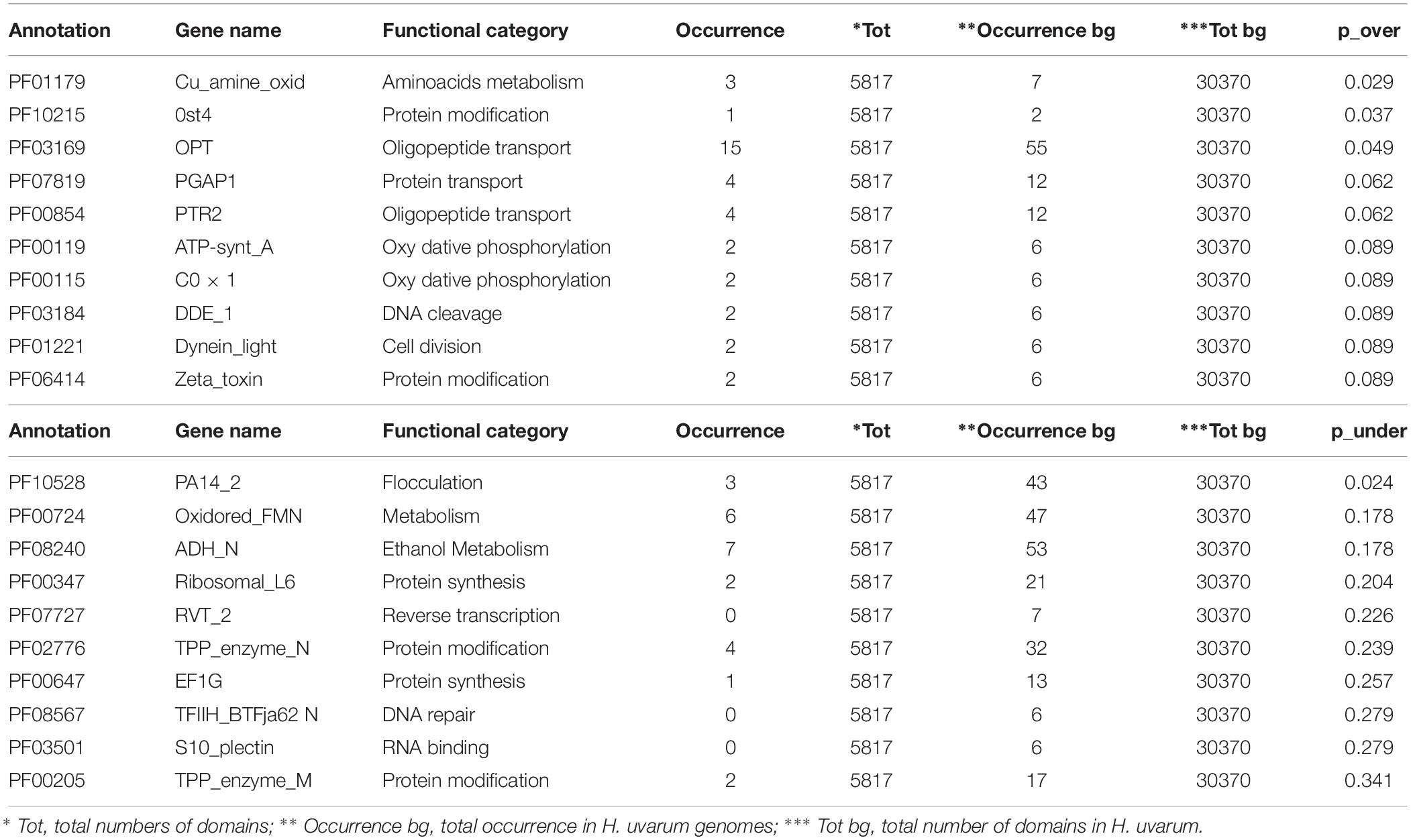
Table 6. The top 10 enriched and under-represented domains of H4 strain.
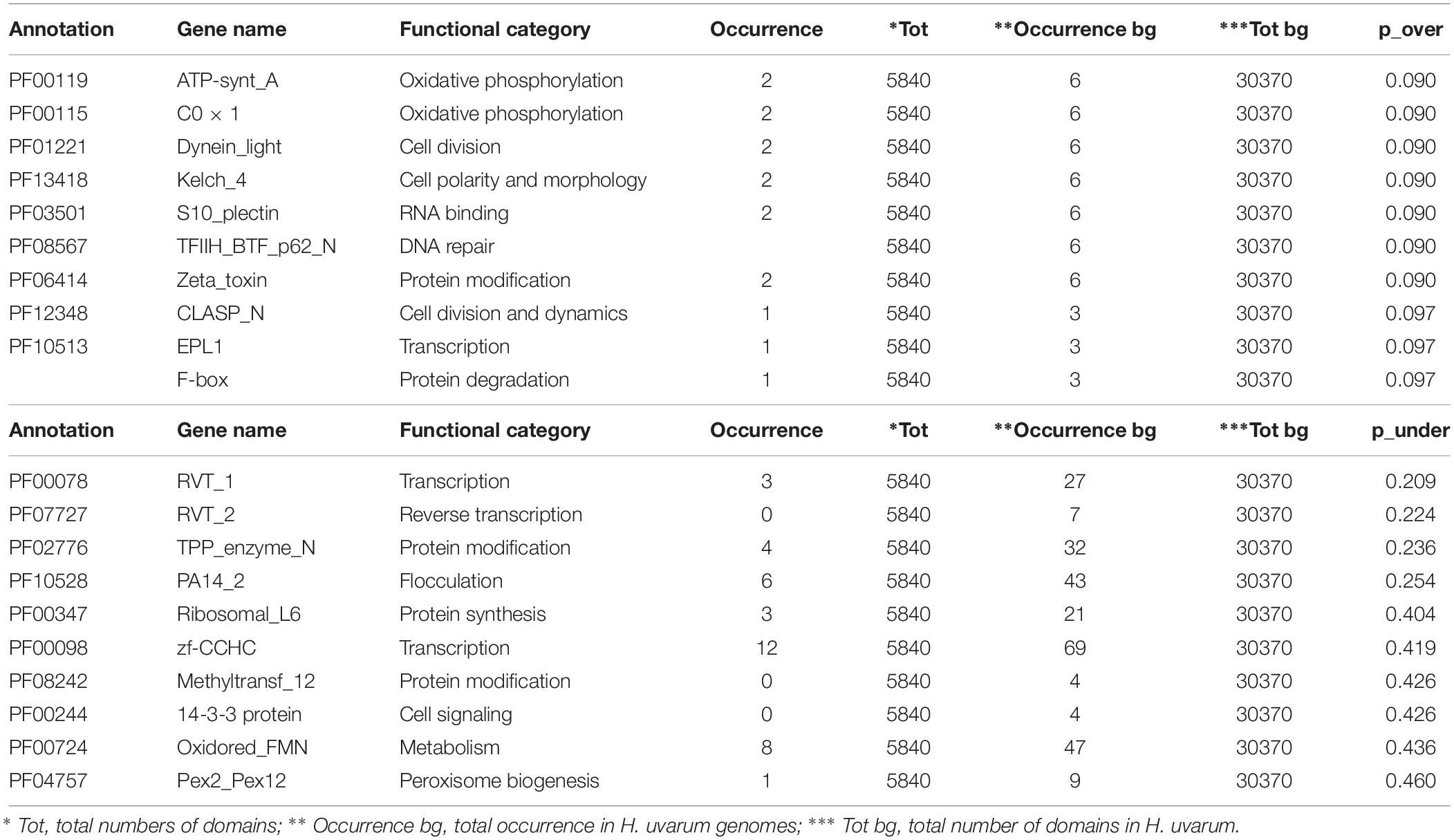
Table 7. The top 10 enriched and under-represented domains of H20 strain.
Over the last years, the beneficial contribution of non-Saccharomyces cerevisiae yeast species to wine characteristics has been recognized, making the exploitation of non-conventional yeasts as a new source of biodiversity with potential biotechnological significance (Masneuf-Pomarede et al., 2015). Among these yeasts, the genus Hanseniaspora, which can play a critical role in the modulation of the wine sensory profile by increasing its complexity and organoleptic richness, is attracting a significant interest (Fleet, 2003). So far, the knowledge on genetics and physiology of Hanseniaspora species remains limited, notwithstanding some recent significant studies open new perspectives in the field, revealing species-specific properties to be explored (Langenberg et al., 2017; Seixas et al., 2019). In this context, genomics analysis may enable a correlation between genetics and useful traits, which could provide a roadmap for biotechnological exploitations (Hittinger et al., 2015; Riley et al., 2016).
Here we present de novo genome sequencing of three Hanseniaspora uvarum indigenous wine strains and comparative genomic analyses of Hanseniaspora at species and genus level. Among 26 isolates from various geographical locations or sources (Table 1), three of them, H2, H4, and H20, isolated from spontaneous grape must fermentation were selected and subjected to further characterization. H2, H4, and H20 showed heterogeneity for relevant genotypic and phenotypic features of oenological interest, such as β-glucosidase activity and the resistance to SO2 under fermentative conditions at laboratory scale (Figure 1 and Table 2) (Fleet and Heard, 1993; Rodriguez et al., 2004; Jolly et al., 2014).
Whole-genome sequencing revealed comparable genome size (∼9 Mb), GC content (∼31%) and number of genes (∼4000) for the three strains (Table 3). These data also converge on the genomic features of the other H. uvarum strains analyzed in this work (Table 4) and by other authors (Langenberg et al., 2017). Hierarchical clustering of Hanseniaspora species indicates that the H. uvarum strains included in this study form a well-supported monophyletic clade (Figure 2A), with high levels of genomic identity and a relatively compact pan-genome, where core genes account for more than 75% of the average number of genes that are annotated in any individual strain (Figure 2C). Unsurprisingly these considerations can be extended also to all members of the genus Hanseniaspora, which display relatively compact genomes containing a moderate number of genes. Consistent with these observations we notice that core genes constitute a consistent proportion (between 28 and 36%) of the average gene content of any Hanseniaspora species (Figure 2B), suggesting that the currently available data provide an almost complete representation of the core- genome of Hanseniaspora. This notwithstanding our results indicate also that accessory genome of Hanseniaspora remains relatively open, and that all in all currently available data offer only a partial representation of the pan-genome of Hanseniaspora.
On the other hand, intra-species analyses of patterns of functional annotation show distinct patterns of enrichment for several PFAM protein domains in the three strains. In particular, the significant difference with respect to terms related to flocculin repeats, found in lectin-like proteins, and flocculation (Flo11) observed in H2 and H4 strains is an intriguing and industrially relevant trait (Tables 5, 6). Flocculation, the process by which yeast cells spontaneously aggregate to form flocs with sediment in the culture, has been observed in different yeast species, including non-Saccharomyces isolates and H. uvarum (Rossouw et al., 2015). Beyond its physiological relevance as a protective mechanism to enhance the survival under environmental stresses (Marika et al., 1993), flocculation is a desirable technological feature allowing the separation of cells from media in fermentation processes such as brewing and winemaking (Pretorius, 2000; Soares, 2011), in particular sparkling wine obtained by the so-called Method Champenoise. This characteristic allows the rapid clarification and reduction of the handling of final products, with a significant decrease in production costs (Tofalo et al., 2016b; Vigentini et al., 2017). Moreover, yeast flocculation seems to be associated with the enhancement of ester production (Pretorius, 2000). Although the interest in non- Saccharomyces yeasts for use in sparkling wine production has increased only in recent years, different studies demonstrated that these yeasts can influence the aromas of sparkling wines through production of enzymes and metabolites during aging in contact with yeast lees (Ivit and Kemp, 2018). It is of note that lab-scale tests performed in this work on the three strains showed a degree of flocculation which mirrors the genetic prediction with a strong flocculation phenotype for H2 and a gradual decrease in the capacity to flocculate for H4 and H20 (data not shown). This confirms that flocculation capacity, as other physiological properties of oenological interest, is strain-dependent in H. uvarum (Romano et al., 1992, 1997; Ciani and Maccarelli, 1998; Caridi and Ramondino, 1999). In this context, the strain H2 characterized by high flocculation ability and high β-glucosidase activity might be considered as a suitable candidate for the production of traditional method sparkling wine with specific sensory attributes and distinctive characters. Additionally, its high resistance to SO2 might favor the persistence of this non-Saccharomyces strain during fermentation as reported in Grangeteau et al. (2016).
Another genetic trait of biotechnological significance is the enrichment in domains involved in oligopeptides transport: the OPT domain (PF03169) shared by H2 and H4 strains and the PTR2 domain (PF00854) found only in H4 (Tables 5, 6). In general, oligopeptides transport affects both fermentation and the formation of wine aroma by mediating nitrogen utilization, storage and mobilization in yeasts. Particularly, it has been demonstrated that the performance and fitness of S. cerevisiae cells using a higher amount of oligopeptides from grape must is due to metabolic effects (Marsit et al., 2016). The relevance of metabolic control in the organoleptic profile of the wine could be also related to the over-represented domains PLA2_B (PF01735) and Cu_amine_oxidase (PF01179), found in H2 and H4 strains and involved in lipid and aminoacids metabolism, respectively (Tables 5, 6). These identified genetic features reveal a promising potential of these indigenous strains to be applied in fermentation processes and modulation of wine flavor and aroma.
The over-representation of the two domains COX1 (PF00115) and ATP-synt_A (PF00119) encoding for putative proteins involved in oxidative phosphorylation clearly indicates conserved regions deriving from the mitochondrial genome (Tables 5–7). Since the mitochondrial genome of H. uvarum exhibits unique features in terms of organization and molecular architect, this point could become an object for further investigation (Pramateftaki et al., 2006).
Overall these data contribute to increase the catalog of publicly available genomes from H. uvarum strains isolated from natural grape samples and provide a good starting point for unraveling the biodiversity and the biotechnological potential of this non-Saccharomyces yeast species at the genus, species and strain levels in oenological applications (Martin et al., 2018).
Whole Genome Assemblies of Hanseniaspora uvarum strains H4, H20, and H2 have been deposited at GenBank under the following accession numbers: WEHR00000000, WEHS00000000 and WEHT00000000. Complete annotations of PFAM domains and inferred clusters of orthologous genes are available at: https://github.com/matteo14c/supplementary_dataset_Guaragnella_et_al.
All authors significantly contributed to this manuscript. RP, GS, and CM performed the experiments. MC performed the bioinformatics analyses. NG, MC, and AC performed the data curation. NG, AC, PR, and GP designed and supervised the different parts of the study. NG, MC, and AC wrote the first draft of the manuscript. All authors contributed to revisions of the manuscript and approved the final version. GP, AC, and PR contributed to the funding acquisition.
This work was supported by POR Puglia FESR-FSE 2014-2020 INNONETWORK – “Miglioramento nei processi produttivi di alimenti freschi prodotti da farine mediante approcci basati su tecnologie omiche ed informazioni complesse, elaborate da un sistema informativo progettato e sviluppato in ambiente Cloud – OMICS4FOOD,” using facilities provided by ELIXIR-IIB and CNRbiomics (PIR01_00017) and by the project PSR Regione Basilicata 2014–2020 Sottomisura 16.1 GO Vite&Vino PROduttività e Sostenibilità in vITIvinicoltura – (PROSIT)-N. 54250365779.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Laura Marra (IBIOM-CNR) for kind assistance with manuscript preparation.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.03133/full#supplementary-material
FIGURE S1 | Heatmap of the number of species-specific genes associated with selected PFAM domains. The heatmap displays the number of species-specific genes associated with biotechnologically relevant PFAM domains. Dark blue indicates high values. Light blue low values. Unity based normalization is applied to the columns of the heatmap (i.e., the domains) to facilitate the comparison.
TABLE S1 | Annotation of heterozygous sites and availability of raw sequencing data. Full names of all the Hanseniaspora species included in this study are reported in the first column. In the second column, “yes” indicates that annotation of heterozygous sites is provided in the reference assembly, a “no” indicates that this information is not available. Availability of raw sequencing data (yes available, no not available) are reported in the 3rd column.
TABLES S2–S4 | Over and under representation of protein PFAM domains in the 3 H. uvarum genomes assembled in this study. Names of annotated PFAM domains are reported in the first column. The 2nd and 3rd column report, respectively the total number of occurrences of each domain, and the total number of annotated PFAM domains identified for that species. The 4th and the 5th column contain the equivalent figures (occurrences each domain and total number of domains annotated) for the H. uvarum pan-genome. p-values for under and over-representation are reported in the 6th and 7th column respectively.
TABLE S5 | Occurrence of selected PFAM domains in species-specific genes. Domains are indicated in the rows and species in the columns.
Albergaria, H., Francisco, D., Gori, K., Arneborg, N., and Girio, F. (2010). Saccharomyces cerevisiae CCMI 885 secretes peptides that inhibit the growth of some non-Saccharomyces wine-related strains. Appl. Microbiol. Biotechnol. 86, 965–972. doi: 10.1007/s00253-009-2409-2406
Albertin, W., Setati, M. E., Miot-Sertier, C., Mostert, T. T., Colonna-Ceccaldi, B., Coulon, J., et al. (2016). Hanseniaspora uvarum from winemaking environments show spatial and temporal genetic clustering. Front. Microbiol. 6:1569. doi: 10.3389/fmicb.2015.01569
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-80362
Andrade, M. J., Rodriguez, M., Sanchez, B., Aranda, E., and Cordoba, J. J. (2006). DNA typing methods for differentiation of yeasts related to dry-cured meat products. Int. J. Food Microbiol. 107, 48–58. doi: 10.1016/j.ijfoodmicro.2005.08.011
Batista, N. N., Ramos, C. L., Dias, D. R., Pinheiro, A. C., and Schwan, R. F. (2016). The impact of yeast starter cultures on the microbial communities and volatile compounds in cocoa fermentation and the resulting sensory attributes of chocolate. J. Food Sci. Technol. 53, 1101–1110. doi: 10.1007/s13197-015-2132-2135
Belda, I., Ruiz, J., Alastruey-Izquierdo, A., Navascués, E., Marquina, D., and Santos, A. (2016). Unraveling the enzymatic basis of Wine “Flavorome”: a Phylo functional study of wine related yeast species. Front. Microbiol. 7:12. doi: 10.3389/fmicb.2016.00012
Berbegal, C., Spano, G., Tristezza, M., Grieco, F., and Capozzi, V. (2017). Microbial resources and innovation in the wine production sector. Afr. J. Enol. Vitic. 38:2.
Bezerra-Bussoli, C., Baffi, M. A., Gomes, E., and Da-Silva, R. (2013). Yeast diversity isolated from grape musts during spontaneous fermentation from a Brazilian winery. Curr. Microbiol. 67, 356–361. doi: 10.1007/s00284-013-0375-379
Boetzer, M., and Pirovano, W. (2014). SSPACE-LongRead: scaffolding bacterial draft genomes using long read sequence information. BMC Bioinform. 15:211. doi: 10.1186/1471-2105-15-211
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Cadez, N., Raspor, P., de Cock, A. W., Boekhout, T., and Smith, M. T. (2002). Molecular identification and genetic diversity within species of the genera Hanseniaspora and Kloeckera. FEMS Yeast Res. 1, 279–289. doi: 10.1111/j.1567-1364.2002.tb00046.x
Cai, Z., Yang, R., Xiao, H., Qin, X., and Si, L. (2015). Effect of preharvest application of Hanseniaspora uvarum on postharvest diseases in strawberries. Postharvest. Biol. Technol. 100, 52–58. doi: 10.1016/j.postharvbio.2014.09.004
Capece, A., Fiore, C., Maraz, A., and Romano, P. (2005). Molecular and technological approaches to evaluate strain biodiversity in Hanseniaspora uvarum of wine origin. J. Appl. Microbiol. 98, 136–144. doi: 10.1111/j.1365-2672.2004.02434.x
Capece, A., Pietrafesa, R., and Romano, P. (2011). Experimental approach for target selection of wild wine yeasts from spontaneous fermentation of “Inzolia” grapes. World J. Microbiol. Biotechnol. 27, 2775–2783. doi: 10.1007/s11274-011-0753-z
Capece, A., and Romano, P. (2019). “Yeasts and their metabolic impact on wine flavour,” in Yeasts in the Production of Wine, eds P. Romano, M. Ciani, and G. H. Fleet (Berlin: Springer).
Capozzi, V., Berbegala, C., Tufariello, M., Grieco, F., Spano, G., and Grieco, F. (2019). Impact of co-inoculation of Saccharomyces cerevisiae, Hanseniaspora uvarum and Oenococcus oeni autochthonous strains in controlled multi starter grape must fermentations. LWT 109, 241–249. doi: 10.1016/j.lwt.2019.04.045
Caridi, A., and Ramondino, D. (1999). Biodiversità fenotipica in ceppi di Hanseniaspora di origine enologica. Enotecnico 45, 71–74.
Ciani, M., and Maccarelli, F. (1998). Enological properties of non-Saccharomyces yeasts associated with wine-making. World J. Microbiol. Biotechnol. 14, 199–203. doi: 10.1023/A:1008825928354
Comitini, F., and Ciani, M. (2010). The zymocidial activity of Tetrapisispora phaffii in the control of Hanseniaspora uvarum during the early stages of winemaking. Lett. Appl. Microbiol. 50, 50–56. doi: 10.1111/j.1472-765X.2009.02754.x
Esteve-Zarzoso, B., Peris-Toran, M. J., Ramon, D., and Quero, A. (2001). Molecular characterisation of Hanseniaspora species. Antonie Van Leeuwenhoek 80, 85–92. doi: 10.1023/A:1012268931569
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., Mitchell, A. L., et al. (2016). The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285. doi: 10.1093/nar/gkv1344
Fleet, G. H. (2003). Yeast interactions and wine flavour. Int. J. Food Microbiol. 86, 11–22. doi: 10.1016/s0168-1605(03)00245-9
Fleet, G. H., and Heard, G. M. (1993). “Yeasts-growth during fermentation,” in Wine, Microbiology and Biotechnology, ed. G. H. Fleet (Lausanne: Harwood Academic), 27–54.
Garrison, E., and Marth, G. (2012). Haplotype-based variant detection from short-read sequencing. arXiv. [Preprint]. Available at: https://www.cs.umd.edu/class/spring2016/cmsc702/public/FreeBayesDraft2015Jan12.pdf (accessed October 10, 2018).
Giorello, F. M., Berna, L., Greif, G., Camesasca, L., Salzman, V., Medina, K., et al. (2014). Genome Sequence of the native apiculate wine yeast Hanseniaspora vineae T02/19AF. Genom. Announc. 2:e0530-14. doi: 10.1128/genomeA.00530-514
Grangeteau, C., Gerhards, D., von Wallbrunn, C., Alexandre, H., Rousseaux, S., and Guilloux-Benatier, M. (2016). Persistence of two non-Saccharomyces yeasts (Hanseniaspora and Starmerella) in the cellar. Front. Microbiol. 7:268. doi: 10.3389/fmicb.2016.00268
Hittinger, C. T., Rokas, A., Bai, F. Y., Boekhout, T., Goncalves, P., Jeffries, T. W., et al. (2015). Genomics and the making of yeast biodiversity. Curr. Opin. Genet. Dev. 35, 100–109. doi: 10.1016/j.gde.2015.10.008
Illeghems, K., De Vuyst, L., Papalexandratou, Z., and Weckx, S. (2012). Phylogenetic analysis of a spontaneous cocoa bean fermentation metagenome reveals new insights into its bacterial and fungal community diversity. PLoS One 7:e38040. doi: 10.1371/journal.pone.0038040
Ivit, N. N., and Kemp, B. (2018). The impact of non-Saccharomyces yeast on traditional method sparkling Wine. Fermentation 4:73. doi: 10.3390/fermentation4030073
Jolly, N. P., Varela, C., and Pretorius, I. S. (2014). Not your ordinary yeast: non-Saccharomyces yeasts in wine production uncovered. FEMS Yeast Res. 14, 215–237. doi: 10.1111/1567-1364.12111
Kosse, D., Seiler, H., Amann, R., Ludwig, W., and Scherer, S. (1997). Identification of yoghurt-spoiling yeasts with 18S rRNA-targeted oligonucleotide probes. Syst. Appl. Microbiol. 20, 468–480. doi: 10.1016/S0723-2020(97)80016-80011
Lachance, M. A. (1995). Yeast communities in a natural tequila fermentation. Antonie Van Leeuwenhoek 68, 151–160. doi: 10.1007/bf00873100
Langenberg, A. K., Bink, F. J., Wolff, L., Walter, S., von Wallbrunn, C., Grossmann, M., et al. (2017). Glycolytic functions are conserved in the genome of the wine yeast Hanseniaspora uvarum, and Pyruvate Kinase limits its capacity for alcoholic fermentation. Appl. Environ. Microbiol. 83:e001580-17. doi: 10.1128/AEM.01580-1517
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Long, C. A., Wu, Z., and Deng, B. X. (2005). Biological control of Penicillium italicum of citrus and botrytis cinerea of grape by strain 34–9 of Kloeckera apiculata. Eur. Food Res.Technol. 221, 197–201. doi: 10.1007/s00217-005-1199-z
Maechler, M., Rousseeuw, P., Struyf, A., Hubert, M., and Hornik, K. (2019). cluster: Cluster Analysis Basics and Extensions. R package version 2.1.0.
Marika, H. S., Kijne, J. W., and Smit, G. (1993). Cause and control of flocculation in yeast. Trends Biotechnol. 11, 228–232. doi: 10.1016/0167-7799(93)90133-t
Marsit, S., Sanchez, I., Galeote, V., and Dequin, S. (2016). Horizontally acquired oligopeptide transporters favour adaptation of Saccharomyces cerevisiae wine yeast to oenological environment. Environ. Microbiol. 18, 1148–1161. doi: 10.1111/1462-2920.13117
Martin, V., Valera, M. J., Medina, K., Boido, E., and Carrau, F. (2018). Oenological impact of the Hanseniaspora/Kloeckera yeast genus on wines–a review. Fermentation 4:76. doi: 10.3390/fermentation4030076
Masneuf-Pomarede, I., Bely, M., Marullo, P., and Albertin, W. (2015). The genetics of non-conventional wine yeasts: current knowledge and future challenges. Front. Microbiol. 6:1563. doi: 10.3389/fmicb.2015.01563
Masoud, W., Cesar, L. B., Jespersen, L., and Jakobsen, M. (2004). Yeast involved in fermentation of Coffea arabica in East Africa determined by genotyping and by direct denaturating gradient gel electrophoresis. Yeast 21, 549–556. doi: 10.1002/yea.1124
Mendes Ferreira, A., Climaco, M. C., and Mendes Faia, A. (2001). The role of non-Saccharomyces species in releasing glycosidic bound fraction of grape aroma components–a preliminary study. J. Appl. Microbiol. 91, 67–71. doi: 10.1046/j.1365-2672.2001.01348.x
Morais, P. B., Rosa, C. A., Linardi, V. R., Pataro, C., and Maia, A. B. (1997). Short communication: characterization and succession of yeast populations associated with spontaneous fermentations during the production of Brazilian sugar-cane aguardente. World J. Microbiol. Biotechnol. 13, 241–243. doi: 10.1023/A:1018558302062
Owuama, C. I., and Saunders, J. R. (1990). Physiological variants of Saccharomyces cerevisiae and Kloeckera apiculata from palm wine and cashew juice. J. Appl. Bacteriol. 68, 491–494. doi: 10.1111/j.1365-2672.1990.tb02901.x
Petruzzi, L., Capozzi, V., Berbegal, C., Corbo, M. R., Bevilacqua, A., Spano, G., et al. (2017). Microbial Resources and enological significance: opportunities and benefits. Front. Microbiol. 8:995. doi: 10.3389/fmicb.2017.00995
Pramateftaki, P. V., Kouvelis, V. N., Lanaridis, P., and Typas, M. A. (2006). The mitochondrial genome of the wine yeast Hanseniaspora uvarum: a unique genome organization among yeast/fungal counterparts. FEMS Yeast Res. 6, 77–90. doi: 10.1111/j.1567-1364.2005.00018.x
Pretorius, I. S. (2000). Tailoring wine yeast for the new millennium: novel approaches to the ancient art of winemaking. Yeast 16, 675–729. doi: 10.1002/1097-0061
Prjibelski, A. D., Vasilinetc, I., Bankevich, A., Gurevich, A., Krivosheeva, T., Nurk, S., et al. (2014). ExSPAnder: a universal repeat resolver for DNA fragment assembly. Bioinformatics 30, i293–i301. doi: 10.1093/bioinformatics/btu266
Pulvirenti, A., Giudici, P., Zucchi, P., Desalvo, F., Marcazzan, G., and Sabatini, A. G. (2009). Qualitative aspects of spoiled honey. Ingredienti Alimentari 8, 6–14.
R Core Team (2018). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available at: https://www.R-project.org/.
Renard, A., Gomez, di Marco, P., Egea-Cortines, M., and Weiss, J. (2008). Application of whole genome amplification and quantitative PCR for detection and quantification of spoilage yeasts in orange juice. Int. J. Food Microbiol. 126, 195–201. doi: 10.1016/j.ijfoodmicro.2008.05.021
Riley, R., Haridas, S., Wolfe, K. H., Lopes, M. R., Hittinger, C. T., Goker, M., et al. (2016). Comparative genomics of biotechnologically important yeasts. Proc. Natl. Acad. Sci. U.S.A. 113, 9882–9887. doi: 10.1073/pnas.1603941113
Rodriguez, M. E., Lopes, C. A., van Broock, M., Valles, S., Ramon, D., and Caballero, A. C. (2004). Screening and typing of Patagonian wine yeasts for glycosidase activities. J. Appl. Microbiol. 96, 84–95. doi: 10.1046/j.1365-2672.2003.02032.x
Romano, P., Suzzi, G., Comi, G., and Zironi, R. (1992). Higher alcohol and acetic acid production by apiculate wine yeasts. J. Appl. Bacteriol. 73, 126–130. doi: 10.1111/j.1365-2672.1992.tb01698.x
Romano, P., Suzzi, G., Comi, G., Zironi, R., and Maifreni, M. (1997). Glycerol and other fermentation products of apiculate wine yeasts. J. Appl. Microbiol. 82, 615–618. doi: 10.1111/j.1365-2672.1997.tb03592.x
Rossouw, D., Bagheri, B., Setati, M. E., and Bauer, F. F. (2015). Co-flocculation of yeast species, a new mechanism to govern population dynamics in microbial ecosystems. PLoS One 10:e0136249. doi: 10.1371/journal.pone.0136249
Roudil, L., Russo, P., Berbegal, C., Albertin, W., Spano, G., and Capozzi, V. (2019). Non-Saccharomyces Commercial starter cultures: scientific trends, recent patents and innovation in the wine sector. Recent Pat Food Nutr. Agric. doi: 10.2174/2212798410666190131103713 [Epub ahead of print].
Schmitt, M. J., and Neuhausen, F. (1994). Killer toxin-secreting double-stranded RNA mycoviruses in the yeasts Hanseniaspora uvarum and Zygosaccharomyces bailii. J. Virol. 68, 1765–1772.
Seixas, I., Barbosa, C., Mendes-Faia, A., Guldener, U., Tenreiro, R., Mendes-Ferreira, A., et al. (2019). Genome sequence of the non-conventional wine yeast Hanseniaspora guilliermondii UTAD222 unveils relevant traits of this species and of the Hanseniaspora genus in the context of wine fermentation. DNA Res. 26, 67–83. doi: 10.1093/dnares/dsy039
Seixas, I., Barbosa, C., Salazar, S. B., Mendes-Faia, A., Wang, Y., Guldener, U., et al. (2017). Genome sequence of the nonconventional wine yeast Hanseniaspora guilliermondii UTAD222. Genom. Announc. 26, 67–83. doi: 10.1128/genomeA.01515-1516
Soares, E. V. (2011). Flocculation in Saccharomyces cerevisiae: a review. J. Appl. Microbiol. 110, 1–18. doi: 10.1111/j.1365-2672.2010.04897.x
Stanke, M., Tzvetkova, A., and Morgenstern, B. (2006). AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol. 7, 11–18. doi: 10.1186/gb-2006-7-s1-s11
Steenwyk, J. L., Opulente, D. A., Kominek, J., Shen, X. X., Zhou, X., Labella, A. L., et al. (2019). Extensive loss of cell-cycle and DNA repair genes in an ancient lineage of bipolar budding yeasts. PLoS Biol. 17:e3000255. doi: 10.1371/journal.pbio.3000255
Sternes, P. R., Lee, D., Kutyna, D. R., and Borneman, A. R. (2016). Genome Sequences of Three Species of Hanseniaspora Isolated from Spontaneous Wine Fermentations. Genom. Announc. 4:e1287-16. doi: 10.1128/genomeA.01287-1216
Suzzi, G., and Romano, P. (1991). Flocculent phenotypes in wine yeasts. Lett. Appl. Microbiol. 13, 7–10. doi: 10.1111/j.1472-765x.1991.tb00556.x
Suzzi, G., Schirone, M., Sergi, M., Marianella, R. M., Fasoli, G., Aguzzi, I., et al. (2012). Multistarter from organic viticulture for red wine Montepulciano d’Abruzzo production. Front. Microbiol. 3:135. doi: 10.3389/fmicb.2012.00135
Tofalo, R., Patrignani, F., Lanciotti, R., Perpetuini, G., Schirone, M., Di Gianvito, P., et al. (2016a). Aroma profile of montepulciano d’abruzzo wine fermented by single and co-culture starters of autochthonous Saccharomyces and non-Saccharomyces yeasts. Front. Microbiol. 7:610. doi: 10.3389/fmicb.2016.00610
Tofalo, R., Perpetuini, G., Di Gianvito, P., Arfelli, G., Schirone, M., Corsetti, A., et al. (2016b). Characterization of specialized flocculent yeasts to improve sparkling wine fermentation. J. Appl. Microbiol. 120, 1574–1584. doi: 10.1111/jam.13113
Tristezza, M., Tufariello, M., Capozzi, V., Spano, G., Mita, G., and Grieco, F. (2016). The oenological potential of Hanseniaspora uvarum in simultaneous and sequential co-fermentation with Saccharomyces cerevisiae for industrial wine production. Front. Microbiol. 7:670. doi: 10.3389/fmicb.2016.00670
Valles, B. S., Bedrinana, R. P., Tascon, N. F., Simon, A. Q., and Madrera, R. R. (2007). Yeast species associated with the spontaneous fermentation of cider. Food Microbiol. 24, 25–31. doi: 10.1016/j.fm.2006.04.001
Vigentini, I., Barrera Cardenas, S., Valdetara, F., Faccincani, M., Panont, C. A., Picozzi, C., et al. (2017). Use of native yeast strains for in-bottle fermentation to face the uniformity in sparkling wine production. Front. Microbiol. 8:1225. doi: 10.3389/fmicb.2017
Wang, C., Garcia-Fernandez, D., Mas, A., and Esteve-Zarzoso, B. (2015a). Fungal diversity in grape must and wine fermentation assessed by massive sequencing, quantitative PCR and DGGE. Front. Microbiol. 6:1156. doi: 10.3389/fmicb.2015.01156
Wang, C., Mas, A., and Esteve-Zarzoso, B. (2015b). Interaction between Hanseniaspora uvarum and Saccharomyces cerevisiae during alcoholic fermentation. Int. J. Food Microbiol. 206, 67–74. doi: 10.1016/j.ijfoodmicro.2015.04.022
Wiles, A. E. (1950). Studies of some yeasts causing spoilage of draught beers. J. Inst. Brew. 56, 183–193. doi: 10.1002/j.2050-0416.1950.tb01531.x
Keywords: non-Saccharomyces yeasts, Hanseniaspora uvarum, genome sequencing and annotation, Hanseniaspora species, comparative genomics, flocculation
Citation: Guaragnella N, Chiara M, Capece A, Romano P, Pietrafesa R, Siesto G, Manzari C and Pesole G (2020) Genome Sequencing and Comparative Analysis of Three Hanseniaspora uvarum Indigenous Wine Strains Reveal Remarkable Biotechnological Potential. Front. Microbiol. 10:3133. doi: 10.3389/fmicb.2019.03133
Received: 30 July 2019; Accepted: 26 December 2019;
Published: 21 January 2020.
Edited by:
Rosanna Tofalo, University of Teramo, ItalyReviewed by:
Vasileios Englezos, University of Turin, ItalyCopyright © 2020 Guaragnella, Chiara, Capece, Romano, Pietrafesa, Siesto, Manzari and Pesole. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicoletta Guaragnella, bmljb2xldHRhLmd1YXJhZ25lbGxhQHVuaWJhLml0
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.