
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 23 January 2020
Sec. Evolutionary and Genomic Microbiology
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.03124
This article is part of the Research TopicBioinformatics Applications In Molecular Plant PathologyView all 13 articles
 Andrew D. Armitage1,2*
Andrew D. Armitage1,2* Helen M. Cockerton1
Helen M. Cockerton1 Surapareddy Sreenivasaprasad3James Woodhall4Charles R. Lane5
Surapareddy Sreenivasaprasad3James Woodhall4Charles R. Lane5 Richard J. Harrison1
Richard J. Harrison1 John P. Clarkson6
John P. Clarkson6The Alternaria section alternaria (Alternaria alternata species group) represents a diverse group of saprotroph, human allergens, and plant pathogens. Alternaria taxonomy has benefited from recent phylogenetic revision but the basis of differentiation between major phylogenetic clades within the group is not yet understood. Furthermore, genomic resources have been limited for the study of host-specific pathotypes. We report near complete genomes of the apple and Asian pear pathotypes as well as draft assemblies for a further 10 isolates representing Alternaria tenuissima and Alternaria arborescens lineages. These assemblies provide the first insights into differentiation of these taxa as well as allowing the description of effector and non-effector profiles of apple and pear conditionally dispensable chromosomes (CDCs). We define the phylogenetic relationship between the isolates sequenced in this study and a further 23 Alternaria spp. based on available genomes. We determine which of these genomes represent MAT1-1-1 or MAT1-2-1 idiomorphs and designate host-specific pathotypes. We show for the first time that the apple pathotype is polyphyletic, present in both the A. arborescens and A. tenuissima lineages. Furthermore, we profile a wider set of 89 isolates for both mating type idiomorphs and toxin gene markers. Mating-type distribution indicated that gene flow has occurred since the formation of A. tenuissima and A. arborescens lineages. We also developed primers designed to AMT14, a gene from the apple pathotype toxin gene cluster with homologs in all tested pathotypes. These primers allow identification and differentiation of apple, pear, and strawberry pathotypes, providing new tools for pathogen diagnostics.
Species within the genus Alternaria encompass a range of lifestyles, acting as saprotroph, opportunistic pathogens, and host-adapted plant pathogens (Thomma, 2003). Large spored species include Alternaria solani, a major pathogen of potato, whereas small spored taxa include the Alternaria alternata species group (Alternaria sect. alternaria), which are found ubiquitously in the environment acting as saprotrophs and opportunistic necrotrophs. This species group is responsible for opportunistic human infections and a range of host adapted plant diseases.
Taxonomy within this presumed asexual genus has been subject to recent revision (Lawrence et al., 2013). Large spored species can be clearly resolved by standard phylogenetic markers such as ITS and are supported by morphological characters. However, small spored species within the A. alternata species group overlap in morphological characters, possess the same ITS haplotype (Kusaba and Tsuge, 1995), and show low variation in other commonly used barcoding markers (Lawrence et al., 2013; Armitage et al., 2015; Woudenberg et al., 2015). Highly variable phylogenetic markers have provided resolution between groups of isolates that possess morphological patterns typical of descriptions for Alternaria gaisen, Alternaria tenuissima, and Alternaria arborescens (Armitage et al., 2015).
The taxonomy of the species group is complicated by designation of isolates as pathotypes, each able to produce polyketide host-selective toxins (HST) adapted to apple, Asian pear, tangerine, citrus, rough lemon, or tomato (Tsuge et al., 2013). Genes involved in the production of these HSTs are located on conditionally dispensable chromosomes (CDCs) (Hatta et al., 2002). CDCs have been estimated to be 1.05 Mb in the strawberry pathotype (Hatta et al., 2002), 1.1–1.7 Mb in the apple pathotype (Johnson et al., 2001), 1.1–1.9 Mb in the tangerine pathotype (Masunaka et al., 2000, 2005), and 4.1 Mb in the pear pathotype (Tanaka et al., 1999; Tanaka and Tsuge, 2000). These CDCs are understood to have been acquired through horizontal gene transfer and as such, the evolutionary history of CDCs may be distinct from the core genome.
The polyketide synthase genes responsible for the production of the six HSTs are present in clusters. Some genes within these clusters are conserved between pathotypes (Hatta et al., 2002; Miyamoto et al., 2009), while genes are also present within these clusters that are unique to particular pathotypes (Ajiro et al., 2010; Miyamoto et al., 2010). This is reflected in structural similarities between the pear (AKT) and strawberry (AFT) and tangerine (ACTT) toxins with each containing a 9,10-epoxy-8-hydroxy-9-methyl-decatrienoic acid moiety. In contrast, the toxin produced by the apple pathotype (AMT) does not contain this moiety and is primarily cyclic in structure (Tsuge et al., 2013).
Studies making use of bacterial artificial chromosomes (BAC) have led to the sequencing of toxin gene cluster regions from three apple pathotype isolates (GenBank accessions: AB525198, AB525199, AB525200; unpublished). These sequences are 100–130 kb in size and contain 17 genes that are considered to be involved in synthesis of the AMT apple toxin (Harimoto et al., 2007). AMT1, AMT2, AMT3, and AMT4 have been demonstrated to be involved in AMT synthesis, as gene disruption experiments have led to loss of toxin production and pathogenicity (Johnson et al., 2000; Harimoto et al., 2007, 2008). However, experimental evidence has not been provided to show that the remaining 13 AMT genes have a role in toxin production. Four genes present in the CDC for the pear pathotype have been identified and have been named AKT1, AKT2, AKT3, AKTR-1 (Tanaka et al., 1999; Tanaka and Tsuge, 2000; Tsuge et al., 2013) and a further two genes (AKT4, AKTS1) have been reported (Tsuge et al., 2013).
The toxicity of an HST is not restricted to the designated host for that pathotype. All or some of the derivatives of a toxin may induce necrosis on “non-target” host leaves. For example, AMT from the apple pathotype can induce necrosis on the leaves of Asian pear (Kohmoto et al., 1976). Therefore, non-host resistance may be triggered by recognition of non-HST avirulence genes.
Alternaria spp. are of phytosanitary importance, with apple and pear pathotypes subject to quarantine regulations in Europe under Annex IIAI of Directive 2000/29/EC as A. alternata (non-European pathogenic isolates). As such, rapid and accurate diagnostics are required for identification. Where genes on essential chromosomes can be identified that phylogenetically resolve taxa, then these can be used for identification of quarantine pathogens (Bonants et al., 2010; Quaedvlieg et al., 2012). However, regulation and management strategies also need to consider the potential for genetic exchange between species. The Alternaria sect. alternaria are presumed asexual but evidence has been presented for either the presence of sexuality or a recent sexual past. Sexuality or parasexuality provides a mechanism for reshuffling the core genome associated with CDCs of a pathotype. It is currently unknown whether pathotype identification can be based on sequencing of phylogenetic loci, or whether the use of CDC-specific primers is more appropriate. This is of particular importance for the apple and Asian pear pathotypes due to the phytosanitary risk posed by their potential establishment and spread in Europe.
Twelve genomes were sequenced, selected from a collection of isolates whose phylogenetic identity was determined in previous work (Armitage et al., 2015). These 12 isolates were three A. arborescens clade isolates (FERA 675, RGR 97.0013, and RGR 97.0016), four A. tenuissima clade isolates (FERA 648, FERA 1082, FERA 1164, and FERA 24350), three A. tenuissima clade apple pathotype isolates (FERA 635, FERA 743, FERA 1166, and FERA 1177), and one A. gaisen clade pear pathotype isolate (FERA 650).
Apple pathotype isolate FERA 1166 and Asian pear pathotype isolate FERA 650 were sequenced using both Illumina and nanopore MinION sequencing technologies and the remaining 10 isolates were sequenced using Illumina sequencing technology. For both illumina and MinION sequencing, DNA extraction was performed on freeze dried mycelium grown in PDB for 14 days.
High molecular weight DNA was extracted for MinION sequencing using the protocol of Schwessinger and McDonald (2017), scaled down to a starting volume of 2 ml. This was followed by phenol-chloroform purification and size selection to a minimum of 30 kb using a Blue Pippin. The resulting product was concentrated using ampure beads before library preparation was performed using a Rapid Barcoding Sequencing Kit (SQK-RBK001) modified through exclusion of LLB beads. Sequencing was performed on an Oxford Nanopore GridION generating 40 and 34 times coverage of sequence data for isolates FERA 1166 and FERA 650, respectively.
gDNA for illumina sequencing of isolate FERA 1166 was extracted using a modified CTAB protocol (Li et al., 1994). gDNA for illumina sequencing of the eleven other isolates was extracted using a Genelute Plant DNA Miniprep Kit (Sigma) using the manufacturer’s protocol with the following modifications: the volume of lysis solutions (PartA and PartB) were doubled; an RNase digestion step was performed as suggested in the manufacturer’s protocol; twice the volume of precipitation solution was added; elution was performed using elution buffer EB (Qiagen). A 200 bp genomic library was prepared for isolate FERA 1166 using a TrueSeq protocol (TrueSeq Kit, Illumina) and sequenced using 76 bp paired-end reads on an Illumina GA2 Genome Analyzer. Genomic libraries were prepared for the other eleven isolates using a Nextera Sample Preparation Kit (Illumina) and libraries sequenced using a MiSeq Benchtop Analyzer (Illumina) using 250 bp, paired-end reads.
RNAseq was performed to aid training of gene models. mRNA was extracted from isolates FERA 1166 and FERA 650 grown in full strength PDB, 1% PDB, Potato Carrot Broth (PCB), and V8 juice broth (V8B). The protocol for making PCB and V8B was as described in Simmons (2007) for making Potato Carrot Agar and V8 juice agar, with the exception that agar was not added to the recipe. Cultures were grown in conical flasks containing 250 ml of each liquid medium for 14 days. mRNA extraction was performed on freeze dried mycelium using the RNeasy Plant RNA extraction Kit (Qiagen). Concentration and quality of mRNA samples were assessed using a Bioanalyzer (Agilent Technologies). mRNA from the sample grown in 1% PDB for isolate FERA 650 showed evidence of degradation and was not used further. Samples were pooled from growth mediums for each isolate and 200 bp cDNA libraries prepared using a TrueSeq Kit (Illumina). These libraries were sequenced in multiplex on a MiSeq (Illumina) using 200 bp paired-end reads.
De novo genome assembly was performed for all 12 isolates. Assembly for isolate FERA 650 was generated using SMARTdenovo1 (February 26, 2017 github commit), whereas assembly for isolate FERA 1166 was generated by merging a SMARTdenovo assembly with a MinION-Illumina hybrid SPAdes v.3.9.0 assembly using quickmerge v.0.2 (Antipov et al., 2016; Chakraborty et al., 2016). Prior to assembly, adapters were removed from MinION reads using Porechop v.0.1.0 and reads were further trimmed and corrected using Canu v.1.6 (Ruan, 2016; Koren et al., 2017). Following initial assembly, contigs were corrected using MinION reads through ten rounds of Racon (May 29, 2017 github commit) correction (Vaser et al., 2017)and one round of correction using MinION signal information with nanopolish (v0.9.0) (Loman et al., 2015). Final correction was performed through ten rounds of Pilon v.1.17 (Walker et al., 2014) using Illumina sequence data. Assemblies for the ten isolates with Illumina-only data were generated using SPAdes v.3.9.0 (Bankevich et al., 2012). Assembly quality statistics were summarized using Quast v.4.5 (Gurevich et al., 2013). Single copy core Ascomycete genes were identified within the assembly using BUSCO v.3 and used to assess assembly completeness (Simão et al., 2015). RepeatModeler, RepeatMasker, and TransposonPSI were used to identify repetitive and low complexity regions2,3. Visualization of whole genome alignments between FERA 1166 and FERA 650 was performed using circos v.0.6 (Krzywinski et al., 2009), following whole genome alignment using the nucmer tool as part of the MUMmer package v.4.0 (Marçais et al., 2018).
Gene prediction was performed on softmasked genomes using Braker1 v.2 (Hoff et al., 2016), a pipeline for automated training and gene prediction of AUGUSTUS v.3.1 (Stanke and Morgenstern, 2005). Additional gene models were called in intergenic regions using CodingQuarry v.2 (Testa et al., 2015). Braker1 was run using the “fungal” flag and CodingQuarry was run using the “pathogen” flag. RNAseq data generated from FERA 1166 and FERA 650 were aligned to each genome using STAR v.2.5.3a (Dobin et al., 2013), and used in the training of Braker1 and CodingQuarry gene models. Orthology was identified between the 12 predicted proteomes using OrthoMCL v.2.0.9 (Li et al., 2003) with an inflation value of 5.
Draft functional genome annotations were determined for gene models using InterProScan-5.18-57.0 (Jones et al., 2014) and through identifying homology (BLASTP, e-value >1 × 10–100) between predicted proteins and those contained in the March 2018 release of the SwissProt database (Bairoch and Apweiler, 2000). Putative secreted proteins were identified through prediction of signal peptides using SignalP v.4.1 and removing those predicted to contain transmembrane domains using TMHMM v.2.0 (Käll et al., 2004; Krogh et al., 2001). Additional programs were used to provide evidence of effectors and pathogenicity factors. EffectorP v.1.0 was used to screen secreted proteins for characteristics of length, net charge and amino acid content typical of fungal effectors (Sperschneider et al., 2016). Secreted proteins were also screened for carbohydrate active enzymes using HMMER3 (Mistry et al., 2013) and HMM models from the dbCAN database (Huang et al., 2018). DNA binding domains associated with transcription factors (Shelest, 2017) were identified along with two additional fungal-specific transcription factors domains (IPR007219 and IPR021858). Annotated assemblies were submitted as Whole Genome Shotgun projects to DDBJ/ENA/GenBank (Table 1). This included passing assemblies through the NCBI contamination screen, which did not identify presence of contaminant organisms.
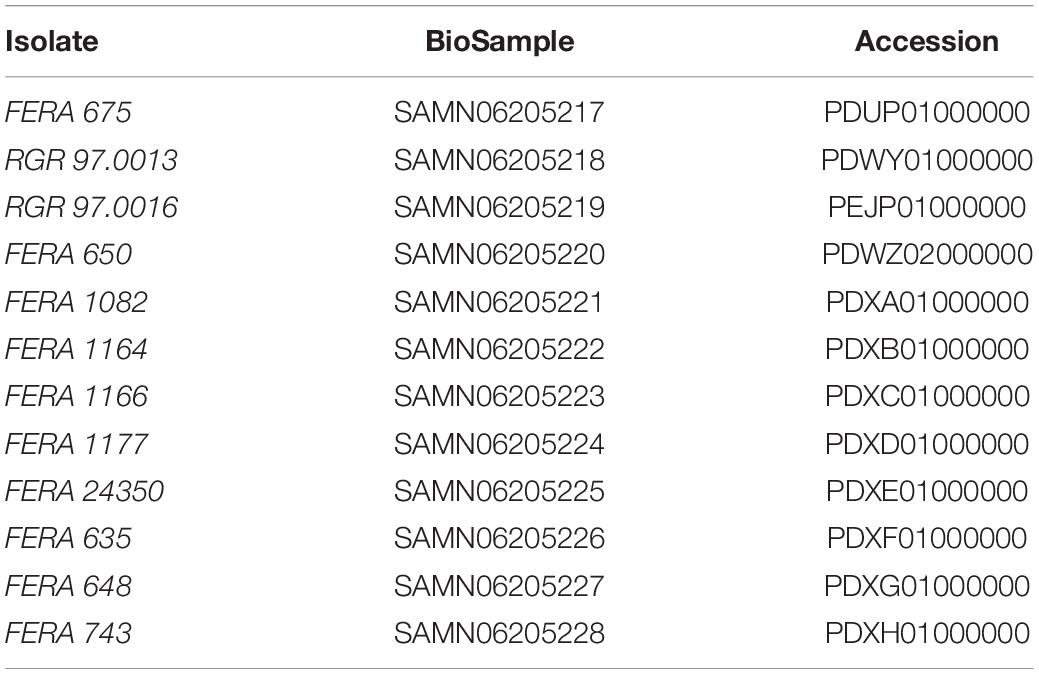
Table 1. NCBI biosample and genome accession numbers of data generated in this study.
BUSCO hits of single copy core ascomycete genes to assemblies were extracted and retained if a single hit was found in all of the 12 sequenced genomes and 23 publicly available Alternaria spp. genomes from the Alternaria genomes database (Dang et al., 2015). Nucleotide sequences from the resulting hits of 500 loci were aligned using MAFFT v6.864b (Katoh and Standley, 2013), before alignments were trimmed using trimAl v.1.4.1 (Capella-Gutiérrez et al., 2009), and trees calculated for each locus using RAxML v.8.1.17 (Liu et al., 2011). The most parsimonious tree from each RAxML run was used to determine a single consensus phylogeny of the 500 loci using ASTRAL v.5.6.1 (Zhang et al., 2018). The resulting tree was visualized using the R package GGtree v.1.12.4 (Yu et al., 2016).
Contigs unique to apple and pear pathotypes were identified through read alignment to assembled genomes. Short read alignment was performed using Bowtie2 (Langmead and Salzberg, 2012), returning a single best alignment for each paired read, whereas long read alignments were performed using Minimap2 v.2.8-r711-dirty (Li, 2018). Read coverage was quantified from these alignments using Samtools (Li et al., 2009).
Sequence data for 40 genes located in A. alternata HST gene clusters were downloaded from GenBank. BLASTn searches were performed for all 40 gene sequences against one another to identify homology between these sequences. Genes were considered homologous where they had >70% identical sequences over the entire query length, and an e-value of 1 × 10–30. tBLASTx was used to search for the presence of these genes in assemblies.
Mating type idiomorphs present in publicly available genomes were identified using BLASTn searches. A wider assessment within 89 characterized Alternaria isolates (Armitage et al., 2015) was undertaken using specific PCR primers (Arie et al., 2000). PCR primers (AAM1-3: 5′-TCCCAAACTCGCAGTG GCAAG-3′; AAM1-3: 5′-GATTACTCTTCTCCGCAGTG-3; M2F: 5′-AAGGCTCCTCGACCGATGAA-3; M2R: 5′-CTGG GAGTATACTTGTAGTC-3) were run in multiplex with PCR reaction mixtures consisting of 10 μl redtaq (REDTaq ReadyMix PCR Reaction Mix, Sigma-Aldrich), 2 μl DNA, 1 μl of each primer (20 μM), and 4 μl purified water (Sigma-Aldrich). PCR reaction conditions comprised of an initial 60 s denaturing step at 94°C followed by 30 cycles of a melting step of 94°C for 30 s, an annealing step at 57°C for 30 s, and an extension step at 72°C for 60 s, these cycles were followed by a final extension step at 72°C for 420 s. MAT1-1-1 or MAT1-2-1 idiomorphs were determined through presence of a 271 or 576 bp product following gel electrophoresis, respectively.
A set of 90 previously characterized isolates was used to further investigate the distribution of pathotypes throughout the A. alternata species group. PCR primers were designed for the amplification of three genes (AMT4, AMT14, AKT3) located within CDC gene clusters involved in toxin synthesis. Primers for AMT4 were designed to amplify apple pathotype isolates, AKT3 to amplify pear pathotype isolates and AMT14 to identify both apple and pear pathotype isolates. These primers were then used to screen isolates for the presence of these genes in 30 cycles of PCR using 0.25 μl Dream taq, 1 μl of 10x PCR buffer, 1 μl of dNTPs, 1 μl of gDNA, 1 μl of each primer (5 μM), and 4.75 μl purified water (Sigma-Aldrich). PCR products were visualized using gel electrophoresis and amplicon identity confirmed through Sanger sequencing. Primers AMT4-EMR-F (5′-CTCGACGACGGTTTGGAGAA-3) and AMT4-EMR-R (5′-TTCCTTCGCATCAATGCCCT-3) were used for amplification of AMT4. Primers AKT3-EMR-F (5′-GCAATGGACGCAGACGATTC-3) and AKT3-EMR-R (5′- CTTGGAAGCCAGGCCAACTA-3) were used for amplification of AKT3. Primers AMT14-EMR-F (5′-TTTCTGCAACGGCG KCGCTT-3) and AMT14-EMR-R (5′-TGAGGAGTYAGACCR GRCGC-3) were used for amplification of AMT14. PCR reaction conditions were the same as described above for mating type loci, but with annealing performed at 66°C for all primer pairs.
Pathogenicity assays were performed on apple cv. Spartan and cv. Bramley’s seedling to determine differences in isolate virulence between A. tenuissima isolates possessing the apple pathotype CDC (FERA 635, FERA 743 or FERA 1166) and non-pathotype isolates lacking the CDC (FERA 648, FERA 1082 or FERA 1164). Briefly, leaves were inoculated with 10 μl of 1 × 105 spore suspensions at six points and the number of leaf spots counted at 14 days post inoculation. One isolate was infected per leaf, with 10 replicates per cultivar. Binomial regression using a generalized linear model (GLM) was used to analyse the number of resulting lesions per leaf.
Unfolded adult apple leaves, less than 10 cm in length were cut from young (less than 12 months old) apple cv. Spartan trees or cv. Bramley’s seedling trees. These were quality-checked to ensure that they were healthy and free from disease. Leaves were grouped by similar size and age and organized into ten experimental replicates of nine leaves. Leaves placed in clear plastic containers, with the abaxial leaf surface facing upwards. The base of these boxes was lined with two sheets of paper towel, and wetted with 50 ml of sterile distilled water (SDW). The cultivars were assessed in two independent experiments.
Spore suspensions were made by growing A. alternata isolates on 1% PDA plates for 4 weeks at 23°C before flooding the plate with 2 ml of SDW, scraping the plate with a disposable L-shaped spreader. Each leaf was inoculated with 10 ml of 1 × 105 spores ml–1 A. alternata spore suspension or 10 ml of sterile-distilled water at six points on the abaxial leaf surface. Of the nine leaves in each box, three leaves were inoculated with a spore suspensions from isolates carrying apple pathotype CDC, three leaves were inoculated with non-pathotype isolates lacking the CDC, and three leaves were inoculated with SDW. Following inoculation, each container was sealed and placed in plastic bags to prevent moisture loss. Boxes were then kept at 23°C with a 12 h light/12 h dark cycle.
Assemblies using nanopore long-read sequence data for the apple pathotype isolate FERA 1166, and pear pathotype isolate FERA 650 were highly contiguous, with the former totaling 35.7 Mb in 22 contigs and the latter totaling 34.3 Mb in 27 contigs (Table 2). Whole genome alignments of these assemblies to the 10 chromosomes of A. solani showed an overall macrosynteny between genomes (Figure 1), but with structural rearrangement of apple pathotype chromosomes in comparison to A. solani chromosomes 1 and 10. The Asian pear pathotype had distinct structural rearrangements in comparison to A. solani, chromosomes 1 and 2 (Figure 1). Scaffolded contigs of FERA 1166 spanned the entire length of A. solani chromosomes 2, 3, 6, 8, and 9 and chromosomes 4, 5, 6, and 10 for FERA 650 (Figure 1). Interestingly, sites of major structural rearrangements within A. solani chromosome 1 were flanked by telomere-like TTAGGG sequences.
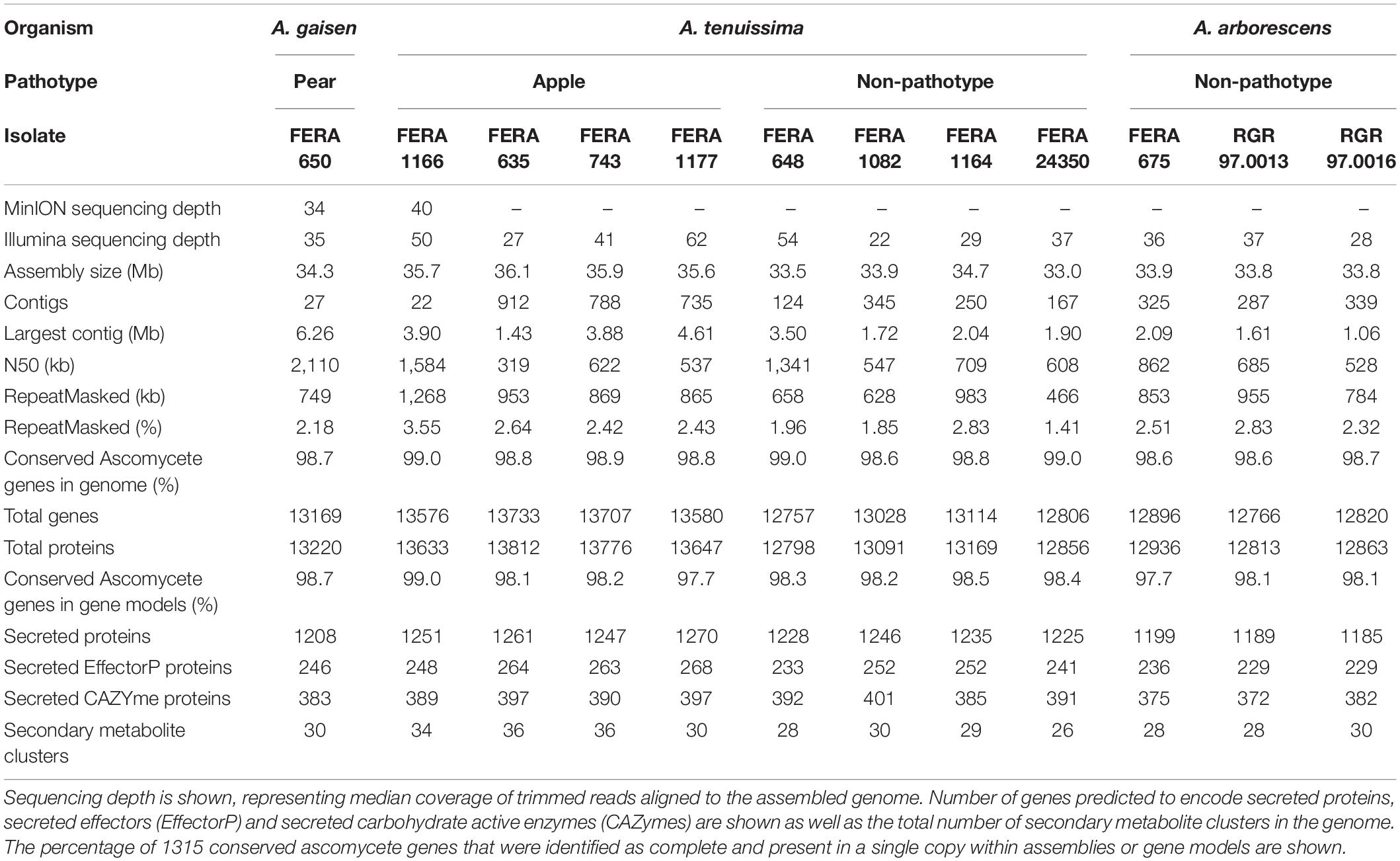
Table 2. Sequence data, assembly and gene prediction statistics for genomes from three A. alternata species group lineages, including apple and Asian pear pathotype isolates.
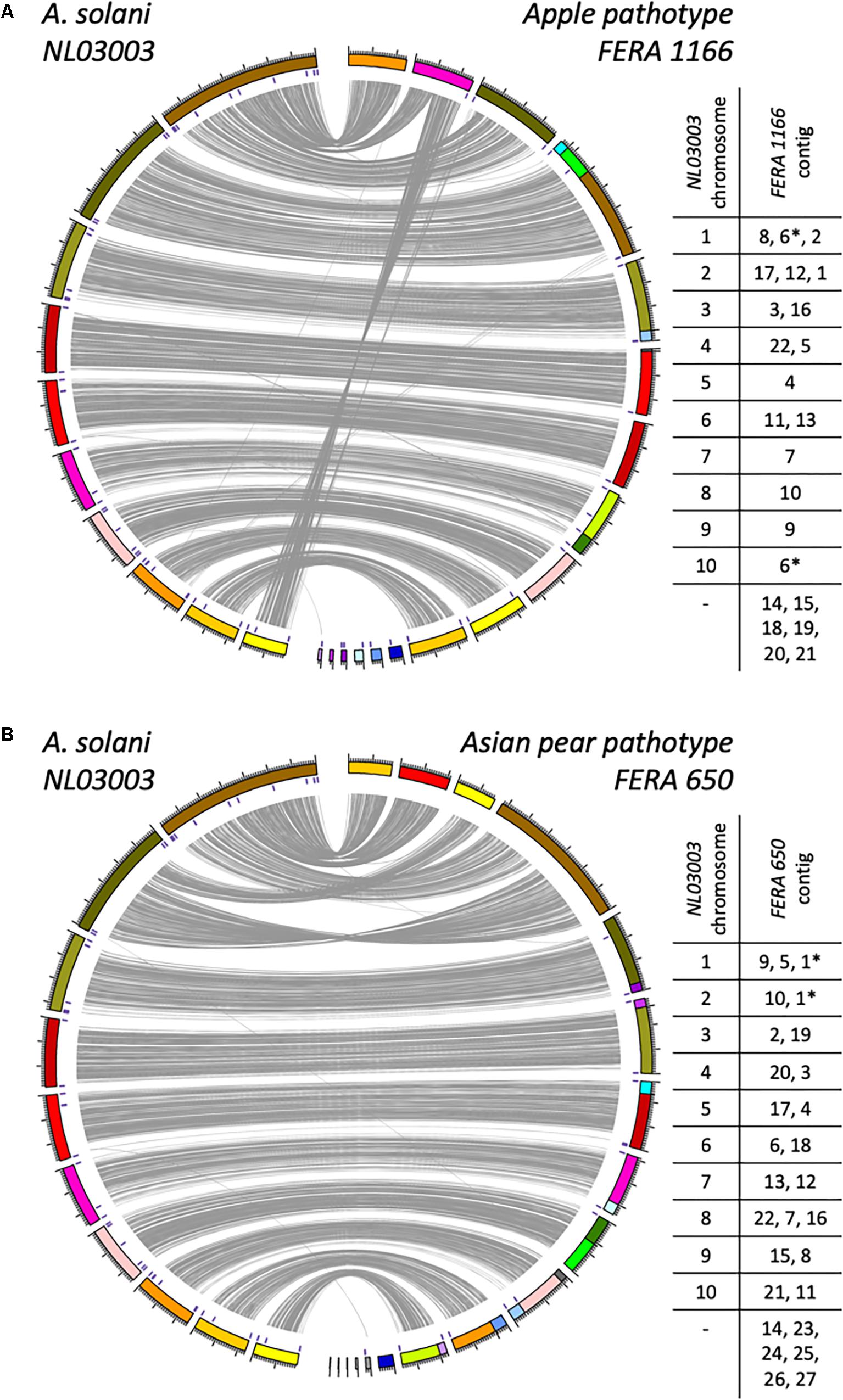
Figure 1. Genome alignment between the reference A. solani genome and long-read assemblies of A. alternata apple (A) and Asian pear (B) pathotypes. Links are shown between aligned regions. Locations of telomere repeat sequences are marked within assembled contigs. Contig order in reference to A. solani chromosomes is summarized, with those contigs displaying evidence of structural rearrangement marked with an asterisk.
Genome assembly of 10 Illumina sequenced isolates yielded assemblies of a similar total size to MinION assemblies (33.9–36.1 Mb) but fragmented into 167-912 contigs. Assembled genomes were repeat sparse, with 1.41–2.83% of genomes repeat masked (Table 2). Genome assemblies of A. arborescens isolates (33.8–33.9 Mb), were of similar total size to non-pathotype A. tenuissima isolates and had similar repetitive content (2.51–2.83 and 1.41–2.83%, respectively). Despite this, identification of transposon families in both genomes showed expansion of DDE (T5df = 5.36, P > 0.01) and gypsy (T5df = 6.35, P > 0.01) families in A. arborescens genomes (Figure 2).
Figure 2. Distinct DDE and gypsy family transposon families between genomes of A. arborescens (arb) and A. tenuissima (ten) clade isolates. Numbers of identified transposons are also shown for hAT, TY1 copia, mariner, cacta, LINE, MuDR/Mu transposases, helitrons, and the Ant1-like mariner elements.
The relationship between the 12 sequenced isolates and 23 Alternaria spp. with publicly available genomes was investigated through phylogenetic analysis of 500 shared core ascomycete genes. A. pori and A. destruens genomes were excluded from the analysis due to low numbers of complete single copy ascomycete genes being found in their assemblies (Supplementary Table S1). The 12 sequenced isolates were distributed throughout A. gaisen, A. tenuissima, and A. arborescens clades (Figure 3). The resulting phylogeny (Figure 3), formed the basis for later assessment of CDC presence and mating type distribution among newly sequenced and publicly available genomes, as discussed below.
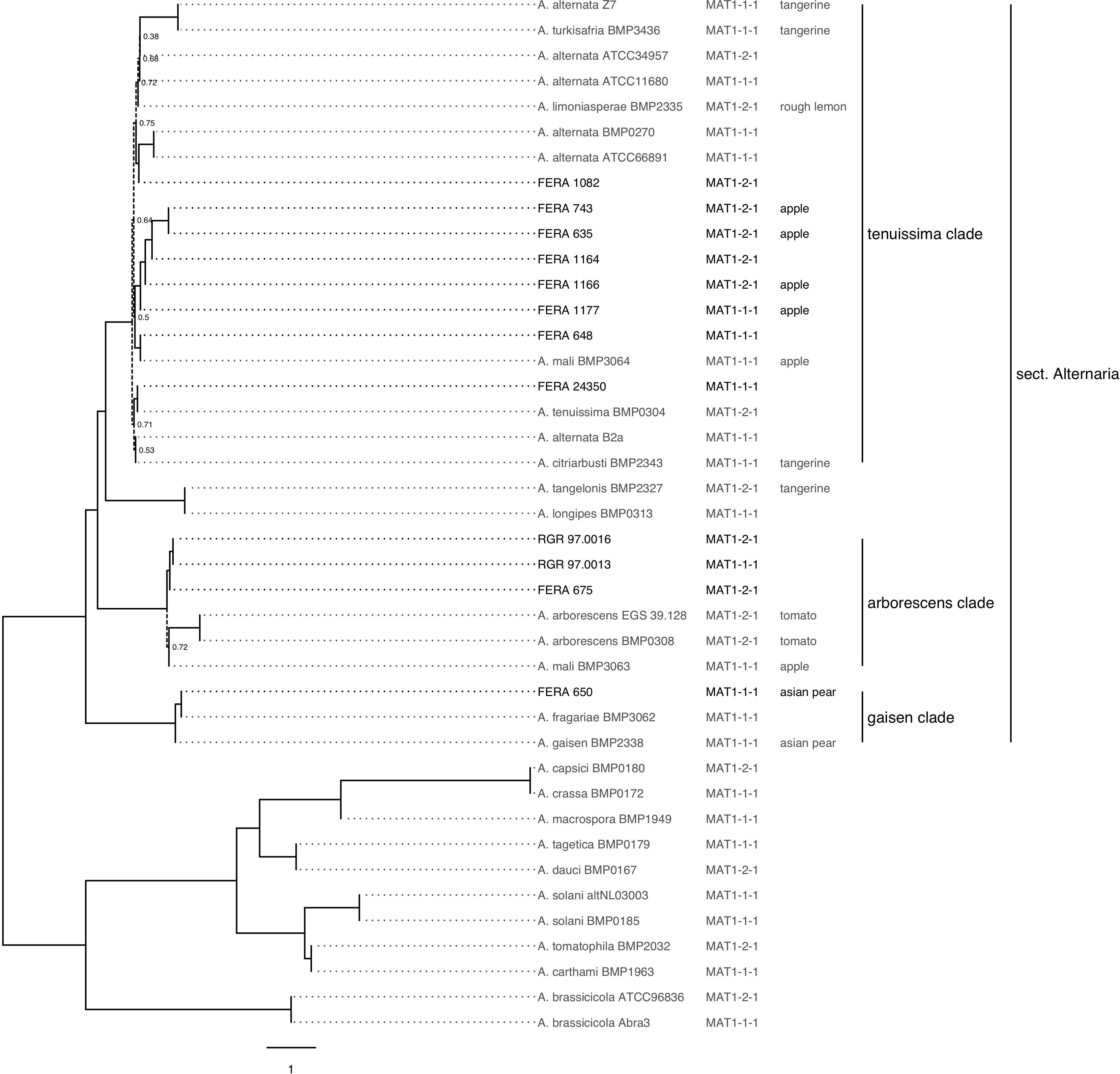
Figure 3. Phylogeny of sequenced and publicly available Alternaria spp. genomes. Maximum parsimony consensus phylogeny of 500 conserved single copy loci. Dotted lines show branches with support from <80% of trees. Mating type idiomorphs MAT1-1-1 and MAT1-2-1 show distribution throughout the phylogeny. Isolates pathotype is labeled following identification of genes involved in synthesis of apple, pear, strawberry, tangerine, rough lemon, and tomato toxins.
Gene prediction resulted in 12757–13733 genes from the our assemblies (Table 2), with significantly more genes observed in the apple pathotype isolates than in A. tenuissima clade non-pathotype isolates (P > 0.01, F2,8df = 51.19). BUSCO analysis identified that gene models included over 97% of the single copy conserved ascomycete genes, indicating well trained gene models. Apple pathotype isolates possessed greater numbers of secondary metabolite clusters (P > 0.01, F2,8df = 8.96) and secreted genes (P > 0.01, F2,8df = 44.21) than non-pathotype A. tenuissima isolates, indicating that CDCs contain additional secreted effectors. Non-pathotype A. tenuissima clade isolates were found to possess greater numbers of genes encoding secreted proteins than A. arborescens isolates (P > 0.01, F2,8df = 44.21), including secreted CAZYmes (P > 0.01, F2,8df = 9.83). The basis of differentiation between these taxa was investigated further.
Orthology analysis was performed upon the combined set of 158,280 total proteins from the 12 sequenced isolates. In total, 99.2% of proteins clustered into 14,187 orthogroups. Of these, 10,669 orthogroups were shared between all isolates, with 10,016 consisting of a single gene from each isolate. This analysis allowed the identification of 239 orthogroups that were either unique to A. arborescens isolates or expanded in comparison to non-pathotype A. tenuissima isolates.
Expanded and unique genes to A. arborescens isolates was further investigated using FERA 675 (Supplementary Table S2). Genes involved in reproductive isolation were in this set, including 21 of the 148 heterokaryon incompatibility (HET) loci from FERA 675. CAZymes were also identified within this set, three of which showed presence of chitin binding activity and the other three having roles in xylan or pectin degradation. In total, 25 genes encoding secreted proteins were within this set, secreted proteins with pathogenicity-associated functional annotations included a lipase, a chloroperoxidase, an aerolysin-like toxin, a serine protease and an aspartic peptidase. A further six secreted genes had an effector-like structure by EffectorP but no further functional annotations. Furthermore, one gene from this set was predicted to encode a fungal-specific transcription factor unique to A. arborescens isolates.
Further to the identification of genes unique or expanded in A. arborescens, 220 orthogroups were identified as unique or expanded in the A. tenuissima. These orthogroups were further investigated using isolate FERA 648 (Supplementary Table S2). This set also contained genes involved in reproductive isolation, including nine of the 153 from FERA 648. CAZymes within the set included two chitin binding proteins, indicating a divergence of LysM effectors between A. tenuissima and A. arborescens lineages. The five additional CAZymes in this set represented distinct families from those expanded/unique in A. arborescens, including carboxylesterases, chitooligosaccharide oxidase, and sialidase. In total, 18 proteins from this set were predicted as secreted, including proteins with cupin protein domains, leucine rich-repeats, astacin family peptidase domains and with four predicted to have effector-like structures but no further annotations. A. tenuissima isolates had their own complement of transcription factors, represented by four genes within this set.
Alignment of Illumina reads to the apple and Asian pear pathotype MinION reference assemblies identified variable presence of some contigs, identifying these as contigs representing CDCs (CDC contigs). Six contigs totaling 1.87 Mb were designated as CDCs in the apple pathotype reference (Table 3) and four contigs totaling 1.47 Mb designated as CDCs in the pear pathotype reference (Table 4). Two A. tenuissima clade non-pathotype isolates (FERA 1082, FERA 1164) were noted to possess apple pathotype CDC contigs 14 and 19 totaling 0.78 Mb (Table 3).
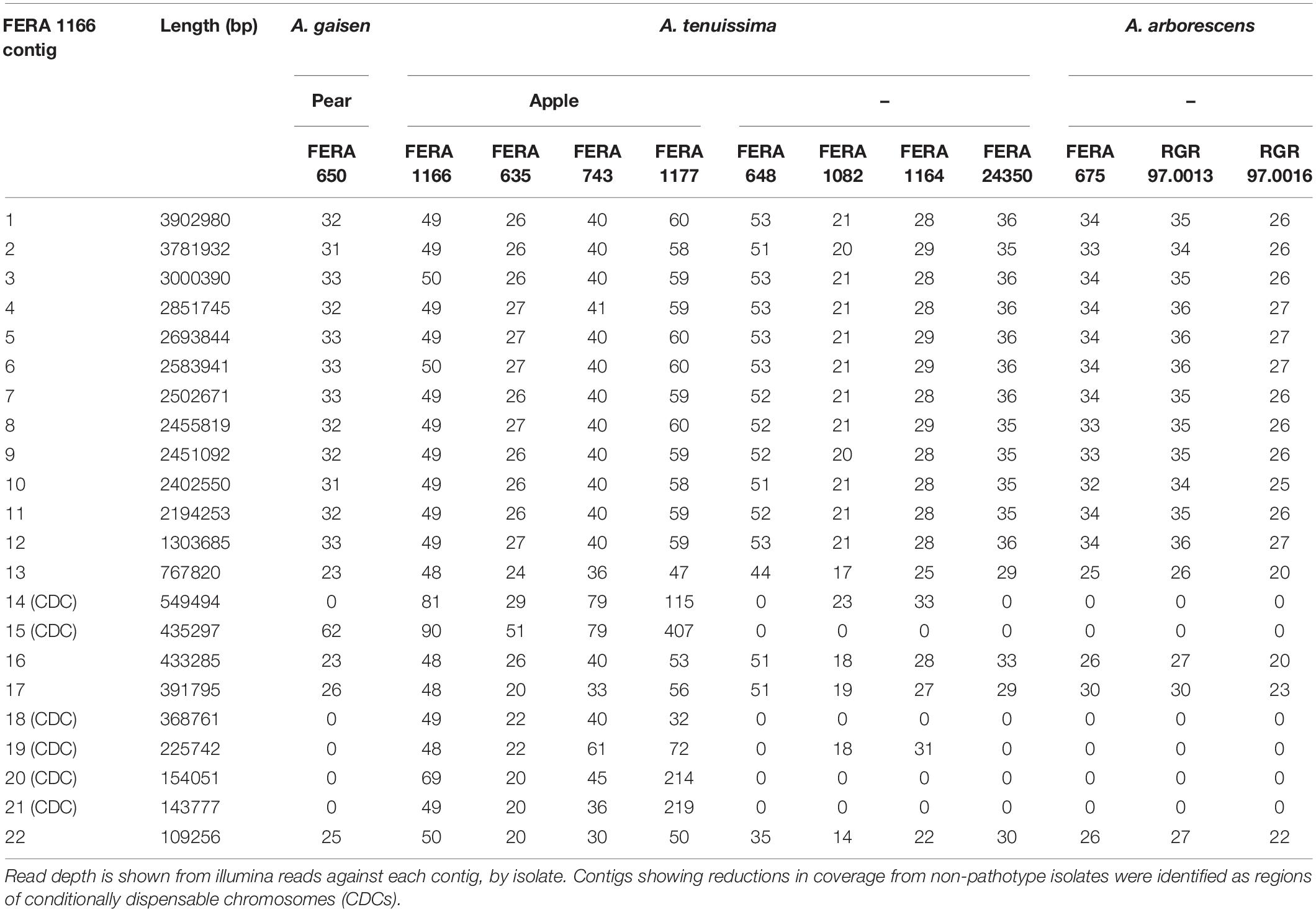
Table 3. Identification of CDC regions in the A. alternata apple pathotype reference genome.
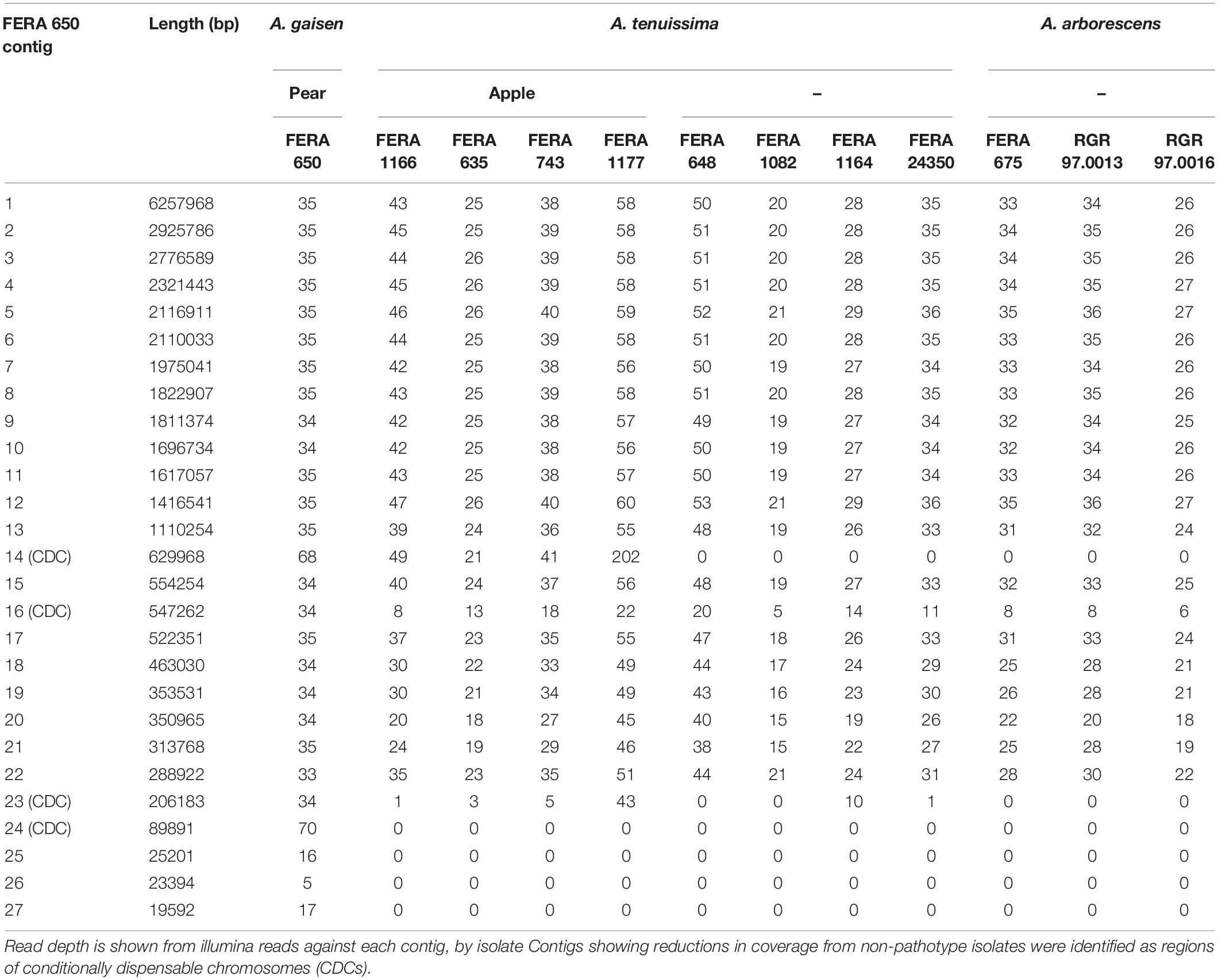
Table 4. Identification of CDC regions in the A. alternata Asian pear pathotype reference genome.
Read alignments showed that CDC contigs were present in multiple copies within A. alternata pathotype isolates. FERA 1166 Illumina reads aligned to its own assembly showed two-fold coverage over contigs 14, 15, 20, and 21 in comparison to core contigs (Table 3). This was more pronounced in isolate FERA 1177 that had between two- and eight-fold coverage of these contigs. The same was observed in pear pathotype CDC regions, with contigs 14 and 24 in isolate FERA 650 showing two-fold coverage from Illumina reads in comparison to core contigs (Table 4).
Homologs to 15 of the 17 AMT cluster genes were located on contigs 20 and 21 in the apple pathotype reference genome (e-value < 1 × 10–30, > 70% query alignment), confirming them as CDC-regions (Table 5). Of the remaining two genes, AMT11 had low-confidence BLAST homologs on contigs 18 and 21 (e-value < 1 × 10–30) whereas the best BLAST hit of AMT15 was located on contig 18 (e-value < 1 × 10–30). Duplication of toxin gene regions was observed between CDC contigs, with contig 20 carrying homologs to 16 toxin genes, but with contig 21 also carrying the AMT1 to AMT12 section of the cluster (Table 5). The three other apple pathotype isolates (FERA 635, FERA 743 and FERA 1177) also showed presence of 15 of the 17 AMT genes (e-value < 1 × 10–30, >70% query alignment), and with some AMT genes present in multiple copies within the genome indicating that the AMT toxin region has also been duplicated in these isolates.
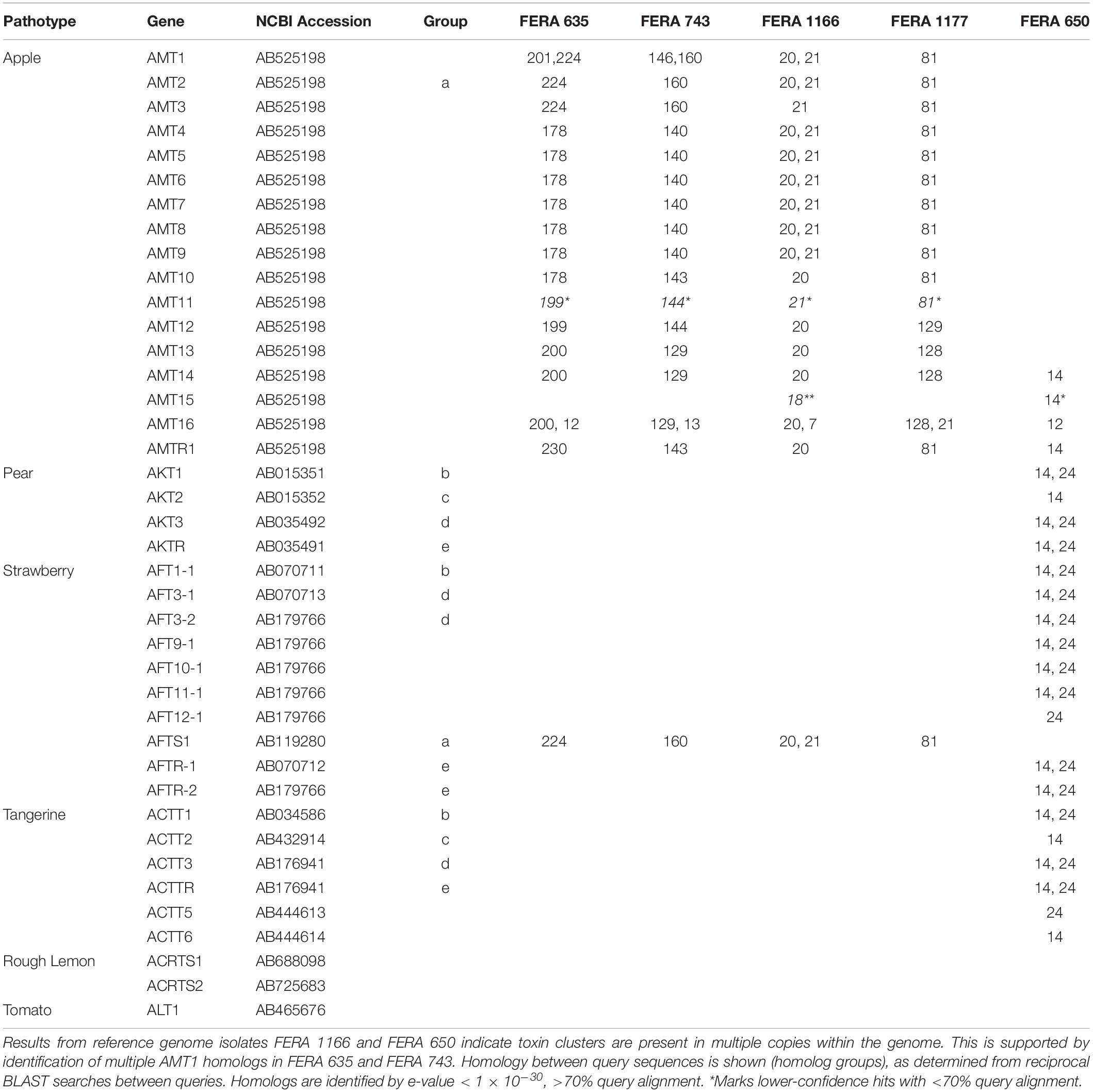
Table 5. Genomic location (contig number) of homologs to genes from apple, pear, strawberry, tangerine, rough lemon, and tomato toxin gene clusters.
The Asian pear pathotype was also found to carry toxin gene clusters in multiple copies, with homologs to the four AKT cluster genes present on contig 14 of the FERA 650 assembly (e-value <1 × 10–30, >70% query alignment), with three of these also present on contig 24 (e-value <1 × 10–30, two with >70% query alignment). BLAST hit results from AKT genes were supported by their homologs from strawberry and tangerine pathotypes also found in these regions (Table 5). The pear pathotype genome was also found to contain additional homologs from apple (AMT14), strawberry (AFT9-1, AFT10-1, AFT11-1, and AFT12-1) and citrus (ACTT5 and ACTT6) located on CDC contigs 14 and 24 (Table 5).
A total of 624 proteins were encoded on the six contigs designated as CDCs in the reference apple pathotype genome, with 502 proteins encoded on the four Asian pear pathotype CDC contigs (Supplementary Table S3). We further investigated the gene complements of these regions.
Approximately a quarter of gene models on apple pathotype CDC contigs were involved in secondary metabolism, with 153 genes present in six secondary metabolite gene clusters. This included AMT toxin gene homologs on contigs 20 and 21, which were located within NRPS secondary metabolite gene clusters. Three other secondary metabolite clusters were located on CDC contigs with two of these involved in the production of T1PKS secondary metabolites and the third with unknown function. A further two secondary metabolite clusters were located on contig 14 shared with two non-pathotype isolates, one of which is involved in the production of a T1PKS. The pear pathotype also carried 153 genes in secondary metabolite gene clusters. These 30% of CDC genes were located in four clusters, with the AKT toxin genes in T1PKS clusters of contigs 14 and 24. A second cluster was present on contig 14 with unknown function and a T1PKS cluster was present on contig 16.
Approximately 5% of the genes on apple CDC contigs encoded secreted proteins, with 32 in isolate FERA 1166 many of which had potential effector functions with six designated as CAZymes and 12 testing positive by EffectorP. Similarly, a total of 41 secreted proteins were predicted on the CDC regions of the Asian pear pathotype, with eight of these designated as secreted CAZymes and 13 testing positive by EffectorP. Further investigation into the 32 secreted proteins from the apple pathotype identified three CAZYmes from the chitin-active AA11 family, two from the cellulose-active GH61 family and one cellulose-active GH3 family protein. Six of the 13 EffectorP proteins also had domains identifiable by interproscan: four carried NTF2-like domains, which are envelope proteins facilitating protein transport into the nucleus; one was a fungal hydrophobin protein; one was a member of an panther superfamily PTHR40845 that shares structural similarity with proteins from the plant pathogens Phaeosphaeria nodorum, Sclerotinia sclerotiorum, and Ustilago maydis. Of the 38 secreted proteins identified from the pear pathotype, two CAZYmes were also identified from the chitin-active AA11 family, two from the AA3 family with single proteins from GH5, CBM67 and AA7 families. Ten of the twelve secreted EffectorP proteins had no functional information as predicted by interproscan, with the other two identified as carrying WSC domains IPR002889, which are cysteine-rich domains involved carbohydrate binding. CDCs may also play important roles in transcriptional regulation with 29 putative transcription factors identified in the apple pathotype CDC contigs (4.6% CDC genes) and 35 identified in pear pathotype CDC contigs (7.0% CDC genes).
The evolutionary relationship between A. alternata pathotypes sequenced in this study and publicly available genomes was analyzed by the core gene phylogeny (Figure 2). We identified four isolates as tangerine pathotypes (Z7, BMP2343, BMP2327, BMP3436) two as tomato pathotypes (BMP0308, EGS39-128), one Asian pear pathotype (MBP2338), one rough-lemon (BMP2335) and two apple pathotypes (BMP3063, BMP3064) through searches for genes from HST-gene clusters (Supplementary Table S4). When plotted on the genome phylogeny, we found the apple and tangerine pathotypes to be polyphyletic (Figure 2). Five of the six sequenced apple pathotype isolates were located in the A. tenuissima clade and one in the A. arborescens clade, whereas the tangerine pathotype was present in both the A. tenuissima clade and in the A. tangelonis/A. longipes clade.
PCR primers for three loci (AMT4, AKT3, and AMT14) were designed to identify the distribution of pathotypic isolates through the A. alternata species group and were screened against a set of 89 previously characterized isolates (Figure 4). Five isolates tested positive for the presence of AMT4, each of which was from the A. tenuissima clade (FERA 635, FERA 743, FERA 1166, FERA 1177). Five isolates tested positive for the presence of AKT3, including the three isolates from Asian pear in the A. gaisen clade and a further two isolates from the A. tenuissima clade that were from strawberry. Sequencing of the AKT3 amplicons from the two isolates ex. strawberry identified them as the AFT3-2 ortholog of AKT3, showing that these isolates were strawberry pathotypes rather than pear pathotypes. Sequencing of PCR products from the other isolates confirmed them to be apple or pear pathotypes as expected. All of the isolates testing positive for AMT4 or AKT3 also tested positive for AMT14, indicating its suitability as a target gene for identification of a range of pathotypes.

Figure 4. Presence/absence of toxin and mating type genes for 89 Alternaria isolates. Results are plotted onto the 5-gene phylogeny of Armitage et al. (2015). MAT1-1-1 and MAT1-2-1 mating type idiomorphs are designated.
Presence of apple pathotype CDCs was confirmed to be associated with pathogenicity through detached apple leaf assays. Apple pathotype isolates showed significantly greater numbers of necrotic lesions when inoculated onto cv. Spartan (F72df = 100.64) and cv. Bramley’s Seedling (F72df = 69.64) leaves than non-pathotype A. tenuissima isolates (Figure 5).
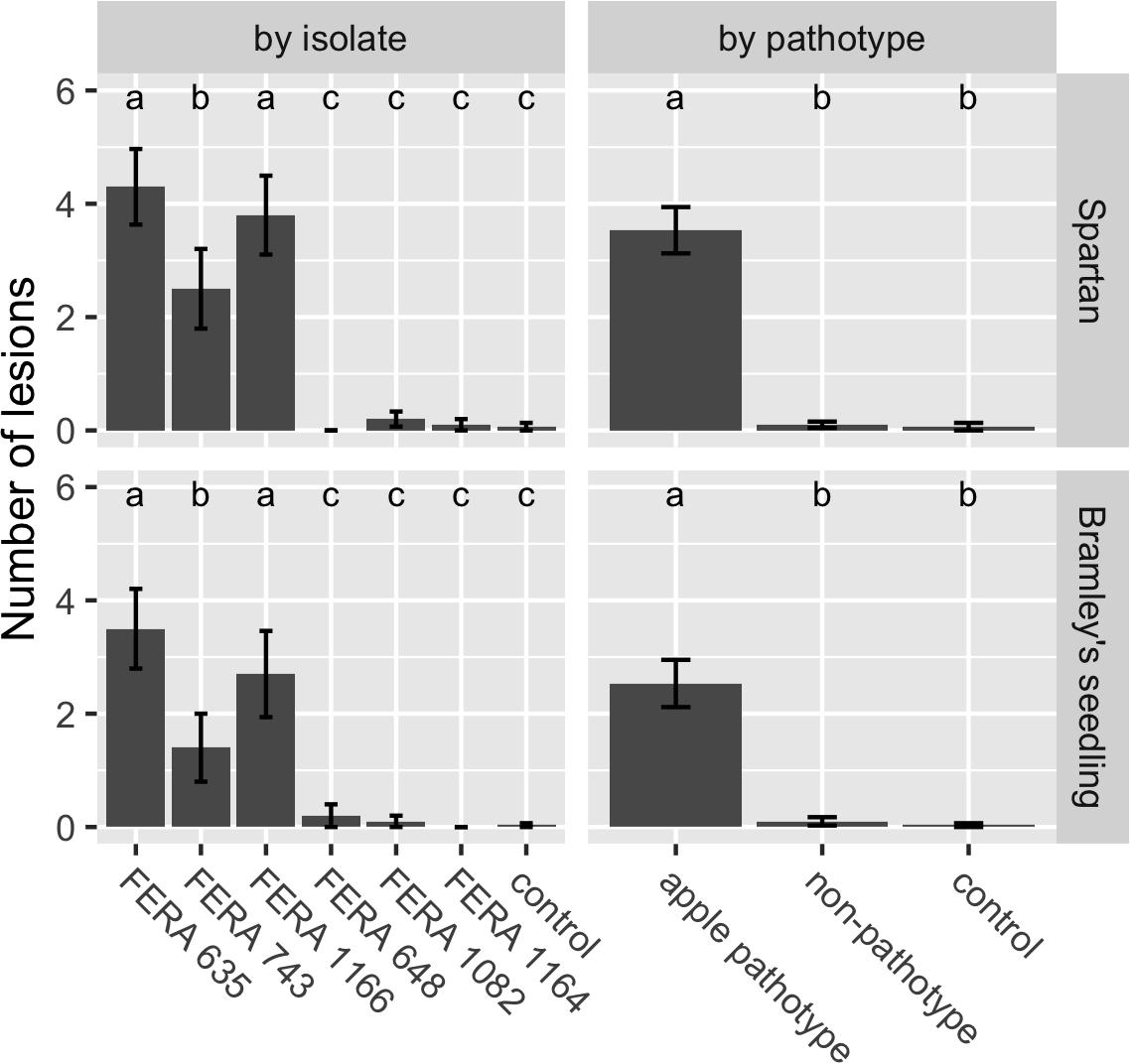
Figure 5. Number of Alternaria lesions per apple leaf at 14 dpi for treatments in virulence assays on cv. Spartan or cv. Bramley’s Seedling leaves. Apple pathotype isolates (FERA 635, FERA 743, and FERA 1166) cause significant disease symptoms in comparison to control leaves, in contrast to non-pathotype isolates (FERA 648, FERA 1082, and FERA 1164). Number of lesions (±SE) are shown with significance at P < 0.05, as determined from a GLM at the isolate and pathotype level.
Of the 12 sequenced isolates, BLAST searches identified five as carrying the MAT1-1-1 idiomorph and seven as carrying MAT1-2-1. Both idiomorphs showed distribution throughout the A. alternata genome phylogeny (Figure 2). These results were supported by PCR assays identifying the mating type of 89 previously characterized isolates (Figure 4). Idiomorphs did not deviate from a 1:1 ratio within A. tenuissima (21 MAT1-1-1: 23 MAT1-2-1; χ2 = 0.09, 1df, P > 0.05) or A. arborescens clades (18 MAT1-1-1: 24 MAT1-2-1; χ2 = 0.86; 1df; P > 0.05), as expected under a random mating population. All three of the A. gaisen clade isolates carried the MAT1-2-1 idiomorph.
This work builds upon the current genomic resources available for Alternaria, including the A. brassicicola and A. solani genomes (Belmas et al., 2018; Wolters et al., 2018), A. alternata from onion (Bihon et al., 2016) the additional 25 Alternaria spp. genomes available on the Alternaria Genomes Database (Dang et al., 2015) as well as recent genomes for other pathotype and non-pathotype A. alternata (Hou et al., 2016; Wang et al., 2016; Nguyen et al., 2016). Of the previously sequenced genomes, A. solani, the citrus pathotype and a non-pathotype A. alternata isolate have benefited from long read sequencing technology with each comprising less than 30 contigs (Wolters et al., 2018; Wang et al., 2016; Nguyen et al., 2016). Total genome sizes in this study (33–36 Mb) were in line with previous estimates for A. alternata, with the tomato pathotype also previously assembled into 34 Mb (Hu et al., 2012). Synteny analysis of our two reference genomes against the chromosome-level A. solani genome revealed structural differences for chromosomes 1 and 10 in the apple pathotype and for chromosomes 1 and 2 in the pear pathotype. These structural differences may represent distinct traits between clades of the A. alternata species group, and may represent a barrier to genetic exchange involved in the divergence of A. gaisen and A. tenuissima lineages. The number of essential chromosomes in our reference genomes is in line with previous findings in A. alternata (Kodama et al., 1998), with 9-11 core.
Species designations within the species group have been subject to recent revision (Woudenberg et al., 2015; Lawrence et al., 2013; Armitage et al., 2015) leading to potential confusion when selecting isolates for study. For example, the available Alternaria fragariae genome (Dang et al., 2015), did not represent a strawberry pathotype isolate and was located in the A. gaisen clade. As such, the phylogenetic context for sequenced Alternaria genomes described in this study, along with pathotype identification provides a useful framework for isolate selection in future work.
A 1:1 ratio of MAT loci was observed within A. arborescens and A. tenuissima clades. This supports previous identification of both idiomorphs within A. alternata, Alternaria brassicae, and A. brassicicola (Berbee et al., 2003). Furthermore, presence of both MAT idiomorphs within apple pathotype isolates indicates that genetic exchange (sexuality or parasexuality) has occurred since the evolution of CDCs, providing a mechanism of transfer of CDCs. Evidence for cryptic sexuality or a parasexual cycle has been previously presented for the citrus pathotype of Alternaria alternata (Stewart et al., 2013). We also show that some recent or historic genetic exchange has occurred between A. tenuissima and A. arborescens clades, with both apple and tangerine pathotypes exhibiting a polyphyletic distribution throughout the phylogeny.
Toxin genes have been proposed to be present in multiple copies within A. sect. alternaria pathotype genomes with AMT2 proposed to be present in at least three copies in the apple pathotype CDC (Harimoto et al., 2008), and multiple copies of AKTR and AKT3 in the pear pathotype (Tanaka et al., 1999; Tanaka and Tsuge, 2000). Through read mapping we demonstrated that this is the case. Furthermore, we show that toxin gene clusters are present on multiple contigs, with differences in the gene complements between these clusters. At this stage, it is unclear whether these different clusters are responsible for the production of the variant R-groups previously characterized in AMT or AKT toxins (Nakashima et al., 1985; Harimoto et al., 2007). Differences were also noted between non-pathotype isolates from the A. tenuissima clade in the presence/absence of contigs 14 and 19, representing a total of 775 kb. Chromosomal loss has been reported in the apple pathotype (Johnson et al., 2001), and it is not clear if this represents chromosomal instability in culture or additional dispensable chromosomes within A. tenuissima clade isolates.
It is now clear that genes on essential chromosomes do not provide reliable targets for identification of different pathotypes and hence loci located directly on CDCs should be used. We found AMT14 homologs to be present in all pathotype genomes and designed primers to this region. These demonstrated specificity to apple, pear and strawberry pathotypes within a set of 86 Alternaria isolates. Furthermore, Sanger sequencing of these amplicons confirmed this to be a single locus that can both identify and discriminate a range of pathotypes. Wider validation of this primer set is now required to test its suitability across other pathotypes.
The divergence of A. tenuissima and A. arborescens lineages was investigated through identification of expanded and unique gene compliments. We identified HET loci unique to A. arborescens or A. tenuissima lineages. HET loci may act as incompatibility barriers to common genetic exchange between these taxa (Glass and Kaneko, 2003). Taxa also showed divergence in effector profiles, including chitin binding effectors, with A. arborescens isolates possessing unique xylan/pectin degradation CAZymes, while A. tenuissima isolates possessed unique carboxylesterase, chitooligosaccharide and sialidase CAZymes. Chitin binding proteins are important in preventing MAMP triggered host recognition by plants and animals during infection, and may also aid persistence of resting bodies outside of the host (Kombrink and Thomma, 2013). Putative transcription factors were also amongst the proteins specific to A. arborescens or A. tenuissima, indicating that these taxa not only possess distinct gene complements but also differ in how they respond to stimuli. Dispersed repeat sequences such as transposable elements have been shown to serve as sites of recombination within and between fungal chromosomes (Zolan, 1995) and we also show distinct transposon profiles between A. arborescens and A. tenuissima. Transposons are known to aid host adaptation in plant pathogens (Faino et al., 2016; Gijzen, 2009; Schmidt et al., 2013) and have been a mechanism for differentiation of these taxa.
Alternaria HSTs are capable of inducing necrosis on non-host leaves (Kohmoto et al., 1976), meaning that non-host resistance must be associated with recognition of other avirulence genes. We investigated the complements of other putative pathogenicity genes and effectors produced by the apple and Asian pear pathotypes and identified additional CAZymes and secondary metabolite profiles on CDC regions, distinct between pathotypes, suggesting additional host-adapted tools for pathogenicity. Additional secondary metabolites clusters were present on both apple and pear pathotype CDCs as well as unique complements of secreted CAZymes. CAZyme families AA3, AA7 and AA9 have previously been reported to be in greater numbers in the citrus pathotype in comparison to non-pathotypes (Wang et al., 2016). Furthermore, putative transcription factor genes were identified in CDCs indicating that these regions may have some level of transcriptional autonomy from the core genome. This has been shown in Fusarium, where effector proteins are regulated by the SGE transcription factor on the core genome but also by FTF and other transcription factor families (TF1-9) located on lineage specific chromosomes (van der Does et al., 2016).
We report near-complete reference genomes for the apple and Asian pear pathotypes of A. sect. alternaria and provide genomic resources for a further ten diverse isolates from this clade. For the first time we show sequenced Alternaria genomes in a phylogenetic context allowing the identification of both mating type idiomorphs present in A. arborescens and A. tenuissima, with a distribution throughout subclades that was indicative of recent genetic exchange. The presence of the apple CDC in isolates of both mating types supports gene flow between isolates. Furthermore, the distribution of isolates from different pathotypes throughout the phylogeny indicated that apple and tangerine pathotypes are polyphyletic. This means that gene flow is not limited to within, but has also occurred between A. tenuissima and A. arborescens lineages. We also developed PCR primers to aid identification of pathotypes, with those targeting the AMT14 locus identifying a range of pathotypes due to its conservation between CDCs. Despite evidence of genetic exchange between A. arborescens and A. tenuissima clades, we show that these taxa are sufficiently isolated to have diverged, with significant differences in core effector profiles and transposon content.
Accession numbers for genomic data are provided in Table 1. Sanger sequence data is deposited on NCBI under accession numbers MK255031–MK255052.
AA, SS, JW, CL, and JC contributed to the conception and design of the study. AA, HC, and RH performed the lab work including library preparation and sequencing. AA performed the bioinformatic analyses and wrote the manuscript. All authors contributed to the manuscript revision, read, and approved the submitted version.
AA was supported by Defra Plant Health Taxonomic fellowship 2010–2014. Bioinformatic pipelines underpinning these analyses were developed by AA as part of BBSRC grants BB/K017071/1 and BB/K020730/1.
CL was employed by FERA Science Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Thanks are given to the FERA Science Ltd., Drs. P. Gannibal, R. Roberts, and E. Simmons for access to Alternaria isolates. Authors are grateful to the BBSRC for supporting associated research on fungal and oomycete pathogens at NIAB EMR, underpinning the advances presented here.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.03124/full#supplementary-material
TABLE S1 | Identification of single copy Ascomycete genes in reference Alternaria spp. genomes.
TABLE S2 | Gene IDs, location, and functional annotations of genes in expanded orthogroups between A. tenuissima and A. arborescens.
TABLE S3 | Gene IDs, location and functional annotations of genes located on CDC contigs from apple and Asian pear pathotype isolates.
TABLE S4 | Toxin gene BLAST hits in publicly available Alternaria spp. genomes, allowing identification of pathotype isolates.
Ajiro, N., Miyamoto, Y., Masunaka, A., Tsuge, T., Yamamoto, M., Ohtani, K., et al. (2010). Role of the host-selective ACT-toxin synthesis gene ACTTS2 encoding an enoyl-reductase in pathogenicity of the tangerine pathotype of Alternaria alternata. Phytopathology 100, 120–126. doi: 10.1094/PHYTO-100-2-0120
Antipov, D., Korobeynikov, A., McLean, J. S., and Pevzner, P. A. (2016). HybridSPAdes: an algorithm for hybrid assembly of short and long reads. Bioinformatics 32, 1009–1015. doi: 10.1093/bioinformatics/btv688
Arie, T., Kaneko, I., Yoshida, T., Noguchi, M., Nomura, Y., and Yamaguchi, I. (2000). Mating-type genes from asexual phytopathogenic ascomycetes Fusarium oxysporum and Alternaria alternata. Mol. PlantMicrobe Int. 13, 1330–1339. doi: 10.1094/mpmi.2000.13.12.1330
Armitage, A. D., Barbara, D. J., Harrison, R. J., Lane, C. R., Sreenivasaprasad, S., Woodhall, J. W., et al. (2015). Discrete lineages within Alternaria alternata species group: identification using new highly variable loci and support from morphological characters. Fungal Biol. 119, 994–1006. doi: 10.1016/j.funbio.2015.06.012
Bairoch, A., and Apweiler, R. (2000). The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48. doi: 10.1093/nar/28.1.45
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Belmas, E., Briand, M., Kwasiborski, A., Colou, J., N’Guyen, G., Iacomi, B., et al. (2018). Genome sequence of the necrotrophic plant pathogen Alternaria brassicicola Abra43. Genome Announc. 6:e01559-17. doi: 10.1128/genomeA.01559-17
Berbee, M. L., Payne, B. P., Zhang, G., Roberts, R. G., and Turgeon, B. G. (2003). Shared ITS DNA substitutions in isolates of opposite mating type reveal a recombining history for three presumed asexual species in the filamentous ascomycete genus Alternaria. Mycol. Res. 107(Pt 2), 169–182. doi: 10.1017/s0953756203007263
Bihon, W., Cloete, M., Gerrano, A. S., Oelofse, D., and Adebola, P. (2016). Draft genome sequence of Alternaria alternata isolated from onion leaves in South Africa. Genome Announc. 4:e01022-16. doi: 10.1128/genomeA.01022-16
Bonants, P., Groenewald, E., Rasplus, J. Y., Maes, M., de Vos, P., Frey, J., et al. (2010). QBOL: a new EU project focusing on DNA barcoding of Quarantine organisms. EPPO Bull. 40, 30–33. doi: 10.1111/j.1365-2338.2009.02350.x
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Chakraborty, M., Baldwin-Brown, J. G., Long, A. D., and Emerson, J. J. (2016). Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 44, e147–e147.
Dang, H. X., Pryor, B., Peever, T., and Lawrence, C. B. (2015). The Alternaria genomes database: a comprehensive resource for a fungal genus comprised of saprophytes, plant pathogens, and allergenic species. BMC Genomics 16:239. doi: 10.1186/s12864-015-1430-7
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Faino, L., Seidl, M. F., Shi-Kunne, X., Pauper, M., van den Berg, G. C. M., Wittenberg, A. H. J., et al. (2016). Transposons passively and actively contribute to evolution of the two-speed genome of a fungal pathogen. Genome Res. 26, 1091–1100. doi: 10.1101/gr.204974.116
Gijzen, M. (2009). Runaway repeats force expansion of the Phytophthora infestans genome. Genome Biol. 10:241. doi: 10.1186/gb-2009-10-10-241
Glass, N. L., and Kaneko, I. (2003). Fatal attraction: nonself recognition and heterokaryon incompatibility in filamentous fungi. Eukaryot. Cell 2, 1–8. doi: 10.1128/ec.2.1.1-8.2003
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Harimoto, Y., Hatta, R., Kodama, M., Yamamoto, M., Otani, H., and Tsuge, T. (2007). Expression profiles of genes encoded by the supernumerary chromosome controlling AM-toxin biosynthesis and pathogenicity in the apple pathotype of Alternaria alternata. Mol. PlantMicrobe Int. 20, 1463–1476. doi: 10.1094/mpmi-20-12-1463
Harimoto, Y., Tanaka, T., Kodama, M., Yamamoto, M., Otani, H., and Tsuge, T. (2008). Multiple copies of AMT2 are prerequisite for the apple pathotype of Alternaria alternata to produce enough AM-toxin for expressing pathogenicity. J. Gen. Plant Pathol. 74, 222–229. doi: 10.1007/s10327-008-0089-1
Hatta, R., Ito, K., Hosaki, Y., Tanaka, T., Tanaka, A., Yamamoto, M., et al. (2002). A conditionally dispensable chromosome controls host-specific pathogenicity in the fungal plant pathogen Alternaria alternata. Genetics 161, 59–70.
Hoff, K. J., Lange, S., Lomsadze, A., Borodovsky, M., and Stanke, M. (2016). BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32, 767–769. doi: 10.1093/bioinformatics/btv661
Hou, Y., Ma, X., Wan, W., Long, N., Zhang, J., Tan, Y., et al. (2016). Comparative genomics of pathogens causing brown spot disease of tobacco: Alternaria longipes and Alternaria alternata. Plos One 11:e0155258. doi: 10.1371/journal.pone.0155258
Hu, J., Chen, C., Peever, T., Dang, H., Lawrence, C., and Mitchell, T. (2012). Genomic characterization of the conditionally dispensable chromosome in Alternaria arborescens provides evidence for horizontal gene transfer. BMC Genomics 13:171. doi: 10.1186/1471-2164-13-171
Huang, L., Zhang, H., Wu, P., Entwistle, S., Li, X., Yohe, T., et al. (2018). dbCAN-seq: a database of carbohydrate-active enzyme (CAZyme) sequence and annotation. Nucleic Acids Res. 46, D516–D521. doi: 10.1093/nar/gkx894
Johnson, L. J., Johnson, R. D., Akamatsu, H., Salamiah, A., Otani, H., Kohmoto, K., et al. (2001). Spontaneous loss of a conditionally dispensable chromosome from the Alternaria alternata apple pathotype leads to loss of toxin production and pathogenicity. Curr. Genet. 40, 65–72. doi: 10.1007/s002940100233
Johnson, R. D., Johnson, L., Itoh, Y., Kodama, M., Otani, H., and Kohmoto, K. (2000). Cloning and characterization of a cyclic peptide synthetase gene from Alternaria alternata apple pathotype whose product is involved in AM-toxin synthesis and pathogenicity. Mol. Plant Microbe Int. 13, 742–753. doi: 10.1094/mpmi.2000.13.7.742
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Käll, L., Krogh, A., and Sonnhammer, E. L. L. (2004). A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338, 1027–1036. doi: 10.1016/j.jmb.2004.03.016
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kodama, M., Akamatsu, H., Itoh, Y., Narusaka, Y., Sanekata, T., Otani, H., et al. (1998). “Host-specific toxin deficient mutants of the tomato pathotype of Alternaria alternata obtained by restriction enzyme-mediated integration,” in Molecular Genetics of Host-Specific Toxins in Plant Disease. Developments in plant pathology, eds K. Kohmoto and O. C. Yoder, (Dordrecht: Springer), 35–42. doi: 10.1007/978-94-011-5218-1_4
Kohmoto, K., Khan, I. D., Renbutsu, Y., Taniguchi, T., and Nishimura, S. (1976). Multiple host-specific toxins of Alternaria mali and their effect on the permeability of host cells. Physiol. Plant Pathol. 8, 141–153. doi: 10.1016/0048-4059(76)90047-3
Kombrink, A., and Thomma, B. P. H. J. (2013). LysM effectors: secreted proteins supporting fungal life. PLoS Pathogens 9:e1003769. doi: 10.1371/journal.ppat.1003769
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E. L. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kusaba, M., and Tsuge, T. (1995). Phylogeny of Alternaria fungi known to produce host-specific toxins on the basis of variation in internal transcribed spacers of ribosomal DNA. Curr. Genet. 28, 491–498. doi: 10.1007/bf00310821
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lawrence, D. P., Gannibal, P. B., Peever, T. L., and Pryor, B. M. (2013). The sections of Alternaria: formalizing species-group concepts. Mycologia 105, 530–546. doi: 10.3852/12-249
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, K. N., Rouse, D. I., and German, T. L. (1994). PCR primers that allow intergeneric differentiation of ascomycetes and their application to Verticillium spp. Appl. Environ. Microbiolo.. 60, 4324–4331.
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Liu, K., Linder, C. R., and Warnow, T. (2011). RAxML and FastTree: comparing two methods for large-scale maximum likelihood phylogeny estimation. Plos One 6:e27731. doi: 10.1371/journal.pone.0027731
Loman, N. J., Quick, J., and Simpson, J. T. (2015). A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 12, 733–735. doi: 10.1038/nmeth.3444
Marçais, G., Delcher, A. L., Phillippy, A. M., Coston, R., Salzberg, S. L., and Zimin, A. (2018). MUMmer4: a fast and versatile genome alignment system. PLoS Comput. Biol. 14:e1005944. doi: 10.1371/journal.pcbi.1005944
Masunaka, A., Ohtani, K., Peever, T. L., Timmer, L. W., Tsuge, T., Yamamoto, M., et al. (2005). An Isolate of Alternaria alternata That Is Pathogenic to Both Tangerines and Rough Lemon and Produces Two Host-Selective Toxins. ACT- and ACR-Toxins. Phytopathology 95, 241–247. doi: 10.1094/PHYTO-95-0241
Masunaka, A., Tanaka, A., Tsuge, T., Peever, T. L., Timmer, L. W., Yamamoto, M., et al. (2000). Distribution and characterization of AKT Homologs in the Tangerine Pathotype of Alternaria alternata. Phytopathology 90, 762–768. doi: 10.1094/PHYTO.2000.90.7.762
Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A., and Punta, M. (2013). Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41:e121. doi: 10.1093/nar/gkt263
Miyamoto, Y., Ishii, Y., Honda, A., Masunaka, A., Tsuge, T., Yamamoto, M., et al. (2009). Function of genes encoding acyl-CoA synthetase and enoyl-CoA hydratase for host-selective act-toxin biosynthesis in the tangerine pathotype of Alternaria alternata. Phytopathology 99, 369–377. doi: 10.1094/PHYTO-99-4-0369
Miyamoto, Y., Masunaka, A., Tsuge, T., Yamamoto, M., Ohtani, K., Fukumoto, T., et al. (2010). ACTTS3 encoding a polyketide synthase is essential for the biosynthesis of ACT-toxin and pathogenicity in the tangerine pathotype of Alternaria alternata. Mol. PlantMicrobe Int. 23, 406–414. doi: 10.1094/MPMI-23-4-0406
Nakashima, T., Ueno, T., Fukami, H., Taga, T., Masuda, H., Osaki, K., et al. (1985). Isolation and structures of AK-toxin I and II. Host-specific phytotoxic metabolites produced by Alternaria alternata Japanese pear pathotype. Agric. Biol. Chem. 49, 807–815. doi: 10.1271/bbb1961.49.807
Nguyen, H. D. T., Lewis, C. T., Lévesque, C. A., and Gräfenhan, T. (2016). Draft Genome Sequence of Alternaria alternata ATCC 34957. Genome Announc. 4:e01554-15. doi: 10.1128/genomeA.01554-15
Quaedvlieg, W., Groenewald, J. Z., de Jesús Yáñez-Morales, M., and Crous, P. W. (2012). DNA barcoding of Mycosphaerella species of quarantine importance to Europe. Persoonia 29, 101–115. doi: 10.3767/003158512X661282
Ruan, J. (2016). Ultra-fast de Novo Assembler using Long Noisy Reads. Available at: https://github.com/ruanjue/smartdenovo (Accessed: 26 June 2018).
Schmidt, S. M., Houterman, P. M., Schreiver, I., Ma, L., Amyotte, S., Chellappan, B., et al. (2013). MITEs in the promoters of effector genes allow prediction of novel virulence genes in Fusarium oxysporum. BMC Genomics 14:119. doi: 10.1186/1471-2164-14-119
Schwessinger, B., and McDonald, M. (2017). High quality DNA From Fungi for Long Read Sequencing e.g. PacBio, Nanopore MinION. Available at: https://www.protocols.io/view/high-quality-dna-from-fungi-for-long-read-sequenci-hadb2a6?version_warning=no (accessed December 07, 2017).
Shelest, E. (2017). Transcription factors in fungi: TFome dynamics. Three major families, and dual-specificity TFs. Front. Genet. 8:53. doi: 10.3389/fgene.2017.00053
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Sperschneider, J., Gardiner, D. M., Dodds, P. N., Tini, F., Covarelli, L., Singh, K. B., et al. (2016). EffectorP: predicting fungal effector proteins from secretomes using machine learning. N Phytol. 210, 743–761. doi: 10.1111/nph.13794
Stanke, M., and Morgenstern, B. (2005). AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–W467.
Stewart, J. E., Thomas, K. A., Lawrence, C. B., Dang, H., Pryor, B. M., Timmer, L. M. P., et al. (2013). Signatures of recombination in clonal lineages of the citrus brown spot pathogen, Alternaria alternata sensu lato. Phytopathology 103, 741–749. doi: 10.1094/PHYTO-08-12-0211-R
Tanaka, A., Shiotani, H., Yamamoto, M., and Tsuge, T. (1999). Insertional mutagenesis and cloning of the genes required for biosynthesis of the host-specific AK-toxin in the Japanese pear pathotype of Alternaria alternata. Mol. PlantMicrobe Int. 12, 691–702. doi: 10.1094/mpmi.1999.12.8.691
Tanaka, A., and Tsuge, T. (2000). Structural and functional complexity of the genomic region controlling AK-toxin biosynthesis and pathogenicity in the Japanese pear pathotype of Alternaria alternata. Mol. PlantMicrobe Int. 13, 975–986. doi: 10.1094/mpmi.2000.13.9.975
Testa, A. C., Hane, J. K., Ellwood, S. R., and Oliver, R. P. (2015). CodingQuarry: highly accurate hidden Markov model gene prediction in fungal genomes using RNA-seq transcripts. BMC Genomics 16:70. doi: 10.1186/s12864-015-1344-4
Thomma, B. P. H. J. (2003). Alternaria spp.: from general saprophyte to specific parasite. Mol. Plant Pathol. 4, 225–236. doi: 10.1046/j.1364-3703.2003.00173.x
Tsuge, T., Harimoto, Y., Akimitsu, K., Ohtani, K., Kodama, M., Akagi, Y., et al. (2013). Host-selective toxins produced by the plant pathogenic fungus Alternaria alternata. FEMS Microbiol. Rev. 37, 44–66. doi: 10.1111/j.1574-6976.2012.00350.x
van der Does, H. C., Fokkens, L., Yang, A., Schmidt, S. M., Langereis, L., Lukasiewicz, J. M., et al. (2016). Transcription factors encoded on core and accessory chromosomes of Fusarium oxysporum induce expression of effector Genes. PLoS Genetics 12:e1006401. doi: 10.1371/journal.pgen.1006401
Vaser, R., Sović, I., Nagarajan, N., and Šikić, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. doi: 10.1101/gr.214270.116
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. Plos One 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, M., Sun, X., Yu, D., Xu, J., Chung, K., and Li, H. (2016). Genomic and transcriptomic analyses of the tangerine pathotype of Alternaria alternata in response to oxidative stress. Sci. Rep. 6:32437. doi: 10.1038/srep32437
Wolters, P. J., Faino, L., van den Bosch, T. B. M., Evenhuis, B., Visser, R. G. F., Seidl, M. F., et al. (2018). Gapless genome assembly of the potato and tomato early blight pathogen Alternaria solani. Mol. PlantMicrobe Int. 31, 692–694. doi: 10.1094/MPMI-12-17-0309-A
Woudenberg, J. H. C., Seidl, M. F., Groenewald, J. Z., de Vries, M., Stielow, J. B., Thomma, B. P. H. J., et al. (2015). Alternaria section Alternaria: species, formae speciales or pathotypes? Stud. Mycol. 82, 1–21. doi: 10.1016/j.simyco.2015.07.001
Yu, G., Smith, D. K., Zhu, H., Guan, Y., and Lam, T. T.-Y. (2016). ggtree : an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol.Br. Ecol. Soc.. 8, 28–36. doi: 10.1111/2041-210x.12628
Zhang, C., Rabiee, M., Sayyari, E., and Mirarab, S. (2018). ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19(Suppl. 6):153. doi: 10.1186/s12859-018-2129-y
Keywords: CDC, host-specific toxin, mating type, Dothideomycetes, Alternaria mali, Alternaria gaisen, nanopore
Citation: Armitage AD, Cockerton HM, Sreenivasaprasad S, Woodhall J, Lane CR, Harrison RJ and Clarkson JP (2020) Genomics Evolutionary History and Diagnostics of the Alternaria alternata Species Group Including Apple and Asian Pear Pathotypes. Front. Microbiol. 10:3124. doi: 10.3389/fmicb.2019.03124
Received: 28 March 2019; Accepted: 24 December 2019;
Published: 23 January 2020.
Edited by:
James Hane, Curtin University, AustraliaReviewed by:
Vasvi Chaudhry, University of Tübingen, GermanyCopyright © 2020 Armitage, Cockerton, Sreenivasaprasad, Woodhall, Lane, Harrison and Clarkson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew D. Armitage, QS5ELkFybWl0YWdlQGdyZS5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.