 Ying Shao1Hongwei Guo1Jianping Zhang1Hui Liu1Kun Wang2Song Zuo2Pengfei Xu2Zhenrong Xia2Qiumei Zhou3Hanghang Zhang4Xiangqing Wang5Anhui Chen1*
Ying Shao1Hongwei Guo1Jianping Zhang1Hui Liu1Kun Wang2Song Zuo2Pengfei Xu2Zhenrong Xia2Qiumei Zhou3Hanghang Zhang4Xiangqing Wang5Anhui Chen1* Yulong Wang6*
Yulong Wang6*- 1Jiangsu Key Laboratory of Food Resource Development and Quality Safe, Xuzhou University of Technology, Xuzhou, China
- 2Jiangsu Konen Biological Engineering Co., Ltd., Nanjing, China
- 3Experimental Center of Clinical Research, The First Affiliated Hospital of Anhui University of Chinese Medicine, Hefei, China
- 4Nanling Forestry Technology Center, Nanling Forestry Bureau, Nanling, China
- 5Department of Neurology, The People’s Hospital of Pingyi County, Pingyi, China
- 6Anhui Provincial Key Laboratory of Microbial Pest Control, Anhui Agricultural University, Hefei, China
The mushroom, Sanghuang is widely used in Asian countries. This medicinal fungus produces diverse bioactive compounds and possesses a potent ability to degrade the wood of the mulberry tree. However, the genes, pathways, and mechanisms that are involved in the biosynthesis of the active compounds and wood degradation by Sanghuang mushroom are still unknown. Here, we report a 34.5 Mb genome—encoding 11,310 predicted genes—of this mushroom. About 16.88% (1909) of the predicted genes have been successfully classified as EuKaryotic Orthologous Groups, and approximately 27.23% (665) of these genes are involved in metabolism. Additionally, a total of 334 genes encoding CAZymes—and their characteristics—were compared with those of the other fungi. Homologous genes involved in triterpenoid, polysaccharide, and flavonoid biosynthesis were identified, and their expression was examined during four developmental stages, 10 and 20 days old mycelia, 1 year old and 3 years old fruiting bodies. Importantly, the lack of chalcone isomerase 1 in the flavonoid biosynthesis pathway suggested that different mechanisms were used in this mushroom to synthesize flavonoids than those used in plants. In addition, 343 transporters and 4 velvet family proteins, involved in regulation, uptake, and redistribution of secondary metabolites, were identified. Genomic analysis of this fungus provides insights into its diverse secondary metabolites, which would be beneficial for the investigation of the medical applications of these pharmacological compounds in the future.
Introduction
Sanghuang is a medically important fungi and has been used as a traditional medicine throughout China, Japan, and Korea. The first use of Sanghuang was reported in the oldest Chinese medicinal book, Shennong’s Compendium of Materia Medica, which reported the use of Sanghuang for the treatment of various disease conditions, such as stomach pain and amenorrhea; the use of this fungus is also correlated with prolonged life, detoxification, and improved digestion after long-term use (Zhu et al., 2008; Lee et al., 2011; Cai et al., 2019). Ikekawa et al. (1968) confirmed that the water extracts of the Sanghuang fruiting bodies had potent biological activities and can be used to inhibit sarcoma cell growth in mice (Shibata et al., 1968). Since then, numerous studies focusing on its medicinal functions have been reported, further characterizing the antitumor and antioxidant properties of this fungi (Zhang et al., 2014; Yan et al., 2016). Extensive studies in this regard have shown that the main components of Sanghuang, as a medicinal fungus, are considered as effective antioxidant and antitumor drugs (Cai et al., 2019).
Because of its large market demand and the limitation of the slow growth of the fruit, natural Sanghuang is in serious short supply; therefore, large scale industrial production of a few Sanghuang species is required to eliminate this shortage. However, the fungal name has been controversial for long time owing to the ambiguity of these fungal circumscription with the other fungi species (Wu et al., 2012; Cui, 2014; Zhou et al., 2015). Sanghuang has been mistakenly reported as Inonotus linteus, Phellinus igniarius, and Phellinus baumii; Zhou et al. (2015) investigated the phylogenetic relationship of Inonotus spp. and defined the three distinct groups as Inonotus s.s., Sanghuangporus, and Tropicoporus. The phylogenetic position, and the morphological and ecological features indicated that the true Sanghuang (I. sanghuang) is a member of Sanghuangporus; thus, the authentic Sanghuang should be renamed as Sanghuangporus sanghuang (S. sanghuang) and characterized as a new species that has been discovered to only grow on the mulberry trees (Wu et al., 2012; Zhou et al., 2015). Thus, it is necessary to clarify the taxonomic position of Sanghuang species that are extensively utilized for industrial production based on the molecular phylogenetic analysis.
Sanghuangporus sanghuang produces hundreds of different compounds, many of which are bioactive fractions and constituents (Lin et al., 2017; Liu et al., 2017; Cai et al., 2019; Wu et al., 2019). Triterpenoids, polysaccharides, and flavonoids are the three major categories of pharmacologically active compounds in S. sanghuang. Polysaccharides, such as water soluble intracellular polysaccharides (IPSWs), homogenous polysaccharides (PIP1), and heteropolysaccharides (PIP60-1), have been reported to exert anti-cancer, anti-diabetes, anti-inflammation and antioxidation effects (Jang et al., 2004; Hwang et al., 2005). Triterpenoids play an important role in antitumor, anti-inflammation, and immune regulatory properties (Taji et al., 2008; Wang et al., 2009). Moreover, S. sanghuang is one of the rare fungi that contains flavone compounds, which have antioxidation and antitumor effects (Feng et al., 2012; Liu et al., 2017). S. sanghuang, as a white rot fungus with wood degradation ability, secretes lots of carbohydrate-active enzymes (CAZymes) for the degradation of the mulberry trees (Zhou et al., 2015).
The availability of fungal genome sequences has facilitated research on gene diversity related to the biosynthesis of the secondary metabolites (Corre and Challis, 2009; Dai, 2019). At present, a large number of medicinal mushroom genomes have been sequenced; these include Ganoderma lucidum, Wolfiporia cocos, and Auricularia heimuer (Chen et al., 2012; Floudas et al., 2012; Dai, 2019). Although S. sanghuang has many important properties, but until now only approximately 120 nucleotide sequences available in the National Center for Biotechnology Information (NCBI) database for S. sanghuang, and most of them are used for phylogenetic analysis. Therefore, the current genomic sequence resources are not sufficient to reveal the pharmacological mechanisms of S. sanghuang at the molecular level.
Here, we report the complete genomic sequence of S. sanghuang Kangneng, which has been used for large scale industrial production in China. The genes and gene clusters involved in secondary metabolism and their regulation have been identified in this study. Information regarding the whole genome sequencing of S. sanghuang contributes to its medicinal application and future studies.
Materials and Methods
Culture Conditions and DNA Isolation
In this study, a total of six S. sanghuang strains that have been used for large scale industrial production in China, labeled as S. sanghuang QianDaoHu, S. sanghuang Korea, S. sanghuang BeiJing, S. sanghuang JinZhai, S. sanghuang KangNeng, and S. sanghuang ZhangJiaJie, were obtained from Jiangsu KONEN Biological Engineering Co. Ltd (Anhui, China). The single spore strains were obtained following the methods of Choi et al. (1999). All the strains were cultured using potato dextrose agar (PDA) at 28°C and are available upon request. The single spore mycelia were used for genomic DNA extraction by the cetyltrimethyl ammonium bromide (CTAB) method.
Phylogenetic Analysis
The internal transcribed spacer (ITS) region was amplified using a primer pair of ITS4 and ITS5, and was sequenced from six strains as reported previously (Tsai et al., 2017). The sequences were aligned using the ClustalW application included in BioEdit1. The alignment was converted to MEGA file and a phylogenetic tree was constructed and produced by Maximum likelihood using MEGA X2 software of the bootstrap values (1000 replicates).
Genome Sequencing and Assembly
Sanghuangporus sanghuang KangNeng was selected for genome sequencing as it is the most widely cultured strain among the six S. sanghuang strains. The DNA extracted from S. sanghuang KangNeng was used for genome sequencing. Illumina HiSeq and the PacBio sequencing approach were combined for the whole genome sequencing to obtain better sequencing. Genome assembly was performed as previously reported (Zhang et al., 2018). Briefly, raw reads from PacBio were filtered out, and Proovread 2.14.0 was used for the subread correction (>1 Kb); then, SMRT Analysis v.2.3.0 was used for assembly. The completeness of this assembly was assessed using the Benchmarking Universal Single-Copy Orthologs v3 (BUSCO), as described previously (Seppey et al., 2015).
Genome Annotations
Tandem Repeats Finder (TRF 4.04) was used to search for tandem repeats in all scaffolds. Transposable element (TE) annotation was performed with RepeatMasker 4.06. The tRNAscan-SE 1.3.1 and RNAmmer 1.2 were used for the identification of the tRNAs and rRNAs, and Genemark-ES 4.21 was used for de novo gene predictions (Ter-Hovhannisyan et al., 2008).
Functional Annotation
The functional annotations of the genes were performed according to databases, including CAZymes (Carbohydrate-Active enZYmes Database), P450, and KOG (Eukaryotic Orthologous Groups) (Tatusov et al., 2003; Cantarel et al., 2009; Magrane and UniProt Consortium, 2011). Fungal Transcription Factor Database was used for transcription factors annotation (Wilson et al., 2008). The antiSMASH analyses3 were run with default parameters (relaxed) to predict the secondary metabolite biosynthesis gene clusters in the fungi.
Quantitative PCR (qPCR)
For the collection of different samples in asexual development, the strain was inoculated onto PDA at 28°C in the dark; all mycelia on the plate were collected and grinded into powder using liquid nitrogen for RNA extraction. For fruiting body production, the mycelia of this strain were inoculated into a 250 ml flask containing 100 ml liquid PDS (20% potato, 2% Mulberry tree sawdust) medium. The flask was then incubated at 28°C in a 200 rpm shaker for 10 days. Sterilized short log sections were inoculated with the fungal culture from the same flask by the top spawning method (Hur, 2008), cultivated in the dark at 30°C with relative humidity of 96% for 2 months, and then maintained at 30°C under a 17:7 h dark/light cycle until collection. Whole fresh fruiting bodies were cut into small pieces and pulverized to powder using a grinder, then further grinded into powder using liquid nitrogen for RNA extraction. Total RNA was extracted at different asexual developmental stages of S. sanghuang, i.e., 10 days old (M10d) and 20 days old (M20d), as well as from different sexual developmental stages, i.e., one year old (OY) and three year old (TY). The cDNA was synthesized and quantitative PCR was performed as reported previously (Shao et al., 2019). qPCR amplification was performed with a SYBR Green kit (Takara) and Bio-Rad CFX96TM system (Hercules, CA, United States). The stability of gene expression of actin, β-tubulin, α-tubulin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was determined in M10d, M20d, OY, and TY, and both α-tubulin and GAPDH were found to be stable in different fungal stages. Therefore, GAPDH was used as the internal control in this study. The expression values were obtained by normalizing to that of the control gene and the 2–Δ Δ Ct method was used for calculating the relative gene expression levels. All the experiments were performed in six replicates, and data are presented as the mean ± SE of six replicates. Primers are listed in Supplementary Table S1.
Data Availability Statement
The genome assembly of S. sanghuang Kangneng has been deposited at NCBI with the accession no. VVIT00000000.
Results
Molecular Phylogeny of Six Selected Strains
Sanghuang has long been misleadingly referred to by some incorrect binomial names, and the species concept of sanghuang has only recently been established by molecular phylogenetic studies; thus, it is necessary to clarify the taxonomic position of sanghuang strains that are extensively utilized in China. Therefore, six S. sanghuang strains that are used for large scale industrial production were selected, and the phylogenetic analysis of their ITS nrDNA sequences was performed. In the phylogenetic analysis (Figure 1), species of Inonotus formed at least three clades (Sanghuangporus, Tropicoporus, and Inonotus sensu stricto), and all the six selected strains were included in the Sanghuangporus clade, suggesting that these commercially available sanghuang mushrooms in China are S. sanghuang as opposed to others (Figure 1). Further analysis showed that the five S. sanghuang species (S. sanghuang QianDaoHu, S. sanghuang Korea, S. sanghuang BeiJing, S. sanghuang JinZhai, and S. sanghuang KangNeng) are more closely related to each other than S. sanghuang Zhangjiajie, revealing that these commercially available sanghuang mushrooms in China are diverse.
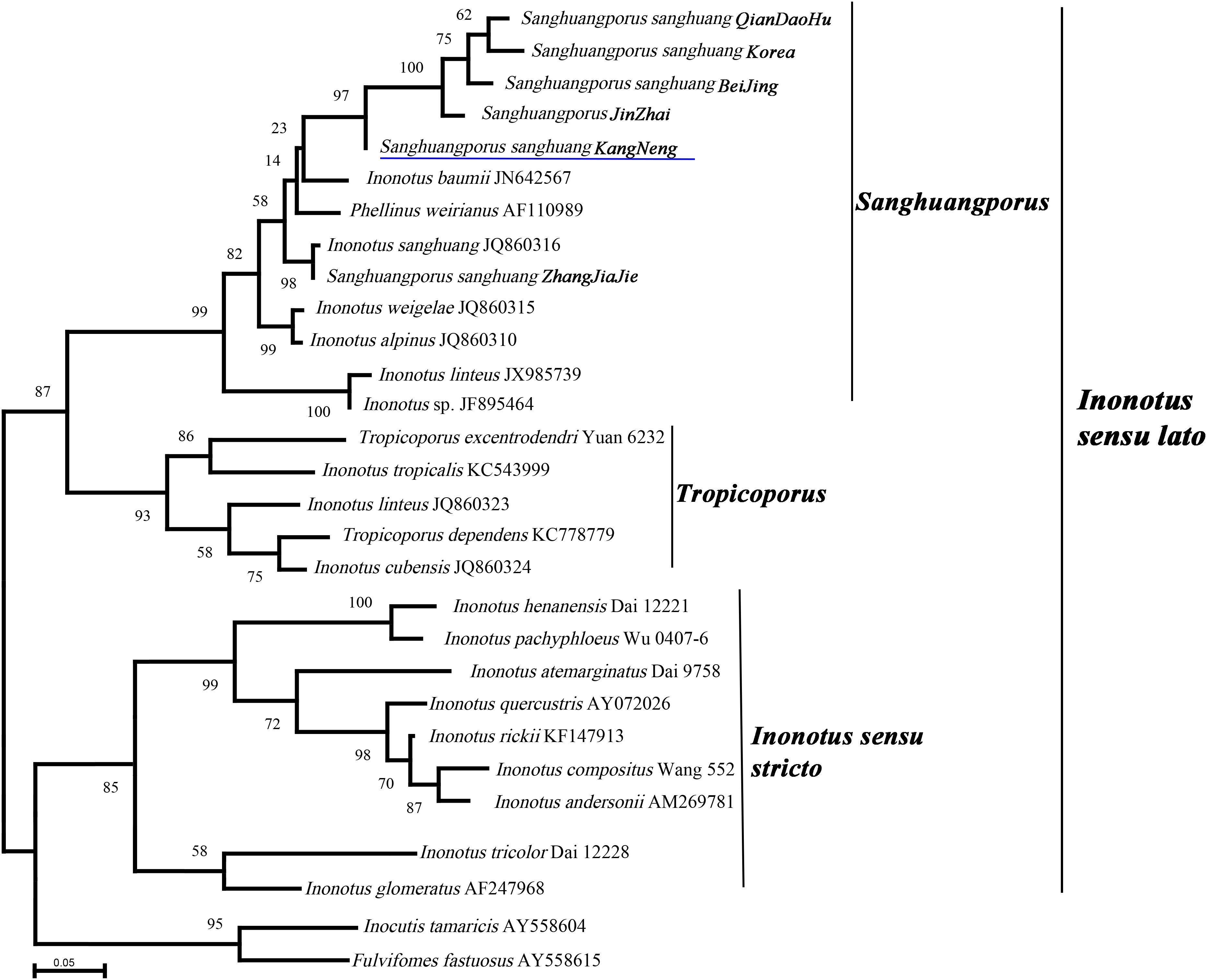
Figure 1. Phylogeny of Sanghuangporus sanghuang strains and selected representative fungi inferred from ITS sequences. Bootstrap values from maximum likelihood analysis. The newly generated sequences for this study have been submitted to GenBank with accession numbers (MN242716–MN242721). The underlined species name was genome-sequenced in this study.
Genome Sequencing, Assembly, and Annotation
Sanghuangporus sanghuang KangNeng was selected for genome sequencing as it is the most widely cultured among the six S. sanghuang strains. First, the characteristics at four different developmental stages (mycelia and fruiting bodies of 1, 2, and 3 year old) were evaluated; the fungi were perennial basidiocarps, effused-reflexed to pileate, and had a brown, dark grayish to black, pileal surface, which is in accordance with the characteristics previously reported (Figure 2A) (Zhou et al., 2015).
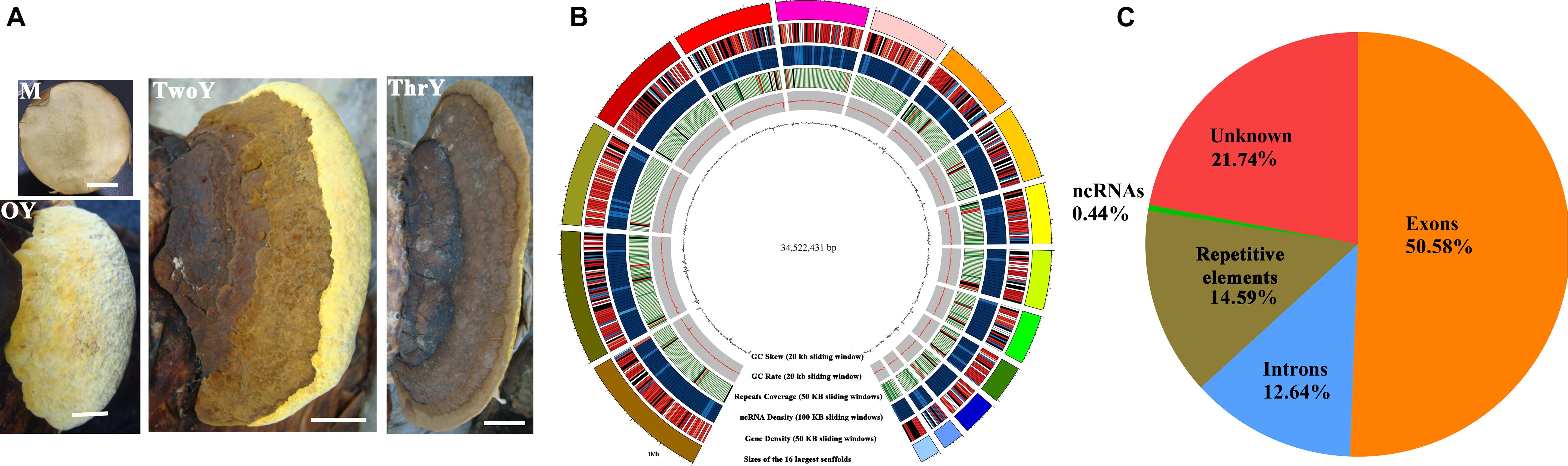
Figure 2. Growth characteristics and general genomic features of S. sanghuang Kangneng. (A) The four developmental stages in the life cycle of the fungus. M, aerial mycelia; OY, one-year-old fruiting bodies; TwoY, two-year-old fruiting bodies; ThrY, three-year-old fruiting bodies. Bar = 3 cm. (B) Characteristics of the de novo assembly genomic features. From the outside to the inside are: I, scaffolds, the different colors represent different scaffolds (scaffolds length > 1 Mbp were chosen); II, gene density; III, the density of non-coding RNA; IV, percentage of coverage of repetitive sequences; V, GC content estimated by the percentage of G + C in 20 kb; VI, GC skew was estimated with (G–C)/(G + C) in 20 kb. (C) Genomic element density.
A 34.5 Mb genome sequence was obtained (∼440 X coverage), and the assembly consisted of 37 scaffolds with 47.95% GC content (Table 1 and Figure 2B). In total, 11,310 genes were predicted, with an average sequence length of 1,930 bp, and accounted for 63.22% of the whole genome sequence length, revealing a high gene density in the genome (Table 1 and Figure 2A). There were 66,421 predicted exons, and the cumulative length accounted for 50.58% of the whole genome sequence length, and 55,111 introns were predicted. There were 251 non-coding RNAs (ncRNAs), such as tRNA, rRNA, and small RNA that were found; these ncRNAs represented 0.45% of the whole genome length, suggesting that ncRNA formed only a small proportion of the overall genome size (Table 1 and Figure 2C).
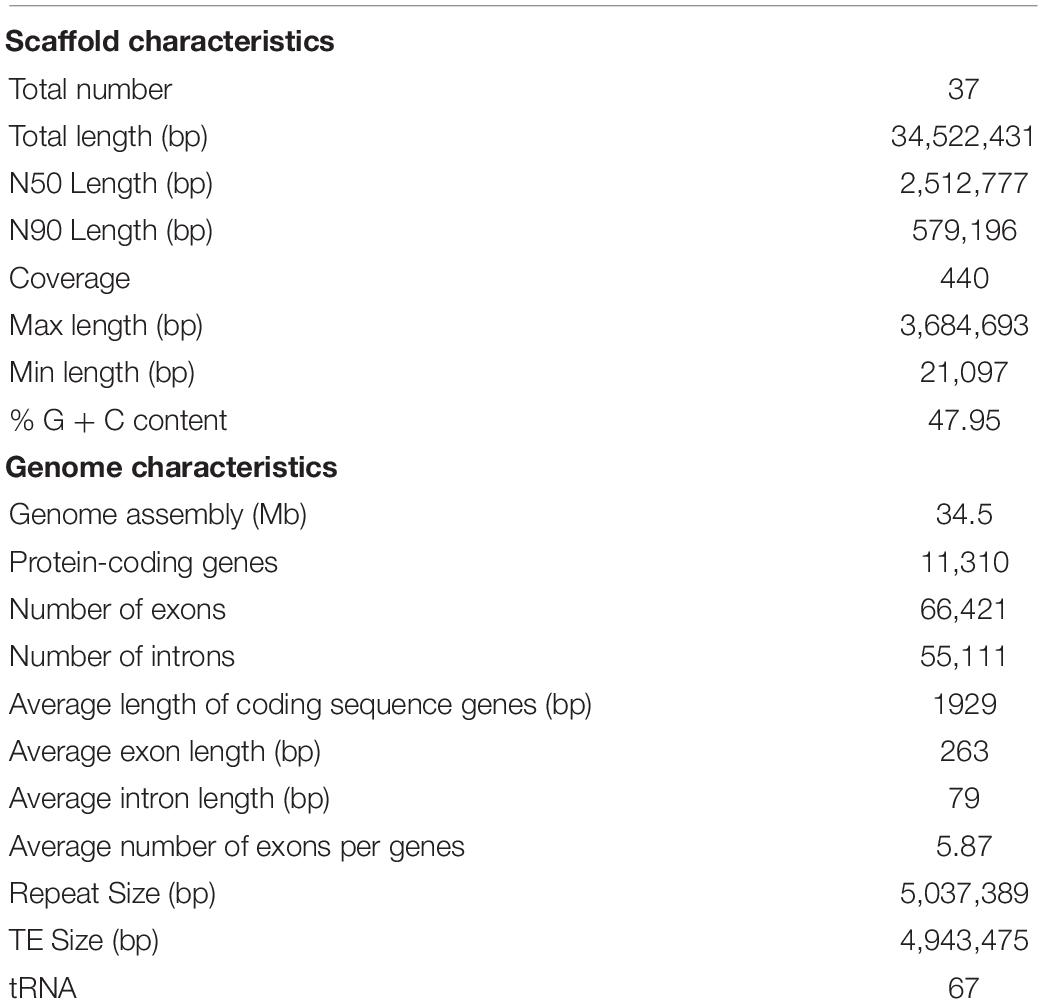
Table 1. Genome assembly and features of S. sanghuang Kangneng.
The total length of the repeat sequences was 5,037,389 bp, covering 14.59% of the genomic length. Tandem repeat sequences only accounted for 0.27%, and transposable elements (TEs) accounted for approximately 14.32% of the genome. Among the TEs, long terminal repeats (LTRs) and non-LTR transposons accounted for 9.84 and 4.48% of the genome, respectively (Table 1).
Gene Function Annotation
All 11,310 protein-encoding genes were annotated in this study (Supplementary Table S1). Among all of the predicted genes, 2,442 genes were annotated by the KEGG pathway, out of which 43.28% (1057) were involved in metabolism, accounting for the major proportion (Supplementary Table S2). Genes classified into functional categories based on COG analysis accounted for 9.45% (1069), and within in these genes, 59.31% (634) of the total predicted genes were involved in metabolism (Supplementary Table S3). A total of 16.88% (1909) of the genes could be classified in the KOG analysis; the results showed that 27.23% (665) of these genes are involved in metabolism, and genes involved in “energy production and conversion,” “amino acid transport and metabolism,” “carbohydrate transport and metabolism” and “lipid transport and metabolism” accounted for the major proportion among these metabolism-associated genes (Figure 3 and Supplementary Table S4).
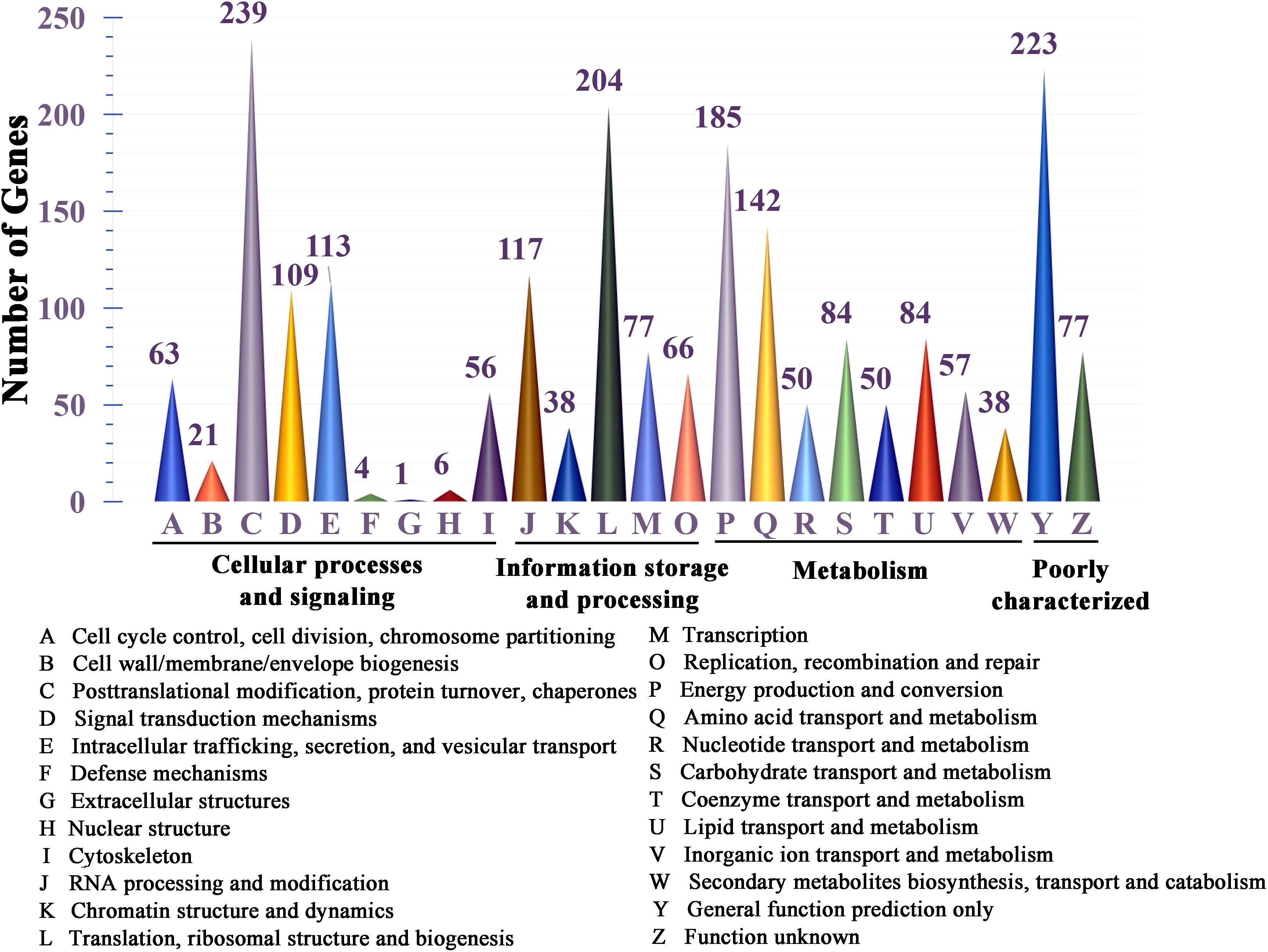
Figure 3. KOG functional classification of proteins in the S. sanghuang Kangneng genome.
Functional Annotation of Putative CAZymes
As a white rot fungus with wood degradation ability, CAZymes were identified in the S. sanghuang genome and were compared with the other fungi. A total of 334 genes could be assigned to CAZyme families as defined in the CAZy database (Supplementary Table S5). All CAZymes were classified into 5 major modules; 185 genes for glycoside hydrolases (GH), 69 genes for glycosyl transferases (GT), 12 genes for polysaccharide lyases (PL), 61 genes for carbohydrate esterases (CE), and 7 genes for carbohydrate-binding modules (CBM) (Supplementary Table S5).
To study the characteristics of CAZymes in S. sanghuang, CAZymes from fungi Ascomycota, Basidiomycota, Chytridiomycota, and Zygomycota were analyzed together, as previously reported (Figure 4A) (Zhao et al., 2013). In the S. sanghuang genome, the number of CAZymes is in the middle among all of the selected fungi but is different from most other fungi, and the number of GHs was remarkably larger than the number of GTs, suggesting that fungal survival is dependent on lignocellulose decomposition. It was concluded that polysaccharide decomposition is more important than polysaccharide construction for the growth and metabolism of S. sanghuang (Figure 4A).
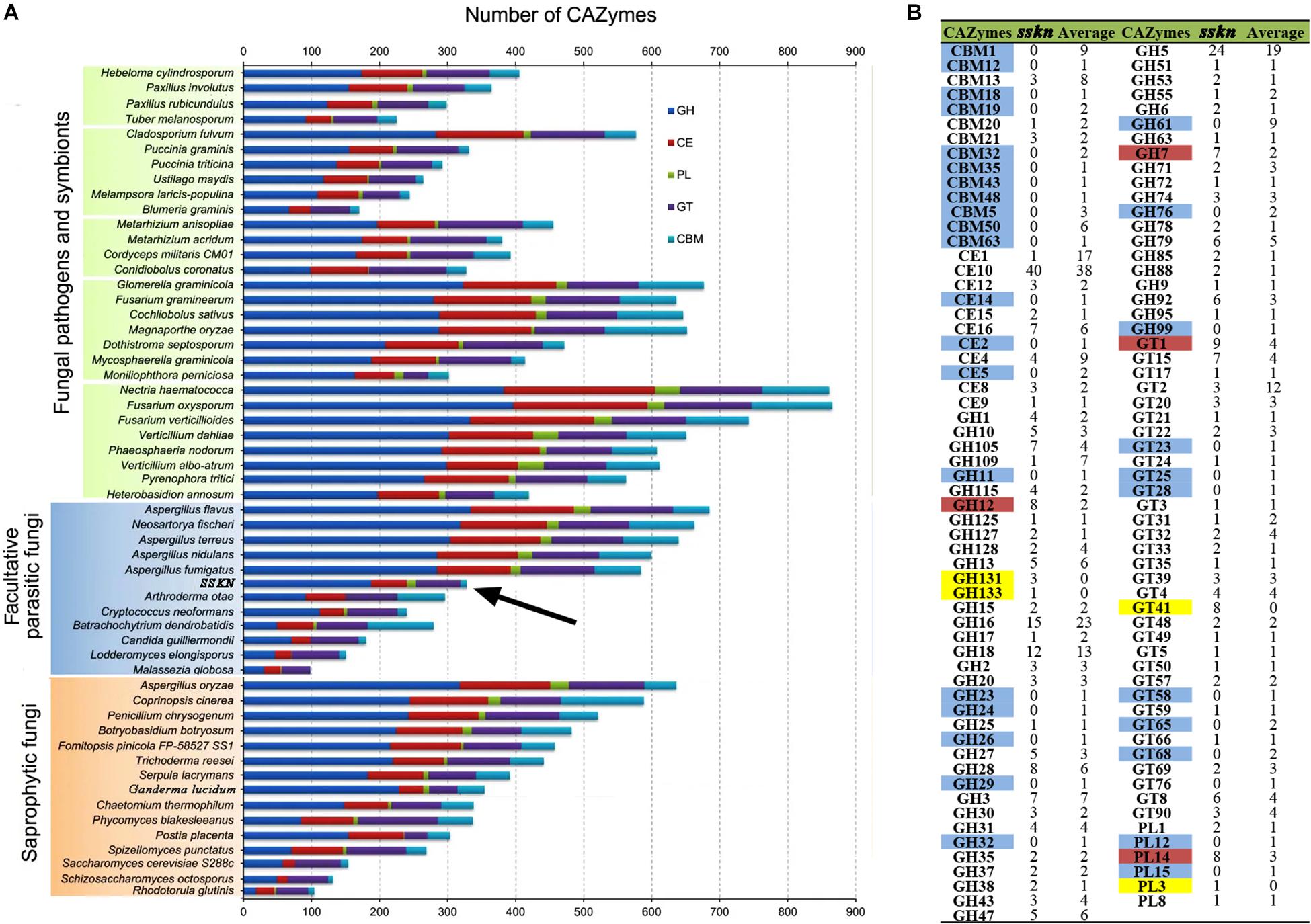
Figure 4. Comparative analysis of CAZymes from S. sanghuang Kangneng and other fungi. (A) Comparative analysis of fungal CAZymes. The numbers of CAZyme modules or domains are represented as horizontal bars. (B) Average number of CAZymes from a total of 30 Basidiomycota fungi. Blue, CAZyme present in other Basidiomycota fungi but not in SSKN; Red, number of CAZymes present in SSKN greater than one-fold the average number of other Basidiomycota fungi; Yellow, CAZymes only present in SSKN. SSKN, S. sanghuang Kangneng.
Moreover, CAZymes from a total of 30 Basidiomycota were identified and compared with those of S. sanghuang (Figure 4B and Supplementary Table S6). Although the results showed that the gene numbers in the 5 major modules of CAZymes were similar in Basidiomycota fungi, GH131, GH133, GT41, and PL3 were specifically present in S. sanghuang, and the copy numbers of GH12, GH7, GT1, and PL14 in the fungi were remarkably higher than those in other Basidiomycota fungi, suggesting that these genes may be involved in mulberry tree cell wall-degradation (Figure 4B and Supplementary Table S6).
The Biosynthesis of Bioactive Compounds in S. sanghuang
Sanghuangporus sanghuang produces diverse secondary bioactive metabolites, such as triterpenoids, polysaccharides, and flavonoids, which are its main components that have immunomodulatory, antioxidant, and antitumor activities (Lin et al., 2017; Liu et al., 2017). Therefore, genes involved in the biosynthesis of these secondary metabolites were searched in the fungal genome.
Triterpenoid Biosynthesis
A total of 18 key enzymes involved in the mevalonate pathway (MVA) pathway were identified in this study (Figure 5A and Table 2). The hydroxymethylglutaryl-CoA (HMG-CoA) synthase, geranyl-diphosphate synthase, diphosphate synthase, and terpenoid cyclase enzymes are each encoded by two or more copies of the respective genes, whereas the other 13 enzymes are encoded by single copy genes (Table 2). A gene (GME6608) encoding lanosterol synthase (LSS) was found, and it catalyzes the cyclization of the triterpenes squalene or 2-3-oxidosqualene to a protosterol cation and finally to lanosterol, the precursor of all steroids (Dai, 2019).
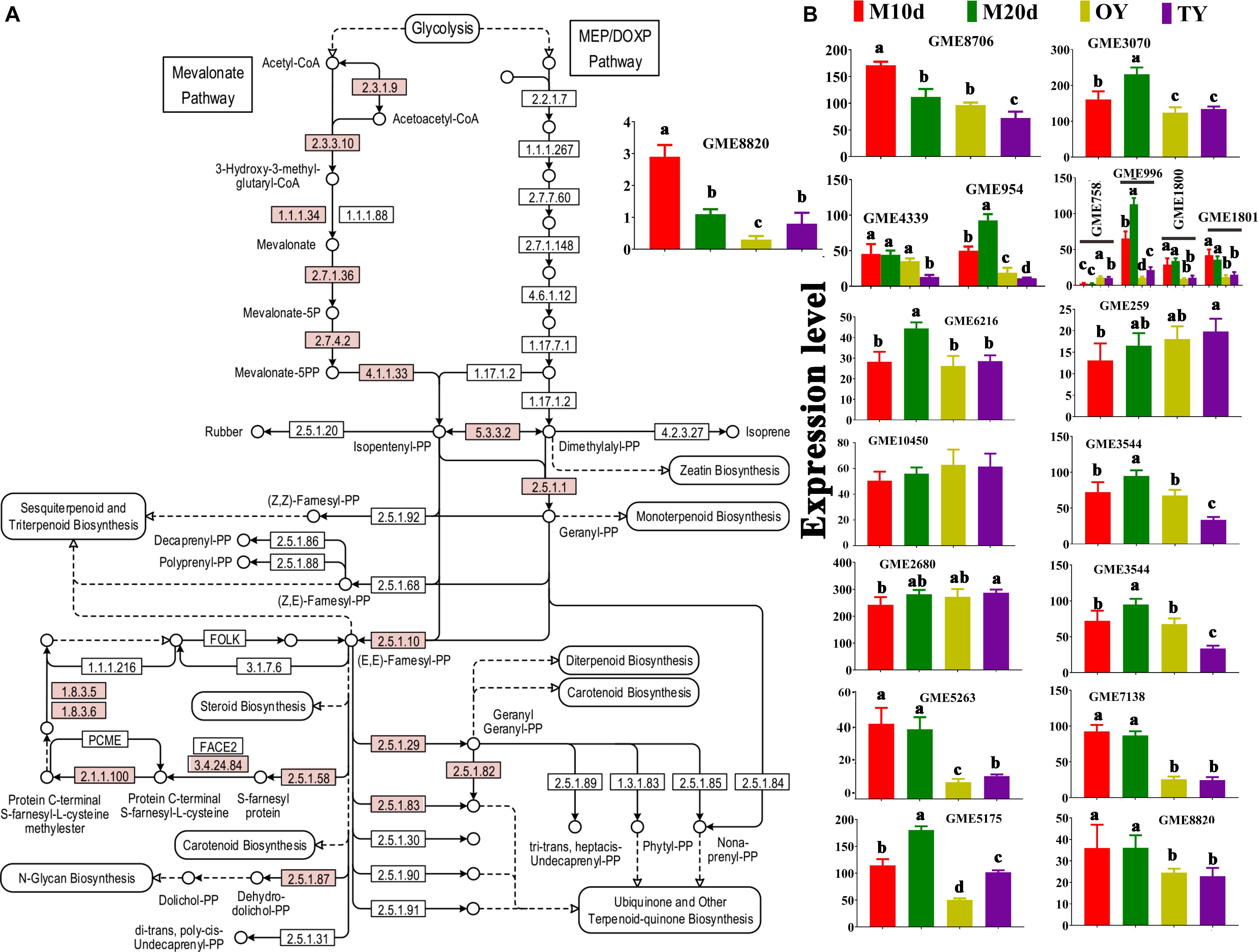
Figure 5. KEGG mapping of the terpenoid backbone biosynthesis pathway and differential expression of the enzymes identified in S. sanghuang Kangneng. (A) The red box indicates existing homologous genes of the enzyme. (B) The expression levels of genes in different developmental stages. Different lowercase letters marked on the bars in each graph denote significant differences (P < 0.05).
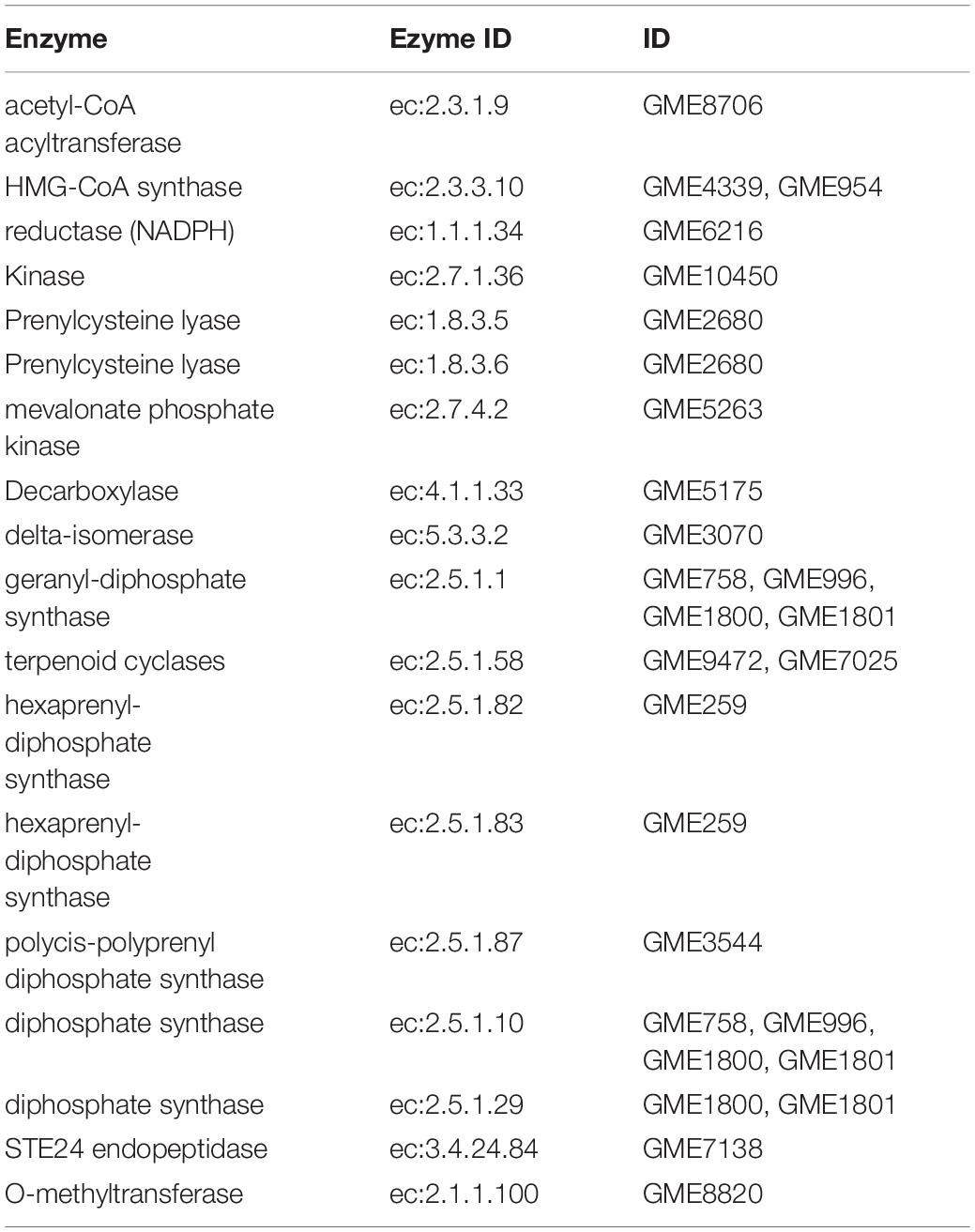
Table 2. Terpenoid backbone biosynthesis (Total = 18).
The expression of the genes in the terpenoid backbone synthesis pathway was detected with qPCR to predict the triterpenoid production at different stages of the fungus (Figure 5B). The results showed that most of these genes were differentially expressed between the mycelium and fruiting body, and most of the genes, LSS included, were expressed at the highest levels in the mycelial stages (M10d or M20d), suggesting that the fungi may produce more triterpenoids in the mycelial stages than in the fruiting body stages (Figure 5B).
Polysaccharide Biosynthesis
Polysaccharides that account for the pharmaceutical potential of S. sanghuang are another kind of highly bioactive compounds. Among all the polysaccharides in the medicinal fungi, the water-soluble 1,3-β-and 1,6-β-glucans are the most active as immunomodulatory and antioxidant compounds (Chen et al., 2012; Mizuno and Nishitani, 2013). The two genes encoding 1,3-β-glucan synthases and seven genes encoding β-glucan biosynthesis-associated proteins were identified in the genome of S. sanghuang; all these proteins contain a SKN1 domain that plays important roles in 1,6-β-glucan biosynthesis, suggesting the same mechanism in fungal polysaccharide biosynthesis (Supplementary Table S7) (Shahinian and Bussey, 2000; Douglas, 2001; Chen et al., 2012). A total of 19 genes involved in polysaccharide biosynthesis and their regulation were found based on the fungal genome; the qPCR results showed that although all of these genes were differentially expressed between the mycelium and fruiting body, approximately half of these genes (10/19) were expressed at the highest levels in the mycelial stages (M10d or M20d), while the others were expressed at their lowest levels during this stage, revealing that further studies should be performed to examine the differences in polysaccharide production at diverse stages of the fungus (Figure 6A).
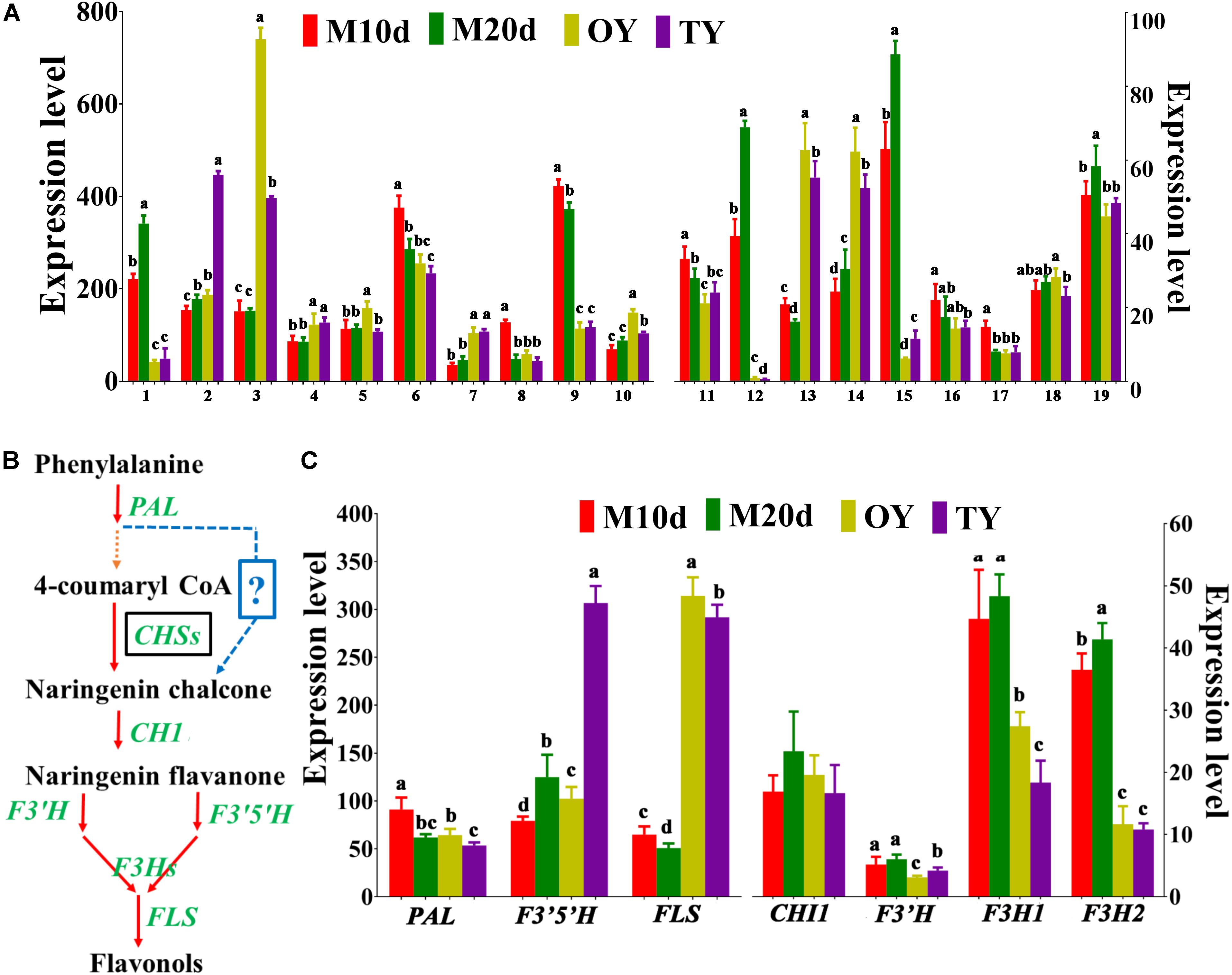
Figure 6. Differential expression of the enzymes involved in polysaccharide and flavonoid biosynthesis identified in S. sanghuang Kangneng. (A) The expression levels of genes involved in polysaccharide biosynthesis in different developmental stages. (B) Putative pathway of flavonoid biosynthesis in S. sanghuang Kangneng. Black box, no homologous gene was found in the fungal genome; ?, unknown reactions in the fungus. (C) The expression levels of genes involved in flavonoid biosynthesis in different developmental stages. Different lowercase letters marked on the bars in each graph denote significant differences (P < 0.05).
Flavonoid Biosynthesis
Sanghuangporus sanghuang is considered as one of the few species of the fungi that is capable of yielding flavonoids, and can produce various flavonoid compounds that have antioxidant, antiproliferative, and antimicrobial activities (Liu et al., 2017). Putative genes involved in flavonoid biosynthesis were searched based on a previous study, and 7 core genes involved in flavonoid biosynthesis were found (Supplementary Table S8) (Saito et al., 2013). In plants, chalcone synthase (CHS) is the first enzyme specific for the flavonoid pathway; however, no gene encoding CHS was found in the S. sanghuang genome, suggesting a different catalytic mechanism for naringenin chalcone production in this fungus (Figure 6B) (Falcone Ferreyra et al., 2012). The other key genes, chalcone isomerase 1 (CHI1), flavonoid-3′, 5-hydroxylase (F3′5′H), F3′H -flavonoid-3′-hydroxylase (F3′H), flavanone-3-hydroxylase 1 (F3H1), flavanone-3-hydroxylase 2 (F3H2), and flavonol synthase (FLS), were present in the fungus, revealing that the central pathway for flavonoid biosynthesis is conserved between this fungus and the plants (Figure 6B and Supplementary Table S9).
The expression levels of genes involved in flavonoid biosynthesis (PAL, CHI1, F3′H, F3′5′H, F3H1, F3H2, and FLS) were examined, and the results showed that six genes (except F3′H) were differentially expressed at different fungal development stages, four genes (F3′5′H, F3H1, F3H2, and FLS) that are downstream of the pathway were expressed most highly in the mycelial stages (M10d or M20d), than the others, revealing that the fungi may produce more flavonoids in the mycelial stages than in the fruiting body stages (Figure 6C).
Prediction of Gene Clusters Involved in Bioactive Secondary Metabolite Biosynthesis of S. sanghuang
In fungi, genes involved in secondary metabolite biosynthesis are always organized in discrete clusters, thus secondary metabolite biosynthesis gene clusters in the fungus were identified using the antiSMASH software (Supplementary Table S10). A total of 21 gene clusters located in different scaffolds were detected. One non-ribosomal peptide synthase (NRPS) and three NRPS-like gene clusters were identified in the S. sanghuang genome, and further analysis showed that the fungal genome encodes 14 terpene synthase (TS), 5 NRPS, and 43 polyketide synthase (PKS) genes (Supplementary Table S11). These data suggested that unlike the medicinal fungi G. lucidum and A. heimuer, which include a few putative NRPS and PKS genes but may not produce NRP and polyketides, NRPS and PKS are numerous and necessary for S. sanghuang (Chen et al., 2012; Dai, 2019). The terpene synthase family is a mid-sized family working on the biosynthesis of monoterpene, sesquiterpene, and diterpene backbones (Chen et al., 2011). A total of 14 TS genes and 14 TS gene clusters were identified in the fungal genome, suggesting that diverse secondary metabolites can be produced in the fungus (Supplementary Tables S10, S11).
Regulation of Secondary Metabolism in S. sanghuang
Velvet family proteins could regulate fungal secondary metabolite production in response to different environmental conditions. In this study, four genes encoding velvet domain containing proteins (VelB, VelA, VosA, and VelC) and the methyltransferase domain containing protein (LaeA) were also identified in the S. sanghuang genome (Supplementary Table S12). VelA and VelB interact with LaeA and regulate the secondary metabolism and development in other fungi, suggesting that the pathway for secondary metabolism and development is conserved in fungi (Bayram et al., 2008).
Transporters in the S. sanghuang Genome
In total, 368 transport proteins from 98 families were found in S. sanghuang, including 51 ATP dependent transporters, 18 ion channels, and 299 secondary transporters (Supplementary Table S13). It has been reported that MFS transporters play important roles in secondary metabolism, and the ATP-binding cassette (ABC) contribute to polysaccharide transportation (Platta and Erdmann, 2007). In the S. sanghuang genome, a total of 299 secondary transporters were found, which are the most abundant among these transporters, 43 of them belonging to the MFS family, indicating MFS family proteins play roles in the secondary metabolite biosynthesis.
Discussion
Limited genetic information on S. sanghuang has hampered its pharmacological applications, although the fungus has antioxidation and antitumor effects (Feng et al., 2012; Liu et al., 2017). Thus, genome sequencing and gene annotation are a critical tool for the research community. Here, we present the genome sequence of S. sanghuang Kangneng generated by Illumina and the PacBio RSII long-read sequencing technologies. Because the species concept of sanghuang has long been misleading, six of the S. sanghuang strains used for large scale industrial production were subjected to phylogenetic analysis, which confirmed that these six strains are the true sanghuang, according to recent molecular phylogenetic studies (Zhou et al., 2015; Han et al., 2016). S. sanghuang Kangneng was selected for genome sequencing as it is the most widely cultured among these six strains in China.
The total assembly size was 34.5 Mb, which was lower than that of the other white rot fungi, such as G. lucidum and Lentinus tigrinus (Chen et al., 2012; Wu et al., 2018). The assembly genome consisted of 37 scaffolds with a 47.95% GC content, which is similar to Penicillium subrubescens but contrary to that of the most fungi with a >50% GC content (Peng et al., 2017; Wu et al., 2018). In total, 11,310 gene models were predicted, which accounted for 63.22% of the whole genome sequence length, while only ∼21.74% formed the “Unknown” regions, including promoter, terminator, and other regulatory regions of the genes, suggesting high gene density and low intergenic regions in the genome. The majority of the repetitive content in the S. sanghuang genome was made up of TEs (14.32%), and among these TEs, the proportion of long terminal repeats (LTRs) was 9.84%, suggesting LTRs may play critical roles in fungal asexual and sexual development.
Main bioactive components identified from S. sanghuang are polysaccharides, flavonoids, and triterpenoids (Cai et al., 2019). Genes involved in polysaccharide, flavonoid, and triterpenoid biosynthesis were identified in this study. As other eukaryotes, S. sanghuang triterpenoids are synthesized by the MVA pathway (Chen et al., 2012). For polysaccharide biosynthesis, 1,3-β-glucan synthases and seven β-glucan biosynthesis associated proteins were found in the fungus; these genes play key roles in the biosynthesis of 1,6-β-glucans and are well conserved in different fungi (Shahinian and Bussey, 2000; Douglas, 2001; Chen et al., 2012). S. sanghuang was considered to be capable of yielding flavonoids; 7 putative core genes for flavonoid biosynthesis were found based on a previous study of the plants because there have been few reports on fungal flavonoid biosynthesis pathways. CHS enzyme was not found in the S. sanghuang genome, suggesting that there is a different catalytic mechanism for naringenin chalcone production in the fungus, which needs to be further elucidated. Moreover, the expression of the genes involved in the biosynthesis of these main components was detected at different stages of the fungus (mycelium and fruiting body), and the results show that the fungi may produce more flavonoids and polysaccharides in the mycelial stages than in the fruiting body stages. Furthermore, some NRPs, PKs, and terpenes biosynthesis associated genes were found in this paper, suggesting that these compounds exist in S. sanghuang and their synthesis might be tightly regulated.
Conclusion
We have sequenced and analyzed the genome of the medicinal macrofungus, S. sanghuang Kangneng, which has been widely cultured in China. The exposition of the S. sanghuang genome has allowed us to study the biosynthesis of the pharmacologically active compounds produced by these medicinal fungi. Research on the secondary metabolite biosynthesis genes in the S. sanghuang genome will accelerate the industrial application of the bioactive molecules. Therefore, information on the S. sanghuang genome could contribute to its medicinal applications.
Data Availability Statement
The datasets generated for this study can be found in the genome assembly of Sanghuangporus sanghuang Kangneng has been deposited at NCBI with accession no. PRJNA564179.
Author Contributions
YW and AC conceived and designed the study. YS, YW, AC, HG, and JZ wrote the manuscript. YS, HG, JZ, HL, KW, SZ, PX, QZ, HZ, ZX, and XW performed the experiments. QZ, XW, and HZ analyzed the data.
Funding
This work was supported by the Natural Science Foundation of Anhui Province (1808085QH250), the Nature Science Foundation of Anhui University of Chinese Medicine (2017fyzd006), the Science and Technology Project of Jiangsu Province (Grant No. BE2017352), and the Major Science and Technology Projects of Anhui Province (Grant No. 17030801019).
Conflict of Interest
KW, SZ, PX, and ZX were employed by Jiangsu Konen Biological Engineering Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.03035/full#supplementary-material
Footnotes
- ^ http://www.mbio.ncsu.edu/bioedit/bioedit.html
- ^ https://www.megasoftware.net/download_form
- ^ https://fungismash.secondarymetabolites.org/#!/start
References
Bayram, O., Krappmann, S., Ni, M., Bok, J. W., Helmstaedt, K., Valerius, O., et al. (2008). VelB/VeA/LaeA complex coordinates light signal with fungal development and secondary metabolism. Science 320, 1504–1506. doi: 10.1126/science.1155888
Cai, C., Ma, J., Han, C., Jin, Y., Zhao, G., and He, X. (2019). Extraction and antioxidant activity of total triterpenoids in the mycelium of a medicinal fungus, Sanghuangporus sanghuang. Sci. Rep. 9:7418. doi: 10.1038/s41598-019-43886-43880
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2009). The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238. doi: 10.1093/nar/gkn663
Chen, F., Tholl, D., Bohlmann, J., and Pichersky, E. (2011). The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 66, 212–229. doi: 10.1111/j.1365-313X.2011.04520.x
Chen, S., Xu, J., Liu, C., Zhu, Y., Nelson, D. R., Zhou, S., et al. (2012). Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 3:913. doi: 10.1038/ncomms1923
Choi, Y. W., Hyde, K. D., and Ho, W. (1999). Single spore isolation of fungi. Fungal. Divers. 3, 29–38.
Corre, C., and Challis, G. L. (2009). New natural product biosynthetic chemistry discovered by genome mining. Nat. Prod. Rep. 26, 977–986. doi: 10.1039/B713024B
Cui, B. (2014). Progress on the species of medicinal fungus Inonotus sanghuang. J. Beijing For. Univ. 36, 1–6.
Dai, Y.-C. (2019). Whole genome sequence of Auricularia heimuer (Basidiomycota. Fungi), the third most important cultivated mushroom worldwide. Genomics 111, 50–58. doi: 10.1016/j.ygeno.2017.12.013
Douglas, C. M. (2001). Fungal beta(1,3)-D-glucan synthesis. Sabouraudia 39, 55–66. doi: 10.1080/mmy.39.1.55.66
Falcone Ferreyra, M. L., Rius, S. P., and Casati, P. (2012). Flavonoids: biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 3:222. doi: 10.3389/fpls.2012.00222
Feng, Y., Zhang, D. Q., Zhou, N., Jiang, B., and Kang, X. L. (2012). Five anthraquinone derivatives in siji sanghuang tablets determined by HPLC. Chin. J. Exp. Trad. Med. Formulae 7, 96–99.
Floudas, D., Binder, M., Riley, R., Barry, K., Blanchette, R. A., Henrissat, B., et al. (2012). The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 336, 1715–1719. doi: 10.1126/science.1221748
Han, J. G., Hyun, M. W., Kim, C. S., Jo, J. W., Cho, J. H., Lee, K. H., et al. (2016). Species identity of Phellinus linteus (sanghuang) extensively used as a medicinal mushroom in Korea. J. Microbiol. 54, 290–295. doi: 10.1007/s12275-016-5520-5522
Hur, H. (2008). Cultural characteristics and log-mediated cultivation of the medicinal mushroom, Phellinus linteus. Mycobiology 36, 81–87. doi: 10.4489/MYCO.2008.36.2.081
Hwang, H. J., Kim, S. W., Lim, J. M., Joo, J. H., Kim, H. O., Kim, H. M., et al. (2005). Hypoglycemic effect of crude exopolysaccharides produced by a medicinal mushroom Phellinus baumii in streptozotocin-induced diabetic rats. Life Sci. 76, 3069–3080. doi: 10.1016/j.lfs.2004.12.019
Ikekawa, T., Nakanishi, M., Uehara, N., Chihara, G., and Fukuoka, F. (1968). Antitumor action of some basidiomycetes, especially Phellinus linteus. GANN Jap. J. Cancer Res. 59, 155–157.
Jang, B. S., Kim, J. C., Bae, J. S., Rhee, M. H., Jang, K. H., Song, J. C., et al. (2004). Extracts of Phellinus gilvus and Phellinus baumii inhibit pulmonary inflammation induced by lipopolysaccharide in rats. Biotechnol. Lett. 26, 31–33. doi: 10.1023/b:bile.0000009456.63616.32
Lee, D., Kim, S. C., Kim, D., Kim, J. H., Park, S. P., Ryu, Y. C., et al. (2011). Screening of Phellinus linteus, a medicinal mushroom, for anti-viral activity. J. Korean Soc. Appl. Bi 54, 475–478. doi: 10.3839/jksabc.2011.073
Lin, W. C., Deng, J. S., Huang, S. S., Wu, S. H., Chen, C. C., Lin, W. R., et al. (2017). Anti-inflammatory activity of Sanghuangporus sanghuang mycelium. Int. J. Mol. Sci. 18:E347. doi: 10.3390/ijms18020347
Liu, K., Xiao, X., Wang, J., Chen, C. Y. O., and Hu, H. (2017). Polyphenolic composition and antioxidant, antiproliferative, and antimicrobial activities of mushroom Inonotus sanghuang. LWT Food Sci. Technol. 82, 154–161. doi: 10.1016/j.lwt.2017.04.041
Magrane, M. UniProt Consortium. (2011). UniProt knowledgebase: a hub of integrated protein data. Database 2011:bar009. doi: 10.1093/database/bar009
Mizuno, M., and Nishitani, Y. (2013). Immunomodulating compounds in Basidiomycetes. J. Clin. Biochem. Nutr. 52, 202–207. doi: 10.3164/jcbn.13-13
Peng, M., Dilokpimol, A., Makela, M. R., Hilden, K., Bervoets, S., Riley, R., et al. (2017). The draft genome sequence of the ascomycete fungus Penicillium subrubescens reveals a highly enriched content of plant biomass related CAZymes compared to related fungi. J. Biotechnol. 246, 1–3. doi: 10.1016/j.jbiotec.2017.02.012
Platta, H. W., and Erdmann, R. (2007). The peroxisomal protein import machinery. FEBS Lett. 581, 2811–2819. doi: 10.1016/j.febslet.2007.04.001
Saito, K., Yonekura-Sakakibara, K., Nakabayashi, R., Higashi, Y., Yamazaki, M., Tohge, T., et al. (2013). The flavonoid biosynthetic pathway in Arabidopsis: structural and genetic diversity. Plant Physiol. Biochem. 72, 21–34. doi: 10.1016/j.plaphy.2013.02.001
Seppey, M., Manni, M., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:btv351. doi: 10.1093/bioinformatics/btv351
Shahinian, S., and Bussey, H. (2000). beta-1,6-glucan synthesis in Saccharomyces cerevisiae. Mol. Microbiol. 35, 477–489. doi: 10.1046/j.1365-2958.2000.01713.x
Shao, Y., Tang, J., Chen, S., Wu, Y., Wang, K., Ma, B., et al. (2019). milR4 and milR16 mediated fruiting body development in the medicinal fungus Cordyceps militaris. Front. Microbiol. 10:83. doi: 10.3389/fmicb.2019.00083
Shibata, S., Nishikawa, Y., Cheng, F. M., Fukuoka, F., and Nakanishi, M. (1968). Antitumor studies on some extracts of Basidiomycetes. GANN Jap. J. Cancer Res. 59, 159–161.
Taji, S., Yamada, T., Wada, S. I., Tokuda, H., Sakuma, K., and Tanaka, R. (2008). Lanostane-type triterpenoids from the sclerotia of Inonotus obliquus possessing anti-tumor promoting activity. Eur. J. Med. Chem. 43, 2373–2379. doi: 10.1016/j.ejmech.2008.01.037
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinform. 4:41. doi: 10.1186/1471-2105-4-41
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, Y. O., and Borodovsky, M. (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 18, 1979–1990. doi: 10.1101/gr.081612.108
Tsai, J. N., Ann, P. J., Liou, R. F., Hsieh, W. H., and Ko, W. H. (2017). Phellinus noxius: molecular diversity among isolates from Taiwan and its phylogenetic relationship with other species of Phellinus based on sequences of the ITS region. Bot. Stud. 58:9. doi: 10.1186/s40529-017-0162-161
Wang, G. J., Tsai, T. H., Chang, T. T., Chou, C. J., and Lin, L. C. (2009). Lanostanes from Phellinus igniarius and their iNOS inhibitory activities. Planta. Med. 75, 1602–1607. doi: 10.1055/s-0029-1185813
Wilson, D., Charoensawan, V., Kummerfeld, S. K., and Teichmann, S. A. (2008). DBD-taxonomically broad transcription factor predictions: new content and functionality. Nucleic Acids Res. 36, D88–D92. doi: 10.1093/nar/gkm964
Wu, B., Xu, Z., Knudson, A., Carlson, A., Chen, N., Kovaka, S., et al. (2018). Genomics and development of Lentinus tigrinus: a white-rot wood-decaying mushroom with dimorphic fruiting bodies. Genome Biol. Evol. 10, 3250–3261. doi: 10.1093/gbe/evy246
Wu, M. D., Cheng, M. J., Chen, Y. L., Chan, H. Y., Hsieh, S. Y., Chen, I. C., et al. (2019). Secondary metabolites from the fermented whole broth of fungal strain Sanghuangporus sanghuang. Chem. Nat. Compd. 55, 36–40. doi: 10.1007/s10600-019-02610-2610
Wu, S. H., Dai, Y. C., Hattori, T., Yu, T. W., Wang, D. M., Parmasto, E., et al. (2012). Species clarification for the medicinally valuable ‘sanghuang’ mushroom. Bot. Stud. 53, 135–149.
Yan, J. K., Wang, Y. Y., Ma, H. L., and Wang, Z. B. (2016). Ultrasonic effects on the degradation kinetics, preliminary characterization and antioxidant activities of polysaccharides from Phellinus linteus mycelia. Ultrason. Sonochem. 29, 251–257. doi: 10.1016/j.ultsonch.2015.10.005
Zhang, C., Deng, W., Yan, W., and Li, T. (2018). Whole genome sequence of an edible and potential medicinal fungus. Cordyceps. guangdongensis. G3, 8, 1863–1870. doi: 10.1534/g3.118.200287
Zhang, H., Ma, H., Liu, W., Pei, J., Wang, Z., Zhou, H., et al. (2014). Ultrasound enhanced production and antioxidant activity of polysaccharides from mycelial fermentation of Phellinus igniarius. Carbohyd. Polym. 113, 380–387. doi: 10.1016/j.carbpol.2014.07.027
Zhao, Z., Liu, H., Wang, C., and Xu, J. R. (2013). Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics 14:274. doi: 10.1186/1471-2164-14-274
Zhou, L. W., Vlasák, J., Decock, C., Assefa, A., Stenlid, J., Abate, D., et al. (2015). Global diversity and taxonomy of the Inonotus linteus complex (Hymenochaetales, Basidiomycota): Sanghuangporus gen. nov., Tropicoporus excentrodendri and T. guanacastensis gen. et spp. nov., and 17 new combinations. Fungal Divers. 77, 335–347. doi: 10.1007/s13225-015-0335-338
Keywords: Sanghuang, genomics, CAZymes, bioactive compound biosynthesis, transporters
Citation: Shao Y, Guo H, Zhang J, Liu H, Wang K, Zuo S, Xu P, Xia Z, Zhou Q, Zhang H, Wang X, Chen A and Wang Y (2020) The Genome of the Medicinal Macrofungus Sanghuang Provides Insights Into the Synthesis of Diverse Secondary Metabolites. Front. Microbiol. 10:3035. doi: 10.3389/fmicb.2019.03035
Received: 07 September 2019; Accepted: 17 December 2019;
Published: 14 January 2020.
Edited by:
Rakesh Sharma, Institute of Genomics and Integrative Biology (CSIR), IndiaReviewed by:
John G. Gibbons, University of Massachusetts Amherst, United StatesZheng Wang, Yale University, United States
Copyright © 2020 Shao, Guo, Zhang, Liu, Wang, Zuo, Xu, Xia, Zhou, Zhang, Wang, Chen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anhui Chen, Y2hlbmFoMjAxQDE2My5jb20=; Yulong Wang, eXVsb25nd2FAbXR1LmVkdQ==