 Jayanti Saha1
Jayanti Saha1 Ayon Pal
Ayon Pal- 1Microbiology & Computational Biology Laboratory, Department of Botany, Raiganj University, Raiganj, India
- 2Mycology & Plant Pathology Laboratory, Department of Botany, Raiganj University, Raiganj, India
Soil is a diversified and complex ecological niche, home to a myriad of microorganisms particularly bacteria. The physico-chemical complexities of soil results in a plethora of physiological variations to exist within the different types of soil dwelling bacteria, giving rise to a wide variation in genome structure and complexity. This serves as an attractive proposition to analyze and compare the genome of a large number soil bacteria to comprehend their genome complexity and evolution. In this study a combination of codon usage and molecular phylogenetics of the whole genome and key housekeeping genes like infB (translation initiation factor 2), trpB (tryptophan synthase, beta subunit), atpD (ATP synthase, beta subunit), and rpoB (RNA polymerase, beta subunit) of 92 soil bacterial species spread across the entire eubacterial domain and residing in different soil types was performed. The results indicated the direct relationship of genome size with codon bias and coding frequency in the studied bacteria. The codon usage profile demonstrated by the gene trpB was found to be relatively different from the rest of the housekeeping genes with a large number of bacteria having a greater percentage of genes with Nc values less than the Nc of trpB. The results from the overall codon usage bias profile also depicted that the codon usage bias in the key housekeeping genes of soil bacteria was majorly due to selectional pressure and not mutation. The analysis of hydrophobicity of the gene product encoded by the rpoB coding sequences demonstrated tight clustering across all the soil bacteria suggesting conservation of protein structure for maintenance of form and function. The phylogenetic affinities inferred using 16S rRNA gene and the housekeeping genes demonstrated conflicting signals with trpB gene being the noisiest one. The housekeeping gene atpD was found to depict the least amount of evolutionary change in the soil bacteria considered in this study except in two Clostridium species. The phylogenetic and codon usage analysis of the soil bacteria consistently demonstrated the relatedness of Azotobacter chroococcum with different species of the genus Pseudomonas.
Introduction
Soil, from the biological point of view, can be considered as one of the great ecological component or a system where a vast majority of microorganisms exists, albeit unexplored (Daniel, 2005; Stefanis et al., 2013; Jansson and Hofmockel, 2018). Soil hosts a wide range of microbial community that has a profound effect on the properties of soil itself (Handelsman et al., 1998; Aislabie et al., 2013), and exerts a great influence on the organism's survival and gradual evolution while bestowing unique biological properties (Delgado and Gómez, 2016).
Soil has always been an enigma, a terra incognita with diversified habitat and diverse array of interactions (Whitman et al., 1998; Huang et al., 2001; Fierer et al., 2014; Andújar et al., 2017) but scientific understanding of the microbial world in soil is somewhat poor, particularly that of soil bacteria (Fierer and Jackson, 2006; Nesme et al., 2016; Jansson and Hofmockel, 2018). There exists a great variability in the microbial population of soil and the adeptness to explore the soil microbial communities could provide answers related to different functional and phylogenetic aspects of soil microorganisms (Nesme et al., 2016). In general, soil biomass is occupied by about 70% microorganisms which helps in the decomposition of the soil organic matter and releasing the essential minerals on the soil surface (Hayat et al., 2010; Jacoby et al., 2017). Microbes from three different domains of life are found in soil (Rao, 1995), and the largest one to dominate the soil habitat are members of the domain Eubacteria. Each of the different types of bacteria residing in the soil owns distinct morphological, physiological, biochemical, and ecological characteristics, and the variation in the structure and composition of different soil types impart a great influence on the diversity of the microbial community that the soil retain (Fierer and Jackson, 2006; Berg and Smalla, 2009). Hence, microbes from different soil habitat must possess a variation in the genome structure and function (Martin-Laurent et al., 2001; Roesch et al., 2007) to cope with this variability. That's why the ancestry and evolution of the microbial population residing in different types of soil needs to be better understood (Mocali and Benedetti, 2010). Many studies relating to soil microbial ecology has already revealed the immense diversity of soil bacteria, and now it is a very common hypothesis that plant species as well as the soil type have a substantial influence on the structure and function of the rhizosphere associated bacterial populations (Berg and Smalla, 2009; Pérez-Valera et al., 2015). Some of the major soil inhabiting bacterial genera which are of great interest include different species of the genera Bacillus, Clostridium, Pseudomonas, Streptomyces, Micromonospora, Nitrosomonas, Nitrobacter, Vibrio, and Thiobacillus (Shimp et al., 1993; Lehman et al., 2015).
Among the different types of microbes studied, soil dwelling bacteria offers a lot of promise but a lot of it still remains unexplored. A considerable large scale and robust comparative genomic and phylogenetic analysis of such bacteria is still deficient. Now-a-days the accessibility of the complete genome sequences of many soil borne bacteria provides a scope to undertake various bioinformatics based approaches (Chen et al., 2002; Mardis, 2008). With the rapid and continuous advancements in sequencing technology and data analysis, the complexity of the microbial world is becoming understandable, albeit slowly (Winsley et al., 2012). The recent widespread availability and affordability of automated sequencing has gained much more attention for cataloging soil bacterial diversity (Will et al., 2010) in the era of Next Generation Sequencing (Clooney et al., 2016). Simultaneously the dramatic progress in computing power provides greater opportunities for a large scale and robust in silico based study. The acquisition of large amount of sequence data from genes as well as proteins from several repositories (Kaminuma et al., 2010; Chen et al., 2017) is paving the way for comparative genomics in a diverse group of microorganisms (Rosselli et al., 2016) whose major objective is to decipher the genetic variation among different species, and determine the genetic relatedness between closely related organisms. This paves the way to reveal the evolutionary relationship between organisms with the help of conserved sequences that are generally found within the last common ancestors of those organisms (Ivanova et al., 2003; Herring et al., 2006).
Phylogenetic inference using sequence data is considered an important and reliable tool (Ludwig and Schleifer, 1994). To focus on bacterial phylogeny, ribosomal RNA is often envisaged as one of the best parameter due to its ubiquitous and informative nature (Morales et al., 2009) but it's not without its pitfalls. Phylogenetic analysis based on the 16S rRNA gene is generally used to articulate the evolutionary history (Rajendhran and Gunasekaran, 2011) but, several discrepancies have been observed in 16S rRNA based phylogeny (Vásquez-Ponce et al., 2018). Some of the major drawbacks include mosaicism due to horizontal gene transfer and recombination events (Eardly et al., 1996; Schouls et al., 2003), presence of polymorphic genes (Janda and Abbott, 2007; Lang et al., 2013), intra-genomic heterogeneity of the 16S rRNA genes, presence of multiple copies of the rRNA operon (Wang et al., 1997; Klappenbach et al., 2000), etc. To overcome these, the housekeeping genes are now considered to be better molecular markers and as a result these have been widely applied to infer proper phylogenetic relationships (Lai et al., 2014; Tyler et al., 2018). Due to the evolutionary conserved nature, some housekeeping genes like atpD, infB, gyrB, rpoB, trpB have been increasingly utilized to improve the discriminatory power in determining the phylogenetic relationships (Martens et al., 2008). Each and every house keeping gene possess the functional constancy and conservation as they encode core metabolic enzymes, and are more or less universally present in a plethora of organisms. An excellent feature demonstrated by the housekeeping genes is the power to overcome conflicting signals from horizontal gene transfer and recombination during phylogeny inference (Case et al., 2007). Therefore, it is a very resourceful proposition to compare the sequences of several housekeeping genes as well as the 16S rRNA genes portraying as a reference for evaluating a comprehensive bacterial phylogeny, and also for investigating the genetic as well as physiological relatedness within the species. A technique which combines analysis of several housekeeping genes called multilocus sequence analysis (MLSA) has emerged as a powerful and pragmatic molecular method to assess pertinent phylogenetic information in the field of polyphasic taxonomy (Das et al., 2014; Vásquez-Ponce et al., 2018). Several studies have been carried out using MLSA in order to get better resolution regarding the phylogenetic analysis and inter-species relatedness in a host of soil bacterial genera like Arthrobacter (Liu et al., 2018), Borrelia (Margos et al., 2009), Burkholderia (Estrada-De Los Santos et al., 2013), Ensifer (Martens et al., 2008), Micromonospora (Carro et al., 2012), Streptomyces (Rong and Huang, 2010), Nocardia (Barcellos et al., 2007; McTaggart et al., 2010; Serrano et al., 2010; Busse and Wieser, 2014), and some other rhizospheric bacteria like Mesorhizobium (Degefu et al., 2011) and Bradyrhizobium (Delamuta et al., 2012).
In addition to molecular phylogenetics, codon usage bias (CUB) within and across the genome also stands as an increasingly demanding factor contributing significantly to gene and genome evolution. CUB is the phenomenon of usage of certain specific codons more frequently than other synonymous codons (Quax et al., 2015), and universally affects the genome of all living beings (Yannai et al., 2018). This is observed mainly due to the degenerate property of the genetic code (Salim and Cavalcanti, 2008; Sharp et al., 2010). It represents balance between translational selection and mutational forces leading to translational efficiency of genes (Sharp et al., 2010; Plotkin and Kudla, 2011). Evidences suggest that strong CUB is generally observed in highly expressed genes (Bennetzen and Hall, 1982; Gouy and Gautier, 1982; Hershberg and Petrov, 2008; Salim and Cavalcanti, 2008; Frumkin et al., 2018), and it is also a means to fine tune the expression of genes (Quax et al., 2015; Sahoo et al., 2019).
Soil is a crucial but diversified habitat, and a wide variety of bacteria lives in soil, demonstrating massive variation in terms of morphology, physiology and biochemistry. Depending on the soil composition and physical attributes the biotic components of soil are going to be diverse but, there is a possibility that every soil dwelling bacteria might have a unifying signature within their genome indicating their origin and evolution in the soil habitat. In this study, we have made an extensive comparative codon usage and phylogenetic analysis of 92 soil dwelling bacterial species distributed within 36 genera spanning the entire eubacterial domain. These 92 bacterial species are unified by the fact that they dwell in soil, albeit of diverse types. The key objectives of this study was to find out if there exists any signature codon usage profile within the genome and major housekeeping genes of soil bacteria. The housekeeping genes are part of core or minimal set of genes primarily responsible for maintaining critical cellular functions (Wei et al., 2018), mainly located on chromosomes with orthologs in related species (Bittner et al., 2007; Villaseñor et al., 2011). The four key housekeeping genes considered in this study are atpD, infB, rpoB, and trpB. These genes play a significant role in genetic information processing and metabolic activities of bacteria (Wen et al., 2016). The atpD gene encodes the beta subunit of ATP synthase that produces ATP from ADP in the presence of a proton gradient across the membrane, whereas, the infB gene encodes the translation initiation factor II, which is essential for prokaryotic protein synthesis initiation. The rpoB gene encodes the highly conserved portion of the bacterial beta subunit in DNA-dependent RNA polymerase enzyme whereas the trpB gene encodes the beta subunit of tryptophan synthase enzyme. In this study, we have also tried to deduce the phylogenetic affinities of the 92 bacterial species considered in this study both at the organismal as well as gene level. We have tried to capture whether the organismal phylogeny based on 16S rRNA gene is corroborated by the individual housekeeping genes or there lies any conflicting signal between them. In addition to this, we have also tried to find out whether the phylogenetic affinities demonstrated by the individual housekeeping genes are in sync with each other. In order to achieve this we have deliberately avoided the use of concatenated housekeeping gene sequences in constructing phylogenies (Thiergart et al., 2014), which have been a popular choice in constructing bacterial phylogeny (Baldauf et al., 2000; Brown et al., 2001; Wu and Eisen, 2008; Tambong et al., 2014). Studies utilizing codon usage parameters like codon adaptation index have been used in the past to detect environmental signatures in bacteria (Willenbrock et al., 2006) but, our study is the first one of its kind where we have adopted a bipartite combinational codon usage and phylogenetic analysis of whole genome as well as key housekeeping genes. This was done to obtain a better resolution into the intraspecific diversity and evolutionary relationships among the different soil dwelling bacteria.
Materials and Methods
Whole Genome Sequence Retrieval
The whole genome sequence of 92 soil bacteria were obtained from Integrated Microbial Genome Database (Markowitz et al., 2012) and NCBI genome database (Benson et al., 2006). Selection of these bacterial species were carried out after thoroughly confirming their soil dwelling nature in consultation with available literature including Bergey's Manual of Systematic Bacteriology (Garrity et al., 2001; Brenner et al., 2005; Vos et al., 2009; Krieg et al., 2010; Whitman et al., 2012), and the metadata associated with the respective genome data. Genomes of bacterial species with improper metadata regarding habitat and source of isolation, and incomplete annotation of the coding sequences were deliberately kept out of the analysis. Draft genomes of bacterial species were also not considered for the analysis. Species without strain demarcation were also left out since ‘strain' forms an integral component of the bacterial species concept (Rosselló-Mora and Amann, 2001). The selected 92 bacterial species considered in this study is spread across the entire Eubacterial domain belonging to the five different taxonomic classes (namely Proteobacteria, Firmicutes, Chlorobi, Actinobacteria, and Acidobacteria) spread over 20 different orders and 27 different families. A list of these organisms is given in Supplementary Table 1. Genes with correct initiation and termination codons were considered for every genome to minimize sampling error.
16S rRNA Gene and Housekeeping Gene Sequences Retrieval
The nucleotide sequences of 16S rRNA gene and that of the housekeeping genes atpD, infB, rpoB, and trpB of the 92 bacterial genera were mined from the whole genome sequences to study codon usage profile and construct multiple sequence alignment (MSA) to infer phylogenetic relationships.
Analysis of CUB
The parameters like effective number of codons or Nc (Wright, 1990), GC content at the third position of the codon or GC3 (Wright, 1990), hydrophobicity and gene length was calculated. Nc is one of the best and widely used measure that quantifies the extent to which the usage of a gene departs from the equal usage of synonymous codons (Fuglsang, 2006; Liu, 2013). It ranges from 20 to 61. A Nc value lower than 40 indicates codon usage bias and higher than 40 indicates equal likely usage of all synonymous codons (Botzman and Margalit, 2011; Pal et al., 2015; Wang et al., 2018; Khandia et al., 2019). GC3 has been reported to be linked with DNA flexibility and codon bias across prokaryotes (Babbitt et al., 2014) and also plays a regulatory role in gene expression and methylation (Tatarinova et al., 2013). In the present study, GC3 was calculated for both the whole genome and every housekeeping gene. Hydrophobicity is actually used for the prediction of the nature of cellular protein coded by the corresponding gene present within the genome. A value below zero indicates the presence of hydrophilic protein and above zero represents the presence of hydrophobic protein (Magdeldin et al., 2012). In our study, we have used this parameter to estimate the physical attribute hydrophobicity of the housekeeping gene products coded by rpoB, atpD, infB, and trpB gene. The Nc-plot depicting the correlation between Nc and GC3 (Wright, 1990) was constructed to determine the variation in inter-specific as well as inter genic synonymous codon usage pattern existing within the whole genome and the housekeeping genes of the soil bacteria. Nc plot is used extensively to elucidate the mechanistic forces shaping CUB, and heterogeneity of the gene using codon bias and base composition (Sun et al., 2016; Wang et al., 2018; Khandia et al., 2019). In this study, the above mentioned parameters were estimated for the whole genomes and housekeeping genes using INCA (Supek and Vlahovicek, 2004) and CodonW (Peden, 1999).
Generation of Multiple Sequence Alignments (MSAs)
MSAs of the 16S rRNA genes and the four housekeeping genes from the 92 bacterial species were prepared using the web interface of Clustal Omega hosted at the EMBL-EBI website (https://www.ebi.ac.uk/Tools/msa/clustalo/). Clustal Omega is a MSA tool that produces alignments between multiple sequences employing seeded guide trees and HMM profile-profile techniques (Sievers et al., 2011).
Phylogenetic Analysis
Phylogenetic inferences using the 16S rRNA gene and the four housekeeping genes were deduced using the maximum likelihood (ML) method. In terms of accuracy, time, and convenience, the ML method is regarded as one of the most appropriate tree estimating method providing information regarding evolutionary relationships (Mulet et al., 2010). Besides, this method utilizes likelihood function and can be used easily to detect the pair wise distances in the phylograms (Rong and Huang, 2014). Since evolutionary models (or models of nucleotide substitution) is a key component of molecular phylogenetics using methods, such as ML, for each gene, the optimal model of evolution was selected using the model test function of MEGA 6.0 (Kumar et al., 2008). The Bayesian Information Criterion (BIC) value has been regarded as an essential criteria for model selection and the models depicting the lowest BIC value were selected as optimal models (Posada and Buckley, 2004; Posada, 2009; Luo et al., 2010). The phylogenetic trees were constructed using MEGA 6.0 (Kumar et al., 2008). For the four housekeeping genes considered in this study, the General Time Reversible model (Nei and Kumar, 2000) with gamma (G) distributed rate variation among sites along with significant proportion of invariable sites (I) or GTR + G + I model was found to be the optimal one based on the BIC score. The 16S rRNA gene based phylogenetic tree was inferred based on the Kimura 2-parameter model (Kimura, 1980). The bootstrap consensus tree inferred from 1,000 replicates (Felsenstein, 1985) was taken to represent the evolutionary history of the analyzed taxa. The visualization and annotation of the phylogenetic trees were done using the online open source tool Interactive Tree of Life (iTOL) ver. 4.4.2 available at https://itol.embl.de/ (Letunic and Bork, 2007, 2019). The raw data containing the phylogenetic trees along with bootstrap support is provided as Supplementary Files in Newick format.
Results and Discussion
In this study, the codon usage profile of the whole genome along with the housekeeping gene of 92 soil dwelling bacterial species belonging to 36 different genera were thoroughly analyzed. Simultaneously, molecular phylogenetic analysis was carried out by comparing the standard 16S rRNA gene based tree in the backdrop with the phylograms obtained using four key housekeeping genes. A detailed list of the 92 selected bacterial species is given in Supplementary Table 1.
Analysis of CUB Pattern of Whole Genome
CUB analysis is a fitting proposition for understanding the functional evolution of genome within and between species. The CUB phenomenon has been found to control a wide range of cellular processes, such as translational efficiency, differential protein production and its subsequent folding (Quax et al., 2015; Song et al., 2017a; Zhang et al., 2018), and is being influenced by a plethora of factors but not restricted to GC content, gene length, gene hydropathy, gene function and expression, protein structure, mutational bias and compositional bias (Tuller et al., 2010; Plotkin and Kudla, 2011; Xu et al., 2011, 2013; Chithambaram et al., 2014). In this study, the primary codon usage parameters like Nc, GC3, gene length and hydrophobicity of 391,415 gene sequences, representing the whole genome, of 92 soil dwelling bacterial species were estimated. It was found that the average genomic Nc (Ncavg) of all the studied taxa lies between 29 and 54. The highest Ncavg value of 53.58 with a standard deviation (SD) of ±4.32 was depicted by Nitrosomonas communis Nm2, a Gram negative Mediterranean soil dwelling organism. About 52 species from different soil types were found to demonstrate Ncavg above 40. This is suggestive of a relatively lower codon bias existing within the genome of these species (Botzman and Margalit, 2011; Pal et al., 2015; Khandia et al., 2019). The lowest Ncavg value (29.99, SD = ±3.91) was observed in Micrococcus luteus NCTC 2665, a Gram positive soil bacterium indicating high level of codon bias within its genome. Other organisms that demonstrated higher genomic codon usage bias includes species of Micromonospora, Clostridium, Streptomyces, Achromobacter, Acidiphilium, Nocardia, etc. The standard deviation within the genomic Nc of each organism was also estimated. The lowest deviation of Nc within the genome was observed in Clostridium butyricum JKY6D1 (SD = ±3.34), a Gram positive bacteria isolated from pit mud whereas the highest deviation in Nc was observed in Azotobacter chroococcum NCIMB 8003 (SD = ±7.17), a Gram negative soil bacterium. The results of the whole genome CUB analysis is given in Table 1.
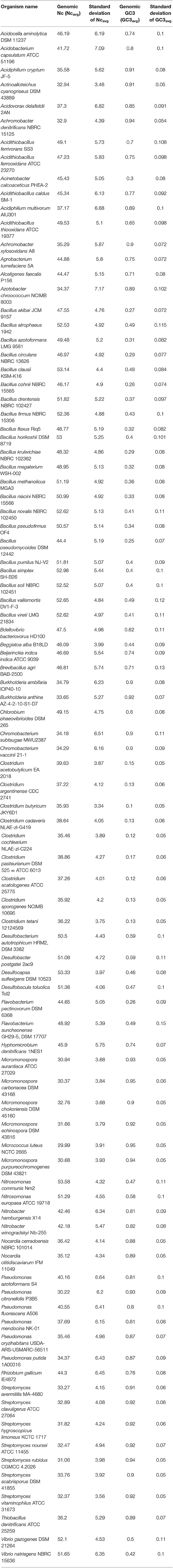
Table 1. List of genomic Nc (Ncavg) and genomic GC3 (GC3avg) along with standard deviation in the 92 species of soil bacteria analyzed in this study.
The average genomic GC3 content (GC3avg) of the organisms under study was found to range between 10 and 95%. Clostridium butyricum JKY6D1 with a GC3avg value of 10% demonstrated the lowest average genomic GC3 content. On the other hand, Micrococcus luteus NCTC 2665 and Micromonospora carbonacea DSM 43168 depicted the highest GC3avg score of 95%. Both these organisms are Gram positive in nature.
In this study, we have tried to determine if the Ncavg is related to the genome size (measured in terms of base count), coding frequency and GC content of the genome (data shown in Supplementary Table 2). To explore this relationship Spearman's rank order correlation was performed. Our results demonstrated that the Ncavg is significantly negatively correlated with genome size (ρ = −0.442, p < 0.01), coding bases (ρ = −0.46, p < 0.01) and GC content (ρ = −0.577, p < 0.01), respectively in the soil bacteria. Alternatively, it implies that the genomic codon usage bias bears a positive correlation with the genome size. We also observed that in these 92 bacterial species there is a significant positive correlation between the genome size and the coding frequency of the genome (ρ = 0.992, p < 0.01). This simultaneously suggests that for these species, there is no significant superfluity of nucleotides in the genome, since majority are engaged in coding function. Coding frequency was also found to correlate significantly in a positive manner with the GC content of the genome (ρ = 0.84, p < 0.01). The increasing GC content within the genome is thus suggestive of better genomic coding efficiency and increased codon bias within these 92 bacterial species. Our results thus reflect a unified trend in the soil dwelling species where genome size is found to be proportional to the codon bias and the coding frequency.
In most of the species a significant negative correlation between Nc and GC3 was depicted, a trend well in line with the standard notion of Nc-GC3 relationship (Hassan et al., 2009; Pandit and Sinha, 2011; Prabha et al., 2012; Malakar et al., 2016, 2019; Song et al., 2017b). The results of the correlation analysis is shown in Supplementary Table 3. Out of the 92 species, 51 were found to depict such a trend, whereas 32 species exhibited substantial positive correlation between Nc and GC3. An interesting observation was the lack of substantial relationship between Nc and GC3 in the two organisms Bdellovibrio bacteriovorus HD100 and Nitrosomonas communis Nm2. Both these bacteria are terrestrial Gram negative soil dwellers, aerobic, free living and isolated from Mediterranean soil. The dispersion within the GC3 of the 92 organisms showed that the GC3 value deviated from 5 to 15% within the 92 organisms with most of the organisms depicting about 5% deviation. The highest deviation in GC3 was demonstrated by Flavobacterium suncheonense GH29-5, DSM 17707 (15%) which is a Gram negative, greenhouse soil living bacterium. But, F. pectinovorum DSM 6368 though a member of the genus Flavobacterium demonstrated only 9% deviation in its GC3 content. Genera like Clostridium, Pseudomonas, Micromonospora, Bacillus, and Streptomyces exhibited similar trend.
Comparative CUB Profile Analysis of Key Housekeeping Genes in Soil Bacteria
To overcome several discrepancies and conflicting signals like mosaicism due to horizontal gene transfer, instances of recombination and presence of polymorphic genes in 16S rRNA based phylogenetic analysis, several housekeeping genes are considered as potent tool in determining bacterial taxonomy (Soler et al., 2004). Each and every housekeeping gene possess the functional constancy and conservation as they encode core metabolic enzymes and are generally present in all common members (Naser et al., 2005). In this study, the housekeeping genes atpD, infB, rpoB, and trpB have been utilized to improve the discriminatory power in determining the phylogenetic relationships and resolve the phylogenetic discrepancies cropping out while using 16SrRNA genes. All these housekeeping genes are closely involved with the core metabolic pathways and genetic information processing pathways found in bacteria. The findings of this study have been detailed in the succeeding section.
Comparative CUB Analysis of rpoB Gene From Soil Bacteria
The CUB profile of rpoB gene was found to reflect the genomic CUB profile. The lowest Nc value of rpoB (Nc = 26) was depicted by the organism Micrococcus luteus NCTC 2665 which also depicted the lowest Ncavg of 29.99. A similar trend was also observed in the genus Nitrosomonas which exhibited the highest genic Nc value for rpoB (N. europaea ATCC 19718, Nc = 51.58) and genome (N. communis Nm2, Ncavg = 53.58). The lowest GC3 content for rpoB was demonstrated by the genus Clostridium which also had the lowest genomic GC3 content. The organism Micrococcus luteus NCTC 2665 did not show the highest GC3 value for rpoB although it possessed a significantly high mean genomic GC3 content (96.33%). The rpoB of Acidiphilium multivorum was found to possess the highest GC3 content (97%) which is somewhat incongruent with respect to its genomic GC3 pattern. The CUB profile of rpoB have been illustrated in Tables 2, 3. In case of hydrophobicity of rpoB gene product, the lowest hydrophobicity (−0.437) was reported by Bacillus akibai JCM 9157 (Gram positive, free living soil bacteria) while the Gram negative Pseudomonads like Pseudomonas citronellolis P3B5, P. putida 1A00316, and P. fluorescens A506 depicted the maximum hydrophobicity ranging from −0.269 to −0.284. A scattered plot showing the distribution of hydrophobicity values of the protein encoded by rpoB gene is given in Figure 1.
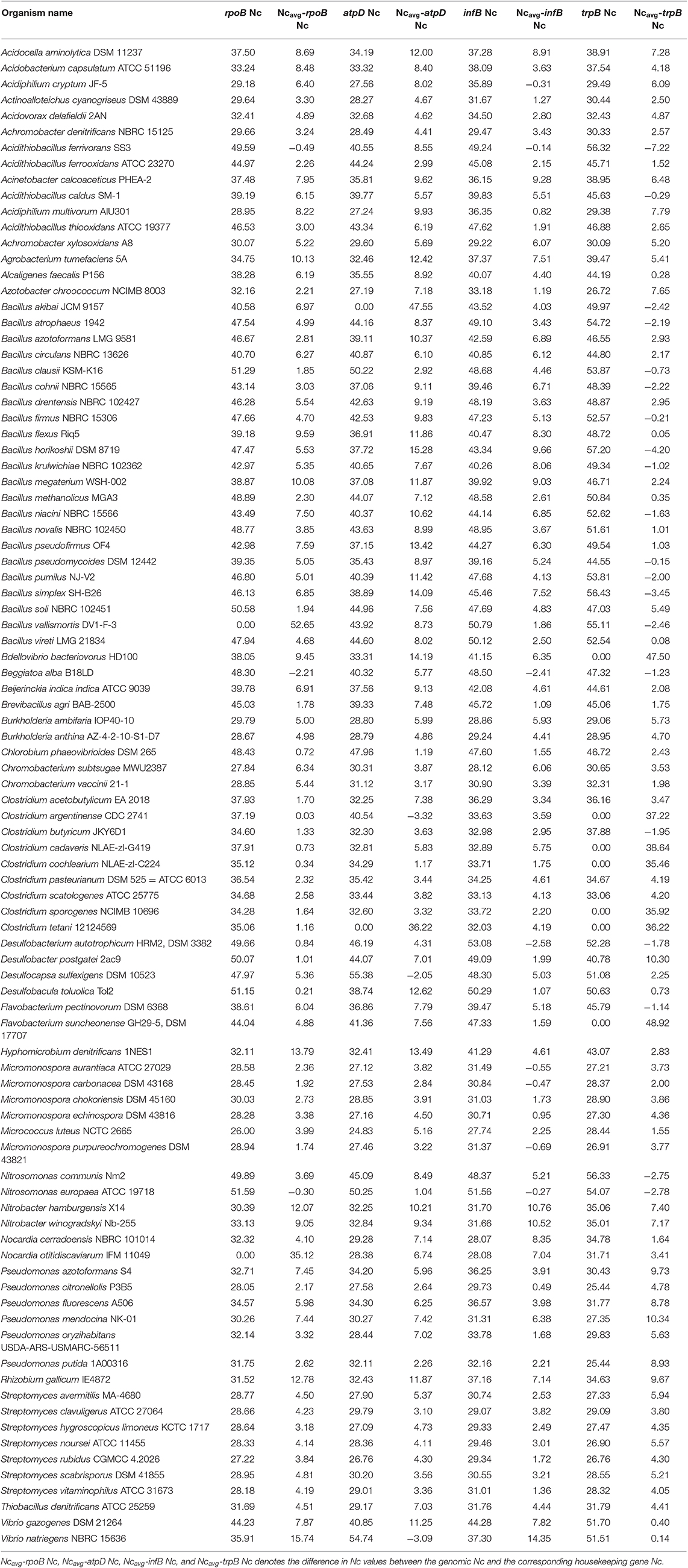
Table 2. Nc profile of the four housekeeping genes in 92 soil bacterial species.
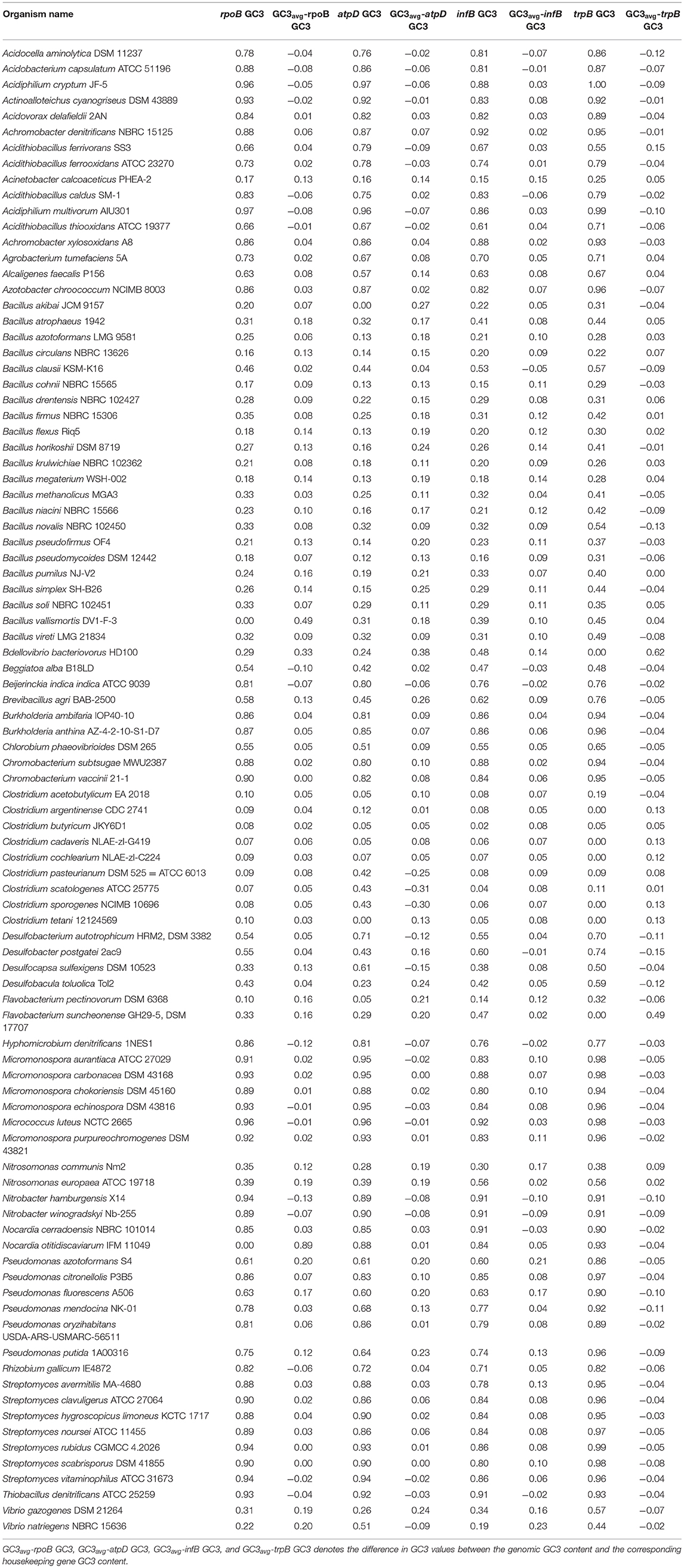
Table 3. GC3 profile of the four housekeeping genes in 92 soil bacterial species.
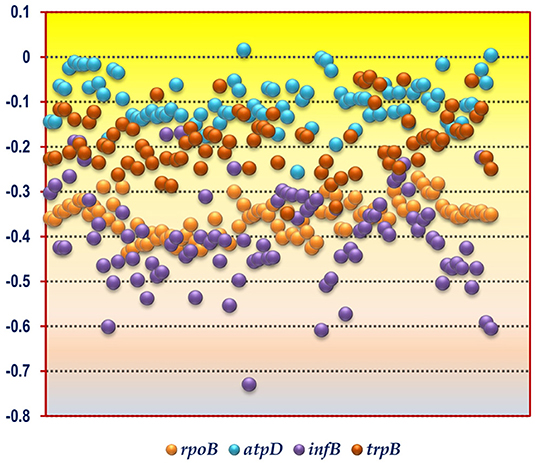
Figure 1. A scattered plot depicting the hydrophobicity profile of the gene products encoded by the four housekeeping genes rpoB, atpD, infB, and trpB from the soil bacterial species considered in this study. The y-axis corresponds to the hydrophobicity value whereas the x-axis corresponds to the bacterial species sorted in alphabetical order as given in Table 1.
Comparative CUB Analysis of atpD Gene From Soil Bacteria
The codon usage parameters for the atpD gene was also found to be similar to that of the rpoB gene in terms of Nc value; the lowest Nc value (24.83) of atpD was demonstrated by the same organism Micrococcus luteus NCTC 2665. M. luteus NCTC 2665 thus display a consistency in terms of Nc pattern. The highest Nc value (55.37) of atpD was exhibited by Desulfocapsa sulfexigens DSM 10523. In this regard Nitrosomonas also exhibited a significantly higher Nc value (50.24) for atpD gene. While Flavobacterium pectinovorum DSM 6368 and Acidiphilium cryptum JF-5 were found to reflect the lowest (4%) and the highest (97%) GC3 value, respectively, Micrococcus luteus NCTC 2665 demonstrated a significantly high GC3 content (96%). The CUB profile of atpD of all the studied taxa is tabulated in Tables 2, 3. The lowest hydrophobicity value for the atpD gene product (−0.256) was observed in Clostridium cochlearium NLAE-zl-C224 and the highest value (0.015) was found to be demonstrated by Beijerinckia indica indica ATCC 9039, a Gram negative free living soil dweller. A scattered plot showing the distribution of hydrophobicity values of the protein encoded by atpD gene is given in Figure 1.
Comparative CUB Analysis of infB Gene From Soil Bacteria
In accordance with the findings of rpoB and atpD, Micrococcus luteus NCTC 2665 was found to display the lowest Nc and highest GC3 content for infB. Similar to atpD, the lowest GC3 content was demonstrated by Clostridium. But highest Nc for infB was demonstrated by Desulfobacterium autotrophicum HRM2, DSM 3382. The lowest and most negative hydrophobicity value of infB gene product (−0.729) was observed in Brevibacillus agri BAB-2500. The CUB profile of infB of all the studied bacteria is given in Tables 2, 3. In terms of hydrophobicity, Bacillus megaterium WSH-002 was found to display the highest hydrophobicity value (−0.168). A scattered plot showing the distribution of hydrophobicity values of infB gene products is given in Figure 1.
Comparative CUB Analysis of trpB Gene From Soil Bacteria
The CUB profile demonstrated by trpB was found to be relatively different from the rest of the housekeeping genes. The lowest Nc value (Nc = 25.44) was exhibited by Pseudomonas putida 1A00316 whereas the highest Nc value (Nc = 57.20) was depicted in Bacillus horikoshii DSM 8719. In terms of GC3 content, the lowest and highest GC3 was depicted by Clostridium butyricum JKY6D1 (GC3 = 5%) and Acidiphilium cryptum JF5 (GC3 = 99%). The CUB profile of trpB of the 92 bacterial species is given in Tables 2, 3. The lowest hydrophobicity of (−0.348) was detected in Clostridium butyricum JKY6D1 and the highest in different species of the genus Micromonospora, Nocardia cerradoensis NBRC 101014 and Streptomyces scabrisporus DSM 41855. A scattered plot showing the distribution of hydrophobicity values of the protein encoded by trpB gene is given in Figure 1.
Out of the four housekeeping genes, trpB was found to demonstrate relatively greater fluctuation in terms of codon bias. The genic Nc of trpB were found to be higher than the mean genomic Nc, in 20 out of the 92 soil bacteria considered in this study. Moreover, the reverse case scenario of lower genic Nc of trpB in comparison to the genomic Nc was found in about 70% of the organisms. Several species of Bacillus, Nitrosomonas, Acidithiobacillus and others displayed this trend. In addition, the genic Nc of infB was found to be greater than the mean genomic Nc in almost 8% of the studied organisms. The rpoB and atpD gene in a small number of organisms was found to display Nc value higher than the mean genomic Nc. On the contrary, for each and every housekeeping gene from the different soil bacteria considered in this study, the genic Nc was found to be much lower than the mean genomic Nc. The range of such deviation was found to be almost 30%. One such example is Vibrio natriegens NBRC 15636 which showed 30% deviation in rpoB, 27% in infB, and negligible deviation in trpB. For each of the four housekeeping genes, we have also calculated the percentage of genes in the genome of each organism which is below the genic Nc value of the housekeeping genes (data shown in Supplementary Table 4). Our results show that for the rpoB and infB gene, Beggiatoa alba B18LD had the highest percentage of genes (75.07% for rpoB and 76.63% for infB) below the genic Nc. In case of the atpD gene, Clostridium argentinense CDC 2741 had 83.64% of its genes below the genic Nc whereas for the trpB gene, Acidithiobacillus ferrivorans SS3 demonstrated a remarkably higher number of genes (86.88%) having Nc less than that of trpB. The results of trpB gene further shows that a large number of bacteria have a greater percentage of genes with Nc values less than the Nc of trpB. This finding indicates the aberrant nature of codon bias in trpB which being a key housekeeping gene demonstrates a significantly reduced codon usage bias. We observed that in most of the soil bacteria having fewer number of genes below the genic Nc of the rpoB gene, also had lesser percentage of genes below the genic Nc of the housekeeping genes infB and atpD. In the case of trpB gene, barring a few organisms, such as Bdellovibrio bacteriovorus HD100, Clostridium cadaveris NLAE-zl-G419, Streptomyces vitaminophilus ATCC 31673, Rhizobium gallicum IE4872, Acidiphilium multivorum AIU301, and Nitrobacter winogradskyi Nb-255 most of the bacteria did not have a large percentage of genes in their genome which is below the genic Nc of the remaining housekeeping genes.
In terms of GC3 content, trpB exhibited a relatively greater fluctuation. In almost 70% of the soil bacterial species higher genic GC3 content was observed in comparison to the genomic GC3 value. The gene atpD showed similar type of deviation in 25 species. These include Clostridium scatologenes ATCC 25775, C. sporogenes NCIMB 10696, Desulfocapsa sulfexigens DSM 10523, Desulfobacterium autotrophicum HRM2, DSM 3382, Vibrio natriegens NBRC 15636, Acidithiobacillus ferrivorans SS3, Nitrobacter winogradskyi Nb-255, N. hamburgensis X14, Acidiphilium cryptum JF-5, and Hyphomicrobium denitrificans 1NES1. The common features shared by these organisms are that these are Gram negative (except Clostridium), free living soil bacteria. Out of these Acidithiobacillus ferrivorans SS and Acidiphilium cryptum JF-5 are from acidic environment. Both the infB and rpoB gene also reflected similar trend. On the contrary, in some species in which genomic GC3 content was found to be much higher than the GC3 content of the housekeeping genes, and in all the concerned housekeeping genes such a deviation was found to exist on a massive scale. For example, the atpD codon profile of Flavobacterium pectinovorum DSM 6368 was found to display a whopping 81% deviation.
Relative Hydrophobicity Analysis of Housekeeping Gene Products From Soil Bacteria
A comparison of the hydrophobicity of the different housekeeping gene products revealed an interesting pattern in the 92 soil bacterial species (Figure 1). We observed that the hydrophobicity of the protein coded by rpoB gene demonstrates a tight clustering compared to the products of the other three housekeeping genes suggesting that in the soil bacteria the hydrophobicity of the housekeeping gene product rpoB is relatively conserved in comparison to atpD, infB and trpB. This is understandable since rpoB is responsible for coding the β subunit of bacterial RNA polymerase which is a key component of the genetic information processing pathway, and regions of the protein susceptible to mutations are characteristically safeguarded (Vos et al., 2012) for preserving function of the protein. In contrast to rpoB, atpD, and trpB, the infB gene products was found to demonstrate a relatively fluctuating degree of hydrophobicity with values ranging from −0.168 to −0.729 within the soil bacteria. In spite of the diversity of the soil bacteria and the dissimilar functional nature of the housekeeping gene products a greater fraction of the coding sequences in the 92 species were found to display a hydrophilic nature as depicted in Figure 1. This clearly demonstrates that the 92 soil bacterial species are unified by a relatively hydrophilic character of their housekeeping gene products although they grow and survive in soil types having different physico-chemical properties.
A comprehensive Spearman's rank-order correlation study between Nc, GC3, length of the housekeeping gene, and hydrophobicity of each of the housekeeping gene product was thoroughly carried out. Our objective was to understand the underlying codon usage trend and ORF structuring of each of the housekeeping gene considered in this study, and comprehend whether each gene carries an underlying signature in terms of nucleotide structuring of the coding sequences. For all the four housekeeping genes, Nc was found to be significantly anti-correlated with GC3 (for rpoB ρ = −0.649; for atpD ρ = −0.633; infB ρ = −0.54; for trpB ρ = −0.766; at p < 0.01 level). The negative correlation between Nc and length was demonstrated by trpB (ρ = −0.457, p < 0.01), infB (ρ = −0.257, p < 0.01), and atpD (ρ = −0.197, p < 0.01). These suggest that in the later three housekeeping genes, the codon bias is positively correlated with increasing gene length. Similarly, the Nc and hydrophobicity was observed to be significantly anti-correlated for the gene rpoB (ρ = −0.303, p < 0.01) and trpB (ρ = −0.418, p < 0.01) in the soil bacteria. Except rpoB, the rest of the three housekeeping genes were found to show significant positive correlation between GC3 and length suggesting the fact that G and C ending codons are preferentially more favored with increasing ORF length. GC3 was also found to display a significant positive correlation with hydrophobicity for the gene rpoB (ρ = 0.509, p < 0.01) and trpB (ρ = 0.467, p < 0.01). This is expected as the presence of G or C residue at the 3′ position of a codon is a feature of hydrophobic amino acids. The correlation between length and hydrophobicity was also found to be significantly positive in case of rpoB (ρ = 0.32, p < 0.01) and trpB (ρ = 0.308, p < 0.01), whereas infB (ρ = −0.337, p < 0.01) was found to depict a negative association. Among the four housekeeping genes, trpB was found to depict a significant correlation between all the four parameters studied. This appears to be a completely atypical profile when compared to the other three housekeeping genes considered in this analysis. This aberrant profile of trpB could be further validated by studying its phylogenetic affinities which has been discussed in the succeeding sections of this study.
Kruskal-Wallis one way analysis of variance on ranks (Daniel, 1990) was performed to scrutinize whether the different codon usage parameters in the housekeeping genes selected in this study have a signature trend utilizing Nc, GC3, gene length and hydrophobicity. GC3, gene length, and hydrophobicity were all found to display a unique trend making it possible to delineate the four genes in terms of their signature GC3 content (H = 7.903, df = 3, p < 0.01), gene length (H = 321.508, df = 3, p < 0.01), and hydrophobicity (H = 268.193, df = 3, p < 0.01). However, the housekeeping genes did not demonstrate any signature trend in terms of their Nc values (H = 7.166, df = 3, p = 0.067).
Mann-Whitney Rank sum test utilizing Nc, GC3, gene length and hydrophobicity was carried out to find out whether the Gram nature of the bacterial species have a significant effect on the codon usage profile leading to a genic signature pattern. The test results suggested that in terms of GC3 (U = 700; p < 0.01), gene length (U = 82; p < 0.01) and hydrophobicity (U = 551; p < 0.01), it is possible to substantially delineate the rpoB gene between Gram negative and Gram positive bacteria. The results further indicate that the GC3 (U = 663; p < 0.01) and gene length (U = 469.50; p < 0.01) of the housekeeping gene infB is also clearly distinguishable between the Gram negative and Gram positive bacterial species. The GC3 (U = 770; p < 0.05) and hydrophobicity (U = 353; p < 0.01) of the protein encoded by the housekeeping gene atpD also appeared to be distinct and depict a genic signature among the two bacterial groups. The Nc of all the four housekeeping genes did not show any characteristic delineation based on the results of the Mann Whitney rank sum test between the Gram negative and Gram positive bacterial species. Apart from trpB, the GC3 profile of all the remaining housekeeping genes were found to be distinct among the Gram positive and Gram negative bacteria. The Mann-Whitney rank sum test results demonstrated that out of the four housekeeping genes, the Nc (U = 893; p >> 0.05), GC3 (U = 844.50; p >> 0.05), gene length (U = 883.50; p >> 0.05) of trpB and hydrophobicity (U = 881; p >> 0.05) of trpB gene product did not depict any statistically significant difference between Gram positive and Gram negative bacterial species. Our results thus further corroborates the finding that the housekeeping gene trpB demonstrates an aberrant codon usage profile in comparison to the rest of the housekeeping genes considered in this study. The trpB gene encodes the β subunit of the enzyme tryptophan synthase, which is a pyridoxal 5′-phosphate-dependent αββα multi enzyme complex, responsible for catalyzing the final two steps of tryptophan biosynthesis (Yanofsky, 2001; Ishida et al., 2002; Dunn et al., 2008; Raboni et al., 2009). This enzyme is bestowed with certain unique features, such as dual catalytic ability, substrate channeling (Leopoldseder et al., 2006; Raboni et al., 2009) and the trpB gene might have been subjected to gene duplication, fusion, loss as well as parallel evolution in some archaea and eubacteria (Xie et al., 2001; Leopoldseder et al., 2006). Therefore, it can be construed that the trpB gene with its inconsistent codon usage pattern may be deemed as an odd gene in terms of its codon usage pattern residing within the genome of many soil dwelling bacteria.
Analysis of Nc Plot
The correlation between Nc and GC3 was plotted on a graph called Nc plot (Wright, 1990) for the 92 species considered in this study. Our previous study has shown that the Nc plots can be used as a valuable tool to detect intra- and inter-specific/genic synonymous codon usage patterns (Pal et al., 2015). Analyzing the Nc plots, three types of gene clustering on the Nc plot was evident—left centric, mid centric and right centric aggregation of coding sequences as shown in Figure 2. Several genera like Acidiphilium, Alcaligenes, Agrobacterium, Actinoalloteichus, Acidobacterium, Achromobacter, Acidovorax, Azotobacter, Burkholderia, Hyphomicrobium, Micrococcus, Micromonospora, Nocardia, Streptomyces, Rhizobium, Thiobacillus, and Nitrobacter exhibited right centric aggregation of the coding sequences on the Nc plot. But some members of the genera Acidithiobacillus, Acidocella, Chromobacterium, and some species, such as Achromobacter xylosoxidans A8, Beijerinckia indica indica ATCC 9039 showed an aberrant mid centric aggregation with right shift (Supplementary Figure 1). It was observed that the left centric Nc plot is the general feature of the genera Clostridium and Bacillus. The organism Nitrosomonas communis Nm2, Nitrosomonas europaea ATCC 19718, Desulfobacter postgatei 2ac9, Chlorobium phaeovibrioides DSM 265, Desulfobacterium autotrophicum HRM2 DSM 3382, Desulfobacula toluolica Tol2, Vibrio gazogenes DSM 21264 demonstrated a mid-centric aggregation of the coding sequences. All these species are free living, Gram negative soil dwellers. An aberrant pattern was found to be displayed by the genus Flavobacterium where two species were found to display two distinct type of clustering on the Nc plot. F. suncheonense GH29-5, DSM 17707 demonstrated a mid to left centric aggregation of the coding sequences whereas F. pectinovorum DSM 6368 exhibited a left centric aggregation (Supplementary Figure 2). The members of the genus Pseudomonas like P. azotoformans S4, P. fluorescens A506, P. putida demonstrated some variation in clustering pattern on the Nc plot with majority of the species characterized by a right centric aggregation on the Nc plot. The coding sequences of Acinetobacter calcoaceticus PHEA-2 was found to demonstrate a typical left centric aggregation on the Nc plot.
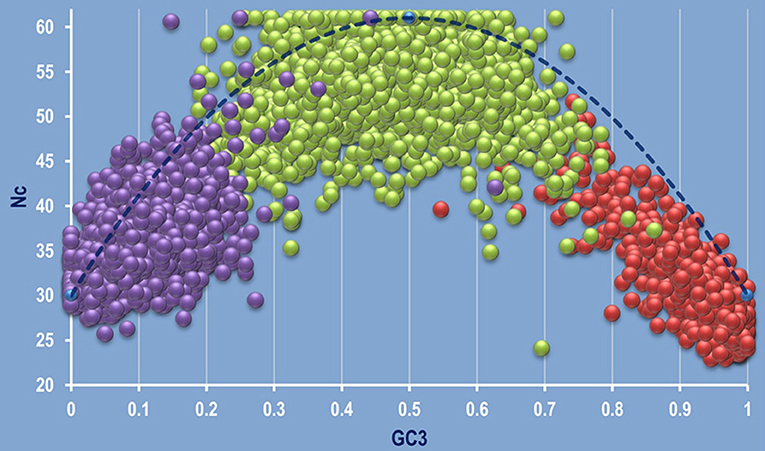
Figure 2. A combined genomic Nc plot utilizing all the coding sequences of the whole genomes depicting the three typical mode of aggregation of coding sequences. Left centric aggregation represented by Clostridium butyricum JKY6D1 (in purple), mid centric aggregation shown in green by Nitrosomonas communis Nm2, and right centric aggregation depicted by Micrococcus luteus NCTC 2665, shown in red. The dashed blue line represents the null hypothesis curve which suggests that codon usage bias is solely due to mutation and not selection (Wright, 1990).
The Nc plots of the four housekeeping genes atpD, infB, rpoB, and trpB revealed a different scenario as a whole. In case of the rpoB gene, which codes for the β-subunit of DNA dependent RNA polymerase enzyme in bacteria, most of the organisms were found to form scattered clusters all throughout the Nc plot (Figure 3). The Nc plots based on atpD, infB and trpB coding sequences exhibited a comparable aggregation pattern as shown in Figure 3. The mechanistic forces shaping codon usage can also be detected utilizing an Nc plot (Wright, 1990; Khandia et al., 2019). Barring a mere 1.4% out of the total 360 coding sequences of the four housekeeping genes, all the remaining coding sequences of the rpoB, atpD, infB, and trpB gene were found to fall below the null hypothesis curve of the Nc plot in Figure 3 indicating selection pressure as a key element shaping codon usage pattern in the housekeeping genes of a majority of the soil bacterial species considered in this study.
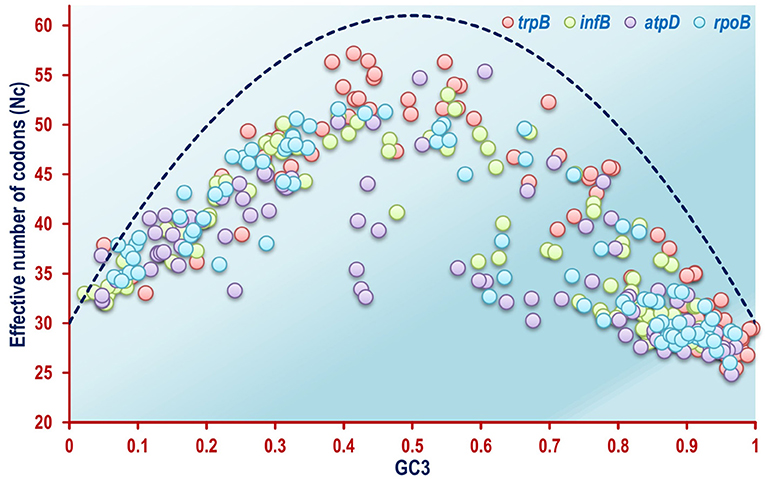
Figure 3. A combined Nc plot of the four housekeeping genes rpoB, atpD, infB, and trpB from the 92 soil bacterial species included in this study, depicting selectional pressure as a major unifying force in shaping codon usage pattern. The dashed blue line represents the null hypothesis curve which suggests that codon usage bias is solely due to mutation and not selection (Wright, 1990).
Molecular Phylogenetic Analysis
To further comprehend the potential relationships between the different soil dwelling bacteria, a comprehensive molecular phylogenetic analysis was carried out using sequences of complete 16S rRNA gene and the four housekeeping genes.
Phylogenetic Analysis Based on 16S rRNA Gene Sequences
The phylogram based on 16S rRNA gene sequences (Figure 4), depicted that species belonging to the same genera share more than 95% sequence similarity and cluster nearly together. This is in line with the traditional taxonomic standing. Bootstrap values indicate that most branches in the dendrogram are highly significant, although some exception exists (data provided as Supplementary File in Newick format). A prominent and close association of the organisms belonging to the same taxonomic classes, were also visible, but with certain exceptions. The genus Bdellovibrio belonging to the class Proteobacteria showed proximity with the genus Flavobacterium under the class Bacteroidetes. On the contrary, at the sub-class level too, some variations were detected. Some genera belonging to the subclass Gammaproteobacteria like Vibrio, Beggiatoa, Acinetobacter, Pseudomonas, and Azotobacter formed cluster with species belonging to the sub-class Betaproteobacteria. Above all, the class Firmicutes, Actinobacteria, Chlorobi, Proteobacteria showed similar ancestral origin but not through the dichotomous branching rather through a polytomy as seen in the Figure 4. The annotated phylogenetic tree inferred utilizing the 16S rRNA gene sequences from the 92 soil bacteria is shown in Figure 4. The raw tree file with bootstrap support in Newick format is provided as Supplementary File.
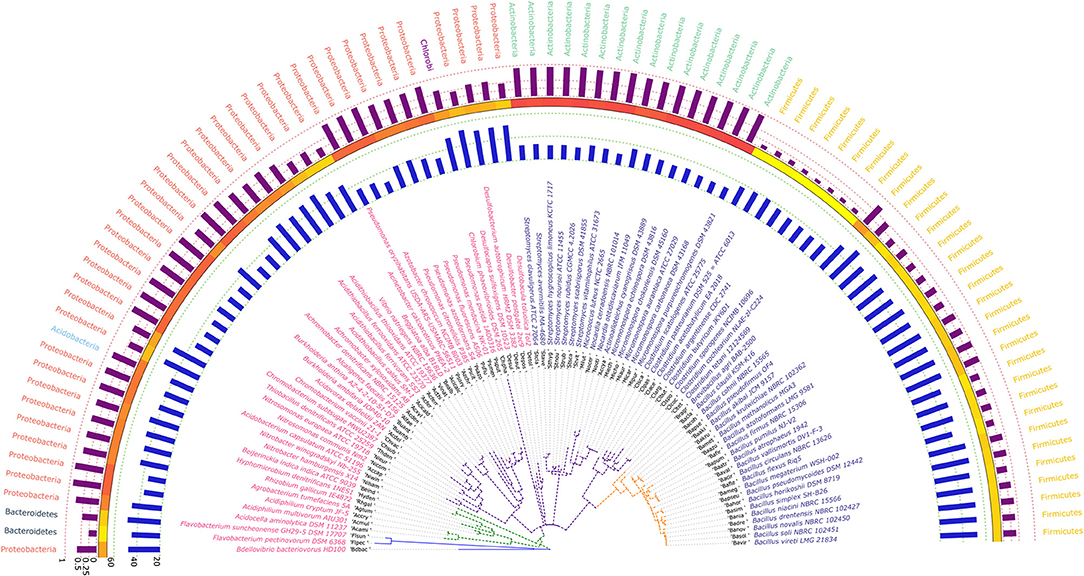
Figure 4. A phylogenetic tree showing the relationship between the soil bacterial species considered in this study based on 16S rRNA gene sequences along with Gram nature, taxonomic position and codon usage annotation data. The name of the species have been depicted in color corresponding to its Gram nature with magenta and blue representing Gram negative and positive nature, respectively. The outermost semicircle with magenta bars represents the genomic GC3 while the innermost semicircle with blue bars represents the genomic Nc. The middle strip with yellow to red color gradient depicts the genomic GC content with red representing maximum GC content. The evolutionary history was inferred by using the Maximum Likelihood method based on the Kimura 2-parameter model (Kimura, 1980). The bootstrap consensus tree inferred from 1,000 replicates is taken to represent the evolutionary history of the taxa analyzed (Felsenstein, 1985). The tree with the highest log likelihood (−6,331.0306) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites [five categories (+G, parameter = 0.5623)]. The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 40.6240% sites). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. All positions containing gaps and missing data were eliminated. There were a total of 357 positions in the final dataset. Evolutionary analyses were conducted in MEGA6 (Kumar et al., 2008). The visualization and annotation of the phylogenetic tree was done using iTOL ver. 4.4.2 (Letunic and Bork, 2007).
Phylogenetic Analysis of Soil Bacteria Based on Housekeeping Genes
Housekeeping genes constitute an integral component of an organism's genome. Functioning in a coordinated fashion the housekeeping genes are involved with either metabolic functions, genetic information processing or environmental signal processing and are generally organized into operational units or operons in prokaryotes like bacteria. The antiquity of these housekeeping genes predates their function and hence these can be used as excellent markers to deduce the relationship between organisms on the functional basis. The use of 16S rRNA as a molecular chronometer, and inference of phylogeny based on it have been a major milestone in molecular taxonomy (Ludwig and Schleifer, 1994). The use of 16S rRNA based phylogeny as a backdrop against the phylogeny constructed from housekeeping genes thus, should be extremely vital in shedding new light on the functional relationship amongst soil dwelling bacteria.
Phylogenetic Analysis Based on rpoB Housekeeping Gene
The phylogenetic tree inferred using rpoB demonstrated a polytomy from inception but the aggregation of the species was found to be quite in congruence with that of the 16S rRNA gene based tree suggesting the relative absence of horizontal gene transfer and homologous recombination in course of evolution in these bacteria. The gene rpoB codes for a fundamental component of bacterial RNA polymerase, the β-subunit. This is a key component of the genetic information processing pathway and may be also considered as one of the most antique nucleic acid polymerizing enzyme. Hence, the congruence of the 16S rRNA and rpoB gene is quite justified looking at the antiquity of both these genes. When compared with the 16S rRNA gene based phylogenetic tree, we found that the group containing Betaproteobacteria in the 16S rRNA based tree ruptures into separate clusters based on their rpoB sequence suggesting diverse phylogenetic affinities. Azotobacter chroococcum was found to share clade with species of Pseudomonas having similar Nc, GC3 and hydrophobicity profile. The degree of codon usage bias variation in rpoB as evident from the Nc values of the genus Bacillus was found to be significant in comparison to the genera Clostridium, Pseudomonas, Streptomyces, and Micromonospora, all of which have a codon biased rpoB. The annotated phylogenetic tree inferred utilizing the rpoB coding sequences along with codon usage and taxonomic data from the studied soil bacteria is shown in Figure 5.
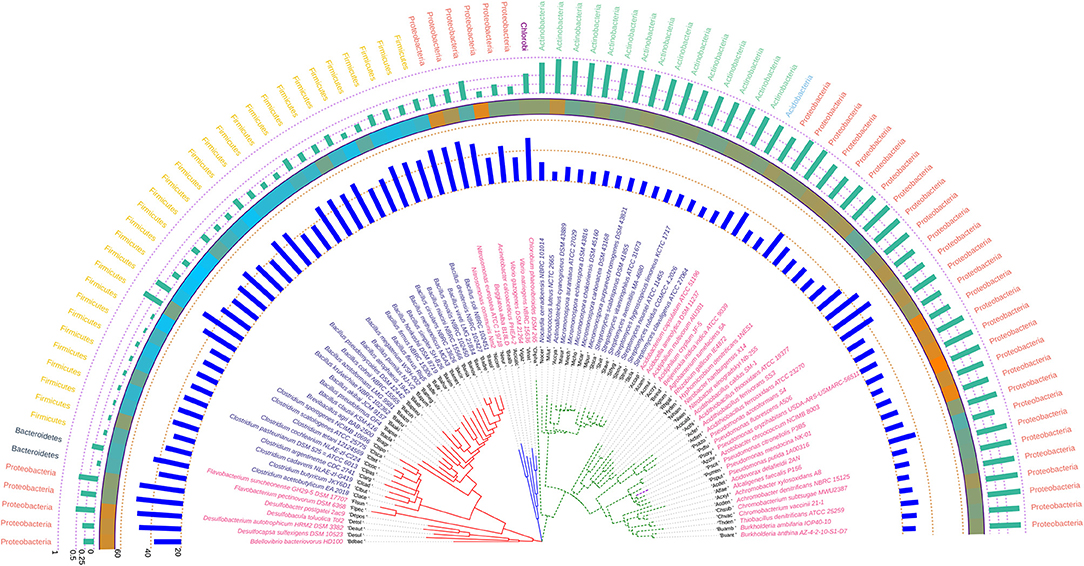
Figure 5. Phylogenetic tree showing the relationship between the soil bacterial species considered in this study based on rpoB gene sequences along with Gram nature, taxonomic position and codon usage annotation data. The name of the species have been depicted in color corresponding to the Gram nature with magenta and blue representing Gram negative and positive, respectively. The outermost semicircle with green bars represents the GC3 content of rpoB sequences while the innermost semicircle with blue bars represents the Nc of the rpoB coding sequences. The middle strip with cyan to orange color gradient depicts the variation in hydrophobicity of the protein encoded by rpoB coding sequences. The evolutionary history was inferred by using the Maximum Likelihood method based on the General Time Reversible model (Nei and Kumar, 2000). The bootstrap consensus tree inferred from 1,000 replicates is taken to represent the evolutionary history of the taxa analyzed (Felsenstein, 1985). The tree with the highest log likelihood (−73,689.4674) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites [five categories (+G, parameter = 0.7988)]. The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 22.2913% sites). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. All positions containing gaps and missing data were eliminated. There were a total of 1,726 positions in the final dataset. Evolutionary analyses were conducted in MEGA6 (Kumar et al., 2008). The visualization and annotation of the phylogenetic tree was done using iTOL ver. 4.4.2 (Letunic and Bork, 2007).
Phylogenetic Analysis Based on atpD Housekeeping Gene
The phylogeny inferred utilizing atpD was found to be quite unique with respect to the positioning of the two Clostridia species, C. cochlearium NLAE-zl-C224 and C. tetani 12124569 in relation to the other seven species of the genera. Though the phylogeny of the atpD gene in the soil bacteria suggests least amount of evolutionary divergence as suggested by the branch lengths in comparison to the other three housekeeping genes but, C. cochlearium NLAE-zl-C224 and C. tetani 12124569 were found to be the most evolutionary distant ones in comparison to the other bacterial species. Similar to what we have seen in rpoB, Azotobacter chroococcum was found to share clade with species of Pseudomonas with similar Nc and hydrophobicity profile. The species of Micromonospora and Streptomyces were found to be consistent both in terms of their codon usage and phylogenetic relatedness. The codon usage profile of members of the genus Bacillus was found to be much more uniform in comparison to rpoB. The atpD coding sequence of Bdellovibrio bacteriovorous HD100 was found to be peculiar since it made the sole Gram negative bacterium to share clade with a host of Gram positive bacterial genera, such as Clostridium and Bacillus. The degree of codon bias of atpD sequence of Bdellovibrio as depicted by Nc was also found to be similar to some of the Clostridium species. In the atpD based evolutionary tree, bacteria belonging to Firmicutes closely resembled the members of Proteobacteria and hence appears to intermingle with each other forming a cluster. The annotated phylogenetic tree inferred utilizing the atpD coding sequences along with codon usage and taxonomic data from the studied soil bacteria is shown in Figure 6.
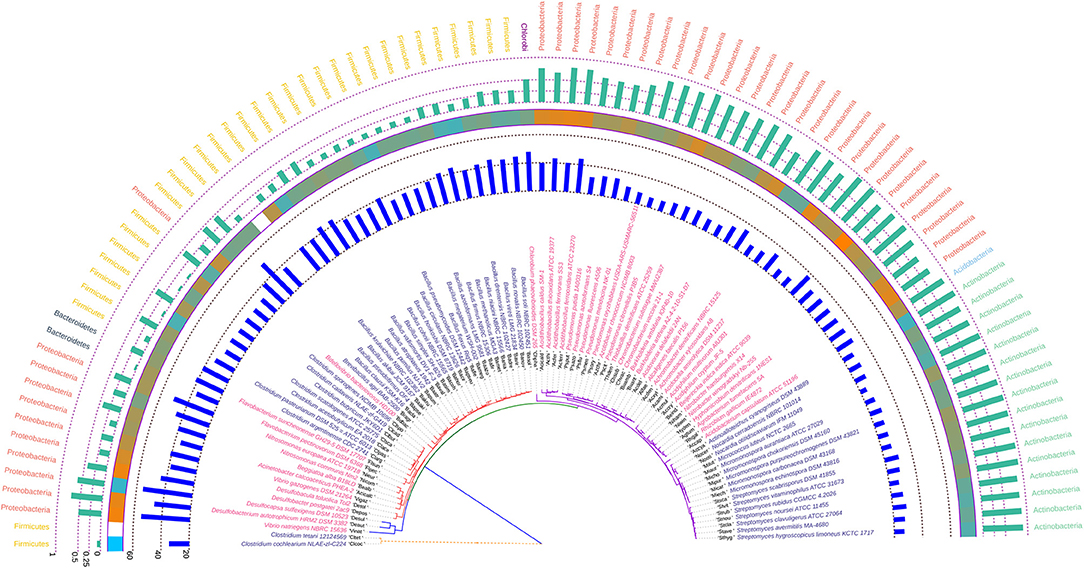
Figure 6. Phylogenetic tree showing the relationship between the soil bacterial species considered in this study based on atpD gene sequences along with Gram nature, taxonomic position and codon usage annotation data. The name of the species have been depicted in color corresponding to its Gram nature with magenta and blue representing Gram negative and positive, respectively. The outermost semicircle with green bars represents the GC3 content of atpD sequences while the innermost semicircle with blue bars represents the Nc of the atpD coding sequences. The middle strip with cyan to orange color gradient depicts the variation in hydrophobicity of the protein encoded by the atpD coding sequences. The evolutionary history was inferred by using the Maximum Likelihood method based on the General Time Reversible model (Nei and Kumar, 2000). The bootstrap consensus tree inferred from 1,000 replicates is taken to represent the evolutionary history of the taxa analyzed (Felsenstein, 1985). Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites [five categories (+G, parameter = 0.9877)]. The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 6.2680% sites). All positions containing gaps and missing data were eliminated. There were a total of 1,123 positions in the final dataset. Evolutionary analyses were conducted in MEGA6 (Kumar et al., 2008). The visualization and annotation of the phylogenetic tree was done using iTOL ver. 4.4.2 (Letunic and Bork, 2007).
Phylogenetic Analysis Based on infB Housekeeping Gene
The phylogenetic tree inferred utilizing the infB coding sequences of the soil bacteria demonstrated absence of polytomy and consistency in the clustering pattern of the different species of the genera particularly Pseudomonas, Streptomyces, Micromonospora, Clostridium, and Bacillus. All the clades of the infB based tree were found to be dichotomous with a relatively short evolutionary distance indicating relatedness and uniformity. The codon usage profile of the infB coding sequences in these genera was also found to be consistent except in some species of Bacillus like B. vallismortis DV1-F-3, B. pumilus NJ-V2, B. atrophaeus 1942, B. vireti LMG 21834, and B. novalis NBRC 102450 which depicted a higher Nc value for atpD suggesting relatively lower codon bias. Similar to the phylogenetic trees based on the housekeeping genes rpoB and atpD, Azotobacter chroococcum was found to cluster with species of Pseudomonas on the basis of the infB coding sequence and show similar codon usage profile comparable to some species of Pseudomonas. The annotated phylogenetic tree inferred utilizing the infB coding sequences along with codon usage and taxonomic data from the studied soil bacteria is shown in Figure 7.
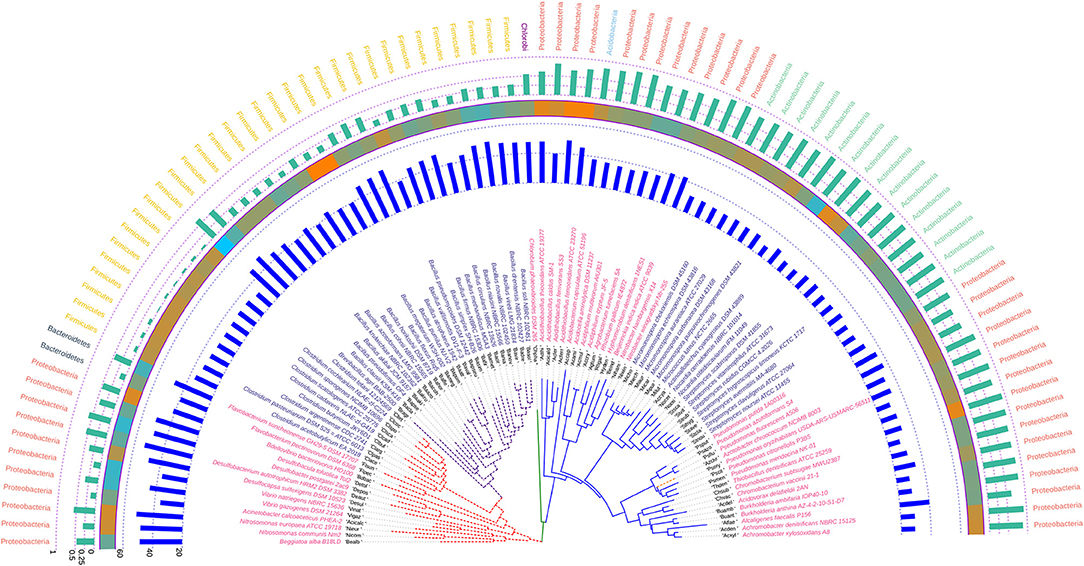
Figure 7. Phylogenetic tree showing the relationship between the soil bacterial species considered in this study based on infB gene sequences along with Gram nature, taxonomic position and codon usage annotation data. The name of the species has been depicted in color corresponding to its Gram nature with magenta and blue representing Gram negative and positive, respectively. The outermost semicircle with green bars represents the GC3 content of infB sequences while the innermost semicircle with blue bars represents the Nc of the infB coding sequences. The middle strip with cyan to orange color gradient depicts the variation in hydrophobicity of the protein encoded by infB coding sequences. The evolutionary history was inferred by using the Maximum Likelihood method based on the General Time Reversible model (Nei and Kumar, 2000). The bootstrap consensus tree inferred from 1,000 replicates is taken to represent the evolutionary history of the taxa analyzed (Felsenstein, 1985). The tree with the highest log likelihood (−84,778.7972) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites [five categories (+G, parameter = 1.1105)]. The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 16.2594% sites). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. All positions containing gaps and missing data were eliminated. There were a total of 1,617 positions in the final dataset. Evolutionary analyses were conducted in MEGA6 (Kumar et al., 2008). The visualization and annotation of the phylogenetic tree was done using iTOL ver. 4.4.2 (Letunic and Bork, 2007).
Phylogenetic Analysis Based on trpB Housekeeping Gene
The odd and inconsistent nature of the trpB gene as evident from the codon usage study is further corroborated by phylogenetic analysis. A conflicting signal was clearly evident from the phylogeny inferred utilizing trpB. Utilizing the 16S rRNA gene tree at the backdrop, significant differences were observed. All the species of the genus Bacillus that were grouped together in the 16S rRNA based tree were found to cluster in three separate groups. Some species of Clostridium, Vibrio along with Flavobacterium pectinovorum DSM 6368 were found to coexist in these clusters. The organism Bacillus firmus NBRC 15306 was found to be distantly related from all the other species of Bacillus considered in this study. The two species of Vibrio, V. gazogenes DSM 21264 and V. natriegens NBRC 15636 was also found to segregate into two separate clades. Similarly the species Streptomyces scabrisporus DSM 41855 formed a separate clade from the other Streptomyces species considered in our study. Besides, the organism Streptomyces scabrisporus DSM 41855 was also found to reside with all of the Micromonospora species on the trpB phylogram indicating monophyletic ancestral origin. The comparative analysis of hydrophobicity of the trpB gene gene products demonstrated a similar sort of relationship where S. scabrisporus DSM 41855 was found to display hydrophobicity profile in line with Micromonospora species. In spite of the conflicting signals and inconsistencies of trpB, species belonging to Clostridium, Pseudomonas, and Micromonospora were found to cluster together on the tree suggesting their phylogenetic closeness. Similar to the other three housekeeping gene based trees, Azotobacter chroococcum was found to cluster together with species of Pseudomonas sharing similar Nc and hydrophobicity profile. The annotated phylogenetic tree inferred utilizing the trpB coding sequences along with codon usage and taxonomic data is shown in Figure 8.
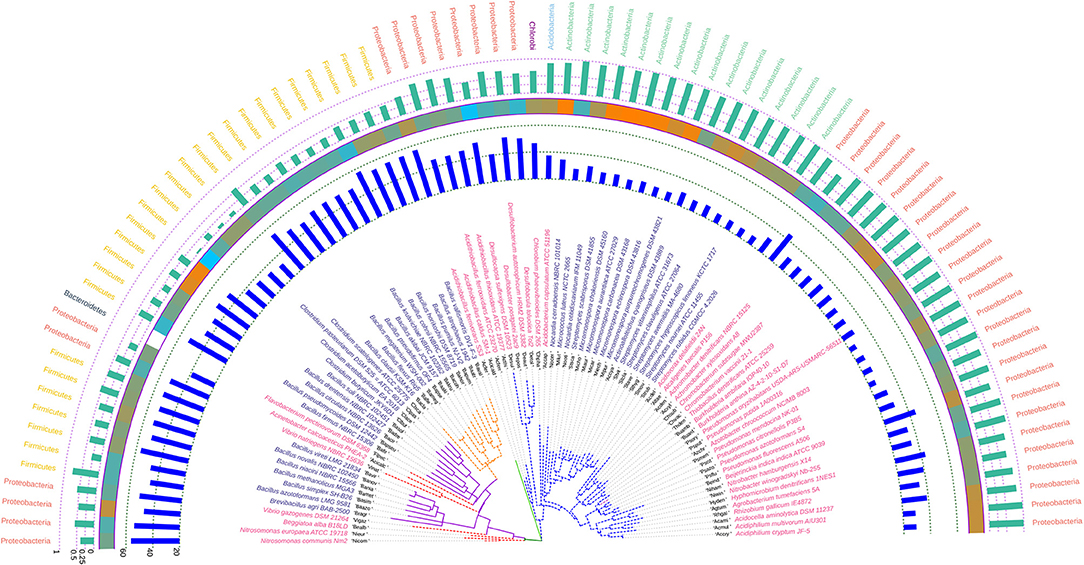
Figure 8. Phylogenetic tree showing the relationship between the soil bacterial species considered in this study based on trpB gene sequences along with Gram nature, taxonomic position and codon usage annotation data. The name of the species has been depicted in color corresponding to its Gram nature with magenta and blue representing Gram negative and positive, respectively. The outermost semicircle with green bars represents the GC3 content of trpB sequences while the innermost semicircle with blue bars represents the Nc of the trpB coding sequences. The middle strip with cyan to orange color gradient depicts the variation in hydrophobicity of the protein encoded by trpB coding sequences. The evolutionary history was inferred by using the Maximum Likelihood method based on the General Time Reversible model (Nei and Kumar, 2000). The bootstrap consensus tree inferred from 1,000 replicates is taken to represent the evolutionary history of the taxa analyzed (Felsenstein, 1985). The tree with the highest log likelihood (−57,296.2790) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites [five categories (+G, parameter = 1.2054)]. The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 14.7706% sites). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. All positions containing gaps and missing data were eliminated. There were a total of 1,098 positions in the final dataset. Evolutionary analyses were conducted in MEGA6 (Kumar et al., 2008). The visualization and annotation of the phylogenetic tree was done using iTOL ver. 4.4.2 (Letunic and Bork, 2007).
Significant topological differences were observed between the phylogenetic trees based on the housekeeping genes and the standard 16Sr RNA gene based one, suggesting conflicting evolutionary signals. In all of the scenarios, the Proteobacteria was found to be the worst affected phylum. Based on rpoB, the members of Proteobacteria were found to be scattered among all the three clades originating from the root whereas the phylogeny inferred utilizing the housekeeping gene infB puts the member of the Proteobacteria dispersed between the phyla Firmicutes and Actinobacteria. Consistent with its fluctuations as evident from the codon usage study, the trpB based phylogeny dispersed the members of the phylum Proteobacteria to a large extent. The atpD based phylogeny also pointed toward the evolutionary distant nature of the Proteobacteria, particularly in the case of Bdellovibrio which was found to be the only Gram negative bacterium residing with the Firmicutes like Bacillus and Clostridium. In almost all the phylograms, species belonging to the same genus revealed a more or less tight clustering with some degree of heterogeneity. The members of the phylum Actinobacteria and Firmicutes were found to be closely associated with each other in all the phylogenetic trees inferred utilizing the housekeeping gene sequences pointing toward the relative absence of conflicting evolutionary signals. In all the phylograms based on both the 16S rRNA and housekeeping genes, the organism Azotobacter chroococcum NCIMB 8003 consistently clustered tightly with a variety of Pseudomonas species. P. citronellolis P3B5 was mostly found to exhibit close proximity with Azotobacter chroococcum NCIMB 8003 with significant bootstrap support. Only in case of trpB and infB this was found to be incongruent. We also observed that for the housekeeping genes atpD, rpoB, and trpB, the bacteria Azotobacter chroococcum NCIMB8003 shared 80–98% identity with various Pseudomonas species. In case of the gene infB, the nucleotide sequences of Azotobacter chroococcum NCIMB was found to share almost 75% identity. The hydrophobicity profile of the protein encoded by the housekeeping gene rpoB also demonstrated the proximity between Azotobacter chroococcum NCIMB and species of Pseudomonas.
Conclusion
This exhaustive study conducted on 92 bacterial species is one of its kind where a large scale comparative genomic analysis of soil dwelling bacteria utilizing a combination of codon usage analyses and molecular phylogenetics emphasizing on key housekeeping genes was carried out. The soil may be regarded as a treasure trove of microorganisms with a lot remaining to be explored. Our study revealed signature codon usage trend in the 92 soil bacteria where all the housekeeping genes were found to be under selectional pressure. An irregular codon usage profile and conflicting phylogenetic profile was consistently visible in case of the key housekeeping gene trpB encoding the beta subunit of the tryptophan synthase enzyme. The presence of conflicting signals with regard to the housekeeping genes in the bacterial phylum Proteobacteria pointed appreciably to the enormous genetic heterogeneity present within the group which was further corroborated by the codon usage analyses study. The taxonomic positioning of the organism Azotobacter chroococcum NCIMB with the Pseudomonads was also a major taxonomic deviation but the signal was consistent and further amplified by all the housekeeping genes measured in this study. Bacterial phylogeny is a controversial issue abetted by the presence of lateral gene transfer and recombination events, and among the soil bacteria, the members of Proteobacteria were found to be the most affected in contrast to that of the members of the phylum Actinobacteria and Firmicutes, a fact also supported by codon usage analysis.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
AP conceived and designed the subject with substantial contribution from JS and PM in analysis and interpretation of data. AP and JS wrote the first draft of the manuscript and carried out the statistical analysis. JS, BS, MP, and VR retrieved and analyzed the data and generated the tables and figures under supervision of AP and PM. BS, MP, and VR assisted in writing. All the authors approved final version of the manuscript, and agreed to be accountable for its contents.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge and thank their University for its support. The authors are also grateful to all the reviewers for their insightful suggestions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02896/full#supplementary-material
Supplementary Figure 1. A genomic Nc plot demonstrating aberrant mid centric aggregation of coding sequences with right shift in the bacterial species Achromobacter xylosoxidans A8 (shown in red) and Beijerinckia indica indica ATCC 9039 (shown in green). The dashed cyan line represents the null hypothesis curve which suggests that codon usage bias is solely due to mutation and not selection (Wright, 1990).
Supplementary Figure 2. A genomic Nc plot demonstrating aberrant inter specific variation in codon usage pattern in the genus Flavobacterium. F. suncheonense (shown in red) demonstrates a mid to left centric aggregation of the coding sequences whereas F. pectinovorum exhibits a left centric aggregation of the coding sequences (shown in green). The dashed black line represents the null hypothesis curve which suggests that codon usage bias is solely due to mutation and not selection (Wright, 1990).
Supplementary Table 1. A detailed list of the 92 soil bacterial species studied along with their NCBI reference sequence number, abbreviated name, Gram nature, habitat and source of isolation.
Supplementary Table 2. A detailed list showing the genome size (DNA total bases), coding bases, G+C bases, percentage of coding bases and GC content of the genome in the 92 soil bacterial species considered in this study.
Supplementary Table 3. Results of the correlation analysis between the different codon usage parameters at the whole genome level in the 92 soil bacterial species considered in this study.
Supplementary Table 4. A list showing the genomic gene count along with the percentage (%age) of genes in the genome of the 92 soil bacterial species considered in this study that is below the genic Nc value of the housekeeping genes rpoB, atpD, infB, and trpB.
References
Aislabie, J., Deslippe, J., and Dymond, J. (2013). Soil Microbes and Their Contribution to Soil Services. Lincoln, OR: Manaaki Whenua Press.
Andújar, C., Arribas, P., and Vogler, A. (2017). Terra incognita of soil biodiversity: unseen invasions under our feet. Mol. Ecol. 26, 3087–3089. doi: 10.1111/mec.14112
Babbitt, G. A., Alawad, M. A., Schulze, K. V., and Hudson, A. O. (2014). Synonymous codon bias and functional constraint on GC3-related DNA backbone dynamics in the prokaryotic nucleoid. Nucleic Acids Res. 42, 10915–10926. doi: 10.1093/nar/gku811
Baldauf, S. L., Roger, A. J., Wenk-Siefert, I., and Doolittle, W. F. (2000). A kingdom-level phylogeny of eukaryotes based on combined protein data. Science 290, 972–977. doi: 10.1126/science.290.5493.972
Barcellos, F. G., Menna, P., Da Silva Batista, J. S., and Hungria, M. (2007). Evidence of horizontal transfer of symbiotic genes from a Bradyrhizobium japonicum inoculant strain to indigenous diazotrophs Sinorhizobium (Ensifer) fredii and Bradyrhizobium elkanii in a Brazilian Savannah Soil. Appl. Environ. Microbiol. 73, 2635–2643. doi: 10.1128/AEM.01823-06
Benson, D. A., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., and Wheeler, D. L. (2006). Genbank. Nucleic Acids Res. 35, D21–D25. doi: 10.1093/nar/gkj157
Berg, G., and Smalla, K. (2009). Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol. Ecol. 68, 1–13. doi: 10.1111/j.1574-6941.2009.00654.x
Bittner, A. N., Foltz, A., and Oke, V. (2007). Only one of five groEL genes is required for viability and successful symbiosis in Sinorhizobium meliloti. J. Bacteriol. 189, 1884–1889. doi: 10.1128/JB.01542-06
Botzman, M., and Margalit, H. (2011). Variation in global codon usage bias among prokaryotic organisms is associated with their lifestyles. Genome Biol. 12:R109. doi: 10.1186/gb-2011-12-10-r109
Brenner, D. J., Krieg, N. R., Staley, J. T., and Garrity, G. M. (2005). Bergey's Manual of Systematic Bacteriology. New York, NY: Springer-Verlag.
Brown, J. R., Douady, C. J., Italia, M. J., Marshall, W. E., and Stanhope, M. J. (2001). Universal trees based on large combined protein sequence data sets. Nat. Genet. 28, 281–285. doi: 10.1038/90129
Busse, H.-J., and Wieser, M. (2014). “The genus arthrobacter,” in The Prokaryotes: Actinobacteria, eds E. Rosenberg, E. F. Delong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin: Springer Berlin Heidelberg), 105–132. doi: 10.1007/978-3-642-30138-4_204
Carro, L., Spröer, C., Alonso, P., and Trujillo, M. E. (2012). Diversity of micromonospora strains isolated from nitrogen fixing nodules and rhizosphere of pisum sativum analyzed by multilocus sequence analysis. Syst. Appl. Microbiol. 35, 73–80. doi: 10.1016/j.syapm.2011.11.003
Case, R. J., Boucher, Y., Dahllöf, I., Holmström, C., Doolittle, W. F., and Kjelleberg, S. (2007). Use of 16S rRNA and rpoB Genes as molecular markers for microbial ecology studies. Appl. Environ. Microbiol. 73, 278–288. doi: 10.1128/AEM.01177-06
Chen, I-M. A., Markowitz, V. M., Chu, K., Palaniappan, K., Szeto, E., Pillay, M., et al. (2017). IMG/M: integrated genome and metagenome comparative data analysis system. Nucleic Acids Res. 45, D507–D516. doi: 10.1093/nar/gkw929
Chen, S., Lesnik, E. A., Hall, T. A., Sampath, R., Griffey, R. H., Ecker, D. J., et al. (2002). A bioinformatics based approach to discover small RNA genes in the Escherichia coli genome. Biosystems 65, 157–177. doi: 10.1016/S0303-2647(02)00013-8
Chithambaram, S., Prabhakaran, R., and Xia, X. (2014). The effect of mutation and selection on codon adaptation in Escherichia coli bacteriophage. Genetics 197, 301–315. doi: 10.1534/genetics.114.162842
Clooney, A. G., Fouhy, F., Sleator, R. D., O'Driscoll, A., Stanton, C., Cotter, P. D., et al. (2016). Comparing apples and oranges?: Next generation sequencing and its impact on microbiome analysis. PLoS ONE 11:e0148028. doi: 10.1371/journal.pone.0148028
Daniel, R. (2005). The metagenomics of soil. Nat. Rev. Microbiol. 3, 470–478. doi: 10.1038/nrmicro1160
Das, S., Dash, H. R., Mangwani, N., Chakraborty, J., and Kumari, S. (2014). Understanding molecular identification and polyphasic taxonomic approaches for genetic relatedness and phylogenetic relationships of microorganisms. J. Microbiol. Methods 103, 80–100. doi: 10.1016/j.mimet.2014.05.013
Degefu, T., Wolde-Meskel, E., and Frostegård, Å. (2011). Multilocus sequence analyses reveal several unnamed mesorhizobium genospecies nodulating acacia species and sesbania sesban trees in Southern regions of Ethiopia. Syst. Appl. Microbiol. 34, 216–226. doi: 10.1016/j.syapm.2010.09.006
Delamuta, J. R. M., Ribeiro, R. A., Menna, P., Bangel, E. V., and Hungria, M. (2012). Multilocus sequence analysis (MLSA) of bradyrhizobium strains: revealing high diversity of tropical diazotrophic symbiotic bacteria. Braz. J. Microbiol. 43, 698–710. doi: 10.1590/S1517-83822012000200035
Delgado, A, and Gómez, J.A. (2016) “The soil. Physical chemical biological properties,” in Principles of Agronomy for Sustainable Agriculture, eds F. Villalobos E. Fereres (Cham: Springer).
Dunn, M. F., Niks, D., Ngo, H., Barends, T. R., and Schlichting, I. (2008). Tryptophan synthase: the workings of a channeling nanomachine. Trends Biochem. Sci. 33, 254–264. doi: 10.1016/j.tibs.2008.04.008
Eardly, B. D., Wang, F.-S., and Van Berkum, P. (1996). Corresponding 16S rRNA gene segments in rhizobiaceae and aeromonas yield discordant phylogenies. Plant Soil 186, 69–74. doi: 10.1007/BF00035057
Estrada-De Los Santos, P., Vinuesa, P., Martínez-Aguilar, L., Hirsch, A. M., and Caballero-Mellado, J. (2013). Phylogenetic analysis of burkholderia species by multilocus sequence analysis. Curr. Microbiol. 67, 51–60. doi: 10.1007/s00284-013-0330-9
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Fierer, N., Barberán, A., and Laughlin, D. C. (2014). Seeing the forest for the genes: using metagenomics to infer the aggregated traits of microbial communities. Front. Microbiol. 5:614. doi: 10.3389/fmicb.2014.00614
Fierer, N., and Jackson, R. B. (2006). The diversity and biogeography of soil bacterial communities. Proc. Nat. Acad. Sci. U.S.A. 103:626. doi: 10.1073/pnas.0507535103
Frumkin, I., Lajoie, M. J., Gregg, C. J., Hornung, G., Church, G. M., and Pilpel, Y. (2018). Codon usage of highly expressed genes affects proteome-wide translation efficiency. Proc. Natl. Acad. Sci. U.S.A. 115, E4940–E4949. doi: 10.1073/pnas.1719375115
Fuglsang, A. (2006). Estimating the effective number of codons: the wright way of determining codon homozygosity leads to superior estimates. Genetics 172, 1301–1307. doi: 10.1534/genetics.105.049643
Garrity, G. M., Boone, D. R., and Castenholz, R. W. (2001). Bergey's Manual of Systematic Bacteriology. New York, NY: Springer-Verlag.
Gouy, M., and Gautier, C. (1982). Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res. 10, 7055–7074. doi: 10.1093/nar/10.22.7055
Handelsman, J., Rondon, M. R., Brady, S. F., Clardy, J., and Goodman, R. M. (1998). Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem. Biol. 5, R245–R249. doi: 10.1016/S1074-5521(98)90108-9
Hassan, S., Mahalingam, V., and Kumar, V. (2009). Synonymous codon usage analysis of thirty two mycobacteriophage genomes. Adv. Bioinform. 2009:316936. doi: 10.1155/2009/316936
Hayat, R., Ali, S., Amara, U., Khalid, R., and Ahmed, I. (2010). Soil beneficial bacteria and their role in plant growth promotion: a review. Ann. Microbiol. 60, 579–598. doi: 10.1007/s13213-010-0117-1
Herring, C. D., Raghunathan, A., Honisch, C., Patel, T., Applebee, M. K., Joyce, A. R., et al. (2006). Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale. Nat. Genet. 38, 1406–1412. doi: 10.1038/ng1906
Hershberg, R., and Petrov, D. A. (2008). Selection on codon bias. Annu. Rev. Genet. 42, 287–299. doi: 10.1146/annurev.genet.42.110807.091442
Huang, P. M., Bollag, J., and Senesi, N. (2001). Interactions Between Soil Particles and Microorganisms: Impact on the Terrestrial Ecosystem. New York, NY: John Wiley & Sons.
Ishida, M., Oshima, T., and Yutani, K. (2002). Overexpression in Escherichia coli of the AT-rich trpA and trpB genes from the hyperthermophilic archaeon Pyrococcus furiosus. FEMS Microbiol. Lett. 216, 179–183. doi: 10.1111/j.1574-6968.2002.tb11433.x
Ivanova, N., Sorokin, A., Anderson, I., Galleron, N., Candelon, B., Kapatral, V., et al. (2003). Genome sequence of bacillus cereus and comparative analysis with bacillus anthracis. Nature 423, 87–91. doi: 10.1038/nature01582
Jacoby, R., Peukert, M., Succurro, A., Koprivova, A., and Kopriva, S. (2017). The role of soil microorganisms in plant mineral nutrition—current knowledge and future directions. Front. Plant Sci. 8:1617. doi: 10.3389/fpls.2017.01617
Janda, J. M., and Abbott, S. L. (2007). 16S rRNA Gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J. Clin. Microbiol. 45:2761. doi: 10.1128/JCM.01228-07
Jansson, J. K., and Hofmockel, K. S. (2018). The soil microbiome—from metagenomics to metaphenomics. Curr. Opin. Microbiol. 43, 162–168. doi: 10.1016/j.mib.2018.01.013
Kaminuma, E., Mashima, J., Kodama, Y., Gojobori, T., Ogasawara, O., Okubo, K., et al. (2010). DDBJ launches a new archive database with analytical tools for next-generation sequence data. Nucleic Acids Res. 38, D33–D38. doi: 10.1093/nar/gkp847
Khandia, R., Singhal, S., Kumar, U., Ansari, A., Tiwari, R., Dhama, K., et al. (2019). Analysis of nipah virus codon usage and adaptation to hosts. Front. Microbiol. 10:886. doi: 10.3389/fmicb.2019.00886
Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. doi: 10.1007/BF01731581
Klappenbach, J. A., Dunbar, J. M., and Schmidt, T. M. (2000). rRNA Operon copy number reflects ecological strategies of bacteria. Appl. Environ. Microbiol. 66:1328. doi: 10.1128/AEM.66.4.1328-1333.2000
Krieg, N. R., Ludwig, W., Whitman, W. B., Hedlund, B. P., Paster, B. J., Staley, J. T., et al. (2010). Bergey's Manual of Systematic Bacteriology. New York, NY: Springer-Verlag.
Kumar, S., Nei, M., Dudley, J., and Tamura, K. (2008). MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 9, 299–306. doi: 10.1093/bib/bbn017
Lai, Q., Liu, Y., Yuan, J., Du, J., Wang, L., Sun, F., et al. (2014). Multilocus sequence analysis for assessment of phylogenetic diversity and biogeography in thalassospira bacteria from diverse marine environments. PLoS ONE 9:e106353. doi: 10.1371/journal.pone.0106353
Lang, J. M., Darling, A. E., and Eisen, J. A. (2013). Phylogeny of bacterial and archaeal genomes using conserved genes: supertrees and supermatrices. PLoS ONE 8:e62510. doi: 10.1371/journal.pone.0062510
Lehman, M. R., Cambardella, A. C., Stott, E. D., Acosta-Martinez, V., Manter, K. D., Buyer, S. J., et al. (2015). Understanding and enhancing soil biological health: the solution for reversing soil degradation. Sustainability 7, 988–1027. doi: 10.3390/su7010988
Leopoldseder, S., Hettwer, S., and Sterner, R. (2006). Evolution of multi-enzyme complexes: the case of tryptophan synthase. Biochemistry 45, 14111–14119. doi: 10.1021/bi061684b
Letunic, I., and Bork, P. (2007). Interactive tree of life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. doi: 10.1093/bioinformatics/btl529
Letunic, I., and Bork, P. (2019). Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Liu, Q., Xin, Y. H., Zhou, Y. G., and Chen, W. X. (2018). Multilocus sequence analysis of homologous recombination and diversity in arthrobacter sensu lato named species and glacier-inhabiting strains. Syst. Appl. Microbiol. 41, 23–29. doi: 10.1016/j.syapm.2017.08.002
Liu, X. (2013). A more accurate relationship between effective number of codons and GC3s under assumptions of no selection. Comput. Biol. Chem. 42, 35–39. doi: 10.1016/j.compbiolchem.2012.11.003
Ludwig, W., and Schleifer, K. H. (1994). Bacterial phylogeny based on 16S and 23S rRNA sequence analysis. FEMS Microbiol. Rev. 15, 155–173. doi: 10.1111/j.1574-6976.1994.tb00132.x
Luo, A., Qiao, H., Zhang, Y., Shi, W., Ho, S. Y., Xu, W., et al. (2010). Performance of criteria for selecting evolutionary models in phylogenetics: a comprehensive study based on simulated datasets. BMC Evol. Biol. 10, 242–242. doi: 10.1186/1471-2148-10-242
Magdeldin, S., Yoshida, Y., Li, H., Maeda, Y., Yokoyama, M., Enany, S., et al. (2012). Murine colon proteome and characterization of the protein pathways. BioData Min. 5:11. doi: 10.1186/1756-0381-5-11
Malakar, A. K., Halder, B., Paul, P., and Chakraborty, S. (2016). Cytochrome P450 genes in coronary artery diseases: codon usage analysis reveals genomic GC adaptation. Gene 590, 35–43. doi: 10.1016/j.gene.2016.06.011
Malakar, A. K., Halder, B., Paul, P., Deka, H., and Chakraborty, S. (2019). Genetic evolution and codon usage analysis of NKX-2.5 gene governing heart development in some mammals. Genomics S0888–S7543, 30333–30337. doi: 10.1016/j.ygeno.2019.07.023
Mardis, E. R. (2008). The impact of next-generation sequencing technology on genetics. Trends Gene. 24, 133–141. doi: 10.1016/j.tig.2007.12.007
Margos, G., Vollmer, S. A., Cornet, M., Garnier, M., Fingerle, V., Wilske, B., et al. (2009). A new Borrelia Species defined by multilocus sequence analysis of housekeeping Genes. Appl. Environ. Microbiol. 75, 5410–5416. doi: 10.1128/AEM.00116-09
Markowitz, V. M., Chen, I. M. A., Palaniappan, K., Chu, K., Szeto, E., Grechkin, Y., et al. (2012). IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 40, D115–D122. doi: 10.1093/nar/gkr1044
Martens, M., Dawyndt, P., Coopman, R., Gillis, M., De Vos, P., and Willems, A. (2008). Advantages of multilocus sequence analysis for taxonomic studies: a case study using 10 housekeeping genes in the genus ensifer (including former sinorhizobium). Int. J. Syst. Evol. Microbiol. 58, 200–214. doi: 10.1099/ijs.0.65392-0
Martin-Laurent, F., Philippot, L., Hallet, S., Chaussod, R., Germon, J. C., Soulas, G., et al. (2001). DNA extraction from soils: old bias for new microbial diversity analysis methods. Appl. Environ. Microbiol. 67:2354. doi: 10.1128/AEM.67.5.2354-2359.2001
McTaggart, L. R., Richardson, S. E., Witkowska, M., and Zhang, S. X. (2010). Phylogeny and identification of nocardia species on the basis of multilocus sequence analysis. J. Clin. Microbiol. 48, 4525–4533. doi: 10.1128/JCM.00883-10
Mocali, S., and Benedetti, A. (2010). Exploring research frontiers in microbiology: the challenge of metagenomics in soil microbiology. Res. Microbiol. 161, 497–505. doi: 10.1016/j.resmic.2010.04.010
Morales, S. E., Cosart, T. F., Johnson, J. V., and Holben, W. E. (2009). Extensive phylogenetic analysis of a soil bacterial community illustrates extreme taxon evenness and the effects of amplicon length, degree of coverage, and dna fractionation on classification and ecological parameters. Appl. Environ. Microbiol. 75, 668–75. doi: 10.1128/AEM.01757-08
Mulet, M., Lalucat, J., and Garcia-Valdes, E. (2010). DNA sequence-based analysis of the Pseudomonas species. Environ. Microbiol. 12, 1513–1530. doi: 10.1111/j.1462-2920.2010.02181.x
Naser, S. M., Thompson, F. L., Hoste, B., Gevers, D., Dawyndt, P., Vancanneyt, M., et al. (2005). Application of multilocus sequence analysis (MLSA) for rapid identification of Enterococcus species based on rpoA and pheS Genes. Microbiology 151, 2141–2150. doi: 10.1099/mic.0.27840-0
Nei, M., and Kumar, S. (2000). Molecular Evolution and Phylogenetics. New York, NY: Oxford University Press.
Nesme, J., Achouak, W., Agathos, S. N., Bailey, M., Baldrian, P., Brunel, D., et al. (2016). Back to the future of soil metagenomics. Front. Microbiol. 7:73. doi: 10.3389/fmicb.2016.00073
Pal, A., Banerjee, R., Mondal, U. K., Mukhopadhyay, S., and Bothra, A. K. (2015). Deconstruction of Archaeal genome depict strategic consensus in core pathways coding sequence assembly. PLoS ONE 10:e0118245. doi: 10.1371/journal.pone.0118245
Pandit, A., and Sinha, S. (2011). Differential Trends in the codon usage patterns in HIV-1 Genes. PLoS ONE 6:e28889. doi: 10.1371/journal.pone.0028889
Peden, J. F. (1999). Analysis of codon usage (Ph.D. doctoral thesis), University of Nottingham, Nottingham, United Kingdom.
Pérez-Valera, E., Goberna, M., and Verdú, M. (2015). Phylogenetic structure of soil bacterial communities predicts ecosystem functioning. FEMS Microbiol. Ecol. 91:fiv031. doi: 10.1093/femsec/fiv031
Plotkin, J. B., and Kudla, G. (2011). Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 12, 32–42. doi: 10.1038/nrg2899
Posada, D. (2009). “Selecting models of evolution,” in The Phylogenetic Handbook: A Practical Approach to Phylogenetic Analysis and Hypothesis Testing, 2nd Edn. eds A.-M. Vandamme, M. Salemi, and P. Lemey (Cambridge: Cambridge University Press, 345–361.
Posada, D., and Buckley, T. R. (2004). Model selection and model averaging in phylogenetics: advantages of akaike information criterion and bayesian approaches over likelihood ratio tests. Syst. Biol. 53, 793–808. doi: 10.1080/10635150490522304
Prabha, R., Singh, D. P., Gupta, S. K., Farooqi, S., and Rai, A. (2012). Synonymous codon usage in thermosynechococcus elongatus (cyanobacteria) identifies the factors shaping codon usage variation. Bioinformation 8, 622–628. doi: 10.6026/97320630008622
Quax, T. E., Claassens, N. J., Soll, D., and Van Der Oost, J. (2015). Codon bias as a means to fine-tune gene expression. Mol. Cell 59, 149–161. doi: 10.1016/j.molcel.2015.05.035
Raboni, S., Bettati, S., and Mozzarelli, A. (2009). Tryptophan synthase: a mine for enzymologists. Cell Mol. Life Sci. 66, 2391–2403. doi: 10.1007/s00018-009-0028-0
Rajendhran, J., and Gunasekaran, P. (2011). Microbial phylogeny and diversity: small subunit ribosomal RNA sequence analysis and beyond. Microbiol. Res. 166, 99–110. doi: 10.1016/j.micres.2010.02.003
Roesch, L. F. W., Fulthorpe, R. R., Riva, A., Casella, G., Hadwin, A. K. M., Kent, A. D., et al. (2007). Pyrosequencing enumerates and contrasts soil microbial diversity. Isme J. 1:283. doi: 10.1038/ismej.2007.53
Rong, X., and Huang, Y. (2010). Taxonomic evaluation of the streptomyces griseus clade using multilocus sequence analysis and DNA–DNA hybridization, with proposal to combine 29 species and three subspecies as 11 genomic species. Int. J. Syst. Evol. Microbiol. 60, 696–703. doi: 10.1099/ijs.0.012419-0
Rong, X., and Huang, Y. (2014). “Chapter 11–Multi-locus sequence analysis: taking prokaryotic systematics to the next level,” in Methods in Microbiology, eds M. Goodfellow, I. Sutcliffe, and J. Chun (London; San Diego, CA; Waltham, MA; Oxford: Academic Press, 221–251.
Rosselli, R., Romoli, O., Vitulo, N., Vezzi, A., Campanaro, S., De Pascale, F., et al. (2016). Direct 16S rRNA-seq from bacterial communities: a PCR-independent approach to simultaneously assess microbial diversity and functional activity potential of each taxon. Sci. Rep. 6:32165. doi: 10.1038/srep32165
Rosselló-Mora, R., and Amann, R. (2001). The species concept for prokaryotes. FEMS Microbiol. Rev. 25, 39–67. doi: 10.1111/j.1574-6976.2001.tb00571.x
Sahoo, S., Das, S. S., and Rakshit, R. (2019). Codon usage pattern and predicted gene expression in arabidopsis thaliana. Gene 2:100012. doi: 10.1016/j.gene.2019.100012
Salim, H. M. W., and Cavalcanti, A. R. O. (2008). Factors influencing codon usage bias in genomes. J. Braz. Chem. Soc. 19, 257–262. doi: 10.1590/S0103-50532008000200008
Schouls, L. M., Schot, C. S., and Jacobs, J. A. (2003). Horizontal transfer of segments of the 16S rRNA Genes between species of the Streptococcus anginosus group. J. Bacteriol. 185:7241. doi: 10.1128/JB.185.24.7241-7246.2003
Serrano, W., Amann, R., Rosselló-Mora, R., and Fischer, U. (2010). Evaluation of the use of multilocus sequence analysis (MLSA) to resolve taxonomic conflicts within the genus marichromatium. Syst. Appl. Microbiol. 33, 116–121. doi: 10.1016/j.syapm.2009.12.003
Sharp, P. M., Emery, L. R., and Zeng, K. (2010). Forces that influence the evolution of codon bias. Philos. Trans. Soc. Lond. B Biol. Sci. 365, 1203–1212. doi: 10.1098/rstb.2009.0305
Shimp, J. F., Tracy, J. C., Davis, L. C., Lee, E., Huang, W., Erickson, L. E., et al. (1993). Beneficial effects of plants in the remediation of soil and groundwater contaminated with organic materials. Crit. Rev. Environ. Sci. Technol. 23, 41–77. doi: 10.1080/10643389309388441
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Soler, L., Yáñez, M. A., Chacon, M. R., Aguilera-Arreola, M. G., Catalán, V., Figueras, M. J., et al. (2004). Phylogenetic analysis of the genus aeromonas based on two housekeeping genes. Int. J. Syst. Evol. Microbiol. 54, 1511–1519. doi: 10.1099/ijs.0.03048-0
Song, H., Gao, H., Liu, J., Tian, P., and Nan, Z. (2017a). Comprehensive analysis of correlations among codon usage bias, gene expression, and substitution rate in arachis duranensis and arachis ipaënsis orthologs. Sci. Rep. 7:14853. doi: 10.1038/s41598-017-13981-1
Song, H., Liu, J., Song, Q., Zhang, Q., Tian, P., and Nan, Z. (2017b). Comprehensive analysis of codon usage bias in seven epichloe species and their peramine-coding Genes. Front. Microbiol. 8:1419. doi: 10.3389/fmicb.2017.01419
Stefanis, C., Alexopoulos, A., Voidarou, C., Vavias, S., and Bezirtzoglou, E. (2013). Principal methods for isolation and identification of soil microbial communities. Fol. Microbiol. 58, 61–68. doi: 10.1007/s12223-012-0179-5
Sun, S., Xiao, J., Zhang, H., and Zhang, Z. (2016). Pangenome evidence for higher codon usage bias and stronger translational selection in core Genes of Escherichia coli. Front. Microbiol. 7:1180. doi: 10.3389/fmicb.2016.01180
Supek, F., and Vlahovicek, K. (2004). INCA: synonymous codon usage analysis and clustering by means of self-organizing map. Bioinformatics 20, 2329–2330. doi: 10.1093/bioinformatics/bth238
Tambong, J. T., Xu, R., Kaneza, C.-A., and Nshogozabahizi, J.-C. (2014). An in-depth analysis of a multilocus phylogeny identifies leus as a reliable phylogenetic marker for the genus pantoea. Evol. Bioinform. Online 10, 115–125. doi: 10.4137/EBO.S15738
Tatarinova, T., Elhaik, E., and Pellegrini, M. (2013). Cross-species analysis of genic GC3 content and DNA methylation patterns. Genome Biol. Evol. 5, 1443–1456. doi: 10.1093/gbe/evt103
Thiergart, T., Landan, G., and Martin, W. F. (2014). Concatenated alignments and the case of the disappearing tree. BMC Evol. Biol. 14:266. doi: 10.1186/s12862-014-0266-0
Tuller, T., Waldman, Y. Y., Kupiec, M., and Ruppin, E. (2010). Translation efficiency is determined by both codon bias and folding energy. Proc. Nat. Acad. Sci. U.S.A. 107:3645. doi: 10.1073/pnas.0909910107
Tyler, S., Tyson, S., Dibernardo, A., Drebot, M., Feil, E. J., Graham, M., et al. (2018). Whole genome sequencing and phylogenetic analysis of strains of the agent of lyme disease borrelia burgdorferi from Canadian emergence zones. Scient. Rep. 8:10552. doi: 10.1038/s41598-018-28908-7
Vásquez-Ponce, F., Higuera-Llantén, S., Pavlov, M. S., Marshall, S. H., and Olivares-Pacheco, J. (2018). Phylogenetic MLSA and phenotypic analysis identification of three probable novel Pseudomonas species isolated on King George Island, South Shetland, Antarctica. Braz. J. Microbiol. 49, 695–702. doi: 10.1016/j.bjm.2018.02.005
Villaseñor, T., Brom, S., Dávalos, A., Lozano, L., Romero, D., and Los Santos, A. G.-D. (2011). Housekeeping genes essential for pantothenate biosynthesis are plasmid-encoded in rhizobium etli and rhizobium leguminosarum. BMC Microbiol. 11:66. doi: 10.1186/1471-2180-11-66
Vos, M., Quince, C., Pijl, A. S., De Hollander, M., and Kowalchuk, G. A. (2012). A comparison of rpoB and 16S rRNA as markers in pyrosequencing studies of bacterial diversity. PLoS ONE 7:e30600–e30600. doi: 10.1371/journal.pone.0030600
Vos, P., Garrity, G., Jones, D., Krieg, N. R., Ludwig, W., Rainey, F. A., et al. (2009). Bergey's Manual of Systematic Bacteriology. New York, NY: Springer-Verlag.
Wang, L., Xing, H., Yuan, Y., Wang, X., Saeed, M., Tao, J., et al. (2018). Genome-wide analysis of codon usage bias in four sequenced cotton species. PLoS ONE 13:e0194372. doi: 10.1371/journal.pone.0194372
Wang, Y., Zhang, Z., and Ramanan, N. (1997). The actinomycete thermobispora bispora contains two distinct types of transcriptionally active 16S rRNA Genes. J. Bacteriol. 179:3270. doi: 10.1128/jb.179.10.3270-3276.1997
Wei, K., Zhang, T., and Ma, L. (2018). Divergent and convergent evolution of housekeeping genes in human-pig lineage. PeerJ 6, e4840–e4840. doi: 10.7717/peerj.4840
Wen, S., Chen, X., Xu, F., and Sun, H. (2016). Validation of reference genes for real-time quantitative PCR (qPCR) analysis of avibacterium paragallinarum. PLoS ONE 11:e0167736. doi: 10.1371/journal.pone.0167736
Whitman, W. B., Coleman, D. C., and Wiebe, W. J. (1998). Prokaryotes: the unseen majority. Proc. Natl. Acad. Sci. U.S.A. 95, 6578–6583. doi: 10.1073/pnas.95.12.6578
Whitman, W. B., Goodfellow, M., Kämpfer, P., Busse, H.-J., Trujillo, M. E., Ludwig, W., et al. (2012). Bergey's Manual of Systematic Bacteriology. New York, NY: Springer-Verlag.
Will, C., Thurmer, A., Wollherr, A., Nacke, H., Herold, N., Schrumpf, M., et al. (2010). Horizon-specific bacterial community composition of German grassland soils, as revealed by pyrosequencing-based analysis of 16S rRNA Genes. Appl. Environ. Microbiol. 76, 6751–6759. doi: 10.1128/AEM.01063-10
Willenbrock, H., Friis, C., Juncker, A. S., and Ussery, D. W. (2006). An environmental signature for 323 microbial genomes based on codon adaptation indices. Genome Biol. 7:R114. doi: 10.1186/gb-2006-7-12-r114
Winsley, T., Van Dorst, J. M., Brown, M. V., and Ferrari, B. C. (2012). Capturing greater 16S rRNA gene sequence diversity within the domain bacteria. Appl. Environ. Microbiol. 78, 5938–5941. doi: 10.1128/AEM.01299-12
Wright, F. (1990). The effective number of codons used in a gene. Gene 87, 23–29. doi: 10.1016/0378-1119(90)90491-9
Wu, M., and Eisen, J. A. (2008). A simple, fast, and accurate method of phylogenomic inference. Genome Biol. 9, R151–R151. doi: 10.1186/gb-2008-9-10-r151
Xie, G., Forst, C., Bonner, C., and Jensen, R. A. (2001). Significance of two distinct types of tryptophan synthase beta chain in bacteria, archaea and higher plants. Genome Biol. 3:RESEARCH0004. doi: 10.1186/gb-2001-3-1-research0004
Xu, C., Cai, X., Chen, Q., Zhou, H., Cai, Y., and Ben, A. (2011). Factors affecting synonymous codon usage bias in chloroplast genome of oncidium gower ramsey. Evol. Bioinform. Online 7, 271–278. doi: 10.4137/EBO.S8092
Xu, C., Dong, J., Tong, C., Gong, X., Wen, Q., and Zhuge, Q. (2013). Analysis of synonymous codon usage patterns in seven different citrus species. Evol. Bioinform. 9:S11930. doi: 10.4137/EBO.S11930
Yannai, A., Katz, S., and Hershberg, R. (2018). The Codon usage of lowly expressed genes is subject to natural selection. Genome Biol. Evol. 10, 1237–1246. doi: 10.1093/gbe/evy084
Yanofsky, C. (2001). Advancing our knowledge in biochemistry, genetics, and microbiology through studies on tryptophan metabolism. Annu. Rev. Biochem. 70, 1–37. doi: 10.1146/annurev.biochem.70.1.1
Keywords: codon usage bias (CUB), molecular phylogenetics, soil bacteria, housekeeping genes, atpD gene, infB gene, trpB gene, rpoB gene
Citation: Saha J, Saha BK, Pal Sarkar M, Roy V, Mandal P and Pal A (2019) Comparative Genomic Analysis of Soil Dwelling Bacteria Utilizing a Combinational Codon Usage and Molecular Phylogenetic Approach Accentuating on Key Housekeeping Genes. Front. Microbiol. 10:2896. doi: 10.3389/fmicb.2019.02896
Received: 25 September 2019; Accepted: 02 December 2019;
Published: 17 December 2019.
Edited by:
Frank T. Robb, University of Maryland, Baltimore, United StatesReviewed by:
Martha Helena Ramírez-Bahena, University of Salamanca, SpainLuis Rey, Polytechnic University of Madrid, Spain
Copyright © 2019 Saha, Saha, Pal Sarkar, Roy, Mandal and Pal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ayon Pal, YXlvbnBhbC5ydWNAZ21haWwuY29t