 Christian A. Devaux
Christian A. Devaux Soraya Mezouar
Soraya Mezouar Jean-Louis Mege
Jean-Louis Mege
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 08 November 2019
Sec. Microbial Immunology
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.02598
Once bound to the epithelium, pathogenic bacteria have to cross epithelial barriers to invade their human host. In order to achieve this goal, they have to destroy the adherens junctions insured by cell adhesion molecules (CAM), such as E-cadherin (E-cad). The invasive bacteria use more or less sophisticated mechanisms aimed to deregulate CAM genes expression or to modulate the cell-surface expression of CAM proteins, which are otherwise rigorously regulated by a molecular crosstalk essential for homeostasis. Apart from the repression of CAM genes, a drastic decrease in adhesion molecules on human epithelial cells can be obtained by induction of eukaryotic endoproteases named sheddases or through synthesis of their own (prokaryotic) sheddases. Cleavage of CAM by sheddases results in the release of soluble forms of CAM. The overexpression of soluble CAM in body fluids can trigger inflammation and pro-carcinogenic programming leading to tumor induction and metastasis. In addition, the reduction of the surface expression of E-cad on epithelia could be accompanied by an alteration of the anti-bacterial and anti-tumoral immune responses. This immune response dysfunction is likely to occur through the deregulation of immune cells homing, which is controlled at the level of E-cad interaction by surface molecules αE integrin (CD103) and lectin receptor KLRG1. In this review, we highlight the central role of CAM cell-surface expression during pathogenic microbial invasion, with a particular focus on bacterial-induced cleavage of E-cad. We revisit herein the rapidly growing body of evidence indicating that high levels of soluble E-cad (sE-cad) in patients’ sera could serve as biomarker of bacterial-induced diseases.
Cadherins (cad) belong to the superfamily of cell-adhesion molecules (CAMs) (Takeichi, 1977; Nollet et al., 2000; Angst et al., 2001; Hulpiau and van Roy, 2009). Characterized by their adhesion properties mediated through repeated extracellular cad domains (ECs) under Ca2+ control, cad play a key role in cell-to-cell interactions (Hyafil et al., 1981). Several subtypes of cad are encoded in the human genome (Oda and Takeichi, 2011). These molecules were classified according to their tissue distribution: for example, the P prefix is used in P-cad (encoded by CDH3) to define placental cad, N-cad (encoded by CDH2 and CDH12) for neural cad, VE-cad (encoded by CDH5) for vascular endothelial cad, and E-cadherin (E-cad) (encoded by CDH1) for epithelial cad. The CDH1 gene, located on chromosome 16q22.1, comprises 16 exons and 15 introns (Berx et al., 1995), and it is transcribed into a 4.5Kb pre-mRNA that is spliced to generate the E-cad mRNA. Transcriptional repression of CDH1 gene is achieved by a range of transcriptional repressors that bind its promoter, including members of the SNAIL and ZEB gene families of zinc-finger transcription factors (Cano et al., 2000; Bolós et al., 2003; Cadigan and Waterman, 2012). Repression of CDH1 gene can also be the result of CpG-island hypermethylation of its promoter, loss of heterozygosis at 16q22.1, and inactivating mutations (Berx et al., 1998; Lombaerts et al., 2006).
Initially described as liver cell adhesion molecule (L-CAM) and uvomorulin (Gallin et al., 1983; Schuh et al., 1986), E-cad is a single-pass type I transmembrane glycoprotein of 120 kDa that plays a major role in cell polarity, intercellular adhesion, and tissue integrity (Ogou et al., 1983; Niessen et al., 2011; van Roy, 2014). It possesses five EC repeats with binding sites for Ca2+ (Shapiro et al., 1995). These predominantly homophilic E-cad dimerize in cis at the cell’s surface and the homodimer can then interact in trans with an adjacent E-cad homodimer on a neighboring epithelial cell to form adherens junctions (Boggon, 2002). However, E-cad can also exhibit heterophilic interactions in trans with the αEβ7 integrin, also called CD103 antigen of T-lymphocytes, which generally lacks E-cad cell surface expression (Cepek et al., 1994; Sheridan and Lefrançois, 2011) as well as it can bind the killer cell lectin receptor G1 (KLRG1) expressed on T-lymphocytes and natural killer (NK) cells (Kilshaw, 1999; Ito et al., 2006). Over-expression of E-cad can delay the rate of cell migration (Hermiston et al., 1996). Loss of E-cad can reduce CD103+ T-cell antitumor activity (Shields et al., 2019). Under physiological conditions, E-cad interacts with p120-ctn and β-catenin (β-cat) via its intracytoplasmic tail (Nagafuchi and Takeichi, 1988; McCrea and Gumbiner, 1991; Kourtidis et al., 2013). The cytoplasmic tail of E-cad consists of the juxta membrane domain (JMD), which allows the clustering of cad and contributes to the adhesive strength via p120-ctn, and the cat-binding domain (CBD), which interacts with β-cat and γ-cat (Kemler, 1993; Yap et al., 1998). The α-cat links the bound β-cat and the actin cytoskeleton. Signaling through E-cad cytoplasmic tail is a complex process which involves multiple contacts with intracytoplasmic partners, whose diversity is just beginning to be elucidated by the characterization of the E-cad interactome (Guo et al., 2014). E-cad is a tumor suppressor acting through intracytoplasmic retention of β-catenin stocks and suppresses inflammatory signaling pathways (Figure 1).
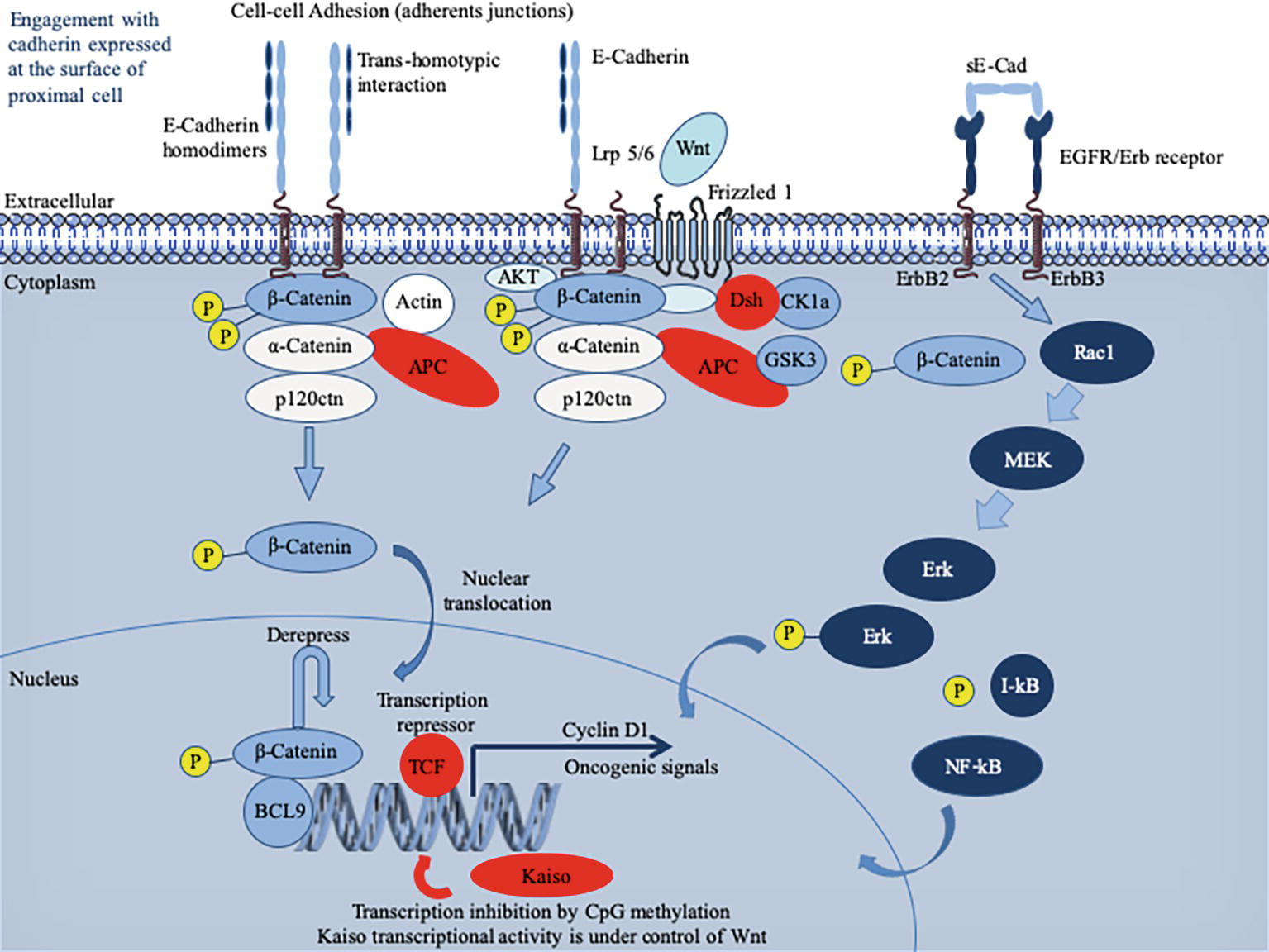
Figure 1. Schematic representation of the E-cadherin (E-cad) interactions and signaling pathway. Newly synthesized E-cad are transported from the Golgi apparatus to the cell surface where they are available to engagement in intercellular interactions. The model presented reflects evidence that E-cad homodimers are involved in adherens’ junctions. Loss of E-cad expression in epithelia results in loosening of intercellular contacts. E-cad regulates the intracytoplasmic pool of α-cat and β-cat acts as a signal transducer molecule in response to upstream Wnt pathway (Fagotto, 2013). Briefly, the Wnt pathway is initiated by the binding of an extracellular Wnt ligand to a surface receptor composed of Frizzled, a seven transmembrane (7TM) molecule and low-density lipoprotein receptor-related protein 5 or 6 (Lrp5/6). As a result of the Wnt pathway activation that mobilizes several intracytoplasmic molecules (including disheveled, adenomatous polyposis coli – APC binds axin and β-cat and inhibits glycogen synthase kinase 3β – and ΑΚΤ kinase) (Fang et al., 2007; MacDonald et al., 2009; Wu et al., 2009), free cytoplasmic β-cat destruction is inhibited and β-cat translocates to the nucleus. Once in the nucleus, β-cat activates expression of genes such as cyclin D1 or c-MYC, otherwise repressed by the T cell factor/lymphoid enhancer factor (TCF/LEF) (Gumbiner, 1995). To achieve trans-activation, β-cat recruits a range of nuclear co-factors including CBP/p300, Brg-1, and the adaptor protein BCL9 (Hecht et al., 2000; Barker et al., 2001; Kramps et al., 2002; de la Roche et al., 2008). Cad-free p120ctn can also trigger the nuclear translocation of β-cat through its association with the Vav2 small GTPase and activation of JNK kinase (Wu et al., 2009; Valls et al., 2012). It is worth noting that TCF/LEF-binding sites have been identified in the CDH1 gene promoter, which may permit a feedback control loop where β-cat might activate E-cad mRNA production (Huber et al., 1996). E-cad expression can also be regulated by microRNAs (not shown), such as miR200, which favor E-cad mRNA expression (Gregory et al., 2008). Cells with greater E-cad abundance can sequester and thereby inhibit the ability of β-catenin to translocate to the cell nucleus to derepress the activity of its DNA-binding factor TCF/LEF. When unable to engage in interactions, E-cad enters an endocytic uptake and is directed to early sorting endosomes from which they can either be recycled back to the cell surface by recycling endosomes or routed from late endosomes to proteasome where they undergo degradation (for details see the reviews by (Niessen et al., 2011; Nava et al., 2013; McCrea et al., 2015). During infection with bacteria, the pathogen can either regulate the expression of the CDH1 gene and thereby the cell-surface abundance of E-cad or trigger the catalytic cleavage of E-cad. This can result in the release of a cytosolic pool of β-catenin acting as a downstream effector in the Wnt signaling pathway and induction of oncogenic signals. The sE-cad can associate with intact E-cad present on other cells to alter Cad-dependent cellular behavior. It can also interact with the EGFR molecule to activate the ERK signaling pathway.
Fusobacterium nucleatum, a pathogen associated with oral plaque formation and colorectal cancers, binds E-cad through its FadA adhesin (Rubinstein et al., 2013). This interaction up-regulates the Annexin A1 and activates β-cat signaling (Zhou et al., 2018; Rubinstein et al., 2019). The F. nucleatum was reported to be associated with a specific epigenetic pattern of tumor cells characterized by hypermethylation of CpG islands, high MSI and MLH1 hypermethylation (epigenetic silencing), and up-regulation of microRNA-21 (Tahara et al., 2014; Yang et al., 2017). Listeria monocytogenes, the causative agent of severe food poisoning, which sometimes lead to meningitis, internalizes when internalin A (InlA) and InlB bind to E-cad and the hepatocyte growth factor receptor on the basolateral surface of epithelial cells (Cossart and Sansonetti, 2004; Barbuddhe and Chakraborty, 2009; Ortega et al., 2017). Streptococcus pneumoniae can cause pneumonia, meningitis, and bacteremia. The flamingo cadherin was reported to serve as receptor for the S. pneumoniae fructose bisphosphate aldolase (Blau et al., 2007), and E-cad was found to act as adherence receptor for the pneumococcal surface adhesin A (PsaA) of S. pneumoniae during the colonization of nasopharyngeal epithelial cells (Anderton et al., 2007). Helicobacter pylori, a bacterium responsible for severe gastric disease, adhere to target cells through interaction with CEACAM cell-surface receptor via its HopQ adhesin (Javaheri et al., 2016). Then, the bacterial HtrA sheddase cleaves the gastric epithelial cell-to-cell junctions through endoproteolysis of E-cad, occludin, and claudin-8 (Tegtmeyer et al., 2017). After transmigration of H. pylori to the basolateral membrane of gastric epithelial cells, the T4SS pilus is activated and injects the CagA cytotoxin into the target cell where the release of β-cat is stimulated (Suzuki et al., 2005; Murata-Kamiya et al., 2007) (Zhao et al., 2018; Tegtmeyer et al., 2019).
In the world of bacteria, other cadherin and CAM play a role during the phase of attachment and invasion (Table 1). The attachment of Leptospira interrogans to host cells was found to be mediated through its interaction with VE-cad, which triggers the process leading to different symptoms, including liver dysfunction, kidney failure, myocarditis, and sometimes the pulmonary hemorrhagic manifestations of leptospirosis (Evangelista et al., 2014). Human host colonization by Haemophilus influenzae began with the binding of the bacteria to I-CAM1 on the surface of the respiratory tract epithelial cells through its Type IV pilus (Tfp), a process leading to respiratory diseases such as cystic fibrosis or chronic obstructive pulmonary disease (Novotny and Bakaletz, 2016). H. influenzae invasion is made even more effective as humans carry adenovirus or respiratory syncytial virus, which are known to increase cell-surface expression of I-CAM1. H. influenzae can also bind CEACAM through outer membrane protein OMP-1 (Tchoupa et al., 2015). CEACAM was also reported to serve as a receptor for the Opa protein of Neisseria gonorrhoeae during the colonization of urogenital mucosal surfaces in humans. It can progress toward acute urethritis with purulent urethral discharge in men, while the infection can remain asymptomatic in women or evolve toward an inflammation of the endocervix or an infection of fallopian tubes (Sintsova et al., 2015).
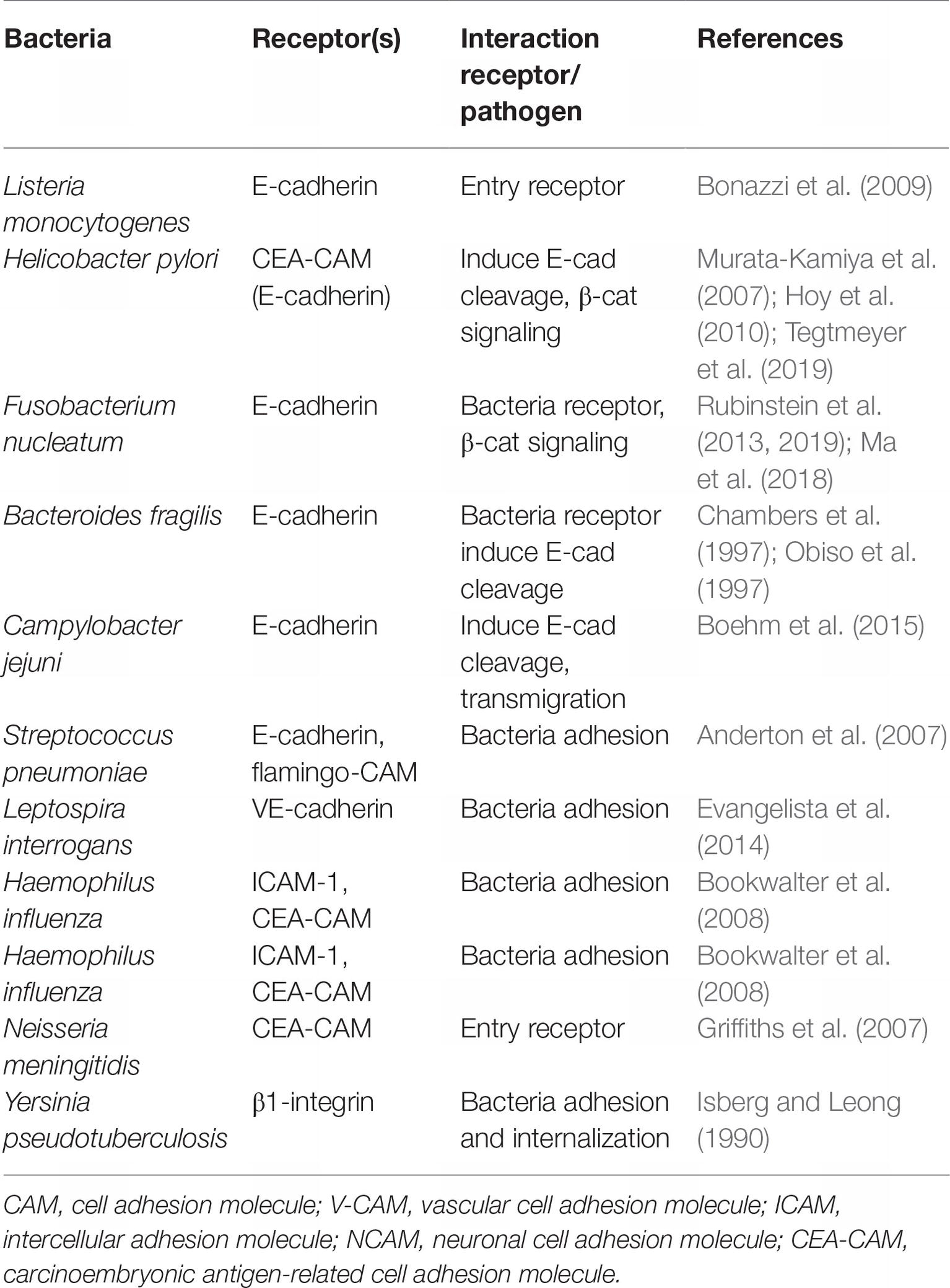
Table 1. Function of E-cad and other CAM molecules in bacteria-mediated infectious diseases.
Besides bacteria, many human pathogens also use E-cad and/or other CAM during the human host colonization, indicating the ubiquitous nature of this process. For example, a fungus, Aspergillus fumigatus, which is responsible for the majority of invasive mold infections in patients undergoing chemotherapy or organ transplantation, was found to bind E-cad and to use it as a receptor for adhesion and endocytosis of blastopores in epithelial cells (Xu et al., 2012; Yan et al., 2015), and Candida albicans, the causative agent of hematogenously disseminated and oropharyngeal candidiasis, internalizes through direct interactions between its surface adhesin Als3 and E-cad on the target cell (Egusa et al., 2005; Phan et al., 2007). Regarding viruses, it has been reported that E-cad, together with claudin 1 and occludin, plays a role in the hepatitis C virus entry into hepatocytes (Colpitts et al., 2016; Li et al., 2016). Several CAM, such as I-CAM, V-CAM, and N-CAM have also been identified as viral receptors for viruses, such as coxsackie A virus; rhinovirus, Enterovirus D68, encephalomyocarditis virus, and rabies virus, respectively (Thoulouze et al., 1998; Bhella, 2015; Wei et al., 2016).
Usually, the commensal microbiota, which supplies the host with molecules essential to life and shapes gene expression in eukaryotic cells (Devaux and Raoult, 2018), together with epithelial cell barriers and appropriate immune responses, efficiently protects the internal body against pathogenic microbial invasion (Abt and Pamer, 2014). Pathogenic bacteria have engineered different strategies to get around these natural defenses by the transcellular route, by acting on cell-to-cell junctions, or by taking advantage of damaged tissues.
Clostridium perfringens, which is the causative agent of gas gangrene and food poisoning, produces a pore-forming delta toxin, which was found capable of reducing cell surface expression of E-cad by enhancing ADAM-10 sheddase activity (Figure 2; Seike et al., 2018). Similarly, the alpha toxin of Staphylococcus aureus binds to and up-regulates ADAM-10 metalloprotease activity in alveolar epithelial cells. This activity results in the cleavage of E-cad and contributes to the pathogenesis of lethal pneumonia (Inoshima et al., 2011). Clostridium botulinum produces the botulinum neurotoxin (BoNT), which provokes flaccid paralysis known as botulism, by inhibiting neurotransmitter release at the neuro-muscular junctions (Carter and Peck, 2015). To allow BoNT complex to pass through the epithelial barrier of the intestinal tract and act on the neurotransmission process, one compound of the BoNT complex termed hemagglutinin (HA), binds E-cad and disrupts the tight junctions (Sugawara and Fujinaga, 2011). The prokaryotic high temperature requirement A (HtrA) protease-mediated cleavage of E-cad that precedes the process of transmigration has been described for gastrointestinal pathogens, such as enteropathogenic Escherichia coli, Shigella flexneri, and Campylobacter jejuni (Boehm et al., 2012; Hoy et al., 2012; Elmi et al., 2016). The HtrA sheddase of Helicobacter pylori was found to open adherens junctions by cleaving E-cad and claudin-8 occludin (Tegtmeyer et al., 2017). HtrA sheddase has been found in most of the bacterial genomes studied to date and is associated with pathogenicity. An opportunistic pathogen like Serratia marcescens produces a pore-forming toxin (ShlA) responsible for the tissues damage required to cross cellular barriers (Hertle and Schwarz, 2004). Another opportunistic bacterium, Pseudomonas aeruginosa, which causes aggressive infections in patients compromised by respiratory diseases such as cystic fibrosis, also encodes a pore-forming toxin (exolysin A) that induces major injuries of tissues (Reboud et al., 2017). Both SHlA and ExlA influence ADAM-10 activation triggering E-cad and VE-cad cleavage in epithelial and endothelial cells, respectively, as well as soluble CAMs shedding, and intercellular junction rupture (Reboud et al., 2017). Leptospira interrogans, which crosses host tissue barriers and causes leptospirosis, secretes a protein named LIC10831 that binds E-cad and VE-cad and plays a role during bacterial invasion (Eshghi et al., 2019). The genome of Porphyromonas gingivalis, a bacterium associated with adult periodontitis (Katz et al., 2000), encodes three cysteine proteases named Gingipains (HRgpA, RgpB, and Kgp). The Kgp protease was found capable to disrupt adherens junction by cleavage of E-cad (Katz, 2002). P. gingivalis also cleaves N- and VE-cads (Sheets et al., 2005). With the L. monocytogenes, the infectious process starts with the interaction between the invasion proteins internalin and InlB and their cellular receptor E-cad and hepatocyte growth factor receptor (HGF-R)/Met (Seveau et al., 2004). E-cad also constitutes a target for L. monocytogenes in order to disrupt the blood brain barrier and facilitate the invasion of the brain (Al-Obaidi and Desa, 2018). In Chlamydia trachomatis infections, a bacterium responsible for acute salpingitis and cervicitis, which can also induce scarring disease of the ocular mucosa, a DNA methylation of the CDH1 promoter and downregulation of E-cad expression, was reported (Rajić et al., 2017). It can be hypothesized that the list of pathogenic bacteria shown to cleave the E-cad during the invasion process will increase rapidly.
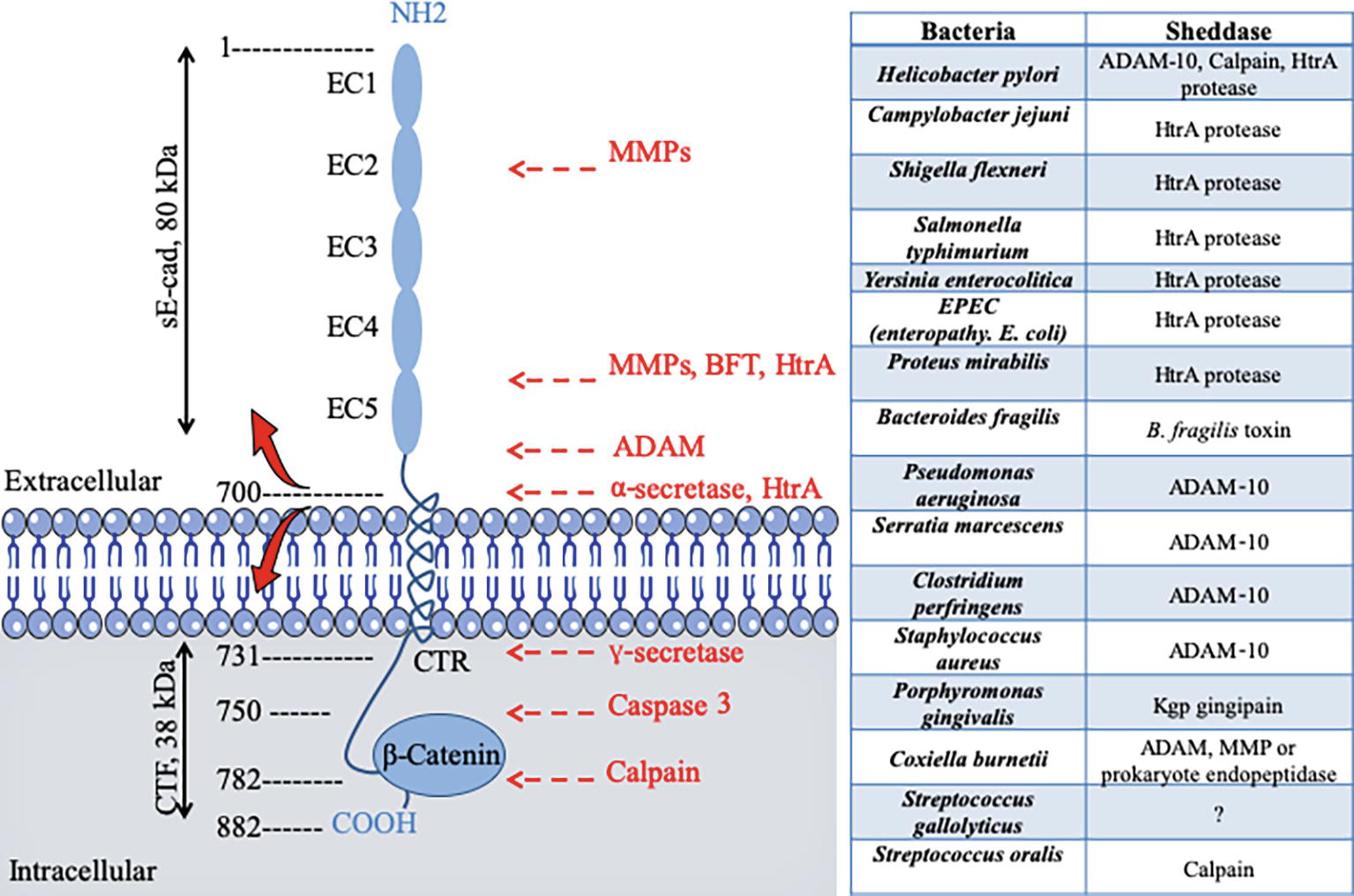
Figure 2. Schematic diagram of E-cad and its cleavage sites by proteases after pathogenic bacteria infection. E-cad is a transmembrane protein containing five extracellular repeated domains (EC1 to EC5), a transmembrane region, and an intracytoplasmic C-terminal region (CTR). The extracellular portion of E-cad forms junction with CAM on proximal cell (see Figure 1), whereas the CTR binds β-catenin and other signaling molecules. Left panel, catalytic enzymes have the capacity to cleave E-cad at specific sites indicated by red arrows. Cleavages occur either in the intracellular CTR of the molecule (e.g., caspase 3 or calpain) generating polypeptides that are capable of triggering signals, or in the extracellular portion of the molecule (e.g., the eukaryotic sheddases MMP and ADAM or the prokaryotic sheddases: BTF or HtrA), leading to soluble extracellular E-cad fragment release. For instance, the eukaryotic ADAM sheddases catalyze a cleavage of E-cad that results in the release of the 80-kDa soluble ectodomain form sE-cad and a 38-kDa C-terminal fragment (CTF). The prokaryotic HtrA serine protease can also cleave E-cad at different extracellular sites. Right panel bacteria reported to trigger E-cad cleavage (and release of sE-cad) and identification of sheddases involved in this process.
For other pathogens, such a requirement to achieve transmigration for colonizing their host can also be illustrated with a few examples. Tissues invasion by C. albicans is associated with degradation of E-cad mediated by the fungus aspartyl proteinase Sap5p under the control of the transcription factor Rim101p (Villar et al., 2007). C. albicans was also reported capable of reducing E-cad mRNA expression (Rouabhia et al., 2012). More recently, it was reported that C. albicans synergized with Streptococcus oralis to increase the proteolytic degradation of E-cad by μ-calpain, which facilitates fungal invasion (Xu et al., 2016). It illustrates the fact that the modulation of cell-surface expression of E-cad is not limited to bacteria, but is rather a general mechanism used by infectious pathogens for invasion and transmigration (Grabowska and Day, 2012).
As described above in this paper, pathogenic bacteria such as H. pylori, Pseudomonas aeruginosa, Serratia marcescens, Clostridium perfringens, and S. aureus have developed takeover stratagems to use eukaryotic sheddases of the human host (e.g., ADAM-10) in order to modulate the host cell surface expression of E-cad.
The cleavage of adhesion molecules is far from being limited to the pathogens' invasion processes. Apart dysbiosis, proteolysis is a common physiological mechanism of post-translational regulation that affects 2–4% of the proteins expressed on the surface of cells (Pandiella et al., 1992; Arribas and Merlos-Suárez, 2003). E-cad is one of the molecules that can undergo proteolytic cleavage (both intracellular and extracellular), providing an alternative regulatory mechanism to reduce its cell surface expression (Noë et al., 2001; Marambaud, 2002; van Roy and Berx, 2008). It is essential to regulate the balance between adhesion and migration of cells. The human genome encodes almost 600 proteases, which control a wide range of processes essential to life (Turk et al., 2012). Proteases can be organized into five main classes, including cysteine proteases, serine proteases, metalloproteases, threonine proteases, and aspartic proteases, with approximately one half being extracellular and the other half intracellular. Quantitative cell surface expression of E-cad is therefore determined by the balance between biosynthesis, trafficking, transfer to cell-surface, intracellular and extracellular proteolytic cleavage, and intracellular degradation, and these processes are considered crucial determinants for cell behavior (Godt and Tepass, 1998). Endoproteases (which cleave internal peptides bonds), which are capable of extracellular E-cad cleavage, belong to the large family of sheddases (Grabowska and Day, 2012). The human sheddases (Figure 3) include zinc-dependent matrix metalloproteases (matrilysin/MMP-2, 3, 7, 9, and 14) (Lee et al., 2007; Symowicz et al., 2007; Klein and Bischoff, 2011), members of the disintegrin metalloproteases family (adamalysin/ADAM-10 and -15) (Maretzky et al., 2008; Najy et al., 2008; Giebeler and Zigrino, 2016), cysteine cathepsins (B, L, S) (Jordans et al., 2009), serine protease kallikrein (KLK-6 and -7) (Johnson et al., 2007; Klucky et al., 2007), plasmin serine protease (Ryniers et al., 2002), and the membrane-bound aspartic proteinases BACE-1 and BACE-2 (Wakabayashi and De Strooper, 2008; Grabowska and Day, 2012). Sheddases are secreted as proenzymes and become mature after processing of the propeptide; for example, the matrix metalloprotease 3 is synthesized as a zymogen (proMMP-3) which is converted to full activity (two active forms of 45-kDa and 25-kDa) by limited proteolysis mediated by elastase and cathepsin G (Okada and Nakanishi, 1989; Becker et al., 1995). Similarly, the MMP-9 is synthesized as a 92-kDa proMMP-9, and then the active MMP-3 initially cleaves proMMP-9 to generate a 86-kDa intermediate ultimately cleaved to convert the intermediate form into a 82-kDa catalytic form (Ogata et al., 1992). The catalytic domains of MMP-9 alone can hydrolyze non-collagenous proteins and synthetic substrates, but cannot cleave triple helix collagens without the hemopexin domain (Nagase et al., 2006). Its fibronectin type II domains are important to cleave type IV collagen, elastin, and gelatins. The hemopexin domain is required for collagenolytic activity of the collagenase. The catalytic activity of sheddases triggers the extracellular release of a soluble E-cad (sE-cad) fragment of about 80-kDa from the cell surface. This process is accompanied by the simultaneous delivery of free β-cat into the cell cytosol, which then translocates into the cell nucleus where it contributes to the modulation of gene expression. It is worth noting that sE-cad might also behave as a signaling molecule through ErbB receptor activation (Najy et al., 2008). As already mentioned above, some pathogenic bacteria can enslave eukaryotic sheddase to get E-cad cleaved.
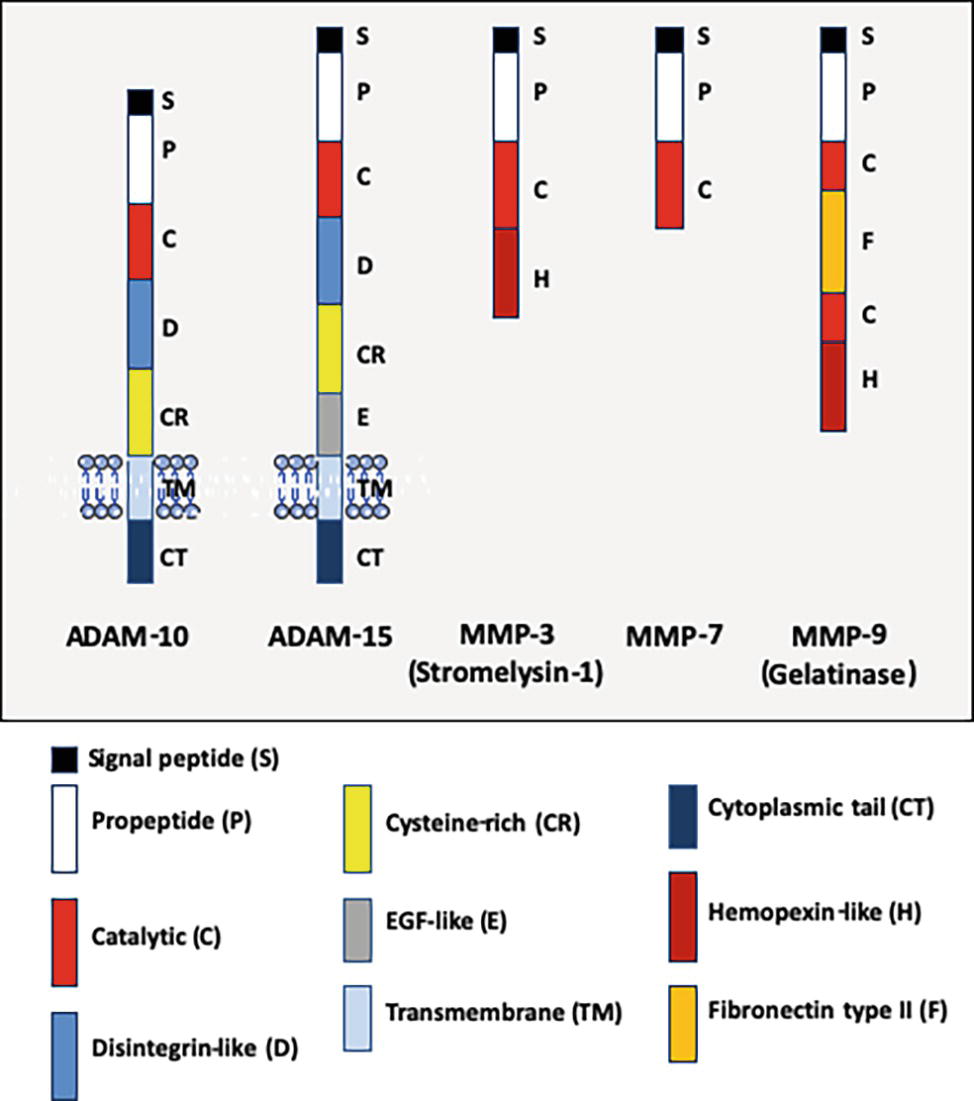
Figure 3. Schematic representation of the structural organization of some human ADAM and MMP pro-enzyme sheddases. The ADAM molecules (ADAM-10 and ADAM-15) are transmembrane proteins, whereas the MMP (MMP-3, MMP-7, and MMP-9) are soluble proteins. It should be noted (not shown) that several MMP are anchored at the cell-surface by a transmembrane domain (e.g., MMP-14 or MMP-15), by a glycosylphosphatidylinositol anchor (e.g., MMP-17 or MMP-25) or by an amino-terminal signal anchor (e.g., MMP-23). From the NH2-terminal extremity to the COOH-terminal extremity, ADAM-10 (748 a.a. residues) and ADAM-15 (772 a.a. to 863 a.a. residues depending the isoform) contain a signal peptide, a pro-peptide, a metalloprotease domain with catalytic activity, a disintegrin-like domain, a cysteine-rich domain, a transmembrane domain, and a cytoplasmic tail. ADAM-15 is characterized by the presence of an EGF-like domain taking place between the cysteine-rich domain and the transmembrane polypeptide. MMP-3 (or stromelysin-1, a 477 a.a. residues pro-enzyme protein of 51-kDa which can be activated in the 43-kDa mature catalytic form by removal of the pro-domain) has a basic MMP structure with a signal peptide, a pro-peptide, a catalytic domain, a hinge region, and an hemopexin domain. MMP9 (or gelatinase B, type IV collagenase, a 707 a.a. residues pro-enzyme protein of 92-kDa which can be activated as a 83-kDa mature enzyme following removal of the pro-domain), contains a signal peptide, a pro-peptide, a catalytic domain, a fibronectin type II domain, a second catalytic domain, an hinge region, and an hemopexin domain.
Several pathogenic bacteria were found to control the CDH1 gene expression at the chromatin level through activation of signaling cascade leading to modulation of DNA-binding proteins or directly in the nucleus through epigenetic modifications (Bierne et al., 2012). Inactivation of the CDH1 gene leads to the down-modulates of E-cad protein expression.
It was reported that methylation of the CDH1 gene promoter is a frequent event in samples from H. pylori infected patients with chronic gastritis, suggesting that CDH1 inactivation is an early step toward gastric tumorigenesis (Kague et al., 2010). Methylation of the CDH1 gene promoter was also reported in about 30–40% of patients with H. pylori-associated gastric carcinoma (Bahnassy et al., 2018). The CDH1 promoter methylation was reduced after H. pylori eradication (Perri et al., 2007). Chlamydia trachomatis infection inducing scarring disease of the ocular mucosa was found to be associated with CDH1 promoter DNA methylation and down-regulation of E-cad (Rajić et al., 2017). Acinetobacter baumannii is an opportunistic pathogen causing severe diseases in patients with mechanical ventilation. The nuclear targeting of Acinetobacter baumannii transposase (Tnp) induces DNA methylation of CpG regions in the promoter of the CDH1 gene resulting in down-regulation of gene expression (Moon et al., 2012). To date, the CDH1 gene inactivation has not been systematically explored for pathogenic bacteria and should be questioned as part of the exploration of the molecular mechanisms by which pathogenic bacteria deregulate E-cad expression to colonize the human host.
The epigenetic modulation of the CDH1 gene during pathogenic bacteria infection mimics processes well-described in the studies regarding the embryologic development. Repression of the CDH1 gene and cad-switching from E-cad to N-cad were considered essential for transitioning away from pluripotency (Sauka-Spengler and Bronner-Fraser, 2008; Li et al., 2010; Pieters and van Roy, 2014). Such cad-switching (Thiery, 2002) as well as reduced expression of E-cad have also been observed in cancer cases (including breast cancer, gastric cancer, colorectal cancer, and hepatocellular carcinomas), indicating that inhibition of E-cad surface expression can play a central role in the progression toward cancer (Jeanes et al., 2008; Gall and Frampton, 2013). However, in lung cancer cells, E-cad was found induced by WNT7a (Ohira et al., 2003), and E-cad expression is increased in both ovarian cancer malignant effusions and solid metastases (Davidson et al., 2000; Elloul et al., 2006). E-cad is generally considered a tumor suppressor through inhibition of β-cat nuclear translocation (Gottardi et al., 2001) and can act as oncogene through binding to EGFR/Erb receptor that triggers ERK and AKT signaling pathways providing advantage for cancer development and metastasis (Wells et al., 2008; Rodriguez et al., 2012).
With regard to viruses as examples of other pathogens modulating CDH1 gene expression, it has been reported that hepatitis B virus (HBV) represses E-cad at the transcriptional level by hypermethylation of the CDH1 promoter on CpG Island 1 with possible consequences on hepatocellular carcinogenesis by promoting detachment of surrounding cells and their migration to the primary tumor site (Lee et al., 2005). Alternatively, loss of E-cad in HBV infected cells can also be regulated at a post-translational level by proteases and SUMOylation (Ha et al., 2016). Hypermethylation that triggers CDH-1 gene repression was also found with the human papillomavirus (HPV) that increases cellular methyltransferase 1 (Dnmt1) activity via its viral E7 protein (Laurson et al., 2010).
Among the gene silencing molecular mechanisms underlying E-cad loss, one involves the expression of nonfunctional truncated CDH1 gene transcript. E-cadherin mRNA with premature termination codon mutation was reported in chronic lymphocytic leukemia cells (Sharma and Lichtenstein, 2009). This transcript (which lacks the exon 11) plays a role in silencing the production of E-cad. The amounts of wild-type E-cad mRNA inversely correlated with the amounts of aberrant transcript resulting in the up-regulation of the Wnt-β catenin pathway. The production of E-cad mRNA variant by alternative splicing has also been linked to a decrease in cell-cell adhesion and an increase in cell migration (Matos et al., 2017). The novel E-cadherin variant, a truncated soluble form of E-cad resulting from a deletion of the first 34 nt in the exon 14 of the CDH1 mRNA, induces changes characteristic of the Epithelial to Mesenchymal Transition (EMT) process, a key event in tumor progression (Matos et al., 2017). Moreover, the over-expression of this novel E-cad truncated form in transfected cells resulted in downregulation of wild-type E-cad expression. This truncated CDH1 gene transcript was recently found in breast cancer cells (Rosso et al., 2019). Aberrant splicing of CDH1 gene transcript (exon 8 or exon 11 skipped aberrant transcripts) was also reported in gastric carcinoma (Garziera et al., 2013; Li et al., 2015). Although this is a possibility, it has not yet been demonstrated that the expression of a CDH1 aberrant transcript in gastric cancers is controlled by H. pylori. However, it is known that aberrant splicing of cellular gene transcripts can occur during bacterial invasion (Sun, 2017). Indeed, massive alterations in the pattern of cellular mRNA splicing were reported upon infection with bacteria such as Anaplasma phagocytophilum, the etiologic agent of the human granulocytic anaplasmosis (Dumler et al., 2018) and Mycobacterium tuberculosis the etiologic agent of tuberculosis (Kalam et al., 2018). This may be consistent with the observation that Mycobacterium tuberculosis induced an epithelial mesenchymal transition in a pulmonary adenocarcinoma epithelial cell line (Gupta et al., 2016), a phenomenon orchestrated at the level of E-cad cell-surface expression. It was also reported that the EMT of mesothelial cells occurred in Mycobacterium tuberculosis-associated pleurisy, together with a reduction in E-cad expression (Kim et al., 2011). Moreover, expression level of E-cad was reported to differ between pulmonary tuberculosis patients and latent tuberculosis individuals (Sun et al., 2018).
Aside from the transcriptional repression of the CDH1 gene, another interesting mechanism, which could interfere with the anti-bacterial defenses of the host, is the release of sE-cad in body fluids following a sheddase-mediated cleavage of E-cad. The combination of sE-cad release together with other pro-inflammatory factors was highlighted as a triggering signal that promotes gastric adenocarcinoma or colorectal tumors in patients infected with H. pylori, B. fragilis, or Streptococcus gallolyticus (Wu et al., 2003; O’Connor et al., 2011; Kumar et al., 2017; Chung et al., 2018).
H. pylori, a bacterium that colonizes the gastric mucosa, is known as a risk factor for the development of chronic atrophic gastritis, gastroduodenal ulcers, and adenocarcinoma. An in vitro experimental model of cell transfection has demonstrated that H. pylori triggers β-cat activation through the interaction of virulence factor CagA with E-cad (Murata-Kamiya et al., 2007). O’Connor et al. (2011) reported that H. pylori induced the activation of host protease calpain via the toll-like receptor 2 (TLR2) and disruption of gastric epithelium. Another report suggests that ADAM-10 is induced in H. pylori infection and contributes to the shedding of E-cad (Schirrmeister et al., 2009). It was also reported that a serine protease HtrA from H. pylori mediate direct cleavage of E-cad (Hoy et al., 2010; Schmidt et al., 2016). Bacterium S. gallolyticus has a strong association with colorectal cancer with increased levels of β-cat and c-Myc (Kumar et al., 2017). The genome of another bacterium, Bacteroides fragilis, encodes a sheddase, the B. fragilis toxin (BFT) also termed fragilis (FRA), which cleaves E-cad and is associated with C-Myc expression and cellular proliferation (Wu et al., 1998, 2003; Remacle et al., 2014; Chung et al., 2018). Chung and co-workers uncovered a complex mechanism whereby the B. fragilis toxin (BFT)-mediated cleavage of E-cad initiates a multi-step inflammatory cascade requiring β-cat, IL-17R, NF-B, and Stat3 signaling in colonic epithelial cells. IL-17 dependent NF-κB activation in colic epithelial cells induces a mucosal gradient of C-X-C chemokines that initiates pro-tumoral myeloid cell infiltration to the distal colon and colon cancer. All these cancers are solid tumors. However, a role for sE-cad in the initiation of a pro-carcinogenic process might also be considered. Over the past 3 years, we have reported results suggesting that the bacterium Coxiella burnetii, responsible of Q fever, was associated with a higher frequency of Non-Hodgkin Lymphoma (NHL) in C. burnetii-infected patients compared to the general population (Melenotte et al., 2016). Recently, we investigated the transcriptional signature that could be associated with the development of NHL in Q fever patients and found an over-expression of genes involved in anti-apoptotic process and a repression of pro-apoptotic genes (Melenotte et al., 2019). Since cell surface expression of E-cad and release of sE-cad have been associated to various pathogenic bacteria known for inducing solid tumors, we have also investigated the levels of expression of these molecules in Q fever patients and observed a significant release of sE-cad in their sera and a down-regulation of E-cad mRNA expression. The sE-cad levels were found increased in the sera of acute and persistent Q fever patients, whereas they remained at the baseline in the control groups of healthy donors, people cured of Q fever, patients suffering from unrelated inflammatory diseases, and past Q fever patients who developed NHL (Table 2). Consequently, sE-cad could be considered a new biomarker of C. burnetii infection rather than a marker of NHL-associated to Q fever (Mezouar et al., 2019). We do not yet know which eukaryotic or prokaryotic sheddase could be responsible for the cleavage of E-cad in patients infected with C. burnetii. Preliminary studies on peripheral blood mononuclear cells exposed to heat-inactivated C. burnetii suggested variations in ADAM-10 and MMP-9 expression. Microarray performed on samples from macrophages and dendritic cells (DC) infected in vitro by C. burnetii revealed an over-expression of MMP-3 in C. burnetii-infected DC. Preliminary investigations in search of sheddase have also been conducted. We tried to blast the protein sequences of more than 20 sheddases known to catalyze the cleavage of E-cad against the hypothetical proteins of four strains of C. burnetii. We found that three eukaryotic sheddases (MMP-3, MMP-9, and ADAM-15) presented sequence similarities with bacterial proteins. Experiments are under progress to identify whether an ADAM or MMP eukaryotic protease or a prokaryotic protease encoded by C. burnetii could be responsible for sE-cad release in Q fever patients.
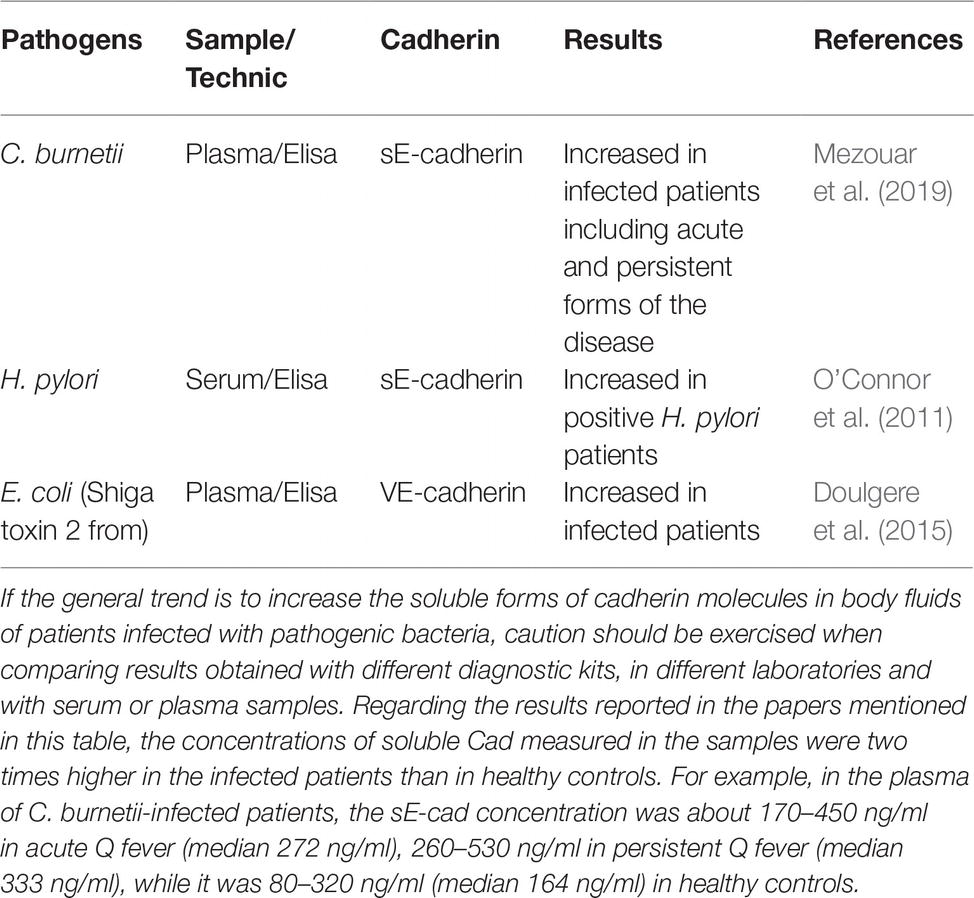
Table 2. Soluble Cad molecules released in body fluids during bacteria-associated infectious diseases.
Care must be taken not to overinterpret the significance of sE-cad increase in body fluids during bacterial infections. It is possible that sE-cad release in body fluids corresponds to a ubiquitous phenomenon induced during the process of colonization of the host by a pathogenic bacterium. We already know that it is not limited to bacteria since there is an apparent correlation between the levels of sE-cad in human immunodeficiency virus (HIV)-positive patients and their viral titers (Streeck et al., 2011). Moreover, abnormal concentrations of sE-cad in patients’ sera has been observed in several metabolic and inflammatory diseases (Pittard et al., 1996; Jiang et al., 2009; Shirahata et al., 2018; Sato et al., 2019). High concentration of sE-cad has also been found associated with cancer progression (Grabowska and Day, 2012; Salama et al., 2013; Repetto et al., 2014). For example, high concentration of sE-cad was described in prostate cancer patients and was associated with an over-expression of MMP-2 and MMP-9 (Kuefer et al., 2005; Biswas et al., 2010; Tsaur et al., 2015). Similarly, increased expression of sE-cad was reported in gastric cancer that was associated with the over-expression of MMP-7 (Lee et al., 2006, 2007). Serum levels of sE-cad were increased in patients with ovarian carcinoma the cleavage of E-cad being mediated by MMP-9 (Gadducci et al., 1999; Symowicz et al., 2007). Altogether, these data suggest that high sE-cad concentration in body fluids could simply behave as a factor that predisposes to inflammation and development of cancers, as described for H. pylori-infected patients.
The immune system naturally provides an anti-infectious surveillance at the epithelium level via cells that express cell-surface molecules, such as CD103 and KLRG1, able to bind the E-cad found at the surface of epithelial cells. These immune cells are not heavily engaged, unless the other defenses have failed. For example, under normal conditions, the intestinal epithelium is protected by a mucus layer that acts as host defense against microbial attachment (Kim and Ho, 2010; Desai et al., 2016). Upon infection, the specialized intestinal Paneth cells secretes antimicrobial proteins and the commensal intestinal microbiota competes with the infectious pathogens, thereby acting as first line of innate defense to fight against pathogenic bacteria (Cash et al., 2006; Mason and Huffnagle, 2009; Vazeille et al., 2011; Bel et al., 2017). The epithelium integrity controlled by the homotypic interaction of E-cad in trans represents a second barrier protecting the host against intruder transmigration (Kim et al., 2010). When pathogenic bacteria had evaded epithelial cell autophagic clearance and dead cells renewal (Yoshikawa et al., 2009; Benjamin et al., 2013), the host’s immune system represents the last rampart before the pathogens can breach the epithelium and disseminate deeper.
The intestinal villous microfold cells (M cells) are specialized epithelial cells of the gut-associated lymphoid tissues (GALT) that deliver luminal antigens to the underlying immune system after their transport to the basolateral membrane of M cells (Miller et al., 2007). An efficient immune response against a microbial attack requires the migration of cells of the host immune system in the microenvironment where infection occurs and the sequential detection of stress signals, tissue damages and conserved bacterial molecules termed pathogen-associated molecular patterns (PAMPs) (Broz and Monack, 2013). PAMPS include molecules as diverse as lipopolysaccharide, flagellin, peptidoglycan, lipoproteins, and unique bacterial nucleic acid structures. Upon detection of bacterial invasion, host cell receptors such as toll-like receptors and C-type lectin receptors, activate signaling pathways that govern the production of inflammatory cytokines including the IL-1β and IL-18 that can restrict bacterial replication. Causative agents of infectious diseases are therefore characterized by their capacity to elaborate mechanisms aimed to damage the protective cellular barriers and/or to modulate immune responses of the host to achieve invasion.
We can question the place of the E-cad in this process of mobilization of immunity cells via trans homotypic (e.g., E-cad) or trans heterotypic (e.g., CD103 and KLRG1) interactions under normal and pathological conditions (Figure 4). Although E-cad is expressed by immature CD4+CD8+ thymocytes (Munro et al., 1996; Müller et al., 1997), after the thymocytes have left the thymus, E-cad is generally absent from most mature lymphocytes (Lee et al., 1994). Yet, under certain pathological conditions, cell-surface expression of E-cad was reported on mature T-lymphocytes (CD3+) subsets, as well as B cells (CD19+), NK cells (DX5+), and monocyte/macrophages (CD11b+) subpopulations (Esch et al., 2000; Sakai et al., 2008). E-cad expression was also confirmed for subpopulations of epithelial γδ T-cells (Lee et al., 1994) and memory CD8+ T-cells in intestinal mucosa (Hofmann and Pircher, 2011). Moreover, we have recently observed that 30% of CD16+ monocytes expressed E-cad after C. burnetii infection (Mezouar et al., 2019). In the intestinal epithelium, it is generally accepted that the immune host defense is mainly mediated by effector cells that express the αE integrin (CD103), an E-cad ligand (Banh and Brossay, 2009; Van den Bossche et al., 2012). CD103 is expressed on 40–50% of CD4+ T-lymphocytes and 90% of CD8+ T-lymphocytes that reside in the intestinal mucosa as well as on the surface of intraepithelial lymphocytes (Cerf-Bensussan et al., 1987; Kilshaw and Murant, 1990; Hadley et al., 1997; Uchida et al., 2011). It is also present on T cells of the intestinal lamina propria (Agace et al., 2000) and a subset of CD4+CD25+ Foxp3+ Treg-cells (Stephens et al., 2007). It was demonstrated that after epithelial damages, the intraepithelial CD103+ γδ T-lymphocytes that reside on the surface of epithelium promote mucosal repair through antibacterial factors (e.g., Reg3γ) and immunomodulatory molecules (e.g., IL1β, CXCL9) (Cepek et al., 1994; Ismail et al., 2009, 2011). It is worth noting that the intestinal CD103+ intraepithelial lymphocytes adherence to epithelial cells is inhibited by antibodies against CD103 (Cepek et al., 1993). Another cell-surface receptor named KLRG1 that is encountered on subsets of immune cells including mature NK cells, memory CD4+T cells, effector CD8+ T cells, and FoxP3+ Treg cells, is known to bind E-cad (Schwartzkopff et al., 2007; Tessmer et al., 2007; Banh and Brossay, 2009; Van den Bossche et al., 2012). It has been reported that high levels of sE-cad could be sufficient to inhibit CD8+ T-cell function in a KLRG1-dependent manner (Streeck et al., 2011).
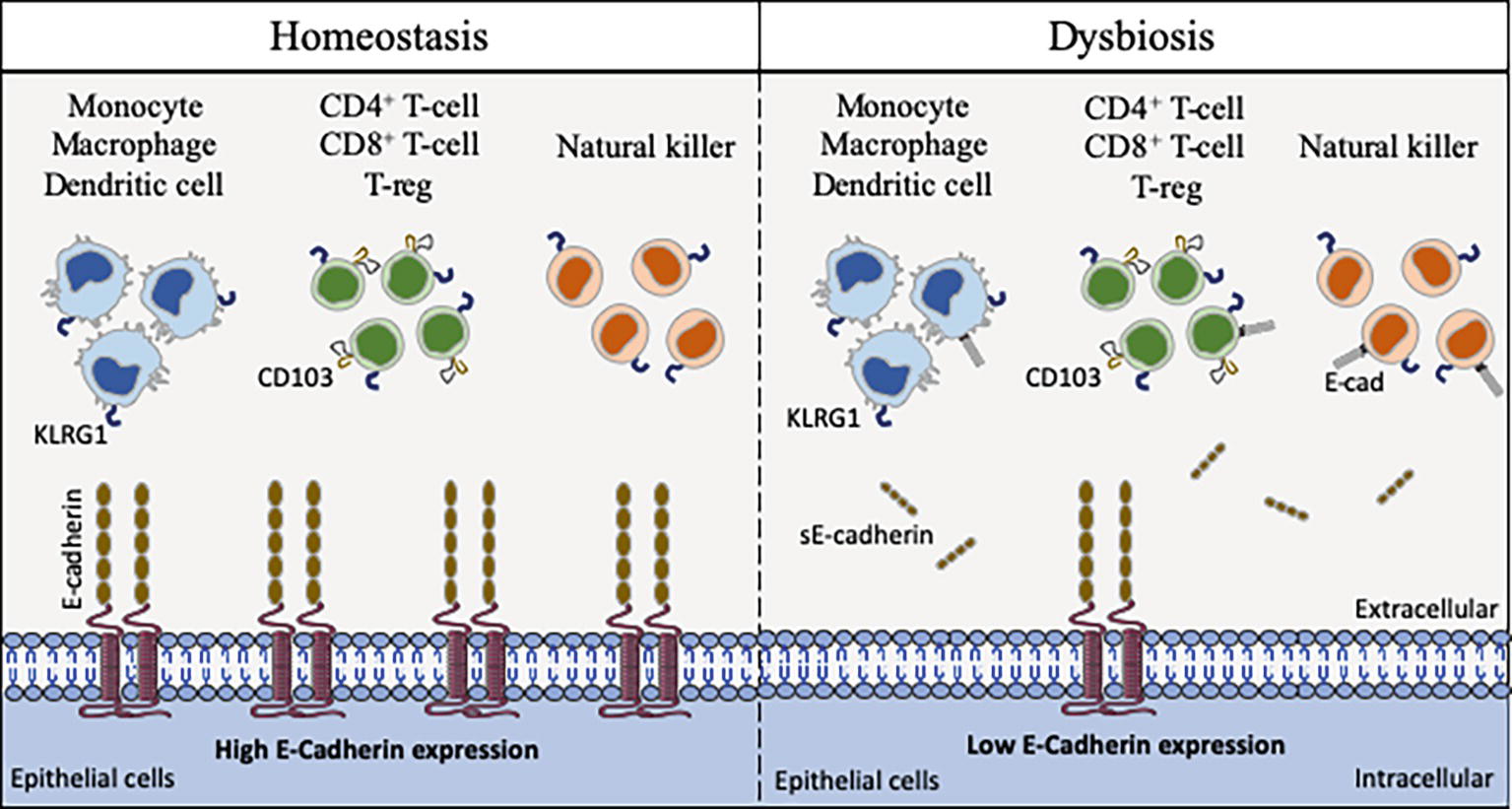
Figure 4. Schematic model of interaction between cells that express CD103, KLRG1, and E-cad. E-cad expressed on epithelial cells (as well as dendritic cells, Langerhans cells, and macrophages) can potentially interact with CD103, KLRG1 or both molecules expressed on the surface of immune cells. In the absence of bacterial infection, the epithelial cells express E-cad at high level, allowing immune cells to ensure the immune surveillance of epithelia (left panel). After pathogenic bacteria invasion, the cell-surface expression of E-cad is weak on epithelial cells due to the activation of sheddases and sE-cad is released in the epithelium microenvironment. The abnormal expression of E-cad on subpopulations of immune cells, the release of sE-cad, the weak expression of E-cad on epithelial cells likely contribute to immune system dysfunction at the bacterial invasion site (right panel).
The E-cad induction on subpopulations of immune response cells in pathological situations remain to be elucidated. It can be hypothesized that the aberrant expression of E-cad under pathological conditions reflects changes in the transmigration and homing capacity of these cells (Reyat et al., 2017). Because several pathogenic bacteria reduce the epithelium surface expression of E-cad at the site of infection, it might be speculated that the decreased expression or the lack of expression of E-cad on epithelial cells is likely to trigger the rerouting of immune cells far from the infection site. As previously shown by Streeck et al. (2011) for the KLRG1+ CD8+ T-cells subpopulation, the release of sE-cad might also serve as a decoy for diverting from their function the immune cells expressing E-cad, CD103 or KLRG1 after engagement of such receptors with sE-cad. Modulation of E-cad expression on the host epithelial cells and sE-cad release could therefore be considered a very efficient stratagem to prevent the immune system from behaving as a line of defense against invaders. In addition, for bacteria that induce cancer, reducing the expression of E-cad on certain tumor cells as previously reported (Shields et al., 2019) and disrupting the migration and attachment capabilities of immune survey cells could be a way of promoting the development of bacteria-induced cancers.
This review highlights how the E-cad can be diverted from its function of maintenance of tissues integrity and prevention of cell migration/differentiation during pathogenic bacterial infections. Pathogenic bacteria can use E-cad for their attachment to epithelial cells. Indeed, they can cleave E-cad to ensure their transmigration and can modulate the responsiveness of immune cells through modulation of cell-surface expression of E-cad and sE-cad release in body fluids. Some bacteria use E-cad to enter their target cells (e.g., F. nucleatum, L. monocytogenes, S. pneumoniae). Several bacteria act on the cell-surface expression of this molecule, either by modulating the CDH1 gene transcription (e.g., C. trachomatis, H. pylori) or by inducing the cleavage of the E-cad molecule (e.g., C. perfringens, S. aureus, C. burnetii) via proteases (sheddases). This process can favor achievement of the trans-epithelial host invasion or modulate host-pathogen molecular crosstalk. Currently, the best studied models are those that refer to intestinal infections that can lead to cancer (Figure 5).
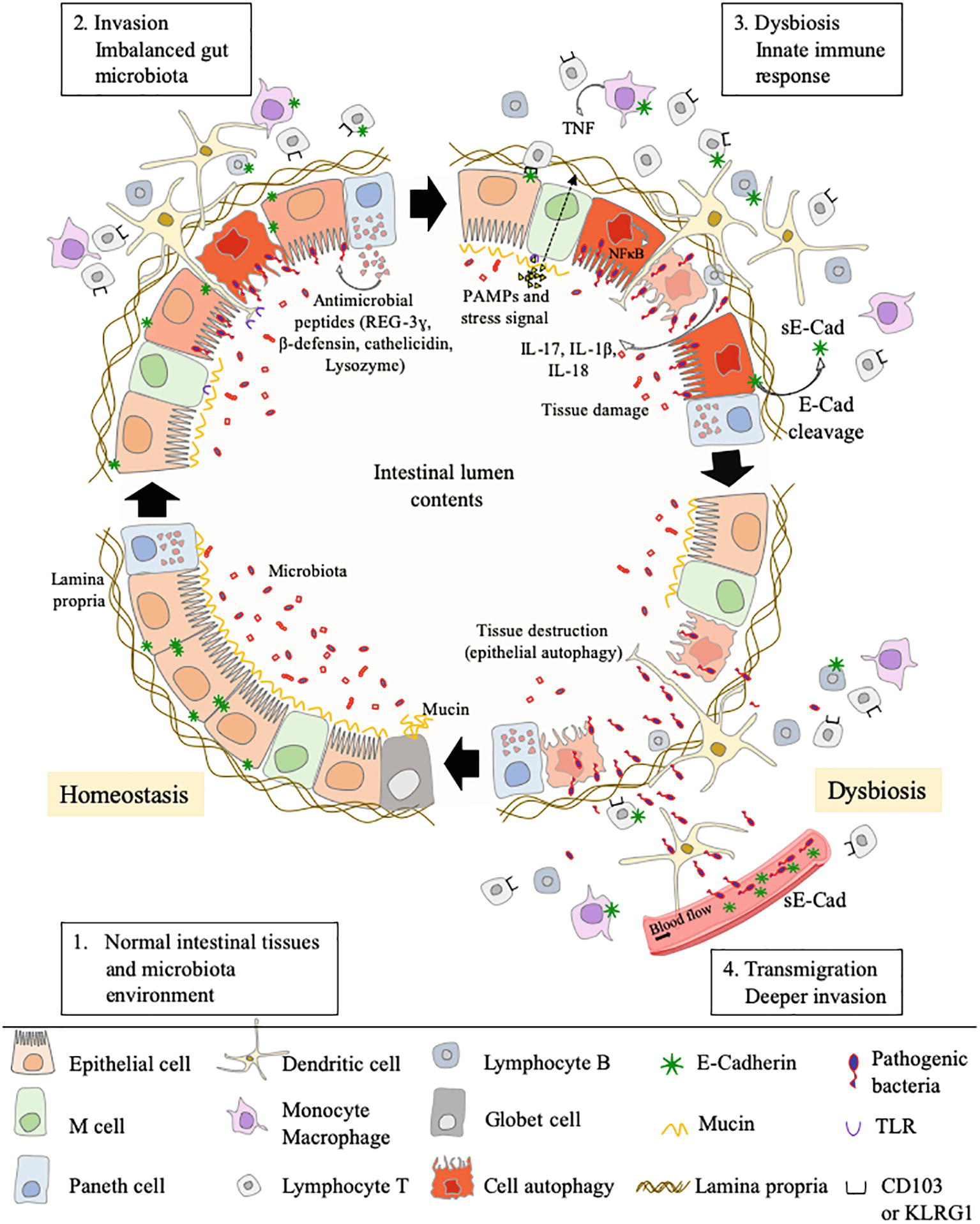
Figure 5. This cartoon illustrates how, by simply acting on the E-cad expression, an intestinal pathogenic bacterium can both bypass the physical defense system represented by the epithelial barrier and confuse the cells of the immune system intended to defend the host. Step 1: under homeostasis, the epithelium (composed of epithelial cells) is protected by a mucus layer synthesized by goblet cells which secrete the mucins (e.g, MUC2 – mucin gel). The mucus layer prevents microbial attachment without interference with the transport of nutrients. The commensal intestinal microbiota is limited to the epithelium-distal mucus layer, while the epithelium-proximal mucus is largely devoid of bacteria. Step 2: upon infection, the commensal intestinal microbiota competes with the infectious pathogens and the Paneth cells produce antimicrobial proteins (e.g., C-type lectin REG3γ, β-defensins, cathelicidins, and lysozyme), to fight the invasion. A regulation of infection is also achieved by epithelial cell autophagic clearance and dead cells renewal. At the same time, the villous microfold cells (M cells) expressing TLR deliver luminal antigens to the underlying immune system to set up a whole arsenal of anti-bacterial actions. In case this response proves sufficient, the invader is destroyed, and the microenvironment returns to homeostasis (Step 1). If not, the conflict is prolonged. Step 3: an efficient immune response against the pathogens requires the migration of cells of the host immune system in the microenvironment where the infection occurs and the sequential detection of stress signals, tissue damages, and PAMPs (e.g., lipopolysaccharide, flagellin, peptidoglycan, lipoproteins, and unique bacterial nucleic acid structures). KLRG1+ dendritic cells and monocytes/macrophages, CD103+ T-cells, KLRG1+ T-cells, and other immune cell subpopulations, colonize the lamina propria. Upon detection of bacterial invasion, host cell receptors, such as TLR and C-type lectin receptors, activate signaling pathways that govern the production of inflammatory cytokines, including the IL-1β and IL-18 that can restrict bacterial replication. Step 4: the pathogenic bacteria reduce the epithelium surface expression of E-cad at the site of infection, resulting in the destruction of adherent’s junctions and allowing transmigration. Moreover, it might be speculated that the induction of E-cad on subpopulation of immune response cells (E-cad+ T-cells and CD16+/E-cad+ monocytes) redirects those cells far from the infection site in microenvironments where they have a higher probability to interact with E-cad+ epithelial cells. The release of sE-cad might also serve as a decoy for diverting immune cells from their function through interaction with E-cad, CD103, or KLRG1 at the surface of immune cells.
Proteases (cysteine proteases, serine proteases, aspartate proteases, and metalloproteases) are ubiquitously encountered in the microbial world and are essential for their survival and replication cycle (Häse and Finkelstein, 1993). Proteases, such as collagenase (Bond and Van Wart, 1984), elastase (Bever and Iglewski, 1988), or metalloprotease (Schiavo et al., 1992) (Domann et al., 1992) were associated with bacterial pathogenesis. Some pathogenic bacteria can activate the production of eukaryotic proteases such as ADAM-10 via signaling (e.g., this was reported for P. aeruginosa, S. marcescens, C. perfringens, S. aureus), whereas others use a portion of their genome to encode their own sheddase, including the HtrA protease (encoded by H. pylori, C. jejuni, S. flexneri, enteropathogenic E. coli) or B. fragilis toxin (encoded by B. fragilis).
As described above in this paper, evidence emphasizing that cleavage of E-cad by sheddases and release of sE-cad into the body fluids are factors that contribute to the progression of cancer (sometimes it was demonstrated). Within the group of bacteria that modulate the expression of E-cad, the association with carcinogenic processes has been investigated, in particular with H. pylori, F. nucleatum, S. gallolyticus, and B. fragilis. H. pylori is known to be a risk factor for the development of gastric adenocarcinoma and the progression toward cancer is likely related to E-cad cleavage and β-cat activation (Murata-Kamiya et al., 2007), F. nucleatum promotes colorectal cancer by modulating the E-cad and Wnt/β-cat signaling via its FadA adhesin and up-regulating annexin A1 (Rubinstein et al., 2013, 2019). S. gallolyticus is also associated with colorectal cancer with known increased level of β-cat and c-Myc activation (Kumar et al., 2017). B. fragilis toxin (BFT)-mediated cleavage of E-cad initiates a multi-step inflammatory cascade requiring β-cat nuclear translocation, activation of NF-κB and Stat3 signaling pathways in colonic epithelial cells as early events leading to pro-tumoral myeloid cell infiltration to the distal colon and colon cancer (Chung et al., 2018). Our recent data indicate that C. burnetii, reported as associated with occurrence of non-Hodgkin lymphoma, is also capable of triggering cleavage of E-cad and release of sE-cad in the sera of Q fever patients (Figure 6; Mezouar et al., 2019). Yet, further experiments are required to formerly demonstrate the association between sE-cad release in sera from Q fever patients and the initiation of a pro-carcinogenic inflammatory process leading to lymphoma development.
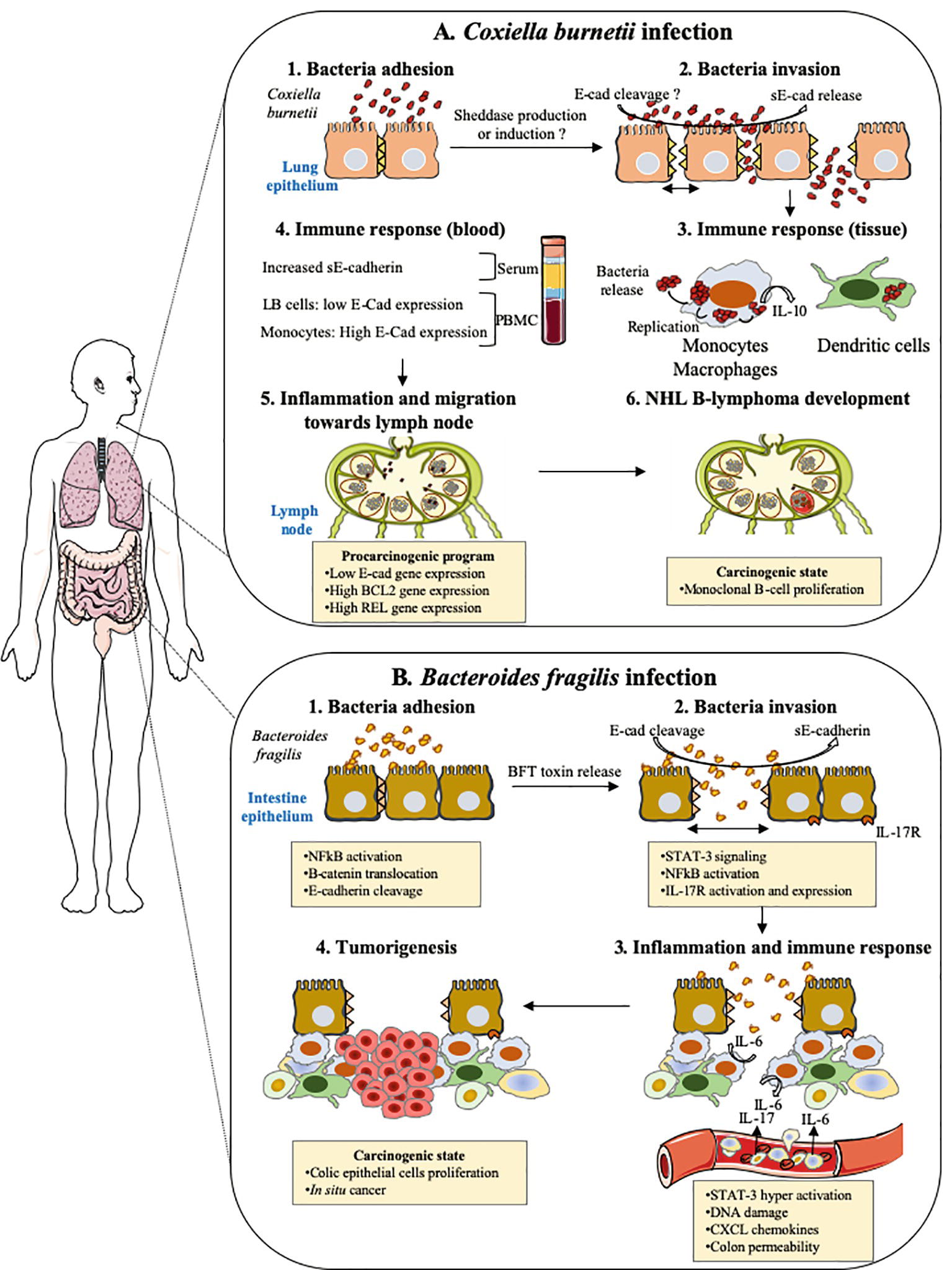
Figure 6. Cellular host E-cad as target during both pulmonary and intestinal bacterial infections. This drawing summarizes the hypothetical models of tumorigenesis associated with Coxiella burnetii and enterotoxigenic Bacteroides fragilis. (A) Coxiella burnetii. Although rare, the incidence of NHL B-lymphoma in patients infected by C. burnetii in France was significantly higher (25-fold) than within the general population. In Q fever, the overproduction of IL-10 by infected monocytes was found critical in both sustaining replication of the C. burnetii and preventing the macrophages microbicidal activity. Moreover, specific genes involved in anti-apoptotic process are over-expressed, whereas pro-apoptotic genes are repressed. Recently, we found elevated concentrations of sE-cad in Q fever patients, along with an increase in cell-surface expression of E-cad in more than 30% of HLADR+/CD16+ monocytes and a decrease in E-cad expression that concerned about 3% of the E-cad+/CD20+ B-cells subpopulation (LB cells). We speculate that the release of sE-cad might participate to the molecular crosstalk, which takes place in the microenvironment of the lymph node during persistent Q fever and might possibly trigger a pro-carcinogenic program required for the initiation of NHL lymphoma. (B) Enterotoxigenic Bacteroides fragilis. The enterotoxigenic Bacteroides fragilis synthesizes a toxin, BFToxin, which damages the protective intestinal epithelial barriers of the host by cleavage of E-cad to achieve invasion. This process leads to epithelial cells activation through nuclear translocation of β-cat and NF-κB, inducing the transcription of genes such as IL-17 receptor (IL17R). A pro-inflammatory immune response is initiated against the pathogen characterized by the in-situ production of IL-17. IL-17R positive cells are induced to produce STAT3 and a gradient of chemokines in the microenvironment favors the recruitment of pro-tumoral myeloid cells that accumulate in the distal colon producing growth factors triggering the proliferation of colic epithelial cells. These cells progressively accumulate DNA damages and form a solid tumor in the colon.
The accumulation of data showing that sE-cad is produced in many pathological processes that can lead to cancer development raises the question of what value can be attributed to this compound as a biomarker of disease severity. At the moment, we do not have enough information to conclude. The presence of sE-cad in body fluids was considered a possible biomarker in H. pylori (O’Connor et al., 2011) and C. burnetii (Mezouar et al., 2019) infections, and soluble VE-cad was also regarded as possible biomarker in E. coli infections (Doulgere et al., 2015). We are currently implementing a test protocol to assess whether sE-cad could be a biomarker for tuberculosis and severe C. difficile infections. CD68+ granuloma macrophages from M. tuberculosis patients were reported to express E-cad (Cronan et al., 2016). Moreover, using M. marinum as model, Cronan and collaborators reported that the mycobacterial granuloma formation is accompanied by macrophage reprogramming that parallels E-cad-dependent mesenchymal-epithelial transitions and alters immune response. E-cadherin induction was found in the granuloma of both M. marinum-infected and uninfected granuloma macrophages, while macrophages residing outside the granuloma remained negative for E-cad. Concerning C. difficile, the etiologic agent of pseudomembranous colitis and severe diarrhea, it was reported that C. difficile TcdB toxin induces the redistribution of occludin and ZO-1without influencing the subjacent E-cad (Nusrat et al., 2001).
For the management of H. pylori gastrointestinal disorders and H. pylori-associated gastric cancer, it is recommended to use a combination of ranitidine bismuth citrate, clarithromycin, and amoxicillin (Goderska et al., 2018). In a near future, it will not be surprising to consider the possibility of proposing therapeutic approaches that will combine antibiotics, probiotics, and sheddase inhibitors to regulate E-cad as well as β-cat inhibitors. Probiotics (e.g., the yeast Saccharomyces boulardii CNCMI-745) have been used to restore intestinal barrier integrity in patients with inflammatory bowel disease, by regulation of E-cad recycling (Terciolo et al., 2017). Intensive research is aimed at developing inhibitors of MMPs (Antczak et al., 2008; Tuccinardi et al., 2008; Schmidt et al., 2016; Katoh, 2017) and ADAMs (Madoux et al., 2016). MMP-9 inhibitors that abrogate E-cad cleavage are considered a promising tool for therapeutic of colorectal cancers (Biswas et al., 2010; Marshall et al., 2015). This opens up new avenues of research for therapeutic purposes.
CD, SM and J-LM conceived the paper. SM and CD designed the tables and figures. CD wrote the paper.
SM was first supported by a “Fondation pour la Recherche Médicale” postdoctoral fellowship (reference: SPF20151234951) and then by the “Fondation Méditerranée Infection”. This work was supported by the French Government under the “Investissements d’avenir” (Investments for the Future) program managed by the “Agence Nationale de la Recherche” (reference: Méditerranée Infection 10-IAHU-03). Art supply of images available under a Creative Commons CCBY 3.0 license.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Magdalen Lardière for English editing.
Abt, M. C., and Pamer, E. G. (2014). Commensal bacteria mediated defenses against pathogens. Curr. Opin. Immunol. 29, 16–22. doi: 10.1016/j.coi.2014.03.003
Agace, W. W., Higgins, J. M., Sadasivan, B., Brenner, M. B., and Parker, C. M. (2000). T-lymphocyte-epithelial-cell interactions: integrin alpha(E)(CD103)beta(7), LEEP-CAM and chemokines. Curr. Opin. Cell Biol. 12, 563–568. doi: 10.1016/S0955-0674(00)00132-0
Al-Obaidi, M. M. J., and Desa, M. N. M. (2018). Mechanisms of blood brain barrier disruption by different types of bacteria, and bacterial-host interactions facilitate the bacterial pathogen invading the brain. Cell. Mol. Neurobiol. 38, 1349–1368. doi: 10.1007/s10571-018-0609-2
Anderton, J. M., Rajam, G., Romero-Steiner, S., Summer, S., Kowalczyk, A. P., Carlone, G. M., et al. (2007). E-cadherin is a receptor for the common protein pneumococcal surface adhesin A (PsaA) of Streptococcus pneumoniae. Microb. Pathog. 42, 225–236. doi: 10.1016/j.micpath.2007.02.003
Angst, B. D., Marcozzi, C., and Magee, A. I. (2001). The cadherin superfamily: diversity in form and function. J. Cell Sci. 114, 629–641.
Antczak, C., Radu, C., and Djaballah, H. (2008). A profiling platform for the identification of selective metalloprotease inhibitors. J. Biomol. Screen. 13, 285–294. doi: 10.1177/1087057108315877
Arribas, J., and Merlos-Suárez, A. (2003). Shedding of plasma membrane proteins. Curr. Top. Dev. Biol. 54, 125–144.
Bahnassy, A. A., Helal, T. E.-A., El-Ghazawy, I. M., Samaan, G. F., Galal El-Din, M. M., Abdellateif, M. S., et al. (2018). The role of E-cadherin and Runx3 in Helicobacter pylori-associated gastric carcinoma is achieved through regulating P21waf and P27 expression. Cancer Genet. 228–229, 64–72. doi: 10.1016/j.cancergen.2018.08.006
Banh, C., and Brossay, L. (2009). Immune receptors, cadherins and their interactions. Curr. Immunol. Rev. 5, 2–9. doi: 10.2174/157339509787314440
Barbuddhe, S. B., and Chakraborty, T. (2009). Listeria as an enteroinvasive gastrointestinal pathogen. Curr. Top. Microbiol. Immunol. 337, 173–195. doi: 10.1007/978-3-642-01846-6_6
Barker, N., Hurlstone, A., Musisi, H., Miles, A., Bienz, M., and Clevers, H. (2001). The chromatin remodelling factor Brg-1 interacts with beta-catenin to promote target gene activation. EMBO J. 20, 4935–4943. doi: 10.1093/emboj/20.17.4935
Becker, J. W., Marcy, A. I., Rokosz, L. L., Axel, M. G., Burbaum, J. J., Fitzgerald, P. M., et al. (1995). Stromelysin-1: three-dimensional structure of the inhibited catalytic domain and of the C-truncated proenzyme. Protein Sci. 4, 1966–1976. doi: 10.1002/pro.5560041002
Bel, S., Pendse, M., Wang, Y., Li, Y., Ruhn, K. A., Hassell, B., et al. (2017). Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science 357, 1047–1052. doi: 10.1126/science.aal4677
Benjamin, J. L., Sumpter, R., Levine, B., and Hooper, L. V. (2013). Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe 13, 723–734. doi: 10.1016/j.chom.2013.05.004
Berx, G., Becker, K. F., Höfler, H., and van Roy, F. (1998). Mutations of the human E-cadherin (CDH1) gene. Hum. Mutat. 12, 226–237. doi: 10.1002/(SICI)1098-1004(1998)12:4<226::AID-HUMU2>3.0.CO;2-D
Berx, G., Staes, K., van Hengel, J., Molemans, F., Bussemakers, M. J., van Bokhoven, A., et al. (1995). Cloning and characterization of the human invasion suppressor gene E-cadherin (CDH1). Genomics 26, 281–289. doi: 10.1016/0888-7543(95)80212-5
Bever, R. A., and Iglewski, B. H. (1988). Molecular characterization and nucleotide sequence of the Pseudomonas aeruginosa elastase structural gene. J. Bacteriol. 170, 4309–4314. doi: 10.1128/jb.170.9.4309-4314.1988
Bhella, D. (2015). The role of cellular adhesion molecules in virus attachment and entry. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 370:20140035. doi: 10.1098/rstb.2014.0035
Bierne, H., Travier, L., Mahlakõiv, T., Tailleux, L., Subtil, A., Lebreton, A., et al. (2012). Activation of type III interferon genes by pathogenic bacteria in infected epithelial cells and mouse placenta. PLoS One 7:e39080. doi: 10.1371/journal.pone.0039080
Biswas, M. H. U., Du, C., Zhang, C., Straubhaar, J., Languino, L. R., and Balaji, K. C. (2010). Protein kinase D1 inhibits cell proliferation through matrix metalloproteinase-2 and matrix metalloproteinase-9 secretion in prostate cancer. Cancer Res. 70, 2095–2104. doi: 10.1158/0008-5472.CAN-09-4155
Blau, K., Portnoi, M., Shagan, M., Kaganovich, A., Rom, S., Kafka, D., et al. (2007). Flamingo cadherin: a putative host receptor for Streptococcus pneumoniae. J. Infect. Dis. 195, 1828–1837. doi: 10.1086/518038
Boehm, M., Hoy, B., Rohde, M., Tegtmeyer, N., Bæk, K. T., Oyarzabal, O. A., et al. (2012). Rapid paracellular transmigration of Campylobacter jejuni across polarized epithelial cells without affecting TER: role of proteolytic-active HtrA cleaving E-cadherin but not fibronectin. Gut Pathog. 4:3. doi: 10.1186/1757-4749-4-3
Boehm, M., Lind, J., Backert, S., and Tegtmeyer, N. (2015). Campylobacter jejuni serine protease HtrA plays an important role in heat tolerance, oxygen resistance, host cell adhesion, invasion, and transmigration. Eur. J. Microbiol. Immunol. 5, 68–80. doi: 10.1556/EuJMI-D-15-00003
Boggon, T. J. (2002). C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science 296, 1308–1313. doi: 10.1126/science.1071559
Bolós, V., Peinado, H., Pérez-Moreno, M. A., Fraga, M. F., Esteller, M., and Cano, A. (2003). The transcription factor slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with snail and E47 repressors. J. Cell Sci. 116, 499–511. doi: 10.1242/jcs.00224
Bonazzi, M., Lecuit, M., and Cossart, P. (2009). Listeria monocytogenes internalin and E-cadherin: from bench to bedside. Cold Spring Harb. Perspect. Biol. 1:a003087. doi: 10.1101/cshperspect.a003087
Bond, M. D., and Van Wart, H. E. (1984). Characterization of the individual collagenases from Clostridium histolyticum. Biochemistry 23, 3085–3091. doi: 10.1021/bi00308a036
Bookwalter, J. E., Jurcisek, J. A., Gray-Owen, S. D., Fernandez, S., McGillivary, G., and Bakaletz, L. O. (2008). A carcinoembryonic antigen-related cell adhesion molecule 1 homologue plays a pivotal role in nontypeable Haemophilus influenzae colonization of the chinchilla nasopharynx via the outer membrane protein P5-homologous adhesin. Infect. Immun. 76, 48–55. doi: 10.1128/IAI.00980-07
Broz, P., and Monack, D. M. (2013). Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 13, 551–565. doi: 10.1038/nri3479
Cadigan, K. M., and Waterman, M. L. (2012). TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb. Perspect. Biol. 4:a007906. doi: 10.1101/cshperspect.a007906
Cano, A., Pérez-Moreno, M. A., Rodrigo, I., Locascio, A., Blanco, M. J., del Barrio, M. G., et al. (2000). The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2, 76–83. doi: 10.1038/35000025
Carter, A. T., and Peck, M. W. (2015). Genomes, neurotoxins and biology of Clostridium botulinum group I and group II. Res. Microbiol. 166, 303–317. doi: 10.1016/j.resmic.2014.10.010
Cash, H. L., Whitham, C. V., Behrendt, C. L., and Hooper, L. V. (2006). Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 313, 1126–1130. doi: 10.1126/science.1127119
Cepek, K. L., Parker, C. M., Madara, J. L., and Brenner, M. B. (1993). Integrin alpha E beta 7 mediates adhesion of T lymphocytes to epithelial cells. J. Immunol. 150, 3459–3470.
Cepek, K. L., Shaw, S. K., Parker, C. M., Russell, G. J., Morrow, J. S., Rimm, D. L., et al. (1994). Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the alpha E beta 7 integrin. Nature 372, 190–193. doi: 10.1038/372190a0
Cerf-Bensussan, N., Jarry, A., Brousse, N., Lisowska-Grospierre, B., Guy-Grand, D., and Griscelli, C. (1987). A monoclonal antibody (HML-1) defining a novel membrane molecule present on human intestinal lymphocytes. Eur. J. Immunol. 17, 1279–1285. doi: 10.1002/eji.1830170910
Chambers, F. G., Koshy, S. S., Saidi, R. F., Clark, D. P., Moore, R. D., and Sears, C. L. (1997). Bacteroides fragilis toxin exhibits polar activity on monolayers of human intestinal epithelial cells (T84 cells) in vitro. Infect. Immun. 65, 3561–3570.
Chung, L., Orberg, E. T., Geis, A. L., Chan, J. L., Fu, K., DeStefano Shields, C. E., et al. (2018). Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe 23, 203–214.e5. doi: 10.1016/j.chom.2018.01.007
Colpitts, C. C., Lupberger, J., and Baumert, T. F. (2016). Multifaceted role of E-cadherin in hepatitis C virus infection and pathogenesis. Proc. Natl. Acad. Sci. USA 113, 7298–7300. doi: 10.1073/pnas.1607732113
Cossart, P., and Sansonetti, P. J. (2004). Bacterial invasion: the paradigms of enteroinvasive pathogens. Science 304, 242–248. doi: 10.1126/science.1090124
Cronan, M. R., Beerman, R. W., Rosenberg, A. F., Saelens, J. W., Johnson, M. G., Oehlers, S. H., et al. (2016). Macrophage epithelial reprogramming underlies mycobacterial granuloma formation and promotes infection. Immunity 45, 861–876. doi: 10.1016/j.immuni.2016.09.014
Davidson, B., Berner, A., Nesland, J. M., Risberg, B., Berner, H. S., Tropè, C. G., et al. (2000). E-cadherin and alpha-, beta-, and gamma-catenin protein expression is up-regulated in ovarian carcinoma cells in serous effusions. J. Pathol. 192, 460–469. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH726>3.0.CO;2-M
de la Roche, M., Worm, J., and Bienz, M. (2008). The function of BCL9 in Wnt/beta-catenin signaling and colorectal cancer cells. BMC Cancer 8:199. doi: 10.1186/1471-2407-8-199
Desai, M. S., Seekatz, A. M., Koropatkin, N. M., Kamada, N., Hickey, C. A., Wolter, M., et al. (2016). A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 167, 1339–1353.e21. doi: 10.1016/j.cell.2016.10.043
Devaux, C. A., and Raoult, D. (2018). The microbiological memory, an epigenetic regulator governing the balance between good health and metabolic disorders. Front. Microbiol. 9:1379. doi: 10.3389/fmicb.2018.01379
Domann, E., Wehland, J., Rohde, M., Pistor, S., Hartl, M., Goebel, W., et al. (1992). A novel bacterial virulence gene in Listeria monocytogenes required for host cell microfilament interaction with homology to the proline-rich region of vinculin. EMBO J. 11, 1981–1990. doi: 10.1002/j.1460-2075.1992.tb05252.x
Doulgere, J., Otto, B., Nassour, M., Wolters-Eisfeld, G., Rohde, H., Magnus, T., et al. (2015). Soluble plasma VE-cadherin concentrations are elevated in patients with STEC infection and haemolytic uraemic syndrome: a case-control study. BMJ Open 5:e005659. doi: 10.1136/bmjopen-2014-005659
Dumler, J. S., Sinclair, S. H., and Shetty, A. C. (2018). Alternative splicing of differentiated myeloid cell transcripts after infection by Anaplasma phagocytophilum impacts a selective group of cellular programs. Front. Cell. Infect. Microbiol. 8:14. doi: 10.3389/fcimb.2018.00014. eCollection 2018.
Egusa, H., Nikawa, H., Makihira, S., Jewett, A., Yatani, H., and Hamada, T. (2005). Intercellular adhesion molecule 1-dependent activation of interleukin 8 expression in Candida albicans-infected human gingival epithelial cells. Infect. Immun. 73, 622–626. doi: 10.1128/IAI.73.1.622-626.2005
Elloul, S., Silins, I., Tropé, C. G., Benshushan, A., Davidson, B., and Reich, R. (2006). Expression of E-cadherin transcriptional regulators in ovarian carcinoma. Virchows Arch. 449, 520–528. doi: 10.1007/s00428-006-0274-6
Elmi, A., Nasher, F., Jagatia, H., Gundogdu, O., Bajaj-Elliott, M., Wren, B., et al. (2016). Campylobacter jejuni outer membrane vesicle-associated proteolytic activity promotes bacterial invasion by mediating cleavage of intestinal epithelial cell E-cadherin and occludin. Cell. Microbiol. 18, 561–572. doi: 10.1111/cmi.12534
Esch, T. R., Jonsson, M. V., Levanos, V. A., Poveromo, J. D., and Sorkin, B. C. (2000). Leukocytes infiltrating the submandibular glands of NOD mice express E-cadherin. J. Autoimmun. 15, 387–393. doi: 10.1006/jaut.2000.0451
Eshghi, A., Gaultney, R. A., England, P., Brûlé, S., Miras, I., Sato, H., et al. (2019). An extracellular Leptospira interrogans leucine-rich repeat protein binds human E- and VE-cadherins. Cell. Microbiol. 21:e12949. doi: 10.1111/cmi.12949
Evangelista, K., Franco, R., Schwab, A., and Coburn, J. (2014). Leptospira interrogans binds to cadherins. PLoS Negl. Trop. Dis. 8:e2672. doi: 10.1371/journal.pntd.0002672
Fagotto, F. (2013). Looking beyond the Wnt pathway for the deep nature of β-catenin. EMBO Rep. 14, 422–433. doi: 10.1038/embor.2013.45
Fang, D., Hawke, D., Zheng, Y., Xia, Y., Meisenhelder, J., Nika, H., et al. (2007). Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J. Biol. Chem. 282, 11221–11229. doi: 10.1074/jbc.M611871200
Gadducci, A., Ferdeghini, M., Cosio, S., Annicchiarico, C., Ciampi, B., Bianchi, R., et al. (1999). Preoperative serum E-cadherin assay in patients with ovarian carcinoma. Anticancer Res. 19, 769–772.
Gall, T. M. H., and Frampton, A. E. (2013). Gene of the month: E-cadherin (CDH1). J. Clin. Pathol. 66, 928–932. doi: 10.1136/jclinpath-2013-201768
Gallin, W. J., Edelman, G. M., and Cunningham, B. A. (1983). Characterization of L-CAM, a major cell adhesion molecule from embryonic liver cells. Proc. Natl. Acad. Sci. USA 80, 1038–1042.
Garziera, M., Canzonieri, V., Cannizzaro, R., Geremia, S., Caggiari, L., De Zorzi, M., et al. (2013). Identification and characterization of CDH1 germline variants in sporadic gastric cancer patients and in individuals at risk of gastric cancer. PLoS One 8:e77035. doi: 10.1371/journal.pone.0077035
Giebeler, N., and Zigrino, P. (2016). A Disintegrin and Metalloprotease (ADAM): historical overview of their functions. Toxins 8:122. doi: 10.3390/toxins8040122
Goderska, K., Agudo Pena, S., and Alarcon, T. (2018). Helicobacter pylori treatment: antibiotics or probiotics. Appl. Microbiol. Biotechnol. 102, 1–7. doi: 10.1007/s00253-017-8535-7
Godt, D., and Tepass, U. (1998). Drosophila oocyte localization is mediated by differential cadherin-based adhesion. Nature 395, 387–391. doi: 10.1038/26493
Gottardi, C. J., Wong, E., and Gumbiner, B. M. (2001). E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesion-independent manner. J. Cell Biol. 153, 1049–1060. doi: 10.1083/jcb.153.5.1049
Grabowska, M. M., and Day, M. L. (2012). Soluble E-cadherin: more than a symptom of disease. Front. Biosci. 17, 1948–1964. doi: 10.2741/4031
Gregory, P. A., Bert, A. G., Paterson, E. L., Barry, S. C., Tsykin, A., Farshid, G., et al. (2008). The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 10, 593–601. doi: 10.1038/ncb1722
Griffiths, N. J., Bradley, C. J., Heyderman, R. S., and Virji, M. (2007). IFN-gamma amplifies NFkappaB-dependent Neisseria meningitidis invasion of epithelial cells via specific upregulation of CEA-related cell adhesion molecule 1. Cell. Microbiol. 9, 2968–2983. doi: 10.1111/j.1462-5822.2007.01038.x
Gumbiner, B. M. (1995). Signal transduction of beta-catenin. Curr. Opin. Cell Biol. 7, 634–640. doi: 10.1016/0955-0674(95)80104-9
Guo, Z., Neilson, L. J., Zhong, H., Murray, P. S., Zanivan, S., and Zaidel-Bar, R. (2014). E-cadherin interactome complexity and robustness resolved by quantitative proteomics. Sci. Signal. 7:rs7. doi: 10.1126/scisignal.2005473
Gupta, P. K., Tripathi, D., Kulkarni, S., and Rajan, M. G. (2016). Mycobacterium tuberculosis H37Rv infected THP-1 cells induce epithelial mesenchymal transition in lung adenocarcinoma epithelial cell line (A549). Cell. Immunol. 300, 33–40. doi: 10.1016/j.cellimm.2015.11.007
Ha, H.-L., Kwon, T., Bak, I. S., Erikson, R. L., Kim, B. Y., and Yu, D.-Y. (2016). IGF-II induced by hepatitis B virus X protein regulates EMT via SUMO mediated loss of E-cadherin in mice. Oncotarget 7, 56944–56957. doi: 10.18632/oncotarget.10922
Hadley, G. A., Bartlett, S. T., Via, C. S., Rostapshova, E. A., and Moainie, S. (1997). The epithelial cell-specific integrin, CD103 (alpha E integrin), defines a novel subset of alloreactive CD8+ CTL. J. Immunol. 159, 3748–3756.
Häse, C. C., and Finkelstein, R. A. (1993). Bacterial extracellular zinc-containing metalloproteases. Microbiol. Rev. 57, 823–837.
Hecht, A., Vleminckx, K., and Stemmler, M. P., van Roy, F., and Kemler, R. (2000). The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J. 19, 1839–1850. doi: 10.1093/emboj/19.8.1839
Hermiston, M. L., Wong, M. H., and Gordon, J. I. (1996). Forced expression of E-cadherin in the mouse intestinal epithelium slows cell migration and provides evidence for nonautonomous regulation of cell fate in a self-renewing system. Genes Dev. 10, 985–996. doi: 10.1101/gad.10.8.985
Hertle, R., and Schwarz, H. (2004). Serratia marcescens internalization and replication in human bladder epithelial cells. BMC Infect. Dis. 4:16. doi: 10.1186/1471-2334-4-16
Hofmann, M., and Pircher, H. (2011). E-cadherin promotes accumulation of a unique memory CD8 T-cell population in murine salivary glands. Proc. Natl. Acad. Sci. USA 108, 16741–16746. doi: 10.1073/pnas.1107200108
Hoy, B., Geppert, T., Boehm, M., Reisen, F., Plattner, P., Gadermaier, G., et al. (2012). Distinct roles of secreted HtrA proteases from gram-negative pathogens in cleaving the junctional protein and tumor suppressor E-cadherin. J. Biol. Chem. 287, 10115–10120. doi: 10.1074/jbc.C111.333419
Hoy, B., Löwer, M., Weydig, C., Carra, G., Tegtmeyer, N., Geppert, T., et al. (2010). Helicobacter pylori HtrA is a new secreted virulence factor that cleaves E-cadherin to disrupt intercellular adhesion. EMBO Rep. 11, 798–804. doi: 10.1038/embor.2010.114
Huber, O., Korn, R., McLaughlin, J., Ohsugi, M., Herrmann, B. G., and Kemler, R. (1996). Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech. Dev. 59, 3–10. doi: 10.1016/0925-4773(96)00597-7
Hulpiau, P., and van Roy, F. (2009). Molecular evolution of the cadherin superfamily. Int. J. Biochem. Cell Biol. 41, 349–369. doi: 10.1016/j.biocel.2008.09.027
Hyafil, F., Babinet, C., and Jacob, F. (1981). Cell-cell interactions in early embryogenesis: a molecular approach to the role of calcium. Cell 26, 447–454. doi: 10.1016/0092-8674(81)90214-2
Inoshima, I., Inoshima, N., Wilke, G. A., Powers, M. E., Frank, K. M., Wang, Y., et al. (2011). A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 17, 1310–1314. doi: 10.1038/nm.2451
Isberg, R. R., and Leong, J. M. (1990). Multiple beta 1 chain integrins are receptors for invasin, a protein that promotes bacterial penetration into mammalian cells. Cell 60, 861–871. doi: 10.1016/0092-8674(90)90099-Z
Ismail, A. S., Behrendt, C. L., and Hooper, L. V. (2009). Reciprocal interactions between commensal bacteria and gamma delta intraepithelial lymphocytes during mucosal injury. J. Immunol. 182, 3047–3054. doi: 10.4049/jimmunol.0802705
Ismail, A. S., Severson, K. M., Vaishnava, S., Behrendt, C. L., Yu, X., Benjamin, J. L., et al. (2011). Gammadelta intraepithelial lymphocytes are essential mediators of host-microbial homeostasis at the intestinal mucosal surface. Proc. Natl. Acad. Sci. USA 108, 8743–8748. doi: 10.1073/pnas.1019574108
Ito, M., Maruyama, T., Saito, N., Koganei, S., Yamamoto, K., and Matsumoto, N. (2006). Killer cell lectin-like receptor G1 binds three members of the classical cadherin family to inhibit NK cell cytotoxicity. J. Exp. Med. 203, 289–295. doi: 10.1084/jem.20051986
Javaheri, A., Kruse, T., Moonens, K., Mejías-Luque, R., Debraekeleer, A., Asche, C. I., et al. (2016). Helicobacter pylori adhesin HopQ engages in a virulence-enhancing interaction with human CEACAMs. Nat. Microbiol. 2:16189. doi: 10.1038/nmicrobiol.2016.243
Jeanes, A., Gottardi, C. J., and Yap, A. S. (2008). Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene 27, 6920–6929. doi: 10.1038/onc.2008.343
Jiang, H., Guan, G., Zhang, R., Liu, G., Cheng, J., Hou, X., et al. (2009). Identification of urinary soluble E-cadherin as a novel biomarker for diabetic nephropathy. Diabetes Metab. Res. Rev. 25, 232–241. doi: 10.1002/dmrr.940
Johnson, S. K., Ramani, V. C., Hennings, L., and Haun, R. S. (2007). Kallikrein 7 enhances pancreatic cancer cell invasion by shedding E-cadherin. Cancer 109, 1811–1820. doi: 10.1002/cncr.22606
Jordans, S., Jenko-Kokalj, S., Kühl, N. M., Tedelind, S., Sendt, W., Brömme, D., et al. (2009). Monitoring compartment-specific substrate cleavage by cathepsins B, K, L, and S at physiological pH and redox conditions. BMC Biochem. 10:23. doi: 10.1186/1471-2091-10-23
Kague, E., Thomazini, C. M., Pardini, M. I. de C. M., Carvalho, F. de, Leite, C. V., and Pinheiro, N. A. (2010). Methylation status of CDH1 gene in samples of gastric mucous from Brazilian patients with chronic gastritis infected by Helicobacter pylori. Arq. Gastroenterol. 47, 7–12. doi: 10.1590/S0004-28032010000100002
Kalam, H., Singh, K., Chauhan, K., Fontana, M. F., and Kumar, D. (2018). Alternate splicing of transcripts upon Mycobacterium tuberculosis infection impacts the expression of functional protein domains. IUBMB Life 70, 845–854. doi: 10.1002/iub.1887
Katoh, M. (2017). Canonical and non-canonical WNT signaling in cancer stem cells and their niches: cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (review). Int. J. Oncol. 51, 1357–1369. doi: 10.3892/ijo.2017.4129
Katz, J. (2002). Hydrolysis of epithelial junctional proteins by porphyromonas gingivalis gingipains. Infect. Immun. 70, 2512–2518. doi: 10.1128/IAI.70.5.2512-2518.2002
Katz, J., Sambandam, V., Wu, J. H., Michalek, S. M., and Balkovetz, D. F. (2000). Characterization of porphyromonas gingivalis-induced degradation of epithelial cell junctional complexes. Infect. Immun. 68, 1441–1449. doi: 10.1128/IAI.68.3.1441-1449.2000
Kemler, R. (1993). From cadherins to catenins: cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet. 9, 317–321.
Kilshaw, P. J., and Murant, S. J. (1990). A new surface antigen on intraepithelial lymphocytes in the intestine. Eur. J. Immunol. 20, 2201–2207. doi: 10.1002/eji.1830201008
Kim, M., Ashida, H., Ogawa, M., Yoshikawa, Y., Mimuro, H., and Sasakawa, C. (2010). Bacterial interactions with the host epithelium. Cell Host Microbe 8, 20–35. doi: 10.1016/j.chom.2010.06.006
Kim, Y. S., and Ho, S. B. (2010). Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr. Gastroenterol. Rep. 12, 319–330. doi: 10.1007/s11894-010-0131-2
Kim, C., Kim, D. G., Park, S. H., Hwang, Y. I., Jang, S. H., Kim, C. H., et al. (2011). Epithelial to mesenchymal transition of mesothelial cells in tuberculous pleurisy. Yonsei Med. J. 52, 51–58. doi: 10.3349/ymj.2011.52.1.51
Klein, T., and Bischoff, R. (2011). Physiology and pathophysiology of matrix metalloproteases. Amino Acids 41, 271–290. doi: 10.1007/s00726-010-0689-x
Klucky, B., Mueller, R., Vogt, I., Teurich, S., Hartenstein, B., Breuhahn, K., et al. (2007). Kallikrein 6 induces E-cadherin shedding and promotes cell proliferation, migration, and invasion. Cancer Res. 67, 8198–8206. doi: 10.1158/0008-5472.CAN-07-0607
Kourtidis, A., Ngok, S. P., and Anastasiadis, P. Z. (2013). p120 catenin: an essential regulator of cadherin stability, adhesion-induced signaling, and cancer progression. Prog. Mol. Biol. Transl. Sci. 116, 409–432. doi: 10.1016/B978-0-12-394311-8.00018-2
Kramps, T., Peter, O., Brunner, E., Nellen, D., Froesch, B., Chatterjee, S., et al. (2002). Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear beta-catenin-TCF complex. Cell 109, 47–60. doi: 10.1016/S0092-8674(02)00679-7
Kuefer, R., Hofer, M. D., Zorn, C. S. M., Engel, O., Volkmer, B. G., Juarez-Brito, M. A., et al. (2005). Assessment of a fragment of e-cadherin as a serum biomarker with predictive value for prostate cancer. Br. J. Cancer 92, 2018–2023. doi: 10.1038/sj.bjc.6602599
Kumar, R., Herold, J. L., Schady, D., Davis, J., Kopetz, S., Martinez-Moczygemba, M., et al. (2017). Streptococcus gallolyticus sub sp. gallolyticus promotes colorectal tumor development. PLoS Pathog. 13:e1006440. doi: 10.1371/journal.ppat.1006440
Laurson, J., Khan, S., Chung, R., Cross, K., and Raj, K. (2010). Epigenetic repression of E-cadherin by human papillomavirus 16 E7 protein. Carcinogenesis 31, 918–926. doi: 10.1093/carcin/bgq027
Lee, K. H., Choi, E. Y., Hyun, M. S., Jang, B. I., Kim, T. N., Kim, S. W., et al. (2007). Association of extracellular cleavage of E-cadherin mediated by MMP-7 with HGF-induced in vitro invasion in human stomach cancer cells. Eur. Surg. Res. 39, 208–215. doi: 10.1159/000101452
Lee, H.-J., Lee, H.-J., Lim, E.-S., Ahn, K.-S., Shim, B.-S., Kim, H.-M., et al. (2005). Cambodian Phellinus linteus inhibits experimental metastasis of melanoma cells in mice via regulation of urokinase type plasminogen activator. Biol. Pharm. Bull. 28, 27–31. doi: 10.1248/bpb.28.27
Lee, K. H., Shin, S. J., Kim, K. O., Kim, M. K., Hyun, M. S., Kim, T. N., et al. (2006). Relationship between E-cadherin, matrix metalloproteinase-7 gene expression and clinicopathological features in gastric carcinoma. Oncol. Rep. 16, 823–830. doi: 10.3892/or.16.4.823
Lee, M. G., Tang, A., Sharrow, S. O., and Udey, M. C. (1994). Murine dendritic epidermal T cells express the homophilic adhesion molecule E-cadherin. Epithelial Cell Biol. 3, 149–155.
Li, X.-W., Shi, B.-Y., Yang, Q.-L., Wu, J., Wu, H.-M., Wang, Y.-F., et al. (2015). Epigenetic regulation of CDH1 exon 8 alternative splicing in gastric cancer. BMC Cancer 15:954. doi: 10.1186/s12885-015-1983-5
Li, Q., Sodroski, C., Lowey, B., Schweitzer, C. J., Cha, H., Zhang, F., et al. (2016). Hepatitis C virus depends on E-cadherin as an entry factor and regulates its expression in epithelial-to-mesenchymal transition. Proc. Natl. Acad. Sci. USA 113, 7620–7625. doi: 10.1073/pnas.1602701113
Li, L., Wang, S., Jezierski, A., Moalim-Nour, L., Mohib, K., Parks, R. J., et al. (2010). A unique interplay between Rap1 and E-cadherin in the endocytic pathway regulates self-renewal of human embryonic stem cells. Stem Cells 28, 247–257. doi: 10.1002/stem.289
Lombaerts, M., van Wezel, T., Philippo, K., Dierssen, J. W. F., Zimmerman, R. M. E., Oosting, J., et al. (2006). E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Br. J. Cancer 94, 661–671. doi: 10.1038/sj.bjc.6602996
Ma, C., Luo, H., Gao, F., Tang, Q., and Chen, W. (2018). Fusobacterium nucleatum promotes the progression of colorectal cancer by interacting with E-cadherin. Oncol. Lett. 16, 2606–2612. doi: 10.3892/ol.2018.8947
MacDonald, B. T., Tamai, K., and He, X. (2009). Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26. doi: 10.1016/j.devcel.2009.06.016
Madoux, F., Dreymuller, D., Pettiloud, J.-P., Santos, R., Becker-Pauly, C., Ludwig, A., et al. (2016). Discovery of an enzyme and substrate selective inhibitor of ADAM10 using an exosite-binding glycosylated substrate. Sci. Rep. 6:11. doi: 10.1038/s41598-016-0013-4
Marambaud, P. (2002). A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 21, 1948–1956. doi: 10.1093/emboj/21.8.1948
Maretzky, T., Scholz, F., Köten, B., Proksch, E., Saftig, P., and Reiss, K. (2008). ADAM10-mediated E-cadherin release is regulated by proinflammatory cytokines and modulates keratinocyte cohesion in eczematous dermatitis. J. Invest. Dermatol. 128, 1737–1746. doi: 10.1038/sj.jid.5701242
Marshall, D. C., Lyman, S. K., McCauley, S., Kovalenko, M., Spangler, R., Liu, C., et al. (2015). Selective allosteric inhibition of MMP9 is efficacious in preclinical models of ulcerative colitis and colorectal cancer. PLoS One 10:e0127063. doi: 10.1371/journal.pone.0127063
Mason, K. L., and Huffnagle, G. B. (2009). Control of mucosal polymicrobial populations by innate immunity. Cell. Microbiol. 11, 1297–1305. doi: 10.1111/j.1462-5822.2009.01347.x
Matos, M. L., Lapyckyj, L., Rosso, M., Besso, M. J., Mencucci, M. V., Briggiler, C. I., et al. (2017). Identification of a novel human E-cadherin splice variant and assessment of its effects upon EMT-related events. J. Cell. Physiol. 232, 1368–1386. doi: 10.1002/jcp.25622
McCrea, P. D., and Gumbiner, B. M. (1991). Purification of a 92-kDa cytoplasmic protein tightly associated with the cell-cell adhesion molecule E-cadherin (uvomorulin). Characterization and extractability of the protein complex from the cell cytostructure. J. Biol. Chem. 266, 4514–4520.
McCrea, P. D., Maher, M. T., and Gottardi, C. J. (2015). Nuclear signaling from cadherin adhesion complexes. Curr. Top. Dev. Biol. 129–196. doi: 10.1016/bs.ctdb.2014.11.018
Melenotte, C., Mezouar, S., Ben Amara, A., Benatti, S., Chiaroni, J., Devaux, C., et al. (2019). A transcriptional signature associated with non-Hodgkin lymphoma in the blood of patients with Q fever. PLoS One 14:e0217542. doi: 10.1371/journal.pone.0217542
Melenotte, C., Million, M., Audoly, G., Gorse, A., Dutronc, H., Roland, G., et al. (2016). B-cell non-Hodgkin lymphoma linked to Coxiella burnetii. Blood 127, 113–121. doi: 10.1182/blood-2015-04-639617
Mezouar, S., Omar Osman, I., Melenotte, C., Slimani, C., Chartier, C., Raoult, D., et al. (2019). High concentrations of serum soluble E-cadherin in patients with Q fever. Front. Cell. Infect. Microbiol. 9:219. doi: 10.3389/fcimb.2019.00219
Miller, H., Zhang, J., Kuolee, R., Patel, G. B., and Chen, W. (2007). Intestinal M cells: the fallible sentinels? World J. Gastroenterol. 13, 1477–1486. doi: 10.3748/wjg.v13.i10.1477
Moon, D. C., Choi, C. H., Lee, S. M., Lee, J. H., Kim, S. I., Kim, D. S., et al. (2012). Nuclear translocation of Acinetobacter baumannii transposase induces DNA methylation of CpG regions in the promoters of E-cadherin gene. PLoS One 7:e38974. doi: 10.1371/journal.pone.0038974
Müller, K. M., Luedecker, C. J., Udey, M. C., and Farr, A. G. (1997). Involvement of E-cadherin in thymus organogenesis and thymocyte maturation. Immunity 6, 257–264. doi: 10.1016/S1074-7613(00)80328-3
Munro, S. B., Duclos, A. J., Jackson, A. R., Baines, M. G., and Blaschuk, O. W. (1996). Characterization of cadherins expressed by murine thymocytes. Cell. Immunol. 169, 309–312. doi: 10.1006/cimm.1996.0123
Murata-Kamiya, N., Kurashima, Y., Teishikata, Y., Yamahashi, Y., Saito, Y., Higashi, H., et al. (2007). Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene 26, 4617–4626. doi: 10.1038/sj.onc.1210251
Nagafuchi, A., and Takeichi, M. (1988). Cell binding function of E-cadherin is regulated by the cytoplasmic domain. EMBO J. 7, 3679–3684. doi: 10.1002/j.1460-2075.1988.tb03249.x
Nagase, H., Visse, R., and Murphy, G. (2006). Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 69, 562–573. doi: 10.1016/j.cardiores.2005.12.002
Najy, A. J., Day, K. C., and Day, M. L. (2008). The ectodomain shedding of E-cadherin by ADAM15 supports ErbB receptor activation. J. Biol. Chem. 283, 18393–18401. doi: 10.1074/jbc.M801329200
Nava, P., Kamekura, R., and Nusrat, A. (2013). Cleavage of transmembrane junction proteins and their role in regulating epithelial homeostasis. Tissue Barriers 1:e24783. doi: 10.4161/tisb.24783
Niessen, C. M., Leckband, D., and Yap, A. S. (2011). Tissue organization by cadherin adhesion molecules: dynamic molecular and cellular mechanisms of morphogenetic regulation. Physiol. Rev. 91, 691–731. doi: 10.1152/physrev.00004.2010
Noë, V., Fingleton, B., Jacobs, K., Crawford, H. C., Vermeulen, S., Steelant, W., et al. (2001). Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. J. Cell Sci. 114, 111–118.
Nollet, F., Kools, P., and van Roy, F. (2000). Phylogenetic analysis of the cadherin superfamily allows identification of six major subfamilies besides several solitary members. J. Mol. Biol. 299, 551–572. doi: 10.1006/jmbi.2000.3777
Novotny, L. A., and Bakaletz, L. O. (2016). Intercellular adhesion molecule 1 serves as a primary cognate receptor for the type IV pilus of nontypeable Haemophilus influenzae: ICAM1 serves as the cognate receptor for NTHI type IV pilus. Cell. Microbiol. 18, 1043–1055. doi: 10.1111/cmi.12575
Nusrat, A., von Eichel-Streiber, C., Turner, J. R., Verkade, P., Madara, J. L., and Parkos, C. A. (2001). Clostridium difficile toxins disrupt epithelial barrier function by altering membrane microdomain localization of tight junction proteins. Infect. Immun. 69, 1329–1336. doi: 10.1128/IAI.69.3.1329-1336.2001
O’Connor, P. M., Lapointe, T. K., Jackson, S., Beck, P. L., Jones, N. L., and Buret, A. G. (2011). Helicobacter pylori activates calpain via toll-like receptor 2 to disrupt adherens junctions in human gastric epithelial cells. Infect. Immun. 79, 3887–3894. doi: 10.1128/IAI.05109-11
Obiso, R. J., Azghani, A. O., and Wilkins, T. D. (1997). The Bacteroides fragilis toxin fragilysin disrupts the paracellular barrier of epithelial cells. Infect. Immun. 65, 1431–1439.
Oda, H., and Takeichi, M. (2011). Structural and functional diversity of cadherin at the adherens junction. J. Cell Biol. 193, 1137–1146. doi: 10.1083/jcb.201008173
Ogata, Y., Enghild, J. J., and Nagase, H. (1992). Matrix metalloproteinase 3 (stromelysin) activates the precursor for the human matrix metalloproteinase 9. J. Biol. Chem. 267, 3581–3584.
Ogou, S. I., Yoshida-Noro, C., and Takeichi, M. (1983). Calcium-dependent cell-cell adhesion molecules common to hepatocytes and teratocarcinoma stem cells. J. Cell Biol. 97, 944–948. doi: 10.1083/jcb.97.3.944
Ohira, T., Gemmill, R. M., Ferguson, K., Kusy, S., Roche, J., Brambilla, E., et al. (2003). WNT7a induces E-cadherin in lung cancer cells. Proc. Natl. Acad. Sci. USA 100, 10429–10434. doi: 10.1073/pnas.1734137100
Okada, Y., and Nakanishi, I. (1989). Activation of matrix metalloproteinase 3 (stromelysin) and matrix metalloproteinase 2 (‘gelatinase’) by human neutrophil elastase and cathepsin G. FEBS Lett. 249, 353–356. doi: 10.1016/0014-5793(89)80657-X
Ortega, F. E., Rengarajan, M., Chavez, N., Radhakrishnan, P., Gloerich, M., Bianchini, J., et al. (2017). Adhesion to the host cell surface is sufficient to mediate Listeria monocytogenes entry into epithelial cells. Mol. Biol. Cell 28, 2945–2957. doi: 10.1091/mbc.E16-12-0851
Pandiella, A., Bosenberg, M. W., Huang, E. J., Besmer, P., and Massagué, J. (1992). Cleavage of membrane-anchored growth factors involves distinct protease activities regulated through common mechanisms. J. Biol. Chem. 267, 24028–24033.
Perri, F., Cotugno, R., Piepoli, A., Merla, A., Quitadamo, M., Gentile, A., et al. (2007). Aberrant DNA methylation in non-neoplastic gastric mucosa of H. pylori infected patients and effect of eradication. Am. J. Gastroenterol. 102, 1361–1371. doi: 10.1111/j.1572-0241.2007.01284.x
Phan, Q. T., Myers, C. L., Fu, Y., Sheppard, D. C., Yeaman, M. R., Welch, W. H., et al. (2007). Als3 is a Candida albicans invasin that binds to cadherins and induces endocytosis by host cells. PLoS Biol. 5:e64. doi: 10.1371/journal.pbio.0050064
Pieters, T., and van Roy, F. (2014). Role of cell-cell adhesion complexes in embryonic stem cell biology. J. Cell Sci. 127, 2603–2613. doi: 10.1242/jcs.146720
Pittard, A. J., Banks, R. E., Galley, H. F., and Webster, N. R. (1996). Soluble E-cadherin concentrations in patients with systemic inflammatory response syndrome and multiorgan dysfunction syndrome. Br. J. Anaesth. 76, 629–631. doi: 10.1093/bja/76.5.629
Rajić, J., Inic-Kanada, A., Stein, E., Dinić, S., Schuerer, N., Uskoković, A., et al. (2017). Chlamydia trachomatis infection is associated with E-cadherin promoter methylation, downregulation of E-cadherin expression, and increased expression of fibronectin and α-SMA—implications for epithelial-mesenchymal transition. Front. Cell. Infect. Microbiol. 7:253. doi: 10.3389/fcimb.2017.00253
Reboud, E., Basso, P., Maillard, A. P., Huber, P., and Attrée, I. (2017). Exolysin shapes the virulence of Pseudomonas aeruginosa clonal outliers. Toxins 9, pii: E364. doi: 10.3390/toxins9110364
Remacle, A. G., Shiryaev, S. A., and Strongin, A. Y. (2014). Distinct interactions with cellular E-cadherin of the two virulent metalloproteinases encoded by a Bacteroides fragilis pathogenicity island. PLoS One 9:e113896. doi: 10.1371/journal.pone.0113896
Repetto, O., De Paoli, P., De Re, V., Canzonieri, V., and Cannizzaro, R. (2014). Levels of soluble E-cadherin in breast, gastric, and colorectal cancers. Biomed. Res. Int. 2014:408047. doi: 10.1155/2014/408047
Reyat, J. S., Chimen, M., Noy, P. J., Szyroka, J., Rainger, G. E., and Tomlinson, M. G. (2017). ADAM10-interacting tetraspanins Tspan5 and Tspan17 regulate VE-cadherin expression and promote T lymphocyte transmigration. J. Immunol. 199, 666–676. doi: 10.4049/jimmunol.1600713
Rodriguez, F. J., Lewis-Tuffin, L. J., and Anastasiadis, P. Z. (2012). E-cadherin’s dark side: possible role in tumor progression. Biochim. Biophys. Acta 1826, 23–31. doi: 10.1016/j.bbcan.2012.03.002
Rosso, M., Lapyckyi, L., Besso, M. L., Monge, M., Reventós, J., Canals, F., et al. (2019). Characterization of the molecular changes associated with the overexpression of a novel epithelial cadherin splice variant mRNA in a breast cancer model using proteomics and bioinformatics approaches: identification of changes in cell metabolism and an increased expression of lactate dehydrogenase B. Cancer Metab. 7:5. doi: 10.1186/s40170-019-0196-9
Rouabhia, M., Semlali, A., Audoy, J., and Chmielewski, W. (2012). Antagonistic effect of Candida albicans and IFNγ on E-cadherin expression and production by human primary gingival epithelial cells. Cell. Immunol. 280, 61–67. doi: 10.1016/j.cellimm.2012.11.008
Rubinstein, M. R., Baik, J. E., Lagana, S. M., Han, R. P., Raab, W. J., Sahoo, D., et al. (2019). Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/β-catenin modulator Annexin A1. EMBO Rep. 20, pii: e47638. doi: 10.15252/embr.201847638
Rubinstein, M. R., Wang, X., Liu, W., Hao, Y., Cai, G., and Han, Y. W. (2013). Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 14, 195–206. doi: 10.1016/j.chom.2013.07.012
Ryniers, F., Stove, C., Goethals, M., Brackenier, L., Noë, V., Bracke, M., et al. (2002). Plasmin produces an E-cadherin fragment that stimulates cancer cell invasion. Biol. Chem. 383, 159–165. doi: 10.1515/BC.2002.016
Sakai, A., Sugawara, Y., Kuroishi, T., Sasano, T., and Sugawara, S. (2008). Identification of IL-18 and Th17 cells in salivary glands of patients with Sjogren’s syndrome, and amplification of IL-17-mediated secretion of inflammatory cytokines from salivary gland cells by IL-18. J. Immunol. 181, 2898–2906. doi: 10.4049/jimmunol.181.4.2898
Salama, R. H. M., Selem, T. H., El-Gammal, M., Elhagagy, A. A., and Bakar, S. M. (2013). Urinary tumor markers could predict survival in bladder carcinoma. Indian J. Clin. Biochem. 28, 265–271. doi: 10.1007/s12291-012-0266-z
Sato, T., Shibata, W., and Maeda, S. (2019). Adhesion molecules and pancreatitis. J. Gastroenterol. 54, 99–107. doi: 10.1007/s00535-018-1500-0
Sauka-Spengler, T., and Bronner-Fraser, M. (2008). A gene regulatory network orchestrates neural crest formation. Nat. Rev. Mol. Cell Biol. 9, 557–568. doi: 10.1038/nrm2428
Schiavo, G., Benfenati, F., Poulain, B., Rossetto, O., Polverino de Laureto, P., DasGupta, B. R., et al. (1992). Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 359, 832–835. doi: 10.1038/359832a0
Schirrmeister, W., Gnad, T., Wex, T., Higashiyama, S., Wolke, C., Naumann, M., et al. (2009). Ectodomain shedding of E-cadherin and c-met is induced by Helicobacter pylori infection. Exp. Cell Res. 315, 3500–3508. doi: 10.1016/j.yexcr.2009.07.029
Schmidt, T. P., Perna, A. M., Fugmann, T., Böhm, M., Jan, H., Haller, S., et al. (2016). Identification of E-cadherin signature motifs functioning as cleavage sites for Helicobacter pylori HtrA. Sci. Rep. 6:23264. doi: 10.1038/srep23264
Schuh, R., Vestweber, D., Riede, I., Ringwald, M., Rosenberg, U. B., Jäckle, H., et al. (1986). Molecular cloning of the mouse cell adhesion molecule uvomorulin: cDNA contains a B1-related sequence. Proc. Natl. Acad. Sci. USA 83, 1364–1368.
Schwartzkopff, S., Gründemann, C., Schweier, O., Rosshart, S., Karjalainen, K. E., Becker, K.-F., et al. (2007). Tumor-associated E-cadherin mutations affect binding to the killer cell lectin-like receptor G1 in humans. J. Immunol. 179, 1022–1029. doi: 10.4049/jimmunol.179.2.1022
Seike, S., Takehara, M., Takagishi, T., Miyamoto, K., Kobayashi, K., and Nagahama, M. (2018). Delta-toxin from Clostridium perfringens perturbs intestinal epithelial barrier function in Caco-2 cell monolayers. Biochim. Biophys. Acta Biomembr. 1860, 428–433. doi: 10.1016/j.bbamem.2017.10.003
Seveau, S., Bierne, H., Giroux, S., Prévost, M.-C., and Cossart, P. (2004). Role of lipid rafts in E-cadherin- and HGF-R/met-mediated entry of Listeria monocytogenes into host cells. J. Cell Biol. 166, 743–753. doi: 10.1083/jcb.200406078
Shapiro, L., Fannon, A. M., Kwong, P. D., Thompson, A., Lehmann, M. S., Grübel, G., et al. (1995). Structural basis of cell-cell adhesion by cadherins. Nature 374, 327–337. doi: 10.1038/374327a0
Sharma, S., and Lichtenstein, A. (2009). Aberrant splicing of the E-cadherin transcript is a novel mechanism of gene silencing in chronic lymphocytic leukemia cells. Blood 114, 4179–4185. doi: 10.1182/blood-2009-03-206482
Sheets, S. M., Potempa, J., Travis, J., Casiano, C. A., and Fletcher, H. M. (2005). Gingipains from Porphyromonas gingivalis W83 induce cell adhesion molecule cleavage and apoptosis in endothelial cells. Infect. Immun. 73, 1543–1552. doi: 10.1128/IAI.73.3.1543-1552.2005
Sheridan, B. S., and Lefrançois, L. (2011). Regional and mucosal memory T cells. Nat. Immunol. 12, 485–491. doi: 10.1038/ni.2029
Shields, B. D., Koss, B., Taylor, E. M., Storey, A. J., West, K. L., Byrum, S. D., et al. (2019). Loss of E-cadherin inhibits CD103 antitumor activity and reduces checkpoint blockade responsiveness in melanoma. Cancer Res. 79, 1113–1123. doi: 10.1158/0008-5472.CAN-18-1722
Shirahata, T., Nakamura, H., Nakajima, T., Nakamura, M., Chubachi, S., Yoshida, S., et al. (2018). Plasma sE-cadherin and the plasma sE-cadherin/sVE-cadherin ratio are potential biomarkers for chronic obstructive pulmonary disease. Biomarkers 23, 414–421. doi: 10.1080/1354750X.2018.1434682
Sintsova, A., Wong, H., MacDonald, K. S., Kaul, R., Virji, M., and Gray-Owen, S. D. (2015). Selection for a CEACAM receptor-specific binding phenotype during Neisseria gonorrhoeae infection of the human genital tract. Infect. Immun. 83, 1372–1383. doi: 10.1128/IAI.03123-14
Stephens, G. L., Andersson, J., and Shevach, E. M. (2007). Distinct subsets of FoxP3+ regulatory T cells participate in the control of immune responses. J. Immunol. 178, 6901–6911. doi: 10.4049/jimmunol.178.11.6901
Streeck, H., Kwon, D. S., Pyo, A., Flanders, M., Chevalier, M. F., Law, K., et al. (2011). Epithelial adhesion molecules can inhibit HIV-1-specific CD8+ T-cell functions. Blood 117, 5112–5122. doi: 10.1182/blood-2010-12-321588
Sugawara, Y., and Fujinaga, Y. (2011). The botulinum toxin complex meets E-cadherin on the way to its destination. Cell Adhes. Migr. 5, 34–36. doi: 10.4161/cam.5.1.13574
Sun, H. (2017). Deciphering alternative splicing and nonsense-mediated decay modulate expression in primary lymphoid tissues of birds infected with avian pathogenic E. coli (APEC). BMC Genet. 18:21. doi: 10.1186/s12863-017-0488-4
Sun, H., Pan, L., Jia, H., Zhang, Z., Gao, M., Huang, M., et al. (2018). Label-free quantitative proteomics identifies novel plasma biomarkers for distinguishing pulmonary tuberculosis and latent infection. Front. Microbiol. 9:1267. doi: 10.3389/fmicb.2018.01267
Suzuki, M., Mimuro, H., Suzuki, T., Park, M., Yamamoto, T., and Sasakawa, C. (2005). Interaction of CagA with Crk plays an important role in Helicobacter pylori-induced loss of gastric epithelial cell adhesion. J. Exp. Med. 202, 1235–1247. doi: 10.1084/jem.20051027
Symowicz, J., Adley, B. P., Gleason, K. J., Johnson, J. J., Ghosh, S., Fishman, D. A., et al. (2007). Engagement of collagen-binding integrins promotes matrix metalloproteinase-9–dependent E-cadherin ectodomain shedding in ovarian carcinoma cells. Cancer Res. 67, 2030–2039. doi: 10.1158/0008-5472.CAN-06-2808
Tahara, T., Yamamoto, E., Suzuki, H., Maruyama, R., Chung, W., Garriga, J., et al. (2014). Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res. 74, 1311–1318. doi: 10.1158/0008-5472.CAN-13-1865
Takeichi, M. (1977). Functional correlation between cell adhesive properties and some cell surface proteins. J. Cell Biol. 75, 464–474. doi: 10.1083/jcb.75.2.464
Tchoupa, A. K., Lichtenegger, S., Reidl, J., and Hauck, C. R. (2015). Outer membrane protein P1 is the CEACAM-binding adhesin of Haemophilus influenzae: P1 binds human CEACAMs to colonize mucosa. Mol. Microbiol. 98, 440–455. doi: 10.1111/mmi.13134
Tegtmeyer, N., Harrer, A., Schmitt, V., Singer, B. B., and Backert, S. (2019). Expression of CEACAM1 or CEACAM5 in AZ-521 cells restores the type IV secretion deficiency for translocation of CagA by Helicobacter pylori. Cell. Microbiol. 21:e12965. doi: 10.1111/cmi.12965
Tegtmeyer, N., Wessler, S., Necchi, V., Rohde, M., Harrer, A., Rau, T. T., et al. (2017). Helicobacter pylori employs a unique basolateral type IV secretion mechanism for CagA delivery. Cell Host Microbe 22, 552–560.e5. doi: 10.1016/j.chom.2017.09.005
Terciolo, C., Dobric, A., Ouaissi, M., Siret, C., Breuzard, G., Silvy, F., et al. (2017). Saccharomyces boulardii CNCM I-745 restores intestinal barrier integrity by regulation of E-cadherin recycling. J. Crohns Colitis 11, 999–1010. doi: 10.1093/ecco-jcc/jjx030
Tessmer, M. S., Fugere, C., Stevenaert, F., Naidenko, O. V., Chong, H. J., Leclercq, G., et al. (2007). KLRG1 binds cadherins and preferentially associates with SHIP-1. Int. Immunol. 19, 391–400. doi: 10.1093/intimm/dxm004
Thiery, J. P. (2002). Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2, 442–454. doi: 10.1038/nrc822
Thoulouze, M. I., Lafage, M., Schachner, M., Hartmann, U., Cremer, H., and Lafon, M. (1998). The neural cell adhesion molecule is a receptor for rabies virus. J. Virol. 72, 7181–7190.
Tsaur, I., Thurn, K., Juengel, E., Gust, K. M., Borgmann, H., Mager, R., et al. (2015). sE-cadherin serves as a diagnostic and predictive parameter in prostate cancer patients. J. Exp. Clin. Cancer Res. 34:43. doi: 10.1186/s13046-015-0161-6
Tuccinardi, T., Nuti, E., Ortore, G., Rossello, A., Avramova, S. I., and Martinelli, A. (2008). Development of a receptor-based 3D-QSAR study for the analysis of MMP2, MMP3, and MMP9 inhibitors. Bioorg. Med. Chem. 16, 7749–7758. doi: 10.1016/j.bmc.2008.07.004
Turk, B., Turk, D., and Turk, V. (2012). Protease signalling: the cutting edge. EMBO J. 31, 1630–1643. doi: 10.1038/emboj.2012.42
Uchida, Y., Kawai, K., Ibusuki, A., and Kanekura, T. (2011). Role for E-cadherin as an inhibitory receptor on epidermal T cells. J. Immunol. 186, 6945–6954. doi: 10.4049/jimmunol.1003853
Valls, G., Codina, M., Miller, R. K., Del Valle-Pérez, B., Vinyoles, M., Caelles, C., et al. (2012). Upon Wnt stimulation, Rac1 activation requires Rac1 and Vav2 binding to p120-catenin. J. Cell Sci. 125, 5288–5301. doi: 10.1242/jcs.101030
Van den Bossche, J., Malissen, B., Mantovani, A., De Baetselier, P., and Van Ginderachter, J. A. (2012). Regulation and function of the E-cadherin/catenin complex in cells of the monocyte-macrophage lineage and DCs. Blood 119, 1623–1633. doi: 10.1182/blood-2011-10-384289
van Roy, F. (2014). Beyond E-cadherin: roles of other cadherin superfamily members in cancer. Nat. Rev. Cancer 14, 121–134. doi: 10.1038/nrc3647
van Roy, F., and Berx, G. (2008). The cell-cell adhesion molecule E-cadherin. Cell. Mol. Life Sci. 65, 3756–3788. doi: 10.1007/s00018-008-8281-1
Vazeille, E., Bringer, M.-A., Gardarin, A., Chambon, C., Becker-Pauly, C., Pender, S. L. F., et al. (2011). Role of meprins to protect ileal mucosa of Crohn’s disease patients from colonization by adherent-invasive E. coli. PLoS One 6:e21199. doi: 10.1371/journal.pone.0021199
Villar, C. C., Kashleva, H., Nobile, C. J., Mitchell, A. P., and Dongari-Bagtzoglou, A. (2007). Mucosal tissue invasion by Candida albicans is associated with E-cadherin degradation, mediated by transcription factor Rim101p and protease Sap5p. Infect. Immun. 75, 2126–2135. doi: 10.1128/IAI.00054-07
Wakabayashi, T., and De Strooper, B. (2008). Presenilins: members of the gamma-secretase quartets, but part-time soloists too. Physiology (Bethesda) 23, 194–204. doi: 10.1152/physiol.00009.2008
Wei, W., Guo, H., Chang, J., Yu, Y., Liu, G., Zhang, N., et al. (2016). ICAM-5/Telencephalin is a functional entry receptor for enterovirus D68. Cell Host Microbe 20, 631–641. doi: 10.1016/j.chom.2016.09.013
Wells, A., Yates, C., and Shepard, C. R. (2008). E-cadherin as an indicator of mesenchymal to epithelial reverting transitions during the metastatic seeding of disseminated carcinomas. Clin. Exp. Metastasis 25, 621–628. doi: 10.1007/s10585-008-9167-1
Wu, G., Huang, H., Garcia Abreu, J., and He, X. (2009). Inhibition of GSK3 phosphorylation of beta-catenin via phosphorylated PPPSPXS motifs of Wnt coreceptor LRP6. PLoS One 4:e4926. doi: 10.1371/journal.pone.0004926
Wu, S., Lim, K.-C., Huang, J., Saidi, R. F., and Sears, C. L. (1998). Bacteroides fragilis enterotoxin cleaves the zonula adherens protein, E-cadherin. Proc. Natl. Acad. Sci. USA 95, 14979–14984.
Wu, S., Morin, P. J., Maouyo, D., and Sears, C. L. (2003). Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology 124, 392–400. doi: 10.1053/gast.2003.50047
Xu, X., Shi, Y., Zhang, P., Zhang, F., Shen, Y., Su, X., et al. (2012). E-cadherin mediates adhesion and endocytosis of Aspergillus fumigatus blastospores in human epithelial cells. Chin. Med. J. 125, 617–621. doi: 10.3760/cma.j.issn.0366-6999.2012.04.011
Xu, H., Sobue, T., Bertolini, M., Thompson, A., and Dongari-Bagtzoglou, A. (2016). Streptococcus oralis and Candida albicans synergistically activate μ-calpain to degrade E-cadherin from oral epithelial junctions. J. Infect. Dis. 214, 925–934. doi: 10.1093/infdis/jiw201
Yan, T., Han, J., and Yu, X. (2015). E-cadherin mediates adhesion of Aspergillus fumigatus to non-small cell lung cancer cells. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 37, 15593–15599.doi: 10.1007/s13277-015-4195-3
Yang, Y., Weng, W., Peng, J., Hong, L., Yang, L., Toiyama, Y., et al. (2017). Fusobacterium nucleatum increases proliferation of colorectal cancer cells and tumor development in mice by activating toll-like receptor 4 signaling to nuclear factor-κB, and up-regulating expression of MicroRNA-21. Gastroenterology 152, 851–866.e24. doi: 10.1053/j.gastro.2016.11.018
Yap, A. S., Niessen, C. M., and Gumbiner, B. M. (1998). The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering, adhesive strengthening, and interaction with p120ctn. J. Cell Biol. 141, 779–789. doi: 10.1083/jcb.141.3.779
Yoshikawa, Y., Ogawa, M., Hain, T., Yoshida, M., Fukumatsu, M., Kim, M., et al. (2009). Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat. Cell Biol. 11, 1233–1240. doi: 10.1038/ncb1967
Zhao, Q., Busch, B., Jiménez-Soto, L. F., Ishikawa-Ankerhold, H., Massberg, S., Terradot, L., et al. (2018). Integrin but not CEACAM receptors are dispensable for Helicobacter pylori CagA translocation. PLoS Pathog. 14:e1007359. doi: 10.1371/journal.ppat.1007359
Keywords: bacterial invasion, bacteria-inducing cancer, pathophysiology, E-cadherin, sheddases
Citation: Devaux CA, Mezouar S and Mege J-L (2019) The E-Cadherin Cleavage Associated to Pathogenic Bacteria Infections Can Favor Bacterial Invasion and Transmigration, Dysregulation of the Immune Response and Cancer Induction in Humans. Front. Microbiol. 10:2598. doi: 10.3389/fmicb.2019.02598
Edited by:
Christoph Hölscher, Research Center Borstel (LG), GermanyReviewed by:
Manja Boehm, University of Erlangen Nuremberg, GermanyCopyright © 2019 Devaux, Mezouar and Mege. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian A. Devaux, Y2hyaXN0aWFuLmRldmF1eEBtZWRpdGVycmFuZWUtaW5mZWN0aW9uLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.