 Yi Li1Qian-Qian Cha1
Yi Li1Qian-Qian Cha1 Yan-Ru Dang1
Yan-Ru Dang1 Xiu-Lan Chen1,2Min Wang3
Xiu-Lan Chen1,2Min Wang3 Andrew McMinn3,4
Andrew McMinn3,4 Giannina Espina5
Giannina Espina5 Yu-Zhong Zhang1,2,3
Yu-Zhong Zhang1,2,3 Jenny M. Blamey5,6*
Jenny M. Blamey5,6* Qi-Long Qin1*
Qi-Long Qin1*- 1State Key Laboratory of Microbial Technology, Marine Biotechnology Research Center, Shandong University, Qingdao, China
- 2Laboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
- 3College of Marine Life Sciences, Institute for Advanced Ocean Study, Ocean University of China, Qingdao, China
- 4Institute for Marine and Antarctic Studies, University of Tasmania, Hobart, TAS, Australia
- 5Fundación Científica y Cultural Biociencia, Santiago, Chile
- 6Faculty of Chemistry and Biology, Universidad de Santiago de Chile, Santiago, Chile
Antarctica is covered by multiple larger glaciers with diverse extreme conditions. Microorganisms in Antarctic regions are primarily responsible for diverse biogeochemical processes. The identity and functionality of microorganisms from polar glaciers are defined. However, little is known about microbial communities from the high elevation glaciers. The Union Glacier, located in the inland of West Antarctica at 79°S, is a challenging environment for life to survive due to the high irradiance and low temperatures. Here, soil and rock samples were obtained from three high mountains (Rossman Cove, Charles Peak, and Elephant Head) adjacent to the Union Glacier. Using metagenomic analyses, the functional microbial ecosystem was analyzed through the reconstruction of carbon, nitrogen and sulfur metabolic pathways. A low biomass but diverse microbial community was found. Although archaea were detected, bacteria were dominant. Taxa responsible for carbon fixation were comprised of photoautotrophs (Cyanobacteria) and chemoautotrophs (mainly Alphaproteobacterial clades: Bradyrhizobium, Sphingopyxis, and Nitrobacter). The main nitrogen fixation taxa were Halothece (Cyanobacteria), Methyloversatilis, and Leptothrix (Betaproteobacteria). Diverse sulfide-oxidizing and sulfate-reducing bacteria, fermenters, denitrifying microbes, methanogens, and methane oxidizers were also found. Putative producers provide organic carbon and nitrogen for the growth of other heterotrophic microbes. In the biogeochemical pathways, assimilation and mineralization of organic compounds were the dominant processes. Besides, a range of metabolic pathways and genes related to high irradiance, low temperature and other stress adaptations were detected, which indicate that the microbial communities had adapted to and could survive in this harsh environment. These results provide a detailed perspective of the microbial functional ecology of the Union Glacier area and improve our understanding of linkages between microbial communities and biogeochemical cycling in high Antarctic ecosystems.
Introduction
The Union Glacier, located in the Ellsworth Mountains of West Antarctica inland, is a large and heavily crevassed glacier (Cordero et al., 2014). Due to the high altitude (around 700 m), low aerosols and high surface albedo, it experiences very high irradiances. It also experiences seasonally high incident ultraviolet (UV) radiation due to seasonal ozone losses (Flemming et al., 2010). The ambient temperature between April and September is around −26 to −28°C and around −6°C in January (Cordero et al., 2014; Rivera et al., 2014). Under the extreme freezing condition, there is almost no liquid-water with most snowfall losses due to direct sublimation. Although climate warming has had a substantial impact on polar regions elsewhere (e.g., the Antarctic Peninsula and West Antarctica) and the cryosphere is rapidly shrinking (Rivera et al., 2010; Boetius et al., 2015), the Union Glacier has remained stable and still has a thickness of 1450 m without any obvious ice velocity changes in recent decades (Rivera et al., 2010, 2014).
Most previous studies of polar environments have focused on the less extreme sub-Antarctic and maritime Antarctic Peninsula locations, which have fundamentally different biomes (Chan et al., 2013). Aquatic microorganisms, for instance, have been found in diverse habitats including Antarctic lakes, ponds and coastal fringes (Chan et al., 2013; Cavicchioli, 2015; Llorens-Marès et al., 2015). In the hyper-arid polar desert of the Dry Valleys, functional traits of specialized microbial communities from distinct lithic and soil niches have been identified (Pointing et al., 2009; Chan et al., 2013). Glaciers and ice sheets, which make up a significant proportion of the earth’s cryosphere, contain unique biomes inhabited by microbial communities of all three domains of life (Boetius et al., 2015; Garcia-Lopez and Cid, 2017). High biodiversities of prokaryotes and eukaryotes from other terrestrial glaciers have already been found (Varin et al., 2012; Garcia-Lopez and Cid, 2017). In addition, the identity and functional capacity of microorganisms from polar coastal glaciers have been defined (Boetius et al., 2015). However, little is known about microbial communities from the high elevation glaciers such as the Union Glacier and almost nothing is known about the stress tolerance mechanisms for climatic extremes in edaphic Antarctic microorganisms (Chan et al., 2013; Barahona et al., 2016). Metagenomic analyses, through the assembly of phylogenetic and biological functional inventories, have made studies in microbial community structure, metabolic potential and ecology (e.g., carbon, nitrogen and sulfur cycling) more straightforward and accurate and allow in situ ecological processes and microbial interactions to be inferred (Lauro et al., 2011; Prosser, 2012; Li et al., 2018).
Here, using a combination of both 16S rRNA gene amplicon and direct metagenomic analyses, the soil microorganisms from mountain surroundings of the Union Glacier were explored. After phylogenetic and functional identification, linkages between microbial community structures and ecological processes for carbon, nitrogen and sulfur cycling were studied in detail. The relative abundance and distribution of marker genes acted as an agent of the potential in situ biogeochemical metabolic pathways. Although the glacier ecosystem experiences extreme climatic conditions, which are usually considered to be hostile to life (Cordero et al., 2014), several metabolic pathways and genes related to stress resistance were analyzed and diverse microbial communities could colonize. Here we reconstruct the ecosystem of the Union Glacier to explore the metabolic potential with respect to carbon, nitrogen and sulfur biogeochemical cycling.
Materials and Methods
Location Description and Sample Collection
Sample sites were located around the Union Glacier (79°45′S, 82°30′W), Antarctica (Figure 1A). A total of 6 samples were obtained from three mountains (namely Rossman Cove, Charles Peak and Elephant Head) during Antarctic summer (Figures 1B–D). Sampling locations, sample descriptions and chemical parameters are given in Table 1. Samples were classified into 4 groups based on their locations and layers, namely CP, EH, GU_S, and GU_D, respectively. All soil and rock samples were stored in sterile tubes (Corning Inc., United States) and then instantly frozen and stored at −80°C (Thermo Fisher Scientific, United States) in the lab until nucleic acid extraction.

Figure 1. Union Glacier site and photos of sampled glacier mountains. (A) Map of Antarctica illustrating the location of Union Glacier. (B) Photograph of the Elephant Head. (C) Photograph of the Charles Peak. (D) Photograph of the Rossman Cove.

Table 1. Summary of the environmental samples from the Union Glacier area.
DNA Extraction and Real-Time Quantitative PCR
Total genomic DNA from each sample was extracted with a PowerSoil® DNA Isolation Kit (MOBIO, United States). The purity and concentration of extracted DNA were checked by agarose gel electrophoresis and micro-spectrophotometer (Nano-100), respectively. Then DNA was prepared for real-time quantitative PCR (qPCR), 16S rRNA gene amplicon and metagenomic sequencing. The qPCR amplifications were performed using LightCycler® 480 II (Roche, Switzerland) following the instruction of SYBR® Premix Ex TaqTM (Takara, Japan). Archaeal 16S rRNA genes were quantified using the primer Arc915R (5′-GTGCTCCCCCGCCAATTCCT-3′) and Arc344F (5′-ACGGGGYGCAGCAGGCGCGA-3′). Analogously, bacterial 16S rRNA genes were quantified using the primer Bac338F (5′-ACTCCTACGGGAGGCAGCA-3′) and Bac806R (5′-GGACTACHVGGGTWTCTAAT-3′) (Li et al., 2018). All standard curves were conducted using a dilution series (between 1 × 102 and 1 × 107 copies/μL) of a known amount of plasmids with cloned partial 16S rRNA genes separately for archaea and bacteria.
16S rRNA Gene Amplicon, Statistical, and Co-occurrence Analysis
Due to low biomass, Archaeal 16S rRNA gene sequencing could not be implemented. Bacterial 16S rRNA genes were amplified using barcoded sequencing primers 338F and 806R. The pooled PCR products were prepared as Illumina MiSeq paired-end libraries and sequenced (Majorbio Bio-Pharm Technology Co., Ltd., China). The obtained raw reads were trimmed to remove primer sequences and barcode sequences using Trimmomatic (Bolger et al., 2014). The trimmed reads were processed following the pipeline of Usearch (Edgar, 2018). Pair-wise reads were merged, then sequence reads from 6 samples were pooled and filtered with a maximum expected error threshold. After standard quality control, remaining reads were assigned to each OTU using the UPARSE-OTU algorithm with a 97% identity (Edgar, 2013). Meanwhile, Chimeric sequences were detected and removed. Obtained reads finally were used to produce an OTUs table. All taxonomic assignments were based on representative OTU sequences. These representative sequences were aligned and annotated through RDP Naive Bayesian rRNA Classifier at an 80% confidence threshold (Wang et al., 2007). The similarity threshold of OTUs assigned to phylum and genera were 100 and 50%, respectively.
Datasets of the OTUs table were normalized for alpha and beta diversity comparisons through the pipeline of qiime 2 (Hall and Beiko, 2018). The OTUs table was transformed into relative abundances for NMDS (non-metric multidimensional scaling) analyses by Primer 6 (Plymouth Marine Laboratory, United Kingdom). Bacterial community compositions and comparisons to relative abundances in different samples were performed by R software. Additionally, potential interactions between microbes were investigated through modeling community structure networks. The correlation network pattern was performed by python and was visualized by Gephi (Bastian et al., 2009). For a valid network analysis, a SparCC correlation coefficient >|0.65| was calculated with statistically significance (P < 0.01) (Barberán et al., 2012).
Metagenomic Assembly and Functional Assignment
Due to insufficient sample size at some sites, metagenomic sequencing was only completed on EH7 soils. After removal of joint sequences by SeqPrep, raw Illumina reads with a length less than 50 bp, with an average base quality of less than 20 and those with ambiguous base “N” were filtered out using Sickle1. Screened paired-end reads were assembled using the IDBA-UD (Peng et al., 2011) algorithm with a kmer size iterated from 67 to 97. Based on the number of contigs (>500 bp), N50/N80 values, longest contig and the total length of the assembly, the kmer size 87 was selected for all further analysis (White et al., 2015). The set of open reading frames (ORFs) was predicted and each ORF was translated into amino acid sequences by Prodigal (Hyatt et al., 2010). Predicted genes were functionally and taxonomically annotated using GhostKOALA (Kanehisa et al., 2016). Functional categorizations of genes were identified through orthology assignment by eggNOG-mapper (Huerta-Cepas et al., 2017). In the metagenomic data, raw reads were matched back to the predicted gene set by Salmon to calculate the abundance of each gene (Patro et al., 2017).
As previously reported (Lauro et al., 2011), in this study carbon, nitrogen and sulfur marker genes were inferred through KEGG pathways and BRITE hierarchies with a few modifications (Supplementary Table S1). Also, the taxonomic identity assigned to each functional gene was determined. The genetic potential of microbial communities in biogeochemical cycles was assessed using a combination of marker genes. Each gene combination (the sum of multiple enzyme abundances in the same conversion step) was summed, which consisted of single or multiple metabolic subprocesses. Different combinations were comparatively analyzed among C, N, and S cycling as agents of the potential in situ relevance of these pathways. Because the marker genes aprA and dsrA could both mediate sulfide oxidation and dissimilatory sulfate reduction, they were assigned to the sulfate reduction or sulfide oxidation step according to phylogeny, i.e., they had a best match to an ortholog from a sulfate-reducing or sulfur-oxidizing clade (Llorens-Marès et al., 2015). Additionally, the functional analyses focused on environmental adaption to conditions found in the vicinity of the glacier (Supplementary Tables S2, S3). Bar-plots and heat-plots were generated using the library “gplots” of R software.
Data Availability Statement
Trimmed 16S rRNA gene reads of all 6 samples and the raw metagenomic reads of EH7 have been deposited in the NCBI Sequence Read Archive (SRA) under the accession number SRP158342 and SRP158636, respectively.
Results
Determination of Bacterial and Archaeal Abundance
Real-time qPCR was performed to estimate the bacterial and archaeal 16S rRNA gene copy numbers that can be considered as an agent for bacterial and archaeal biomass (Table 2). Each mountain site supported both bacterial and archaeal phylotypes. The total number in CP14 (archaea plus bacteria) was 1.88 × 107 16S rRNA gene copies/g soil (wet weight), which was 1–2 orders of magnitude greater than the values for EH7 soils (5.93 × 106 copies/g soil) and GU7 soils (4.68 × 105 copies/g soil). The archaeal 16S rRNA gene copy numbers made up a relatively minor proportion (0.4–8.6%) of the total number of 16S rRNA gene copies in each soil sample. Hence, the microbial populations at the Union Glacier sites were all bacteria-dominated communities.
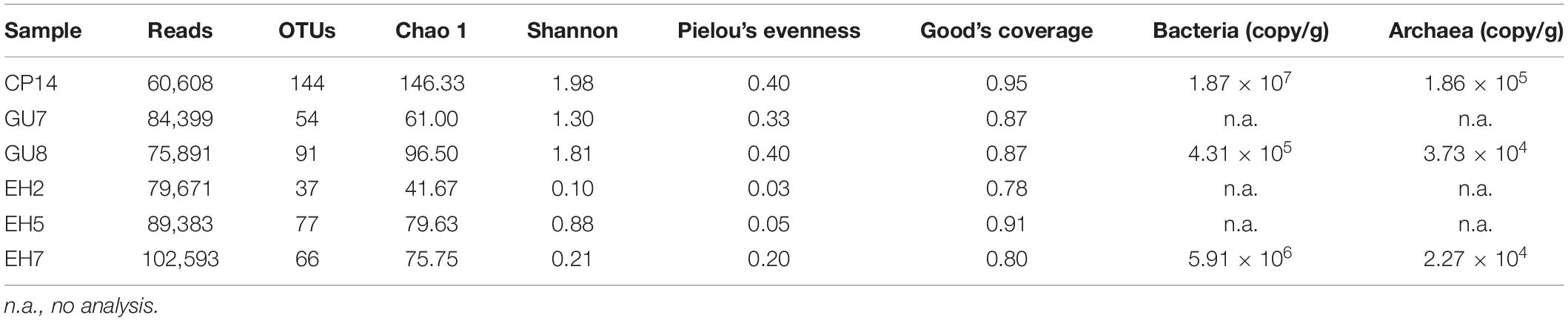
Table 2. Biodiversity indicies of bacterial communities and16S rRNA gene copies from the Union Glacier area.
Taxonomic Structure of Microbial Communities
A total of 492,545 high-quality reads were retained from all samples after filtering. In total, 212 OTUs were assigned with an average length of approximately 420 bp (Table 2). The number of OTUs changed from 37 to 144 across all samples, with the highest OTU numbers in CP14. There were strong differences in Shannon diversity and Pielou’s evenness indicies between samples. Furthermore, the bacterial diversity in Elephant Head sample was significantly lower than that in those from Rossman Cove and Charles Peak.
The 212 OTUs could be grouped into 127 genera, representing 11 different phyla. Proteobacteria were predominant throughout the soil and rock samples (66.0%, on average), therein, the class Gammaproteobacteria dominated. Two other phyla, Firmicutes and Actinobacteria, were also distributed in all samples. Firmicutes showed a much higher abundance in subsurface soils relative to surface soils. The relative abundance of Actinobacteria, ranging from 1.2 to 39.9%, was the highest in GU7. The phylum Bacteroidetes was mainly distributed in surface soils of GU8 (11.7%) but its relative abundance varied greatly. Acidobacteria and Gemmatimonadetes were also found in high abundance across most samples (Figure 2).
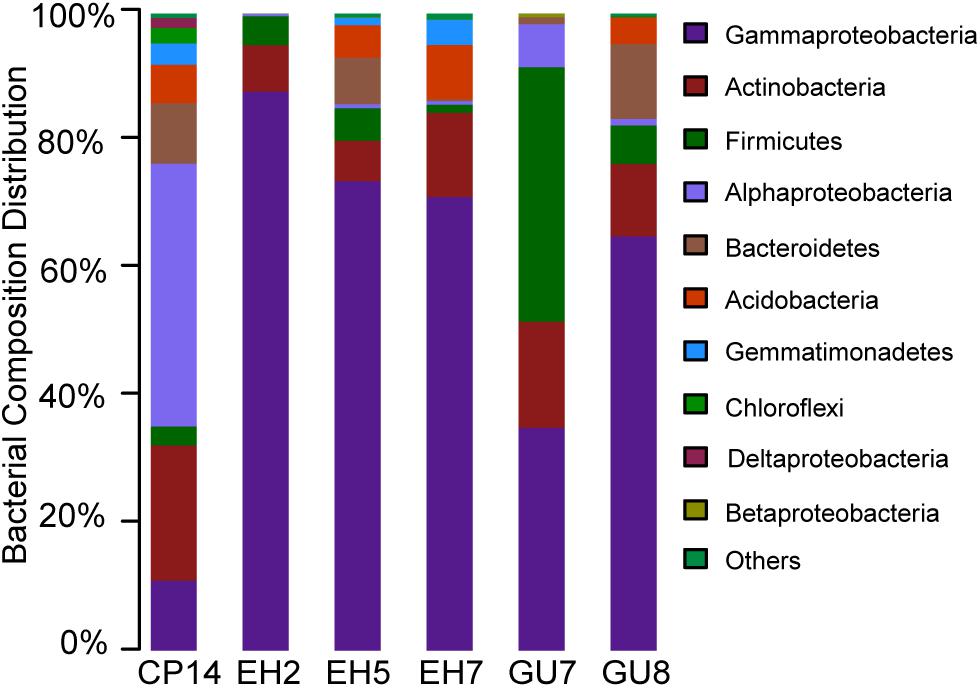
Figure 2. Bacterial composition distributions at the phylum level across all samples in the Union Glacier area. Sequences were assigned in the RDP reference database by using a 80% confidence cut-off.
To identify phylogenetic differences at a finer taxonomic level, bacterial abundance variations were analyzed at genus level (Supplementary Figure S1). Less than 7.9% of all observed genera were present in all samples and their abundances varied greatly (Supplementary Table S4). As most abundant genus, Halomonas was mainly present in the EH group (52.4%, average relative abundance), which may be related to physical and chemical properties (e.g., salinity) of the soil and rock from the Elephant Head region. At the Rossman Cove summit, the genera Halomonas and Lysobacter were abundant in surface soils, accounting for 19.0 and 11.5%, respectively. Anoxybacillus, belonging to Firmicutes, made up 32.1% of the total bacterial communities in the subsurface soils. As a widespread member of the Bacteroidetes, Flavobacterium was abundant in GU8.
Phylogenetic annotations of metagenomic reads were used to assess the overall taxonomic community structure. Bacteria clearly dominated the composition of the microbial communities. More than 91% of all taxonomically assigned metagenomic reads matched bacteria, with a few representatives of archaea (1%) and eukaryotes (8%). The low abundance of archaeal reads was in agreement with the qPCR result where 16S rRNA gene copies of archaeal cells were numerically two orders of magnitude lower than bacterial cells. Within bacterial taxa, Proteobacteria still constituted a major fraction (52.9%), followed by Actinobacteria (11.1%), Acidobacteria (9.7%), Bacteroidetes (9.1%) and Firmicutes (4.9%). The bacterial taxonomic composition, provided by the metagenomic determined functional genes, was in agreement with the 16S rRNA gene amplicon taxonomic profiling of EH7 (Figure 2 and Supplementary Figure S1). Most of the archaeal metagenomic reads matched Euryarchaeota members (80.5%). Methanosarcina was found to be the most abundant with 13.3% of the sequences assigned, followed by Methanocella and Methanothrix (<5%). Additionally, representatives of Crenarchaeota and Thaumarchaeota were also abundant (8.0 and 6.7%). Here, differences in bacterial abundance between amplicon and metagenomic taxonomic profiling were detected. These differences probably arose because an amplicon in complex DNA mixtures can lead to different biases and distort original 16S rRNA gene ratios (Vavourakis et al., 2016).
Beta-Diversity and Network Analyses of Microbial Communities
The unconstrained non-metric MDS analysis was performed on the relative abundance of species in bacterial communities (Figure 3). The six samples divided into four groups, which showed significant community differences and there was a clear difference in community structures between groups (Supplementary Figure S2). At a 40% similarity level, four groups were clustered into three larger groups, which respectively were located in the three mountain sites. The microbial network, which was mainly comprised of two community groups, showed 65 nodes and 57 edges (Figure 4). Most of the positive correlations were detected between members of Proteobacteria, Actinobacteria and Firmicutes. The high abundances of Gp4, Coxiella, Devosia, Faecalibacterium, Gp16, and Ralstonia indicated a high incidence of inter- and intra-phylum correlations. Interestingly, chemoautotrophic Methylobacterium members exhibited interactions with other heterotrophic bacteria. In addition, Flavobacteriales communities had wider positive correlations with members from other phyla, likely illustrating some synergistic heterotrophic microorganisms potentially grew partly due to Flavobacteriia-derived organic products (Bunse and Pinhassi, 2017).
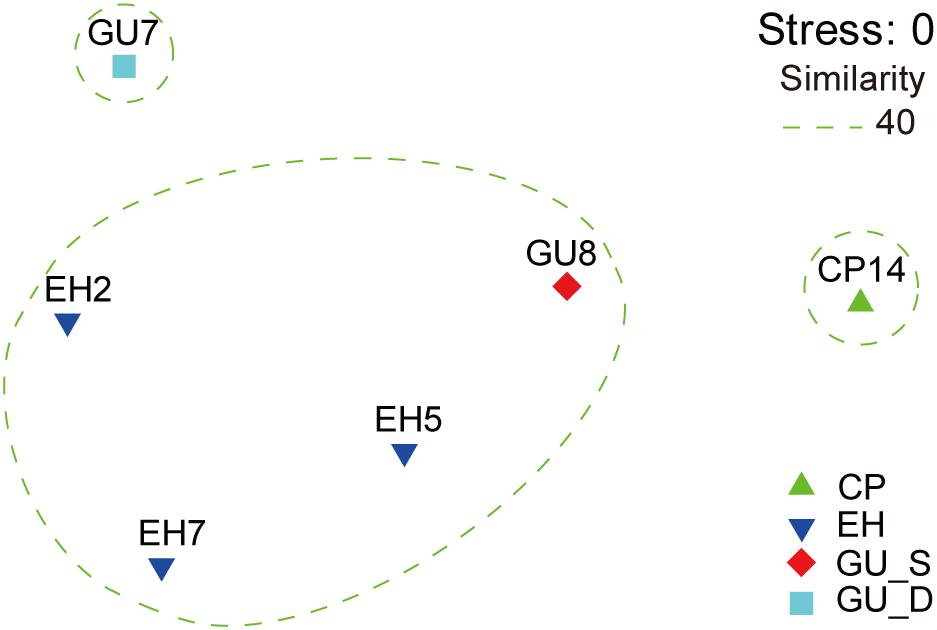
Figure 3. The non-metric multidimensional scaling (NMDS) of community similarities with the relative abundance of OTUs in the Union Glacier area. The ordination was built based on the rank order of bacterial Bray-Curtis similarity. All samples were divided into four groups: CP, EH, GU_S, and GU_D.
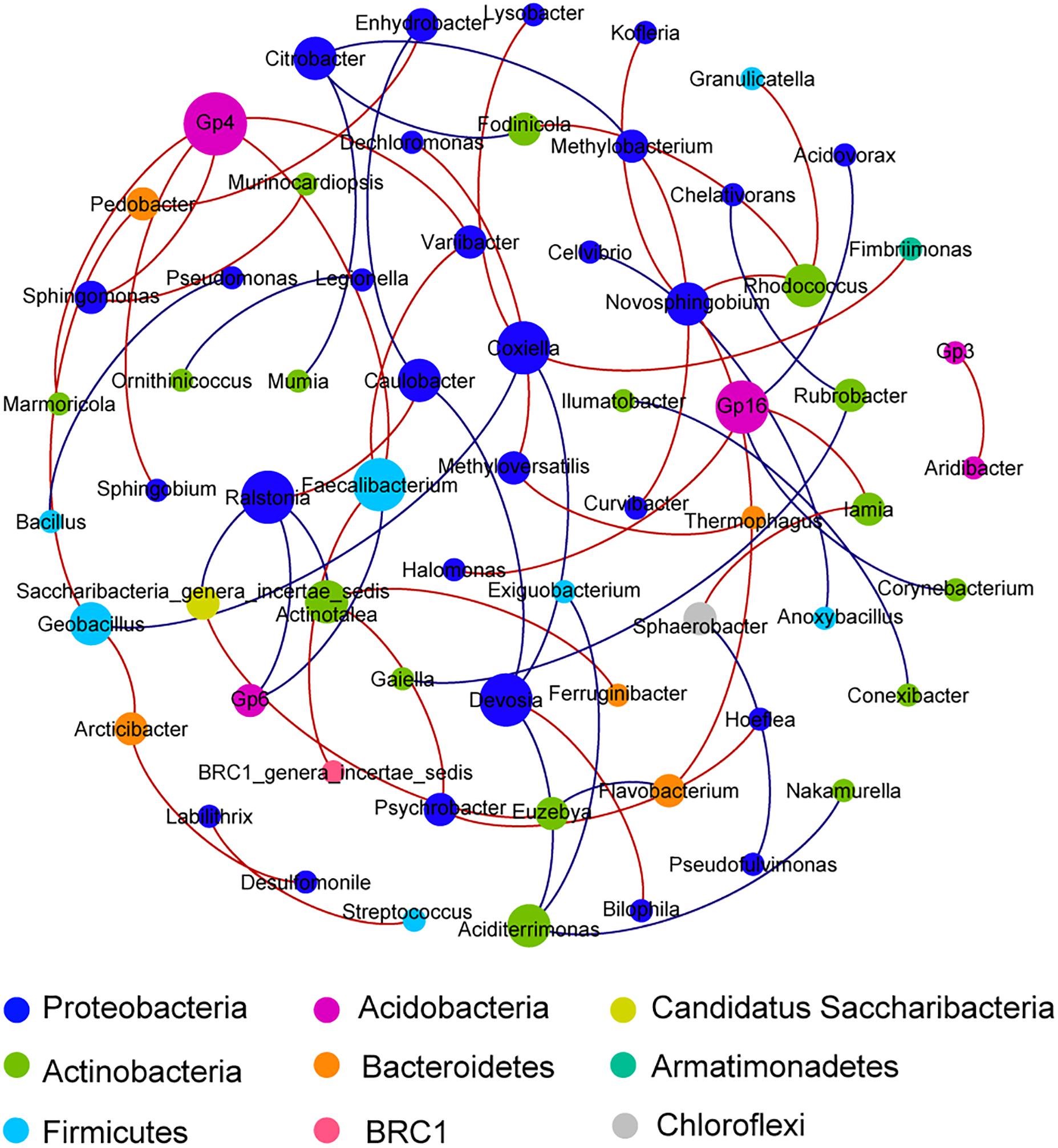
Figure 4. A network of co-occurrence pattern at genus level from all samples. Each line represents a significant correlation between two nodes; the red lines represent positive correlations, while the blue lines represent negative correlations. The size of each node is proportional to the number of connections. Correlations were identified by SparCC’s coefficient >0.65 or <−0.65 and P < 0.01, with each node representing a bacterial genus and the color representing the phylum that the genus is affiliated with.
Functional Structure of the Microbial Communities
The raw metagenomic reads were assembled into 167,519 contigs, which comprised 418,938 predicted genes. Based on the GhostKOALA analysis, 99.9% of metagenomic data were taxonomically assigned to Eukaryota and Prokaryote, while 45.0% could be assigned a KO number and used for subsequent functional analyses. Here, the marker genes of prokaryotes from identified KOs were focused on for a detailed study of carbon, nitrogen and sulfur cycling and stress responses (Supplementary Tables S1–S3). 285,522 (68.2%) of all predicted genes were assigned through an EggNOG database (Supplementary Figure S3). Among the known functional categories, the replication, recombination and repair (L) was the most abundant functional category, with 7.8% relative abundance. The second most abundant functional category was amino acid transport and metabolism (E) with a relative abundance of 7.0%, followed by cell wall/membrane/envelope biogenesis (M) (6.3%) and signal transduction mechanisms (T) (5.6%), respectively.
Carbon Metabolism
The main pathway for the carbon cycle was aerobic respiration, primarily associated with the heterotrophic genus Luteitalea (Acidobacteria) and the genus Bradyrhizobium (Alphaproteobacteria) (Figures 5A,B). In the aerobic carbon fixation, RuBisCO and Phosphoribulokinase (PRK) genes were found and affiliated to phototrophic and chemoautotrophic bacteria. Phototrophic Cyanobacteria communities [Trichormus and Nostoc (Nostocales) and Cyanobium (Synechococcales)] were found and these potentially used light energy to fix CO2 via the Calvin cycle. Thermomonospora, Nitrobacter, and Bradyrhizobium taxa comprised a predominant proportion of potential chemolithotrophic aerobic carbon fixers. By contrast, anaerobic carbon fixation was abundant with different types of pathway, including anoxygenic phototrophy by the Calvin cycle (Chromatiales), probably anaplerotic (Bacteroidales) and reductive citric acid cycle [sulfate-reducing bacteria (SRB)] (Lauro et al., 2011; Llorens-Marès et al., 2015). Another anoxygenic pathway, fermentation, was found to mineralize organic carbon into small molecules, and ultimately to CO2, this was mostly undertaken by the Actinobacteria and Firmicutes communities. However, these organic compounds can be broken into CO through incomplete biotic oxidation. Consistent with this, CO dehydrogenase genes were significantly present (20% of selected marker genes for carbon cycle). The phylogenetic affiliation of these marker genes was primarily to the genus Bradyrhizobium. In addition, both methanogenesis and methane oxidation marker genes were present at low abundances. Methanogenic archaea (e.g., Methanosphaerula and Methanococcus) were detected, while methanogenic bacteria were not found. The genes involved in methane oxidation were affiliated with the genera Methylocystis, Nitrosospira, and Nitrosomonas.
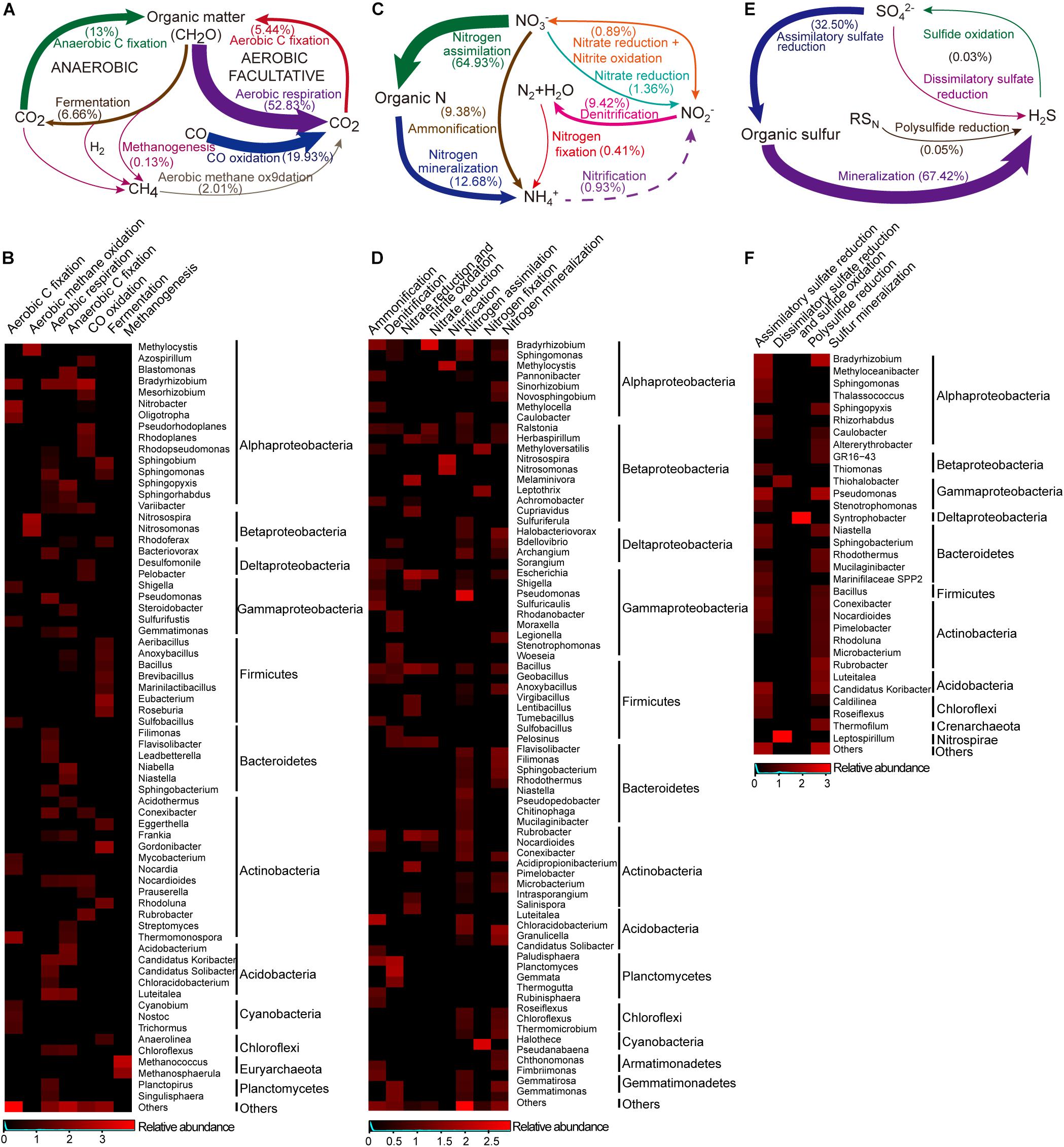
Figure 5. The carbon, nitrogen and sulfur cycles and their major taxa in the Union Glacier area. The genetic potential for each conversion step was estimated using a combination of normalized marker genes (Supplementary Table S1). Arrow size is proportional to the total potential flux (Supplementary Table S6). Heatmap presented the relative abundances of major microbes (only for those that contributed >1% of the marker genes mixture) potentially driving each conversion step. Color bars represent the row-scaled value, in which a blue curve illustrates the distribution density percentage. (A) Carbon cycle and (B) its major taxa. (C) Nitrogen cycle and (D) its major taxa. Dotted lines represent not detected marked genes but putative presence of the pathway. (E) Sulfur cycle and (F) its major taxa.
Nitrogen Metabolism
For the nitrogen cycle, most of detected marker genes were related to nitrogen assimilation, followed by mineralization (Figure 5C). In the pathways related to exogenous ammonia input, high abundances of GS (glutamine synthetase) and gdh (glutamate dehydrogenase) were detected (Supplementary Figure S4). A complete set of denitrification genes [nosZ (nitrous oxide reductase), norB (nitric-oxide reductase) and nirK (nitrite reductase)] were found (9.4%) in the selected nitrogen functional reads. Rhodanobacter and Bradyrhizobium comprised a large proportion of potential denitrifiers (55.8%) (Figures 5D, 6). However, a potentially complete nitrification pathway was also found, in spite of lacking the hao (hydroxylamine dehydrogenase) gene. Aerobic ammonia oxidation made up a low percentage (0.9%) of overall nitrogen pathways. Potential ammonia oxidizers were assigned to Nitrosomonas and Nitrosospira (Nitrosomonadales, ammonia-oxidizing bacteria, AOB) and Methylocysti (Alphaproteobacteria). A low abundance (0.07% of total metagenomic reads) of ammonia-oxidizing archaea (AOA) was found, based on phylogenetic annotation of metagenomic functional reads. However, ammonia-oxidizing bacteria (Nitrobacter-, Nitrosospira-, Nitrosomonas-, and Nitrosococcus-like) and nitrite-oxidizing bacteria (Nitrospirae-like) metagenomic reads were detected at a 10 times higher abundance (1% of total reads). Unlike marker genes for aerobic ammonia oxidation, anammox marker genes coding for hydrazine synthase and dehydrogenase were not found. Additionally, with a low abundance (0.4% of the total selected nitrogen functional pathways), nitrogen fixation genes was associated with Halothece (Cyanobacteria), Methyloversatilis, and Leptothrix (Betaproteobacteria) and Methanococcus (Euryarchaeota) (Figures 5D,6).

Figure 6. Heatmap showing relative abundances for main microbial taxa of selected key genes related to nitrogen metabolism in the Union Glacier area. (A) At the phylum level. (B) At the genus level. Color bars represent the row-scaled value, in which a blue curve illustrates the distribution density percentage.
Sulfur Metabolism
Sulfur mineralization and assimilatory sulfate reduction marker genes were abundant (67.4 and 32.5%) among sulfur metabolic pathways, and 34.7 and 23.8% of marker genes in both pathways potentially originated from heterotrophic Bradyrhizobium and Pseudomonas, respectively (Figures 5E,F). The marker genes associated with dissimilatory sulfate reduction were found and attached to the genus Leptospirillum (Nitrospirae). Sulfide oxidation marker genes were assigned to Thiohalobacter (Gammaproteobacteria). A complete sulfur oxidation (Sox) system, by which thiosulfate was converted to sulfate, was also detected. Based on the metagenomic taxonomic profiling, a large diversity of potential heterotrophic sulfur oxidizers were identified and classified as green sulfur bacteria (GSB), within the order Chlorobiales, photosynthetic sulfur bacteria (PSB), within the family Chromatiaceae, and purple non-sulfur bacteria (PNSB), within the family Rhodobacteraceae (Schütte et al., 2016). Additionally, sulfur-reducing Geobacter was the most abundant of the sulfur reducers, followed by sulfur-reducing Desulfuromonas, sulfate-reducing Desulfobulbus, and sulfate-, sulfite-, and sulfur-reducing Desulfobacteraceae (Schütte et al., 2016). Interestingly, Desulfomonile (Syntrophobacterales), which not only acts as a heterotrophic sulfate reducer but also as a sulfide oxidizer was found (Llorens-Marès et al., 2015). Polysulfide reduction, which is driven by Syntrophobacter (Deltaproteobacteria) was rare.
Adaption to Extreme Environment Stress
Different metabolic pathways which are related to adaptation to Antarctic extreme environments were observed in the metagenomic data (Figure 7). For adaptation to high solar radiation, the complete DNA repair pathway was detected in abundance (1.46% of total prokaryotic gene abundance). Different DNA repair forms, such as base excision repair, nucleotide excision repair, mismatch excision repair, homologous recombination and non-homologous end-joining, were all detected. Superoxide dismutase, catalase, and catalase-peroxidase genes have been found to play important roles in antioxidation. With regard to the adaptation to low temperature, cold-related proteins, cold-adapted enzymes and unsaturated fatty acid biosynthesis were observed in the metagenomic data. The genes related to adaptation to high radiation and low temperature were found in diverse range of microorganisms, indicating that the adaptation to extreme conditions is prevalent in Antarctic mountain ecosystems. Taxa related to acid and osmotic adaptations were also potentially identified (Supplementary Table S5).

Figure 7. Heatmap showing relative abundances for main microbes (only for those that contributed >1% of the marker genes mixture) potentially driving DNA repair, antioxidation and cold adaption to an extremely low temperature in the Union Glacier area. Color bars represent the row-scaled value, in which a blue curve illustrates the distribution density percentage.
Discussion
Though Baeza et al. reported the microfungal diversity in the terrestrial habitats of the Union Glacier regions (Baeza et al., 2017). In this study, we further explored the microbial community structure and their potential functions in high altitude mountain soils of this region. A large variety of putative phototrophic and chemolithotrophic organisms, including sulfide-oxidizing and sulfate-reducing bacteria, fermenters, denitrifiers, methanogens, methane oxidizers, carbon and nitrogen fixers were found. The linkage between important biogeochemical metabolic pathways and their phylogenetic identities revealed how putative functional ecology was driven by these microorganisms.
Low Abundance and Diversity in Microbial Communities
Antarctic soils typically contain low numbers of microbes (Boetius et al., 2015). Interestingly, cell numbers in soils sampled in this study were around 105–107 per gram. Compared with this, biomass in the Antarctic Dry Valleys was about 106–108 cells g–1 soil (Cowan, 2009; Stomeo et al., 2012). Other Antarctica terrestrial niches, such as hypolith, chasmolith, and endolith, have higher biomass (107–109 cells g–1) than soils we sampled (Pointing et al., 2009). Totally 212 OTUs were found in the samples examined herein, which suggested a relatively low biodiversity compared to microbial communities of temperate and tropical environments (Fierer and Jackson, 2006). The Union Glacier area is exposed to high levels of ultraviolet radiation and very low temperatures (Madigan, 2012; Boetius et al., 2015) and these are likely to be responsible for the lower biomass and biodiversity (Pearce et al., 2012).
Bacteria numerically dominated in this soil habitat, as reported in the other cryospheres (sea ice, supraglacial, and subglacial habitats) (Boetius et al., 2015). These bacterial communities harbored primarily Proteobacteria, along with Actinobacteria, Firmicutes, and Bacteroidetes. The similar observation was found in other extreme environments, e.g., Antarctic supraglacial habitats, high Alpine mountains, and Arctic soils (Boetius et al., 2015; Malard and Pearce, 2018). Inconsistently, in the McMurdo Dry Valleys, Acidobacteria, and Actinobacteria dominated soil bacterial communities, which may be related to soil characteristics (Pointing et al., 2009; Niederberger et al., 2019). Gammaproteobacteria constituted a major fraction of Proteobacteria in this study, which also were often found in Arctic and Antarctic sea ice (Boetius et al., 2015). However, high Alpine snow microbes were dominated by Alphaproteobacteria and Betaproteobacteria (Wunderlin et al., 2016; Azzoni et al., 2018). In this study, occurrence of Cyanobacteria and Chloroflexi members in soils indicated a true soil community may indeed exist in this barren region (Pointing et al., 2009). Flavobacteriales members were found to degrade high molecular weight organic matter into smaller molecules for the benefit of other heterotrophic microorganisms (Figure 4; Bunse and Pinhassi, 2017). These interactions between trophic microbes, resulting in the transformation of organic matter, could be likely to contribute to the formation of a complete ecosystem in this area.
Carbon and Nitrogen Fixation by Autotrophic Microorganisms
In this study, Cyanobacteria, an important primary producer in different niches of cryosphere (Boetius et al., 2015), was found to participate in aerobic carbon fixation. Some Cyanobacteria members have the capacity to upregulate carbon concentrating mechanisms (CCM) to enhance the acquisition of inorganic carbon (CO2 or bicarbonate) (Price, 2011). Carboxysomes use the paradigm of metabolic channeling to enhance the local CO2 concentrations and increase the efficiency of RuBisCO (Lin et al., 2014). In this study, four genes related to carboxysome structure were identified. Additionally, a nitrifier, Nitrobacter, which uses energy from the oxidation of nitrite to fix CO2 via the Calvin cycle (Fiencke et al., 2005), was observed in this study. Another chemolitoautotrophic nitrifier, Nitrolancea (Chloroflexi), which participates in aerobic carbon fixation and interacts with other bacteria, was identified in the network analysis (Figure 4). As reported previously, these photoautotrophs and chemoautotrophs functioned during sunlight or dark conditions and are the basis for complex food webs in extreme environments where plants barely exist (Boetius et al., 2015; Malard and Pearce, 2018).
The reverse tricarboxylic acid (rTCA) cycle and anaerobic C1-pathway have been reported to be the main pathways for anaerobic carbon fixation in the hydrothermal chimney (He et al., 2015). In this study, a high percentage of anaerobic carbon fixation compared to aerobic carbon fixation was observed. The most likely organisms responsible for chemotrophic CO2 fixation were Chromatiales, sulfate-reducing bacteria (SRB) and Chlorobiales (GSB), which were also found in Antarctic Ace Lake (Cavicchioli, 2015). Desulfobacterales, which were found here, undertake formate assimilation and CO2 fixation through anaerobic C1-pathway (Fuchs, 2011). Sulfate-reducing bacteria (e.g., Pelobacter and Desulfomonile) were detected herein, which have the potential to fix carbon and degrade a wide variety of carbon compounds (Llorens-Marès et al., 2015). Therefore, in combination with sulfur and nitrogen metabolism, the data presented here suggests that inorganic carbon fixation provided a carbon source and energy for the survival of these polar microbial populations.
A key process in nitrogen cycling is nitrogen fixation, which were found to be primarily driven by diazotrophic cyanobacterial taxa in this study. Cyanobacteria from Antarctic hypolithic subsurface soils have also been found to be the dominant contributors to nitrogen fixation (Cowan et al., 2011). Allen et al. (2009) further reported that methanogens, such as M. burtonii, could benefit from nitrogen fixation. Consistently, nif genes of the methanogen, Methanococcus, were detected in the metagenomic data reported herein. Hence, evidence for symbiotic relationships between diazotrophic archaea and methanogens was tentatively found, as reported previously (Lauro et al., 2011). Methyloversatilis (Nitrosomonadales) was detected among diazotrophic taxa herein, although it commonly acted as a methylotroph in the sediments (Kumaresan et al., 2018). It was reported that Methylotrophs can function in methane oxidation and carbon fixation through the C1-pathway (Kumaresan et al., 2018). This result suggests that the methylotroph Methyloversatilis could provide nitrogen and carbon for other microbes, this was demonstrated here in the interaction network analyses (Figure 4). Bradyrhizobium, from which a large number of denitrification genes were found in this study, has been identified as a chemolithoautotroph that can fix atmospheric nitrogen to form ammonia or ammonium (Hennecke, 1990).
Other Metabolic Processes for Carbon, Nitrogen, and Sulfur
The incomplete biological oxidation of organic carbon compounds can lead to CO production. Several studies have found that the use of CO as an alternative energy and carbon source is widespread among heterotrophs (Cunliffe, 2010). For example, the heterotrophic species, Halobacteria, benefits from metabolizing CO in surface waters (Vavourakis et al., 2016). In this study, CO oxidation accounted for a large proportion (19.9%) in selected carbon pathways (Figure 5A and Supplementary Table S6). Interestingly, 2.9% of CO dehydrogenase genes were related to Sulfate-reducing bacteria Pelobacter and Desulfomonile. This may be because CO oxidation was probably coupled to sulfate reduction or CO2 reduction to produce energy-yielding substrates such as sulfide, H2 and acetate (Llorens-Marès et al., 2015). It was also found that only a low proportion of genes (0.1%) were associated with methanogenic archaea. Aerobic methane oxidation was also rare. Surprisingly, typical ammonia oxidizers, such as Nitrosospira and Nitrosomonas, were found to participate in methane oxidation. Fiencke et al. (2005) have suggested that Nitrosomonas can use energy gained from the oxidation of ammonia to fix CO2. This illustrates that CO2 fixation, methane oxidation and ammonia oxidation may function together in polar high altitude soils.
In this study most of the genetic nitrogen pathways were associated with nitrogen assimilation and remineralization. Glutamine synthetase, glutamate synthase and glutamate dehydrogenase were detected herein and have been recorded through the metaproteome in Antarctic Ace Lake, which implied that active nitrogen assimilation and remineralization happened (Lauro et al., 2011). The rate of nitrification was restricted by the first step, i.e., oxidation of ammonia to nitrite, which was driven by AOA and AOB. A low abundance of ammonia oxidizers (both AOA and AOB) was found in the metagenomic pool. Additionally, a significantly higher abundance of AOB with respect to AOA was found. Unlike known ammonia-oxidizing bacteria, several ammonia-oxidizing archaea were also reported that could potentially sustain high specific oxidation rates under extreme oligotrophic ammonium limitation (Martens-Habbena et al., 2009). The high affinity of AOA for oligotrophic ammonium could compensate for their low abundance and further illustrates that AOA could potentially compete with heterotrophic bacteria in this extreme environment.
The main turnover of sulfur compounds occurred within the sulfur mineralization process, suggesting that numerous heterotrophic microorganisms potentially degraded organic sulfur compounds into small inorganic molecules. Thiohalobacter was detected to probably oxidize sulfide in the polar mountain soils examined herein, which agreed with the research that the Thiohalobacter group was a moderately halophilic, sulfur-oxidizing community and some species of this could degrade thiocyanate aerobically (Sorokin et al., 2010). Genes for dissimilatory sulfate reduction were present in the genus Leptospirillum. Previous studies have shown that the Leptospirillum group is typical acidophilic and iron-oxidizing communities, which play a key role in bioleaching and biooxidation (Smith and Johnson, 2018). Also, Leptospirillum is strictly chemolithoautotrophic and able to fix carbon using ferrous iron as its electron donor and oxygen as the electron acceptor (Mi et al., 2011). Some phototrophic microorganisms, such as the GSB and PSB groups, which are potential sulfur oxidizers, were found here. It has been suggested that PSB can oxidize a variety of reduced sulfur species (e.g., sulfide, thiosulfate, and elemental sulfur) (Schütte et al., 2016). Similarly, GSB typically oxidize sulfide and thiosulfate but has lost essential genes for elemental sulfur oxidation (Gregersen et al., 2011). Both PSB and GSB carry out anoxygenic photosynthesis using reduced sulfur, implying that they were able to coordinate the sulfur and carbon cycles (Llorens-Marès et al., 2015).
Extreme Environments Adaptation
Some microorganisms have adapted to living in environments exposed to high levels of solar irradiation by a combination of mechanisms, e.g., efficient DNA repair, high resistance to oxygen-free radicals through production of superoxide dismutase and other special survival modes (Martín-Cerezo et al., 2015). In this study, several complete metabolic pathways of DNA repair and antioxidation genes were detected in a range of microorganisms. These phyla were common and occurred generally in different niches (Chan et al., 2013). Extreme low temperatures, such as experienced in the Union Glacier area, is one of the most challenging stresses for life on Earth (Garcia-Lopez and Cid, 2017). However, microorganisms have developed complex metabolic strategies for survival at low temperatures (Madigan, 2012; Boetius et al., 2015). Genes for cold adaptions were found in this study. In addition, microorganisms from the Union Glacier area were expected to synthesize glycine/betaine/proline compounds, osmoprotectants and various osmotic proteins to improve their adaption at high salinity. These taxa mainly included Proteobacteria and Actinobacteria, which was also found in Antarctic rocky substrates (Chan et al., 2013). Sometimes, unique microorganisms (e.g., xerophiles and acidophiles) can tolerate desiccation and extremely acidic conditions (Imshenetsky et al., 1973; Cid et al., 2010). Here, it is clearly shown that the microbial communities had abundant and diverse stress response pathways. The similar observations have been detected for osmotic, radiation, desiccation and cold stress from soils, hypoliths, chasmoendoliths and cryptoendolith in Dry Valley niches (Chan et al., 2013). These genes and pathways would have equipped the communities with a diverse potential “arsenal” of responses to environmental extremes allowing them to survive in this very inhospitable terrain.
Conclusion
The metagenomic analysis demonstrated the relationships between microbial communities and biogeochemical cycling in this Antarctic mountain soils. A low biomass and diversity was observed, probably due to the severe local environment. Although archaea were detected, bacteria dominated. A diverse range of producers were found and the ecosystem was reconstructed based on analyses of carbon, nitrogen and sulfur metabolic pathways. Carbon fixation taxa were comprised of photoautotrophs (Cyanobacteria) and chemoautotrophs (mainly Alphaproteobacterial clades: Bradyrhizobium, Sphingopyxis, and Nitrobacter). The main nitrogen fixation taxa were Halothece (Cyanobacteria), Methyloversatilis, and Leptothrix (Betaproteobacteria). These putative producers would have been able to provide organic carbon and nitrogen and interact with other heterotrophic microbes. In addition, a number of complete metabolic pathways and genes associated with high radiation, low temperature and other stress adaptations were detected. These indicate that the microbial communities had adapted to this harsh environment and could survive. Next, together with in situ geochemical parameter measurements, more efforts will be spent on metatranscriptome and metaproteome analyses, which could further confirm and estimate metabolic potentials of the microbial community at an active level.
Data Availability Statement
The datasets generated for this study can be found in the NCBI Sequence Read Archive, SRP158342 and SRP158636.
Author Contributions
YL, Q-QC, and Y-RD performed the laboratory work. GE and JB collected and processed the environmental samples to obtain the genetic material and took the landscape photographies shown in Figure 1. YL wrote the manuscript. Q-LQ helped in data analysis. Q-LQ, X-LC, MW, and AM helped to revise the manuscript. Y-ZZ and JB designed the research.
Funding
This work was supported by the National Key R&D Program of China (2018YFC1406703 and 2018YFC1406702), Chilean Antarctic Institute (INACH), Fundacion Biociencia, the National Natural Science Foundation of China (U1706207, 31630012, 91851205, and 31870101), the Program of Shandong for Taishan Scholars (tspd20181203), AoShan Talents Cultivation Program supported by Qingdao National Laboratory for Marine Science and Technology (2017ASTCP-OS14), Young Scholars Program of Shandong University (2016WLJH36), and the Key Research Project of Shandong Province (2017GSF21106).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02408/full#supplementary-material
Footnotes
References
Allen, M. A., Lauro, F. M., Williams, T. J., Burg, D., Siddiqui, K. S., De Francisci, D., et al. (2009). The genome sequence of the psychrophilic archaeon, Methanococcoides burtonii: the role of genome evolution in cold adaptation. ISME J. 3, 1012–1035. doi: 10.1038/ismej.2009.45
Azzoni, R. S., Tagliaferri, I., Franzetti, A., Mayer, C., Lambrecht, A., Compostella, C., et al. (2018). Bacterial diversity in snow from mid-latitude mountain areas: alps, Eastern Anatolia, Karakoram and Himalaya. Ann. Glaciol. 59, 10–20. doi: 10.1017/aog.2018.18
Baeza, M., Barahona, S., Alcaíno, J., and Cifuentes, V. (2017). Amplicon-metagenomic analysis of fungi from antarctic terrestrial habitats. Front. Microbiol. 8:2235. doi: 10.3389/fmicb.2017.02235
Barahona, S., Yuivar, Y., Socias, G., Alcaíno, J., Cifuentes, V., and Baeza, M. (2016). Identification and characterization of yeasts isolated from sedimentary rocks of Union Glacier at the Antarctica. Extremophiles 20, 479–491. doi: 10.1007/s00792-016-0838-6
Barberán, A., Bates, S. T., Casamayor, E. O., and Fierer, N. (2012). Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 6, 343–351. doi: 10.1038/ismej.2011.119
Bastian, M., Heymann, S., and Jacomy, M. (2009). “Gephi: an Open Source Software for Exploring and Manipulating Networks,” in Proceedings of the Third International Conference on Weblogs and Social Media, ICWSM 2009, San Jose, CA.
Boetius, A., Anesio, A. M., Deming, J. W., Mikucki, J. A., and Rapp, J. Z. (2015). Microbial ecology of the cryosphere: sea ice and glacial habitats. Nat. Rev. Microbiol. 13, 677–690. doi: 10.1038/nrmicro3522
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bunse, C., and Pinhassi, J. (2017). Marine bacterioplankton seasonal succession dynamics. Trends Microbiol. 25, 494–505. doi: 10.1016/j.tim.2016.12.013
Cavicchioli, R. (2015). Microbial ecology of Antarctic aquatic systems. Nat. Rev. Microbiol. 13, 691–706. doi: 10.1038/nrmicro3549
Chan, Y., Van Nostrand, J. D., Zhou, J., Pointing, S. B., and Farrell, R. L. (2013). Functional ecology of an Antarctic Dry Valley. Proc. Natl. Acad. Sci. U.S.A. 110, 8990–8995. doi: 10.1073/pnas.1300643110
Cid, C., Garcia-Descalzo, L., Casado-Lafuente, V., Amils, R., and Aguilera, A. (2010). Proteomic analysis of the response of an acidophilic strain of Chlamydomonas sp. (Chlorophyta) to natural metal-rich water. Proteomics 10, 2026–2036. doi: 10.1002/pmic.200900592
Cordero, R. R., Damiani, A., Ferrer, J., Jorquera, J., Tobar, M., Labbe, F., et al. (2014). UV Irradiance and Albedo at Union Glacier Camp (Antarctica): a case study. PLoS One 9:e90705. doi: 10.1371/journal.pone.0090705
Cowan, D. A. (2009). Cryptic microbial communities in Antarctic deserts. Proc. Natl. Acad. Sci. U.S.A. 106, 19749–19750. doi: 10.1073/pnas.0911628106
Cowan, D. A., Sohm, J. A., Makhalanyane, T. P., Capone, D. G., Green, T. G. A., Cary, S. C., et al. (2011). Hypolithic communities: important nitrogen sources in Antarctic desert soils. Environ. Microbiol. Rep. 3, 581–586. doi: 10.1111/j.1758-2229.2011.00266.x
Cunliffe, M. (2010). Correlating carbon monoxide oxidation with cox genes in the abundant marine roseobacter clade. ISME J. 5, 685–691. doi: 10.1038/ismej.2010.170
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C. (2018). Accuracy of taxonomy prediction for 16S rRNA and fungal ITS sequences. PeerJ 6:e4652. doi: 10.7717/peerj.4652
Fiencke, C., Spieck, E., and Bock, E. (2005). “Nitrifying Bacteria,” in Nitrogen Fixation in Agriculture, Forestry, Ecology, and the Environment, eds D. Werner and W. E. Newton, (Dordrecht: Springer Netherlands), 255–276.
Fierer, N., and Jackson, R. B. (2006). The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. U.S.A. 103, 626–631. doi: 10.1073/pnas.0507535103
Flemming, J., Inness, A., Jones, L., Eskes, H., Huijnen, V., Schultz, M. G., et al. (2010). Forecasts and Assimilation Experiments of the Antarctic Ozone Hole 2008. Reading: ECMWF.
Fuchs, G. (2011). Alternative pathways of carbon dioxide fixation: insights into the early evolution of life? Annu. Rev. Microbiol. 65, 631–658. doi: 10.1146/annurev-micro-090110-102801
Garcia-Lopez, E., and Cid, C. (2017). Glaciers and ice sheets as analog environments of potentially habitable icy worlds. Front. Microbiol. 8:1407. doi: 10.3389/fmicb.2017.01407
Gregersen, L., Bryant, D., and Frigaard, N.-U. (2011). Mechanisms and evolution of oxidative sulfur metabolism in green sulfur bacteria. Front. Microbiol. 2:116. doi: 10.3389/fmicb.2011.00116
Hall, M., and Beiko, R. G. (2018). “16S rRNA Gene Analysis with QIIME2,” in Microbiome Analysis: Methods and Protocols, eds R. G. Beiko, W. Hsiao, and J. Parkinson, (New York, NY: Springer), 113–129. doi: 10.1007/978-1-4939-8728-3_8
He, Y., Feng, X., Fang, J., Zhang, Y., and Xiao, X. (2015). Metagenome and metatranscriptome revealed a highly active and intensive sulfur cycle in an oil-immersed hydrothermal chimney in guaymas basin. Front. Microbiol. 6:1236. doi: 10.3389/fmicb.2015.01236
Hennecke, H. (1990). Nitrogen fixation genes involved in the Bradyrhizobium japonicum-soybean symbiosis. FEBS Lett. 268, 422–426. doi: 10.1016/0014-5793(90)81297-2
Huerta-Cepas, J., Forslund, K., Szklarczyk, D., Jensen, L. J., von Mering, C., and Bork, P. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Hyatt, D., Chen, G.-L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Imshenetsky, A. A., Kouzyurina, L. A., and Jakshina, V. M. (1973). On the multiplication of xerophilic micro-organisms under simulated Martian conditions. Life Sci. Space Res. 11, 63–66.
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kumaresan, D., Stephenson, J., Doxey, A. C., Bandukwala, H., Brooks, E., Hillebrand-Voiculescu, A., et al. (2018). Aerobic proteobacterial methylotrophs in Movile Cave: genomic and metagenomic analyses. Microbiome 6:1. doi: 10.1186/s40168-017-0383-2
Lauro, F. M., DeMaere, M. Z., Yau, S., Brown, M. V., Ng, C., Wilkins, D., et al. (2011). An integrative study of a meromictic lake ecosystem in Antarctica. ISME J. 5, 879–895. doi: 10.1038/ismej.2010.185
Li, Y., Jing, H., Xia, X., Cheung, S., Suzuki, K., and Liu, H. (2018). Metagenomic insights into the microbial community and nutrient cycling in the western subarctic pacific ocean. Front. Microbiol. 9:623. doi: 10.3389/fmicb.2018.00623
Lin, M. T., Occhialini, A., Andralojc, P. J., Parry, M. A. J., and Hanson, M. R. (2014). A faster rubisco with potential to increase photosynthesis in crops. Nature 513, 547–550. doi: 10.1038/nature13776
Llorens-Marès, T., Yooseph, S., Goll, J., Hoffman, J., Vila-Costa, M., Borrego, C. M., et al. (2015). Connecting biodiversity and potential functional role in modern euxinic environments by microbial metagenomics. ISME J. 9, 1648–1661. doi: 10.1038/ismej.2014.254
Madigan, M. (2012). Brock Biology of Microorganisms, 13th Edn. Upper Saddle River, NJ: Benjamin Cummings, 271–282.
Malard, L. A., and Pearce, D. A. (2018). Microbial diversity and biogeography in Arctic soils. Environ. Microbiol. Rep. 10, 611–625. doi: 10.1111/1758-2229.12680
Martens-Habbena, W., Berube, P. M., Urakawa, H., de la Torre, J. R., and Stahl, D. A. (2009). Ammonia oxidation kinetics determine niche separation of nitrifying Archaea and Bacteria. Nature 461, 976–979. doi: 10.1038/nature08465
Martín-Cerezo, M. L., García-López, E., and Cid, C. (2015). Isolation and identification of a red pigment from the Antarctic bacterium Shewanella frigidimarina. Protein Peptide Lett. 22, 1076–1082. doi: 10.2174/0929866522666150915122247
Mi, S., Song, J., Lin, J., Che, Y., Zheng, H., and Lin, J. (2011). Complete genome of Leptospirillum ferriphilum ML-04 provides insight into its physiology and environmental adaptation. J. Microbiol. 49, 890–901. doi: 10.1007/s12275-011-1099-9
Niederberger, T. D., Bottos, E. M., Sohm, J. A., Gunderson, T., Parker, A., Coyne, K. J., et al. (2019). Rapid microbial dynamics in response to an induced wetting event in antarctic dry valley soils. Front. Microbiol. 10:621. doi: 10.3389/fmicb.2019.00621
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. doi: 10.1038/nmeth.4197
Pearce, D., Newsham, K., Thorne, M., Calvo-Bado, L., Krsek, M., Laskaris, P., et al. (2012). Metagenomic analysis of a southern maritime antarctic soil. Front. Microbiol. 3:403. doi: 10.3389/fmicb.2012.00403
Peng, Y., Leung, H. C. M., Yiu, S. M., and Chin, F. Y. L. (2011). Meta-IDBA: a de Novo assembler for metagenomic data. Bioinformatics 27, 94–101. doi: 10.1093/bioinformatics/btr216
Pointing, S. B., Chan, Y., Lacap, D. C., Lau, M. C. Y., Jurgens, J. A., and Farrell, R. L. (2009). Highly specialized microbial diversity in hyper-arid polar desert. Proc. Natl. Acad. Sci. U.S.A. 106, 19964–19969. doi: 10.1073/pnas.0908274106
Price, G. D. (2011). Inorganic carbon transporters of the cyanobacterial CO2 concentrating mechanism. Photosynth. Res. 109, 47–57. doi: 10.1007/s11120-010-9608-y
Prosser, J. I. (2012). Ecosystem processes and interactions in a morass of diversity. FEMS Microbiol. Ecol. 81, 507–519. doi: 10.1111/j.1574-6941.2012.01435.x
Rivera, A., Zamora, R., Rada, C., Walton, J., and Proctor, S. (2010). Glaciological investigations on union glacier, ellsworth mountains, West Antarctica. Ann. Glaciol. 51, 91–96. doi: 10.3189/172756410791392772
Rivera, A., Zamora, R., Uribe, J. A., Jaña, R., and Oberreuter, J. (2014). Recent ice dynamic and surface mass balance of Union Glacier in the West Antarctic Ice Sheet. Cryosphere 8, 1445–1456. doi: 10.5194/tc-8-1445-2014
Schütte, U. M. E., Cadieux, S. B., Hemmerich, C., Pratt, L. M., and White, J. R. (2016). Unanticipated geochemical and microbial community structure under seasonal ice cover in a dilute, dimictic arctic lake. Front. Microbiol. 7:1035. doi: 10.3389/fmicb.2016.01035
Smith, S. L., and Johnson, D. B. (2018). Growth of Leptospirillum ferriphilum in sulfur medium in co-culture with Acidithiobacillus caldus. Extremophiles 22, 327–333. doi: 10.1007/s00792-018-1001-3
Sorokin, D. Y., Kovaleva, O. L., Tourova, T. P., and Muyzer, G. (2010). Thiohalobacter thiocyanaticus gen. nov., sp. nov., a moderately halophilic, sulfur-oxidizing gammaproteobacterium from hypersaline lakes, that utilizes thiocyanate. Int. J. Syst. Evol. Microbiol. 60, 444–450. doi: 10.1099/ijs.0.012880-0
Stomeo, F., Makhalanyane, T. P., Valverde, A., Pointing, S. B., Stevens, M. I., Cary, C. S., et al. (2012). Abiotic factors influence microbial diversity in permanently cold soil horizons of a maritime-associated Antarctic Dry Valley. FEMS Microbiol. Ecol. 82, 326–340. doi: 10.1111/j.1574-6941.2012.01360.x
Varin, T., Lovejoy, C., Jungblut, A. D., Vincent, W. F., and Corbeil, J. (2012). Metagenomic analysis of stress genes in microbial mat communities from antarctica and the high arctic. Appl. Environ. Microbiol. 78, 549–559. doi: 10.1128/AEM.06354-11
Vavourakis, C. D., Ghai, R., Rodriguez-Valera, F., Sorokin, D. Y., Tringe, S. G., Hugenholtz, P., et al. (2016). Metagenomic insights into the uncultured diversity and physiology of microbes in four hypersaline soda lake brines. Front. Microbiol. 7:211. doi: 10.3389/fmicb.2016.00211
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
White, R. III, Power, I., Dipple, G., Southam, G., and Suttle, C. (2015). Metagenomic analysis reveals that modern microbialites and polar microbial mats have similar taxonomic and functional potential. Front. Microbiol. 6:966. doi: 10.3389/fmicb.2015.00966
Keywords: microorganism community, biogeochemical cycling, extreme environmental adaption, metagenomic analysis, antarctic glacier
Citation: Li Y, Cha Q-Q, Dang Y-R, Chen X-L, Wang M, McMinn A, Espina G, Zhang Y-Z, Blamey JM and Qin Q-L (2019) Reconstruction of the Functional Ecosystem in the High Light, Low Temperature Union Glacier Region, Antarctica. Front. Microbiol. 10:2408. doi: 10.3389/fmicb.2019.02408
Received: 08 March 2019; Accepted: 07 October 2019;
Published: 18 October 2019.
Edited by:
Thulani Peter Makhalanyane, University of Pretoria, South AfricaReviewed by:
Marc Warwick Van Goethem, Lawrence Berkeley National Laboratory, United StatesAnne D. Jungblut, Natural History Museum, United Kingdom
Copyright © 2019 Li, Cha, Dang, Chen, Wang, McMinn, Espina, Zhang, Blamey and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jenny M. Blamey, amJsYW1leUBiaW9zY2llbmNlLmNs; Qi-Long Qin, cWlucWlsb25nQHNkdS5lZHUuY24=