 Hai-Wei Lou1,2
Hai-Wei Lou1,2 Jun-Fang Lin
Jun-Fang Lin Li-Qiong Guo
Li-Qiong Guo- 1Department of Bioengineering, College of Food Science, South China Agricultural University, Guangzhou, China
- 2College of Food Science and Technology, Henan University of Technology, Zhengzhou, China
- 3Research Center for Micro-Ecological Agent Engineering and Technology of Guangdong Province, Guangzhou, China
Cordyceps militaris, a valuable edible and medicinal fungus, has attracted increasing attention because of its various bioactive ingredients. However, the biosynthetic pathway of C. militaris carotenoids is still unknown due to lack of transcriptome information. To uncover genes related to the biosynthesis of C. militaris carotenoids, the transcriptomes of mycelia CM10_D cultured under dark conditions and mycelia CM10_L cultured under light exposure conditions were sequenced. Compared with mycelia CM10_D, 866 up-regulated genes and 856 down-regulated genes were found in mycelia CM10_L. Gene ontology (GO) analysis of differentially expressed genes (DEGs) indicated that DEGs were mainly classified into the “metabolic process,” “membrane,” and “catalytic activity” terms. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of DEGs suggested that DEGs were mainly enriched in “metabolic pathways,” “MAPK signaling pathway-yeast,” and “biosynthesis of secondary metabolites.” In addition, the carotenoid content of the Cmtns gene deletion mutant (ΔCmtns) was significantly lower than that of the wild-type C. militaris CM10, while the carotenoid content of the complementary strain (ΔCmtns-c) of the Cmtns gene was not significantly different from that of C. militaris CM10, suggesting that the Cmtns gene significantly affected the biosynthesis of carotenoids in C. militaris. These results potentially pave the way for revealing the biosynthetic pathway of carotenoids and improving carotenoids production in C. militaris.
Introduction
Cordyceps militaris, as a model species of Cordyceps fungi, is widely used as a nourishing food and used in traditional Chinese medicine in East Asia (Zhang et al., 2018; Kunhorm et al., 2019). C. militaris is also often used as a substitute for Ophiocordyceps sinensis because it contains similar bioactive components (including cordycepic acid, polysaccharides, adenosine, ergosterol, etc.) (Zhu et al., 2016; Wang et al., 2017; Yin et al., 2018). It is noteworthy that C. militaris contains cordycepin and pentostatin which O. sinensis does not contain (Xia et al., 2017). These bioactive ingredients endow C. militaris with anti-inflammatory, anti-tumor, anti-cancer, anti-oxidation, and hypoglycemic activities (Zhang et al., 2006; Lee et al., 2015; Kim et al., 2017; Hu et al., 2019; Liu et al., 2019). Based on this, the market demand of C. militaris is increasing year by year.
Fungi often produce a variety of pigments that give them different color phenotypes (Lu et al., 2013). Similarly, C. militaris produces pigments that make it yellow or orange (Yang et al., 2016). The pigments contained in C. militaris were first considered to be carotenoids and water-soluble (Friederichsen and Engel, 1958). In 2013, four novel water-soluble carotenoids were isolated and identified from C. militaris (Dong et al., 2013b). Traditional carotenoids are fat-soluble pigments and insoluble in water, while C. militaris carotenoids can be dissolved in water, which will expand the application field of C. militaris. However, the content of carotenoids in C. militaris is low, which cannot meet the market demand. Up to now, the biosynthetic pathway of C. militaris carotenoids is still unknown, and there are few reports on the related genes of carotenoid biosynthesis. Therefore, it is urgent to study the biosynthetic pathway and related genes of C. militaris carotenoids in order to improve the content of carotenoids by genetic engineering.
Light is an essential environmental factor for the production of carotenoids in C. militaris. After exposure to light, the colony color of C. militaris changes from white to yellow or orange (Yang et al., 2016; Sun et al., 2017; Zhang et al., 2017). This indicates that light can induce the expression of genes synthesizing C. militaris carotenoids. Therefore, the gene expression profiles of C. militaris cultured under different light exposure conditions should be different. RNA sequencing (RNA-seq) technology is an efficient, rapid, and sensitive method to deeply study and analyze biological transcriptomes, which provides a whole perspective for the study of gene expression at RNA levels, whether low-expression genes or differentially expressed genes (DEGs; Zhao et al., 2019). Based on this, the aim of this study was to identify genes involved in the synthesis of carotenoids and the biosynthetic pathway of carotenoids by comparing the transcriptomes of two C. militaris mycelial samples cultured under different light exposure conditions. To further validate transcriptome data, gene expression analysis using quantitative real-time PCR analysis (qRT-PCR) was performed. In addition, the terpenoid synthase gene (Cmtns) of C. militaris was complemented to its deleted mutant (ΔCmtns) to identify the function of the Cmtns gene. This study provides a better understanding of the biosynthetic mechanism of carotenoids in C. militaris under light exposure.
Materials and Methods
Strains and Vectors
A laboratory and commercial strain of C. militaris, CM10 (Ningyang County Haixin Biological Technology Co., Ltd., Shandong, China), was used in this study. Agrobacterium tumefaciens strain AGL-1 (purchased from Shanghai Weidi Biotechnology Co., Ltd., Shanghai, China) carrying the vector pCAMBIA0390-Ben-Comtns was used to perform Agrobacterium tumefaciens-mediated transformation (ATMT) of the ΔCmtns strain to achieve complementation of the Cmtns gene. The vector pCAMBIA0390-Ben-Comtns, containing a benomyl (ben) resistant gene cassette and a Cmtns gene (GenBank: NW_006271971.1) expression cassette, was constructed based on pCAMBIA0390-Ben in our laboratory (Supplementary Figure S1).
Preparation of Mycelial Samples for Transcriptome Sequencing
Cordyceps militaris strain CM10 was inoculated on potato dextrose agar (PDA, 200.0 g potato, 10.0 g dextrose, 3.0 g KH2PO4, 1.0 g MgSO4, 20.0 g agar, 1000 mL distilled water) plates for activation and cultured at 25°C for 28 days (including 21 days of cultivation under dark conditions in the first phase and 7 days of cultivation under light exposure in the second phase). The CM10 strain activated on the PDA medium was cut into pieces of 1 cm × 1 cm size, and three pieces were inoculated into 100 mL of potato dextrose broth (PDB, 200.0 g potato, 10.0 g dextrose, 3.0 g KH2PO4, 1.0 g MgSO4, 1000 mL distilled water), and cultured at 25°C on a shaker at 150 rpm for 4 days in the dark. 5 mL PDB culture was inoculated into solid medium (14.6 g wheat powder, 4.0 g wheat bran powder, 1.4 g corn powder, 25 mg MgSO4, 25 mg KH2PO4, 1 mg vitamin B1, 25 mL distilled water) and cultured at 25°C. The mycelial sample CM10_D was obtained after 18 days of cultivation under dark conditions. The mycelial sample CM10_L was obtained by first culturing for 10 days under dark conditions and then culturing for 8 days under light exposure conditions (750 Lux). These two mycelial samples cultured under different light exposure conditions were frozen in liquid nitrogen and immediately stored in a −80°C freezer for RNA extraction.
cDNA Library Construction and Transcriptome Sequencing
The C. militaris mycelia CM10_D from six biologically repeated petri dishes were mixed together to obtain the mycelial sample CM10_D. Similarly, the C. militaris mycelia CM10_L from six biologically repeated petri dishes were mixed together to obtain the mycelial sample CM10_L. The total RNA was extracted from mycelial samples using TRIzol reagent (Invitrogen, Carlsbad, CA, United States) according to the manufacturer’s instructions. RNA quality and concentration were evaluated using an Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA, United States) and NanoDrop Spectrophotometer (NanoDrop Technologies, Wilmington, DE, United States). The mRNA of each sample was then isolated according to the polyA selection method using oligo (dT) beads. Two cDNA libraries were generated using a NEB Next Ultra RNA Library Prep Kit for Illumina (NEB, Ipswich, MA, United States) following the manufacturer’s recommendations. Each cDNA library was sequenced once from both ends on an Illumina HiSeq 4000 platform at Beijing Genomics Institute (BGI, Shenzhen, China). Based on the sequencing quality assessment, ambiguous reads that had more than 5% unknown bases (“N”) or a sequencing quality was less than 20% per read were filtered out using in-house software developed by BGI. The remaining reads with a length of 100 base pairs (bps), called clean reads, were then proceeded through the Trinity reconstruction pipeline to generate full-length transcripts (Haas et al., 2013). All subsequent analyses in this study were based on the clean data. The clean data were deposited in NCBI Sequence Read Archive (SRA) under the BioProject accession number PRJNA541819.
Genome Mapping and Analysis of Differentially Expressed Genes
The clean reads were mapped to the C. militaris genomic data (BioProject accession no. PRJNA225510) using HISAT (v0.1.6) (Zheng P. et al., 2011; Kim et al., 2015). The number of reads mapped to genes was normalized for RNA length according to FPKM (fragments per kilobase of exon per million mapped reads). After mapping to the reference genome, we used StringTie to reconstruct transcripts (Pertea et al., 2015), and with genome annotation information we could identify novel transcripts exist in our samples use cuffcompare, a tool of cufflinks (Trapnell et al., 2012). After novel transcript detection, we merged novel coding transcripts with reference transcripts to get the complete reference, then we mapped clean reads to it use Bowtie2 (Langmead and Salzberg, 2012) and calculated gene expression level for each sample with RSEM (Li and Dewey, 2011).
Gene expression levels were normalized using the FPKM method. Differential expression analysis was performed using the Deseq2 tool (Love et al., 2014). The p-values were adjusted using the method of Benjamini and Hochberg (1995). Unigenes with a false discovery rate (FDR) ≤0.001 and a fold change ≥2 in a comparison (CM10_D vs CM10_L) were identified as significantly DEGs.
Functional Annotation of DEGs
For functional annotation and analysis, all DEGs derived from RNA-seq data were annotated by searching against the NCBI non-redundant protein sequence database (NR), gene ontology (GO) database1, and Kyoto Encyclopedia of Genes and Genomes (KEGG) database2 (Mao et al., 2005; Young et al., 2010). Furthermore, the annotated data of C. militaris strain CM01 were also used for functional annotation by BLASTN and BLASTX, respectively (Zheng P. et al., 2011).
Validation of Transcriptome Data by Quantitative Real-Time PCR Analysis
To validate the quality of transcriptome data, five genes (CCM_04119, CCM_05246, CCM_06728, CCM_08263, and CCM_09155) were selected for qRT-PCR analysis using the elongation factor 1-alpha (tef1) gene (GenBank: DQ070019) as the internal control gene (Lian et al., 2014b). Total RNA of C. militaris mycelial samples was extracted using the Fungal RNA Kit (Omega, Stamford, CT, United States), and then reverse-transcribed using TransScript All-in-One First-Strand cDNA Synthesis SuperMix (TransGen Biotech, Beijing, China). qRT-PCR was performed using TransStart Tip Green qPCR SuperMix (TransGen Biotech, Beijing, China) with a QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, United States). The reaction system and reaction conditions of the qRT-PCR were performed as previously described (Chen et al., 2018). The relative expression levels of genes were calculated using the 2–ΔΔCT method (Livak and Schmittgen, 2001). All the primers used in this study are shown in Supplementary Table S1.
Complementation of the Cmtns Disruption Mutant
The only terpenoid synthase gene Cmtns in DEGs was obtained by transcriptome analysis. In order to study the mechanism of carotenoid biosynthesis in C. militaris, we performed functional identification of the Cmtns gene. In our previous studies, we had knocked out the Cmtns gene by homologous recombination and obtained the target gene mutant ΔCmtns (Lou et al., 2018). In this study, the Cmtns gene was complemented to the ΔCmtns mutant by the ATMT method. The entire Cmtns gene with a 2290-bp upstream region as its putative promoter (Ptns) and a 520-bp downstream region as the putative terminator (Ttns) was amplified from the C. militaris genome with primers CmtnsC-F/R and inserted into the binary vector pCAMBIA0390-Ben to generate the binary vector pCAMBIA0390-Ben-Comtns (Supplementary Figure S1) for C. militaris transformantion. The binary vector pCAMBIA0390-Ben-Comtns was transformed into A. tumefaciens strain AGL-1 to obtain the strain AGL1-pCAMBIA0390-Ben-Comtns. The A. tumefaciens strain AGL1-pCAMBIA0390-Ben-Comtns and conidia of the ΔCmtns mutant were co-cultured to achieve complementation of the Cmtns gene by the previously described ATMT method (Zheng Z. et al., 2011). Transformants were selected on PDA plates supplemented with 3 μg/mL of benomyl at 25°C. Positive complemented transformants (ΔCmtns-c) were confirmed by PCR and qRT-PCR.
Determination of Carotenoid Content
The fruiting bodies of all strains (wild-type strain CM10, ΔCmtns strain, and ΔCmtns-c strain) were cultured on rice medium according to the method described in previous report (Jiang and Han, 2015). The fruiting bodies of C. militaris were vacuum freeze-dried, and then dried fruiting bodies were used for the determination of carotenoid content according to previously reported methods (Yang et al., 2014; Zhang et al., 2018).
Statistical Analysis
The qRT-PCR experiment and the determination of carotenoid content were performed in triplicate. Data were analyzed by SPSS 20.0 software (SPSS Inc., Chicago, IL, United States). One-way analysis of variance (ANOVA) and Duncan’s multiple-range tests were used to determine significant differences. The values were shown as the mean ± standard error (SE). P-values less than 0.05 were considered significant.
Results
Preparation of C. militaris Mycelia and Summary of RNA-Seq Data Sets
Mycelial samples of C. militaris cultured under different light exposure conditions are shown in Figure 1. The mycelia (CM10_D) of C. militaris cultured under dark conditions was pure white, and the mycelia (CM10_L) of C. militaris cultured under light exposure conditions was orange. It can be concluded that light is an essential factor for the production of C. militaris pigments.
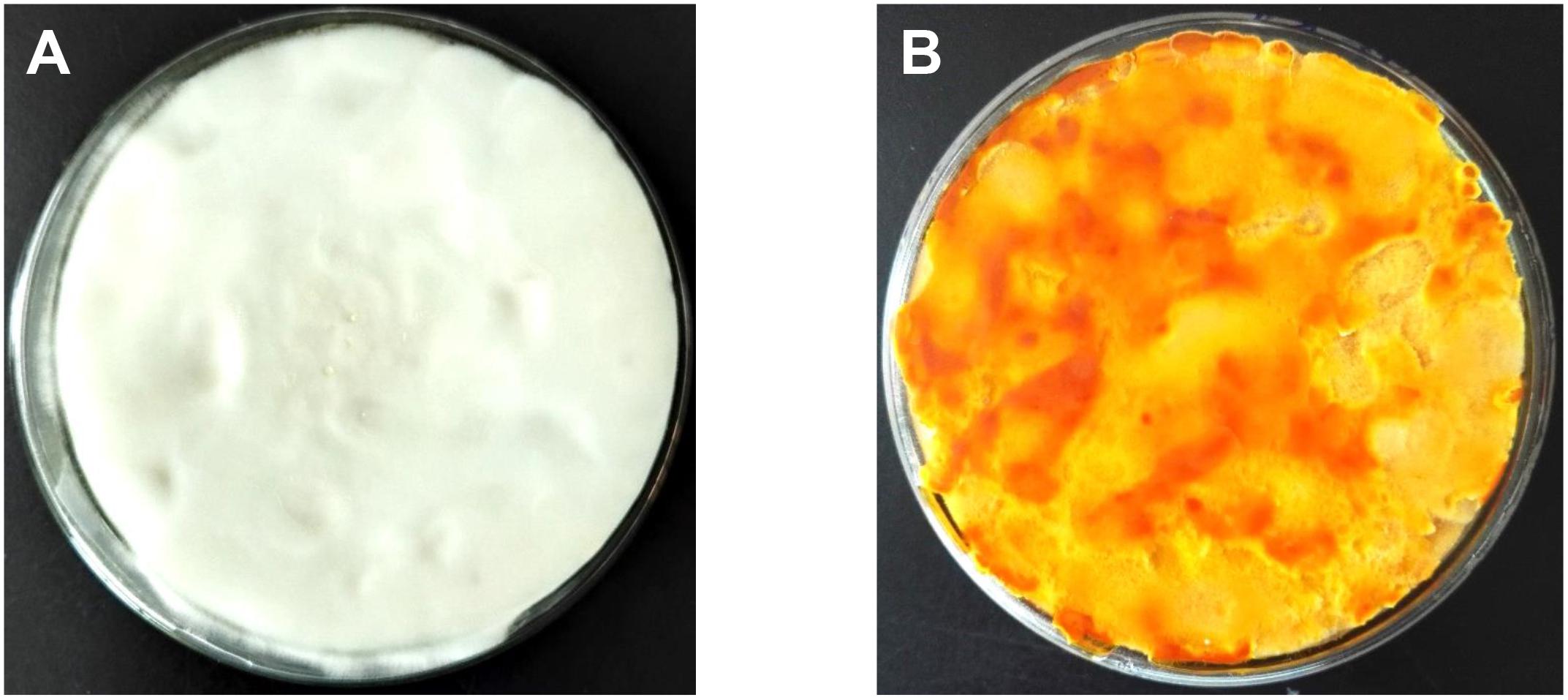
Figure 1. Cordyceps militaris mycelial samples CM10_D (A) and CM10_L (B).
To compare the gene expression profiles of the white mycelia and the orange mycelia, the cDNA libraries were built from cultivated mycelia (CM10_D and CM10_L) and subjected to Illumina deep sequencing. As shown in Table 1, mycelia CM10_D and mycelia CM10_L produced raw reads of 30,769,028 reads and 32,387,910 reads, respectively. After quality filtering, 29,635,460 clean reads were obtained from mycelia CM10_D and 30,261,506 clean reads were obtained from mycelia CM10_L. The Q30 values of each sample were more than 94%, indicating that the sequence data was accurate. 84.70% of CM10_D reads and 79.94% of CM10_L reads could be mapped to the C. militaris genome with 9684 protein-coding genes (Zheng P. et al., 2011). In addition, 8793 genes (including 8655 known genes and 138 novel genes) were detected from CM10_D and 8807 genes (including 8670 known genes and 137 novel genes) were detected from CM10_L (Supplementary Table S2).

Table 1. Summary of transcriptome sequencing.
Functional Annotation and Analysis of DEGs
Transcriptome data were used to identify genes showing significant changes in expression of C. militaris cultured under different light exposure conditions (CM10_D and CM10_L). Compared with CM10_D, 1722 DEGs were identified in CM10_L (866 up-regulated DEGs and 856 down-regulated DEGs).
Furthermore, GO and KEGG pathway analysis were carried out to study the functions of DEGs. All DEGs were mapped to the GO terms in three main categories of GO database. The GO classification results showed that 2341 DEGs were categorized into 34 GO terms (Figure 2). In the “biological process” category, it is worth noting that the term with the most DEGs was the metabolic process. In the “cellular component” category, it is clear that the term with the most DEGs was the membrane. In the “molecular function” category, it is obvious that the term with most DEGs was the catalytic activity. Therefore, DEGs classified as the “metabolic process,” “membrane,” and “catalytic activity” terms may be responsible for the biosynthesis of C. militaris pigments. Different genes often cooperate to play roles in different biological processes. KEGG pathway enrichment analysis helps us to understand more about the biological functions of genes (Zhao et al., 2019). The results suggested that the DEGs were specifically located in the pathways of “metabolic pathways,” “MAPK signaling pathway-yeast,” and “biosynthesis of secondary metabolites,” which indicated that these DEGs were mainly involved in cell metabolism and signal transduction (Figure 3).
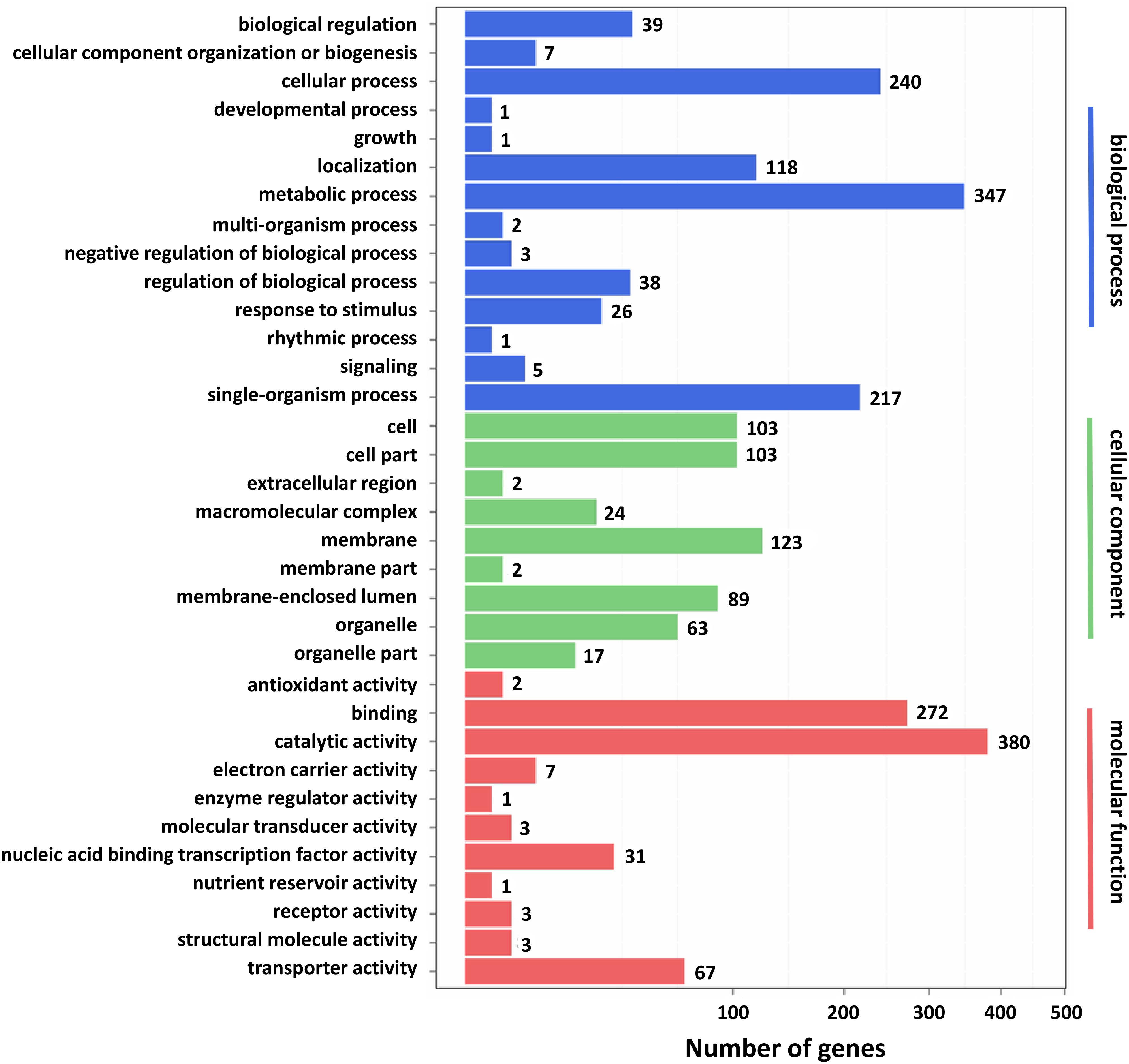
Figure 2. Gene ontology (GO) classification of differentially expressed genes (DEGs). X axis represents the number of DEGs. Y axis represents the GO term.
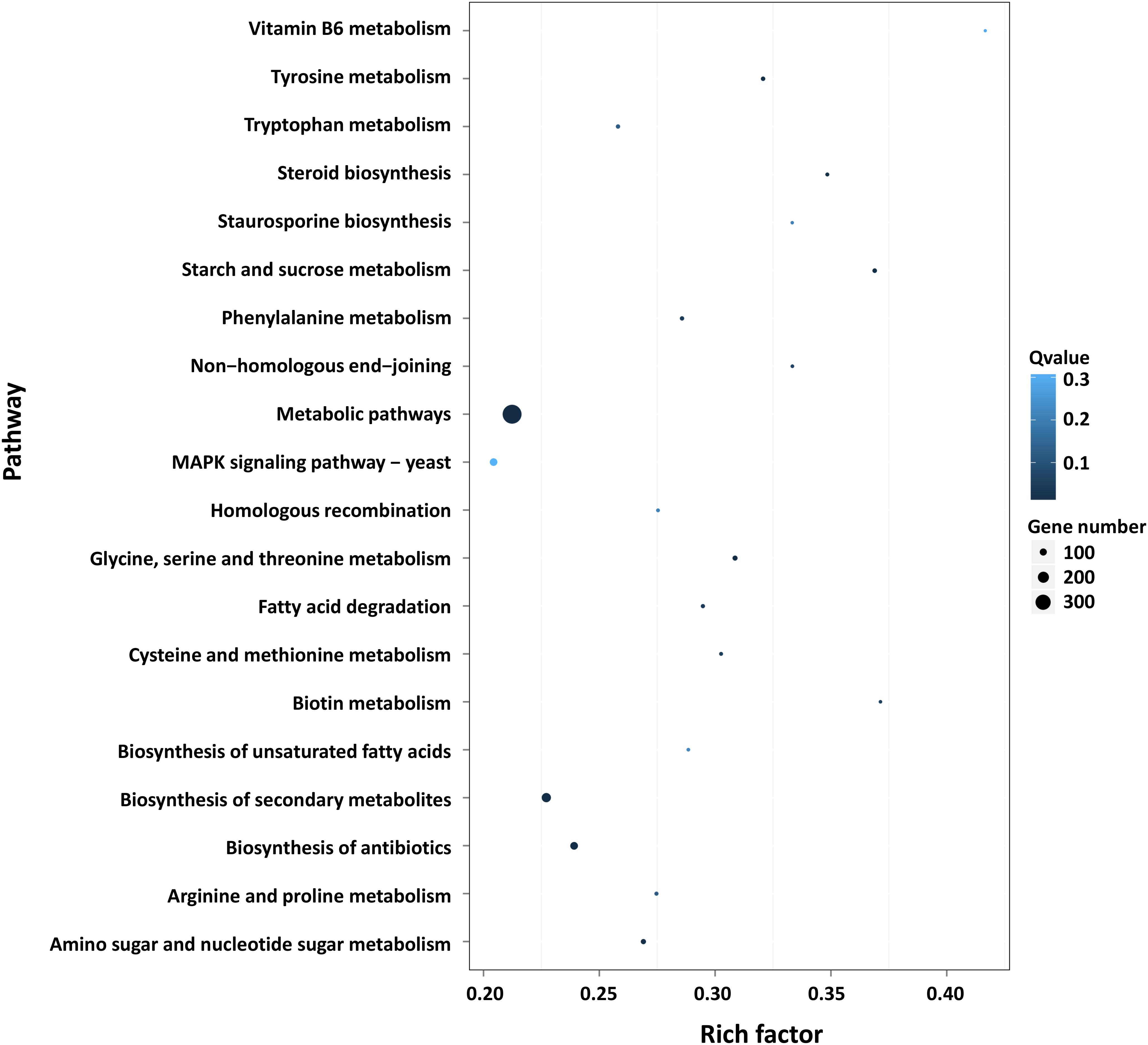
Figure 3. Pathway functional enrichment of DEGs. X axis represents the enrichment factor. Y axis represents the pathway name. Coloring indicates the q-value (high: white; low: blue), and the lower q-value indicates the more significant enriched. The point size indicates the number of DEGs (more: big; less: small).
It has been reported that C. militaris pigments were carotenoids (Dong et al., 2013a; Yi et al., 2014). Therefore, all unigenes derived from RNA-seq data were searched against the carotenoid biosynthetic pathway in the KEGG database, and the results showed that only the CCM_06728 (CAO-2) and CCM_09155 (YLO-1) genes were found to be involved in the biosynthesis of carotenoids (Supplementary Figure S2). Unexpectedly, the expression levels of these two genes (CCM_06728 and CCM_09155) were not significantly different between mycelia CM10_D and mycelia CM10_L. Because carotenoids belong to terpenoids, the only terpenoid synthase gene Cmtns in DEGs was significantly up-regulated. We believe that the Cmtns gene might be involved in the biosynthesis of carotenoids in C. militaris.
Validation of Transcriptome Data by qRT-PCR
To validate the transcriptome data, five genes were selected to analyze their expression levels by qRT-PCR. With mycelia CM10_D as a control sample, CCM_04119 (Cmtns), CCM_05246, and CCM_08263 genes were significantly up-regulated in mycelia CM10_L, while CCM_06728 and CCM_09155 genes were not significantly changed (Table 2). The results of qRT-PCR revealed that the expression levels of the five genes were consistent with the results of RNA-seq, indicating that the transcriptome data were accurate.
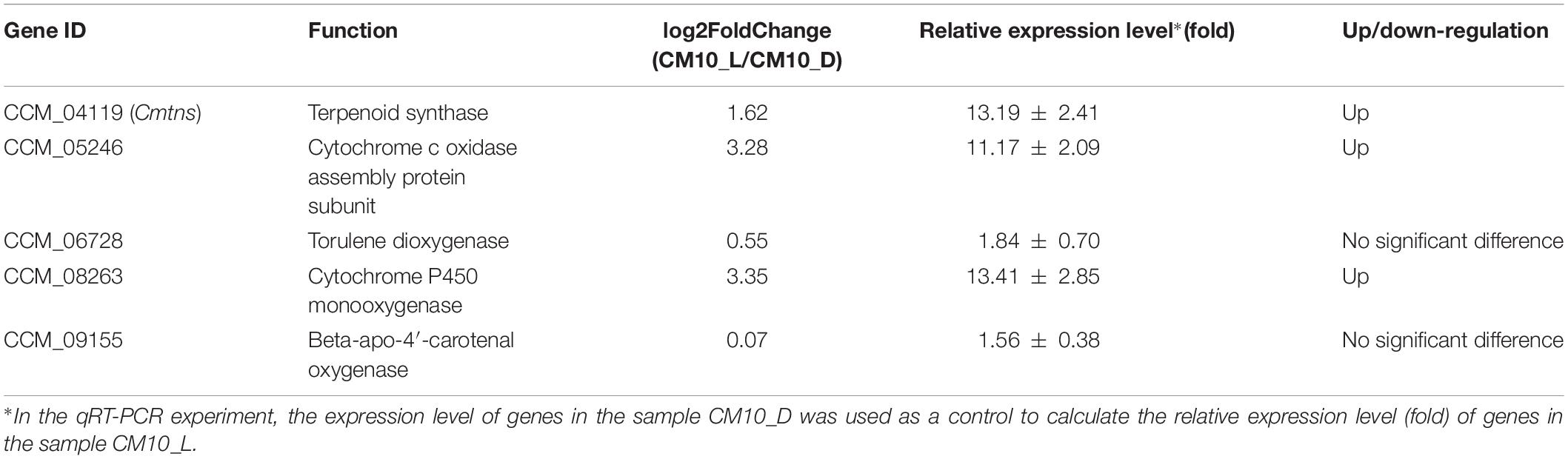
Table 2. The relative expression levels of five genes by qRT-PCR.
Complementation of the ΔCmtns Mutant
To identify the function of the Cmtns gene, the complementary experiment of the Cmtns gene was carried out. The co-cultured PDA plate (containing 3 μg/mL of benomyl) of complementary the Cmtns gene by ATMT method was shown in Figure 4A, and some resistant transformants clearly appeared on the resistant plate. Primer pairs of JCBen-F/JCBen-R and CmtnsC-F/CmtnsC-R were used to amplify the ben gene and the Cmtns gene in transformants, respectively (Figures 4B,C). The PCR products were sequenced and confirmed that the ben gene and the Cmtns cassette were successfully integrated into the genome of the ΔCmtns mutant. The results of qRT-PCR analysis demonstrated that the expression of the Cmtns gene in mycelia CM10_L was significantly up-regulated compared with mycelia CM10_D. However, no expression of the Cmtns gene was detected in the ΔCmtns mutant, which might be due to the deletion of the Cmtns gene. When the Cmtns gene was complemented to the ΔCmtns mutant, there was no significant difference in the expression level of the Cmtns gene between the ΔCmtns-c strain and the wild-type strain (CM10), which indicated that the Cmtns gene was successfully complemented to the ΔCmtns mutant and could be expressed in the ΔCmtns-c strain (Figure 4D).
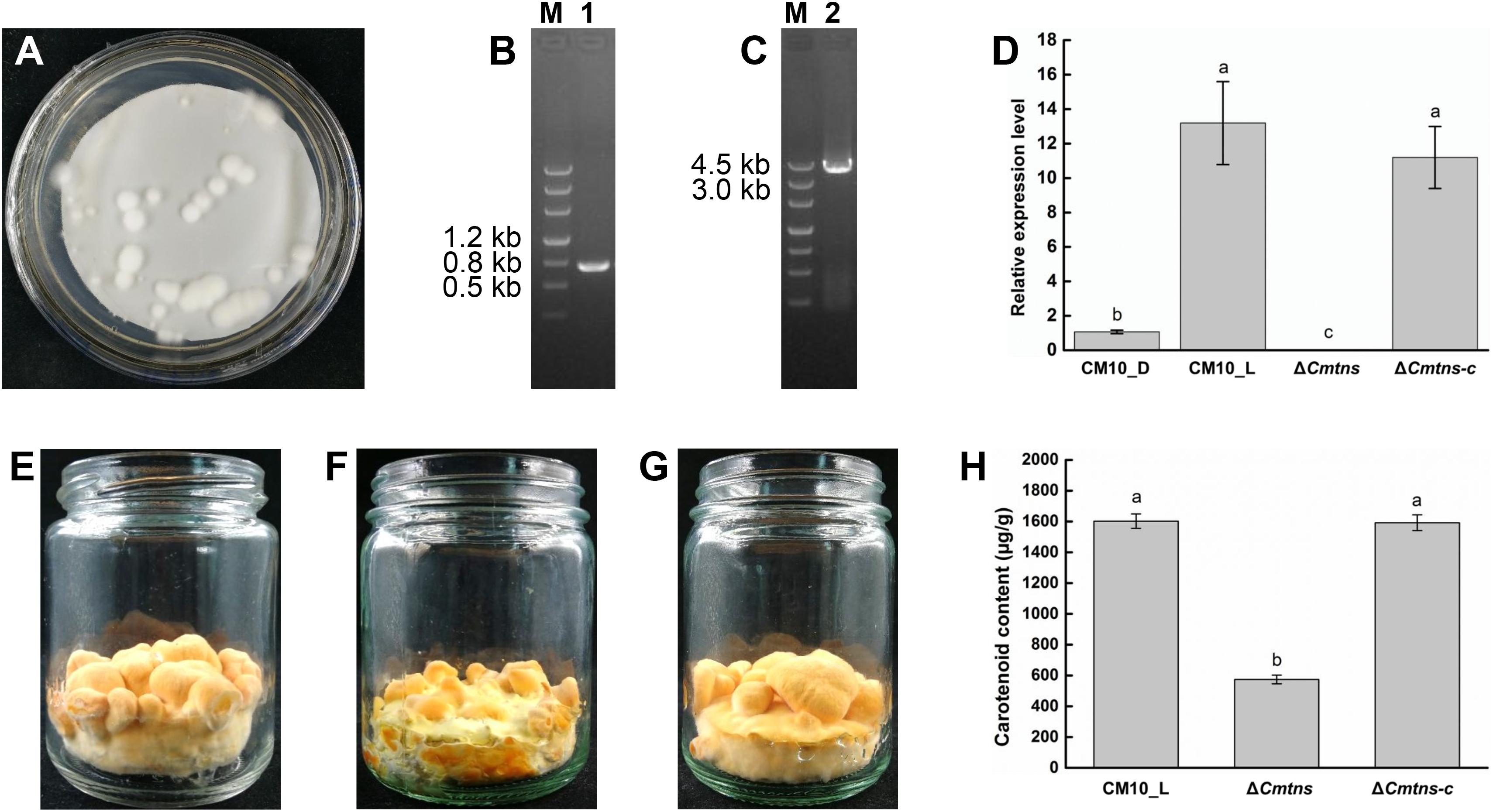
Figure 4. Study on the function of the Cmtns gene. (A) The conidia of the ΔCmtns mutant were co-cultured with the Agrobacterium tumefaciens strain AGL1-pCAMBIA0390-Ben-Comtns on PDA medium supplemented with 3 μg/mL of benomyl, and the co-culture plate for complementation of the Cmtns gene was obtained. (B) PCR amplification of the ben gene in the ΔCmtns-c strain. (C) PCR amplification of the Cmtns gene in the ΔCmtns-c strain. (D) qRT-PCR analysis of the Cmtns gene expression. (E) C. militaris CM10 cultured on rice medium. (F) C. militaris ΔCmtns cultured on rice medium. (G) C. militarisΔCmtns-c cultured on rice medium. (H) Analysis of carotenoid content in C. militaris. Data are means ± standard error from three independent experiments. Different letters (a–c) above the bars indicated significant difference (ANOVA followed by Duncan’s multiple-range tests, P < 0.05).
Effect of the Cmtns Gene on Carotenoid Content of C. militaris
The color phenotypes of the fruiting bodies of all C. militaris strains (CM10, ΔCmtns, ΔCmtns-c) were orange (Figures 4E–G). The carotenoid content of C. militaris ΔCmtns was significantly lower than that of C. militaris CM10, which might be attributed to the knockout of the Cmtns gene. However, when the Cmtns gene was complemented to the ΔCmtns mutant, the carotenoid content of the complementary strain ΔCmtns-c was not significantly different from that of wild-type C. militaris strain CM10, which indicated that the Cmtns gene significantly affected the biosynthesis of carotenoids in C. militaris (Figure 4H).
Discussion
Cordyceps militaris contains a variety of secondary metabolites which are beneficial to human body, and water-soluble carotenoids in C. militaris have attracted more and more attention (Dong et al., 2013b). Light is a necessary condition for the production of carotenoids in C. militaris (Yang et al., 2016). To identify the genes involved in the biosynthesis of carotenoids in C. militaris, the transcriptome differences between mycelia CM10_D cultured under dark conditions and mycelia CM10_L cultured under light exposure conditions were compared and analyzed. Moreover, the function of the up-regulated gene Cmtns was identified and significantly affected the biosynthesis of carotenoids in C. militaris.
There were 1722 DEGs (866 up-regulated DEGs and 856 down-regulated DEGs) between mycelia CM10_D and mycelia CM10_L. Yang et al. (2016) found that there were 1042 DEGs (545 up-regulated DEGs and 497 down-regulated DEGs) between mycelia Wt_L cultured under light exposure conditions and mycelia Wt_D cultured under dark conditions. Wang et al. (2018) found that there were only 697 DEGs (471 up-regulated DEGs and 226 down-regulated DEGs) between mycelia 498L cultured under light exposure conditions and mycelia 498D cultured under dark conditions. The number of their DEGs was significantly less than the number of DEGs in this study, which might be due to different culture conditions, different strains, and different media.
Cordyceps militaris pigments are secondary metabolites induced by light and were considered to be carotenoids (Wang et al., 2017). However, the KEGG pathway analysis of this study indicated that only two genes (CCM_06728 and CCM_09155) were involved in the biosynthesis of carotenoids in C. militaris, and the expression levels of these two genes were not significantly different between mycelia CM10_D and mycelia CM10_L, which was consistent with previous studies (Yang et al., 2016; Wang et al., 2018). Three types of geranylgeranyl diphosphate synthase (GGPPS) genes (CCM_03059, CCM_03697, and CCM_06355) had been cloned from C. militaris, and CCM_03059 was thought to be involved in the biosynthesis of carotenoids (Lian et al., 2014a; Figure 5). In this study, the expression level of CCM_03059 gene was not significantly different between mycelia CM10_D and mycelia CM10_L, which was consistent with previous studies (Yang et al., 2016). Therefore, the CCM_03059 gene may not be directly involved in the biosynthesis of carotenoids in C. militaris. Nevertheless, the expression level of the CCM_03697 gene in mycelia CM10_L was significantly higher than that in mycelia CM10_D, so we believe that the CCM_03697 gene is probably responsible for the biosynthesis of carotenoids in C. militaris, which is different from previous studies (Lian et al., 2014a; Yang et al., 2016; Figure 5). In addition, phytoene synthase and phytoene dehydrogenase are two key enzymes in the biosynthesis of carotenoids, but these two key enzymes were not found in this study. Furthermore, Yang et al. (2016) had not found these two key enzymes in C. militaris, and they believed that the biosynthetic pathway of carotenoids between C. militaris and Neurospora crassa were totally different. Based on the above, we found that the biosynthetic pathway of carotenoids in C. militaris might be different from the biosynthetic pathway of carotenoids in the KEGG database.
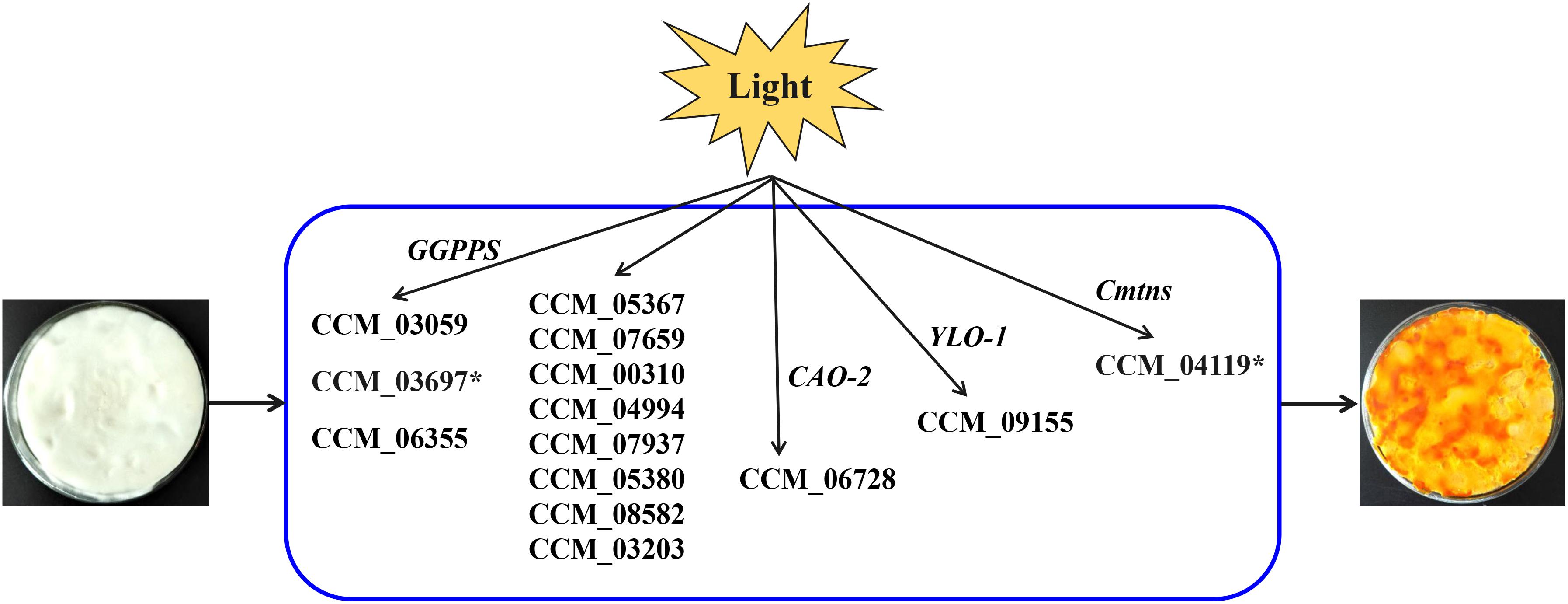
Figure 5. Putative model for light-induced carotenoid formation in C. militaris based on transcriptome analysis and previous studies. ∗Indicates that the gene is up-regulated in a comparison (CM10_D vs CM10_L).
Since the biosynthetic pathway of carotenoids in C. militaris is unknown, Wang et al. (2018) proposed a putative carotenoid biosynthetic pathway for C. militaris. In this putative carotenoid biosynthetic pathway, there were 11 genes involved (CCM_05367, CCM_07659, CCM_00310, CCM_04994, CCM_07937, CCM_05380, CCM_08582, CCM_03203, CCM_03697, CCM_06728, and CCM_09155) (Figure 5). The expression levels of these 11 genes were not significantly different between mycelia 498D (cultured under dark conditions) and mycelia 498L (cultured under light exposure conditions) (Wang et al., 2018). In this study, except for the CCM_03697 gene, the expression levels of the other 10 genes were also not significantly different between mycelia CM10_D and mycelia CM10_L. Hence, the functions of these 11 genes need to be identified.
Since the DEGs involved in carotenoid biosynthesis of C. militaris were not found by KEGG pathway analysis, the function of the only terpenoid synthase gene Cmtns in DEGs was studied. Although the fruiting bodies produced by the CM10, ΔCmtns, and ΔCmtns-c strains all showed orange color, the carotenoid content of the ΔCmtns strain was significantly lower than that of the CM10 strain, which indicated that the deletion of the Cmtns gene could decrease the carotenoid production of C. militaris. In addition, we found that the deletion of the Cmtns gene also reduced the size of C. militaris fruiting bodies, which suggested that the Cmtns gene might be a multifunctional gene. These results were also confirmed by the complementation experiment of the Cmtns gene.
Because the biosynthetic pathway of carotenoids in C. militaris is unknown, and there are few reports on the genes involved in its biosynthesis. Therefore, we believe that the following work will be helpful to clarify the biosynthetic pathway of carotenoids in C. militaris: (1) purifying and identifying carotenoids of C. militaris; (2) identifying the function of genes involved in the biosynthesis of carotenoids in C. militaris; (3) because degeneration of C. militaris can lead to a decrease in the content of carotenoids (Sun et al., 2017), elucidating the molecular mechanism of C. militaris degeneration will be helpful to reveal the biosynthesis mechanism of C. militaris carotenoids; and (4) analyzing the transcriptome of C. militaris cultured under a light pulse or a shorter light exposure condition will facilitate the identification of genes involved in biosynthesis of carotenoids (Navarro et al., 2001; Zhang et al., 2016). The comparative transcriptome analysis used in this study laid the foundation for identifying genes involved in the biosynthesis of carotenoids in C. militaris and revealing its biosynthetic pathway. In addition, the successful application of the CRISPR-Cas9 gene disruption system in C. militaris will accelerate the identification of gene function of C. militaris (Chen et al., 2018).
Conclusion
Cordyceps militaris mycelia CM10_D cultured under dark conditions was pure white, while C. militaris mycelia CM10_L cultured under light exposure conditions was orange. Comparative transcriptome analysis revealed that there were 1722 DEGs between mycelia CM10_D and mycelia CM10_L. KEGG pathway analysis indicated that the CCM_06728 and CCM_09155 genes might be involved in the biosynthesis of carotenoids in C. militaris. What’s more, we identified for the first time that the Cmtns gene significantly affected the biosynthesis of carotenoids in C. militaris. The results will provide a basis for further research on the biosynthetic pathway of carotenoids in C. militaris.
Data Availability
The datasets generated for this study can be found in the clean reads of the Cordyceps militaris transcriptome which has been deposited in NCBI Sequence Read Archive (SRA) under the BioProject accession number PRJNA541819.
Author Contributions
J-FL and L-QG conceived and designed the experiment. YZ cultured C. militaris and performed the transcriptome sequencing. H-BT and Z-WY transformed the Cmtns gene into the genome of the ΔCmtns strain. TW constructed the vector pCAMBIA0390-Ben-Comtns and determined the carotenoid content. H-WL analyzed the transcriptome data, identified complementary transformants, and wrote the manuscript. All authors reviewed and approved the submitted version of the manuscript.
Funding
This study was supported by the Guangdong Province Science and Technology Innovation Strategy Special Fund (Grant Nos. 2018B020205001 and 2018B020205003), the National Natural Science Foundation of China (Grant Nos. 31572178, 31772273, and 31801918), and the Edible Fungus Industrial Technology System of Modern Agriculture of Guangdong Province (Grant Nos. 2018LM1125 and 2018LM1126).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02105/full#supplementary-material
Footnotes
References
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 57, 289–300. doi: 10.2307/2346101
Chen, B. X., Wei, T., Ye, Z. W., Yun, F., Kang, L. Z., Tang, H. B., et al. (2018). Efficient CRISPR-Cas9 gene disruption system in edible-medicinal mushroom Cordyceps militaris. Front. Microbiol. 9:1157. doi: 10.3389/fmicb.2018.01157
Dong, J. Z., Lei, C., Zheng, X. J., Ai, X. R., Wang, Y., and Wang, Q. (2013a). Light wavelengths regulate growth and active components of Cordyceps militaris fruit bodies. J. Food Biochem. 37, 578–584. doi: 10.1111/jfbc.12009
Dong, J. Z., Wang, S. H., Ai, X. R., Yao, L., Sun, Z. W., Lei, C., et al. (2013b). Composition and characterization of cordyxanthins from Cordyceps militaris fruit bodies. J. Funct. Foods 5, 1450–1455. doi: 10.1016/j.jff.2013.06.002
Friederichsen, I., and Engel, H. (1958). The pigment of Cordyceps militaris L. Arch. Mikrobiol. 30, 393–395. doi: 10.1007/BF00411233
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-Seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hu, T., Liang, Y., Zhao, G., Wu, W., Li, H., and Guo, Y. (2019). Selenium biofortification and antioxidant activity in Cordyceps militaris supplied with selenate, selenite, or selenomethionine. Biol. Trace Elem. Res. 187, 553–561. doi: 10.1007/s12011-018-1386-y
Jiang, K., and Han, R. (2015). Rhf1 gene is involved in the fruiting body production of Cordyceps militaris fungus. J. Ind. Microbiol. Biotechnol. 42, 1183–1196. doi: 10.1007/s10295-015-1637-2
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kim, Y., Shin, W., Dong, X., Kim, E., Pahalagedara, W., Srilal, A., et al. (2017). Anti-inflammatory effect of the extract from fermented Asterina pectinifera with Cordyceps militaris mycelia in LPS-induced. Food Sci. Biotechnol. 26, 1633–1640. doi: 10.1007/s10068-017-0233-9
Kunhorm, P., Chaicharoenaudomrung, N., and Noisa, P. (2019). Enrichment of cordycepin for cosmeceutical applications: culture systems and strategies. Appl. Microbiol. Biotechnol. 103, 1681–1691. doi: 10.1007/s00253-019-09623-3
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lee, H. H., Lee, S., Lee, K., Shin, Y. S., Kang, H., and Cho, H. (2015). Anti-cancer effect of Cordyceps militaris in human colorectal carcinoma RKO cells via cell cycle arrest and mitochondrial apoptosis. DARU J. Pharm. Sci. 23:35. doi: 10.1186/s40199-015-0117-6
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Lian, T., Dong, C., Yang, T., and Sun, J. (2014a). Three types of geranylgeranyl diphosphate synthases from the medicinal caterpillar fungus, Cordyceps militaris (Ascomycetes). Int. J. Med. Mushrooms 16, 115–124. doi: 10.1615/IntJMedMushr.v16.i2.20
Lian, T., Yang, T., Liu, G., Sun, J., and Dong, C. (2014b). Reliable reference gene selection for Cordyceps militaris gene expression studies under different developmental stages and media. FEMS Microbiol. Lett. 356, 97–104. doi: 10.1111/1574-6968.12492
Liu, X., Zhu, Z., Liu, Y., and Sun, H. (2019). Comparisons of the anti-tumor activity of polysaccharides from fermented mycelia and cultivated fruiting bodies of Cordyceps militaris in vitro. Int. J. Biol. Macromol. 130, 307–314. doi: 10.1016/j.ijbiomac.2019.02.155
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using Real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lou, H. W., Ye, Z. W., Yun, F., Lin, J. F., Guo, L. Q., Chen, B. X., et al. (2018). Targeted gene deletion in Cordyceps militaris using the split-marker approach. Mol. Biotechnol. 60, 380–385. doi: 10.1007/s12033-018-0080-9
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lu, R., Luo, F., Hu, F., and Huang, B. (2013). Identification and production of a novel natural pigment, cordycepoid A, from Cordyceps bifusispora. Appl. Microbiol. Biotechnol. 97, 6241–6249. doi: 10.1007/s00253-013-4966-y
Mao, X., Cai, T., Olyarchuk, J. G., and Wei, L. (2005). Databases and ontologies automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793. doi: 10.1093/bioinformatics/bti430
Navarro, E., Lorca-Pascual, J. M., Quiles-Rosillo, M. D., Nicolás, F. E., Garre, V., Torres-Martínez, S., et al. (2001). A negative regulator of light-inducible carotenogenesis in Mucor circinelloides. Mol. Gen. Genomics 266, 463–470. doi: 10.1007/s004380100558
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Sun, S. J., Deng, C. H., Zhang, L. Y., and Hu, K. H. (2017). Molecular analysis and biochemical characteristics of degenerated strains of Cordyceps militaris. Arch. Microbiol. 199, 939–944. doi: 10.1007/s00203-017-1359-0
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Wang, F., Liu, Q., Zhang, J., Liu, K., Li, K., and Liu, G. (2018). Comparative transcriptome analysis between a spontaneous albino mutant and its sibling strain of Cordyceps militaris in response to light stress. Front. Microbiol. 9:1237. doi: 10.3389/fmicb.2018.01237
Wang, F., Song, X., Dong, X., Zhang, J., and Dong, C. (2017). DASH-type cryptochromes regulate fruiting body development and secondary metabolism differently than CmWC-1 in the fungus Cordyceps militaris. Appl. Microbiol. Biotechnol. 101, 4645–4657. doi: 10.1007/s00253-017-8276-7
Xia, Y., Luo, F., Shang, Y., Chen, P., Lu, Y., Wang, C., et al. (2017). Fungal cordycepin biosynthesis is coupled with the production of the safeguard molecule pentostatin article fungal cordycepin biosynthesis is coupled with the production of the safeguard molecule pentostatin. Cell Chem. Biol. 24, 1479–1489. doi: 10.1016/j.chembiol.2017.09.001
Yang, T., Guo, M., Yang, H., Guo, S., and Dong, C. (2016). The blue-light receptor CmWC-1 mediates fruit body development and secondary metabolism in Cordyceps militaris. Appl. Microbiol. Biotechnol. 100, 743–755. doi: 10.1007/s00253-015-7047-6
Yang, T., Sun, J., Lian, T., Wang, W., and Dong, C. (2014). Process optimization for extraction of carotenoids from medicinal caterpillar fungus, Cordyceps militaris (Ascomycetes). Int. J. Med. Mushrooms 16, 125–135. doi: 10.1615/IntJMedMushr.v16.i2.30
Yi, Z., Huang, W., Ren, Y., Onac, E., Zhou, G., Peng, S., et al. (2014). LED lights increase bioactive substances at low energy costs in culturing fruiting bodies of Cordyceps militaris. Sci. Hortic. 175, 139–143. doi: 10.1016/j.scienta.2014.06.006
Yin, J., Xin, X., Weng, Y., Li, S., Jia, J., and Gui, Z. (2018). Genotypic analysis of degenerative Cordyceps militaris cultured in the pupa of Bombyx mori. Entomol. Res. 48, 137–144. doi: 10.1111/1748-5967
Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A. (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11:R14. doi: 10.1186/gb-2010-11-2-r14
Zhang, G., Huang, Y., and Bian, Y. (2006). Hypoglycemic activity of the fungi Cordyceps militaris, Cordyceps sinensis, Tricholoma mongolicum, and Omphalia lapidescens in streptozotocin-induced diabetic rats. Appl. Microbiol. Biotechnol. 72, 1152–1156. doi: 10.1007/s00253-006-0411-9
Zhang, J. J., Wang, F., Liu, K. B., Liu, Q., Yang, Y., and Dong, C. H. (2018). Heat and light stresses affect metabolite production in the fruit body of Cordyceps militaris. Appl. Microbiol. Biotechnol. 102, 4523–4533. doi: 10.1007/s00253-018-8899-3
Zhang, X., Dong, X. M., Song, X. H., Wang, F., and Dong, C. H. (2017). Photoperiodic responses and characterization of the Cmvvd gene encoding a blue light photoreceptor from the medicinal caterpillar fungus Cordyceps militaris (Ascomycetes). Int. J. Med. Mushrooms 19, 163–172. doi: 10.1615/intjmedmushrooms.v19.i2.80
Zhang, Y., Navarro, E., Cánovas-Márquez, J. T., Almagro, L., Chen, H., Chen, Y. Q., et al. (2016). A new regulatory mechanism controlling carotenogenesis in the fungus Mucor circinelloides as a target to generate β-carotene over-producing strains by genetic engineering. Microb. Cell Fact. 15:99. doi: 10.1186/s12934-016-0493-8
Zhao, X., Zhang, G., Li, C., and Ling, J. (2019). Cordycepin and pentostatin biosynthesis gene identified through transcriptome and proteomics analysis of Cordyceps kyushuensis Kob. Microbiol. Res. 218, 12–21. doi: 10.1016/j.micres.2018.09.005
Zheng, P., Xia, Y., Xiao, G., Xiong, C., Hu, X., Zhang, S., et al. (2011). Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biol. 12:R116. doi: 10.1186/gb-2011-12-11-r116
Zheng, Z., Huang, C., Cao, L., Xie, C., Han, R., and Spatafora, J. W. (2011). Agrobacterium tumefaciens-mediated transformation as a tool for insertional mutagenesis in medicinal fungus Cordyceps militaris. Fungal Biol. 115, 265–274. doi: 10.1016/j.funbio.2010.12.011
Keywords: Cordyceps militaris, transcriptome, pigment, illumination, carotenoid, terpenoid synthase
Citation: Lou H-W, Zhao Y, Tang H-B, Ye Z-W, Wei T, Lin J-F and Guo L-Q (2019) Transcriptome Analysis of Cordyceps militaris Reveals Genes Associated With Carotenoid Synthesis and Identification of the Function of the Cmtns Gene. Front. Microbiol. 10:2105. doi: 10.3389/fmicb.2019.02105
Received: 02 July 2019; Accepted: 27 August 2019;
Published: 10 September 2019.
Edited by:
Hector Mora Montes, University of Guanajuato, MexicoReviewed by:
Gábor Nagy, University of Szeged, HungaryBernardo Franco, University of Guanajuato, Mexico
Copyright © 2019 Lou, Zhao, Tang, Ye, Wei, Lin and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun-Fang Lin, bGluamZAc2NhdS5lZHUuY24=; Li-Qiong Guo, Z3VvbHFAc2NhdS5lZHUuY24=