
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 28 August 2019
Sec. Microbial Immunology
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.01972
 Esther Merlini1†
Esther Merlini1† Maddalena Cerrone1,2†
Maddalena Cerrone1,2† Bonnie van Wilgenburg3
Bonnie van Wilgenburg3 Leo Swadling3E. Stefania Cannizzo1Antonella d’Arminio Monforte1
Leo Swadling3E. Stefania Cannizzo1Antonella d’Arminio Monforte1 Paul Klenerman3
Paul Klenerman3 Giulia Marchetti1*
Giulia Marchetti1*Both HIV and HCV infections feature increased microbial translocation (MT) and gut dysbiosis that affect immune homeostasis and disease outcome. Given their commitment to antimicrobial mucosal immunity, we investigated mucosal-associated invariant T (MAIT) cells and Vα7.2+CD161- T-cell frequency/function and their possible associations with MT and gut dysbiosis, in chronic HIV and/or HCV infections. We enrolled 56 virally infected (VI) patients (pts): 13 HIV+ on suppressive cART (HIV-RNA < 40cp/ml), 13 HCV+ naive to DAA (direct-acting antiviral) anti-HCV agents; 30 HCV+/HIV+ on suppressive cART and naive to anti-HCV. 13 age-matched healthy controls (HC) were enrolled. For Vα7.2+CD161++ and Vα7.2+CD161-CD8+ T cells we assessed: activation (CD69), exhaustion (PD1/CD39), and cytolytic activity (granzymeB/perforin). Following PMA/ionomycin and Escherichia coli stimulation we measured intracellular IL17/TNFα/IFNγ. Markers of microbial translocation (Plasma LPS, 16S rDNA, EndoCAb and I-FABP) were quantified. In 5 patients per group we assessed stool microbiota composition by 16S targeted metagenomics sequencing (alpha/beta diversity, relative abundance). Compared to controls, virally infected pts displayed significantly lower circulating Vα7.2+CD161++CD8+ MAIT cells (p = 0.001), yet expressed higher perforin (p = 0.004) and granzyme B (p = 0.002) on CD8+ MAIT cells. Upon E. coli stimulation, the residual MAIT cells are less functional particularly those from HIV+/HCV+ patients. Conversely, in virally infected pts, Vα7.2+CD161-CD8+ cells were comparable in frequency, highly activated/exhausted (CD69+: p = 0.002; PD-1+: p = 0.030) and with cytolytic potential (perforin+: p < 0.0001), yet were poorly responsive to ex vivo stimulation. A profound gut dysbiosis characterized virally infected pts, especially HCV+/HIV+ co-infected patients, delineating a Firmicutes-poor/Bacteroidetes-rich microbiota, with significant associations with MAIT cell frequency/function. Irrespective of mono/dual infection, HIV+ and HCV+ patients display depleted, yet activated/cytolytic MAIT cells with reduced ex vivo function, suggesting an impoverished pool, possibly due to continuous bacterial challenge. The MAIT cell ability to respond to bacterial stimulation correlates with the presence of Firmicutes and Bacteroidetes, possibly suggesting an association between gut dysbiosis and MAIT cell function and posing viral-mediated dysbiosis as a potential key player in the hampered anti-bacterial MAIT ability.
Mucosal-associated invariant T (MAIT) cells are unconventional T lymphocytes that are relatively abundant in humans, representing 1–10% of circulating T-cells and which are further enriched in mucosal tissues, such as gut lamina propria and liver (Dusseaux et al., 2011; Tang et al., 2013). MAIT cells are characterized by the expression of CD161 and an invariant T cell receptor α segment (Vα7.2) (Dias et al., 2017) that recognizes and is activated by small vitamin metabolites produced by microbes and presented by the major histocompatibility complex class I-related molecule MR1 (Le Bourhis et al., 2010, 2011; Kjer-Nielsen et al., 2012; Gapin, 2014).
Once activated, MAIT cells respond rapidly by producing cytokines and cytolytic products. Indeed, upon TCR-dependent or TCR-independent activation [mediated by IL-12/IL-18 (Ussher et al., 2014; Sattler et al., 2015)], MAITs produce interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), IL-17, cytolytic products (perforin and granzymes) and degranulate (exposing CD107a to the cell surface) (Dusseaux et al., 2011; Le Bourhis et al., 2013; Kurioka et al., 2015).
Increasing evidence suggests that MAIT cells play a protective role in anti-bacterial immunity at mucosal interfaces (Le Bourhis et al., 2010, 2011; Salou et al., 2017; Wong et al., 2017). While the exact mechanisms by which MAITs exert their function are not fully assessed, it has been shown that they are depleted in many bacterial infections including active tuberculosis (Jiang et al., 2014), vibrio cholera infection (Leung et al., 2014) and septic shock (Grimaldi et al., 2014), possibly supporting a role of MAITs in controlling bacterial threats.
Unexpectedly, despite the fact that viruses lack the metabolic pathways to synthesize riboflavin and therefore do not directly activate MAIT cells via MR1, several authors have reported a massive and irreversible loss of MAIT cells in peripheral blood, as well as in mucosal tissues of HIV and/or HCV chronically infected individuals (Leeansyah et al., 2013; Fernandez et al., 2015; Barathan et al., 2016; Eberhard et al., 2016; Hengst et al., 2016; Saeidi et al., 2016; Spaan et al., 2016; Bolte et al., 2017). The nature of this impairment is not clear. While Cosgrove et al. (2013) reported that MAIT cell depletion was due to activation-induced cell death from over-stimulation secondary to microbial translocation, Leeansyah et al. (2013) suggested that continuous exposure to bacterial products in HIV disease may lead to MAIT cell exhaustion and loss, with concomitant expansion of the CD161neg population originated from the MAIT cells.
In the last few years, the understanding of the interactions between gut mucosal immunity and microbiota has significantly broadened (Powell and MacDonald, 2017; Shi et al., 2017). Intestinal microbiota shapes host immunity and contribute to the maintenance of intestinal homeostasis and to the inhibition of excessive inflammation (Lee and Mazmanian, 2010; McDermott and Huffnagle, 2014; Sun et al., 2015). Indeed, an impaired interaction between intestinal microbiota and the mucosal immune system is associated with the pathogenesis of several inflammatory diseases, such as inflammatory bowel disease, rheumatoid arthritis, systemic lupus erythematosus and ankylosing spondylitis (Mazmanian et al., 2008; Hevia et al., 2014; Wang et al., 2014; Costello et al., 2015; Wright et al., 2015; Zhang et al., 2015). Similarly, during chronic HCV and HIV infections a marked dysbiosis has been described, which is not reverted by anti-HCV or anti-HIV treatments (Bajaj et al., 2016; Williams et al., 2016), highlighting the need to explore the function of microbiota in such diseases.
Given that MAIT cells are predominantly present in the gastrointestinal tract and in the liver, where they exert their antimicrobial function and help fight off bacterial infection by responding to infected cells and producing cytokines, it is plausible to hypothesize a role for MAIT cells in maintaining intestinal homeostasis, through the wide reactivity toward several microbial species, including commensal organisms (Gaardbo et al., 2015; Johansson et al., 2016). Thus, in a cohort of chronically infected HIV+ and/or HCV+ patients, we aimed to investigate MAIT cells and Vα7.2+CD161- T-cell phenotype and function and the associations with microbial translocation and gut dysbiosis.
HIV+, HCV+, HCV+/HIV+ infected patients (age >18 years old) were enrolled from January 2009 to December 2016 at the Clinic of Infectious Diseases, University of Milan – ASST Santi Paolo e Carlo, Milan, after providing written, informed consent (approved by the Institutional Review Board at the ASST Santi Paolo e Carlo, Milan, Italy) in accordance with the Declaration of Helsinki. Inclusion criteria were: (i) HIV infection: patients on virally suppressive cART (HIV-RNA < 40 cp/ml), and any current CD4 level; (ii) chronic HCV infection: patients naïve to direct acting antiviral (DAA)-based anti-HCV therapy, and any detectable HCV-RNA. Thirteen HIV-negative age-matched healthy subjects were enrolled as controls (HC). All patients underwent blood sampling, while in a subgroup of 5 subjects per group we also collected stool samples for metagenomic analyses. For the metagenomic analyses, patients were randomly selected, blood and stool were collected the same day and processed for DNA extraction within 4 months. Liver fibrosis was assessed using liver elastography (FibroScan®, Echosens, Paris, France) and Fib-4 score and categorized as absent to moderate (Metavir-score F0-F2) or severe to liver cirrhosis (Metavir-score F3-F4, Fib-4 score ≥ 3.25). No subject with decompensated liver disease was included in the study.
Lymphocyte surface phenotypes were evaluated by flow cytometry on cryopreserved PBMCs. Cell viability was assessed by live/dead (Invitrogen): only samples with viability greater than 90% were used for the experiments. Surface antibodies were incubated at 4°C for 20 min. For the evaluation of the intracellular markers a fixation/permeabilization step was required. The following fluorochrome-labeled anti-human antibodies were used: CD3 APC-H7/PE-Cy7 (BD Biosciences), CD3 PB (Biolegend), CD8 PE-Cy5 (BD Biosciences), CD8 PerCP.Cy5.5/PerCP (Biolegend), CD161 APC (BD Biosciences), CD161 PE (Miltenyi Biotech), CD4 VioGreen (Miltenyi Biotech), CD39 FITC (BD Biosciences), PD-1 PE/PB (BD Biosciences), CD69 FITC (BD Biosciences), Granzyme B PE (BD Biosciences), Granzyme B APC (Invitrogen, PERFORIN A488 (BD Biosciences), Perforin PB (Invitrogen), LIVE/DEAD (Thermo Fisher Scientific), LIVE/DEAD (Invitrogen), IL18R PE (Biolegend), IL-18Ra APC (eBioscience), TCR Vα7.2 PE-Cy7/APC/FITC(Biolegend). MAIT cells were defined as Vα7.2+CD161++CD3+ for total MAIT cell population and Vα7.2+CD161++CD8+. We evaluated MAIT cell activation (CD69), exhaustion (CD39/PD-1), IL18R expression and cytolytic activity (Granzyme B/perforin). For intracellular staining PBMCs were fixed with PFA 1% for 30 min, then washed and permeabilized with saponin 0.2% for 30 min at room temperature (RT). To verify that different labelled-antibodies gave similar results, we thawed the same cell batch of PBMCs and performed titration curves of the antibodies, evaluating the stain index, the height and the width of the peak and the resolution. Cells were acquired using a FACSVerse flow cytometer (Becton Dickinson) or MACSQuant (Miltenyi Biotech). Five patients were used to standardize the acquisition and analysis of the MAIT cells on two different cytometers. In particular, the same batch of PBMCs was thawed, stained and split in 2 tubes, each tube was run in one cytometer alone. Results were then compared. The axes were set based on positive versus negative.
Escherichia coli (DH5α, Invitrogen) was cultured overnight at 37°C in SOC broth. Bacteria were washed once in PBS and fixed in 2% paraformaldehyde for 20 min, then washed extensively before counting by Flow Cytometry (FACSVerse, BD Biosciences) and added to the THP1 (a human monocytic cell line derived from an acute monocytic leukemia patient) in a bacteria/cell ratio of 100:1 for 24 h. PBMCs were cultured for 1 h in round-bottom 96-well plates in the presence of E. coli-activated THP1 (1:2) or PMA (250 ng/μl) plus ionomycin (1 μg/μl) as positive control. Brefeldin A (10 μg/ml) was added and cells were incubated for further 4 h. Cells were harvested and stained as previously described. For the intracellular quantification of cytokine production, the following antibodies were used: IL-17 PE (Miltenyi Biotech), IFNγ PE and TNFα FITC (BD Biosciences).
The assay setup for both PMA/iono and bacterial stimulation was optimized on 2 HIV– and 1 HIV+ subjects. During each round of experiments, PBMCs from the same batch of the same HIV– subject were included. This allowed us to verify that the culture conditions were reliable and that the stimuli were working as expected.
Endotoxin Core Antibodies (EndoCab) and Intestinal Fatty Acid Binding Protein (I-FABP) were measured by ELISA (R&D systems), in accordance with the manufacturer’s instructions. Circulating lipopolysaccharide (LPS) was assessed using the LAL test (Lonza), as per the manufacturer’s instructions. Samples were diluted 1:150 and preheated at 95°C for 10 min.
Total DNA was extracted as previously described (Paisse et al., 2016). The V3-V4 hypervariable regions of the 16S rDNA were amplified and quantified by qPCR, sequenced with MiSeq technology, and clustered into operational taxonomic units (OTUs) before taxonomic assignment as described (Garidou et al., 2015; Lelouvier et al., 2016; Paisse et al., 2016).
The total 16S rDNA present in the samples was measured by qPCR in triplicate and normalized using a plasmid-based standard scale (Vaiomer SAS, Labége, France). The targeted metagenomic sequences from fecal and plasma microbiota were analyzed using the bioinformatics pipeline established by Vaiomer from the FROGS guidelines. Briefly, after demultiplexing of the barcoded Illumina paired reads, single read sequences were cleaned and paired for each sample independently into longer fragments. After quality-filtering and alignment against a 16S reference database, a clustering into OTU with a 97% identity threshold and a taxonomic assignment were performed in order to determine community profiles. The list of possible species for each unique sequence was further investigated using the BLASTN program from NCBI Blast against the NCBI 16S Microbial database. Only the BLASTN hits covering the full query sequence length with an overall sequence identity of 97% or more were considered as possible species. DNA from plasma (240 mL) was extracted using a DNA isolation kit (NucleoSpin Plasma XS, Macherey-Nagel). All DNA extracts were stored at –80°C until further processing.
Reads obtained from the MiSeq sequencing system have been processed using Vaiomer bioinformatics pipeline. Based on the results, graphical representations were made of the relative proportion of taxa for each taxonomic level (phylum, class, order, family, genus, and species) present in individual study samples. Taxa are identified by name in the plot for abundance >1%. Taxa are merged into the “Other” category only if it exists in any sample with abundance greater than 0.01%. Taxa are merged into the “Multi-affiliation” category when they can correspond to two or more different taxa. Alpha diversity (α-diversity) represents the mean of species diversity per sample in each group/class. Diversity analysis is presented for (1) observed, (2) Chao1, (3) Shannon, (4) Simpson, and (5) inverse Simpson. Finally, the output matrix containing the relative abundance of OTUs per sample was processed with the linear discriminant analysis effect size (LEfSe) algorithm (Segata et al., 2011) using an alpha cut-off of 0.05 and an effect size cut-off of 2.0.
Continuous variables were expressed as median and interquartile range (IQR), whereas categorical variables were expressed as absolute numbers and percentages. The different groups of patients and the different time points were compared using Chi-squared or Fisher’s exact test, Kruskal–Wallis or Wilcoxon matched pairs test as appropriate and the correlations among variables were tested by Spearman Rank correlation. Dunn’s multiple comparison test was performed as appropriate: a line below the p-value showing which groups are being compared and resulted significant has been added in each graph. p-Values < 0.05 were considered statistically significant. Data were analyzed with GraphPad 6.2 Prism (GraphPad Software Inc.).
A total of 13 HIV+ mono-infected, 13 HCV+ mono-infected, 30 HCV+/HIV+ co-infected patients and 13 age-matched HIV- and HCV-uninfected healthy controls (HC) were enrolled in the study. Patients’ epidemiological and clinical characteristics are presented in Table 1. Virally infected patients were preferentially men (p = 0.031, Table 1), with a higher proportion of men who have sex with men (MSM) and intravenous drug users (IVDU) (p < 0.0001). With regard to HIV-related features, HIV+ and HCV+/HIV+ patients were comparable in terms of CD4 nadir and % of CD4 count at time of analysis, duration of infection and suppressive cART (Table 1). With regard to HCV infection, HCV+/HIV+ patients showed significantly higher levels of circulating HCV-RNA (p = 0.007, Table 1) compared to HCV+, yet similar duration of infection, liver fibrosis, as assessed by ultrasound transient elastography and HCV genotype distribution (Table 1). As expected, HCV+ infected patients (mono-infected and HIV+ co-infected) showed higher serum transaminases compared to both healthy controls and HIV+ individuals (p < 0.0001, Table 1).

Table 1. Epidemiological, clinical and immunological characteristics of the study groups.
The CD3+ MAIT cell population comprises CD4+ and CD4/CD8 double negative (DN) subsets, along with the CD8+ subpopulation. Given that CD8+ MAIT cells represent the majority of circulating MAIT, we decided to focus our attention on this specific subset. We first identified circulating Vα7.2+CD161++CD8+ (MAIT) by flow cytometry (gating strategy shown in Figure 1). Compared to healthy individuals, virally infected patients displayed a significantly lower proportion of peripheral MAIT cells (p < 0.0001; Figure 2A), with no differences in IL18R-expressing Vα7.2+CD161++CD8+ (p = 0.061; Figure 2B). No differences in PD-1- (p = 0.236; Figure 2C), CD39-expressing Vα7.2+CD161++CD8+ (p = 0.774; Figure 2D) and activated CD69+Vα7.2+CD161++CD8+ (p = 0.414; Figure 2E) were shown in virally infected subjects when compared to HC. Conversely, virally infected patients displayed significantly higher perforin+ (p = 0.004; Figure 2F) and granzyme B+ MAIT cells (p = 0.002; Figure 2G), with no differences between the three groups of infected patients. Comparable MAIT frequency and phenotypes were shown in patients stratified according to liver fibrosis (F0–F2 vs. F3–F4) (data not shown). Similar data were shown with regard to total CD3+ MAIT cells: virally infected patients were confirmed to display a lower Vα7.2+CD161++CD3+ frequency (p = 0.003) with a similar proportion of activated/exhausted MAIT, yet higher cytolytic activity (granzyme B: p = 0.007) versus healthy controls (Supplementary Figures S1a–g). Furthermore, virally infected patients presented lower frequency of CD4/CD8 double negative MAIT (HIV–: 6.14% vs. HIV+: 0.36% vs. HCV+: 0.22% vs. HCV+/HIV+ 1.56%; p < 0.0001), yet higher level of PD-1-expressing (HIV–: 5.53% vs. HIV+: 4.07% vs. HCV+: 5.52% vs. HCV+/HIV+ 42.1%; p = 0.010), Granzyme B-expressing (HIV–: 0.19% vs. HIV+: 15.4% vs. HCV+: 19.3% vs. HCV+/HIV+ 3.7%; p = 0.003) and Perforin-expressing CD4/CD8 double negative MAIT (HIV–: 0.37% vs. HIV+: 1.12% vs. HCV+: 2.3% vs. HCV+/HIV+ 27.8%; p = 0.0027).
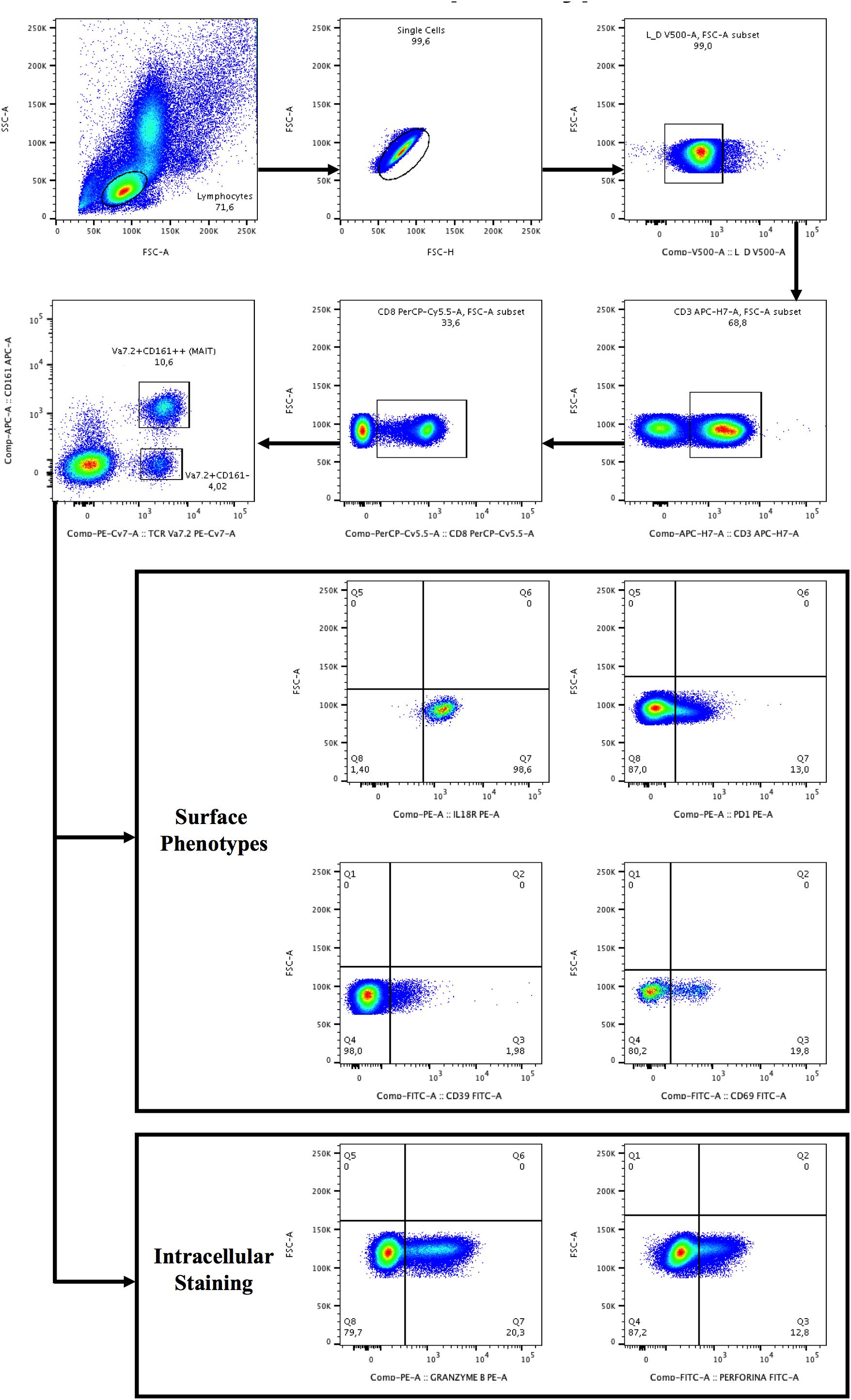
Figure 1. Gating strategy for the identification of Vα7.2+CD161++CD8+ and Vα7.2+CD161-CD8+ T-cell subset and surface/intracellular phenotypes. Lymphocytes were gated from forward (FSC) and side scatters (SSC), doublets were removed and live cells were selected, segregated for CD3+ T-cells and subsequently for CD8+ T-cells. CD8+ T+cells were categorized as CD161- or CD161++ (bright), followed by the selection of TCR Vα7.2. The gates were set up based on positive versus negative peak. Depicted are representative plots from representative subject PBMCs.
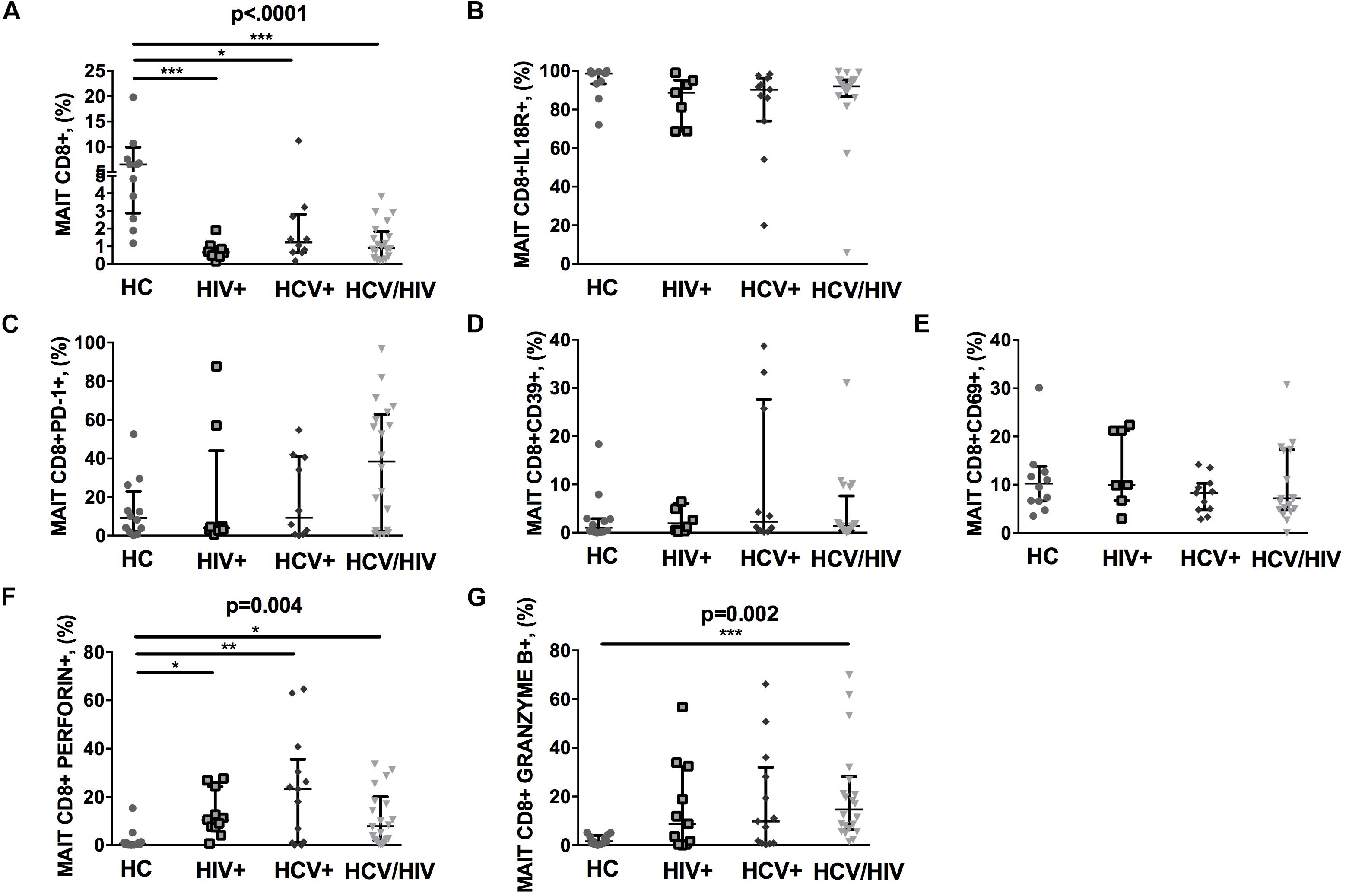
Figure 2. Frequency and phenotypes of Vα7.2+CD161++CD8+ T cells. The lines indicate the significant comparison between two groups. ∗ indicates the p-value for each pair of groups: ∗ p < 0.05, ∗∗ p < 0.01, ∗∗∗ p < 0.001. (A) Compared to healthy individuals (HC), virally infected patients displayed a significant lower proportion of peripheral MAIT cells (HC: 6.47%, HIV+: 0.65%, HCV+: 1.22%, HCV+/HIV+: 0.91%, p < 0.0001). (B) Virally infected individuals showed similar IL18R-expressing MAIT cells (HC: 98.70%, HIV+: 88.8%, HCV+: 90.4%, HCV+/HIV+: 92%, p = 0.061). (C) Similar levels of PD-1-expressing MAIT cells (HC: 9.15%, HIV+: 3.84%, HCV+: 9.28%, HCV+/HIV+: 38.45%, p = 0.236), (D) CD39-expressing MAIT cells (HC: 1.04%, HIV+: 1.91%, HCV+: 2.30%, HCV+/HIV+ 1.38%, p = 0.774), and (E) CD69-expressing MAIT cells (HC: 10.25%, HIV+: 9.96%, HCV+: 8.33%, HCV+/HIV+: 7.15%, p = 0.414) were observed among study groups. (F) Compared to the uninfected controls, virally infected patients featured higher perforin-producing (HC: 0.59%, HIV+: 10.5%, HCV+: 23.2%, HCV+/HIV+: 7.86%, p = 0.004) and (G) granzyme B-producing MAITs (HC: 1.53%, HIV+: 8.78%, HCV+: 9.79%, HCV+/HIV+: 14.6%, p = 0.002).
To further characterize MAIT cells during chronic viral infections, we sought to investigate their ability to produce cytokines following in vitro challenge (Figure 3A shows the gating strategy).
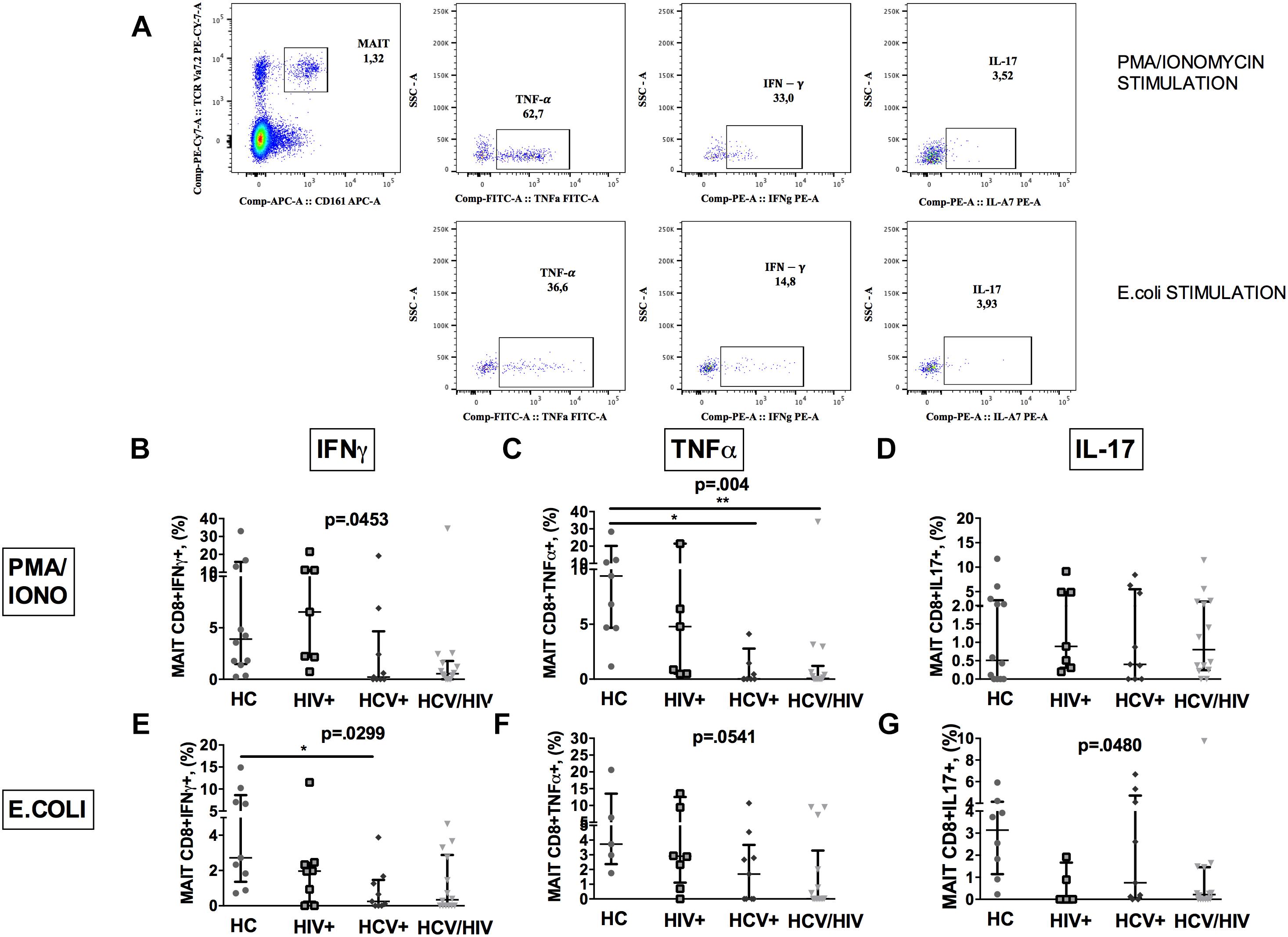
Figure 3. Vα7.2+CD161++CD8+ cytokine production following ex vivo stimulation. The lines indicate the significant comparison between two groups. ∗ indicates the p-value for each pair of groups: ∗ p < 0.05, ∗∗ p < 0.01, ∗∗∗ p < 0.001. (A) Flow cytometry gating strategy for cytokine production. (B) Stimulation with PMA/ionomycin resulted in significantly lower production of IFNγ (HC: 3.57%, HIV+: 4.39%, HCV+: 0.22%, HCV+/HIV+: 0.54%, p = 0.045) in virally infected, particularly co-infected individuals, compared to HC. (C) Co-infected individuals showed the lowest TNFα release after PMA/ionomycin (HC: 8.11%, HIV+: 4.39%, HCV+: 0.02%, HCV+/HIV+: 0.07%, p = 0.004). (D) Similar production of IL-17 between the study groups following PMA/ionomycin exposure (HC: 0.43%, HIV+: 0.70%, HCV+: 0.40%, HCV+/HIV+: 0.80%, p = 0.883). (E) Following E. coli stimulation, MAITs of infected patients produced inadequate quantity of IFNγ (HC: 2.72%, HIV+: 1.96%, HCV+: 0.25%, HCV+/HIV+: 0.33%, p = 0.025). (F) Virally infected patients showed lower TNFα production upon bacterial challenge (HC: 3.73%; HIV+: 0.17%, HCV+: 1.69%, HCV+/HIV+: 0.01%, p = 0.054) (G) E. coli exposure resulted in lower IL-17 production in virally infected patients vs. HC (HC: 3.14%, HIV+: 0%, HCV+: 0.75%, HCV+/HIV+: 0.21%, p = 0.048).
PMA/ionomycin stimulation resulted in significantly lower production of IFNγ (p = 0.0453; Figure 3B) and TNFα (p = 0.004; Figure 3C), but not of IL-17 (p = 0.883; Figure 3D) in virally infected, particularly HCV+/HIV+ co-infected individuals, compared to controls.
Having shown dysfunctional MAIT intracellular cytokine production and given the role of microbial translocation in supporting immune inflammation/activation (Brenchley et al., 2006), we next investigated MAIT response to ex vivo bacterial challenge. Upon E. coli stimulation, a lower percentage of MAIT cells from virally infected patients produced IFNγ (p = 0.0299; Figure 3E), TNFα (p = 0.0541; Figure 3F) and IL-17 (p = 0.048; Figure 3G) when compared to uninfected controls, which overall suggests a defective anti-bacterial MAIT effector function in chronically virally infected patients.
Similarly, total CD3+ MAIT cells of virally infected subjects showed lower production of cytokines following both PMA/ionomycin (IFNγ: p = 0.078; TNFα: p = 0.030) and E. coli exposure (IFNγ: p = 0.028; TNFα: p = 0.048) as compared to healthy individuals (Supplementary Figures S1h–m). CD4/CD8 double negative MAIT cell subset confirmed the same trend (data not shown).
Given the well-recognized role of microbial translocation (MT) in sustaining chronic immune activation and disease progression in HIV infection (Brenchley et al., 2006; Paiardini et al., 2008; Estes et al., 2010; Marchetti et al., 2011, 2013), we explored the possible association between MAIT impairment and markers of damaged intestinal integrity and MT.
We found similar plasma levels of I-FABP, a marker of enterocyte damage, between infected patients and controls (p = 0.158; Figure 4A), as well as direct markers of MT (LPS, p = 0.188; 16S rDNA, p = 0.261; Figures 4B,C). Conversely, virally infected subjects displayed significantly lower endotoxin core antibodies (EndoCAb) (p = 0.026; Figure 4D), supporting inefficient control over translocating bacteria.
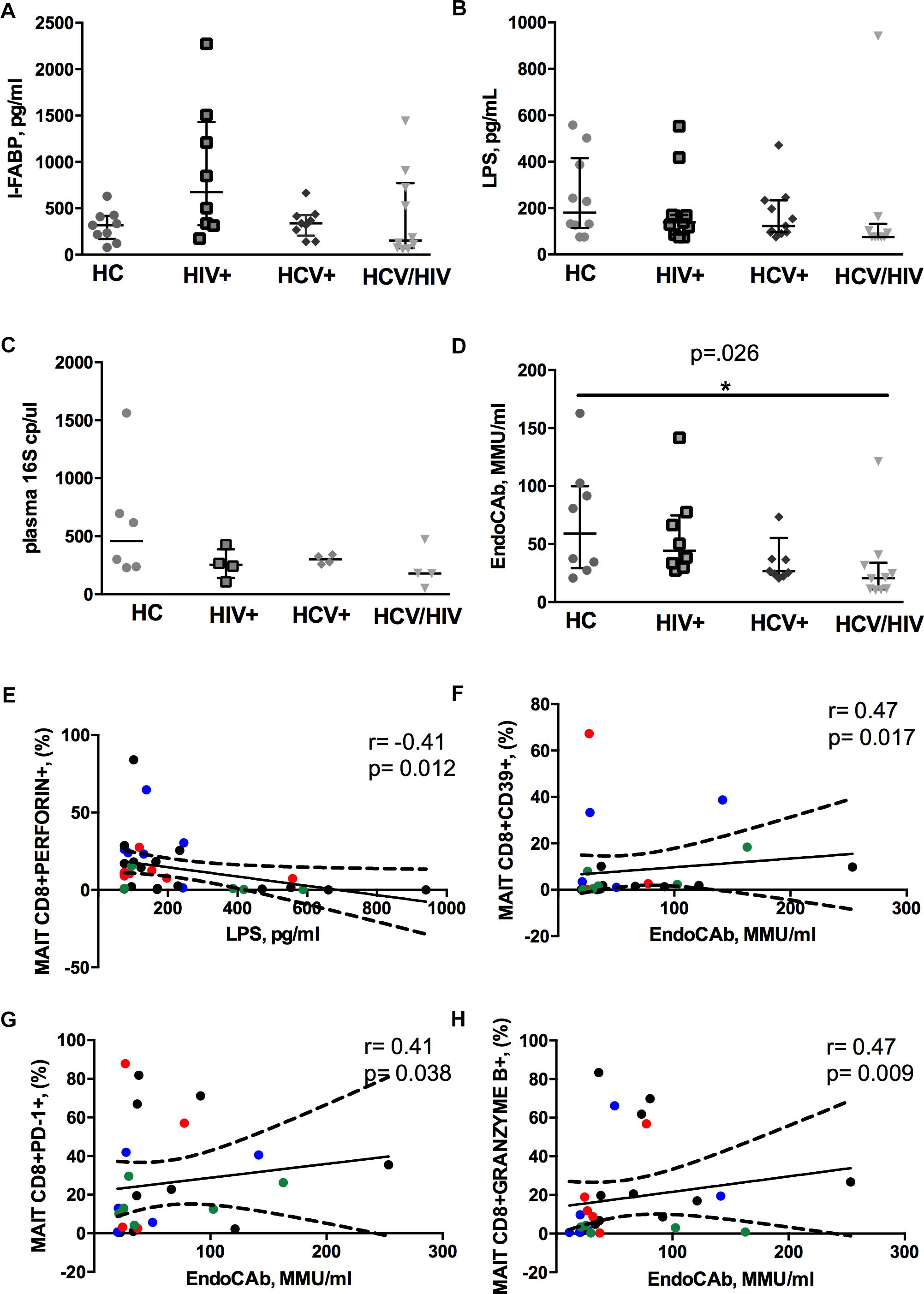
Figure 4. Circulating microbial translocation and gut damage markers and their associations with Vα7.2+CD161++CD8+. The lines indicate the significant comparison between two groups. ∗ indicates the p-value for each pair of groups: ∗ p < 0.05, ∗∗ p < 0.01, ∗∗∗ p < 0.001. (A) The levels of I-FABP, marker of enterocyte damage, were similar between the donor group and the infected patients (HC: 317 pg/ml [169–418], HIV+: 675 pg/ml [319–1431], HCV+: 339 pg/ml [206–426], HCV/HIV: 154 pg/ml [72–771], p = 0.158). (B) We observed similar plasma levels of LPS (HC: 180 pg/ml [114–416], HIV+: 138 pg/ml [86–170], HCV+ 123 pg/ml [96–234], HCV/HIV: 75 pg/ml [75–132], p = 0.188) between the study groups. (C) Circulating 16S rDNA in a subgroup of 5 patients per group was comparable (HC: 458 cp/ml [235–913], HIV+: 253 cp/ml [141–387], HCV+: 301 cp/ml [264–338], HCV/HIV: 179 cp/ml [83–400], p = 0.261). (D) The plasma levels of endotoxin core antibodies (EndoCAb) were significantly lower in virally infected subjects, particularly HCV/HIV co-infected patients (HC: 59 MMU/ml [29–100], HIV+ 44 MMU/ml [31–75], HCV+: 27 MMU/ml [23–55], HCV/HIV: 21 MMU/ml [11–34] p = 0.026). (E–H) Each patients’ cohort is marked by a different color: HC are represented in green, HIV+ in red, HCV+ in blue, HCV/HIV co-infected patients in black. (E) LPS plasma levels negatively correlate with perforin-expressing MAITs (r = –0.41, p = 0.012). (F) EndoCAb levels were positively associated with exhausted MAITs (CD39 +): r = 0.47, p = 0.017. (G) Vα7.2+CD161++CD8+PD1+ T-cells were positively associated with EndoCAb levels (r = 0.41, p = 0.38). (H) Granzyme B-expressing MAITs positively associate with plasma EndoCAb (r = 0.47, p = 0.09).
Interesting correlative patterns were evident between MT and MAIT phenotype: perforin-expressing MAITs negatively correlated with circulating LPS (r = –0.41, p = 0.012; Figure 4E), while activated/exhausted CD39+/PD-1+ and granzyme B+ MAITs positively associated with EndoCAb (CD39+: r = 0.47, p = 0.017; PD1+: r = 0.41, p = 0.038; granzyme B+: r = 0.47, p = 0.009; Figures 4F–H).
Because CD161 cell-surface down-regulation was suggested as a mechanism behind MAIT cell depletion during chronic infections (Leeansyah et al., 2013), we sought to investigate the phenotype and function of Vα7.2+CD161-CD8+ T-cells (gating strategy shown in Figure 1).
HCV+/HIV+ co-infected patients featured similar Vα7.2+CD161-CD8+ cell proportions (p = 0.918; Figure 5A), significantly lower IL18R+ (p = 0.0003; Figure 5B), and higher PD-1+ (p = 0.030; Figure 5C), yet similar CD39+ (p = 0.667; Figure 5D) and higher CD69+Vα7.2+CD161-CD8+ (p = 0.002; Figure 5E). Virally infected patients displayed significantly higher levels of perforin (p < 0.0001; Figure 5F) and similar granzyme B+ expression in Vα7.2+CD161-CD8+ cells (p = 0.105; Figure 5G).
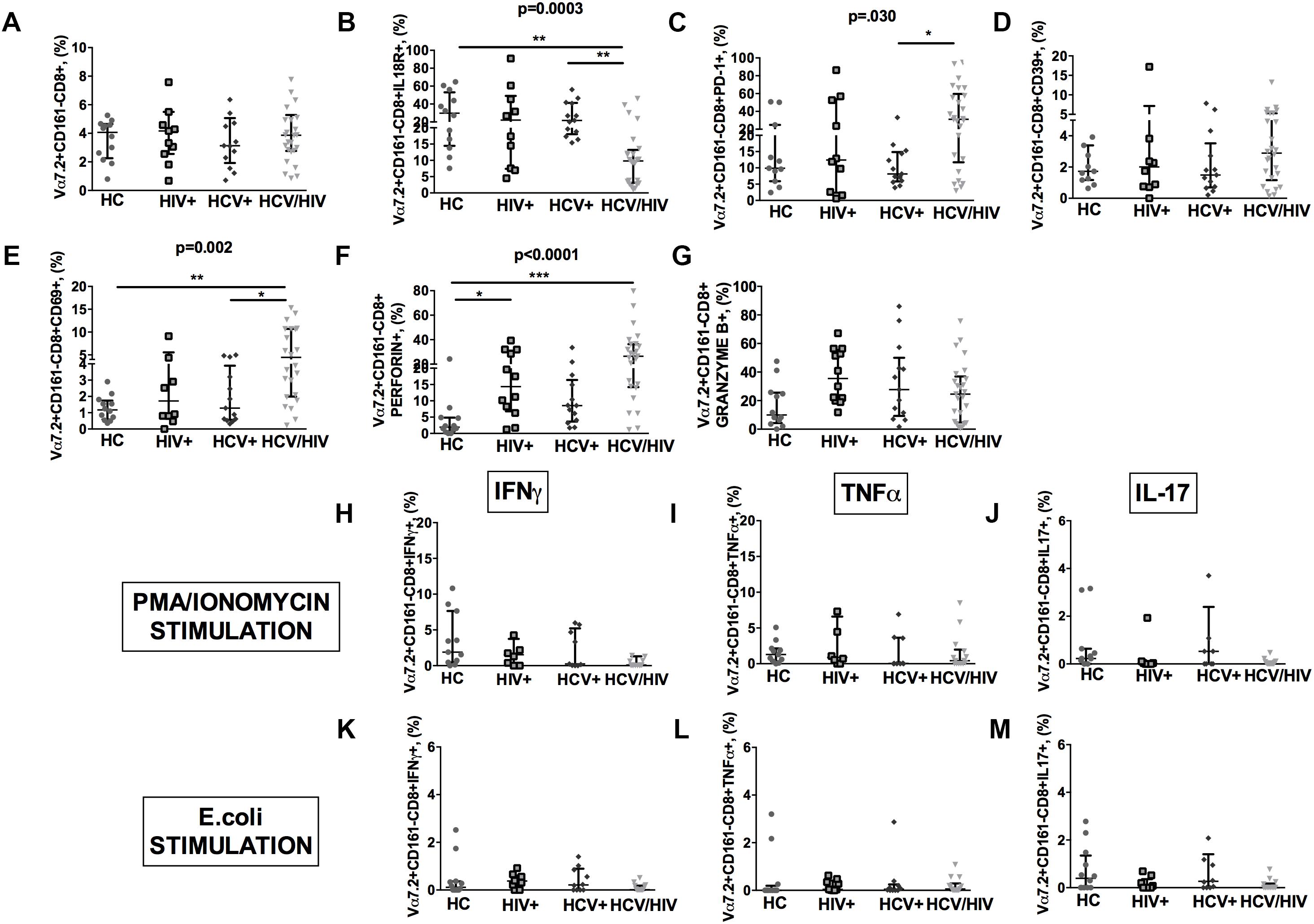
Figure 5. Frequency, phenotypes and cytokine production of Vα7.2+CD161-CD8+ T-cells. The lines indicate the significant comparison between two groups. ∗ indicates the p-value for each pair of groups: ∗ p < 0.05, ∗∗ p < 0.01, ∗∗∗ p < 0.001. (A) The frequency of circulating Vα7.2+CD161-CD8+ subset was similar among the four study groups (HC: 4.07%, HIV+: 4.17%, HCV+: 3.13%, HCV+/HIV+: 3.87%, p = 0.918). (B) HCV+/HIV+ co-infected patients featured significantly lower levels of IL18R (HC: 29.8%, HIV+: 22%, HCV+: 21.7%, HCV+/HIV+: 9.81%, p = 0.0003). (C) Similar levels of CD39 on Vα7.2+CD161-CD8+ were found (HC: 1.73%, HIV+: 2.01%, HCV+: 1.49%, HCV+/HIV+: 2.89%, p = 0.667). (D) Vα7.2+CD161-CD8+PD-1+ T-cells were significantly increased in co-infected subjects (HC: 9.89%, HIV+: 12.4%, HCV+: 8.15%, HCV+/HIV+: 31.2%, p = 0.030). (E) The highest levels of Vα7.2+CD161-CD8+CD69+ were found in HCV/HIV patients (HC: 1.17%, HIV+: 1.72%, HCV+: 1.28%, HCV+/HIV+: 4.46%, p = 0.002). (F) We observed significantly higher levels of perforin (HC: 1.91%, HIV+: 14.3%, HCV+: 8.54%, HCV+/HIV+: 26.5%, p < 0.0001) in virally infected patients. (G) Similar granzyme B levels were found in virally infected patients, as compared to the control group (HC: 10%, HIV+: 35.4%, HCV+: 27.7%, HCV+/HIV+: 24.6%, p = 0.105). Vα7.2+CD161-CD8+ T-cell population produced very limited cytokine amount following both (H–J) PMA/ionomycin and (K–M) E. coli stimulation, with no differences between the study groups.
As to their cytokine-producing profile, Vα7.2+CD161-CD8+ population produced very limited quantities of cytokines following both PMA/ionomycin and E. coli exposure, with no differences between the study groups (Figures 5H–M).
Given the role of MAIT cells in gut immunity and in anti-bacterial response (Napier et al., 2015), we next questioned whether MAIT defects in our cohort of virally infected patients might be linked to gut microbiota dysbiosis.
In a subgroup of 5 subjects per group, we first evaluated the gut microbiota complexity, by calculating richness (observed and Chao1) and evenness (Shannon Entropy and Simpson) indexes and failed to find any differences among the study groups (Figures 6A–D).
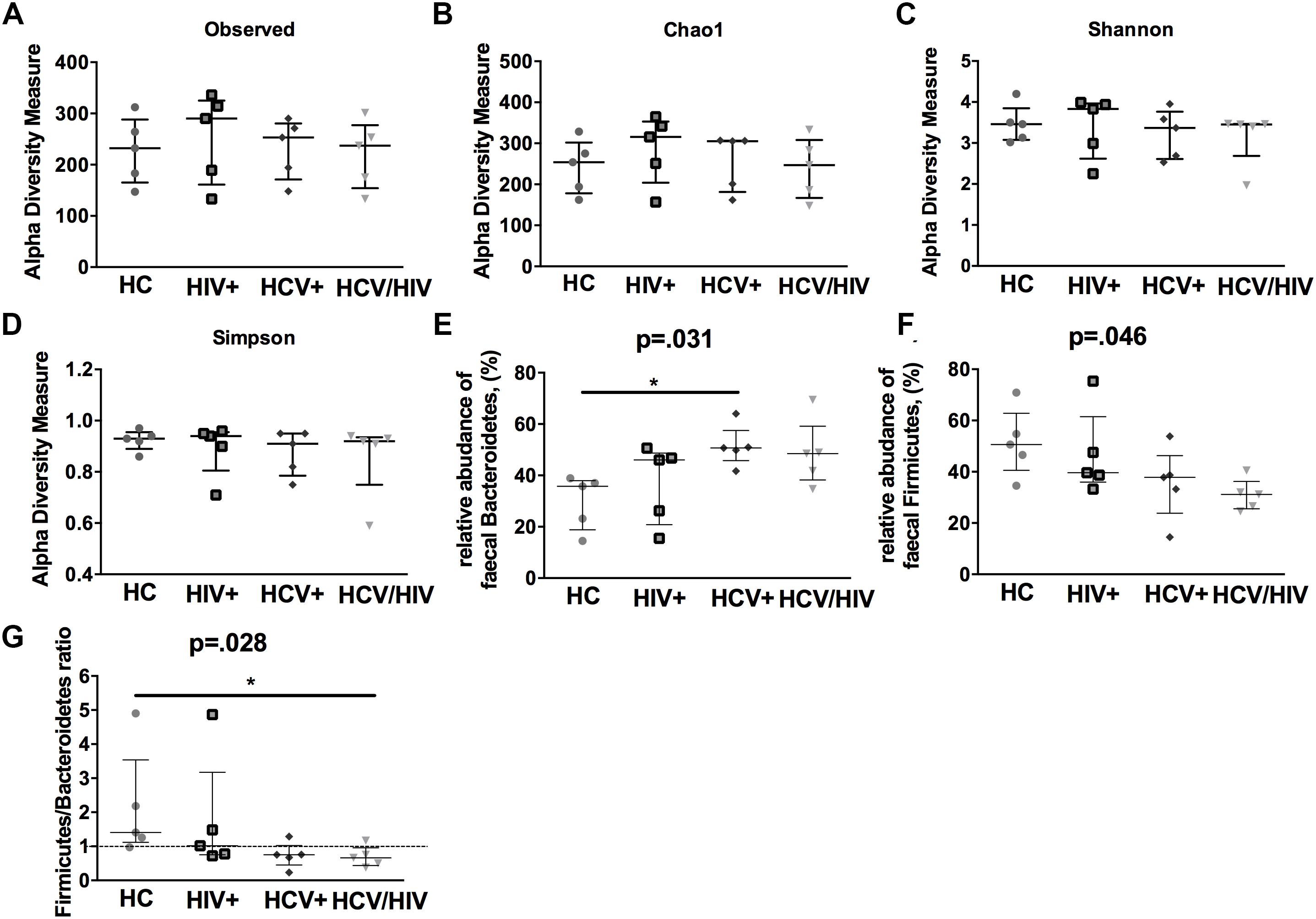
Figure 6. Alpha-diversity indexes and fecal bacteria relative abundance. The lines indicate the significant comparison between two groups. ∗ indicates the p-value for each pair of groups: ∗ p < 0.05, ∗∗ p < 0.01, ∗∗∗ p < 0.001. We calculated (A,B) richness (observed and Chao1) and evenness (C,D) (Shannon and Simpson) indexes, failing to find any differences among the study groups. (E) higher relative abundance of Bacteroidetes (p = 0.031), (F) lower relative abundance of Firmicutes (G) and lower Firmicutes/Bacteroidetes ratio (p = 0.028).
We further characterized the relative proportion of fecal bacteria species at each taxonomic level (phylum, class, order, family, genus and species), finding significant differences between virally infected individuals and healthy controls. Interestingly, virally infected patients and particularly co-infected individuals displayed higher relative abundance of Bacteroidetes (p = 0.031; Figure 6E), lower relative abundance of Firmicutes (p = 0.046; Figure 6F) and lower Firmicutes/Bacteroidetes ratio (p = 0.028; Figure 6G).
Finally, by using the linear discriminant analysis (LDA) effect size (LEfSe) with LDA score >2 as the cut-off, we observed an enrichment of Bacteroidetes phylum in HCV+ and an impoverishment in Firmicutes phylum in HCV+/HIV+ when compared to HC (Figure 7), with no differences among virally infected groups (data not shown).
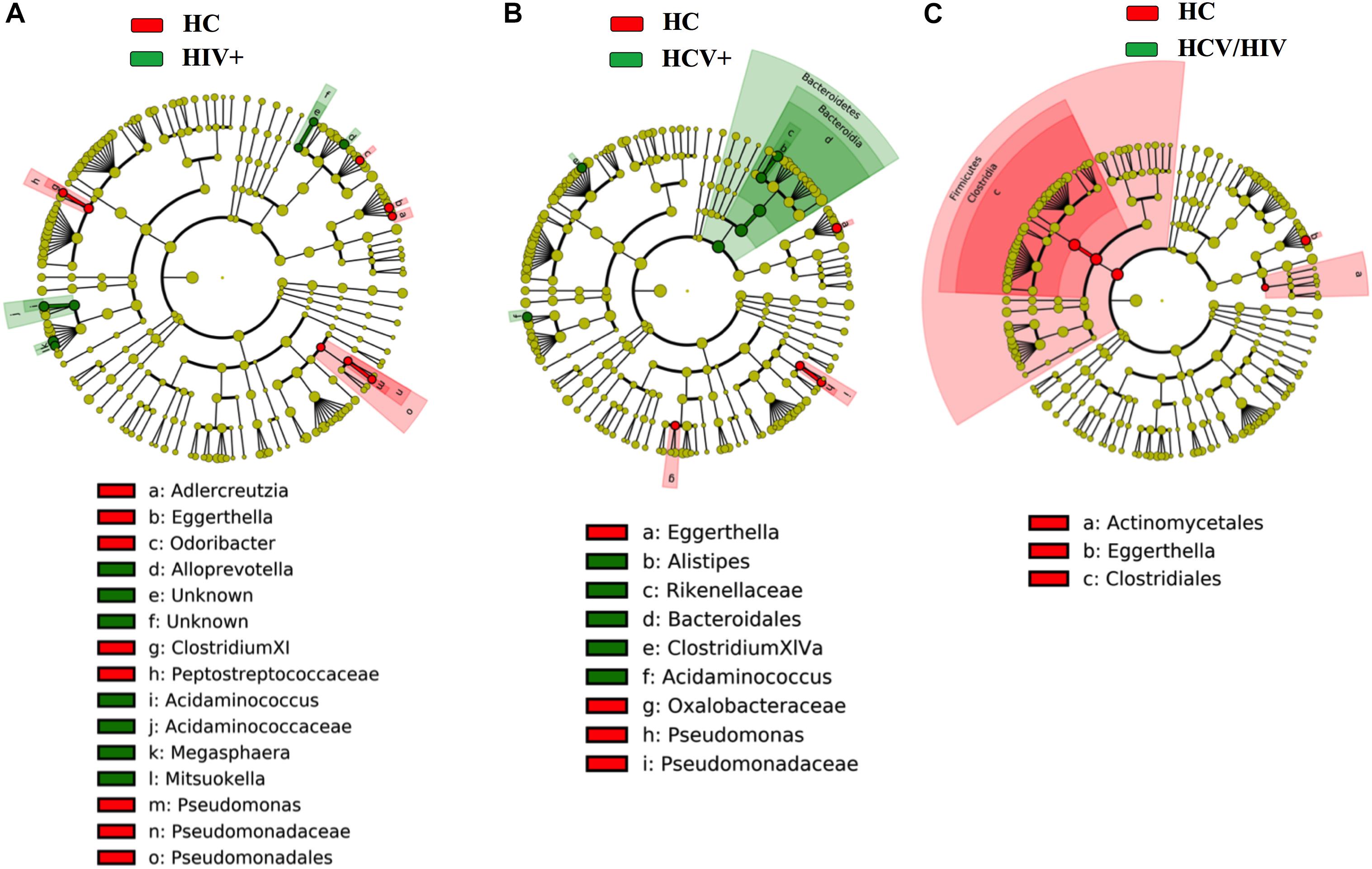
Figure 7. Fecal bacteria beta diversity analysis: LEfSe Analysis. Analysis of differences in the microbiota between the study groups using LEfSe software (LDA, coupled with effect size measurement). (A–C) HCV+ individuals (in green) featured higher content of bacteria belonging to the Bacteroidetes phylum, HCV+/HIV+ loss of bacteria belonging to the Firmicutes phylum as compared to healthy controls (in red).
We next assessed whether differences in gut microbiota composition might be connected to the disrupted MAIT cell and Vα7.2+CD161- T-cell frequency and function.
The frequency of circulating MAIT cells was positively associated with the relative abundance of Bacteroides spp. (r = 0.51, p = 0.046; Figure 8A), some of which are known to be inducers of regulatory T-cell functions (Troy and Kasper, 2010).
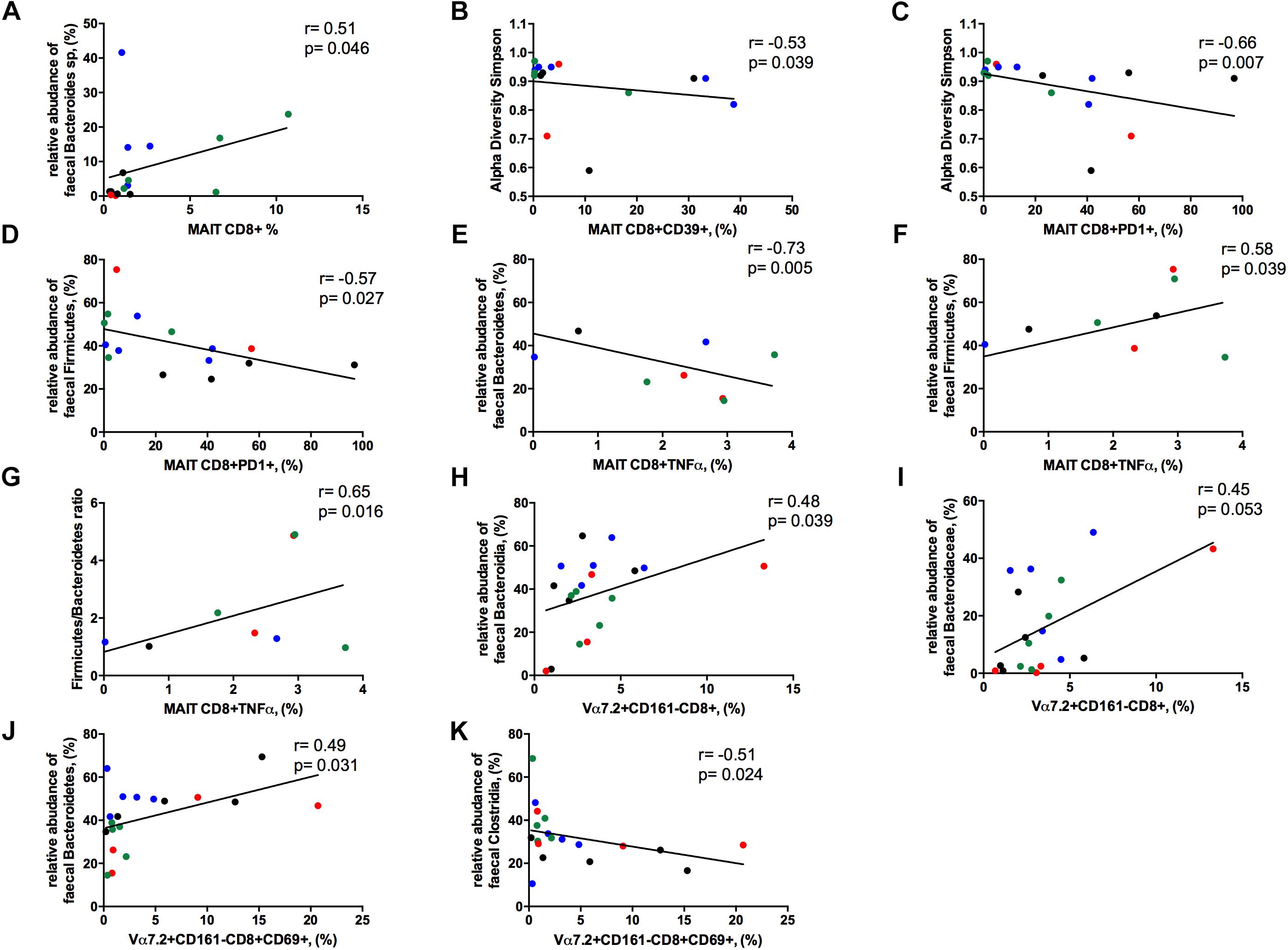
Figure 8. Correlations between Vα7.2+CD161++CD8+ or Vα7.2+CD161-CD8+ and gut dysbiosis. Each patients’ cohort is marked by a different color: HC are represented in green, HIV+ in red, HCV+ in blue, HCV/HIV co-infected patients in black. (A) The frequency of circulating MAITs was positively associated with the relative abundance of Bacteroides spp. (r = 0.51, p = 0.046). (B) The levels of exhausted Vα7.2+CD161++CD8+39+ were negatively correlated with alpha diversity Simpson index (r = –0.53, p = 0.039). (C) Vα7.2+CD161++CD8+PD1+ negatively associated with alpha diversity Simpson index (r = –0.66, p = 0.007). (D) PD-1-expressing MAITs showed an inverse association with the relative abundance of Firmicutes (r = –0.57, p = 0.027). MAIT ability to produce TNFα following bacterial challenge was negatively associated with the (E) relative abundance of Bacteroidetes (r = –0.73, p = 0.005) and positively with the (F) relative abundance of Firmicutes (r = 0.58, p = 0.039) and (G) with Firmicutes/Bacteroidetes ratio (r = 0.65, p = 0.016). (H) The frequency of Vα7.2+CD161-CD8+ T-cells were positively associated with the order Bacteroidia (r = 0.477, p = 0.039). (I) The family of Bacteroidaceae correlates with the proportion of Vα7.2+CD161-CD8+ T-cell subset (r = 0.45, p = 0.053). (J) The activated Vα7.2+CD161-CD8+ were positively associated to the relative abundance of Bacteroidetes (r = 0.49, p = 0.031) (K) CD69-expressing Vα7.2+CD161-CD8+ T-cells negatively correlated with the order Clostridia (r = –0.51, p = 0.024).
The levels of activated/exhausted CD39+ and PD1+ MAIT cells correlated negatively with alpha diversity Simpson index (r = –0.53. p = 0.039; r = –0.66. p = 0.007, respectively; Figures 8B,C). Moreover, PD-1-expressing MAIT showed an inverse association with the relative abundance of Firmicutes (r = –0.57, p = 0.027; Figure 8D). Interestingly, MAIT ability to produce TNFα following bacterial challenge negatively associated with the relative abundance of Bacteroidetes (r = –0.73, p = 0.005; Figure 8E) and positively with the relative abundance of Firmicutes (r = 0.58, p = 0.039; Figure 8F) and with Firmicutes/Bacteroidetes ratio (r = 0.65, p = 0.016; Figure 8G).
Contrarily, Vα7.2+CD161-CD8+ T-cells positively associated with the order Bacteroidia (r = 0.48, p = 0.039; Figure 8H) with a trend toward the family of Bacteroidaceae (r = 0.45, p = 0.053; Figure 8I). Besides, the levels of activation on this subset were positively associated to the relative abundance of Bacteroidetes (r = 0.49, p = 0.031; Figure 8J) and negatively correlated with the order Clostridia (r = –0.51, p = 0.024; Figure 8K).
We found no association between MAIT cells and markers of HIV disease progression (CD4, HIV-RNA levels, duration of infection and of cART), and HCV disease progression (HCV-RNA levels and length of infection).
In chronically HIV- and/or HCV-infected patients, we hereby show a significant loss of circulating MAIT cells, the residual proportion of which display higher production of perforin and granzyme B, and yet an impaired ex vivo function. Further, Vα7.2+CD161-CD8+ T-cells, despite equally frequent in virally infected and healthy individuals, feature an activated phenotype and yet appear functionally impaired. Interestingly, both MAIT cells and Vα7.2+CD161-CD8+ T-cells significantly associate with gut dysbiosis.
In line with literature data (Barathan et al., 2016; Spaan et al., 2016), our study shows a massive depletion of the MAIT compartment in both HIV- and HCV-infected populations, with no differences among mono- and co-infection. The residual MAIT cells are functionally exhausted and display higher cytolysis markers, possibly indicating continuous antigen exposure. Given that microbial translocation may stimulate innate immune cells via TLR pathways, leading to systemic immune activation (Ussher et al., 2016), we explored the association between activated/exhausted MAIT phenotypes and microbial translocation. Interestingly, while LPS plasma levels negatively correlate with perforin-expressing MAIT cells, supporting the role of endotoxin in hampering MAIT function, EndoCAb levels were positively associated with exhausted/activated (CD39/PD1) and granzyme B-expressing MAIT cells, suggesting the effort of the immune system to fight continuous microbial threat. Some studies suggested that rather than being depleted, MAIT cells have an altered phenotype, namely, the down-regulation of CD161; Leeansyah et al. (2013) suggested that continuous exposure to bacterial products in HIV disease may lead to MAIT cell exhaustion and loss, with concomitant expansion of the CD161neg population originated from the MAIT cells. Similarly, Freeman et al. (2017) showed that in immune failure patients, the reduction in peripheral MAIT cells seems to be due, at least in part, to a loss in CD161 expression, and not merely the result of trafficking into mucosal tissues or cell death. By contrast other studies, argued that Vα7.2+CD161-CD8+ are not MAIT cells (Leeansyah et al., 2013; Sandberg et al., 2013; Fergusson et al., 2014), since they do not retain their functions. In our cohort, we describe a Vα7.2+CD161-CD8+ T cell phenotype/function divergent from MAIT T cells, with preserved frequency, inefficient cytokine production, and no association with microbial translocation, altogether supporting the idea of a distinct T-cell population. However, given that the MAIT cell loss is a continuous long-term process, we could not exclude that what we described is the result of different and concomitant mechanisms, such as homing to sites of inflammation, exhaustion, and also loss of CD161 expression late in chronic diseases.
Having shown altered MAIT phenotypes during chronic viral infections, we next asked whether their anti-bacterial properties were also impaired. Interestingly, compared to healthy controls, we found that the ex vivo reactivity of MAIT cells to cells cultured together with bacteria is very weak, with HCV+/HIV+ co-infected patients displaying the lowest cytokine production, implying a defect in MAIT antimicrobial functions, possibly due to tolerance driven by continuous microbial translocation. However, the lack of association between MAIT ability to face bacteria challenge and MT markers, seems to suggest that other mechanisms might be involved, such as continuous MAIT engagement, in turn resulting in functional exhaustion, defects in the triggering and/or in signal transduction or deficiency in cytokine production/release.
Given the lack of direct MAIT stimulation by viruses, our finding of such MAIT impairment in both HIV and HCV chronic infections, raises the question of whether other factors rather than the viruses themselves are involved (van Wilgenburg et al., 2016). In the last few years, both HIV- and HCV-infected patients have been proven to feature a marked dysbiosis, that is not reverted by antiviral treatments (Bajaj et al., 2016; Williams et al., 2016). The homeostasis between microbiota, intestinal epithelium and innate and adaptive immunity favors the predominance of regulatory networks that prevent inflammation or immune-mediated disease (Duerkop et al., 2009; Maynard et al., 2012; Powell and MacDonald, 2017). Recently, some authors have reported a direct link between MAIT cells and gut microbiota. Indeed, MAIT cells have been demonstrated to discriminate and categorize complex human microbiota through computation of TCR signals and to shape their phenotypes and responses according to local environment-driven microbial metabolism (Schmaler et al., 2018; Tastan et al., 2018). Thus, we hypothesized that MAIT disruption might be associated with a decreased frequency of bacteria involved in epithelial barrier health and immune-regulation, and increased abundance of bacteria with known pro-inflammatory potential in the setting of chronic viral infections.
To test this hypothesis, we first investigated gut microbiota composition, confirming a profound dysbiosis in virally infected patients, mainly in HCV+/HIV+ co-infected individuals, delineating a Firmicutes-poor/Bacteroidetes-rich microbiota. Indeed, the Firmicutes phylum comprises some commensal bacteria with immune regulatory properties, while by contrast the Bacteroidetes phylum includes bacteria with proinflammatory properties (Eckburg et al., 2005). In view of this, our finding of a shift toward lower Firmicutes, such as Clostridia, might support the role of gut dysbiosis in sustaining immune activation as a consequence of regulatory function loss, possibly leading to clinical progression. The factors associated with HIV and/or HCV-driven dysbiosis still remain to be fully elucidated, whether it is elicited by the viruses themselves, or rather it is a consequence of differences in lifestyle (i.e., MSM vs. heterosexual), co-morbidities and the use of chronic therapies. Interestingly, our results seem to closely link gut dysbiosis and MAIT compartment. Indeed, we found a positive association between MAIT frequency and Bacteroides spp., some of which are reported to be inducers of regulatory T-cell function (Troy and Kasper, 2010), leading us to hypothesize a role of certain bacteria in modulating MAIT cells, probably improving the anti-bacterial properties of the immune system. A reduced diversity of the gut microbiota has been associated with immune dysfunction and reduced CD4 T-cell counts (Nowak et al., 2015), our observation of an inverse correlation between exhausted MAIT and alpha diversity suggests an important role of low microbiota diversity in weakening the efficiency of the host immune system.
In addition, we have shown an association between TNFα-producing MAIT cells and gut microbiota composition, with bacteria belonging to the Firmicutes phylum positively correlating with MAIT TNFα production following ex vivo E. coli stimulation, while bacteria belonging to the Bacteroidetes phylum negatively associating with MAIT TNFα release. Despite the fact that the small number of samples tested might limit the statistical power of the findings, our data altogether seem to suggest a link between gut dysbiosis and inflammation, supporting the hypothesis of a direct effect of altered microbiota composition on immune function, possibly posing viral-mediated dysbiosis as a central player in the hampered anti-bacterial MAIT capability. As intriguing and interesting as it is, we acknowledge that the few samples tested do not allow definitive conclusions to be drawn and need to be confirmed in larger cohorts.
Lastly, we didn’t find any association between MAIT cells and usual markers of both HIV and HCV diseases, probably as a reflection of the long history of infection, hence supporting the theory of significant and permanent damages of MAIT compartment early in the infection.
To our knowledge this is the first study to investigate the associations between gut microbiota and MAIT cells in chronic HIV and HCV infections. The correlative nature of our study did not allow us to show a causal relationship, however, the observations that certain bacteria may associate to MAIT activation/function support the hypothesis of a reciprocal interaction of MAIT cells and intestinal microbiota, paving the way to possible future research on microbiome-targeted therapy aimed at restoring mucosal immunity in chronic HIV and HCV infections. The next step, i.e., the in vitro stimulations of the MAIT cells using representative members of the identified phyla/genera will clarify the mechanisms and the effects exerted by these bacteria on MAIT cell population.
Several limitations must be acknowledged. The small sample size might have hampered the ability to detect a relationship between MAIT cells and liver fibrosis that has been described in literature (Beudeker et al., 2018; Hegde et al., 2018). Likewise, microbiota analyses on larger cohorts with extensive evaluation of microbial community function, might have provided broader pathogenic insight. Similarly, the study of bacteria composition, gut damage and MAIT cells directly at mucosal sites, might have shed light onto the causal relationship between mucosal adherent microbiota. Furthermore, given the limited sample size we were forced to merge the patient groups in the correlative analyses. Thus, we could not exclude that by mixing cohorts of patients with markedly different viruses we might have lost some differences. Finally, the availability of CD4 count in HIV non-infected groups might have helped us in explaining some of our finding.
In conclusion, the MAIT compartment is profoundly impaired in chronic HIV and HCV, possibly as a consequence of the marked dysbiosis that features both infections. The microbial and environmental signals that lead to the migration, differentiation, expansion and maintenance of unconventional T-cells under physiological and pathological conditions are poorly defined and need to be further studied. An additional imperative is to explore whether differences in microbial communities result into modification in key bacterial functional pathways that drive mucosal immune dysfunction.
All datasets generated for this study are included in the manuscript and the Supplementary Files.
This study was carried out in accordance with the recommendations of “Comitato Etico, ASST Santi Paolo e Carlo” with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the “Comitato Etico, ASST Santi Paolo e Carlo.”
EM and MC designed the study, designed and performed experiments, analyzed and interpreted the data, designed the figures, and wrote the manuscript. BvW, LS, and EC performed the experiments and helped with analyzing the data. AdM helped with interpreting the results and edited the manuscript. PK and GM conceived and designed the study, interpreted the data, and wrote the manuscript.
This study was supported in part by the Italian Ministry of Health, Regione Lombardia, grant “Giovani Ricercatori” (Number GR-2009-1592029) to GM and grant “Ricerca Finalizzata” (Number NET-2013-02355333-3) to GM.
MC was awarded a European Union Lifelong Learning Program LLP/Erasmus studentship to conduct part of the analysis at the University of Oxford. GM received research grant by the Italian Ministry of Health.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all the patients who participated in the study and the staff of the Clinic of Infectious Diseases and Tropical Medicine at “ASST Santi Paolo e Carlo” who cared for the patients. This study was presented in part at CROI 2017, February 13–16, 2017, Seattle, WA, United States. Poster #239.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01972/full#supplementary-material
FIGURE S1 | Frequency, phenotypes and cytokine production of Vα7.2+CD161++CD3+ T-cells. The lines indicate the significant comparison between two groups. ∗ indicates the p-value for each pair of groups: ∗ p < 0.05, ∗∗ p < 0.01, ∗∗∗ p < 0.001. (a) Virally infected patients were confirmed to display lower Vα7.2+CD161++CD3+ (p = 0.003) frequency as compared to healthy controls. (b–e) Similar proportion of activated/exhausted MAIT cells was observed among the study groups. (f) Comparable levels of perforin-producing MAIT cells. (g) Higher granzyme B-producing MAIT cells (p = 0.007) in virally infected patients as compared to healthy controls. (h–j) Upon PMA/ionomycin stimulation, total CD3+ MAIT cells of virally infected subjects showed lower production of cytokines (IFNγ: p = 0.078; TNFα: p = 0.030). (k–m) E. coli exposure resulted in lower cytokine production (IFNγ: p = 0.028; TNFα: p = 0.048) as compared to healthy individuals.
Bajaj, J. S., Sterling, R. K., Betrapally, N. S., Nixon, D. E., Fuchs, M., Daita, K., et al. (2016). HCV eradication does not impact gut dysbiosis or systemic inflammation in cirrhotic patients. Aliment. Pharmacol. Ther. 44, 638–643. doi: 10.1111/apt.13732
Barathan, M., Mohamed, R., Vadivelu, J., Chang, L. Y., Saeidi, A., Yong, Y. K., et al. (2016). Peripheral loss of CD8(+) CD161(++) TCRValpha7.2(+) mucosal-associated invariant T cells in chronic hepatitis C virus-infected patients. Eur. J. Clin. Invest. 46, 170–180. doi: 10.1111/eci.12581
Beudeker, B. J. B., van Oord, G. W., Arends, J. E., Schulze Zur Wiesch, J., van der Heide, M. S., de Knegt, R. J., et al. (2018). Mucosal-associated invariant T-cell frequency and function in blood and liver of HCV mono- and HCV/HIV co-infected patients with advanced fibrosis. Liver Int. 38, 458–468. doi: 10.1111/liv.13544
Bolte, F. J., O’Keefe, A. C., Webb, L. M., Serti, E., Rivera, E., Liang, T. J., et al. (2017). Intra-hepatic Depletion of Mucosal Associated Invariant T cells in Hepatitis C Virus-induced Liver Inflammation. Gastroenterology 153, 1392–1403. doi: 10.1053/j.gastro.2017.07.043
Brenchley, J. M., Price, D. A., Schacker, T. W., Asher, T. E., Silvestri, G., Rao, S., et al. (2006). Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 12, 1365–1371.
Cosgrove, C., Ussher, J. E., Rauch, A., Gartner, K., Kurioka, A., Huhn, M. H., et al. (2013). Early and nonreversible decrease of CD161++/MAIT cells in HIV infection. Blood 121, 951–961. doi: 10.1182/blood-2012-06-436436
Costello, M. E., Ciccia, F., Willner, D., Warrington, N., Robinson, P. C., Gardiner, B., et al. (2015). Brief report: intestinal dysbiosis in ankylosing spondylitis. Arthritis Rheumatol. 67, 686–691. doi: 10.1002/art.38967
Dias, J., Sandberg, J. K., and Leeansyah, E. (2017). Extensive phenotypic analysis, transcription factor profiling, and effector cytokine production of human MAIT cells by flow cytometry. Methods Mol. Biol. 1514, 241–256.
Duerkop, B. A., Vaishnava, S., and Hooper, L. V. (2009). Immune responses to the microbiota at the intestinal mucosal surface. Immunity 31, 368–376. doi: 10.1016/j.immuni.2009.08.009
Dusseaux, M., Martin, E., Serriari, N., Peguillet, I., Premel, V., Louis, D., et al. (2011). Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood 117, 1250–1259. doi: 10.1182/blood-2010-08-303339
Eberhard, J. M., Kummer, S., Hartjen, P., Hufner, A., Diedrich, T., Degen, O., et al. (2016). Reduced CD161+ MAIT cell frequencies in HCV and HIV/HCV co-infection: is the liver the heart of the matter? J. Hepatol. 65, 1261–1263.
Eckburg, P. B., Bik, E. M., Bernstein, C. N., Purdom, E., Dethlefsen, L., Sargent, M., et al. (2005). Diversity of the human intestinal microbial flora. Science 308, 1635–1638.
Estes, J. D., Harris, L. D., Klatt, N. R., Tabb, B., Pittaluga, S., Paiardini, M., et al. (2010). Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. 6:e1001052. doi: 10.1371/journal.ppat.1001052
Fergusson, J. R., Smith, K. E., Fleming, V. M., Rajoriya, N., Newell, E. W., Simmons, R., et al. (2014). CD161 defines a transcriptional and functional phenotype across distinct human T cell lineages. Cell Rep. 9, 1075–1088. doi: 10.1016/j.celrep.2014.09.045
Fernandez, C. S., Amarasena, T., Kelleher, A. D., Rossjohn, J., McCluskey, J., Godfrey, D. I., et al. (2015). MAIT cells are depleted early but retain functional cytokine expression in HIV infection. Immunol. Cell Biol. 93, 177–188. doi: 10.1038/icb.2014.91
Freeman, M. L., Morris, S. R., and Lederman, M. M. (2017). CD161 expression on mucosa-associated invariant t cells is reduced in HIV-infected subjects undergoing antiretroviral therapy who do not recover CD4(+) T Cells. Pathog. Immun. 2, 335–351. doi: 10.20411/pai.v2i3.136
Gaardbo, J. C., Trosied, M., Stiksrud, B., Midttun, O., Ueland, P. M., Ullum, H., et al. (2015). Increased tryptophan catabolism is associated with increased frequency of CD161+Tc17/MAIT cells and lower CD4+ T-cell count in HIV-1 infected patients on cART after 2 years of follow-up. J. Acquir. Immune Defic. Syndr. 70, 228–235. doi: 10.1097/QAI.0000000000000758
Garidou, L., Pomie, C., Klopp, P., Waget, A., Charpentier, J., Aloulou, M., et al. (2015). The gut microbiota regulates intestinal CD4 T cells expressing RORgammat and controls metabolic disease. Cell Metab. 22, 100–112. doi: 10.1016/j.cmet.2015.06.001
Grimaldi, D., Le Bourhis, L., Sauneuf, B., Dechartres, A., Rousseau, C., Ouaaz, F., et al. (2014). Specific MAIT cell behaviour among innate-like T lymphocytes in critically ill patients with severe infections. Intensive Care Med. 40, 192–201. doi: 10.1007/s00134-013-3163-x
Hegde, P., Weiss, E., Paradis, V., Wan, J., Mabire, M., Sukriti, S., et al. (2018). Mucosal-associated invariant T cells are a profibrogenic immune cell population in the liver. Nat. Commun. 9:2146. doi: 10.1038/s41467-018-04450-y
Hengst, J., Strunz, B., Deterding, K., Ljunggren, H. G., Leeansyah, E., Manns, M. P., et al. (2016). Nonreversible MAIT cell-dysfunction in chronic hepatitis C virus infection despite successful interferon-free therapy. Eur. J. Immunol. 46, 2204–2210. doi: 10.1002/eji.201646447
Hevia, A., Milani, C., Lopez, P., Cuervo, A., Arboleya, S., Duranti, S., et al. (2014). Intestinal dysbiosis associated with systemic lupus erythematosus. mBio 5:e1548-14. doi: 10.1128/mBio.01548-14
Jiang, J., Wang, X., An, H., Yang, B., Cao, Z., Liu, Y., et al. (2014). Mucosal-associated invariant T-cell function is modulated by programmed death-1 signaling in patients with active tuberculosis. Am. J. Respir. Crit. Care Med. 190, 329–339. doi: 10.1164/rccm.201401-0106OC
Johansson, M. A., Bjorkander, S., Mata Forsberg, M., Qazi, K. R., Salvany Celades, M., Bittmann, J., et al. (2016). Probiotic Lactobacilli modulate staphylococcus aureus-induced activation of conventional and unconventional T cells and NK cells. Front. Immunol. 7:273. doi: 10.3389/fimmu.2016.00273
Kjer-Nielsen, L., Patel, O., Corbett, A. J., Le Nours, J., Meehan, B., Liu, L., et al. (2012). MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 491, 717–723. doi: 10.1038/nature11605
Kurioka, A., Ussher, J. E., Cosgrove, C., Clough, C., Fergusson, J. R., Smith, K., et al. (2015). MAIT cells are licensed through granzyme exchange to kill bacterially sensitized targets. Mucosal Immunol. 8, 429–440. doi: 10.1038/mi.2014.81
Le Bourhis, L., Dusseaux, M., Bohineust, A., Bessoles, S., Martin, E., Premel, V., et al. (2013). MAIT cells detect and efficiently lyse bacterially-infected epithelial cells. PLoS Pathog. 9:e1003681. doi: 10.1371/journal.ppat.1003681
Le Bourhis, L., Guerri, L., Dusseaux, M., Martin, E., Soudais, C., and Lantz, O. (2011). Mucosal-associated invariant T cells: unconventional development and function. Trends Immunol. 32, 212–218.
Le Bourhis, L., Martin, E., Peguillet, I., Guihot, A., Froux, N., Core, M., et al. (2010). Antimicrobial activity of mucosal-associated invariant T cells. Nat. Immunol. 11, 701–708. doi: 10.1038/ni.1890
Lee, Y. K., and Mazmanian, S. K. (2010). Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 330, 1768–1773. doi: 10.1126/science.1195568
Leeansyah, E., Ganesh, A., Quigley, M. F., Sonnerborg, A., Andersson, J., Hunt, P. W., et al. (2013). Activation, exhaustion, and persistent decline of the antimicrobial MR1-restricted MAIT-cell population in chronic HIV-1 infection. Blood 121, 1124–1135. doi: 10.1182/blood-2012-07-445429
Lelouvier, B., Servant, F., Paisse, S., Brunet, A. C., Benyahya, S., Serino, M., et al. (2016). Changes in blood microbiota profiles associated with liver fibrosis in obese patients: a pilot analysis. Hepatology 64, 2015–2027. doi: 10.1002/hep.28829
Leung, D. T., Bhuiyan, T. R., Nishat, N. S., Hoq, M. R., Aktar, A., Rahman, M. A., et al. (2014). Circulating mucosal associated invariant T cells are activated in Vibrio cholerae O1 infection and associated with lipopolysaccharide antibody responses. PLoS Negl. Trop. Dis. 8:e3076. doi: 10.1371/journal.pntd.0003076
Marchetti, G., Cozzi-Lepri, A., Merlini, E., Bellistrì, G. M., Castagna, A., Galli, M., et al. (2011). Microbial translocation predicts disease progression of HIV-infected antiretroviral-naive patients with high CD4+ cell count. AIDS 25, 1385–1394. doi: 10.1097/QAD.0b013e3283471d10
Marchetti, G., Tincati, C., and Silvestri, G. (2013). Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin. Microbiol. Rev. 26, 2–18. doi: 10.1128/CMR.00050-12
Maynard, C. L., Elson, C. O., Hatton, R. D., and Weaver, C. T. (2012). Reciprocal interactions of the intestinal microbiota and immune system. Nature 489, 231–241. doi: 10.1038/nature11551
Mazmanian, S. K., Round, J. L., and Kasper, D. L. (2008). A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453, 620–625. doi: 10.1038/nature07008
McDermott, A. J., and Huffnagle, G. B. (2014). The microbiome and regulation of mucosal immunity. Immunology 142, 24–31. doi: 10.1111/imm.12231
Napier, R. J., Adams, E. J., Gold, M. C., and Lewinsohn, D. M. (2015). The role of mucosal associated invariant T cells in antimicrobial immunity. Front. Immunol. 6:344. doi: 10.3389/fimmu.2015.00344
Nowak, P., Troseid, M., Avershina, E., Barqasho, B., Neogi, U., Holm, K., et al. (2015). Gut microbiota diversity predicts immune status in HIV-1 infection. AIDS 29, 2409–2418. doi: 10.1097/QAD.0000000000000869
Paiardini, M., Frank, I., Pandrea, I., Apetrei, C., and Silvestri, G. (2008). Mucosal immune dysfunction in AIDS pathogenesis. AIDS Rev. 10, 36–46.
Paisse, S., Valle, C., Servant, F., Courtney, M., Burcelin, R., Amar, J., et al. (2016). Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion 56, 1138–1147. doi: 10.1111/trf.13477
Powell, N., and MacDonald, T. T. (2017). Recent advances in gut immunology. Parasite Immunol. 39:e12430.
Saeidi, A., Ellegard, R., Yong, Y. K., Tan, H. Y., Velu, V., Ussher, J. E., et al. (2016). Functional role of mucosal-associated invariant T cells in HIV infection. J. Leukoc. Biol. 100, 305–314. doi: 10.1189/jlb.4RU0216-084R
Salou, M., Franciszkiewicz, K., and Lantz, O. (2017). MAIT cells in infectious diseases. Curr. Opin. Immunol. 48, 7–14.
Sandberg, J. K., Dias, J., Shacklett, B. L., and Leeansyah, E. (2013). Will loss of your MAITs weaken your HAART [corrected]? AIDS 27, 2501–2504.
Sattler, A., Dang-Heine, C., Reinke, P., and Babel, N. (2015). IL-15 dependent induction of IL-18 secretion as a feedback mechanism controlling human MAIT-cell effector functions. Eur. J. Immunol. 45, 2286–2298. doi: 10.1002/eji.201445313
Schmaler, M., Colone, A., Spagnuolo, J., Zimmermann, M., Lepore, M., Kalinichenko, A., et al. (2018). Modulation of bacterial metabolism by the microenvironment controls MAIT cell stimulation. Mucosal Immunol. 11, 1060–1070. doi: 10.1038/s41385-018-0020-9
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Shi, N., Li, N., Duan, X., and Niu, H. (2017). Interaction between the gut microbiome and mucosal immune system. Mil. Med. Res. 4:14. doi: 10.1186/s40779-017-0122-9
Spaan, M., Hullegie, S. J., Beudeker, B. J., Kreefft, K., van Oord, G. W., Groothuismink, Z. M., et al. (2016). Frequencies of circulating MAIT cells are diminished in chronic HCV, HIV and HCV/HIV co-infection and do not recover during therapy. PLoS One 11:e0159243. doi: 10.1371/journal.pone.0159243
Sun, M., He, C., Cong, Y., and Liu, Z. (2015). Regulatory immune cells in regulation of intestinal inflammatory response to microbiota. Mucosal Immunol. 8, 969–978.
Tang, X. Z., Jo, J., Tan, A. T., Sandalova, E., Chia, A., Tan, K. C., et al. (2013). IL-7 licenses activation of human liver intrasinusoidal mucosal-associated invariant T cells. J. Immunol. 190, 3142–3152. doi: 10.4049/jimmunol.1203218
Tastan, C., Karhan, E., Zhou, W., Fleming, E., Voigt, A. Y., Yao, X., et al. (2018). Tuning of human MAIT cell activation by commensal bacteria species and MR1-dependent T-cell presentation. Mucosal Immunol. 11, 1591–1605. doi: 10.1038/s41385-018-0072-x
Troy, E. B., and Kasper, D. L. (2010). Beneficial effects of Bacteroides fragilis polysaccharides on the immune system. Front. Biosci. 15:25–34.
Ussher, J. E., Bilton, M., Attwod, E., Shadwell, J., Richardson, R., de Lara, C., et al. (2014). CD161++ CD8+ T cells, including the MAIT cell subset, are specifically activated by IL-12+IL-18 in a TCR-independent manner. Eur. J. Immunol. 44, 195–203. doi: 10.1002/eji.201343509
Ussher, J. E., van Wilgenburg, B., Hannaway, R. F., Ruustal, K., Phalora, P., Kurioka, A., et al. (2016). TLR signaling in human antigen-presenting cells regulates MR1-dependent activation of MAIT cells. Eur. J. Immunol. 46, 1600–1614. doi: 10.1002/eji.201545969
van Wilgenburg, B., Scherwitzl, I., Hutchinson, E. C., Leng, T., Kurioka, A., Kulicke, C., et al. (2016). MAIT cells are activated during human viral infections. Nat. Commun. 7:11653. doi: 10.1038/ncomms11653
Wang, Z. K., Yang, Y. S., Chen, Y., Yuan, J., Sun, G., and Peng, L. H. (2014). Intestinal microbiota pathogenesis and fecal microbiota transplantation for inflammatory bowel disease. World J. Gastroenterol. 20, 14805–14820. doi: 10.3748/wjg.v20.i40.14805
Williams, B., Landay, A., and Presti, R. M. (2016). Microbiome alterations in HIV infection a review. Cell Microbiol. 18, 645–651. doi: 10.1111/cmi.12588
Wong, E. B., Ndung’u, T., and Kasprowicz, V. O. (2017). The role of mucosal-associated invariant T cells in infectious diseases. Immunology 150, 45–54.
Wright, E. K., Kamm, M. A., Teo, S. M., Inouye, M., Wagner, J., and Kirkwood, C. D. (2015). Recent advances in characterizing the gastrointestinal microbiome in Crohn’s disease: a systematic review. Inflamm. Bowel Dis. 21, 1219–1228. doi: 10.1097/MIB.0000000000000382
Keywords: mucosal-associated invariant T-cells, HIV infection, HCV infection, microbial translocation, gut microbiota, dysbiosis
Citation: Merlini E, Cerrone M, van Wilgenburg B, Swadling L, Cannizzo ES, d’Arminio Monforte A, Klenerman P and Marchetti G (2019) Association Between Impaired Vα7.2+CD161++CD8+ (MAIT) and Vα7.2+CD161-CD8+ T-Cell Populations and Gut Dysbiosis in Chronically HIV- and/or HCV-Infected Patients. Front. Microbiol. 10:1972. doi: 10.3389/fmicb.2019.01972
Received: 08 April 2019; Accepted: 12 August 2019;
Published: 28 August 2019.
Edited by:
Daniel M. Altmann, Imperial College London, United KingdomReviewed by:
Hazel Marguerite Dockrell, University of London, United KingdomCopyright © 2019 Merlini, Cerrone, van Wilgenburg, Swadling, Cannizzo, d’Arminio Monforte, Klenerman and Marchetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giulia Marchetti, Z2l1bGlhLm1hcmNoZXR0aUB1bmltaS5pdA==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.