 Ulrike Kemmerling
Ulrike Kemmerling Antonio Osuna
Antonio Osuna Alejandro Gabriel Schijman
Alejandro Gabriel Schijman Carine Truyens
Carine Truyens- 1Programa de Anatomía y Biología del Desarrollo, Instituto de Ciencias Biomédicas, Facultad de Medicina, Universidad de Chile, Santiago, Chile
- 2Grupo de Bioquímica y Parasitología Molecular, Departamento de Parasitología, Instituto de Biotecnología, Universidad de Granada, Granada, Spain
- 3Molecular Biology of Chagas Disease Laboratory, Genetic Engineering and Molecular Biology Research Institute Dr. Héctor Torres (INGEBI-CONICET), Buenos Aires, Argentina
- 4Laboratory of Parasitology, Faculty of Medicine, Université Libre de Bruxelles, Brussels, Belgium
Chagas disease (CD), caused by the protozoan parasite Trypanosoma cruzi, is considered a neglected tropical disease by the World Health Organization. Congenital transmission of CD is an increasingly relevant public health problem. It progressively becomes the main transmission route over others and can occur in both endemic and non-endemic countries. Though most congenitally infected newborns are asymptomatic at birth, they display higher frequencies of prematurity, low birth weight, and lower Apgar scores compared to uninfected ones, and some suffer from severe symptoms. If not diagnosed and treated, infected newborns are at risk of developing disabling and life-threatening chronic pathologies later in life. The success or failure of congenital transmission depends on interactions between the parasite, the placenta, the mother, and the fetus. We review and discuss here the current knowledge about these parameters, including parasite virulence factors such as exovesicles, placental tropism, potential placental defense mechanisms, the placental transcriptome of infected women, gene polymorphism, and the maternal and fetal/neonatal immune responses, that might modulate the risk of T. cruzi congenital transmission.
Chagas Disease
Trypanosoma cruzi, a protozoan parasite, is the etiological agent of CD, a neglected tropical disease (WHO, 2015; Lenk et al., 2018) endemic to Latin American countries. Affected countries extend from the United States to Argentina and Chile (Pérez-Molina and Molina, 2018). The parasite is primarily transmitted by insect vectors, but can also be transmitted through blood transfusion, organ transplantation, consumption of parasite-contaminated food, and vertically from mother to child. In Latin America, CD is an important cause of cardiovascular morbidity and premature death (Rassi et al., 2012; WHO, 2015).
The disease evolves in two phases. The acute phase, defined by high parasitemia, lasts 2–3 months. It is often asymptomatic or involves non-specific flu-like symptoms. However, 2–12% of the infected individuals, mostly children under 3 years, die from acute myocardiopathy at times associated with a meningoencephalic compromise (Carlier and Truyens, 2015). The immune response eliminates most but not all parasites. As a consequence, the individual enters the chronic phase and remains infected for the rest of their lives. Although most patients remain asymptomatic for periods ranging in length from several months to decades, 30 – 40% of the infected people develop cardiac (mostly) and/or digestive tract (less frequent) pathologies that may lead to premature death (Rassi et al., 2012; de Oliveira et al., 2018).
Congenital Chagas Disease
Most women of gestational age are chronically infected, having acquired the infection during infancy. Around 5% of them transmit the parasite to their fetus, and thus approximately 9000 newborns infected with CD in Latin America are born every year. Cases of congenital CD have also been reported outside of the naturally endemic area (WHO, 2015; Antinori et al., 2017). Congenital infection is gaining importance as a route of T. cruzi transmission in endemic countries, due to control programs of vectorial and transfusional transmission routes, and is now estimated to account for 22% of new cases (WHO, 2015; Picado et al., 2018). In non-endemic countries, most of the new cases result from congenital transmission (Howard et al., 2014; WHO, 2015). Importantly, since T. cruzi maternal-fetal transmission can be repeated at each pregnancy and observed from one generation to another, this way of transmission can easily expand in time (Messenger et al., 2017).
Congenital CD is characteristically an acute parasite infection. Though most (around 60%) congenitally infected newborns are asymptomatic at birth, they display higher frequencies of low birth weight, prematurity, and lower Apgar scores at birth compared to uninfected newborns, while some infected newborns suffer from severe symptoms that can rapidly lead to death (Torrico et al., 2004; Liempi et al., 2016). Moreover, congenitally infected infants are at risk of disabling and life-threatening chronic pathologies later in life (Cardoso et al., 2012; Requena-Méndez et al., 2014; Carlier et al., 2015; Antinori et al., 2017). It is therefore essential to prevent congenital transmission and to rapidly diagnose and treat congenitally infected newborns.
Importantly, the occurrence of congenital transmission depends on interactions between T. cruzi, the placenta, the maternal immune system, and the developing fetal/neonatal immune response (Carlier and Truyens, 2015; Liempi et al., 2016). Here we will discuss the factors mentioned above that modulate the risk and probability of T. cruzi maternal-fetal transmission.
The Parasite Diversity
Trypanosoma cruzi belongs to the Kinetoplastida order, Trypanosomatidae family and presents a complex life cycle involving invertebrate triatomine hosts. Four main cellular forms can be identified during its life cycle: (i) the blood trypomastigote is the infective extracellular non-replicative form of the parasite, which is found in the bloodstream of the mammalian host, (ii) the metacyclic trypomastigote, also non-replicative, is present in the terminal portion of the digestive and urinary tracts of the vectors, (iii) the epimastigote is the replicative extracellular form of the parasite present in the triatomin insect vector, and (iv) the amastigote is the intracellular replicative form of the parasite in the vertebrate host (Rodrigues et al., 2014).
Trypanosoma cruzi is a paradigmatic case of predominantly clonal evolution (Tibayrenc et al., 1986) with “unequivocal evidence of genetic recombination” (Gaunt et al., 2003). Actually, the different known strains and clones of the parasite are classified into seven discrete typing units (DTUs), that are defined as a “sets of stocks that are genetically closer to each other than to any other stock and are identifiable by common molecular, genetic, biochemical, or immunological markers” (Tibayrenc, 1998). The DTUs ranged from T. cruzi I to T. cruzi VI, and lately, T. cruzi bat was also added (Zingales et al., 2012; Lima et al., 2015). Parasites from these DTUs are distributed differentially among insect and mammalian host species and therefore lives in different geographical areas (Zingales et al., 2012; Brenière et al., 2016). All T. cruzi parasites, regardless of the DTUs or where they belong, can cause CD. However, epidemiological studies suggest that T. cruzi I is associated with anthroponotic as well as sylvatic environments. On the other hand, T. cruzi II, V, and VI are mainly related to human environments and particularly chronic CD patients contrarily to T. cruzi III and IV which are found to be present in sylvatic habitats (Yeo et al., 2005). An association between DTUs and clinical outcomes is suspected (Messenger et al., 2015). These host and geographic specificities have been proposed to determine the probability of transmission and are related to the pathogenesis of CD. Parasites belonging to diverse DTUs have different biological properties, including growth rates in cultures, tropisms to tissue and organs, antigenicity, capacity to infect potential insect vectors, drug susceptibility, number of chromosomes, and DNA content (Macedo and Pena, 1998; Macedo et al., 2004). Characterization of the gene content and genome architecture of T. cruzi, as well as a whole-organism proteomic analysis of its four life cycle stages, has been reported (El-Sayed et al., 2005). Further, comparative genomics of different strains using different sequencing strategies and technologies provide data for the identification of genes associated with host tropism, pathogenicity, and modes of transmission (Berná et al., 2018; Callejas-Hernández et al., 2018; Reis-Cunha et al., 2018).
Whether all T. cruzi DTUs can be congenitally transmitted in utero is a matter of debate. There is presently no clear evidence that particular T. cruzi DTUs, as defined by the currently used molecular markers, would preferentially be transmitted congenitally [reviewed in Carlier and Truyens (2015), Truyens and Carlier (2017)]. We however, recently observed that congenital transmissions in Argentina and Mexico were associated with a maternal portage of “non-TcI” parasites (we did not detect congenital cases harboring TcI DTU) (Buekens et al., 2018). Concordantly, higher congenital transmission rates were reported in Brazilian regions where T. cruzi DTU V predominantly circulates as compared to a country where Tc II predominates (Luquetti et al., 2015).
Parasite Tropism for Placenta
Tissue tropism to the placenta of different T. cruzi strains has been described previously, particularly in the murine model (Andrade, 1982). T. cruzi I (Colombiana strain) parasites present a high incidence of placental parasitism (98%) compared to T. cruzi II (Y strain) parasites that only infect 17% of the placentas. The same strains also present differences regarding the localization of the amastigotes in the placenta; only the Colombian strain could be observed consistently in the vascular part of the placenta. In a more recent work, chronic infected mice with two different T. cruzi strains, one obtained from a congenital CD patient [VD: Tc VI (Risso et al., 2004)] and the other one previously characterized as non-transmissible in mice [K98,Tc I (Solana et al., 2002)] showed, by means of DNA amplification and 18s ribosomal RNA expression studies, that VD displayed stronger placental tropism and lower parasitic loads in peripheral blood than the K98 strain (Juiz et al., 2017). On the other hand, female mice infected with the K98 strain present higher parasitemia than the ones infected with the RA strain (DTU VI). However, in RA infected females, the infected mice produced parasite-infected newborns, while K98 did not.
We also showed recently in human placental explants (HPE), as well as in a placenta-derived epithelial cell line (BeWo), that VD parasites present a higher infection capacity compared to the Y strain (Medina et al., 2018). These studies suggest that the parasite genotype plays a role in tissue tropism toward the placenta and might contribute to the probability of congenital transmission.
Parasite Exovesicles as Virulence Factors
The persistence of T. cruzi parasites at a low level within specific tissues causes chronic pathogenic inflammation in some patients. How T. cruzi manages to persist and what factors released by the parasite influence its dynamics in tissue during chronic infection, remains poorly known (Teixeira et al., 2011). Nearly all cells can release extracellular vesicles, or exovesicles (EVs), which have been recognized as a mode of communication between cells. EVs may also participate in pathogenic processes (van Niel et al., 2006; Trocoli Torrecilhas et al., 2009; Cestari et al., 2012; de Pablos Torró et al., 2018). EVs are small membrane vesicles that are classified according to size, biogenesis, and composition, into exosomes and microvesicles (MVs). Exosomes, of 30 to 100 nm in size, are of endocytic origin, have a lipid bilayer, and are released into the extracellular compartment through the fusion of the multivesicular body (MVB) with the plasma membrane of the cell (Raposo and Stoorvogel, 2013). MVs are also referred to as ectosomes; are more heterogeneous in shape; and can vary between 100 and 300 nm in diameter. They are released as a result of the evagination toward the extracellular space from the plasma membrane. EVs differ not only in origin and size but also in lipid and protein composition. They have been identified in all biological fluids (Villagrasa et al., 2014), and their functions include intercellular communication, host-pathogen interactions, as well as the modulation of the immune response against infectious diseases or afflictions such as cancer (Schorey and Bhatnagar, 2008; Webber et al., 2015). We recently published reviews on the existence of EVs in protozoa and parasitic helminths and on the role of T. cruzi EVs (Marcilla et al., 2014; de Pablos Torró et al., 2018). Moreover, the induction of physiological modifications in cells by EVs derived from T. cruzi trypomastigotes has been recently demonstrated. These alterations involve the blocking of the cell cycle of the host cell, the permeabilization of the cells, and the disorganization of the cytoskeleton, among others (Retana Moreira et al., 2019).
EVs in T. cruzi were first described in 1970 (da Silveira et al., 1979) and found to be rich in glycoconjugates. Surface glycoconjugates are formed from glycoproteins (mucins) (de Lederkremer and Colli, 1995; Acosta-Serrano et al., 2001; Buscaglia et al., 2006; Mendonça-Previato et al., 2013), glycolipids (lipopeptidophosphoglycan-LPPG or glycoinosytolphospholipids-GIPL), and glycopeptides (NETNES) (Macrae et al., 2005). Electron microscopy and cryo-fracture have demonstrated that the vesicles come from the plasma membrane of the protozoan and the flagellar pocket. Recently, several surface proteins of the trans-sialidase, the cruzipain and the mucin-associated surface proteins (MASPs) families have been described in EVs, whose activity had previously been related to the modulation of the immune response by means of activating B cells and inducing a Th17 response or processes of adherence and cell invasion (Bermejo et al., 2011, 2013). Interestingly, MASPs are strongly expressed in the parasite and present a high variability between the different strains (Seco-Hidalgo et al., 2015). In recent works, we demonstrated that EVs, circulating in patients with CD, contained immature MASPs, with the presence of the C-proximal and N-terminal regions (peptide signal) (De Pablos et al., 2016; Díaz Lozano et al., 2017). MASPs are linked to the membrane through glycosylphosphatidylinositol (GPI) anchors. GPI participates in the inflammation processes that characterize CD (Almeida and Gazzinelli, 2001). The immature MASPs present in EVs (De Pablos et al., 2016; Díaz Lozano et al., 2017) are recognized by antibodies from CD patients. They help the parasite, in the presence of anti-T. cruzi antibodies, to evade the immune response by inhibiting the activity of C3 convertase and thereby impairing complement activation (Cestari et al., 2012). Similarly, the EVs isolated from the sera of CD patients are recognized both by the antibodies of the patients as well as by the serum of mice infected with the parasite. Therefore, it has been proposed that the immature MASPs in the outer side of the membrane of these EVs can form immunocomplexes with the IgGs of the patients (Díaz Lozano et al., 2017). In these studies, we demonstrate the involvement of EVs in the inhibition of the complement pathway, in which the terminal regions C- and N- of the immature MASPs participate. We also show how the antigens of the C- and N- immature regions of these MASPs inhibit the complement by antigen competition, in the presence of antibodies from CD patients. Besides, we identified differences in antigen recognition of the EVs by sera from the patients, depending on the pathology manifested by those affected by CD (De Pablos et al., 2016; Díaz Lozano et al., 2017). These results allowed us to consider T. cruzi EVs as markers of pathology, suggesting that they play a role in the pathogeny of CD. Their role in modulating the interactions between the parasite and the placenta and the congenital transmission is under investigation. However, preliminary results show that the parasite increases placenta-derived exosomes in HPE (Castillo et al., 2017a). The placenta is a rich source of exosomes which are related to fetal-placental-maternal communication, fetal allograft survival, and resistance to specific infections, among other functions (Ouyang et al., 2014; Schorey and Harding, 2016). Therefore, it is highly probable that placenta-derived exosomes play essential roles during T. cruzi-host interplay.
Placental Responses to T. cruzi
The placenta is a temporary organ that separates the maternal and fetal compartments throughout pregnancy (Arora et al., 2017). The placenta is responsible for the metabolic exchange between mother and fetus, and fulfills endocrine and immune functions that ensure normal prenatal development of the fetus and pregnancy-related changes in the mother (Liempi et al., 2016; Arora et al., 2017). Mainly due to its regulatory role in the maternal/fetal immune response, this organ is able to protect the fetus against several pathogens (Delorme-Axford et al., 2014; Mor et al., 2017) (see below).
The human placenta is a highly invasive (hemochorial) chorioallantoic placenta. The functional units, where the placental barrier is located, are the free-floating chorionic villi formed by the trophoblast and the villous stroma. Maternal blood surrounds and contacts the trophoblast in the placental intervillous space (IVS). The trophoblast is a bi-stratified covering epithelium composed of a superficial non-proliferative syncytiotrophoblast (ST) and a proliferative germinal layer, the cytotrophoblast (CT). The trophoblast is connected to and separated from the villous stroma (VS), the fetal connective tissue by a basal lamina, a specialized structure of extracellular matrix (ECM) (Benirschke et al., 2012). Therefore, trophoblast, basal laminae and VS, the latter containing fetal capillaries, form the placental barrier that must be crossed by T. cruzi in order to infect the fetus during trans-placental transmission (Figure 1; Duaso et al., 2010; Carlier et al., 2012; Liempi et al., 2016). Of note, T. cruzi displays a high tropism for the decidual part of the placenta as compared to the heart, that is likely related to the local immune environment [see further and (Mjihdi et al., 2002)]. Also, in the placenta of mothers of congenitally infected infants, only a few amastigotes can be found in stromal cells such as macrophages, fibroblasts, and giant cells while they are not or scarcely found in the trophoblast, except in case of severe mortal cases of congenital infection (Carlier and Truyens, 2015).
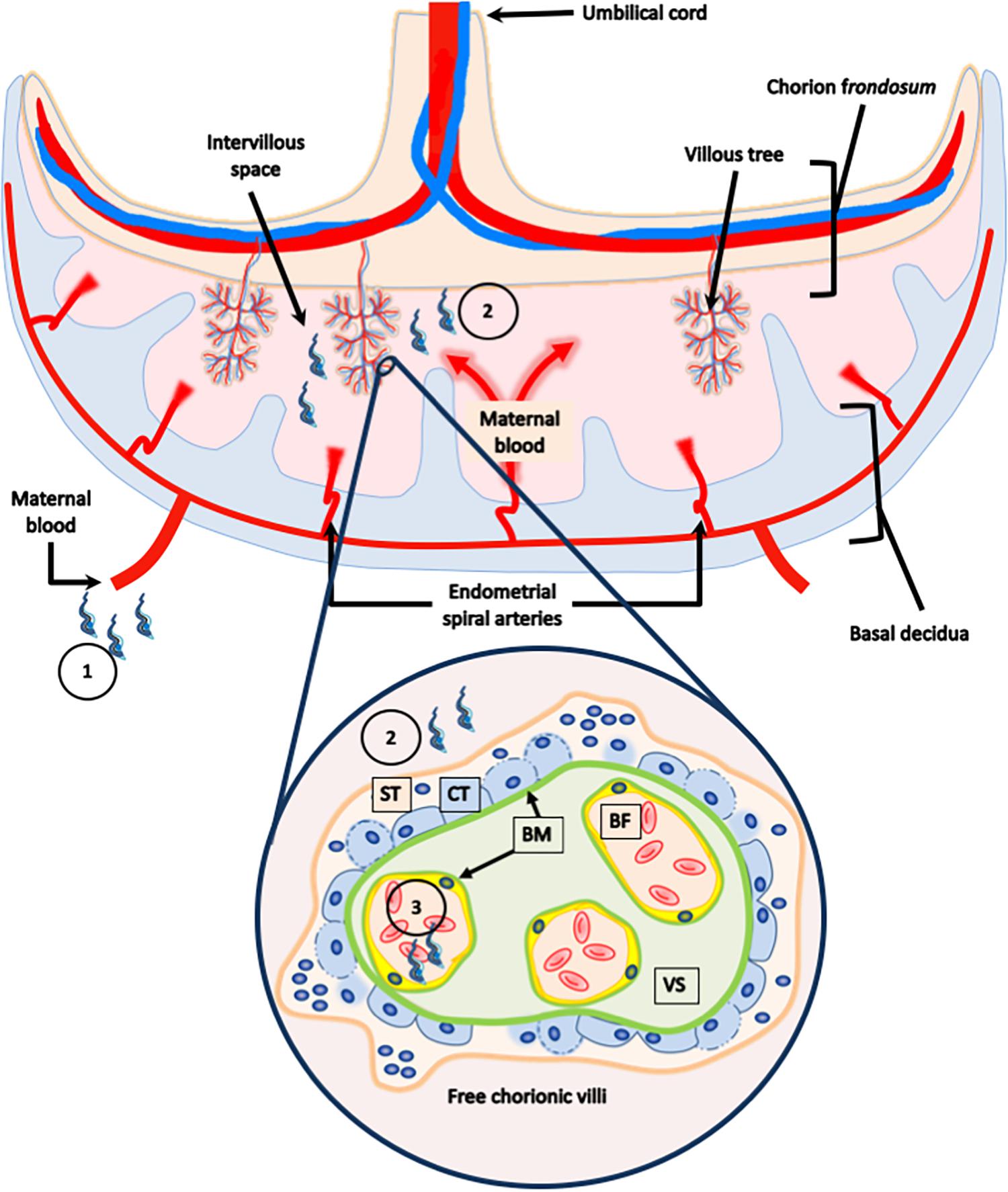
Figure 1. Human placental barrier. The human placenta is classified as a hemochorial chorioallantoic placenta. The placenta is composed of a fetal portion, developed from the chorion frondosum, and a maternal portion, or basal decidua, which originates from the endometrium. The functional units, were the placental barrier is located, are the free-floating chorionic villi formed by the trophoblast, and the villous stroma. Maternal blood surrounds and contacts the trophoblast in the placental intervillous space. The trophoblast is a bi-stratified covering epithelium composed of a superficial non-proliferative syncytiotrophoblast (ST), and a proliferate germinal layer, the cytotrophoblast (CT). The trophoblast is connected to and separated from the villous stroma (VS), the fetal connective tissue, and by a basal lamina. The parasite present in the maternal blood (1), that comes in contact with the trophoblast in the intervillous space (2), must cross the placental barrier in order to reach the fetal capillaries (3), and infect the fetus during transplacental transmission.
Since the trophoblast is the first placental tissue in contact with the maternal blood, the ST is the first fetal cell layer exposed to the parasite. It has been demonstrated that a high concentration of T. cruzi trypomastigotes induces the destruction of the trophoblast in ex vivo infected HPE (Duaso et al., 2010). However, low parasite concentration induces cellular proliferation (Liempi et al., 2016; Droguett et al., 2017) and differentiation (Liempi et al., 2014, 2016) in the same placental tissue (see below). The basal laminae, located between trophoblast and the fetal connective tissue and around the fetal vessels are other structures that T. cruzi needs to overcome (Duaso et al., 2010). T. cruzi can bind to ECM molecules such as glycosaminoglycans, fibronectin, and laminin (Lima et al., 2002) using surface molecules such as gp85 (Maeda et al., 2014) and gp83 (Nde et al., 2006). The VS, the placental fetal connective tissue, is also an important obstacle for the parasite. The parasite presents several proteases, among which the cruzipain has been proposed to be responsible for collagen I destruction and disorganization in placentas from mothers with chronic CD or in ex vivo infected HPE (Scharfstein and Morrot, 1999; Castillo et al., 2012; Duaso et al., 2012; Maeda et al., 2014). Matrix metalloproteinases (MMPs) such as MMP-2 and MMP-9 are activated, and their expression levels are increased in the presence of the parasite. Interestingly, if these MMPs are inhibited, the parasite-induced damage in the placental tissue is prevented, and the presence of parasite DNA is decreased in ex vivo infected HPE (Castillo et al., 2012). Importantly, the ECM forms a complex tridimensional network with different types of elastic fibers and collagen molecules including collagen I fibers, glycoproteins, and proteoglycans (Duaso et al., 2010; Theocharis et al., 2019). Collagen I fibers are the main components of the ECM, and if these molecules are disorganized or destroyed, the 3D network of the ECM is also disrupted, a fact that could facilitate the parasite’s motility in the tissue, and its entrance into cells (Duaso et al., 2010). Moreover, in vitro studies have shown that during parasite-ECM interactions, the parasite changes its cytoskeletal conformation, the state of protease activation and adapts its metabolism (Mattos et al., 2019). In addition, parasite-induced ECM changes modulate the presence of cytokines and chemokines, allowing T. cruzi to manipulate and escape the innate and adaptive immune responses (Marino et al., 2003). Studies performed in CD patients with cardiac alteration have shown that T. cruzi derived-antigens promote a differential expression of MMP-2 and MMP-9. There is a positive correlation between MMP-2 and the immunomodulating cytokine IL-10, and a negative one with the pro-inflammatory cytokine IL-1β, whereas MMP-9 showed a negative correlation with IL-10. Therefore, it has been suggested that MMPs and cytokines produced in the myocardium in patients with CD are essential contributors to cardiac remodeling (Medeiros et al., 2017). It would be interesting to study the interactions between parasite-modulated MMPs and cytokines in the placenta.
Another placental response to the parasite is the trophoblast epithelial turnover. The epithelial turnover is part of the innate immune system. Pathogens adhere to the plasma membrane before invading host cells and, in case of a lining epithelium, they adhere to the cells of the superficial cell layer which is continuously removed. Therefore, the attached pathogens are removed with the superficial epithelial cells (Liempi et al., 2016).
The trophoblast turnover implies precise orchestration of different cellular processes: (i) cell proliferation of the CT, (ii) cellular differentiation (referring to the incorporation of CT cells into a non-replicative ST), and (iii) cell death by forming apoptotic bodies (knots) in the ST that counterbalance the proliferation of the CT cells. The apoptotic knots are released into the IVS where the maternal blood circulates (Benirschke et al., 2012; Mayhew, 2014; Liempi et al., 2016).
We have previously shown that T. cruzi induces the above-mentioned cellular processes related to the epithelial turnover in the trophoblast in both ex vivo infected HPE and in vitro infected BeWo cells (trophoblastic cell line) (Duaso et al., 2011; Liempi et al., 2014, 2016; Carrillo et al., 2016; Droguett et al., 2017). Thus, T. cruzi increases DNA synthesis as well as cellular proliferation markers such as PCNA (proliferating cell nuclear antigen) (Liempi et al., 2016). In BeWo cells, the parasite also increases the percentage of cells in the S and G2/M cell cycle phases and other commonly used proliferation markers (AgNORs and Ki67) (Droguett et al., 2017). It is noteworthy that PCNA modulates other cellular processes including DNA repair, cell cycle, survival, and gene expression (Wang, 2014), and that an increase of PCNA expression might occur in response to T. cruzi-induced damage. Regarding trophoblast differentiation, the parasite increases, in HPE and BeWo cells, the expression and or secretion of the main biochemical markers, including β-human chorionic gonadotropin (β-hCG) and syncytin (Liempi et al., 2014, 2016). Additionally, in BeWo cells T. cruzi induces cell fusion as demonstrated by the analysis of the re-distribution of desmoplakin (intercellular adhesion protein) and by a two-color fusion assay (Liempi et al., 2014). The parasite also activates the ERK1/2 MAPK pathway (Castillo et al., 2013b), one of the MAPK signaling pathways that mediate trophoblast differentiation (Forbes and Westwood, 2010). Finally, the parasite induces apoptotic cell death as part of trophoblast turnover (Liempi et al., 2016). Indeed, T. cruzi activates in HPE a caspase 3 like activity, followed by DNA fragmentation and pyknosis (Duaso et al., 2011). These data indicate that T. cruzi can boost all steps of the turnover of chorionic trophoblasts. Interestingly, a relation exists in trophoblasts between the cellular processes related to apoptosis and their differentiation (Gauster et al., 2009a, b). This is through caspase 8, a caspase operating upstream the pro-apoptotic caspase 3. Caspase 8 is transiently activated in CT cells just before their fusion into ST and is an essential protein in the process of formation of the ST barrier (Huppertz and Borges, 2008). We observed that T. cruzi infection increases the expression and activation of caspase 8 in BeWo cells as well as in HPE (Duaso et al., 2011; Carrillo et al., 2016). This fact might favor the integrity of the ST barrier as well as limit parasite entrance into ST (by favoring its turnover). Also, we showed that caspase 8 can slow down intracellular parasite multiplication (Carrillo et al., 2016), pinpointing caspase-8 activity as part of the trophoblast cell defense mechanisms against T. cruzi infection. Altogether, these results suggest that the trophoblastic response to T. cruzi may be a mechanism that reduces the risk of congenital transmission of the parasite. On the other hand, caspase 8 has recently been disclosed to display pro-inflammatory effects, by optimizing the cytokinic response to TLRs and by being able to activate the cytokine IL-1β (Lawlor et al., 2017). We speculate that such pro-inflammatory action causes placental lesions, favoring the trans-placental passage of T. cruzi.
As mentioned above, the placenta is an immune regulatory organ that modulates fetal as well as maternal immune responses (Mor et al., 2017). Pathogens are recognized by pattern recognition receptors (PRRs), including Toll-like receptors (TLRs). Activation of those PPRs results in the secretion of cytokines and chemokines aiming to fight infections (Janeway and Medzhitov, 2002). The human trophoblast expresses all ten of the known functional TLRs (Koga et al., 2009). T. cruzi is recognized by TLR-2 and TLR-4 (present at the cell surface) as well as the endosomal TLR-7 and TLR-9 (Tarleton, 2007; Gravina et al., 2013; Castillo et al., 2017b). We have shown, in HPE, that T. cruzi infection induces protein expression and activation of TLR-2, but not of TLR-4 and TLR-9 (Castillo et al., 2017b). T. cruzi induces the secretion of IL-1β, IL-6, IL-8, IL-10, and TNF-α in HPE (Castillo et al., 2017b). Of note is that secretion of IL-1β, IL-6, and TNF-α have been associated, within the trophoblast, with cellular proliferation, and differentiation (Haider and Knöfler, 2009; Hamilton et al., 2012). Interestingly, the inhibition of TLR-2 impairs trophoblast turnover and increases parasite infection (Castillo et al., 2017c). However, to date we do not know if cells other than trophoblasts, such as macrophages in the VS, are also involved in TLR activation. However, our results allow us to propose TLR-2 initiated cytokine profile as a local placental defense mechanism.
Transcriptomic Studies in Placentas in Response to T. cruzi Infection
The global placental transcriptomic response to T. cruzi infection has also been studied. This approach allows us to study the gene expression profiles during infection, allowing the identification of new genes and/or pathways implicated in the establishment of the infection and pathogenesis. The RNA-seq study has been carried out in placentas obtained from term deliveries in T. cruzi-infected and non-infected women. Forty-two differentially expressed genes (DEGs) were identified, and gene-set association analysis was performed to detect pathways linked to parasite infection. It showed, not surprisingly, that the inflammatory and the immune responses were upregulated in infected placentas while the anti-inflammatory cytokine IL-38 (formerly IL-1 family member 10-IL1F10) (Garraud et al., 2018) was under-represented (Juiz et al., 2018). Placental inflammation is in line with other reports showing that: (i) newborns of T. cruzi-infected mothers are prone to produce higher levels of pro-inflammatory cytokines in comparison to those born to non-infected mothers (Vekemans et al., 2000), (ii) T. cruzi infection of pregnant women affects the developing immune system of fetuses independently of congenital infection (Dauby et al., 2009), which might be related to placental inflammation (Jennewein et al., 2017), and (iii) the parasite induces, as mentioned above, in ex vivo infected HPE, a highly significant increase of pro-inflammatory and immunomodulatory cytokines (Castillo et al., 2017b,c).
Analysis of DEGs also suggests that the placental inflammation might be counter-regulated in infected mothers. For instance, placentas from infected women displayed lower expression of genes related to exocytosis pathways and neutrophil degranulation, probably limiting damages associated with inflammation. Moreover, the expression of the immunomodulatory HLA-G was increased (Juiz et al., 2018). HLA-G is responsible for the generation of immunological tolerance during gestation by inducing regulatory T cells and tolerogenic dendritic cells (Carosella, 2011; Ferreira et al., 2017) and might increase susceptibility to infections. Hence, in malaria, increased levels of HLA-G in infected mothers are associated with an increased risk to acquire malaria during infancy (Sadissou et al., 2014). S100A14 is another interesting parasite-modulated gene, that is downregulated in placentas from women with CD. This gene codes for a member of the S100 protein family that regulates cell cycle progression, cellular differentiation, and triggers an inflammatory response by engaging the receptor for advanced glycation end products (RAGE) (Jin et al., 2011). Interestingly, this same protein, S100A14, is reported to increase the expression of the collagenase MMP2 (Chen et al., 2012; Qian et al., 2016), which as explained above regulates parasite infection. This might be in line with the observation that mutations on the MMP2 gene favor congenital T. cruzi transmission (see hereunder). On the other hand, PRG2 a gene that encodes the precursor form of the eosinophil major basic protein (proMBP) is an upregulated DEG in T. cruzi-infected placentas. MBP can participate in the extracellular killing of parasites in the absence of antibodies (Nakhle et al., 2018). Another important function of MBP in the placenta is the inhibition of the pregnancy-associated plasma protein A (PAPPA) (Weyer and Glerup, 2011), whose low levels are associated with intrauterine growth restriction (Albu et al., 2014). Therefore, overexpression of the PAPPA inhibitor in the placenta of women with CD might also play a role in the congenital transmission of T. cruzi.
Other, DEGs observed in placentas from seropositive women and related to pregnancy were the genes CGB5 and KISS1, which encode for hCG and kisspeptin, respectively. Both genes were downregulated, and low serum levels of both proteins have been proposed as markers of miscarriage (Jayasena et al., 2014). Low KISS1 expression is associated with recurrent pregnancy loss as well as preeclampsia and intrauterine growth restriction (Armstrong et al., 2009; Park et al., 2012). In contrast, the DEG TAC3 gene [encoding neurokinin B (NKB)] was upregulated in placentas from seropositive women. NKB overexpression in the trophoblast is associated with a decreased blood flow to the placenta and increased vasoconstriction in the endometrium and therefore related to preeclampsia (Familari et al., 2017). Interestingly, NKB and kisspeptin have a role in hCG placental expression in response to estradiol (Oride et al., 2015) and hCG, as described above, is induced by T. cruzi in HPE and BeWo cells (Liempi et al., 2014).
A microarray-based transcriptomics study carried out in ex vivo infected HPE (Castillo et al., 2018), corroborates most data obtained by RNA-seq in whole placentas (Juiz et al., 2017, 2018) as well as the T. cruzi-invasion mechanisms in HPE. Genes that are involved with ECM remodeling are upregulated, in concordance with our previous findings that demonstrate the parasite-induced expression and activation of MMP-2 and MMP-9 (Castillo et al., 2012). The changes in gene expression regarding signal transduction pathways were also confirmed (Castillo et al., 2013b, 2017b). Genes involved in innate immunity were overexpressed, including CD46 and C1q that regulate or form part of the complement system. Upregulation of C1q might have an impact since it binds the T. cruzi calreticulin, thereby enhancing the infection (Castillo et al., 2013a). On the other hand, TLR-7 and TLR-8 are mainly increased while TLR-2, whose inhibition favors infection and tissue damage (Castillo et al., 2017c), appeared not to be upregulated in the microarray analysis. Another contradictory observation was that a high T. cruzi concentration decreased IL-6 expression more than 60-fold as compared to control non-infected explants, whereas no changes were observed when HPE were infected with a low parasite concentration. On the other hand, mRNA expression analysis showed a higher level of transcription, suggesting that regulation of IL-6 could occur at post-transcriptional stages (Castillo et al., 2017c).
Furthermore, similar to the study in whole placentas, the majority of overexpressed genes were related to fetal development and pregnancy-related processes, particularly GH2, CSH1, and CSH2 genes that encode the pregnancy-specific beta-1-glycoproteins growth hormone 2, chorionic somatomammotropin hormone 1 and 2, respectively.
Another microarray-based study was done in placental tissues from C57Bl/6J mice chronically infected with two distinct T. cruzi strains (VD and K98, see above) (Juiz et al., 2017) A total of 247 DEGs were identified between infected and non-infected mice: 140 and 107 genes were up- and genes downregulated, respectively, compared to non-infected mice. Placentas infected with the VD parasites showed a higher number of DEGs compared to the K98 infected ones (211 vs. 89 DEGs). There were DEGs common to both infected groups and specific ones for each infected group; while 69% (59/89) of genes were downregulated by K98 infection, VD infection produced this response only in 39% (83/211) of genes. Analysis of DEG networks by GeneMANIA showed that the “Secretory Granule” pathway was downregulated in both infected groups, whereas “Response to Interferon-gamma” as well as “Innate Immune Response” pathways were upregulated only in placental tissues infected with VD. This is interesting since, as said above, the K98 strain displays poor placental tropism and no congenital transmissibility, contrary to the VD strain. It suggests the role of placental inflammation in the process of congenital transmission. Another analysis that detects small changes in predetermined gene sets (Gene-Set Enrichment Analysis algorithm) showed downregulation of genes involved in transcription, macromolecular transport, and metabolism in infected placentas and upregulation of genes regulating signal transduction pathways. Genes regulating apoptotic cell death were downregulated in the murine placentas infected with both parasite strain.
These transcriptomic analyses identify a gene expression profile in the placentas from T. cruzi- seropositive mothers that are different from those from seronegative women. It globally shows that affected genes are involved in cell adhesion, cellular proliferation and differentiation, apoptosis, vesicle transport processes, ECM organization, lipid and protein metabolism, and the inflammatory/immune responses.
Family Clustering of Congenital Transmission of T. cruzi Infection and SNPs
It is still unknown why some infected mothers transmit the infection in successive gestations to their babies while others do not, leading to family clustering of congenital transmission. A case-control study of single-nucleotide polymorphisms (SNPs) located within human loci encoding different proteins expressed in placental tissues was performed using genomic DNA from clinical samples of 116 non-infected children born to seropositive women and children with congenital T. cruzi infection. Logistic regression analysis showed that susceptibility of congenital infection was associated with SNPs in sites rs243866, rs17859821, and rs2285053from MMP2 gene and rs11244787 and rs1871054 sites from the ADAM 12 locus. In case of MMP2 rs243866 and rs17859821 positions, one or both copies of the mutant allele (Adenine) increased the likelihood of congenital infection, whereas, for rs2285053, both T alleles are needed for susceptibility to infection. Mutation in rs2285053 interrupts a CCACC box promoter site (Sp1-type) causing a weaker activity of the promotor. The rs243866 position is located upstream of a half-palindromic potential estrogen receptor binding site (Harendza et al., 2003). Both rs243866 and rs2285053 mutant alleles reduce the transcription activity of MMP2, which in turn modulate the ECM-remodeling and immune responses, and thus the susceptibility to infection. ADAM12 belongs to the ADAM protein family; it is a membrane-bound MMP-protease with a role in cell-cell and cell-matrix interactions that are associated with muscle development, fertilization, neurogenesis inflammatory, and immune responses. In preeclampsia (Grill et al., 2009) and ectopic pregnancy (Rausch and Barnhart, 2012), among other disorders related to pregnancy, ADAM 12 plasmatic concentrations are altered. Also, ADAM 12 promotes the transforming growth factor β (TGF-β) signaling activation leading to transcriptional activation. Importantly, TGF-β activation increases host cell susceptibility to T. cruzi infection (Ming et al., 1995; Hall and Pereira, 2000).
Various other SNPs have been related to the susceptibility to T. cruzi infection and progression of CD. Particularly, genes encoding cytokines/chemokines involved in inflammatory and immune response have been proposed as biomarkers (Calzada et al., 2009; Torres et al., 2010; Pissetti et al., 2011; Flórez et al., 2012; Nogueira et al., 2015; Furini et al., 2016; Ferreira et al., 2018). Thus, TGF-β1 polymorphism, mainly CT and TT genotypes at position – 509 of the TGFB1 gene have been associated with the susceptibility of acquiring CD in a Brazilian population (Ferreira et al., 2018). The same genotype, in addition to the genotypes −988 C/A; −800 G/A; −10 T/C; and 263 C/T has been studied in the Colombian and Peruvian populations. In this study, the genotype 10 C/C was increased in the group of CD patients of both populations and has been proposed to be involved in differential susceptibility to T. cruzi infection (Torres et al., 2010). Another important pro-inflammatory cytokine involved in CD is TNF-α. The TNF- α gene is located in the MHC locus, and its polymorphism is associated with many infectious diseases, including parasitic ones. TNF-α is increased in the hearts of patients with chronic chagasic cardiomyopathy and is associated with tissue damage. An association was observed between the absence of the TNF-238A allele and negative serology for CD. Seropositive individuals carrying the TNF-238A allele produced significantly higher TNF-alpha levels than healthy ones and therefore the TNF-α polymorphism at position −238 has also been associated to the susceptibility to T. cruzi infection and CD progression (Pissetti et al., 2011). Other SNPs in genes encoding for cytokines and molecules involved in the immune response has been related to CD, including interferon (IFN)-γ (-874 T/A) (Torres et al., 2010) and IL-18 (rs2043055 polymorphism) (Flórez et al., 2012; Nogueira et al., 2015), RANTES, and chemokine receptors CCR2 and CCR5 (Flórez et al., 2012). It would thus be worthwhile to also investigate such polymorphisms in the context of congenital transmission of T. cruzi.
Maternal T. cruzi Systemic Immune Response, Gestation, and Congenital Infection
The immune system during pregnancy is characterized by a subtle balance between immune tolerance and activation. Regulatory T cells are increased during pregnancy in order to avoid fetal rejection. Indeed, the maternal immune cells recognize paternal antigens expressed by the trophoblast and other fetal cells crossing the placenta to the maternal circulation. The mechanisms leading to this regulatory environment have been largely described elsewhere (Aluvihare et al., 2004; Racicot et al., 2014). Maternal immune tolerance is fundamental to a successful pregnancy. It does, however, not mean that the pregnant women are unable to mount immune responses, and trophoblasts possess multiple ways to escape immune attack (Straszewski-Chavez et al., 2004). Besides, immune responses during gestation are, except for the periods of implantation, placentation, and parturition, physiologically oriented toward a Th2-type. It likely aims to protect the uteroplacental unit and the fetus against harmful inflammation (Mor et al., 2017). Whereas the regulatory and Th2-biased environments are particularly pronounced at the level of the UPU, it may also impact peripheral, and systemic immune responses (Reinhard et al., 1998).
Trypanosoma cruzi infection induces a complex immune response. The control of T. cruzi infection relies on diverse effector mechanisms needed to fight both extracellular trypomastigotes and intracellular amastigotes. Therefore, IFN-γ, cytotoxic cells, and antibodies (Abs) all play a role in the control of the infection [reviewed in Truyens and Carlier (2017)]. Antibodies potentially participate in extracellular parasite elimination mainly by inducing complement-dependent and independent lysis of parasites and phagocytosis of opsonized parasites. IFN-γ, initially produced by NK cells, then by both CD4 + Th1 and CD8 + T cells, is a significant player in the immune responses against intracellular pathogens (Kak et al., 2018). It activates among other the microbicidal properties of phagocytes, mainly macrophages, allowing them to limit and/or kill the intracellular parasites. Infected cells can be killed by cytotoxic CD8 + T cells while the cytotoxic action of NK cell on T. cruzi-infected cells is likely not significant (Truyens and Carlier, 2017). The cellular response to T. cruzi is thus mainly Th1-oriented. Our studies in mice show that this inflammatory immune response has serious harmful effects on gestation when the host is in the acute phase of infection. Indeed, acute infection impedes reproduction, related to either infertility induced by the infection (inhibition of cell division of the embryo before implantation) or fetal loss occurring along gestation, due to massive placental invasion by parasites. Chronic maternal infection does not affect reproductive capacity but induces reversible fetal growth retardation (Carlier et al., 1987). Such deleterious effects did not rely on congenital infection but were associated with increased production of TNF-α (Rivera et al., 1995; Mjihdi et al., 2002). In humans, most women of gestational age have a chronic infection (see above). We have similarly observed that T. cruzi-infected women give birth to newborns with low birth weight, in the absence of congenital infection more frequently (Torrico et al., 2004).
Besides, in the absence of congenital transmission, several data indicate bidirectional and opposite interactions between the immune response to T. cruzi and the immune environment of gestation. On one side, pregnancy dampens and modifies the immune response to T. cruzi. Indeed, we found that blood cells from chronically infected pregnant women released, on average, two to threefold less IFN-γ and IL-2 in response to non-specific lymphocyte stimulants (PHA and LPS) than non-pregnant women (Hermann et al., 2004). They also produced, on average, sevenfold less IL-10. Accordingly, lower levels of IL-10 and IFN-γ were detected in the serum of pregnant infected women than in non-pregnant infected ones (Cardoni et al., 2004). This shows that pregnancy reduces the global capacity of their lymphocytes to be activated (though it is not completely inhibited), likely as a result of the relative immunosuppression associated with gestation. Also, the cytokine profile moved toward a more inflammatory environment in infected pregnant women, since the capacity to produce the inflammatory cytokine IFN-γ was less inhibited during pregnancy than the capacity to produce IL-10 (an anti-inflammatory cytokine), being in line with the inflammatory transcriptome detected in the placenta of T. cruzi-infected women [see above (Juiz et al., 2018)]. On the other hand, Egui et al. recently observed in a murine experimental T. cruzi infection model that chronic infection reduced the expression of gestation-induced inhibitory receptors on T cells (CTLA4 on CD4 + and CD8 + T cells and CD160 on CD8 + T cells), suggesting that T. cruzi chronic infection counteracts the immunosuppression associated with gestation but does not impact the gestation outcome (Egui et al., 2017).
Our previous studies in pregnant chronically infected women showed that gestation also reduced the specific IFN-γ response to parasite antigens as compared to the response of non-pregnant infected ones (Hermann et al., 2004). This was the case for women that transmitted (T mothers) as well as those that did not transmit (NT mothers) the parasite to their fetus. Strikingly, this specific IFN-γ response was markedly lower in T than NT mothers and was associated with less activated monocytes (Hermann et al., 2004). On the other hand, we and others have reported that the circulating parasite load is slightly increased during the second and third trimester of pregnancy (Brutus et al., 2010; Siriano et al., 2011). Moreover, T mothers displayed higher parasitemia than NT ones (Hermann et al., 2004; Kaplinski et al., 2015). IFN-γ is well known as a key factor in the control of T. cruzi infection (Truyens and Carlier, 2017). These data strongly suggest that the increase of parasite burden in T mothers results from a reduced control of the infection and points to a central role of parasite burden as a risk factor of congenital transmission. In line with this, several studies underline that congenital transmission occurs mostly in mothers displaying blood parasites amounts that are detectable by PCR (i.e., PCRs are generally negative in NT mothers). The microbial burden is also a key factor of congenital transmission of other pathogens (Lilleri and Gerna, 2017).
The increase of parasite burden during gestation likely results from the relative immunosuppression associated with gestation, while the increased parasite load in T mothers probably relates to their more depressed IFN-γ response. The question is, thus, why T mothers display such feature? Several factors may be considered, concerning the parasite, the host, or epidemiological parameters. Living in endemic countries where vectorial transmission occurs may lead to frequent reinfections. Multiple infections, though leading to transiently increased parasite levels, result in lower parasitemia in the long term, the hypothesis being that repeated antigen exposure reinforces the Th1 response that controls the parasite (Machado et al., 2001; Bustamante et al., 2007; Kaplinski et al., 2015). In line with this, it was observed that higher vector exposure of women living in endemic countries decreases the risk of congenital transmission (Sánchez Negrette et al., 2005; Kaplinski et al., 2015). Congenital transmission rates are also lower in non-endemic countries (2.7%), free of vectorial reinfections, than in endemic countries [2.7% vs. 5%, respectively (Howard et al., 2014)]. Parasite load may also vary according to the parasite genotype/strain (Moreira et al., 2013).
Our data also suggest that host factors modulate the risk of congenital transmission of T. cruzi. Genetic and epigenetic factors may influence the efficacy of immune responses against pathogens and consequently modulate the microbial load (Möller et al., 2018). For instance, polymorphism of genes encoding proteins of the axis IL-12-IFN-γ and genetic variations affecting the modulatory action of long non-coding RNAs or micro-RNAs are reported to impact diseases (Vannberg et al., 2011; Duval et al., 2017; Gao and Wei, 2017; Ellwanger et al., 2018). We compared the ability of T and NT women to produce IFN-γ to T. cruzi and mitogens several months after they had given birth, i.e., in the absence of the immunosuppression of pregnancy (Hermann et al., 2004). We found that T women still displayed a more depressed ability to produce this cytokine than NT women, suggesting that inherent host factors involved might be genetic. Likewise, we have recently reported that several genes related to the immune system were differentially expressed in the placenta of T and NT mothers as well as an association between congenital T. cruzi transmission and the presence of polymorphism among some gene expressed in the placenta (see above) (Juiz et al., 2016, 2018). Of note, differential immune responses associated with the congenital transmission of other pathogens have also been recently reported (Lilleri and Gerna, 2017; Wujcicka et al., 2018). Besides, epigenetic modulation may also have occurred in some women in relation either with the T. cruzi strains present in the host or to their history of infections with other pathogens (Gomez et al., 2013; Smith and Denning, 2014).
Finally, we and others noticed that T mothers were younger and had a lower mean number of previous pregnancies than the NT mothers (Hermann et al., 2004; Kaplinski et al., 2015), which might be related to the immuno-enhancing effect of multiparity on the maternal immune system (Skowron-Cendrzak et al., 1999).
Neonatal Immune Response to T. cruzi
Immune responses in early life are physiologically different from adult responses. Similarly, to what occurs during pregnancy, the neonatal immune system is characterized by a certain degree of immunodeficiency and weak Th1 but excessive Th2 responses, contributing to high susceptibility to pathogens and the development of suboptimal immune responses to vaccines administered in early life. However, the mechanisms leading to immunodeficiency and Th2 bias are completely different from those activated during pregnancy and have been reviewed elsewhere (Levy, 2007; PrabhuDas et al., 2011; Elahi et al., 2013). Mechanisms responsible for the regulatory environment comprises, among others, impaired signaling pathways downstream of Toll-like receptors 3 and 4, the presence of higher circulating levels of adenosine, the presence of higher numbers of regulatory T cells, and the presence of particular inhibitory “erythroid cells.” The Th2 bias results from the hypermethylation of the IFNG gene, while the Th2 locus is hypomethylated. Also, the proliferation of Th1 T cells in early life is strongly hindered due to the expression of a particular receptor for IL-4 produced by Th2 T cells. Engagement of this receptor induces apoptosis of Th1 cells.
Our studies in humans, performed in infants from chagasic mothers, point out that T. cruzi infection triggers neonatal type 1 immune responses, overcoming the physiological immune deficiency associated with early life (Hermann et al., 2002). Indeed, CD8 + T cells, and CD4 + T cells to a lesser extent, were activated in T. cruzi congenitally infected new-borns to an adult-like level and produced IFN-γ. Besides, their NK cells display phenotypic and functional alterations suggestive of a previous in utero activation when parasites were transmitted from the mother (Hermann et al., 2006). It also pinpoints the imprinting of the maternal T. cruzi infection on the neonatal immune system, revealing an immuno-stimulatory/adjuvant property of the parasite, as both congenitally infected and uninfected infants from chagasic mothers responded more strongly to vaccines directly administered during the first 6 months of life, like those against BCG, hepatitis B, tetanus, and diphtheria (Dauby et al., 2009). In trying to decipher the mechanisms allowing the parasite to induce type 1 immune responses in early life, we showed that the parasite could strongly activate neonatal NK cells to produce IFN-γ, known to drive Th1 type responses rapidly. NK cell activation by T. cruzi is indirect and depends on cross-talk with monocytes (and not with dendritic cells), on IL-12 synthesis and engagement of TLR2, 4, 7, and 9 (Guilmot et al., 2013). The parasite also very efficiently licensed neonatal dendritic cells to activate CD4 + and CD8 + T cells. Interestingly, DC and monocyte activation by T. cruzi is reinforced in the presence of maternal IgG isolated from cord blood samples from neonates born to NT infected mothers, i.e., carrying T. cruzi-specific IgG (Rodriguez et al., 2012). These observations allow us to make the hypothesis that monocytes, likely activated in fetuses from infected mothers combined with the maternally transmitted T. cruzi – specific antibodies might allow the fetus to fight parasites and maybe in some cases to eliminate them if only a few parasites were transmitted.
Final Remarks
Some relevant questions regarding the role of parasite diversity, host genetic response, and host immune responses deserve further investigations. The study of gene expression profiles during infection constitutes a powerful tool to analyze global responses of several kinds of cells and tissues, allowing the identification of new genes and/or pathways implicated in the establishment of the infection and pathogenesis as well as possible local tissue responses. Other aspects such as the role of maternal microbiomes in the likelihood of vertical transmission of T. cruzi have not been investigated yet.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the ERANET-LAC grants ELAC2014/HID-0328 and ERANet17/HLH-0142 (to UK, AO, AS, and CT), FONDECYT 1190341 (Conicyt, Chile to UK), and PICT 2015-0074 (FONCyT, Argentina to AS).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Acosta-Serrano, A., Almeida, I. C., Freitas-Junior, L. H., Yoshida, N., and Schenkman, S. (2001). The mucin-like glycoprotein super-family of Trypanosoma cruzi: structure and biological roles. Mol. Biochem. Parasitol. 114, 143–150. doi: 10.1016/s0166-6851(01)00245-6
Albu, A. R., Anca, A. F., Horhoianu, V. V., and Horhoianu, I. A. (2014). Predictive factors for intrauterine growth restriction. J. Med. Life 7, 165–171.
Almeida, I. C., and Gazzinelli, R. T. (2001). Proinflammatory activity of glycosylphosphatidylinositol anchors derived from Trypanosoma cruzi: structural and functional analyses. J. Leukoc. Biol. 70, 467–477.
Aluvihare, V. R., Kallikourdis, M., and Betz, A. G. (2004). Regulatory T cells mediate maternal tolerance to the fetus. Nat. Immunol. 5, 266–271. doi: 10.1038/ni1037
Andrade, S. G. (1982). The influence of the strain of Trypanosoma cruzi in placental infections in mice. Trans. R. Soc. Trop. Med. Hyg. 76, 123–128. doi: 10.1016/0035-9203(82)90036-0
Antinori, S., Galimberti, L., Bianco, R., Grande, R., Galli, M., and Corbellino, M. (2017). Chagas disease in Europe: a review for the internist in the globalized world. Eur. J. Intern. Med. 43, 6–15. doi: 10.1016/j.ejim.2017.05.001
Armstrong, R. A., Reynolds, R. M., Leask, R., Shearing, C. H., Calder, A. A., and Riley, S. C. (2009). Decreased serum levels of kisspeptin in early pregnancy are associated with intra-uterine growth restriction and pre-eclampsia. Prenat. Diagn. 29, 982–985. doi: 10.1002/pd.2328
Arora, N., Sadovsky, Y., Dermody, T. S., and Coyne, C. B. (2017). Microbial vertical transmission during human pregnancy. Cell Host Microbe 21, 561–567. doi: 10.1016/j.chom.2017.04.007
Benirschke, K., Kaufmann, P., and Baergen, P. (2012). Pathology of the Human Placenta, 6 th Edn. New York, NY: Springer Link.
Bermejo, D. A., Amezcua Vesely, M. C., Khan, M., Acosta Rodríguez, E. V., Montes, C. L., Merino, M. C., et al. (2011). Trypanosoma cruzi infection induces a massive extrafollicular and follicular splenic B-cell response which is a high source of non-parasite-specific antibodies. Immunology 132, 123–133. doi: 10.1111/j.1365-2567.2010.03347.x
Bermejo, D. A., Jackson, S. W., Gorosito-Serran, M., Acosta-Rodriguez, E. V., Amezcua-Vesely, M. C., Sather, B. D., et al. (2013). Trypanosoma cruzi trans-sialidase initiates a program independent of the transcription factors RORγt and Ahr that leads to IL-17 production by activated B cells. Nat. Immunol. 14, 514–522. doi: 10.1038/ni.2569
Berná, L., Rodriguez, M., Chiribao, M. L., Parodi-Talice, A., Pita, S., Rijo, G., et al. (2018). Expanding an expanded genome: long-read sequencing of Trypanosoma cruzi. Microb. Genomics 4:e000177. doi: 10.1099/mgen.0.000177
Brenière, S. F., Waleckx, E., and Barnabé, C. (2016). Over six thousand Trypanosoma cruzi strains classified into discrete typing units (DTUs): attempt at an inventory. PLoS Negl. Trop. Dis. 10:e0004792. doi: 10.1371/journal.pntd.0004792
Brutus, L., Castillo, H., Bernal, C., Salas, N. A., Schneider, D., Santalla, J.-A., et al. (2010). Detectable Trypanosoma cruzi parasitemia during pregnancy and delivery as a risk factor for congenital Chagas disease. Am. J. Trop. Med. Hyg. 83, 1044–1047. doi: 10.4269/ajtmh.2010.10-0326
Buekens, P., Cafferata, M. L., Alger, J., Althabe, F., Belizán, J. M., Bustamante, N., et al. (2018). Congenital transmission of Trypanosoma cruzi in Argentina, Honduras, and Mexico: an observational prospective study. Am. J. Trop. Med. Hyg. 98, 478–485. doi: 10.4269/ajtmh.17-0516
Buscaglia, C. A., Campo, V. A., Frasch, A. C. C., and Di Noia, J. M. (2006). Trypanosoma cruzi surface mucins: host-dependent coat diversity. Nat. Rev. Microbiol. 4, 229–236. doi: 10.1038/nrmicro1351
Bustamante, J. M., Novarese, M., Rivarola, H. W., Lo Presti, M. S., Fernández, A. R., Enders, J. E., et al. (2007). Reinfections and Trypanosoma cruzi strains can determine the prognosis of the chronic chagasic cardiopathy in mice. Parasitol. Res. 100, 1407–1410. doi: 10.1007/s00436-006-0425-3
Callejas-Hernández, F., Rastrojo, A., Poveda, C., Gironès, N., and Fresno, M. (2018). Genomic assemblies of newly sequenced Trypanosoma cruzi strains reveal new genomic expansion and greater complexity. Sci. Rep. 8:14631. doi: 10.1038/s41598-018-32877-2
Calzada, J. E., Beraún, Y., González, C. I., and Martín, J. (2009). Transforming growth factor beta 1 (TGFβ1) gene polymorphisms and Chagas disease susceptibility in Peruvian and Colombian patients. Cytokine 45, 149–153. doi: 10.1016/j.cyto.2008.11.013
Cardoni, R. L., García, M. M., and De Rissio, A. M. (2004). Proinflammatory and anti-inflammatory cytokines in pregnant women chronically infected with Trypanosoma cruzi. Acta Trop. 90, 65–72. doi: 10.1016/j.actatropica.2003.09.020
Cardoso, E. J., Valdéz, G. C., Campos, A. C., Sanchez, R., de la, L., Mendoza, C. R., et al. (2012). Maternal fetal transmission of Trypanosoma cruzi: a problem of public health little studied in Mexico. Exp. Parasitol. 131, 425–432. doi: 10.1016/j.exppara.2012.05.013
Carlier, Y., Rivera, M. T., Truyens, C., Puissant, F., and Milaire, J. (1987). Interactions between chronic murine Trypanosoma cruzi infection and pregnancy: fetal growth retardation. Am. J. Trop. Med. Hyg. 37, 534–540. doi: 10.4269/ajtmh.1987.37.534
Carlier, Y., Sosa-Estani, S., Luquetti, A. O., and Buekens, P. (2015). Congenital Chagas disease: an update. Mem. Inst. Oswaldo Cruz 110, 363–368. doi: 10.1590/0074-02760140405
Carlier, Y., and Truyens, C. (2015). Congenital Chagas disease as an ecological model of interactions between Trypanosoma cruzi parasites, pregnant women, placenta and fetuses. Acta Trop. 151, 103–115. doi: 10.1016/j.actatropica.2015.07.016
Carlier, Y., Truyens, C., Deloron, P., and Peyron, F. (2012). Congenital parasitic infections: a review. Acta Trop. 121, 55–70. doi: 10.1016/j.actatropica.2011.10.018
Carosella, E. D. (2011). The tolerogenic molecule HLA-G. Immunol. Lett. 138, 22–24. doi: 10.1016/j.imlet.2011.02.011
Carrillo, I., Droguett, D., Castillo, C., Liempi, A., Muñoz, L., Maya, J. D., et al. (2016). Caspase-8 activity is part of the BeWo trophoblast cell defense mechanisms against Trypanosoma cruzi infection. Exp. Parasitol. 168, 9–15. doi: 10.1016/j.exppara.2016.06.008
Castillo, C., Carrillo, I., Libisch, G., Juiz, N., Schijman, A. G., Robello, C., et al. (2018). Host-parasite interaction: changes in human placental gene expression induced by Trypanosoma cruzi. Parasit. Vectors 11, 1–13. doi: 10.1186/s13071-018-2988-0
Castillo, C., Carrillo, I., Liempi, A., Medina, L., Navarrete, A., López, P., et al. (2017a). Trypanosoma cruzi exosomes increases susceptibility to parasite infection in human placental chorionic villi explants. Placenta 51, 123–124. doi: 10.1016/J.PLACENTA.2017.01.086
Castillo, C., Muñoz, L., Carrillo, I., Liempi, A., Gallardo, C., Galanti, N., et al. (2017b). Ex vivo infection of human placental chorionic villi explants with Trypanosoma cruzi and Toxoplasma gondii induces different Toll-like receptor expression and cytokine/chemokine profiles. Am. J. Reprod. Immunol. 78, 1–8. doi: 10.1111/aji.12660
Castillo, C., Muñoz, L., Carrillo, I., Liempi, A., Medina, L., Galanti, N., et al. (2017c). Toll-like receptor-2 mediates local innate immune response against Trypanosoma cruzi in ex vivo infected human placental chorionic villi explants. Placenta 60, 40–46. doi: 10.1016/j.placenta.2017.10.005
Castillo, C., López-Muñoz, R. A., Duaso, J., Galanti, N., Jaña, F., Ferreira, J., et al. (2012). Role of matrix metalloproteinases 2 and 9 in ex vivo Trypanosoma cruzi infection of human placental chorionic villi. Placenta 33, 991–997. doi: 10.1016/j.placenta.2012.10.004
Castillo, C., Ramírez, G., Valck, C., Aguilar, L., Maldonado, I., Rosas, C., et al. (2013a). The interaction of classical complement component C1 with parasite and host calreticulin mediates Trypanosoma cruzi infection of human placenta. PLoS Negl. Trop. Dis. 7:e2376. doi: 10.1371/journal.pntd.0002376
Castillo, C., Villarroel, A., Duaso, J., Galanti, N., Cabrera, G., Maya, J. D. J. D., et al. (2013b). Phospholipase C gamma and ERK1/2 Mitogen Activated Kinase Pathways are differentially modulated by Trypanosoma cruzi during tissue invasion in human placenta. Exp. Parasitol. 133, 12–17. doi: 10.1016/j.exppara.2012.10.012
Cestari, I., Ansa-Addo, E., Deolindo, P., Inal, J. M., and Ramirez, M. I. (2012). Trypanosoma cruzi immune evasion mediated by host cell-derived microvesicles. J. Immunol. 188, 1942–1952. doi: 10.4049/jimmunol.1102053
Chen, H., Yuan, Y., Zhang, C., Luo, A., Ding, F., Ma, J., et al. (2012). Involvement of S100A14 protein in cell invasion by affecting expression and function of matrix metalloproteinase (MMP)-2 via p53-dependent transcriptional regulation. J. Biol. Chem. 287, 17109–17119. doi: 10.1074/jbc.M111.326975
da Silveira, J. F., Abrahamsohn, P. A., and Colli, W. (1979). Plasma membrane vesicles isolated from epimastigote forms of Trypanosoma cruzi. Biochim. Biophys. Acta 550, 222–232. doi: 10.1016/0005-2736(79)90209-8
Dauby, N., Alonso-Vega, C., Suarez, E., Flores, A., Hermann, E., Córdova, M., et al. (2009). Maternal infection with Trypanosoma cruzi and congenital Chagas disease induce a trend to a type 1 polarization of infant immune responses to vaccines. PLoS Negl. Trop. Dis. 3:e571. doi: 10.1371/journal.pntd.0000571
de Lederkremer, R. M., and Colli, W. (1995). Galactofuranose-containing glycoconjugates in trypanosomatids. Glycobiology 5, 547–552. doi: 10.1093/glycob/5.6.547
de Oliveira, A. B. B., Alevi, K. C. C., Imperador, C. H. L., Madeira, F. F., and de Azeredo-Oliveira, M. T. V. (2018). Parasite-vector interaction of chagas disease: a mini-review. Am. J. Trop. Med. Hyg. 98, 653–655. doi: 10.4269/ajtmh.17-0657
De Pablos, L. M., Díaz Lozano, I. M., Jercic, M. I., Quinzada, M., Giménez, M. J., Calabuig, E., et al. (2016). The C-terminal region of Trypanosoma cruzi MASPs is antigenic and secreted via exovesicles. Sci. Rep. 6:27293. doi: 10.1038/srep27293
de Pablos Torró, L. M., Retana Moreira, L., and Osuna, A. (2018). Extracellular vesicles in Chagas disease: a new passenger for an old disease. Front. Microbiol. 9:1190. doi: 10.3389/fmicb.2018.01190
Delorme-Axford, E., Sadovsky, Y., and Coyne, C. B. (2014). The placenta as a barrier to viral infections. Annu. Rev. Virol. 1, 133–146. doi: 10.1146/annurev-virology-031413-085524
Díaz Lozano, I. M., De Pablos, L. M., Longhi, S. A., Zago, M. P., Schijman, A. G., and Osuna, A. (2017). Immune complexes in chronic Chagas disease patients are formed by exovesicles from Trypanosoma cruzi carrying the conserved MASP N-terminal region. Sci. Rep. 7:44451. doi: 10.1038/srep44451
Droguett, D., Carrillo, I., Castillo, C., Gómez, F., Negrete, M., Liempi, A., et al. (2017). Trypanosoma cruzi induces cellular proliferation in the trophoblastic cell line BeWo. Exp. Parasitol. 173, 9–17. doi: 10.1016/j.exppara.2016.12.005
Duaso, J., Rojo, G., Cabrera, G., Galanti, N., Bosco, C., Maya, J. D. J. D., et al. (2010). Trypanosoma cruzi induces tissue disorganization and destruction of chorionic villi in an ex vivo infection model of human placenta. Placenta 31, 705–711. doi: 10.1016/j.placenta.2010.05.007
Duaso, J., Rojo, G., Jaña, F., Galanti, N., Cabrera, G., Bosco, C., et al. (2011). Trypanosoma cruzi induces apoptosis in ex vivo infected human chorionic villi. Placenta 32, 356–361. doi: 10.1016/j.placenta.2011.02.005
Duaso, J., Yanez, E., Castillo, C., Galanti, N., Cabrera, G., Corral, G., et al. (2012). Reorganization of extracellular matrix in placentas from women with asymptomatic chagas disease: mechanism of parasite invasion or local placental defense? J. Trop. Med. 2012:758357. doi: 10.1155/2012/758357
Duval, M., Cossart, P., and Lebreton, A. (2017). Mammalian microRNAs and long noncoding RNAs in the host-bacterial pathogen crosstalk. Semin. Cell Dev. Biol. 65, 11–19. doi: 10.1016/j.semcdb.2016.06.016
Egui, A., Lasso, P., Thomas, M. C., Carrilero, B., González, J. M., Cuéllar, A., et al. (2017). Expression of inhibitory receptors and polyfunctional responses of T cells are linked to the risk of congenital transmission of T. cruzi. PLoS Negl. Trop. Dis. 11:e0005627. doi: 10.1371/journal.pntd.0005627
Elahi, S., Ertelt, J. M., Kinder, J. M., Jiang, T. T., Zhang, X., Xin, L., et al. (2013). Immunosuppressive CD71+ erythroid cells compromise neonatal host defence against infection. Nature 504, 158–162. doi: 10.1038/nature12675
Ellwanger, J. H., Zambra, F. M. B., Guimarães, R. L., and Chies, J. A. B. (2018). MicroRNA-related polymorphisms in infectious diseases-tiny changes with a huge impact on viral infections and potential clinical applications. Front. Immunol. 9:1316. doi: 10.3389/fimmu.2018.01316
El-Sayed, N. M., Myler, P. J., Bartholomeu, D. C., Nilsson, D., Aggarwal, G., Tran, A.-N., et al. (2005). The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 309, 409–415. doi: 10.1126/science.1112631
Familari, M., Cronqvist, T., Masoumi, Z., and Hansson, S. R. (2017). Placenta-derived extracellular vesicles: their cargo and possible functions. Reprod. Fertil. Dev. 29, 433–447. doi: 10.1071/RD15143
Ferreira, L. M. R., Meissner, T. B., Tilburgs, T., and Strominger, J. L. (2017). HLA-G: at the interface of maternal–fetal tolerance. Trends Immunol. 38, 272–286. doi: 10.1016/j.it.2017.01.009
Ferreira, R. R., Madeira, F., da, S., Alves, G. F., Chambela, M., da, C., et al. (2018). TGF- β polymorphisms are a risk factor for Chagas disease. Dis. Markers 2018, 1–10. doi: 10.1155/2018/4579198
Flórez, O., Martín, J., and González, C. I. (2012). Genetic variants in the chemokines and chemokine receptors in Chagas disease. Hum. Immunol. 73, 852–858. doi: 10.1016/j.humimm.2012.04.005
Forbes, K., and Westwood, M. (2010). Maternal growth factor regulation of human placental development and fetal growth. J. Endocrinol. 207, 1–16. doi: 10.1677/JOE-10-0174
Furini, A. A., da, C., Cassiano, G. C., Petrolini Capobianco, M., Dos Santos, S. E. B., and Dantas Machado, R. L. (2016). Frequency of TNFA, INFG, and IL10 gene polymorphisms and their association with malaria vivax and genomic ancestry. Mediat. Inflamm. 2016:5168363. doi: 10.1155/2016/5168363
Gao, P., and Wei, G.-H. (2017). Genomic insight into the role of lncRNA in cancer susceptibility. Int. J. Mol. Sci. 18:E1239. doi: 10.3390/ijms18061239
Garraud, T., Harel, M., Boutet, M.-A., Le Goff, B., and Blanchard, F. (2018). The enigmatic role of IL-38 in inflammatory diseases. Cytokine Growth Factor Rev. 39, 26–35. doi: 10.1016/j.cytogfr.2018.01.001
Gaunt, M. W., Yeo, M., Frame, I. A., Stothard, J. R., Carrasco, H. J., Taylor, M. C., et al. (2003). Mechanism of genetic exchange in American trypanosomes. Nature 421, 936–939. doi: 10.1038/nature01438
Gauster, M., Moser, G., Orendi, K., and Huppertz, B. (2009a). Factors involved in regulating trophoblast fusion: potential role in the development of preeclampsia. Placenta 30(Suppl. A), S49–S54. doi: 10.1016/j.placenta.2008.10.011
Gauster, M., Siwetz, M., and Huppertz, B. (2009b). Fusion of villous trophoblast can be visualized by localizing active caspase 8. Placenta 30, 547–550. doi: 10.1016/j.placenta.2009.03.007
Gomez, J. A., Wapinski, O. L., Yang, Y. W., Bureau, J.-F., Gopinath, S., Monack, D. M., et al. (2013). The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-γ locus. Cell 152, 743–754. doi: 10.1016/j.cell.2013.01.015
Gravina, H. D., Antonelli, L., Gazzinelli, R. T., and Ropert, C. (2013). Differential use of TLR2 and TLR9 in the regulation of immune responses during the infection with Trypanosoma cruzi. PLoS One 8:e63100. doi: 10.1371/journal.pone.0063100
Grill, S., Rusterholz, C., Zanetti-Dällenbach, R., Tercanli, S., Holzgreve, W., Hahn, S., et al. (2009). Potential markers of preeclampsia – a review. Reprod. Biol. Endocrinol. 7:70. doi: 10.1186/1477-7827-7-70
Guilmot, A., Bosse, J., Carlier, Y., and Truyens, C. (2013). Monocytes play an IL-12-dependent crucial role in driving cord blood NK cells to produce IFN-g in response to Trypanosoma cruzi. PLoS Negl. Trop. Dis. 7:e2291. doi: 10.1371/journal.pntd.0002291
Haider, S., and Knöfler, M. (2009). Human tumour necrosis factor: physiological and pathological roles in placenta and endometrium. Placenta 30, 111–123. doi: 10.1016/j.placenta.2008.10.012
Hall, B. S., and Pereira, M. A. (2000). Dual role for transforming growth factor beta-dependent signaling in Trypanosoma cruzi infection of mammalian cells. Infect. Immun. 68, 2077–2081. doi: 10.1128/iai.68.4.2077-2081.2000
Hamilton, S. T., Scott, G., Naing, Z., Iwasenko, J., Hall, B., Graf, N., et al. (2012). Human cytomegalovirus-induces cytokine changes in the placenta with implications for adverse pregnancy outcomes. PLoS One 7:e52899. doi: 10.1371/journal.pone.0052899
Harendza, S., Lovett, D. H., Panzer, U., Lukacs, Z., Kuhnl, P., and Stahl, R. A. K. (2003). Linked common polymorphisms in the gelatinase a promoter are associated with diminished transcriptional response to estrogen and genetic fitness. J. Biol. Chem. 278, 20490–20499. doi: 10.1074/jbc.M211536200
Hermann, E., Alonso-Vega, C., Berthe, A., Truyens, C., Flores, A., Cordova, M., et al. (2006). Human congenital infection with Trypanosoma cruzi induces phenotypic and functional modifications of cord blood NK cells. Pediatr. Res. 60, 38–43. doi: 10.1203/01.pdr.0000220335.05588.ea
Hermann, E., Truyens, C., Alonso-Vega, C., Even, J., Rodriguez, P., Berthe, A., et al. (2002). Human fetuses are able to mount an adultlike CD8 T-cell response. Blood 100, 2153–2158.
Hermann, E., Truyens, C., Alonso-Vega, C., Rodriguez, P., Berthe, A., Torrico, F., et al. (2004). Congenital transmission of Trypanosoma cruzi is associated with maternal enhanced parasitemia and decreased production of interferon- gamma in response to parasite antigens. J. Infect. Dis. 189, 1274–1281. doi: 10.1086/382511
Howard, E., Xiong, X., Carlier, Y., Sosa-Estani, S., and Buekens, P. (2014). Frequency of the congenital transmission of Trypanosoma cruzi: a systematic review and meta-analysis. BJOG 121, 22–33. doi: 10.1111/1471-0528.12396
Huppertz, B., and Borges, M. (2008). “Placenta trophoblast fusion,” in Methods in molecular biology, ed. E. H. Chen (Totowa, NJ: Humana press), 135–147. doi: 10.1007/978-1-59745-250-2_8
Janeway, C. A., and Medzhitov, R. (2002). Innate immune recognition. Annu. Rev. Immunol. 20, 197–216. doi: 10.1146/annurev.immunol.20.083001.084359
Jayasena, C. N., Abbara, A., Izzi-Engbeaya, C., Comninos, A. N., Harvey, R. A., Gonzalez Maffe, J., et al. (2014). Reduced levels of plasma Kisspeptin during the antenatal booking visit are associated with increased risk of miscarriage. J. Clin. Endocrinol. Metab. 99, E2652–E2660. doi: 10.1210/jc.2014-1953
Jennewein, M. F., Abu-Raya, B., Jiang, Y., Alter, G., and Marchant, A. (2017). Transfer of maternal immunity and programming of the newborn immune system. Semin. Immunopathol. 39, 605–613. doi: 10.1007/s00281-017-0653-x
Jin, Q., Chen, H., Luo, A., Ding, F., and Liu, Z. (2011). S100A14 stimulates cell proliferation and induces cell apoptosis at different concentrations via receptor for advanced glycation end products (RAGE). PLoS One 6:e19375. doi: 10.1371/journal.pone.0019375
Juiz, N. A., Cayo, N. M., Burgos, M., Salvo, M. E., Nasser, J. R., Búa, J., et al. (2016). Human polymorphisms in placentally expressed genes and their association with susceptibility to congenital Trypanosoma cruzi infection. J. Infect. Dis. 213, 1299–1306. doi: 10.1093/infdis/jiv561
Juiz, N. A., Solana, M. E., Acevedo, G. R., Benatar, A. F., Ramirez, J. C., da Costa, P. A., et al. (2017). Different genotypes of Trypanosoma cruzi produce distinctive placental environment genetic response in chronic experimental infection. PLoS Negl. Trop. Dis. 11:e0005436. doi: 10.1371/journal.pntd.0005436
Juiz, N. A., Torrejón, I., Burgos, M., Torres, A. M. F., Duffy, T., Cayo, N. M., et al. (2018). Alterations in placental gene expression of pregnant women with chronic Chagas disease. Am. J. Pathol. 188, 1345–1353. doi: 10.1016/j.ajpath.2018.02.011
Kak, G., Raza, M., and Tiwari, B. K. (2018). Interferon-gamma (IFN-γ): exploring its implications in infectious diseases. Biomol. Concepts 9, 64–79. doi: 10.1515/bmc-2018-0007
Kaplinski, M., Jois, M., Galdos-Cardenas, G., Rendell, V. R., Shah, V., Do, R. Q., et al. (2015). Sustained domestic vector exposure is associated with increased Chagas Cardiomyopathy risk but decreased parasitemia and congenital transmission risk among young women in Bolivia. Clin. Infect. Dis. 61, 918–926. doi: 10.1093/cid/civ446
Koga, K., Aldo, P. B., and Mor, G. (2009). Toll-like receptors and pregnancy: trophoblast as modulators of the immune response. J. Obstet. Gynaecol. Res. 35, 191–202. doi: 10.1111/j.1447-0756.2008.00963.x
Lawlor, K. E., Feltham, R., Yabal, M., Conos, S. A., Chen, K. W., Ziehe, S., et al. (2017). XIAP Loss Triggers RIPK3- and Caspase-8-Driven IL-1β activation and cell death as a consequence of TLR-MyD88-Induced cIAP1-TRAF2 degradation. Cell Rep. 20, 668–682. doi: 10.1016/j.celrep.2017.06.073
Lenk, E. J., Redekop, W. K., Luyendijk, M., Fitzpatrick, C., Niessen, L., Stolk, W. A., et al. (2018). Socioeconomic benefit to individuals of achieving 2020 targets for four neglected tropical diseases controlled/eliminated by innovative and intensified disease management: human African trypanosomiasis, leprosy, visceral leishmaniasis, Chagas disease. PLoS Negl. Trop. Dis. 12:e0006250. doi: 10.1371/journal.pntd.0006250
Levy, O. (2007). Innate immunity of the newborn: basic mechanisms and clinical correlates. Nat. Rev. Immunol. 7, 379–390. doi: 10.1038/nri2075
Liempi, A., Castillo, C., Carrillo, I., Muñoz, L., Droguett, D., Galanti, N., et al. (2016). A local innate immune response against Trypanosoma cruzi in the human placenta: the epithelial turnover of the trophoblast. Microb. Pathog. 99, 123–129. doi: 10.1016/j.micpath.2016.08.022
Liempi, A., Castillo, C., Duaso, J., Droguett, D., Sandoval, A., Barahona, K., et al. (2014). Trypanosoma cruzi induces trophoblast differentiation: a potential local antiparasitic mechanism of the human placenta? Placenta 35, 1035–1042. doi: 10.1016/j.placenta.2014.09.017
Lilleri, D., and Gerna, G. (2017). Maternal immune correlates of protection from human cytomegalovirus transmission to the fetus after primary infection in pregnancy. Rev. Med. Virol. 27:e1921. doi: 10.1002/rmv.1921
Lima, A. P. C. A., Almeida, P. C., Tersariol, I. L. S., Schmitz, V., Schmaier, A. H., Juliano, L., et al. (2002). Heparan sulfate modulates kinin release by Trypanosoma cruzi through the activity of cruzipain. J. Biol. Chem. 277, 5875–5881. doi: 10.1074/jbc.M108518200
Lima, L., Espinosa-Álvarez, O., Ortiz, P. A., Trejo-Varón, J. A., Carranza, J. C., Pinto, C. M., et al. (2015). Genetic diversity of Trypanosoma cruzi in bats, and multilocus phylogenetic and phylogeographical analyses supporting Tcbat as an independent DTU (discrete typing unit). Acta Trop. 151, 166–177. doi: 10.1016/j.actatropica.2015.07.015
Luquetti, A. O., Tavares, S. B., Siriano Lda, R., Oliveira, R. A., Campos, D. E., de Morais, C. A., et al. (2015). Congenital transmission of Trypanosoma cruzi in central Brazil. A study of 1,211 individuals born to infected mothers. Mem. Inst. Oswaldo Cruz 110, 369–376. doi: 10.1590/0074-02760140410
Macedo, A. M., Machado, C. R., Oliveira, R. P., and Pena, S. D. J. (2004). Trypanosoma cruzi: genetic structure of populations and relevance of genetic variability to the pathogenesis of chagas disease. Mem. Inst. Oswaldo Cruz 99, 1–12. doi: 10.1590/s0074-02762004000100001
Macedo, A. M., and Pena, S. D. (1998). Genetic variability of Trypanosoma cruzi: implications for the pathogenesis of Chagas disease. Parasitol. Today 14, 119–124. doi: 10.1016/s0169-4758(97)01179-4
Machado, E. M., Fernandes, A. J., Murta, S. M., Vitor, R. W., Camilo, D. J., Pinheiro, S. W., et al. (2001). A study of experimental reinfection by Trypanosoma cruzi in dogs. Am. J. Trop. Med. Hyg. 65, 958–965. doi: 10.4269/ajtmh.2001.65.958
Macrae, J. I., Acosta-Serrano, A., Morrice, N. A., Mehlert, A., and Ferguson, M. A. J. (2005). Structural characterization of NETNES, a novel glycoconjugate in Trypanosoma cruzi epimastigotes. J. Biol. Chem. 280, 12201–12211. doi: 10.1074/jbc.M412939200
Maeda, F. Y., Cortez, C., Izidoro, M. A., Juliano, L., and Yoshida, N. (2014). Fibronectin-degrading activity of Trypanosoma cruzi cysteine proteinase plays a role in host cell invasion. Infect. Immun. 82, 5166–5174. doi: 10.1128/IAI.02022-14
Marcilla, A., Martin-Jaular, L., Trelis, M., de Menezes-Neto, A., Osuna, A., Bernal, D., et al. (2014). Extracellular vesicles in parasitic diseases. J. Extracell. Vesicles 3:25040. doi: 10.3402/jev.v3.25040
Marino, A. P. M. P., Silva, A. A., Pinho, R. T., and Lannes-Vieira, J. (2003). Trypanosoma cruzi infection: a continuous invader-host cell cross talk with participation of extracellular matrix and adhesion and chemoattractant molecules. Braz. J. Med. Biol. Res. 36, 1121–1133. doi: 10.1590/s0100-879x2003000800020
Mattos, E. C., Canuto, G., Manchola, N. C., Magalhães, R. D. M., Crozier, T. W. M., Lamont, D. J., et al. (2019). Reprogramming of Trypanosoma cruzi metabolism triggered by parasite interaction with the host cell extracellular matrix. PLoS Negl. Trop. Dis. 13:e0007103. doi: 10.1371/journal.pntd.0007103
Mayhew, T. M. (2014). Turnover of human villous trophoblast in normal pregnancy: What do we know and what do we need to know? Placenta 35, 229–240. doi: 10.1016/j.placenta.2014.01.011
Medeiros, N. I., Fares, R. C. G., Franco, E. P., Sousa, G. R., Mattos, R. T., Chaves, A. T., et al. (2017). Differential expression of matrix metalloproteinases 2, 9 and cytokines by neutrophils and monocytes in the clinical forms of Chagas disease. PLoS Negl. Trop. Dis. 11:e0005284. doi: 10.1371/journal.pntd.0005284
Medina, L., Castillo, C., Liempi, A., Herbach, M., Cabrera, G., Valenzuela, L., et al. (2018). Differential infectivity of two Trypanosoma cruzi strains in placental cells and tissue. Acta Trop. 186, 35–40. doi: 10.1016/j.actatropica.2018.07.001
Mendonça-Previato, L., Penha, L., Garcez, T. C., Jones, C., and Previato, J. O. (2013). Addition of α-O-GlcNAc to threonine residues define the post-translational modification of mucin-like molecules in Trypanosoma cruzi. Glycoconj. J. 30, 659–666. doi: 10.1007/s10719-013-9469-7
Messenger, L. A., Gilman, R. H., Verastegui, M., Galdos-Cardenas, G., Sanchez, G., Valencia, E., et al. (2017). Toward improving early diagnosis of congenital Chagas disease in an endemic setting. Clin. Infect. Dis. 65, 268–275. doi: 10.1093/cid/cix277
Messenger, L. A., Miles, M. A., and Bern, C. (2015). Between a bug and a hard place: Trypanosoma cruzi genetic diversity and the clinical outcomes of Chagas disease. Expert Rev. Anti. Infect. Ther. 13, 995–1029. doi: 10.1586/14787210.2015.1056158
Ming, M., Ewen, M. E., and Pereira, M. E. (1995). Trypanosome invasion of mammalian cells requires activation of the TGF beta signaling pathway. Cell 82, 287–296. doi: 10.1016/0092-8674(95)90316-x
Mjihdi, A., Lambot, M.-A., Stewart, I. J., Detournay, O., Noël, J.-C., Carlier, Y., et al. (2002). Acute Trypanosoma cruzi infection in mouse induces infertility or placental parasite invasion and ischemic necrosis associated with massive fetal loss. Am. J. Pathol. 161, 673–680. doi: 10.1016/S0002-9440(10)64223-X
Möller, M., Kinnear, C. J., Orlova, M., Kroon, E. E., van Helden, P. D., Schurr, E., et al. (2018). Genetic resistance to Mycobacterium tuberculosis infection and disease. Front. Immunol. 9:2219. doi: 10.3389/fimmu.2018.02219
Mor, G., Aldo, P., and Alvero, A. B. (2017). The unique immunological and microbial aspects of pregnancy. Nat. Rev. Immunol. 17, 469–482. doi: 10.1038/nri.2017.64
Moreira, O. C., Ramírez, J. D., Velázquez, E., Melo, M. F. A. D., Lima-Ferreira, C., Guhl, F., et al. (2013). Towards the establishment of a consensus real-time qPCR to monitor Trypanosoma cruzi parasitemia in patients with chronic Chagas disease cardiomyopathy: a substudy from the BENEFIT trial. Acta Trop. 125, 23–31. doi: 10.1016/j.actatropica.2012.08.020
Nakhle, M. C., de Menezes M da, C., and Irulegui, I. (2018). Eosinophil levels in the acute phase of experimental Chagas’ disease. Rev. Inst. Med. Trop. Sao Paulo 31, 384–391. doi: 10.1590/s0036-46651989000600004
Nde, P. N., Simmons, K. J., Kleshchenko, Y. Y., Pratap, S., Lima, M. F., and Villalta, F. (2006). Silencing of the laminin gamma-1 gene blocks Trypanosoma cruzi infection. Infect. Immun. 74, 1643–1648. doi: 10.1128/IAI.74.3.1643-1648.2006
Nogueira, L. G., Frade, A. F., Ianni, B. M., Laugier, L., Pissetti, C. W., Cabantous, S., et al. (2015). Functional IL18 polymorphism and susceptibility to chronic Chagas disease. Cytokine 73, 79–83. doi: 10.1016/j.cyto.2015.01.037
Oride, A., Kanasaki, H., Mijiddorj, T., Sukhbaatar, U., Ishihara, T., and Kyo, S. (2015). Regulation of kisspeptin and gonadotropin-releasing hormone expression in rat placenta: study using primary cultures of rat placental cells. Reprod. Biol. Endocrinol. 13:90. doi: 10.1186/s12958-015-0083-3
Ouyang, Y., Mouillet, J. F., Coyne, C. B., and Sadovsky, Y. (2014). Review: placenta-specific microRNAs in exosomes - good things come in nano-packages. Placenta 35, S69–S73. doi: 10.1016/j.placenta.2013.11.002
Park, D.-W., Lee, S.-K., Hong, S. R., Han, A.-R., Kwak-Kim, J., and Yang, K. M. (2012). Expression of Kisspeptin and its receptor GPR54 in the first trimester trophoblast of women with recurrent pregnancy loss. Am. J. Reprod. Immunol. 67, 132–139. doi: 10.1111/j.1600-0897.2011.01073.x
Pérez-Molina, J. A., and Molina, I. (2018). Chagas disease. Lancet 391, 82–94. doi: 10.1016/S0140-6736(17)31612-4
Picado, A., Cruz, I., Redard-Jacot, M., Schijman, A. G., Torrico, F., Sosa-Estani, S., et al. (2018). The burden of congenital Chagas disease and implementation of molecular diagnostic tools in Latin America. BMJ Glob. Heal. 3:e001069. doi: 10.1136/bmjgh-2018-001069
Pissetti, C. W., Correia, D., de Oliveira, R. F., Llaguno, M. M., Balarin, M. A. S., Silva-Grecco, R. L., et al. (2011). Genetic and functional role of TNF-alpha in the development Trypanosoma cruzi infection. PLoS Negl. Trop. Dis. 5:e976. doi: 10.1371/journal.pntd.0000976
PrabhuDas, M., Adkins, B., Gans, H., King, C., Levy, O., Ramilo, O., et al. (2011). Challenges in infant immunity: implications for responses to infection and vaccines. Nat. Immunol. 12, 189–194. doi: 10.1038/ni0311-189
Qian, J., Ding, F., Luo, A., Liu, Z., and Cui, Z. (2016). Overexpression of S100A14 in human serous ovarian carcinoma. Oncol. Lett. 11, 1113–1119. doi: 10.3892/ol.2015.3984
Racicot, K., Kwon, J.-Y., Aldo, P., Silasi, M., and Mor, G. (2014). Understanding the complexity of the immune system during pregnancy. Am. J. Reprod. Immunol. 72, 107–116. doi: 10.1111/aji.12289
Raposo, G., and Stoorvogel, W. (2013). Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol. 200, 373–383. doi: 10.1083/jcb.201211138
Rassi, A., Rassi, A., and Marcondes de Rezende, J. (2012). American trypanosomiasis (Chagas disease). Infect. Dis. Clin. North Am. 26, 275–291. doi: 10.1016/j.idc.2012.03.002
Rausch, M. E., and Barnhart, K. T. (2012). Serum biomarkers for detecting ectopic pregnancy. Clin. Obstet. Gynecol. 55, 418–423. doi: 10.1097/GRF.0b013e31825109f6
Reinhard, G., Noll, A., Schlebusch, H., Mallmann, P., and Ruecker, A. V. (1998). Shifts in the TH1/TH2 balance during human pregnancy correlate with apoptotic changes. Biochem. Biophys. Res. Commun. 245, 933–938. doi: 10.1006/bbrc.1998.8549
Reis-Cunha, J. L., Baptista, R. P., Rodrigues-Luiz, G. F., Coqueiro-dos-Santos, A., Valdivia, H. O., de Almeida, L. V., et al. (2018). Whole genome sequencing of Trypanosoma cruzi field isolates reveals extensive genomic variability and complex aneuploidy patterns within TcII DTU. BMC Genomics 19:816. doi: 10.1186/s12864-018-5198-4
Requena-Méndez, A., Albajar-Viñas, P., Angheben, A., Chiodini, P., Gascón, J., and Muñoz, J. (2014). Health policies to control Chagas disease transmission in European countries. PLoS Negl. Trop. Dis. 8:e3245. doi: 10.1371/journal.pntd.0003245
Retana Moreira, L., Rodríguez Serrano, F., and Osuna, A. (2019). Extracellular vesicles of Trypanosoma cruzi tissue-culture cell-derived trypomastigotes: induction of physiological changes in non-parasitized culture cells. PLoS Negl. Trop. Dis. 13:e0007163. doi: 10.1371/journal.pntd.0007163
Risso, M. G., Garbarino, G. B., Mocetti, E., Campetella, O., Gonzalez Cappa, S. M., Buscaglia, C. A., et al. (2004). Differential expression of a virulence factor, the trans-sialidase, by the main Trypanosoma cruzi phylogenetic lineages. J. Infect. Dis. 189, 2250–2259. doi: 10.1086/420831
Rivera, M. T., Marques de Araujo, S., Lucas, R., Deman, J., Truyens, C., Defresne, M. P., et al. (1995). High tumor necrosis factor alpha (TNF-alpha) production in Trypanosoma cruzi-infected pregnant mice and increased TNF-alpha gene transcription in their offspring. Infect. Immun. 63, 591–595.
Rodrigues, J. C. F., Godinho, J. L. P., and de Souza, W. (2014). Biology of human pathogenic trypanosomatids: epidemiology, lifecycle and ultrastructure. Sub Cell. Biochem. 74, 1–42. doi: 10.1007/978-94-007-7305-9_1
Rodriguez, P., Carlier, Y., and Truyens, C. (2012). Activation of cord blood myeloid dendritic cells by Trypanosoma cruzi and parasite-specific antibodies, proliferation of CD8+ T cells, and production of IFN-γ. Med. Microbiol. Immunol. 201, 157–169. doi: 10.1007/s00430-011-0217-y
Sadissou, I., d’Almeida, T., Cottrell, G., Luty, A., Krawice-Radanne, I., Massougbodji, A., et al. (2014). High plasma levels of HLA-G are associated with low birth weight and with an increased risk of malaria in infancy. Malar. J. 13, 312. doi: 10.1186/1475-2875-13-312
Sánchez Negrette, O., Mora, M. C., and Basombrío, M. A. (2005). High prevalence of congenital Trypanosoma cruzi infection and family clustering in Salta, Argentina. Pediatrics 115, e668–e672. doi: 10.1542/peds.2004-1732
Scharfstein, J., and Morrot, A. (1999). A role for extracellular amastigotes in the immunopathology of Chagas disease. Mem. Inst. Oswaldo Cruz 94(Suppl. 1), 51–63. doi: 10.1590/s0074-02761999000700005
Schorey, J. S., and Bhatnagar, S. (2008). Exosome function: from tumor immunology to pathogen biology. Traffic 9, 871–881. doi: 10.1111/j.1600-0854.2008.00734.x
Schorey, J. S., and Harding, C. V. (2016). Extracellular vesicles and infectious diseases: new complexity to an old story. J. Clin. Invest. 126, 1181–1189. doi: 10.1172/JCI81132
Seco-Hidalgo, V., De Pablos, L. M., and Osuna, A. (2015). Transcriptional and phenotypical heterogeneity of Trypanosoma cruzi cell populations. Open Biol. 5:150190. doi: 10.1098/rsob.150190
Siriano, L., da, R., Luquetti, A. O., Avelar, J. B., Marra, N. L., and de Castro, A. M. (2011). Chagas disease: increased parasitemia during pregnancy detected by hemoculture. Am. J. Trop. Med. Hyg. 84, 569–574. doi: 10.4269/ajtmh.2011.10-0015
Skowron-Cendrzak, A., Rudek, Z., Sajak, A., Kubera, M., Basta-Kaim, A., and Shani, J. (1999). Effect of multiparity on T-cell proliferation response to mitogen stimulation in elderly women. Int. J. Immunopharmacol. 21, 177–183. doi: 10.1016/s0192-0561(98)00078-2
Smith, N. L. D., and Denning, D. W. (2014). Clinical implications of interferon- γ genetic and epigenetic variants. Immunology 143, 499–511. doi: 10.1111/imm.12362
Solana, M. E., Celentano, A. M., Tekiel, V., Jones, M., and González Cappa, S. M. (2002). Trypanosoma cruzi: effect of parasite subpopulation on murine pregnancy outcome. J. Parasitol. 88, 102–106. doi: 10.1645/0022-3395(2002)088%5B0102:tceops%5D2.0.co;2
Straszewski-Chavez, S. L., Abrahams, V. M., Funai, E. F., and Mor, G. (2004). X-linked inhibitor of apoptosis (XIAP) confers human trophoblast cell resistance to Fas-mediated apoptosis. Mol. Hum. Reprod. 10, 33–41. doi: 10.1093/molehr/gah001
Tarleton, R. L. (2007). Immune system recognition of Trypanosoma cruzi. Curr. Opin. Immunol. 19, 430–434. doi: 10.1016/j.coi.2007.06.003
Teixeira, A. R. L., Hecht, M. M., Guimaro, M. C., Sousa, A. O., and Nitz, N. (2011). Pathogenesis of Chagas’ disease: parasite persistence and autoimmunity. Clin. Microbiol. Rev. 24, 592–630. doi: 10.1128/CMR.00063-10
Theocharis, A. D., Manou, D., and Karamanos, N. K. (2019). The extracellular matrix as a multitasking player in disease. FEBS J. doi: 10.1111/febs.14818 [Epub ahead of print].
Tibayrenc, M. (1998). Genetic epidemiology of parasitic protozoa and other infectious agents: the need for an integrated approach. Int. J. Parasitol. 28, 85–104. doi: 10.1016/s0020-7519(97)00180-x
Tibayrenc, M., Ward, P., Moya, A., and Ayala, F. J. (1986). Natural populations of Trypanosoma cruzi, the agent of Chagas disease, have a complex multiclonal structure. Proc. Natl. Acad. Sci. U.S.A. 83, 115–119. doi: 10.1073/pnas.83.1.115
Torres, O. A., Calzada, J. E., Beraún, Y., Morillo, C. A., González, A., González, C. I., et al. (2010). Role of the IFNG +874T/A polymorphism in Chagas disease in a Colombian population. Infect. Genet. Evol. 10, 682–685. doi: 10.1016/j.meegid.2010.03.009
Torrico, F., Alonso-Vega, C., Suarez, E., Rodriguez, P., Torrico, M.-C., Dramaix, M., et al. (2004). Maternal Trypanosoma cruzi infection, pregnancy outcome, morbidity, and mortality of congenitally infected and non-infected newborns in Bolivia. Am. J. Trop. Med. Hyg. 70, 201–209. doi: 10.4269/ajtmh.2004.70.201
Trocoli Torrecilhas, A. C., Tonelli, R. R., Pavanelli, W. R., da Silva, S., Schumacher, R. I., de Souza, W., et al. (2009). Trypanosoma cruzi: parasite shed vesicles increase heart parasitism and generate an intense inflammatory response. Microbes Infect. 11, 29–39. doi: 10.1016/j.micinf.2008.10.003
Truyens, C., and Carlier, Y. (2017). “Protective host response to Trypanosoma cruzi and its limitations”, in American Trypanosomiasis Chagas Disease, 2nd Edn, eds J. Telleria and M. Tibayrenc (London: Elsevier), 579–604. doi: 10.1016/B978-0-12-801029-7.00026-5
van Niel, G., Porto-Carreiro, I., Simoes, S., and Raposo, G. (2006). Exosomes: a common pathway for a specialized function. J. Biochem. 140, 13–21. doi: 10.1093/jb/mvj128
Vannberg, F. O., Chapman, S. J., and Hill, A. V. S. (2011). Human genetic susceptibility to intracellular pathogens. Immunol. Rev. 240, 105–116. doi: 10.1111/j.1600-065X.2010.00996.x
Vekemans, J., Truyens, C., Torrico, F., Solano, M., Torrico, M. C., Rodriguez, P., et al. (2000). Maternal Trypanosoma cruzi infection upregulates capacity of uninfected neonate cells To produce pro- and anti-inflammatory cytokines. Infect. Immun. 68, 5430–5434. doi: 10.1128/iai.68.9.5430-5434.2000
Villagrasa, A., Álvarez, P. J., Osuna, A., Garrido, J. M., Aránega, A., and Rodríguez-Serrano, F. (2014). Exosomes derived from breast cancer cells, small Trojan Horses? J. Mammary Gland Biol. Neoplasia 19, 303–313. doi: 10.1007/s10911-015-9332-5
Wang, S.-C. (2014). PCNA: a silent housekeeper or a potential therapeutic target? Trends Pharmacol. Sci. 35, 178–186. doi: 10.1016/j.tips.2014.02.004
Webber, J., Yeung, V., and Clayton, A. (2015). Extracellular vesicles as modulators of the cancer microenvironment. Semin. Cell Dev. Biol. 40, 27–34. doi: 10.1016/j.semcdb.2015.01.013
Weyer, K., and Glerup, S. (2011). Placental regulation of peptide hormone and growth factor activity by proMBP. Biol. Reprod. 84, 1077–1086. doi: 10.1095/biolreprod.110.090209
WHO (2015). Chagas disease in Latin America: an epidemiological update based on 2010 estimates. Relev. Epidemiol. Hebd. 90, 33–43.
Wujcicka, W., Wilczyński, J., Śpiewak, E., and Nowakowska, D. (2018). Genetic modifications of cytokine genes and Toxoplasma gondii infections in pregnant women. Microb. Pathog. 121, 283–292. doi: 10.1016/j.micpath.2018.05.048
Yeo, M., Acosta, N., Llewellyn, M., Sánchez, H., Adamson, S., Miles, G. A. J., et al. (2005). Origins of Chagas disease: Didelphis species are natural hosts of Trypanosoma cruzi I and armadillos hosts of Trypanosoma cruzi II, including hybrids. Int. J. Parasitol. 35, 225–233. doi: 10.1016/j.ijpara.2004.10.024
Keywords: Trypanosoma cruzi, infection, maternal-fetal interactions, placenta, congenital chagas disease
Citation: Kemmerling U, Osuna A, Schijman AG and Truyens C (2019) Congenital Transmission of Trypanosoma cruzi: A Review About the Interactions Between the Parasite, the Placenta, the Maternal and the Fetal/Neonatal Immune Responses. Front. Microbiol. 10:1854. doi: 10.3389/fmicb.2019.01854
Received: 28 February 2019; Accepted: 29 July 2019;
Published: 14 August 2019.
Edited by:
Guan Zhu, Texas A&M University, United StatesReviewed by:
Antonio Ortega-Pacheco, Universidad Autónoma de Yucatán, MexicoDaniel Adesse, Oswaldo Cruz Foundation (Fiocruz), Brazil
Copyright © 2019 Kemmerling, Osuna, Schijman and Truyens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ulrike Kemmerling, dWtlbW1lcmxpbmdAdWNoaWxlLmNs; Antonio Osuna, YW9zdW5hQHVnci5lcw==; Alejandro Gabriel Schijman, c2NoaWptYW5AZG5hLnViYS5hcg==; Carine Truyens, Y3RydXllbnNAdWxiLmFjLmJl