Erratum: Microbial Community Structures and Important Associations Between Soil Nutrients and the Responses of Specific Taxa to Rice-Frog Cultivation
 Xiaomei Yi
Xiaomei Yi Kai Yi2
Kai Yi2 Linkui Cao
Linkui Cao- 1School of Agriculture and Biology, Shanghai Jiao Tong University, Shanghai, China
- 2China National Cereals, Oils and Foodstuffs Corporation, Beijing, China
Rice-frog cultivation is a traditional farming system in China and has been reintroduced as an agricultural practice in China in recent years. The microbial community in paddy rhizospheric soils has attracted much attention because many microorganisms participate in functional processes in soils. In this study, Illumina MiSeq sequencing-based techniques were used to investigate soil microbial communities and functional gene patterns across samples obtained by conventional rice cultivation (CR) and rice-frog cultivation (RF). The results showed that RF significantly affected the microbial community composition and richness, which indicated that the rhizospheric microorganisms responded to the introduction of tiger frogs into the paddy fields. Operational taxonomic units (OTUs) from Sandaracinaceae, Anaerolineaceae, Candidatus Nitrososphaera, Candidatus Nitrosotalea, Candidatus Nitrosoarchaeum and some unclassified OTUs from Euryarchaeota and Agaricomycetes were significantly enriched by RF. The abiotic parameters soil organic carbon (SOC), nitrate nitrogen (NO3−-N), and available phosphorus (AP) changed under RF treatment and played essential roles in establishing the soil bacterial, archaeal, and fungal compositions. Correlations between environmental factors and microbial communities were described using network analysis. SOC was strongly correlated with Anaerolineaceae, Methanosaeta, and Scutellinia. NO3−-N showed strong positive correlations with Opitutus, Geobacter, and Methanosaeta. NH4+-N was strongly positively associated with Sideroxydans, and TN was strongly positively correlated with Candidatus Nitrotoga. Compared to conventional CR, RF greatly enriched specific microbial taxa. These taxa may be involved in the decomposition of complex organic matter and the transformation of soil nutrients, thus promoting plant growth by improving nutrient cycling. The unique patterns of microbial taxonomic and functional composition in soil profiles suggested functional redundancy in these paddy soils. RF could significantly affect the bacterial, archaeal, and fungal communities though changing SOC and AP levels.
Introduction
In most developing countries, agriculture is the main source of food, employment, income and nutrition (Saiful Islam et al., 2015). Hence, it is necessary to sustainably increase grain production, achieve food self-sufficiency and improve the well-being of small-scale farmers (Berg and Tam, 2018). This growth in food production must be achieved by reducing the use of land, water, labor and chemicals (Doss, 2010; Yuan et al., 2017). As farmers intensify production by increasing the use of chemicals, concerns about the negative effects of those practices on human health and the environment are growing. Therefore, crop management practices need to be improved to increase productivity and minimize adverse impacts on the quality of the natural resource base (Laborte et al., 2009; Berg and Tam, 2018).
Traditional agricultural systems have shown successful adaptation to different environments and high biodiversity (Lu and Li, 2006; Suh, 2014; Teng et al., 2015; Ren et al., 2018). Recognizing the ecological heritage of these traditional agricultural systems and incorporating these unique conditions into future agricultural designs could aid the development of sustainable agricultural practices. Many studies have described the ecosystems of rice-fish, rice-turtle, rice-duck, and rice-frog cultivation and have shown how these practices reduce the use of chemical fertilizers and pesticides (Li et al., 2008; Hu et al., 2016; Zhang et al., 2016; Yang et al., 2018). It was found that rice-duck, rice-fish and rice-turtle cultivation could improve soil microbial abundance and increase the number of functional microbes, but the control of pests in paddy fields by these methods was distinctly inferior to that by rice-frog cultivation (Teng et al., 2015, 2016). In addition, previous studies have indicated that traditional rice-duck, rice-fish and rice-turtle farming methods have focused on the feeding of animals (Furuno, 2001; Luo, 2016, 2018). Frogs have been used to control pests in paddy fields for a long time. The predatory behavior of frogs in paddy fields can reduce the incidence of pests, decrease the application of pesticides and contribute to biological control (Yi et al., 2018). Although rice-frog cultivation is an ancient farming system adopted by Asian farmers for many years, this innovation may have the potential to keep up with the surge in food security demand (Sha et al., 2017).
Microorganisms are core factors affecting the biological characteristics, biogeochemical processes, and ecology of soils (Vigdis and Lise, 2002). Recent research has largely advocated that niche, biogeographic, and neutral/stochastic processes interactively determine the microbiome community composition across spatiotemporal scales in the ecosystem (Saleem et al., 2015). Soil scientists have long noticed that the natural properties of soil, such as pH, texture, and base saturation, determined by parent materials, maintain the biodiversity of soils in nature and greatly affect the basic fertility and productivity of soils during soil formation (Bai et al., 2017). In addition, anthropogenic activities such as tillage, fertilization, irrigation, and tillage have a great impact on the structure and functional properties of soil microbial communities by changing soil properties (Tripathi et al., 2015). Many recent studies have shown that the large-scale diversity and community compositions of soil microorganisms are largely driven by soil pH and some other soil properties, such as organic matter content and salinity (Su et al., 2015; Chen et al., 2016). Certain microbial taxa at high taxonomic levels can exhibit properties of ecological coherence because these taxa respond predictably to environmental variables (Li et al., 2017). Fierer et al. (2012) proposed that certain microbial phyla could be differentiated into ecologically relevant nutritional and barren categories based on substrate preferences and life strategies. Furthermore, these taxa have potential beneficial or adverse effects on crop productivity and even the stability of agro-ecosystems (Francioli et al., 2016). Under the background of exogenous organic decomposition and soil nutrient transformation, there are complex interactions between microbial taxa (Banerjee et al., 2016). Network analysis of taxon co-occurrence, as measured based on correlations between the abundances of microbial taxa, can help decipher complex microbial association patterns and the ecological rules that guide community assembly (Barberan et al., 2012). Network analysis not only reveals intertaxon associations in shared common niche spaces but also links microbial taxa to environmental parameters (Fuhrman, 2009; Ma et al., 2018). Rice frogs can directly change soil properties to influence the abundances of certain microbial phyla (Yi et al., 2018). However, knowledge regarding the changes in soil bacterial, archaeal and fungal taxa at low taxonomic levels (e.g., species, genus) in response to the introduction of tiger frogs into paddy fields remains insufficient, and network analysis of soil metagenomic-related patterns has not been done before.
We hypothesized that rice-frog cultivation systems, especially with high primary production, should be suitable for the growth of bacteria, archaea, and fungi, thus changing the microbial community structures. To test this hypothesis, rhizospheric soils were sampled from a trial field that had been subjected to rice-frog cultivation for 8 years, and Illumina MiSeq sequencing targeting the V4–V5 region of the bacterial 16S rRNA genes, 524F-10-extF/Arch958R of the archaeal 16S rRNA genes, and the fungal SSU 18S rRNA genes were used for identification and quantification of rhizospheric soil bacterial, archaeal and fungal taxa. We used network analysis to explore the potential effects of specific bacterial taxa on the relationship between microorganisms and soil nutrients.
Materials and Methods
Experimental Sites
The experimental base was established in the Qingpu Modern Agricultural Park of Qingpu, Shanghai (121.12° E, 31.15° N) in 2009. The experimental site for rice cultivation patterns is located in the Yangtze River Delta region. Rice is the main crop in this area, which is planted once a year. This region is a subtropical monsoon climate. The mean annual air temperature is 15.5°C, and the mean annual precipitation is 1200 mm. The soil pH of this site is approximately 6.8. Before transplantation of rice plants, the soil pH, electrical conductivity (EC), and available N (AN), available phosphorus (AP), and available potassium (AK) levels of the site were 7.40, 0.13 mS cm–1, 1.70 g kg–1, 0.38 g kg–1, and 0.55 g kg–1, respectively.
Experimental Design
Two rice cultivation treatments were established in the experiment: conventional rice cultivation without frogs (CR) and rice-frog cultivation (RF). In both the CR and RF fields, the same amount (300 kg N ha–1) of nitrogen fertilizer was applied for each treatment. A randomized complete block experimental design was used with three replicates. A total of 12,000 frogs/ha were introduced into the RF paddy fields. Tiger frogs (Rana tigrina rugulosa), which are highly adaptable to the environment, were introduced and raised by the Zizaiyuan Agricultural Development Co., Ltd., Shanghai. At 15 days after rice transplantation, frogs large enough (≥20 g) to prey on pests were released into the paddy fields. In addition, the tiger frogs used in this study were managed in accordance with relevant guidelines and regulations of the Guide for the Care and Use of Laboratory Animals of the Ministry of Health, China. We domesticated and bred the tiger frogs with the permission of the Shanghai Forestry Bureau, and the license permission number was (2008) 419.
Sampling and Measurements
In October 2016, samples were taken from the rhizospheric soil at the rice maturation stage. Five rice plants (with roots) were randomly selected from each plot, collected by an investigator wearing disposable gloves and then pooled. All the rhizospheric soil samples were gently scraped from the roots, and the samples were placed into aseptic sealed plastic bags and transported back to the laboratory in an ice box containing liquid N. Each soil sample was divided into two parts. One part was freeze-dried and stored at −80°C for DNA extraction. Another part was air-dried, ground and passed through a 2 mm sieve to obtain suitable powder for the analysis of soil physicochemical characteristics: soil pH, EC and AP, available potassium (AK), available N (AN), total N (TN), and soil organic carbon (SOC) levels.
DNA Extraction
According to the manufacturer’s protocol, DNA was extracted from 0.2 g of freeze-dried soil using the E.Z.N.A.® Soil DNA Kit (Omega Biotek, Norcross, GA, United States). DNA quality was evaluated by 1% sodium agarose gel electrophoresis. Using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific Inc., Wilmington, DE, United States) to determine DNA purity and concentrations, the DNA was stored at −20°C prior to amplification.
Amplicon Library Preparation and Sequencing
Amplicon library preparation and Illumina® MiSeq sequencing (Illumina, San Diego, CA, United States) were carried out by Majorbio Biopharm Technology Co., Ltd. (Shanghai, China). The 16S rRNA V4–V5 gene fragments were amplified by the primer pair 515F/907R (F: 5′-GTGCCAGCMGCCGCGG-3′ and R: 5′-CCGTCAATTCMTTTRAGTTT-3′) (Edwards et al., 2015). For the archaeal 16S rRNA gene fragments, the primer pair used was 524F/958R (F: 5′-TGYCAGCCGCCGCGGTAA-3′ and R: 5′-YCCGGCGTTGAMTCCAATT-3′) (Besaury et al., 2014). Amplification of fungal 18S rRNA gene fragments was performed by using the forward primers SSU0817F/1196R (5′-TTAGCATGGAATAATRRAATAGGA-3′ and 5′-TCTGGACCTGGTGAGTTTCC-3′) (Xiao et al., 2018; Yuan et al., 2018). The following procedures were used for PCR: denaturation for 3 min at 95°C; 27 cycles of 30 s at 95°C, annealing for 30 s at 55°C, and elongation for 45 s at 72°C; and a final extension step for 10 min at 72°C. The PCR buffers were performed in triplicate in 20 μL mixtures containing 0.8 μL of each primer (5 μM), 2 μL of 2.5 mM dNTPs, 4 μL of 5× FastPfu buffer, 0.4 μL of FastPfu polymerase (TransGen, Beijing, China) and 10 ng of template DNA. The PCR products were extracted from 2% agarose gels, purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, United States) and quantified using QuantiFluorTM-ST (Promega, United States) according to the manufacturer’s protocol. The purified amplicons were pooled in equimolar concentrations and processed with MiSeq Reagent Kit V2, and then 250 bp paired-end dual index sequencing was performed with an Illumina MiSeq instrument (Illumina, San Diego, CA, United States). The original raw sequencing reads were submitted to the short read archives of the National Biotechnology Information Center under registration number PRJNA47103.
Bioinformatic and Statistical Analysis of the Community
The original raw fastq files were demultiplexed, quality-filtered by Trimmomatic and merged using FLASH with the following criteria: (i) the reads were truncated at any site that received an average quality score < 20 over a 50 bp sliding window; (ii) the primers matched perfectly, allowing 2 nucleotide mismatches, and reads containing ambiguous bases were deleted; and (iii) sequences that overlapped by more than 10 bp were merged based on the overlap sequence (Magoc and Salzberg, 2011). Operational taxonomic units (OTUs) were clustered by similarity using Mothur version 1.31.1 with 97% cutoff points, and chimeric sequences were removed by quantitative insights into microbial ecology (QIIME) (Schloss et al., 2009; Caporaso et al., 2010). The taxonomy of each gene sequence was examined against the Silva (SSU123) database with a confidence threshold of 70%. A total of 298,045 high-quality 16S rRNA reads, 291,954 high-quality arch-16S rRNA reads and 300,714 high-quality 18S rRNA reads were obtained.
Statistical analyses were carried out using Statistical Product and Service Solutions (SPSS) 18.0 software and the R vegan package1. The remaining sequences of all the samples were rarefied to the same sequencing depth. Principal coordinates analysis (PCoA) of “the Bray-Curtis distances” was performed using the R package “pcoa.” Venn diagrams were generated with the “vennerable” package in R. Redundancy analysis (RDA) of multiple correlation variations among environmental variables (SOC, TN, AP, pH and community composition at the phylum level) was carried out by using the "rda" function, and the environmental factors were fitted with the ordination plots using the vegan package in R with 999 permutations. The differential OTU abundances were calculated by using the R package “DESeq2.” Differential abundance analysis was performed by fitting the generalized linear model with a negative binomial distribution to the normalized value of each OTU and using a Wald test to test the differential abundance. Enriched and depleted OTUs were defined as OTUs with absolute differential abundance >1.0 and p < 0.05.
Network Analysis
Network analysis was conducted on microbial OTUs and soil properties (AN, NH4+-N, NO3−-N, TN, and SOC) by Pearson correlation analysis. To decrease pairwise comparisons and minimize network complexity, 200 OTUs with the highest abundances from CR and RF (adjusted P < 0.01) were used for network analysis. The OTUs with strongly positive (r > 0.8) and strongly negative (r < −0.8) values were used to calculate the network in Cytoscape v.3.6.1 (Shannon et al., 2003). The NetworkAnalyzer tool in Cytoscape was used to calculate the network topology characteristics.
Results
Soil Biochemical Properties
The soil pH, TN content and NH4+-N content showed no significant differences between treatments. RF significantly decreased the NO3−-N, AP, and AK content by 37.47, 54.30, and 28.41%, respectively, and significantly increased the SOC and AN content by 4.68 and 20.22%, respectively (Table 1).
TABLE 1
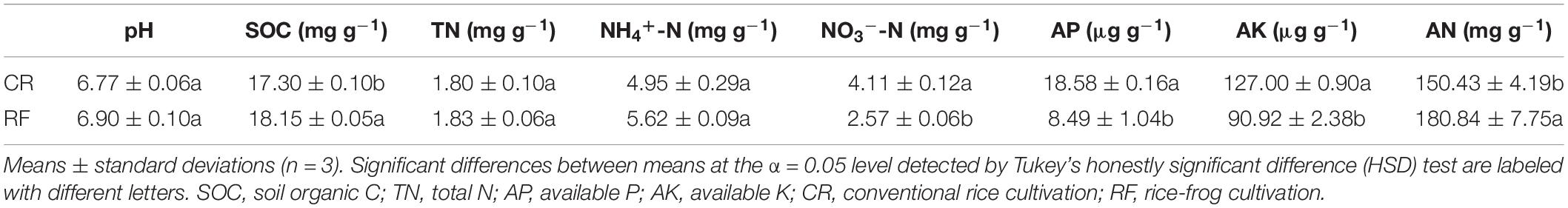
Table 1. Soil biochemical properties between CR and RF.
Within-Habitat Diversity and Relative Abundances of Major Phyla
Measurement of α-diversity revealed differences in community diversity between RF and CR. We calculated the abundance-based coverage estimator (ACE) index to estimate the richness of each soil sample. The Shannon diversity index was used to evaluate the microbial diversity of each soil sample. The mean estimated community diversity was higher in the RF rhizospheric soil than in the CR soil, however, there was no statistically significant difference in the archaeal community between these two treatments (Figure 1A). Compared with the CR treatment, RF significantly increased the ACE richness indexes of the bacterial, archaeal and fungal communities (Figure 1B). There were significant differences in the proportions of various phyla across the treatments. Proteobacteria (31.01–34.52%), Acidobacteria (18.08–18.34%), and Chloroflexi (16.82–17.51%) were the dominant bacterial phyla across treatments (Figure 2A); MCG (Miscellaneous Crenarchaeotal Group, 42.52–59.34%), Thaumarchaeota (16.31–46.32%), and Euryarchaeota (9.92–21.84%) were the dominant archaeal phyla (Figure 2B); and Ascomycota (74.2–59.34%) and Basidiomycota (3.83–7.81%) were the dominant fungal phyla (Figure 2C). The RF soil had a notably greater proportion of MCG and Euryarchaeota than the CR rhizospheric soil, whereas Proteobacteria, Acidobacteria, Thaumarchaeota, Ascomycota, and Basidiomycota were mostly depleted in the RF treatment.
FIGURE 1
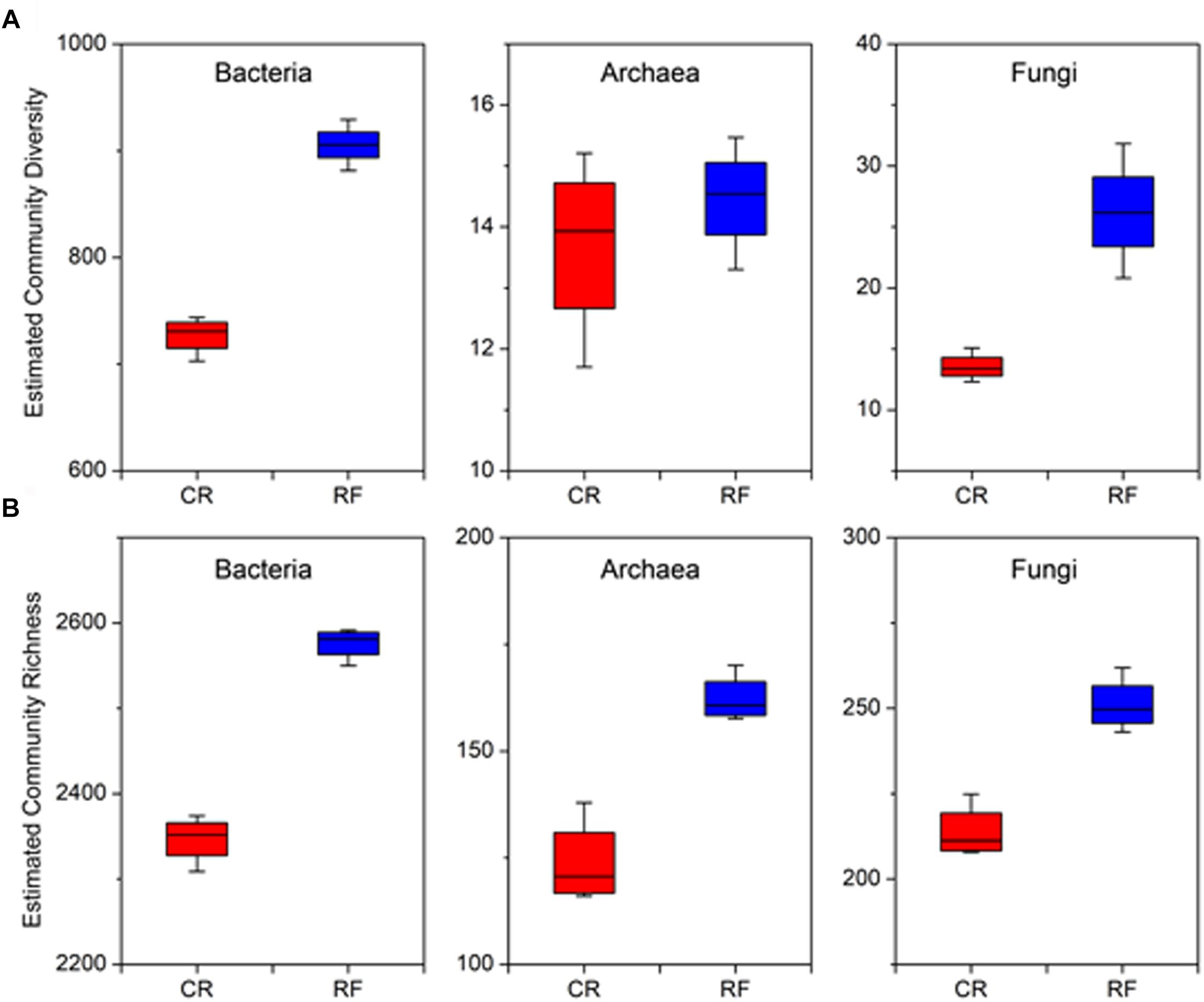
Figure 1. Within-sample (α-diversity) measurements between CR and RF. (A) Estimated species richness was calculated as eShannon_entropy. (B) Abundance-based coverage estimator (ACE) index. The horizontal bars within the boxes represent the median. The tops and bottoms of the boxes represent the 75th and 25th quartiles, respectively. The upper and lower whiskers extend 1.5× the interquartile range from the upper and lower edges of each box, respectively. All outliers are plotted as individual points. CR, conventional rice cultivation; RF, rice-frog cultivation.
FIGURE 2
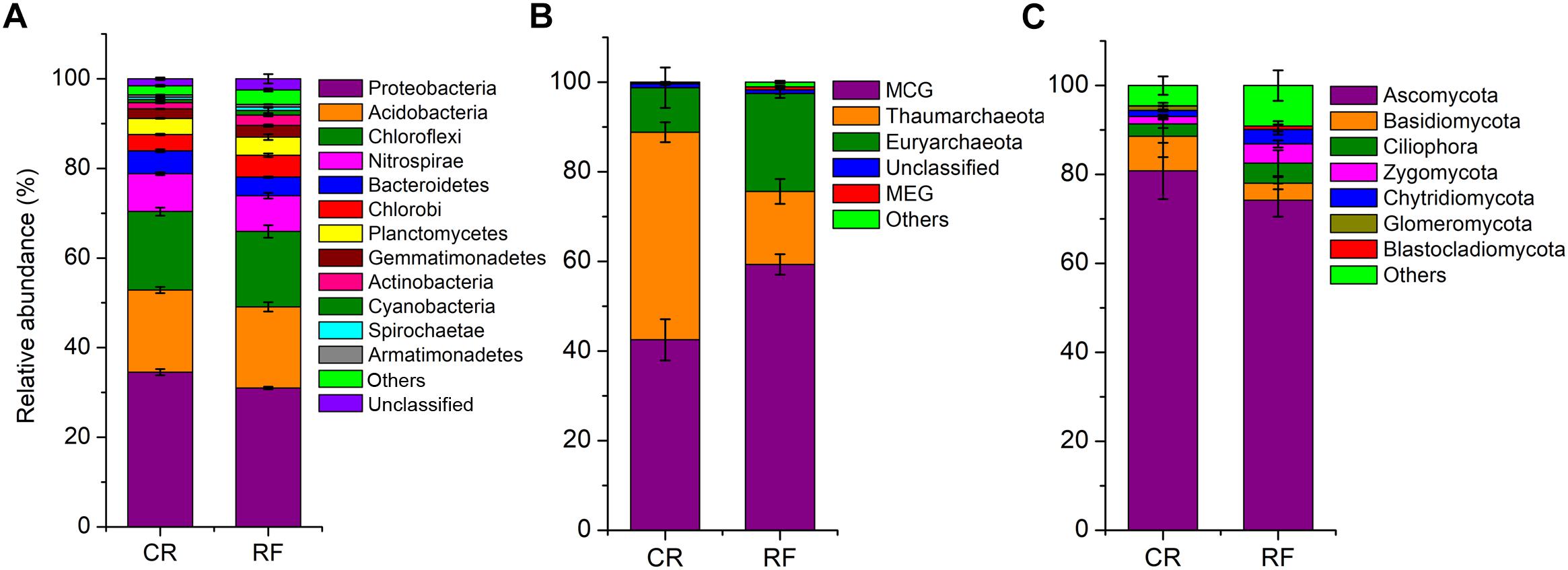
Figure 2. Histograms of the relative abundances of microbial phyla between CR and RF. (A) Relative abundances of bacterial phyla. (B) Relative abundances of archaeal phyla. (C) Relative abundances of fungal phyla. CR, conventional rice cultivation; RF, rice-frog cultivation.
OTUs Enriched and Depleted by Rice-Frog Cultivation
We performed differential abundance analysis to confirm OTUs that were greatly influenced by RF (Figure 3A). Using OTU abundances from CR as a control and no more than 0.05 as an adjusted p-value cutoff, “enriched OTUs (eOTUs)” and “depleted OTUs (dOTUs)” were identified, representing OTUs for which the relative abundance significantly increased and decreased, respectively, by more than twofold in response to RF. There were 483, 25, and 30 eOTUs and 645, 88, and 115 dOTUs in bacteria, archaea and fungi, respectively. Among the top 10 most influential OTUs in bacteria, most eOTUs were identified as a number of unclassified members of Methylophilaceae, Sandaracinaceae, and Anaerolineaceae. Among the top 10 most influential OTUs in archaea and fungi, the eOTUs were chiefly identified as Candidatus Nitrososphaera, Candidatus Nitrosotalea, Candidatus Nitrosoarchaeum, some unclassified OTUs of Euryarchaeota, several unclassified members of Agaricomycetes, and Incertae Sedis of Zygomycota and Glomeromycota. The Venn plot (Figure 3B) showed that the bacterial, archaeal, and fungal communities shared 2202, 119, and 120 OTUs, respectively.
FIGURE 3
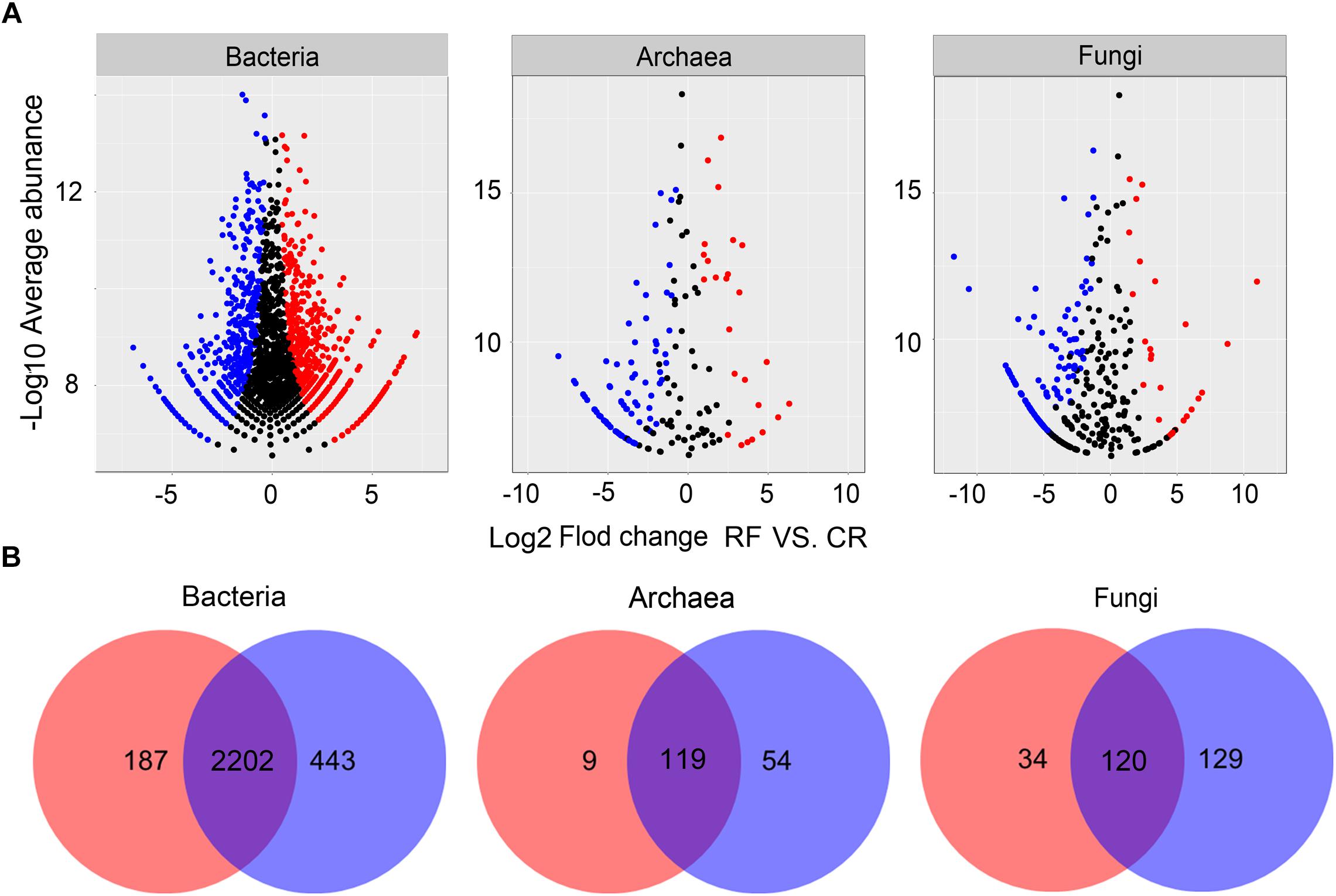
Figure 3. (A) Volcano plots illustrating OTUs that were significantly enriched (red) and depleted (blue) by RF compared with CR as determined by differential abundance analysis. Each point represents an individual OTU, and the Y-axis indicates the fold change in abundance. (B) Number of differential OTUs between each treatment. CR, conventional rice cultivation; RF, rice-frog cultivation.
Community Structure, Variation, and Determinants
PCoA with Bray-Curtis distances showed that the community distinctly separated RF from CR along the first principal coordinate (Figure 4A). This result indicated that RF had a certain influence on bacterial, archaeal and fungal communities. RDA was used to quantify the influences of edaphic factors (i.e., pH, SOC, TN, and AP) on the bacterial, archaeal and fungal community compositions. The four constrained factors considerably contributed to the bacterial community (P = 0.03), archaeal community (P = 0.04), and fungal community (P = 0.03). SOC was the determinant of these factors in the bacterial community, however, AP was the determinant in the archaeal and fungal communities (Figure 4B). These results indicated that the soil bacterial community composition was mostly driven by the SOC and AP levels under long-term fertilization.
FIGURE 4
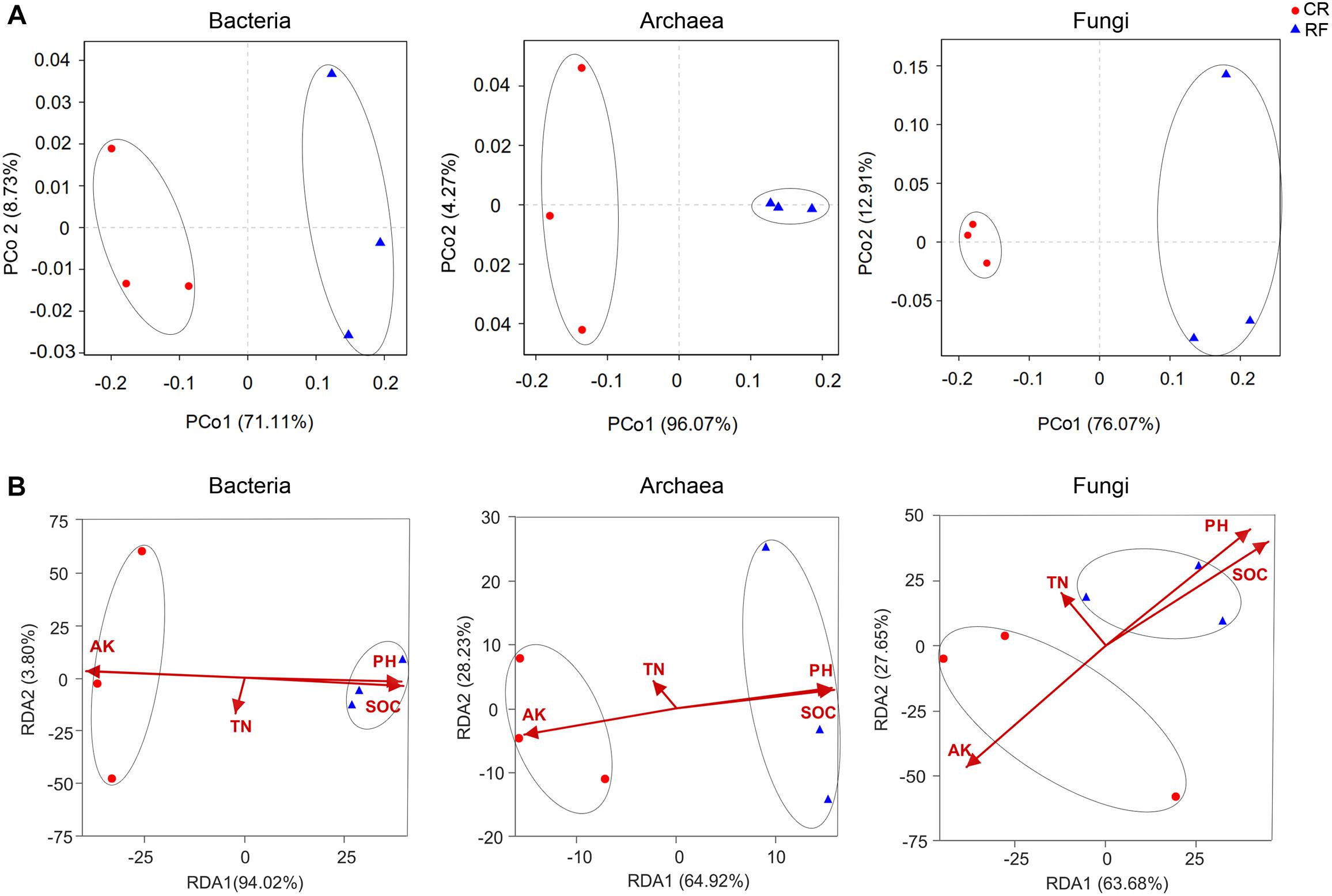
Figure 4. (A) Principal coordinates analysis (PCoA) of bacterial, archaeal, and fungal community variations between CR (conventional rice cultivation) and RF (rice-frog cultivation). (B) Redundancy analysis (RDA) based on Bray-Curtis dissimilarities of bacterial, archaeal and fungal communities and soil properties. AP, available phosphorus; TN, total nitrogen; pH, hydrogen ion concentration; SOC, soil organic C.
Network Associations Among OTUs and Soil Properties
The bacterial network consisted of 3920 significant associations (edges) of 202 nodes, with an average clustering coefficient of 0.56 and a total diameter of 38 edges. The network showed an average number of 44 neighbors and a feature path length of 2.43. The network edges were predominantly composed of strong positive associations, and the dominant identifiable OTUs belonged to Proteobacteria, Acidobacteria, and Nitrospirae (Figure 5A). AN showed strong positive associations with one Chloroflexi KD4-96 member and Acidobacteria subgroup 6, subgroup 17, and subgroup 18 members and a strong negative association with Geobacter. NH4+-N showed strong associations with Sideroxydans, Methylosarcina, etc., and strong negative associations with Acidibacter, Nitrospira, etc. NO3−-N was positively correlated with Opitutus, Geobacter, etc., and negatively correlated with Planctomyces, Geobacter, etc. SOC showed a strong positive association with one uncultured member belonging to Anaerolineaceae and negative associations with Planctomyces, Geobacter, etc. TN was positively correlated with Candidatus Nitrotoga and one member of Syntrophobacteraceae. The archaeal network included 3116 notable associations (edges) of 178 nodes, with an average clustering coefficient of 0.63 and a total diameter of 35 edges. The network showed an average number of neighbors of 39 and a typical path length of 2.31. The network edges were significantly composed of strongly positive correlations, and the identifiable OTUs mainly belonged to the phyla Thaumarchaeota and Euryarchaeota (Figure 5B). AN showed strong positive associations with MCG members and Methanosaeta, etc. NH4+-N was positively correlated with Methanobacterium, etc. NO3−-N showed a strong negative association with Methanosaeta, etc. SOC was positively correlated with one member within Rice Cluster II and Methanosaeta, etc. and negatively correlated with Candidatus Nitrosoarchaeum, etc. The fungal network consisted of 3464 significant associations (edges) of 208 nodes, with an average clustering coefficient of 0.57 and a total diameter of 33 edges. The fungal network showed 38 neighbors and a typical path length of 2.51. The network edges predominantly consisted of strongly positive and negative correlations, and the dominant identifiable OTUs belonged to Ascomycota, Chytridiomycota and Euryarchaeota (Figure 5C). AN showed strong positive associations with Incertae Sedis within Zygomycota and a negative association with Incertae Sedis within Glomeromycota. NH4+-N was strongly positively associated with Cochliobolus, etc., and negatively associated with Trimastix, etc. NO3−-N showed a strong association with Incertae Sedis within Ascomycota, etc., SOC was strongly positively correlated with Scutellinia, etc.
FIGURE 5
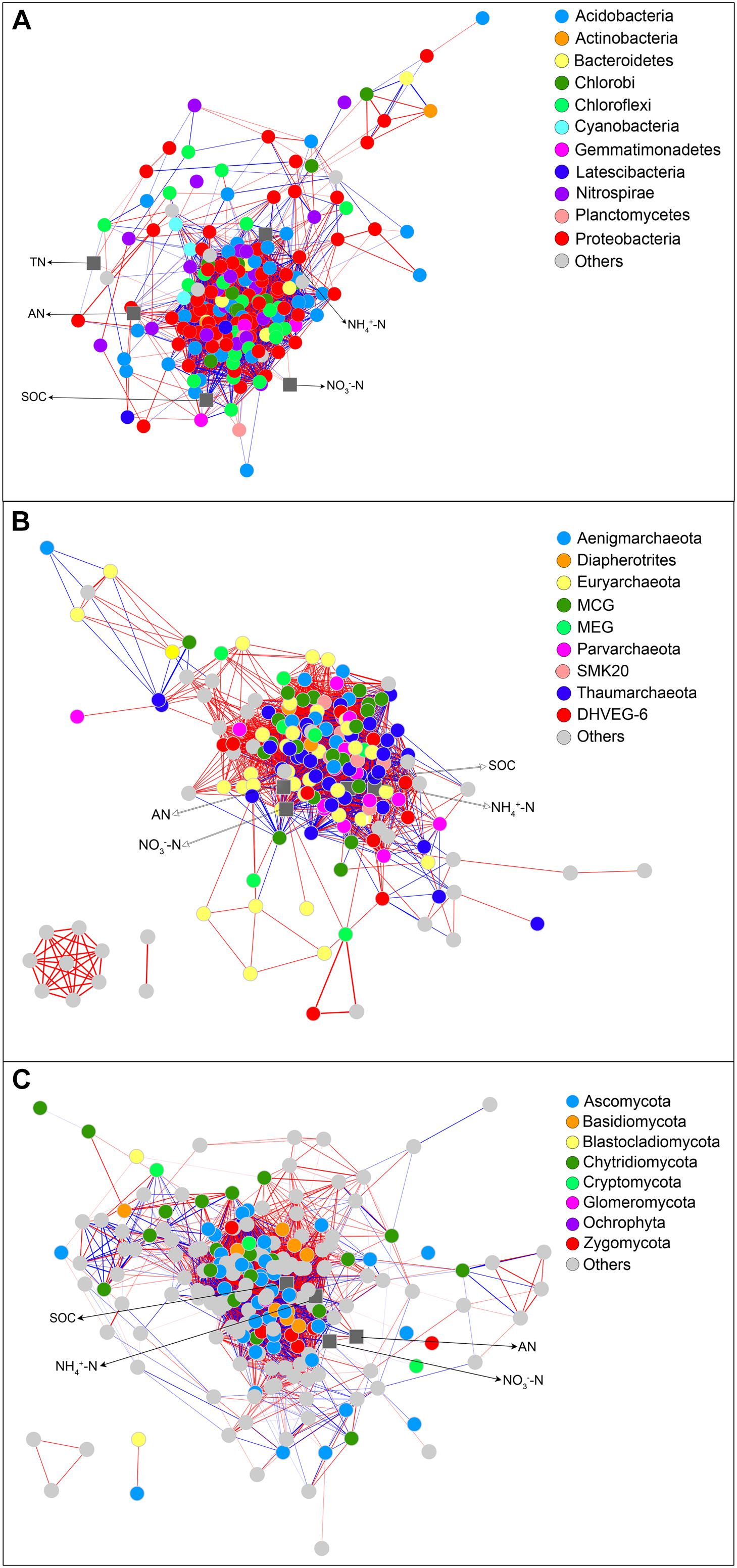
Figure 5. Correlation network between differentially abundant OTUs in bacterial (A), archaeal (B), and fungal (C) communities and soil properties. Blue and red lines represent significantly negative (P < 0.01, r < −0.8) and positive (P < 0.01, r > 0.8) correlations, respectively. Colored nodes signify corresponding OTUs assigned to major phyla. Soil properties are indicated with a rounded rectangle. SOC, soil organic C; TN, total N; NO3−-N, nitrate nitrogen; NH4+-N, ammonium nitrogen. CR, conventional rice cultivation; RF, rice-frog cultivation.
Discussion
Changes in Microbial Community Composition in Response to Rice-Frog Cultivation
To the best of our knowledge, this study was the first comprehensive investigation of the structural and functional differentiation of microbial communities in rice paddy soil under rice-frog cultivation. RF treatment was used as an organic fertilizer and could be considered an organic treatment. The observed Shannon index showed that the RF treatment significantly increased the microbial community diversity compared to that of the CR treatment. The bacterial, archaeal and fungal community richness was significantly enhanced by RF. Similar results have been observed in previous studies for paddy soil (Zhong et al., 2009; Geisseler and Scow, 2014; Wang et al., 2017). RF had an important influence on the structure and function of the rice rhizospheric soil. The phyla Proteobacteria, Acidobacteria, and Chloroflexi dominated bacterial communities across all treatments and roughly corresponded to the results of previous studies on rice rhizospheric soils (Edwards et al., 2015). Most paddy soils have abundant nutritional lifestyles; generally, the phylum Proteobacteria accounts for the largest proportion of the soil bacterial communities in terms of both metabolism and genetics, even in other paddy soils (Yuan et al., 2018). Phylum Chloroflexi is a facultative anaerobic bacterium that plays a recognized role in heterotrophic oligotrophs in soils, showing the viability of recalcitrant plant polymers (Hug et al., 2013).
RF had no significant effect on archaeal community diversity. However, the relative abundances of three dominant phyla, namely, MCG, Thaumarchaeota, and Euryarchaeota, changed significantly. The phylum Thaumarchaeota constituted the largest fraction of the soil bacterial communities in the CR rhizospheric soil, however, the relative abundance of the phylum Thaumarchaeota was lower than that of MCG or Euryarchaeota in RF. Euryarchaeota is the dominant phylum in a large number of soil environments (Lee et al., 2015; Liu et al., 2016). Mesophilic Crenarchaea have recently been considered to play a key role in soil nitrogen and carbon cycling (Lam et al., 2007; Brochier-Armanet et al., 2008). MCG species have been detected in a variety of environmental conditions, including marine and terrestrial environments, as well as oxic and anoxic geochemical zones. MCG species are heterotrophic, including ubiquitously distributed and dominant MCGs in systems with a high content of organic carbon in the sediment (Meador et al., 2015). Euryarchaeota, the second dominant archaeal phylum identified in our study, plays notable roles in anaerobic degradation through nitrate reduction (Cabello et al., 2004), nitrogen fixation (Raymond et al., 2004), organic matter degradation (Ke et al., 2014), methane oxidation (Walter et al., 2002), and the metabolism of sulfur and iron (Edwards, 2000). These species exist in all major habitats of the rice field ecosystem, especially those belonging to the classes Methanobacteria and Methanomicrobia, which are involved in greenhouse gas production, with CH4 as the final product of anaerobic respiration (Hu et al., 2013).
Fungi, as an important component of microbial communities, promote soil organic matter cycling, nutrient transformation, toxic degradation, and crop diseases. Fungal community diversity is a prerequisite for the maintenance of the normal functions of soil ecosystems (Liu et al., 2016). Similar to most other flooded paddy soils (Yuan et al., 2018), the dominant fungi in our samples were members of the phylum Ascomycota, which is consistent with our hypothesis. RF significantly increased the fungal diversity and the relative abundance of Zygomycota but decreased the relative abundances of Ascomycota and Basidiomycota. Based on the results, we can conclude that although Ascomycota was the dominant fungal phylum, the abundance of this phylum was significantly reduced in RF. The frogs introduced into RF produced frog dung, which increased the nutrient content and the fungal abundance. The differences in nutrition, including different quantities of frogs and fertilizers, may also affect the diversity and richness of fungi (Thormann, 2006; Broeckling et al., 2008). Higher species diversity and nutrition reduced the abundance of pathogenic fungi (Wang et al., 2017), which may have significantly decreased the relative abundance of Ascomycota in RF.
Rice Frogs Influenced the Taxonomic Abundances in the Microbial Community
We performed differential abundance analysis to distinguish OTUs that were responsible for the observed microbial community differences between the RF and CR rhizospheric soils. The OTUs from the phyla Betaproteobacteria, Gammaproteobacteria, Chloroflexi, and Acidobacteria were the most significantly enriched by RF. Chen et al. (2015) declared that the phyla Betaproteobacteria, Gammaproteobacteria, and Chloroflexi thrive under conditions with high substrate availability. Although there are many oligotrophic members in the phylum Acidobacteria (Pascault et al., 2013), some members of Acidobacteria were depleted by RF, while others were largely enriched. Our results were consistent with previous studies that reported that several members of Acidobacteria (e.g., subgroups 1 and 7) exhibited very low abundance, while others (e.g., subgroups 4 and 6) were extremely abundant in soils with rich SOC content (Liu et al., 2014). We analyzed the top 10 most OTUs that were influenced at the genus level and observed that the OTUs that were most highly enriched by RF were several unclassified members of the Methylophilaceae, Anaerolineaceae, and Sandaracinaceae families. Soluble simple organic matter can provide electronic donors for aerobic denitrification of Methylophilaceae of Betaproteobacteria under microaerobic conditions, and aerobic denitrification can be performed by using nitrate or nitrite (Cao et al., 2019). Anaerolineaceae species are keystone microbes that participate in the degradation of organic matter (Meng et al., 2019). The species of Sandaracinaceae are heterotrophic consumers of low-molecular-weight organic compounds, such as ethanol, hydrogen, butyrate, and acetate (Probandt et al., 2017). In our study, the specific bacterial taxa substantially enriched by RF play indispensable roles in organic decomposition and soil C, N, and P transformation.
For the archaeal community, the OTUs from Thaumarchaeota were the most significantly enriched by RF. At the genus level, the main enriched OTUs in the rhizospheric soil were Candidatus Nitrososphaera, Candidatus Nitrosotalea, Candidatus Nitrosoarchaeum and several members of SAGMCG-1 (South African Gold Mine Group 1). Candidatus Nitrososphaera, Candidatus Nitrosotalea, and Candidatus Nitrosoarchaeum all belong to ammonia-oxidizing archaea. Ammonia oxidation is the first and rate-limiting step of nitrification and is dominated by ammonia-oxidizing archaea (AOA) and ammonia-oxidizing bacteria (AOB) (Fitzgerald et al., 2015). The abundance of AOA, which dominate soil microbial activity, is usually greater than that of AOB (Lehtovirta-Morley et al., 2016). Previous studies showed that AOA had the capacity to grow mixotrophically by assimilating organics (Hallam et al., 2006; Walker et al., 2010; Liu et al., 2018). A recent study showed that some organics (pyruvate, oxaloacetate) could detoxify intracellular H2O2, functioning as chemical scavengers rather than archaeal membrane lipids, indicating that they have strict autotrophic growth (Kim et al., 2016). These results may explain why the relative abundance of AOA increased in RF.
In terms of fungi, the OTUs from Glomeromycota, Basidiomycota, and Zygomycota were the most significantly enriched by RF. These results were consistent with those of previous studies (Lynch and Thorn, 2006; Wang et al., 2017). At the class level, the main enriched OTU in the rhizospheric soil was Agaricomycetes after RF. Most Agaricomycetes species are saprotrophic species and play key roles in the decomposition of organic matter, such as wood and plant litter (Hibbett, 2007; Wang J. et al., 2018; Wang M.M. et al., 2018), which might explain the high relative abundance of Agaricomycetes in RF. Furthermore, some members of Agaricomycetes are known to be ectomycorrhizal fungi that can mobilize nutrients from organic substrates (Tibbett, 2002) and are conducive to plant growth. In addition, RF could greatly increase the relative abundance of the phylum Zygomycota.
Correlations Between Environmental Factors and the Microbial Community
PCA plots revealed that all the bacterial, archaeal and fungal communities were significantly changed by introducing tiger frogs into the rice paddy, which is roughly consistent with previous studies (Liu et al., 2017; Wang et al., 2017). Further Mantel test analysis revealed that soil AP was significantly correlated with both archaeal and fungal community compositions in the soil. However, SOC was significantly correlated with the bacterial community. SOC and AP levels have been previously identified as key factors that influence microbial community composition (Dong et al., 2014; Liu et al., 2017).
C and N are essential resources for microbial growth (Li et al., 2017). Soil C and N levels are closely associated with specific taxa that are significantly affected by RF. Our hypothesis was confirmed by a network analysis based on co-occurrence, which revealed strong positive correlations of SOC with some taxa, e.g., one uncultured member belonging to Anaerolineaceae from Chloroflexi, Rice Cluster II and Methanosaeta from Euryarchaeota, and Scutellinia from Ascomycota. NO3−-N showed strong positive correlations with Opitutus, Geobacter, and Methanosaeta. NH4+-N was strongly positively associated with Sideroxydans and Methylosarcina from Proteobacteria, Methanobacterium from Euryarchaeota, and Cochliobolus from Ascomycota. TN was strongly positively correlated with Candidatus Nitrotoga from Proteobacteria. Studies have indicated that Anaerolineaceae species are keystone microbes that participate in degrading organic matter (In’t Zandt et al., 2018; Meng et al., 2019). Previously, Methanosaeta species have been believed to be limited to acetate, which is a substrate for methane production, living in wastewater, marshes and wetlands and releasing methane and carbon dioxide by breaking down the acetic salts produced by other microbes in the environment (Barua et al., 2018; Ji et al., 2018). In addition, as the dominant bacteria in the aggregates, Geobacter species were highly connected with NO3−-N and expressed genes related to ethanol metabolism, suggesting that Geobacter and Methanosaeta species exchange electrons through direct interspecific electron transfer (Rotaru et al., 2014). Cocultures of Geobacter and Methanosaeta stoichiometrically convert ethanol to methane. As a genus of cup fungi, Scutellinia belongs to the family Pyronemataceae. The strong correlation between this genus and plant growth indicates possible beneficial effects of Scutellinia in reducing plant disease in soils (Franke-Whittle et al., 2015). Opitutus species are dominant denitrifying bacteria (Tanikawa et al., 2018), so the abundance of Opitutus species was strongly associated with the NO3−-N content. Sideroxydans is a genus of Fe-oxidizing bacteria that is associated with iron cycling in freshwater and marine environments and groundwater, as well as in most sediments and soils (Emerson et al., 2010). Nitrification is an important process in biogeochemical N-cycling and biological wastewater treatment. The second step, oxidation of nitrite to nitrate, is catalyzed by nitrite-oxidizing bacteria (NOB). Uncultured NOB of the genus Candidatus Nitrotoga are widely distributed in both natural and engineered ecosystems (Kitzinger et al., 2018). Candidatus Nitrotoga species exhibit metabolic activity under low-oxygen or anoxic conditions, which expands the environmental niche for Candidatus Nitrotoga, similar to that of other NOB (Boddicker and Mosier, 2018). Our results showing that the relative abundance of Candidatus Nitrotoga was positively associated with the TN content were roughly consistent with previous conclusions.
Conclusion
In the present study, 8 years RF significantly affected the microbial composition and structure. OTUs from Sandaracinaceae, Anaerolineaceae, Candidatus Nitrososphaera, Candidatus Nitrosotalea, and Candidatus Nitrosoarchaeum and some unclassified OTUs of Euryarchaeota and Agaricomycetes were significantly enriched by RF. The abiotic parameters SOC, NO3−-N, and AP changed in the RF treatment, which played important roles in the soil bacterial, archaeal, and fungal compositions. Furthermore, correlations between environmental factors and microbial communities were described using network analysis. SOC was strongly correlated with Anaerolineaceae, Methanosaeta, and Scutellinia. NO3−-N showed strong positive correlations with Opitutus, Geobacter, and Methanosaeta. NH4+-N was strongly positively associated with Sideroxydans, and TN was strongly positively correlated with Candidatus Nitrotoga. Compared to conventional rice cultivation, RF greatly enriched specific microbial taxa. These taxa are involved in the degradation of complex organic matter and the conversion of soil nutrients, thus improving nutrient utilization to promote plant growth.
Data Availability
The datasets generated for this study can be found in NCBI, PRJNA47103.
Author Contributions
XY and LC contributed to the conception and design of the study, acquisition, analysis, interpretation of data, drafting, revising, and final approval of the manuscript. KY, KF, HG, and WD contributed to the editing and final approval of the manuscript.
Funding
This study was financially supported by the National Key Research (No. 31770482), Development Program of the China National Natural Science Foundation of China (No. 2016YFD0801106), and the Domestic Science and Technology Cooperation Project in Shanghai (No. 18295810400).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Shanghai Qingpu Modern Agricultural Park and Shanghai Zizaiyuan Agricultural Development Co., Ltd., for providing an experimental base and for paddy field management.
Footnotes
References
Bai, R., Wang, J. T., Deng, Y., He, J. Z., Feng, K., and Zhang, L. M. (2017). Microbial community and functional structure significantly varied among distinct types of paddy soils but responded differently along gradients of soil depth layers. Front. Microbiol. 8:945. doi: 10.3389/fmicb.2017.00945
Banerjee, S., Kirkby, C. A., Schmutter, D., Bissett, A., Kirkegaard, J. A., and Richardson, A. E. (2016). Network analysis reveals functional redundancy and keystone taxa amongst bacterial and fungal communities during organic matter decomposition in an arable soil. Soil Biol. Biochem. 97, 188–198. doi: 10.1016/j.soilbio.2016.03.017
Barberan, A., Bates, S. T., Casamayor, E. O., and Fierer, N. (2012). Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 6, 343–351. doi: 10.1038/ismej.2011.119
Barua, S., Zakaria, B. S., and Dhar, B. R. (2018). Enhanced methanogenic co-degradation of propionate and butyrate by anaerobic microbiome enriched on conductive carbon fibers. Bioresour. Technol. 266, 259–266. doi: 10.1016/j.biortech.2018.06.053
Berg, H., and Tam, N. T. (2018). Decreased use of pesticides for increased yields of rice and fish-options for sustainable food production in the Mekong Delta. Sci Total Environ. 619-620, 319–327. doi: 10.1016/j.scitotenv.2017.11.062
Besaury, L., Ghiglione, J. F., and Quillet, L. (2014). Abundance, activity, and diversity of archaeal and bacterial communities in both uncontaminated and highly copper-contaminated marine sediments. Mar. Biotechnol. 16, 230–242. doi: 10.1007/s10126-013-9542-z
Boddicker, A. M., and Mosier, A. C. (2018). Genomic profiling of four cultivated Candidatus Nitrotoga spp. predicts broad metabolic potential and environmental distribution. ISME J. 12, 2864–2882. doi: 10.1038/s41396-018-0240-8
Brochier-Armanet, C., Boussau, B., Gribaldo, S., and Forterre, P. (2008). Mesophilic Crenarchaeota: proposal for a third archaeal phylum, the Thaumarchaeota. Nat. Rev. Microbiol. 6, 245–252. doi: 10.1038/nrmicro1852
Broeckling, C. D., Broz, A. K., Bergelson, J., Manter, D. K., and Vivanco, J. M. (2008). Root exudates regulate soil fungal community composition and diversity. Appl. Environ. Microbiol. 74, 738–744. doi: 10.1128/aem.02188-07
Cabello, P., Roldan, M. D., and Moreno-Vivian, C. (2004). Nitrate reduction and the nitrogen cycle in archaea. Microbiology 150, 3527–3546. doi: 10.1099/mic.0.27303-0
Cao, Q., Liu, X. F., Ran, Y., Li, Z. D., and Li, D. (2019). Methane oxidation coupled to denitrification under microaerobic and hypoxic conditions in leach bed bioreactors. Sci Total Environ. 649, 1–11. doi: 10.1016/j.scitotenv.2018.08.289
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336.
Chen, J., Sun, X., Li, L., Liu, X., Zhang, B., Zheng, J., et al. (2016). Change in active microbial community structure, abundance and carbon cycling in an acid rice paddy soil with the addition of biochar. Eur. J. Soil Sci. 67, 857–867. doi: 10.1111/ejss.12388
Chen, L., Zhang, J. B., Zhao, B. Z., Zhou, G. X., and Ruan, L. (2015). Bacterial community structure in maize stubble-amended soils with different moisture levels estimated by bar-coded pyrosequencing. Appl. Soil Ecol. 86, 62–70. doi: 10.1016/j.apsoil.2014.09.011
Dong, W. Y., Zhang, X. Y., Dai, X. Q., Fu, X. L., Yang, F. T., Liu, X. Y., et al. (2014). Changes in soil microbial community composition in response to fertilization of paddy soils in subtropical China. Appl. Soil Ecol. 84, 140–147. doi: 10.1016/j.apsoil.2014.06.007
Doss, C. R. (2010). Analyzing technology adoption using microstudies: limitations, challenges, and opportunities for improvement. Agric. Econ. 34, 207–219. doi: 10.1111/j.1574-0864.2006.00119.x
Edwards, J., Johnson, C., Santos-Medellin, C., Lurie, E., Podishetty, N. K., Bhatnagar, S., et al. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. U.S.A. 112, 911–920. doi: 10.1073/pnas.1414592112
Edwards, K. J. (2000). An archaeal iron-oxidizing extreme acidophile important in acid mine drainage. Science 287, 1796–1799. doi: 10.1126/science.287.5459.1796
Emerson, D., Fleming, E. J., and McBeth, J. M. (2010). Iron-oxidizing bacteria: an environmental and genomic perspective. Annu. Rev. Microbiol. 64, 561–583. doi: 10.1146/annurev.micro.112408.134208
Fierer, N., Lauber, C. L., Ramirez, K. S., Zaneveld, J., Bradford, M. A., and Knight, R. (2012). Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 6, 1007–1017. doi: 10.1038/ismej.2011.159
Fitzgerald, C. M., Camejo, P., Oshlag, J. Z., and Noguera, D.R. (2015). Ammonia-oxidizing microbial communities in reactors with efficient nitrification at low-dissolved oxygen. Water Res. 70, 38–51. doi: 10.1016/j.watres.2014.11.041
Francioli, D., Schulz, E., Lentendu, G., Wubet, T., Buscot, F., and Reitz, T. (2016). Mineral vs. organic amendments: microbial community structure, activity and abundance of agriculturally relevant microbes are driven by long-term fertilization strategies. Front. Microbiol. 7:1446. doi: 10.3389/fmicb.2016.01446
Franke-Whittle, I. H., Manici, L. M., Insam, H., and Stres, B. (2015). Rhizosphere bacteria and fungi associated with plant growth in soils of three replanted apple orchards. Plant Soil 395, 317–333. doi: 10.1007/s11104-015-2562-x
Fuhrman, J. A. (2009). Microbial community structure and its functional implications. Nature 459, 193–199. doi: 10.1038/nature08058
Furuno, T. (2001). The Power of Duck: Integrated Rice and Duck Farming. Sisters Creek, TAS: Tagari Publications.
Geisseler, D., and Scow, K. M. (2014). Long-term effects of mineral fertilizers on soil microorganisms – A review. Soil Biol. Biochem. 75, 54–63. doi: 10.1016/j.soilbio.2014.03.023
Hallam, S. J., Mincer, T. J., Schleper, C., Preston, C. M., Roberts, K., Richardson, P. M., et al. (2006). Pathways of carbon assimilation and ammonia oxidation suggested by environmental genomic analyses of marine Crenarchaeota. PLoS Biol. 4:e95. doi: 10.1371/journal.pbio.0040095
Hibbett, D. S. (2007). A phylogenetic overview of the Agaricomycotina. Mycologia 98, 917–925. doi: 10.1080/15572536.2006.11832621
Hu, H. W., Zhang, L. M., Yuan, C. L., and He, J. Z. (2013). Contrasting Euryarchaeota communities between upland and paddy soils exhibited similar pH-impacted biogeographic patterns. Soil Biol. Biochem. 64, 18–27. doi: 10.1016/j.soilbio.2013.04.003
Hu, L. L., Zhang, J., Ren, W. Z., Guo, L., Cheng, Y. X., Li, J. Y., et al. (2016). Can the co-cultivation of rice and fish help sustain rice production? Sci. Rep. 6:28728. doi: 10.1038/srep28728
Hug, L. A., Castelle, C. J., Wrighton, K. C., Thomas, B. C., Sharon, I., Frischkorn, K. R., et al. (2013). Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome 1:22. doi: 10.1186/2049-2618-1-22
In’t Zandt, M. H., Beckmann, S., Rijkers, R., Jetten, M. S. M., Manefield, M., and Welte, C. U. (2018). Nutrient and acetate amendment leads to acetoclastic methane production and microbial community change in a non-producing Australian coal well. Microb. Biotechnol. 11, 626–638. doi: 10.1111/1751-7915.12853
Ji, Y., Liu, P. F., and Conrad, R. (2018). Response of fermenting bacterial and methanogenic archaeal communities in paddy soil to progressing rice straw degradation. Soil Biol. Biochem. 124, 70–80. doi: 10.1016/j.soilbio.2018.05.029
Ke, X. B., Lu, Y. H., and Conrad, R. (2014). Different behaviour of methanogenic archaea and Thaumarchaeota in rice field microcosms. FEMS Microbiol. Ecol. 87, 18–29. doi: 10.1111/1574-6941.12188
Kim, J. G., Park, S. J., Sinninghe Damsté, J. S., Schouten, S., Rijpstra, W. I., Jung, M. Y., et al. (2016). Hydrogen peroxide detoxification is a key mechanism for growth of ammonia-oxidizing archaea. Proc. Natl. Acad. Sci. U.S.A. 113, 7888–7893. doi: 10.1073/pnas.1605501113
Kitzinger, K., Koch, H., Lücker, S., Sedlacek, C., Herbold, C., Schwarz, J., et al. (2018). Characterization of the first “Candidatus Nitrotoga” isolate reveals metabolic versatility and separate evolution of widespread nitrite-oxidizing bacteria. MBio 9:e1186-18. doi: 10.1128/mBio.01186-18
Laborte, A. G., Schipper, R. A., Van Ittersum, M. K., Van Den Berg, M. M., Van Keulen, H., Prins, A. G., et al. (2009). Farmers’ welfare, food production and the environment: a model-based assessment of the effects of new technologies in the Northern Philippines. NJAS - Wageningen J. Life Sci. 56, 345–373. doi: 10.1016/s1573-5214(09)80004-3
Lam, P., Jensen, M. M., Lavik, G., Mcginnis, D. F., Müller, B., Schubert, C. J., et al. (2007). Linking crenarchaeal and bacterial nitrification to anammox in the Black Sea. Proc. Natl. Acad. Sci. U. S. A. 104, 7104–7109. doi: 10.1073/pnas.0611081104
Lee, H. J., Jeong, S. E., Kim, P. J., Madsen, E. L., and Jeon, C. O. (2015). High resolution depth distribution of Bacteria, Archaea, methanotrophs, and methanogens in the bulk and rhizosphere soils of a flooded rice paddy. Front. Microbiol. 6:639. doi: 10.3389/fmicb.2015.00639
Lehtovirta-Morley, L. E., Sayavedra-Soto, L. A., Gallois, N., Schouten, S., Stein, L. Y., Prosser, J. I., et al. (2016). Identifying potential mechanisms enabling acidophily in the ammonia-oxidizing archaeon "Candidatus nitrosotalea devanaterra". Appl. Environ. Microbiol. 82, 2608–2619. doi: 10.1128/AEM.04031-15
Li, C. F., Cao, C. G., Wang, J. P., Zhan, M., Yuan, W. L., and Ahmad, S. (2008). Nitrogen losses from integrated rice–duck and rice–fish ecosystems in southern China. Plant Soil 307, 207–217. doi: 10.1007/s11104-008-9597-1
Li, F., Chen, L., Zhang, J. B., Yin, J., and Huang, S. M. (2017). Bacterial community structure after long-term organic and inorganic fertilization reveals important associations between soil nutrients and specific taxa involved in nutrient transformations. Front. Microbiol. 8:187. doi: 10.3389/fmicb.2017.00187
Liu, H. Y., Li, J., Zhao, Y., Xie, K. X., Tang, X. J., Wang, S. X., et al. (2018). Ammonia oxidizers and nitrite-oxidizing bacteria respond differently to long-term manure application in four paddy soils of south of China. Sci. Total Environ. 633, 641–648. doi: 10.1016/j.scitotenv.2018.03.108
Liu, J. J., Sui, Y. Y., Yu, Z. H., Shi, Y., Chu, H. Y., Jin, J., et al. (2014). High throughput sequencing analysis of biogeographical distribution of bacterial communities in the black soils of northeast China. Soil Biol. Biochem. 70, 113–122. doi: 10.1016/j.soilbio.2013.12.014
Liu, J. S., Zhang, X., Wang, H., Hui, X. L., Wang, Z. H., and Qiu, W. H. (2017). Long-term nitrogen fertilization impacts soil fungal and bacterial community structures in a dryland soil of Loess Plateau in China. J. Soils Sedim. 18, 1632–1640. doi: 10.1007/s11368-017-1862-6
Liu, M., Liu, J., Chen, X. F., Jiang, C. Y., Wu, M., and Li, Z. P. (2017). Shifts in bacterial and fungal diversity in a paddy soil faced with phosphorus surplus. Biol. Fertil. Soils 54, 259–267. doi: 10.1007/s00374-017-1258-1
Liu, Y. L., Wang, P., Pan, G. X., Crowley, D., Li, L. Q., Zheng, J. W., et al. (2016). Functional and structural responses of bacterial and fungal communities from paddy fields following long-term rice cultivation. J. Soils Sedim. 16, 1460–1471. doi: 10.1007/s11368-015-1343-8
Lu, J. B., and Li, X. (2006). Review of rice–fish-farming systems in China — One of the Globally Important Ingenious Agricultural Heritage Systems (GIAHS). Aquaculture 260, 106–113. doi: 10.1016/j.aquaculture.2006.05.059
Luo, S. M. (2016). Agroecology Development in China: Science, Practice, and Sustainable Management. Boca Raton, FL: CRC Press.
Luo, S. M. (2018). Agroecological Rice Production in China: Restoring Biological Interactions. Rome: FAO.
Lynch, M. D., and Thorn, R. G. (2006). Diversity of basidiomycetes in michigan agricultural soils. Appl. Environ. Microbiol. 72, 7050–7056. doi: 10.1128/aem.00826-06
Ma, B., Zhao, K. K., Lv, X. F., Su, W. Q., Dai, Z. M., Gilbert, J. A., et al. (2018). Genetic correlation network prediction of forest soil microbial functional organization. ISME J. 12, 2492–2505. doi: 10.1038/s41396-018-0232-8
Magoc, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Meador, T. B., Bowles, M., Lazar, C. S., Zhu, C., Teske, A., and Hinrichs, K. U. (2015). The archaeal lipidome in estuarine sediment dominated by members of the Miscellaneous Crenarchaeotal Group. Environ. Microbiol. 17, 2441–2458. doi: 10.1111/1462-2920.12716
Meng, D. L., Li, J., Liu, T. B., Liu, Y. J., Yan, M. L., Hu, J., et al. (2019). Effects of redox potential on soil cadmium solubility: insight into microbial community. J. Environ. Sci. 75, 224–232. doi: 10.1016/j.jes.2018.03.032
Pascault, N., Ranjard, L., Kaisermann, A., Bachar, D., Christen, R., Terrat, S., et al. (2013). Stimulation of different functional groups of bacteria by various plant residues as a driver of soil priming effect. Ecosystems 16, 810–822. doi: 10.1007/s10021-013-9650-7
Probandt, D., Knittel, K., Tegetmeyer, H. E., Ahmerkamp, S., Holtappels, M., and Amann, R. (2017). Permeability shapes bacterial communities in sublittoral surface sediments. Environ. Microbiol. 19, 1584–1599. doi: 10.1111/1462-2920.13676
Raymond, J., Siefert, J. L., Staples, C. R., and Blankenship, R. E. (2004). The natural history of nitrogen fixation. Mol. Biol. Evol. 21, 541–554.
Ren, W. Z., Hu, L. L., Guo, L., Zhang, J., Tang, L., Zhang, E. T., et al. (2018). Preservation of the genetic diversity of a local common carp in the agricultural heritage rice-fish system. Proc. Natl. Acad. Sci. U.S.A. 115, 546–554. doi: 10.1073/pnas.1709582115
Rotaru, A. E., Shrestha, P. M., Liu, F., Shrestha, M., Shrestha, D., Embree, M., et al. (2014). A new model for electron flow during anaerobic digestion: direct interspecies electron transfer to Methanosaeta for the reduction of carbon dioxide to methane. Energy Environ. Sci. 7, 408–415. doi: 10.1039/c3ee42189a
Saiful Islam, A. H. M., Barman, B. K., and Murshed-e-Jahan, K. (2015). Adoption and impact of integrated rice–fish farming system in Bangladesh. Aquaculture 447, 76–85. doi: 10.1016/j.aquaculture.2015.01.006
Saleem, M., Pervaiz, Z. H., and Traw, M. B. (2015). Theories, mechanisms and patterns of microbiome species coexistence in an era of climate change. Microb. Commun. Ecol. 13–15.
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Sha, Z. M., Chu, Q. N., Zhao, Z., Yue, Y. B., Lu, L., Yuan, J., et al. (2017). Variations in nutrient and trace element composition of rice in an organic rice-frog co-culture system. Sci. Rep. 7:15706. doi: 10.1038/s41598-017-15658-1
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Su, J. Q., Ding, L. J., Xue, K., Yao, H. Y., Quensen, J., Bai, S. J., et al. (2015). Long-term balanced fertilization increases the soil microbial functional diversity in a phosphorus-limited paddy soil. Mol. Ecol. 24, 136–150. doi: 10.1111/mec.13010
Suh, J. (2014). Theory and reality of integrated rice–duck farming in Asian developing countries: a systematic review and SWOT analysis. Agric. Syst. 125, 74–81.
Tanikawa, D., Nakamura, Y., Tokuzawa, H., Hirakata, Y., Hatamoto, M., and Yamaguchi, T. (2018). Effluent treatment in an aquaponics-based closed aquaculture system with single-stage nitrification–denitrification using a down-flow hanging sponge reactor. Int. Biodeteriorat. Biodegrad. 132, 268–273. doi: 10.1016/j.ibiod.2018.04.016
Teng, Q., Hu, X. F., Cheng, C., Luo, Z., Luo, F., Xue, Y., et al. (2016). Ecological effects of rice-duck integrated farming on soil fertility and weed and pest control. J. Soils Sedim. 16, 2395–2407. doi: 10.1007/s11368-016-1455-9
Teng, Q., Hu, X. F., Luo, F., Cheng, C., Ge, X., Yang, M., et al. (2015). Influences of introducing frogs in the paddy fields on soil properties and rice growth. J. Soils Sedim. 16, 51–61. doi: 10.1007/s11368-015-1183-6
Thormann, M. N. (2006). Diversity and function of fungi in peatlands: a carbon cycling perspective. Can. J. Soil Sci. 86, 281–293. doi: 10.4141/s05-082
Tibbett, M. (2002). Ectomycorrhizal symbiosis can enhance plant nutrition through improved access to discrete organic nutrient patches of high resource quality. Ann. Bot. 89, 783–789. doi: 10.1093/aob/mcf129
Tripathi, B. M., Kim, M., Tateno, R., Kim, W., Wang, J., Lai-Hoe, A., et al. (2015). Soil pH and biome are both key determinants of soil archaeal community structure. Soil Biol. Biochem. 88, 1–8. doi: 10.1139/cjm-2015-0669
Vigdis, T., and Lise, V. S. (2002). Microbial diversity and function in soil: from genes to ecosystems. Curr. Opin. Microbiol. 5, 240–245. doi: 10.1016/s1369-5274(02)00324-7
Walker, C. B., De La Torre, J. R., Klotz, M. G., Urakawa, H., Pinel, N., Arp, D. J., et al. (2010). Nitrosopumilus maritimus genome reveals unique mechanisms for nitrification and autotrophy in globally distributed marine crenarchaea. Proc. Natl. Acad. Sci. U.S.A. 107, 8818–8823. doi: 10.1073/pnas.0913533107
Walter, M., Richard, S., Katja, N., Tina, T., Volker, T., Martin, B., et al. (2002). Microbial reefs in the Black Sea fueled by anaerobic oxidation of methane. Science 297, 1013–1015. doi: 10.1126/science.1072502
Wang, J., Geoff, R., Qiwei, H., and Qirong, S. (2018). Plant growth stages and fertilization regimes drive soil fungal community compositions in a wheat-rice rotation system. Biol. Fertil. Soils 54, 731–742. doi: 10.1007/s00374-018-1295-4
Wang, J., Song, Y., Ma, T., Raza, W., Li, J., Howland, J. G., et al. (2017). Impacts of inorganic and organic fertilization treatments on bacterial and fungal communities in a paddy soil. Appl. Soil Ecol. 112, 42–50. doi: 10.1016/j.apsoil.2017.01.005
Wang, M. M., Ding, J. J., Sun, B., Zhang, J. Y., Wyckoff, K. N., Yue, H. W., et al. (2018). Microbial responses to inorganic nutrient amendment overridden by warming: consequences on soil carbon stability. Environ. Microbiol. 20, 2509–2522. doi: 10.1111/1462-2920.14239
Xiao, D., Huang, Y., Feng, S., Ge, Y. H., Zhang, W., He, X. Y., et al. (2018). Soil organic carbon mineralization with fresh organic substrate and inorganic carbon additions in a red soil is controlled by fungal diversity along a pH gradient. Geoderma 321, 79–89. doi: 10.1016/j.geoderma.2018.02.003
Yang, H. S., Yu, D. G., Zhou, J. J., Zhai, S. L., Bian, X. M., and Weih, M. (2018). Rice-duck co-culture for reducing negative impacts of biogas slurry application in rice production systems. J. Environ. Manage. 213, 142–150. doi: 10.1016/j.jenvman.2018.02.077
Yi, X. M., Yuan, J., Zhu, Y. H., Yi, X. J., Zhao, Q., Fang, K. K., et al. (2018). Comparison of the abundance and community structure of N-Cycling bacteria in paddy rhizosphere soil under different rice cultivation patterns. Int. J. Mol. Sci. 19:3772. doi: 10.3390/ijms19123772
Yuan, C. L., Zhang, L. M., Hu, H. W., Wang, J. T., Shen, J. P., and He, J. Z. (2018). The biogeography of fungal communities in paddy soils is mainly driven by geographic distance. J. Soils Sedim. 18, 1795–1805. doi: 10.1007/s11368-018-1924-4
Yuan, J., Sha, Z. M., Hassani, D., Zhao, Z., and Cao, L. K. (2017). Assessing environmental impacts of organic and inorganic fertilizer on daily and seasonal greenhouse gases effluxes in rice field. Atmos. Environ. 155, 119–128. doi: 10.1016/j.atmosenv.2017.02.007
Yuan, J., Yuan, Y. K., Zhu, Y. H., and Cao, L. K. (2018). Effects of different fertilizers on methane emissions and methanogenic community structures in paddy rhizosphere soil. Sci. Total Environ. 627, 770–781. doi: 10.1016/j.scitotenv.2018.01.233
Zhang, J., Hu, L. L., Ren, W. Z., Guo, L., Tang, J. J., Shu, M. A., et al. (2016). Rice-soft shell turtle coculture effects on yield and its environment. Agric. Ecosyst. Environ. 224, 116–122. doi: 10.1016/j.agee.2016.03.045
Keywords: bacteria, archaea, fungi, rice-frog cultivation, paddy rhizosphere soil, network analysis
Citation: Yi X, Yi K, Fang K, Gao H, Dai W and Cao L (2019) Microbial Community Structures and Important Associations Between Soil Nutrients and the Responses of Specific Taxa to Rice-Frog Cultivation. Front. Microbiol. 10:1752. doi: 10.3389/fmicb.2019.01752
Received: 05 March 2019; Accepted: 15 July 2019;
Published: 06 August 2019.
Edited by:
Rima Franklin, Virginia Commonwealth University, United StatesReviewed by:
Muhammad Saleem, Alabama State University, United StatesLidong Shen, Nanjing University of Information Science and Technology, China
Copyright © 2019 Yi, Yi, Fang, Gao, Dai and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linkui Cao, clk@sjtu.edu.cn