 Jin-Yan Wu
Jin-Yan Wu Duan-Yong Zhou
Duan-Yong Zhou Ying Zhang
Ying Zhang Fei Mi
Fei Mi Jianping Xu
Jianping Xu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 26 April 2019
Sec. Fungi and Their Interactions
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.00900
Candida tropicalis is a globally distributed human pathogenic yeast, especially prevalent in tropical and sub-tropical regions. Over the last several decades, a large number of studies have been published on the genetic diversity and molecular epidemiology of C. tropicalis from different parts of the world. However, the global pattern of genetic variation remains largely unknown. Here we analyzed the published multilocus sequence data at six loci for 876 isolates from 16 countries representing five continents. Our results showed that 280 of the 2677 (10.5%) analyzed nucleotides were polymorphic, resulting in a mean of 82 (a range of 38–150) genotypes per locus and a total of 633 combined diploid sequence types (DSTs). Among these, 93 combined DSTs were shared by 336 strains, including 10 by strains from different continents. Analysis of Molecular Variance (AMOVA) showed that 89% of the observed genetic variations were found within regional and national populations while < 10% was due to among-country separations. Pairwise geographic population analyses showed overall low but statistically significant genetic differentiation between most geographic populations, with the Singaporean and Indian populations being the most distinct from other populations. However, the Mantel test showed no significant correlation between genetic distance and geographic distance among the geographic populations. Consistent with high genetic variation within and limited variations among geographic populations, results from STRUCTURE analyses showed that the 876 isolates could be grouped into 15 genetic clusters, with each cluster having a broad geographic distribution. Together, our results suggest frequent gene flows among certain regional, national, and continental populations of C. tropicalis, resulting in abundant regional and national genetic diversities of this important human fungal pathogen.
Candida tropicalis is an ascomycete yeast and an important opportunistic human pathogen (Falagas et al., 2010; Zuza-Alves et al., 2017). It’s broadly distributed in a diversity of ecological niches such as soil, aquatic environments, plant materials, and animals, including humans (Vogel et al., 2007; Lord et al., 2010; Lachance et al., 2011; Brilhante et al., 2013, 2015; Cordeiro et al., 2013). In human hosts, C. tropicalis is commonly found in the oral cavity and on certain parts of the skin and mucosal surfaces as a commensal organism. With an increasing number of immunocompromised patients due to cancer treatment, HIV infection, and/or organ transplantation, C. tropicalis has emerged as a major opportunistic pathogen that can cause serious invasive infections such as endocarditis and the bloodstream and urinary tract infections, resulting in significant morbidity and mortality (Falagas et al., 2010; Fesharaki et al., 2013; Dong et al., 2015; Zuza-Alves et al., 2017; Diba et al., 2018). In certain geographic regions, C. tropicalis is ranked as the first or second most prevalent invasive pathogenic yeast species (Adhikary and Joshi, 2011; Dóczi et al., 2012; Wang et al., 2013; Morii et al., 2014; Pahwa et al., 2014; Arrua et al., 2015; Al-Obaid et al., 2017). However, despite its global medical significance, relatively little is known about its global patterns of genetic variation.
Over the past three decades, several molecular methods have been used to identify genotypes and examine the relationships among strains and populations of human pathogenic yeasts (Xu, 2005; Lachance et al., 2011; Zuza-Alves et al., 2017). These included multilocus isozyme electrophoresis (e.g., Brandt et al., 1993), electrophoretic karyotyping (e.g., Fries et al., 1996), PCR fingerprinting (e.g., Xu et al., 2000a), amplified fragment length polymorphisms (e.g., Duarte-Escalante et al., 2013), simple sequence repeats (e.g., Field et al., 1996), and multilocus sequence typing (MLST) (e.g., Xu et al., 2000b; Tavanti et al., 2005; Afsarian et al., 2015). For C. tropicalis, the emerging consensus since 2005 for strain typing is MLST, based on the single nucleotide polymorphisms (SNPs) at six gene fragments (Tavanti et al., 2005). These marker loci were ICL1, MDR1, SAPT2, SAPT4, XYR1, and ZWF1a and they were chosen for MLST of C. tropicalis for several reasons, including: (i) the high success rates of PCR amplification and sequencing using the same primers; (ii) their high-levels of single nucleotide polymorphisms (SNPs) within and among strains; and (iii) when sequences at the six loci are combined, their high genotypic discriminating power among strains (Tavanti et al., 2005). Indeed, these MLST markers have not only been used to analyze the relationships among strains from different hosts and wards within hospitals, but also to monitor strain maintenance, replacement, and microevolution within individual human hosts (Chen et al., 2009; Wu et al., 2012; Magri et al., 2013). Furthermore, the establishment of the MLST database for C. tropicalis has facilitated the comparisons of strains and populations from different laboratories and studies (e.g., Wu et al., 2017; Scordino et al., 2018). As of the end of May 2018, the MLST database for C. tropicalis includes DNA sequence information at these six loci for 876 isolates. However, the global genetic structure of C. tropicalis populations remains poorly known.
The objective of this study was to investigate the global patterns of genetic variation by analyzing the published MLST sequence data of C. tropicalis from around the world. We were specifically interested in the geographic patterns of DNA sequence variation and in where some of our knowledge gaps might be. Because C. tropicalis is commonly found in the environment (Odds, 1988; Vogel et al., 2007; Lachance et al., 2011; Carvalho et al., 2014) and that most hosts likely acquire their commensal strains from their immediate environment, we hypothesized that populations of C. tropicalis from different countries and continents should be genetically different from each other and that geographic distance should contribute a significantly to the overall genetic differentiation among geographic populations.
All DNA sequence data as well as the meta-data associated with each strain were retrieved from the Candida tropicalis multilocus sequence typing database1. The species identification, DNA extraction, PCR amplification, and DNA sequencing for all strains were described in the original publications reporting the strains in the database. The database was curated by Dr. Frank Odds and his colleagues and the retrieval and use of the data were approved and supported by Dr. Frank Odds. For each strain, the deposited DNA sequence at each of the six loci was separately downloaded. All the retrieved sequences for each locus were aligned and imported into GenAlEx 6.5 (Peakall and Smouse, 2012) for analyses. Since C. tropicalis is a diploid and heterozygous nucleotide sites have been frequently found, our analyses of the patterns of DNA sequence variation follow those for diploids. The number of polymorphic nucleotide sites and the number of genotypes at each locus were then obtained through this program, following those described in an earlier study (Wu et al., 2017).
To determine strain relationships, we combined the DNA sequences at the six loci and analyzed the concatenated sequences through cluster analysis using UPGMA (unweighted pair group method using their arithmetic averages) of the MEGA Version 6 software (Tamura et al., 2013). To analyze multilocus genotype distributions and patterns of genetic variations, the concatenated DNA sequences from all six loci for all strains were imported into GenAlEx6.5. The analysis of molecular variance (AMOVA) was conducted at three geographic levels depending on the samples. Specifically, due to the availability and relatively large sample sizes from multiple regions within China, our first-level analysis estimated the within- and among-regional populations to the overall genetic variation in the Chinese C. tropicalis sample. The second and third-level analyses were conducted together and estimated the relative contributions of within countries, between countries within continents, and among continents to the overall genetic variation. All AMOVA analyses were conducted using GenAlEx6.5. To further investigate the relationships between specific geographic populations, we calculated pairwise PhiPT values between pairs of regional populations within China and pairs of national populations. In these analyses, only samples with a sample size > 10 were included. Statistical significance of the observed contributions at various geographic levels to the overall patterns of genetic variation was obtained by 1000 randomizations. In addition, the potential relationship between geographic distance and genetic differentiation was estimated using the Mantel test. The detailed instructions for AMOVA, pairwise PhiPT, and Mantel tests can be found in the GenAlEx program manual (Peakall and Smouse, 2012).
Aside from the above population genetic analyses, we also estimated the putative number of genetic clusters K in the global sample of C. tropicalis using the program STRUCTURE version 2.3.4 (Pritchard et al., 2000). STRUCTURE implements a clustering algorithm based on a Bayesian Monte Carlo Markov Chain (MCMC) approach to assign individuals into K distinct populations. Using the admixture model, 10 replicated runs of K = 1–20 were carried out after a burn-in period of 100,000 generations followed by a run length of 1,000,000 generations. The number of genetically homogeneous clusters (K) was identified by following the method developed by Evanno et al. (2005). STRUCTURE 2.3.4 was also used to identify the assignment of individual isolates to specific clusters (K).
Up to the end of May 2018, a total 876 isolates were deposited in the C. tropicalis MLST database. The metadata and DNA sequences at the six MLST loci of all 876 strains were retrieved from the database. Below we summarize the observed data and the results of our analyses.
The geographic distributions of these strains in the C. tropicalis MLST database are shown in Table 1. These strains came from broad geographic areas, representing 16 countries on five continents. At the continental level, the vast majority of the 876 strains were from Asia (∼74%), followed by Europe (∼16%), South America (∼7%), North America (∼2%), and Australia (0.8%). There was no strain from Africa in the database and over 90% of the countries in the world were not represented. At the country level, 67.58% of the global strains in the MLST database were from China; followed by the United Kingdom (∼14%); Brazil (∼5%); India (2.9%); South Korea (2.5%); the United States (1.9%); Singapore (1.5%); Belgium (1.1%); Colombia (1%); Australia (0.8%); Argentina (0.46%); Germany and Netherlands (0.34% each); and Sweden, Greece, and Spain (one strain from each of the three countries; 0.11%). Within an individual country, China not only contributed the largest number of strains but also that the strains were from broad geographic regions within the country, from tropical Hainan province to temperate Heilongjiang province.

Table 1. Distribution of the geographic populations of Candida tropicalis analyzed in this study.
Among the 876 strains, 760 had information about their ecological niche or anatomic body site while the remaining 106 had no ecological niche or anatomic body site information (Supplementary Table 1). Among the 760 strains, three were from animals (from Belgium and Spain) and 757 were from humans. The human isolates came from several types of human tissues and organs, including the blood (287 strains total; from Argentina, Australia, Brazil, China, Colombia, India, Korea, Singapore, Netherlands, United Kingdom, and United States); feces (39 strains total; from China and United Kingdom); oropharynx (275 strains total; from Belgium, Brazil, China, Germany, India, Korea, United Kingdom, and United States); other sterile body sites (14 strains total, from Brazil, China, Colombia, Singapore, United Kingdom, and United States); skin and other superficial surfaces (40 strains total, from Belgium, China, Korea, and United Kingdom); urine (97 strains total, from China, Colombia, and United Kingdom); and vagina (58 strains total, from Belgium, Brazil, China, Sweden, United Kingdom, and United States).
We retrieved the DNA sequences at the six MLST loci (ICL1, MDR1, SAPT2, SAPT4, XYR1, and ZWFa1) for all 876 isolates. Information about these loci and the levels of DNA sequence polymorphisms within this global sample are summarized in Table 2. These six sequenced DNA fragments ranged in sizes from 370 to 525 bp and they are located on different scaffolds (Tavanti et al., 2005; Butler et al., 2009). Together, the six gene fragments cover a total of 2677 bp, representing < 0.02% of the entire genome. The haploid genome size of C. tropicalis is estimated at about 14.63 Mb, containing 6,441 genes (Butler et al., 2009). All six sequenced gene fragments were highly polymorphic in this global sample. Specifically, the percentages of nucleotides that are polymorphic in the global sample range from 7.11 to 16.41%, with an overall 10.46% (280/2677). At these six loci, there was no clear correlation between the length of sequenced fragment and the number of polymorphic nucleotide sites (Pearson correlation coefficient R = 0.0093; p = 0.986). Indeed, the fragment (i.e., SAPT4) with the highest number of SNPs (at 64) was the second shortest sequenced fragment, at 390 bp. At present, the reason for the different levels of polymorphisms among the sequenced gene fragments is not known but may be related to the different environmental and/or functional constraints that these genes have experienced during the evolution of C. tropicalis.
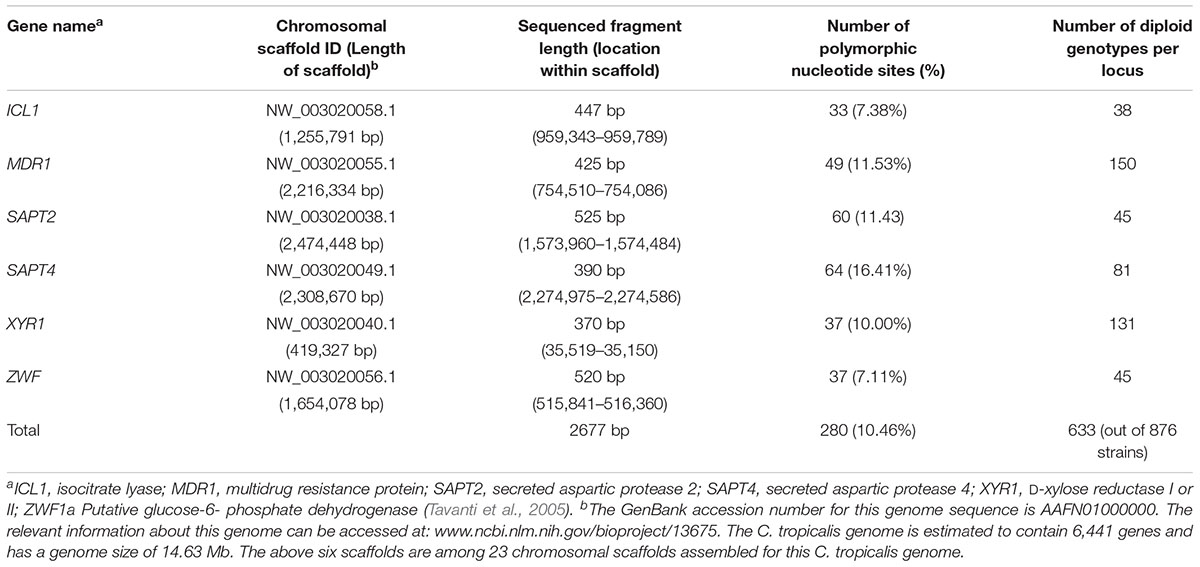
Table 2. Single nucleotide polymorphisms at the six loci in the global sample of C. tropicalis.
The 280 SNPs resulted in the identification of a large number of genotypes at both the individual locus level as well as the combined six loci (Table 2). At the individual locus level, the number of genotypes ranged from 38 (locus ICL1) to 150 (locus MDR1), with a mean of 81.67. Similar to that in SNP density, the number of genotypes per locus was not significantly correlated with either fragment length or SNP density. For example, the Pearson correlation coefficient between the number of SNPs and number of genotypes at the six loci was 0.042, with a P-value of 0.937. In contrast to our expectation, there was a negative correlation between the sequenced fragment length and the number of genotypes among the six loci (Pearson correlation coefficient = -0.688). However, this seemingly negative correlation was statistically insignificant (P = 0.132). Regardless, these results are consistent with the distinctiveness of these six loci in the C. tropicalis genome (Table 2) and that it’s often difficult to predict the level of genotypic polymorphism based on either the fragment length and/or SNP density.
The relationships among all 876 strains are shown in Supplementary Figure 1. Together, the combined analyses of all six gene fragments identified a total of 633 multilocus genotypes out of the 876 strains (Supplementary Table 1). Among these 633 combined genotypes, 93 were shared by a total of 336 strains (Table 3). The remaining 540 multilocus genotypes were each represented by only one strain. Of the 93 shared multilocus genotypes, the majority (56; 60%) were only found within one region of one country each. The remaining 37 multilocus genotypes showed different distributions: 23 were shared by strains from different regions within a country, four were shared among countries within a continent, and 10 were shared among continents. Of these 37 multilocus genotypes, 17 were also shared among strains within a region of a country (Table 3).
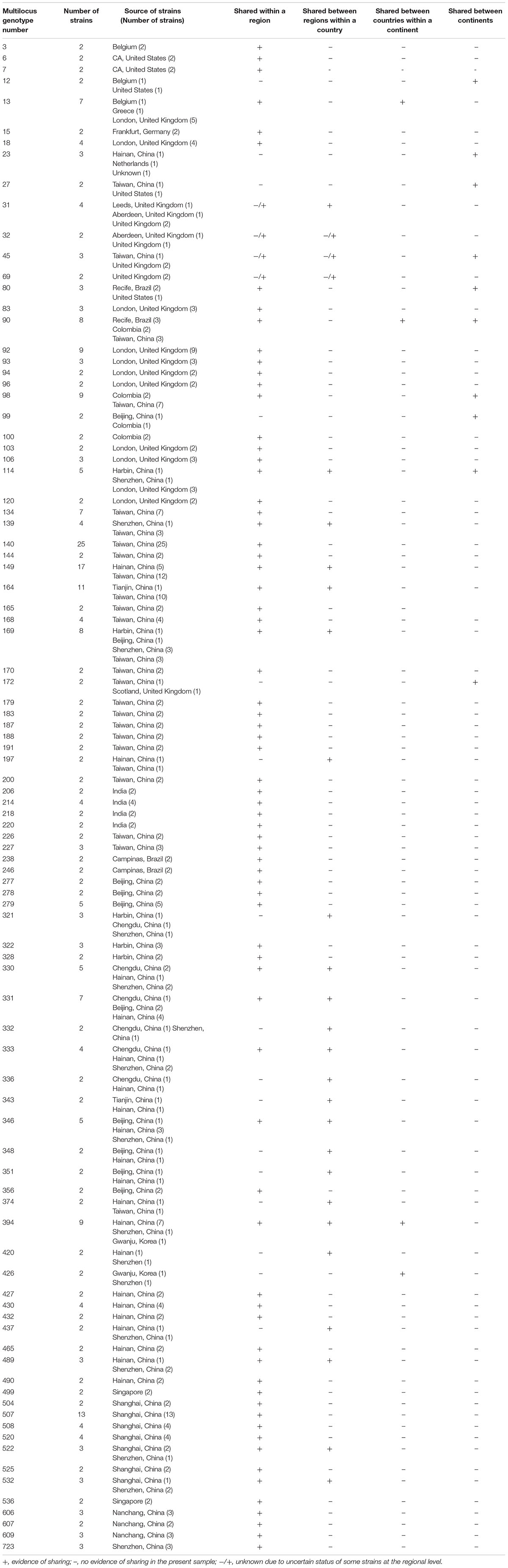
Table 3. Shared multilocus genotypes of Candida tropicalis in the MLST database.
Among the 93 shared multilocus genotypes, the most frequent (genotype #140) was shared by 25 strains, all of which were from Taiwan (Table 3). The second most frequent multilocus genotype (genotype #149) was shared by 17 strains with five from Hainan and 12 from Taiwan, two geographically isolated islands off the south and east coasts of Mainland China respectively. The third most frequently shared multilocus genotype (genotype# 507) had 13 strains, all of which were from Shanghai. The fourth and fifth most frequently shared were multilocus genotypes #164 and #169 that contained strains from different regions of Mainland China and Taiwan. These results are consistent with clonal dispersal of several genotypes of C. tropicalis within and across diverse regions in China.
The 56 multilocus genotypes exclusively shared among strains within a region were distributed in all continents, including most of the countries represented in the database: Belgium, Brazil, China, Colombia, Germany, India, Singapore, United Kingdom, and the United States (Table 3). Most multilocus genotypes that were shared among regions within a country involved strains from China. This result was likely due to the fact that China was the most intensively sampled country for C. tropicalis in terms of the diversity of geographic locations and the number of strains within a country. In contrast to the skewed distribution at the local and within a country-level, the multilocus genotypes shared among countries were geographically more broadly represented, with multilocus genotype #13 from Europe, #90 from South America, and #394 and #426 from Asia.
Ten multilocus genotypes were shared by strains from different continents. Genotype #12 was shared between Belgium and the United States; genotype #23 between China and the Netherlands; genotype #27 between China and the United States; genotypes #45, #114, #172 between China and the United Kingdom; genotype #80 between Brazil and the United States; genotype #90 among Brazil, Colombia, and China, and genotypes #98 and #99 between Colombia and China. Though countries from Asia, Europe, South America and North America were all represented here, there was a bias for strains from China, with eight of the 10 multilocus genotypes having representative strains from China. Again, the bias was most likely related to the differences in sample sizes in the C. tropicalis MLST database.
Together, the above results suggest that short-, medium-, and long- distance dispersals were all possible for C. tropicalis strains.
The sharing of multilocus genotypes between regional, national and continental populations suggests that gene flow might be common in C. tropicalis. To quantify the effects of geographic separation at regional, national and continental levels on the observed genetic variation, we conducted an analysis of molecular variance (AMOVA). At the global level, our analyses revealed that the majority (89%) of the SNP variations was found within individual countries while that among countries contributed the remaining 11% (Table 4). Interestingly, there is almost no contribution at the continental level beyond that at the national level. A similar trend was observed for the samples from China where the majority of the observed genetic variation (94%) was found within the regional populations and the remaining 6% was due to between-regional (i.e., provincial) populations. Though the contributions from among regional populations within China (6%) and among national populations at the global level (11%) were relatively low, such contributions were statistically significant (p-values < 0.001 in both cases). Together, these results are consistent with an overall low but statistically significant differentiation among regional populations within China and among national populations in the world.
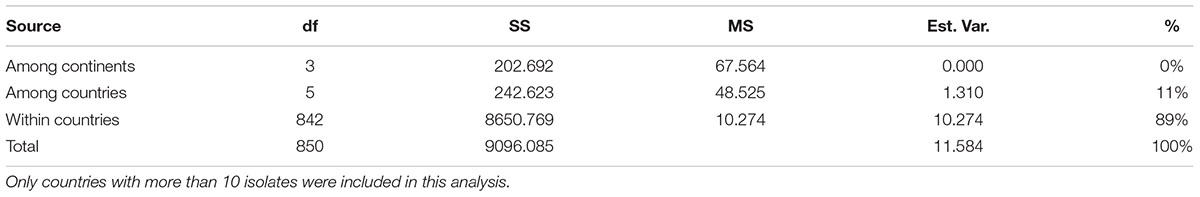
Table 4. Analyses of molecular variance (AMOVA) at different geographic levels.
We further investigated the extent of genetic differentiation between pairs of national populations. The summary results among national populations are shown in Table 5. Among these, the Singapore sample was genetically the most differentiated from others (all with PhiPT values greater than 20%), followed by those between India and the remaining countries (all with PhiPT values between 12 and 15%). Between other pairs of national samples, their PhiPT values were about 10% or less. Statistically speaking, the only pairs of samples that were not significantly differentiated were those between Belgium and the United Kingdom, between Belgium and the United States, and between Brazil and the United States. The remaining pairs of samples were all statistically significantly differentiated at p < 0.05 (Table 5).
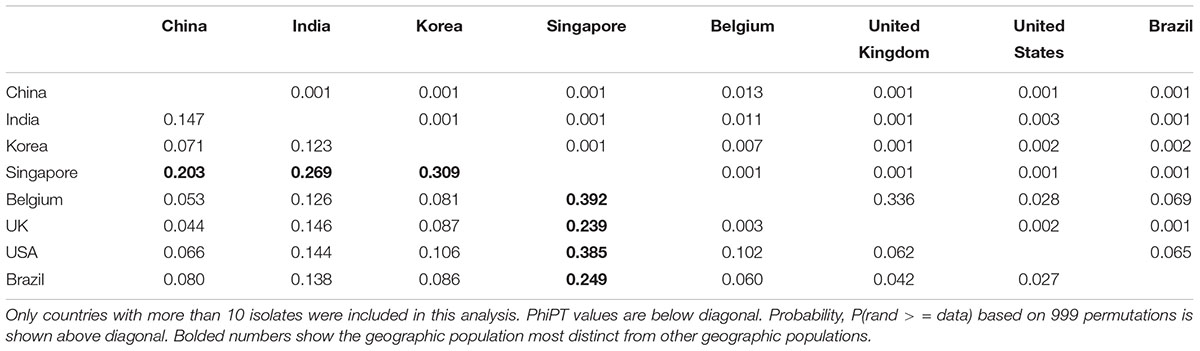
Table 5. Genetic differentiation between pairs of geographic populations of C. tropicalis from different countries.
The extent of genetic differentiation between pairs of regional populations were estimated for those in three countries (Brazil, United Kingdom, and China). Among the three regional samples in Brazil (Campinas, Recife, and São Paulo), that between Recife and São Paulo had a statistically significant pairwise PhiPT value of 0.132 (p = 0.031). The other two pairs had low and statistically insignificant PhiPT values: 0.006 between Campinas and Recife (p = 0.261), and 0.031 between Campinas and São Paulo (p = 0.132). Within the United Kingdom, the three geographic populations (Aberdeen, Leeds, and London) were genetically indistinguishable from each other with a PhiPT value of 0.000 for both between Aberdeen and Leeds (p = 0.398) and between Leeds and London (p = 0.352). The PhiPT value between Aberdeen and London was slightly higher than 0.000, at 0.017 and statistically also insignificant (p = 0.299).
The pairwise genetic differences among regional populations in China are presented in Table 6. Interestingly, different from those from the United Kingdom and to some extent from Brazil, most pairs of regional populations in China (22 of the 28 pairs) were statistically highly significantly differentiated. The only regional sample that showed no differentiation from the rest was that from Sichuan province. In contrast, pairs of populations from the remaining 7 regions were all significantly differentiated. The regional sample showing overall the biggest differences (PhiPT values ranging from 0.112 to 0.269) from other samples was that from Jiangxi province located in south central China, followed by that from Heilongjiang province in far northeastern China.
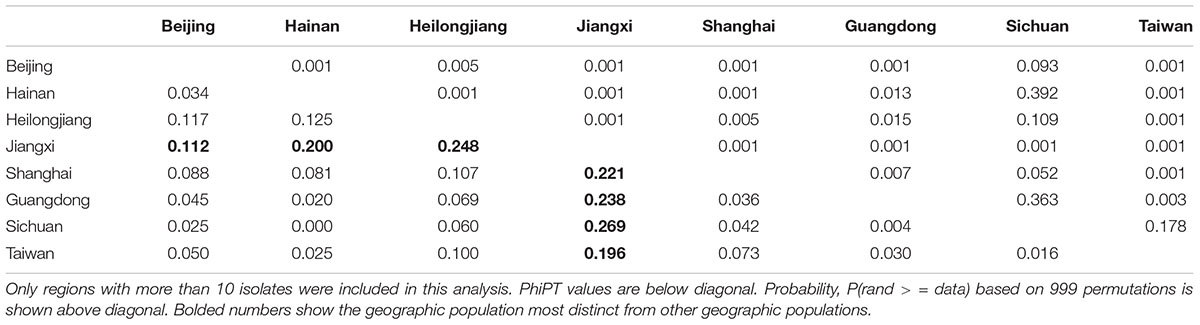
Table 6. Genetic differentiation between pairs of geographic samples of C. tropicalis from different regions in China.
These results suggest that even though there is gene flow among geographic populations (also evidenced by multilocus genotype sharing across geographic scales), the amount of gene flow between most regional and national populations is insufficient to obscure the genetic differences among most of these populations. However, despite the observed significant genetic differentiation between most geographic populations, there is no evidence of genetic isolation by geographic distance (Figure 1).
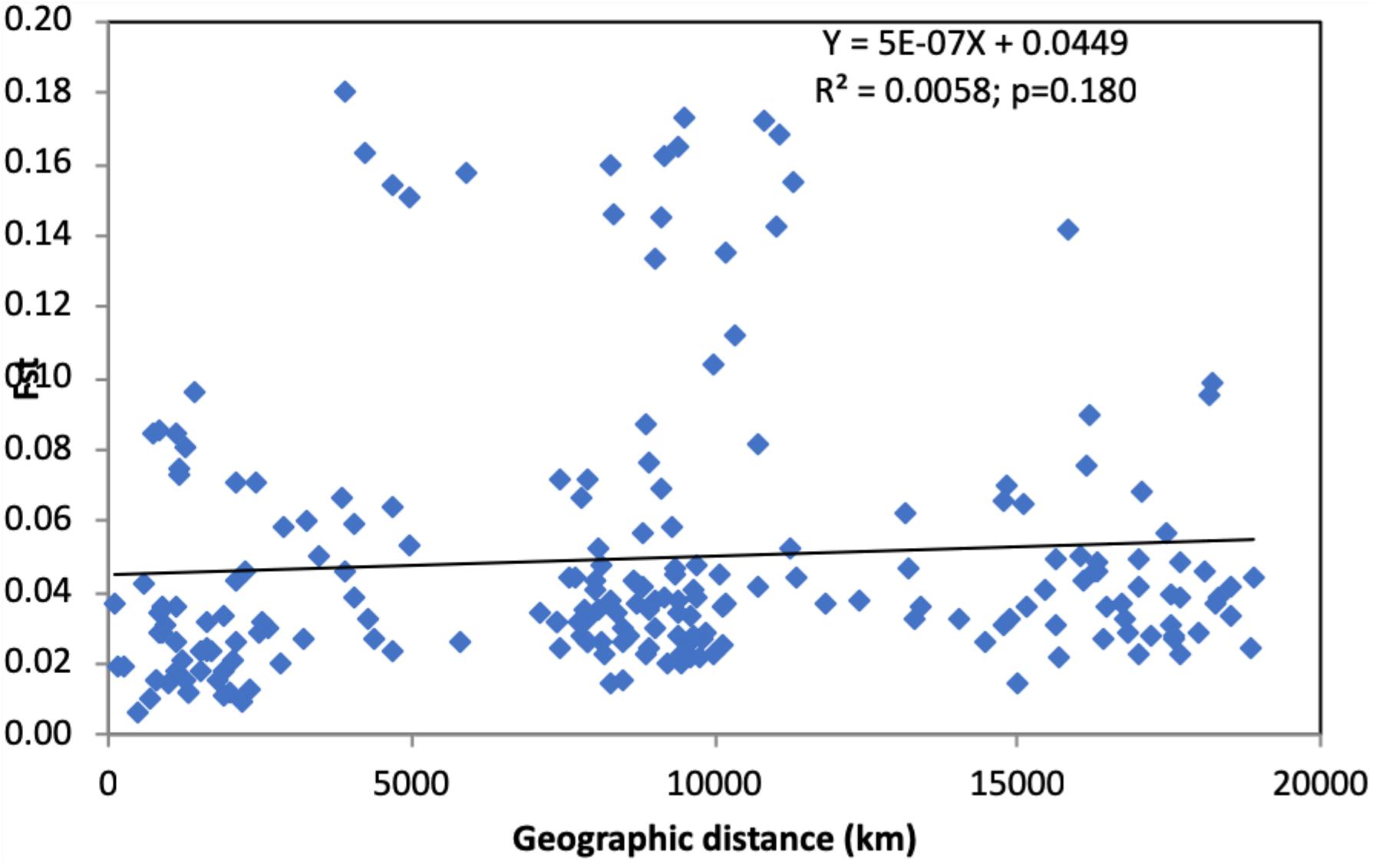
Figure 1. The relationship between genetic differentiation and geographic distance between pairs of regional and national samples of Candida tropicalis. Only regional or national samples with sample sizes greater than 10 were included in this analysis. X-axis: geographic distance, Y-axis: pairwise FST values. No statistically significant correlation was observed between geographic distance and genetic difference in the global sample of C. tropicalis (p = 0.180).
To identify potentially distinct genetic populations within the global C. tropicalis sample, we investigated the likely number of genetic clusters using the STRUCTURE software. Our analyses revealed an optimal number of 15 genetic clusters. The geographic distributions and sample sizes of these genetic clusters are presented in Table 7. The sample sizes of the genetic clusters ranged from 40 (Pop7) to 74 strains (Pop4), with each genetic cluster being relatively well represented in the global sample. Each of the 15 genetic clusters is distributed in at least two countries. Among these 15 genetic clusters, two (Pop6 and Pop14) are so far found only in Asia while each of the remaining 13 have been found in at least two continents. Three genetic clusters (Pop4, Pop7, and Pop12) have been found in four continents and two genetic clusters (Pop2 and Pop9) are found in all five continents from where multilocus sequences of C. tropicalis have been deposited in the MLST database.
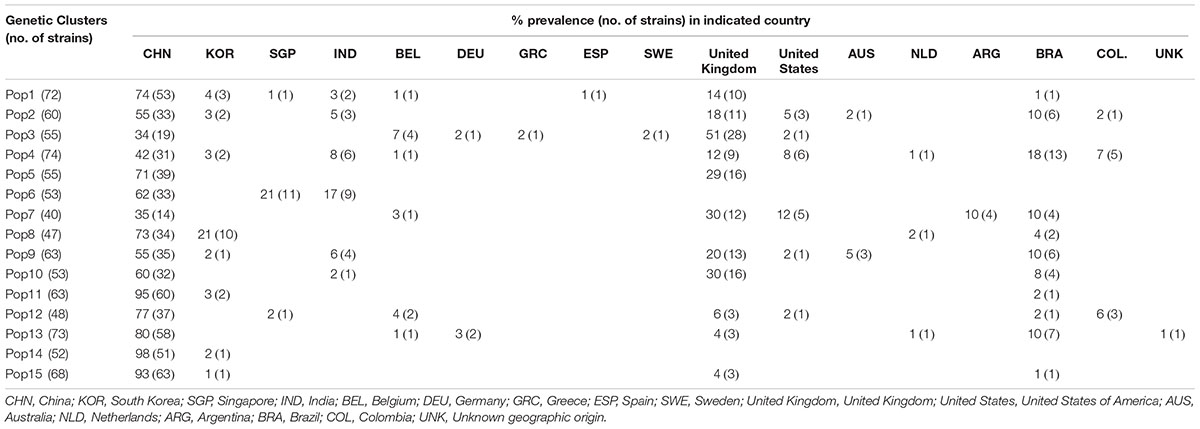
Table 7. Distribution of the 15 inferred genetic clusters of C. tropicalis at the national level.
At the individual country-level, most countries with sample sizes greater than 10 contained strains belonging to at least two different genetic clusters. The two countries with the most strains in the database also had the highest numbers of genetic clusters. Specifically, all 15 genetic clusters are well-represented in China, the country with the highest number of strains in the database. Indeed, over 90% of the strains in three of the 15 genetic clusters (Pop11, Pop14, and Pop15) came from China. Similarly, 12 of the 15 genetic clusters are found in the United Kingdom sample. Taken together, the STRUCTURE analyses indicate that there are multiple distinct genetic clusters in the global sample of C. tropicalis and that most genetic clusters are broadly distributed, with some showing geographic bias. The results are overall consistent with the geographic analyses of multilocus genotype distributions, pairwise PhiPT, and AMOVA.
This study analyzed the geographic patterns of genotype variation in a global sample of C. tropicalis. Our analyses revealed that over 10% of the 2677 nucleotides at the six sequenced gene fragments were polymorphic in this global sample. These SNPs generated a large number of genotypes at each locus. In total, the DNA sequence variation at the six loci allowed the identification of 633 multilocus genotypes among the 876 strains. Among these genotypes, 93 were shared by 336 strains while the remaining 540 were represented by one strain each. The majority of the 93 shared multilocus genotypes were between strains from the same geographic region. However, strains from different regions within a country, from different countries, and from different continents have also been found to share identical multilocus genotypes. These results are consistent with short- medium-, and long- distance dispersals of C. tropicalis strains.
Population genetic analyses revealed that most genetic variations were found within regional and national samples. However, statistically significant genetic differences were found between many regional populations within China and between most national populations. Consistent with the high genetic diversity within most regional and national populations, STRUCTURE results showed that the global C. tropicalis sample belonged to 15 genetic clusters, with most genetic clusters distributed in multiple regions and countries. Overall, these results suggest that the global C. tropicalis population have likely been historically differentiated but recent gene flows have brought strains with divergent genotypes together into the same geographic regions. A similar result has been reported in another human fungal pathogen Aspergillus fumigatus. In that study, the analyses of genotype data at nine microsatellite loci for over 2000 strains revealed eight distinct genetic clusters with most clusters being broadly distributed in Asia, Europe, Australia, and South and North America (Ashu et al., 2017). Similar to that proposed for A. fumigatus, the dispersals observed here for C. tropicalis were likely due to anthropogenic factors such as commercial trade and human travel.
Our study relied on data from previous C. tropicalis MLST studies and the tremendous effort by the curator Dr. Frank Odds and his team in maintaining the database, to ensure the quality of submitted sequence data. For example, even though 742 DSTs were originally assigned by the end of May 2018 based on the initial sequence submissions by investigators, 109 of these DSTs were later invalidated based on critical inspections of submitted sequences (Table 2 and Supplementary Table 1). Due to the diploid nature of C. tropicalis, automatic base calls of sequence chromatographs can produce incorrect sequence reads and heterozygous sites may be mistakenly called homozygous. As a result, close inspection of sequence file is essential for ensuring the accuracy of the sequence information for each strain at each locus.
Previous studies of C. tropicalis MLST variation have mostly focused on local and regional samples and examined the potential relationships between strains with regard to patient characteristics (including age, sex, underlying condition, etc), source of infection, route of transmission, and the potential effects of antifungal drug treatments on genotypic changes within individual patients and hospital environments (e.g., Chen et al., 2009; Silva et al., 2012; Yang et al., 2012; Fan et al., 2014; Al-Obaid et al., 2017; Scordino et al., 2018). In general, these individual hospital-based or geographic region – specific studies revealed limited evidence for significant contribution of host body sites or other host factors to C. tropicalis genetic diversity in humans. For example, there were multiple cases of multilocus genotype sharing among unrelated individuals from different body sites and different geographic regions (e.g., Li et al., 2009; Wang et al., 2016; Supplementary Table 1). In one example, the most commonly shared genotype #140 was shared by strains from different anatomic sites among unrelated people in Taiwan and this result was hypothesized as evidence of a clonal expansion due to the strain’s ability to grow at high fluconazole concentration (Li et al., 2009). Our human anatomic site-based AMOVA of the global C. tropicalis samples identified that the anatomic sites in humans (blood, feces, oropharynx, other sterile body sites, skin and other superficial surfaces, urine, vagina) contributed only 0.5% to the total genetic variation (detailed data not shown). Together, the results suggest that at the global level, there is very little evidence for human anatomic site-specific adaptation of C. tropicalis MLST genotypes and that most genotypes and genetic clusters are capable of colonizing a diversity of human body sites and causing infections. We would like to emphasize that our current study focuses on the geographic patterns of C. tropicalis genetic variation based on the published and publicly available MLST data. Our study differs from previous studies by taking a broad population genetic approach to examine the distributions of genotypes and genetic variations across large geographic scales. While several broad and interesting patterns have emerged through our analyses, the amount of genetic diversity as well as the patterns of variation as revealed here are likely incomplete and biased. Below we discuss two areas of research that could help enhance our understanding of the global C. tropicalis population.
The first is to expand geographic sampling and representation of C. tropicalis in the MLST database. At present, the samples in the MLST database are geographically highly skewed at the regional, national, and continental levels. Less that 10% of the countries are represented in the database and literature. There are few or no isolates from most countries, including those in the entire African continent. Though the total number of C. tropicalis strains analyzed and deposited in the MLST database so far is large (876), over two-thirds of the strains were from one country, China. The second biggest source of the analyzed strains was the United Kingdom. However, over 50% (67/124) of the United Kingdom strains was from the first MLST study of C. tropicalis that established the consensus MLST loci and the database (Tavanti et al., 2005). Indeed, most or all strains in the C. tropicalis MLST database from several countries were deposited by this first C. tropicalis MLST study (Tavanti et al., 2005), including those from Australia (4 of 7 strains), United States (all 17 strains), Belgium (9 of 10 strains), Netherlands and Germany (3 strains from each of the two countries), and Greece and Sweden (1 strain from each country). It should be noted that the subsequent lack of data from some of these and many other developed countries was not due to the reduced clinical importance of C. tropicalis in these and other countries. In fact, C. tropicalis is often the second most common yeast species colonizing mucosal surfaces and causing bloodstream infections in many regions and countries (Capoor et al., 2005; Odds et al., 2007; Falagas et al., 2010; Adhikary and Joshi, 2011; Wang et al., 2013; Vaezi et al., 2017; Wu et al., 2017; Zuza-Alves et al., 2017). In addition, strains of C. tropicalis are often more resistant to the azole and polyene antifungal drugs than those of most other Candida species, including C. albicans, causing complications in disease treatments (Oliver et al., 2005; Chou et al., 2007; Desnos-Ollivier et al., 2008; Wu et al., 2014; Wang et al., 2016). Instead, we believe the skewed sample sizes among regions and countries in the C. tropicalis MLST database most likely reflect the efforts (or lack of) by medical mycologists from different countries and geographic regions over the last decade. As evidenced by a recent study in the journal Mycopathologia, studies of medical fungi have undergone a notable shift, with an increasing number of papers coming from emerging economies such as China, India, and Brazil etc. (Chaturvedi et al., 2018).
The second area of future research is to integrate natural ecological studies of C. tropicalis into clinical investigations and global population genetic analyses. C. tropicalis has been found in a diversity of ecological niches, including multiple anatomical sites in humans (Supplementary Table 1), soil (Yang et al., 2012; Aljohani et al., 2018), trees (Carvalho et al., 2014), aquatic environments (Vogel et al., 2007), and animals such as wild birds (Lord et al., 2010), horses (Cordeiro et al., 2013), rheas (Brilhante et al., 2013) as well as in tortoises and sea turtles (Brilhante et al., 2015). However, as seen in Supplementary Table 1, most geographic populations (at either the regional or national levels) in the current database had only a limited number of analyzed ecological niches (mostly in humans) and that most body sites had relatively few strains of C. tropicalis each. The small sample sizes in most geographic and ecological populations prevented us from conducting a robust analysis on the potential influences of ecological factors on the overall patterns of genetic variation. Even though there was a relatively large number of strains from Mainland China for most of the anatomic body sites (45 from blood, 11 from feces, 172 from the oral mucosal and oropharynx, four from sterile body sites other than blood, 32 from the skin and other superficial surfaces, 30 from urine, and 42 from vaginal tract), these samples were from geographically very diverse and distant provinces within China and most provincial populations only had one or two body sites with reasonable sample sizes (Supplementary Table 1). As a result, it’s difficult to estimate the potential contributions of ecological niches to the overall genetic variation without taking into account of the influences of geography at the same time. Though our preliminary analysis revealed that the global samples of C. tropicalis from different anatomic body sites had very little genetic difference from each other, it’s possible that populations of C. tropicalis from humans differ from those around their environments. Indeed, a previous study based on PCR fingerprinting revealed that the clinical and tree hollow populations of human pathogenic yeasts (including C. tropicalis) in Hamilton, ON, Canada were different (Carvalho et al., 2014). To accurately investigate the potential ecological contributions to C. tropicalis genetic variation and the relationships between environmental and human samples, future studies should focus on obtaining and analyzing diverse ecological samples from each geographic region and from many different regions and countries using the same MLST system.
Our study revealed that the global population of C. tropicalis consists multiple genetic clusters with most of the clusters broadly distributed geographically. Our analyses showed that most observed genetic variations were found at local and regional levels and rejected the hypothesis that the global populations of C. tropicalis were structured based on geographic distances. Evidence for both medium- and long- distance dispersals was found. This study reaffirmed that the C. tropicalis MLST database provided a powerful platform not only for sharing strain and genotype information among investigators but also for helping us identifying broad epidemiological and population genetic patterns. Our analyses revealed several knowledge gaps from which future studies of C. tropicalis ecology, population genetics and molecular epidemiology could be focused on.
J-YW retrieved and analyzed the data. JX guided the data analyses and drafted the manuscript. D-YZ, FM, and YZ participated in data analyses. All authors read and approved the manuscript.
The research in our labs on medical fungi are supported by grants from the National Natural Science Foundation of China (Grant No. 31860035), Natural Science Foundation of Hainan Province (20158374), the Education Department Foundation of Hainan Province (Hjkj2012-36), and the Natural Sciences and Engineering Research Council of Canada (CRDPJ 474638-14).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Dr. Frank Odds and his team for maintaining the Candida tropicalis MLST database and for allowing the public to freely access the data.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00900/full#supplementary-material
FIGURE S1 | Relationships among all 876 strains in the C. tropicalis MLST database as determined by their concatenated sequences at six gene fragments. For each strain, the strain name is followed by continent, country, and (when available) region (province/state/city). Scale bar represents the percent nucleotide base difference (0–12% on the scale bar) between strains, adjusted for diploids.
TABLE S1 | Information about all 876 Candida tropicalis isolates analyzed in this study. Included in this table for each isolate are isolate name, geographic source, ecological niche, and multilocus genotype at six gene loci.
Adhikary, R., and Joshi, S. (2011). Species distribution and anti-fungal susceptibility of Candidaemia at a multi super-specialty center in Southern India. Indian J. Med. Microbiol. 29, 309–311. doi: 10.4103/0255-0857.83920
Afsarian, M. H., Badali, H., Boekhout, T., Shokohi, T., and Katiraee, F. (2015). Multilocus sequence typing of Candida albicans isolates from a burn intensive care unit in Iran. J. Med. Microbiol. 64, 248–253. doi: 10.1099/jmm.0.000015
Aljohani, R., Samarasinghe, H., Ashu, T., and Xu, J. (2018). Diversity and relationships among strains of culturable yeasts in agricultural soils in Cameroon. Sci. Rep. 8:1. doi: 10.1038/s41598-018-34122-2
Al-Obaid, K., Asadzadeh, M., Ahmad, S., and Khan, Z. (2017). Population structure and molecular genetic characterization of clinical Candida tropicalis isolates from a tertiary-care hospital in Kuwait reveal infections with unique strains. PLoS One 12:e0182292. doi: 10.1371/journal.pone.0182292
Arrua, J. M., Rodrigues, L. A., Pereira, F. O., and Lima, E. O. (2015). Prevalence of Candida tropicalis and Candida krusei in onychomycosis in João Pessoa, Paraiba, Brazil from 1999 to 2010. An. Acad. Bras. Cienc. 87, 1819–1822. doi: 10.1590/0001-3765201520130418
Ashu, E. E., Hagen, F., Chowdhary, A., Meis, J. F., and Xu, J. (2017). Global population genetic analysis of Aspergillus fumigatus. mSphere 2:e19-17. doi: 10.1128/mSphere.00019-17
Brandt, M. E., Bragg, S. L., and Pinner, R. W. (1993). Multilocus enzyme typing of Cryptococcus neoformans. J. Clin. Microbiol. 31, 2819–2823.
Brilhante, R. S., de Alencar, L. P., Castelo-Branco, D. S., Lima, D. T., Paiva, M. A., Monteiro, A. J., et al. (2013). Detection of Candida species resistant to azoles in the microbiota of rheas (Rhea americana): possible implications for human and animal health. J. Med. Microbiol. 62, 889–895. doi: 10.1099/jmm.0.055566-0
Brilhante, R. S., Rodrigues, P. H., de Alencar, L. P., Riello, G. B., Ribeiro, J. F., de Oliveira, J. S., et al. (2015). Evidence of fluconazole-resistant Candida species in tortoises and sea turtles. Mycopathologia 180, 421–426. doi: 10.1007/s11046-015-9923-0
Butler, G., Rasmussen, M. D., Lin, M. F., Santos, M. A., Sakthikumar, S., Munro, C. A., et al. (2009). Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 459, 657–662. doi: 10.1038/nature08064
Capoor, M. R., Nair, D., Deb, M., Verma, P. K., Srivastava, L., and Aggarwal, P. (2005). Emergence of non-albicans Candida species and antifungal resistance in a tertiary care hospital. Jpn. J. Infect. Dis. 58, 344–348. doi: 10.1111/j.1756-1051.2009.00484.x
Carvalho, C., Yang, J., Vogan, A., Maganti, H., Yamamura, D., and Xu, J. (2014). Clinical and tree hollow populations of human pathogenic yeasts in Hamilton. Ontario, Canada are different. Mycoses 57, 271–283. doi: 10.1111/myc.12156
Chaturvedi, V., Bouchara, J. P., Hagen, F., Alastruey-Izquierdo, A., Badali, H., Bocca, A. L., et al. (2018). Eighty years of Mycopathologia: a retrospective analysis of progress made in understanding human and animal fungal pathogens. Mycopathologia 183, 859–877. doi: 10.1007/s11046-018-0306-1
Chen, K. W., Chen, Y. C., Lin, Y. H., Chou, H. H., and Li, S. Y. (2009). The molecular epidemiology of serial Candida tropicalis isolates from ICU patients as revealed by multilocus sequence typing and pulsed-field gel electrophoresis. Infect. Genet. Evol. 9, 912–920. doi: 10.1016/j.meegid.2009.06.011
Chou, H. H., Lo, H. J., Chen, K. W., Liao, M. H., and Li, S. Y. (2007). Multilocus sequence typing of Candida tropicalis shows clonal cluster enriched in isolates with resistance or trailing growth of fluconazole. Diagn. Microbiol. Infect. Dis. 58, 427–433. doi: 10.1016/j.diagmicrobio.2007.03.014
Cordeiro, R. D. A., Bittencourt, P. V., Teixeir, C. E. C., Silv, S. T. D. C., Alencar, L. P. D., Souz, E. R. Y., et al. (2013). Species of Candida as a component of the nasal microbiota of healthy horses. Med. Mycol. 51, 731–736. doi: 10.3109/13693786.2013.777858
Desnos-Ollivier, M., Bretagne, S., Bernède, C., Robert, V., Raoux, D., Chachaty, E., et al. (2008). Clonal population of flucytosine-resistant Candida tropicalis from blood cultures. Paris, France. Emerg. Infect. Dis. 14, 557–565. doi: 10.3201/eid1404.07
Diba, K., Makhdoomi, K., Nasri, E., Vaezi, A., Javidnia, J., Gharabagh, D. J., et al. (2018). Emerging Candida species isolated from renal transplant recipients: species distribution and susceptibility profiles. Microb. Pathog. 2018, 240–245. doi: 10.1016/j.micpath.2018.09.026
Dóczi, I., Petõ, Z., Fodor, E., Bereczki, L., Nagy, E., and Hajdú, E. (2012). Evaluation of fungaemia infections in a hungarian university hospital between 1996 and 2009. Acta Microbiol. Immunol. Hung. 59, 29–41. doi: 10.1556/AMicr.59.2012.1.4
Dong,Z., Zhang, L., Jiang, C., Mao, E., Wang, X., et al. (2015). Clinical and microbiological investigation of fungemia from four hospitals in China. Mycopathologia 179, 407–414. doi: 10.1007/s11046-014-9855-0
Duarte-Escalante, E., Zúñiga, G., Frías-De-León, M. G., Canteros, C., Castañón-Olivares, L. R., and Reyes-Montes, M. R. (2013). AFLP analysis reveals high genetic diversity but low population structure in Coccidioides posadasii isolates from Mexico and Argentina. BMC Infect. Dis. 13:411. doi: 10.1186/1471-2334-13-411
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x
Falagas, M. E., Roussos, N., and Vardakas, K. Z. (2010). Relative frequency of albicans and the various non-albicans Candida spp among candidemia isolates from inpatients in various parts of the world: a systematic review. Int. J. Infect. Dis. 14, e954–e966. doi: 10.1016/j.ijid.2010.04.006
Fan, X., Xiao, M., Wang, H., Zhang, L., Kong, F., Lu, J., et al. (2014). Multilocus sequence typing indicates diverse origins of invasive Candida tropicalis isolates in China. Chin. Med. J. 127, 4226–4233.
Fesharaki, S. H., Haghani, I., Mousavi, B., Kargar, M. L., Boroumand, M., Anvari, M. S., et al. (2013). Endocarditis due to a co-infection of Candida albicans and Candida tropicalis in a drug abuser. J. Med. Microbiol. 62, 1763–1767. doi: 10.1099/jmm.0.060954-0
Field, D., Eggert, L., Metzgar, D., Rose, R., and Wills, C. (1996). Use of polymorphic short and clustered coding region microsatellites to distinguish strains of Candida albicans. FEMS Immunol. Med. Microbiol. 15, 73–79. doi: 10.1016/0928-8244(96)00034-x
Fries, B. C., Chen, F. Y., Currie, B. P., and Casadevall, A. (1996). Karyotype instability in Cryptococcus neoformans infection. J. Clin. Microbiol. 34,1531–1534.
Lachance, M. A., Boekhout, T., Scorzetti, G., Fell, J. W., and Kurtzman, C. P. (2011). “Candida berkhout,” in The Yeasts, A Taxonomic Study, eds C. P. Kurtzman, J. W. Fell, and T. Boekhout (Amsterdam: Elsevier Science), 987–1278.
Li, S. Y., Yang, Y. L., Lin, Y. H., Ko, H. C., Wang, A. H., Chen, K. W., et al. (2009). Two closely related Fluconazole-resistant Candida tropicalis clones circulating in Taiwan from 1999 to 2006. Microb. Drug. Resist. 15, 205–210. doi: 10.1089/mdr.2009.0915
Lord, A. T., Mohandas, K., Somanath, S., and Ambu, S. (2010). Multidrug resistant yeasts in synanthropic wild birds. Ann. Clin. Microbiol. Antimicrob. 9:11. doi: 10.1186/1476-0711-9-11
Magri, M. M., GomesGouvêa, M. S., de Freitas, V. L., Motta, A. L., Moretti, M. L., and Shikanaiyasuda, M. A. (2013). Multilocus sequence typing of Candida tropicalis shows the presence of different clonal clusters and fluconazole susceptibility profiles in sequential isolates from Candidemia patients in São Paulo, Brazil. J. Clin. Microbiol. 5, 268–277. doi: 10.1128/JCM.02366-12
Morii, D., Seki, M., Binongo, J. N., Ban, R., Kobayashi, A., Sata, M., et al. (2014). Distribution of Candida species isolated from blood cultures in hospitals in Osaka. Japan. J. Infect. Chemother. 20, 558–562. doi: 10.1016/j.jiac.2014.05.009
Odds, F. C., Hanson, M. F., Davidson, A. D., Jacobsen, M. D., Wright, P., Whyte, J. A., et al. (2007). One-year prospective survey of Candida bloodstream infections in Scotland. J. Med. Microbiol. 56, 1066–1075. doi: 10.1099/jmm.0.47239-0
Oliver, B. G., Silver, P. M., and White, T. C. (2005). “Evolution of drug resistance in pathogenic fungi,” in Evolutionary Genetics of Fungi, ed. J. Xu (England: Horizon Bioscience Press), 253–288.
Pahwa, N., Kumar, R., Nirkhiwale, S., and Bandi, A. (2014). Species distribution and drug susceptibility of Candida in clinical isolates from a tertiary care centre at Indore. Indian J. Med. Microbiol. 32, 44–48. doi: 10.4103/0255-0857.124300
Peakall, R., and Smouse, P. E. (2012). GenAlEx 6.5: genetic analysis in Excel: population genetic software for teaching and research — an update. Bioinformatics 28, 2537–2539. doi: 10.1093/bioinformatics/bts460
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Scordino, F., Giuffrè, L., Barberi, G., Merlo, F. M., Orlando, M. G., Giosa, D., et al. (2018). Multilocus sequence typing reveals a new cluster of closely related Candida tropicalis genotypes in Italian patients with neurological disorders. Front. Microbiol. 9:679. doi: 10.3389/fmicb.2018.00679
Silva, S., Negri, M., Henriques, M., Oliveira, R., Williams, D. W., and Azeredo, J. (2012). Candida glabrata, Candida parapsilosis and Candida tropicalis: biology, epidemiology, pathogenicity and antifungal resistance. FEMS Microbiol. Rev. 36, 288–305. doi: 10.1111/j.1574-6976.2011.00278.x
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30,2725–2729. doi: 10.1093/molbev/mst197
Tavanti, A., Davidson, A. D., Johnson, E. M., Maiden, M. C. J., Shaw, D. J., Gow, N. A. R., et al. (2005). Multilocus sequence typing for differentiation of strains of candida tropicalis. J. Clin. Microbiol. 43, 5593–5600. doi: 10.1128/JCM.43.11.5593-5600.2005
Vaezi, A., Boekhout, T., and Badali, H. (2017). Epidemiological and mycological characteristics of candidemia in Iran: a systematic review and meta-analysis. J Mycol Med. 27, 146–152. doi: 10.1016/j.mycmed.2017.02.007
Vogel, C., Rogerson, A., Schatz, S., Laubach, H., Tallman, A., and Fell, J. (2007). Prevalence of yeasts in beach sand at three bathing beaches in South Florida. Water Res. 41, 1915–1920. doi: 10.1016/j.watres.2007.02.010
Wang, H., Xu, J., Guo, H., Wu, J., Yi, G., Pei, H., et al. (2013). Patterns of human oral yeast species distribution on Hainan Island in China. Mycopathologia 176, 359–368. doi: 10.1007/s11046-013-9703-7
Wang, Y., Shi, C., Liu, J. Y., Li, W. J., Zhao, Y., and Xiang, M. J. (2016). Multilocus sequence typing of Candida tropicalis shows clonal cluster enrichment in azole-resistant isolates from patients in Shanghai, China. Infect. Genet. Evol. 44, 418–424. doi: 10.1016/j.meegid.2016.07.026
Wu, J. Y., Guo, H., Wang, H. M., Yi, G. H., Zhou, L. M., He, X. W., et al. (2017). Multilocus sequence analyses reveal extensive diversity and multiple origins of fluconazole resistance in Candidatropicalis from tropical China. Sci. Rep. 7:42537. doi: 10.1038/srep42537
Wu, J. Y., Guo, H., Yi, G., Zhou, L., He, X., Huang, X., et al. (2014). Prevalent drug resistance among oral yeasts from asymptomatic patients in Hainan, China. Mycopathologia 177, 299–307. doi: 10.1007/s11046-014-9747-3
Wu, Y., Zhou, H., Wang, J., Li, L., Li, W., Cui, Z., et al. (2012). Analysis of the clonality of Candida tropicalis strains from a general hospital in Beijing using multilocus sequence tying. PLoS One 7:e47767. doi: 10.1371/journal.pone.0047767
Xu, J. (2005). “Fundamentals of fungal molecular population genetic analyses,” in Evolutionary Genetics of Fungi, ed. J. Xu (England: Horizon Bioscience Press), 87–116.
Xu, J., Ramos, A., Vilgalys, R., and Mitchell, T. G. (2000a). Clonal and spontaneous origins of fuconazole resistance in Candida albicans. J. Clin. Microbiol. 38, 1214–1220.
Xu, J., Vilgalys, R. J., and Mitchell, T. G. (2000b). Multiple gene genealogies reveal recent dispersion and hybridization in the human pathogenic fungus Cryptococcus neoformans. Mol. Ecol. 9, 1471–1481. doi: 10.1046/j.1365-294x.2000.01021.x
Yang, Y.-L., Lin, C.-C., Chang, T.-P., Lauderdale, T.-L., Chen, H.-T., Lee, C.-F., et al. (2012). Comparison of human and soil Candida tropicalis isolates with reduced susceptibility to fluconazole. PLoS One 7:e34609. doi: 10.1371/journal.pone.0034609
Keywords: multilocus sequence typing, genotype sharing, geographic pattern, genetic clusters, yeast, geographic distribution
Citation: Wu J-Y, Zhou D-Y, Zhang Y, Mi F and Xu J (2019) Analyses of the Global Multilocus Genotypes of the Human Pathogenic Yeast Candida tropicalis. Front. Microbiol. 10:900. doi: 10.3389/fmicb.2019.00900
Received: 04 January 2019; Accepted: 09 April 2019;
Published: 26 April 2019.
Edited by:
Orazio Romeo, University of Messina, ItalyReviewed by:
Guilherme Maranhão Chaves, Federal University of Rio Grande do Norte, BrazilCopyright © 2019 Wu, Zhou, Zhang, Mi and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianping Xu, anB4dUBtY21hc3Rlci5jYQ==; anB4dW1jbWFzdGVyQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.