
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 12 March 2019
Sec. Food Microbiology
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.00472
This article is part of the Research TopicInsights of Fermented Foods and Beverages: Microbiology and Health-Promoting BenefitsView all 19 articles
 Zhuolin Yi1,2,3Yanling Jin2,3Yao Xiao4Lanchai Chen2,3,5Li Tan2,3Anping Du2,3Kaize He2,3Dayu Liu1Huibo Luo6Yang Fang2,3*
Zhuolin Yi1,2,3Yanling Jin2,3Yao Xiao4Lanchai Chen2,3,5Li Tan2,3Anping Du2,3Kaize He2,3Dayu Liu1Huibo Luo6Yang Fang2,3* Hai Zhao1,2,3*
Hai Zhao1,2,3*Jiang-flavor (JF) daqu is a liquor starter used for production of JF baijiu, a well-known distilled liquor in China. Although a high temperature stage (70°C) is necessary for qualifying JF daqu, little is known regarding its active microbial community and functional enzymes, along with its role in generating flavor precursors for JF baijiu aroma. In this investigation, based on metatranscriptomics, fungi, such as Aspergillus and Penicillium, were identified as the most active microbial members and 230 carbohydrate-active enzymes were identified as potential saccharifying enzymes at 70°C of JF daqu. Notably, most of enzymes in identified carbohydrate and energy pathways showed lower expression levels at 70°C of JF daqu than those at the high temperature stage (62°C) of Nong-flavor (NF) daqu, indicating lowering capacities of saccharification and fermentation by high temperature stage. Moreover, many enzymes, especially those related to the degradation of aromatic compounds, were only detected with low expression levels at 70°C of JF daqu albeit not at 62°C of NF daqu, indicating enhancing capacities of generating special trace aroma compounds in JF daqu by high temperature stage. Additionally, most of enzymes related to those capacities were highly expressed at 70°C by fungal genus of Aspergillus, Coccidioides, Paracoccidioides, Penicillium, and Rasamsonia. Therefore, this study not only sheds light on the crucial functions of high temperature stage but also paves the way to improve the quality of JF baijiu and provide active community and functional enzymes for other fermentation industries.
Baijiu (Chinese liquor), one of the oldest known distilled liquors with an approximate 2000-year history, is the largest consumed spirit globally (over 13 billion liters in 2016) (Liu and Sun, 2018). Compared with whisky and brandy, baijiu is well known for its taste with more flavor compounds (>1870 volatile compounds) in liquor, including alcohols, aldehydes, organic acids, esters, phenols, lactones, heterocycles, terpenes, aromatic compounds, amino acids, and peptides, which leads to the final special aroma and health of baijiu (Jin et al., 2017; Liu and Sun, 2018). Thus, based on its distinctive flavor characteristics, baijiu can be divided into three major categories [i.e., Jiang-flavor (JF, also called sauce-flavor) baijiu, Nong-flavor (NF) baijiu and Qing-flavor (QF) baijiu] and nine minor categories, among which JF baijiu is with a full-bodied long-lasting aroma (Zheng and Han, 2016; Liu and Sun, 2018). The representative JF baijiu is moutai, the most famous baijiu, having the distinction as “the national liquor” and largely dominating the market in China (Zheng and Han, 2016; Jin et al., 2017). JF baijiu is fermented and distilled under solid-state conditions with a production process that mainly includes four distinct stages; i.e., daqu preparation (approximately 4 months), stacking fermentation (2–4 days), alcoholic fermentation and distillation processes (Figure 1) (Fan et al., 2012; Zheng and Han, 2016).
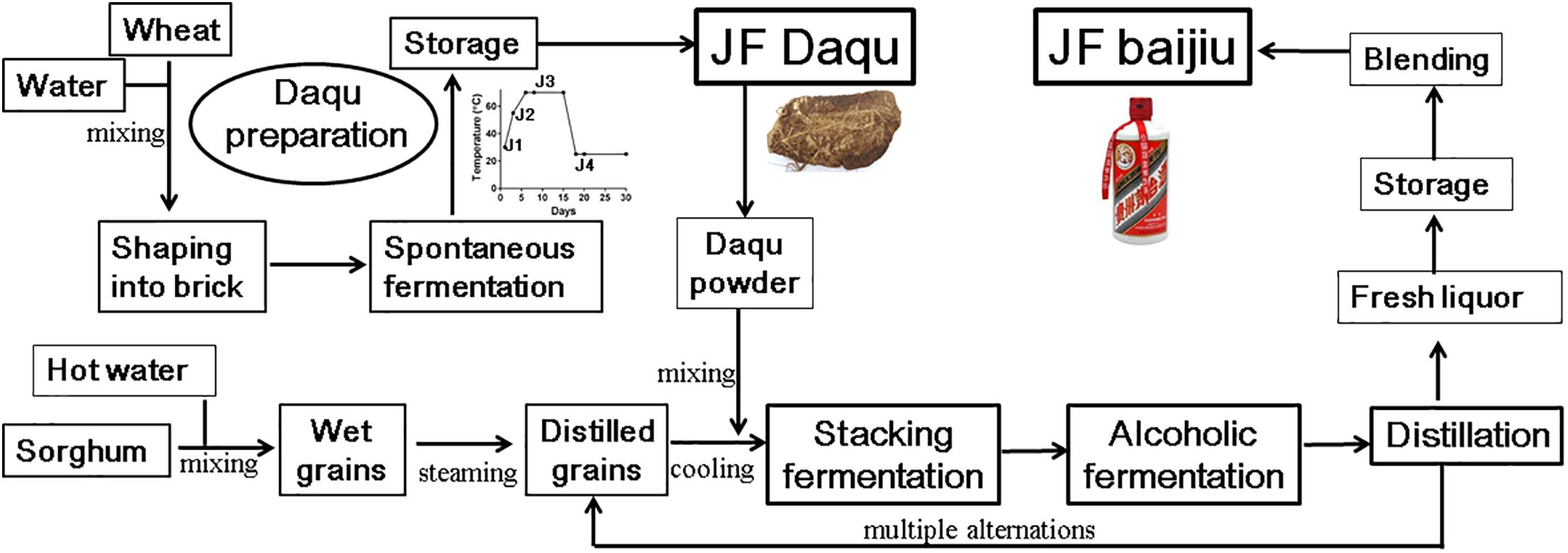
Figure 1. Process diagram of traditional production of Jiang-flavor (JF) baijiu. JF daqu was produced with two steps of spontaneous fermentation in a Qu-room and drying without ventilation in a storage room, and its maximum cultural temperature was usually between 60 and 70°C. Besides stage of daqu preparation, JF baijiu was produced with other three distinct stages of stacking fermentation, multiple alternations of alcoholic fermentation and distillation processes.
Typically, daqu, a liquor starter used to initiate the alcoholic fermentation process, constitutes the most essential component for alcoholic fermentation, not only providing the microbial community and enzymes (as a saccharifying and fermenting agent) for alcoholic fermentation but also significantly contributing to the final liquor flavor (Zheng and Han, 2016; Liu and Sun, 2018). Similar liquor starters can be found in many Asian countries, e.g., xiaoqu/fuqu in China (Zheng and Han, 2016; Jin et al., 2017), meju in Korea (Kim et al., 2011), ragi in Indonesia (Fibri and Frøst, 2019), marcha/thiat/dawdim/hamei/chowan in India (Sha et al., 2018), bubod in Philippines (Tamang et al., 2016), and banh men in Vietnam (Thanh et al., 2008). All those liquor starters are prepared in an open system with starchy materials (wheat, rice, etc), shaped into different sizes and shapes, and cultured under different conditions (temperature and time) (Liu and Sun, 2018; Sha et al., 2018; Waché et al., 2018). Among them, JF daqu is made from wheat, shaped into brick, and produced with two steps of spontaneous fermentation for approximately 1 month in a Qu-room and drying for another 3 months without ventilation in a storage room (Huang et al., 2017b; Jin et al., 2017). During the spontaneous fermentation process, the cultivation of JF daqu is controlled by manually turning over the bricks and opening/closing the windows to change the ventilation and temperature, with the special microbial community being enriched from raw materials and the working environments by environmental variables (temperature and moisture), among which temperature serves as a key driving force (Huang et al., 2017a; Xiao et al., 2017; Liu and Sun, 2018). According to the maximum temperature in the daqu preparation process, JF daqu is grouped into high-temperature (60–70°C) daqu and requires cultivation at the high temperature stage for approximately 7–8 days (Huang et al., 2017b; Liu and Sun, 2018). Owing to this high temperature stage, the thermophilic microbial community may be enriched in JF daqu and various thermostable enzymes (i.e., proteinase, glucoamylase, cellulase, alpha-amylase, and esterase) may also be produced to degrade materials and generate special flavor compounds.
Recently, the daqu microbial community has been studied throughout fermentation by culture-dependent and -independent methods, and their diversity and dynamics are well understood (Yan et al., 2013; Wang and Xu, 2015; Huang et al., 2017b; Xiao et al., 2017). However, little is known regarding the active microbial community and their metabolic functions. In addition, although numerous crude enzymes have been identified in daqu (Li et al., 2015; Liu et al., 2018), active enzymes and their relationships with the microbial community are yet unknown. Metatranscriptomics constitutes an ideal tool for studying daqu microbial ecology, as it directly analyzes mRNA from environments and provides information not only on the microbial community composition but also on active members and their specifically expressed enzymes (Bokulich et al., 2016). This technology has been successfully applied in microbial ecological systems; e.g., compost (Mello et al., 2017), mouse gut (Just et al., 2018), cattle rumen (Pandit et al., 2018), sludge (Xia et al., 2018), ocean (Yoshida et al., 2018), and human feces (Abu-Ali et al., 2018). Nevertheless, owing to the complicated conditions in baijiu brewing systems, such as the high content of starch and fermentation products along with strongly colored materials, it remains challenging to extract high-quality RNA from baijiu fermentation samples, especially from the high temperature stage (70°C) of JF daqu, in which greater amounts of fermentation products were generated with strong colors than in all the other daqus’ making stages. Thus, to our knowledge, only samples from the JF alcoholic fermentation process (42.8°C) (Song et al., 2017) and Nong-flavor (NF) daqu (a medium-temperature daqu) preparation process (62°C) (Huang et al., 2017a) have previously been studied using metatranscriptomics.
The cultivation temperatures in the production process for JF daqu, a typical high-temperature daqu, are largely higher than those in other daqus including NF daqu (Huang et al., 2017b). The high temperature condition constitutes the most striking difference among the daqu production processes of JF daqu and other daqus, as well as their subsequent alcoholic fermentation processes, which results in unique microbial community, enzymes, and aroma compounds being generated in the JF daqu and fermented feedstock (Wu et al., 2009; Wang et al., 2014; Xiao et al., 2016; Liu and Sun, 2018). Compared with NF daqu, JF daqu has a lower capacity for saccharification, liquefaction, and fermentation (Zheng and Han, 2016; Liu and Sun, 2018), thus requiring the use of a large amount of JF daqu (nearly 1:0.9 ratio of daqu versus feedstock) in the alcoholic fermentation process, which is higher than that of NF daqu (approximately 1:2 ratio). Thus, the flavor precursors, enzymes, and microbial community enriched in JF daqu would likely be more strongly associated with the final liquor flavor than those in NF daqu. Recently, we have published breakthrough research wherein significant differences were predictively shown in energy, carbohydrate metabolism, and degradation of aromatic compounds between the JF daqu and NF daqu bacterial community (Huang et al., 2017b), and the active microbial community was found to highly express pivotal enzymes at the high temperature stage of NF daqu making process (Huang et al., 2017a). However, the active microbial community and important enzymes, as well as their functional correlations in JF daqu remain to be identified. More specific understanding regarding differences of the high temperature stage between JF and NF daqu have not been clarified. Therefore, in this study, we first employed metatranscriptomics to explain the structure and function of the actual microbial community and its pivotal enzymes at the high temperature stage of JF daqu making process. Moreover, a comprehensive and global comparison was performed between JF and NF daqu to shed light on functions of the high temperature stage with regard to saccharification and fermentation along with flavor compound generation. This study provides fundamental information related to the active microbial community and functional enzymes and may facilitate a comparative understanding of the pivotal role of the high temperature stage in the JF daqu making process and JF baijiu brewing.
JF daqu samples were collected at different time points from a fermentation workshop of Kweichow Hanwang Group Co., Ltd. in Renhuai, Guizhou, China, as described previously (Huang et al., 2017b). Briefly, Sample J1 was harvested at the beginning of daqu production (30°C); J2 was harvested after 3 days of daqu preparation (55°C); J3 was harvested after 8 days of daqu preparation (70°C); and J4 was harvested from the mature daqu after fermentation for 20 days (25°C) (Figure 1). In addition, all samples were selected and mixed from three locations in the same Qu-room at each time point. For RNA extraction, the daqu samples were frozen in liquid nitrogen immediately after collection, transferred to the Chengdu Biology Institute, Chinese Academy of Sciences on that day and stored in a -80°C freezer. For enzyme analysis, all the samples were suspended in 0.1% (v/v) Tween 80 solution and transferred to the institute at room temperature (Huang et al., 2017a).
A total of 18 polymer analogs of insoluble chromogenic AZurine Cross-Linked (AZCL) polysaccharides (Megazyme, Ireland) were selected for detecting enzyme activities on cellulose, hemicellulose, starch, chitin, and glucan degradation (Table 1). As in our prior study (Huang et al., 2017a), all daqu samples in 0.1% (v/v) Tween 80 solution were incubated at 25°C and 100 rpm overnight, then their supernatants were added directly onto the wells of solid plates with AZCL polysaccharides according to the manufacturer’s protocol. After incubation at 35, 45, or 55°C for 22 h, carbohydrate-degrading enzyme activities were determined by measuring the diameter of the blue haloes, which were recorded in millimeters.
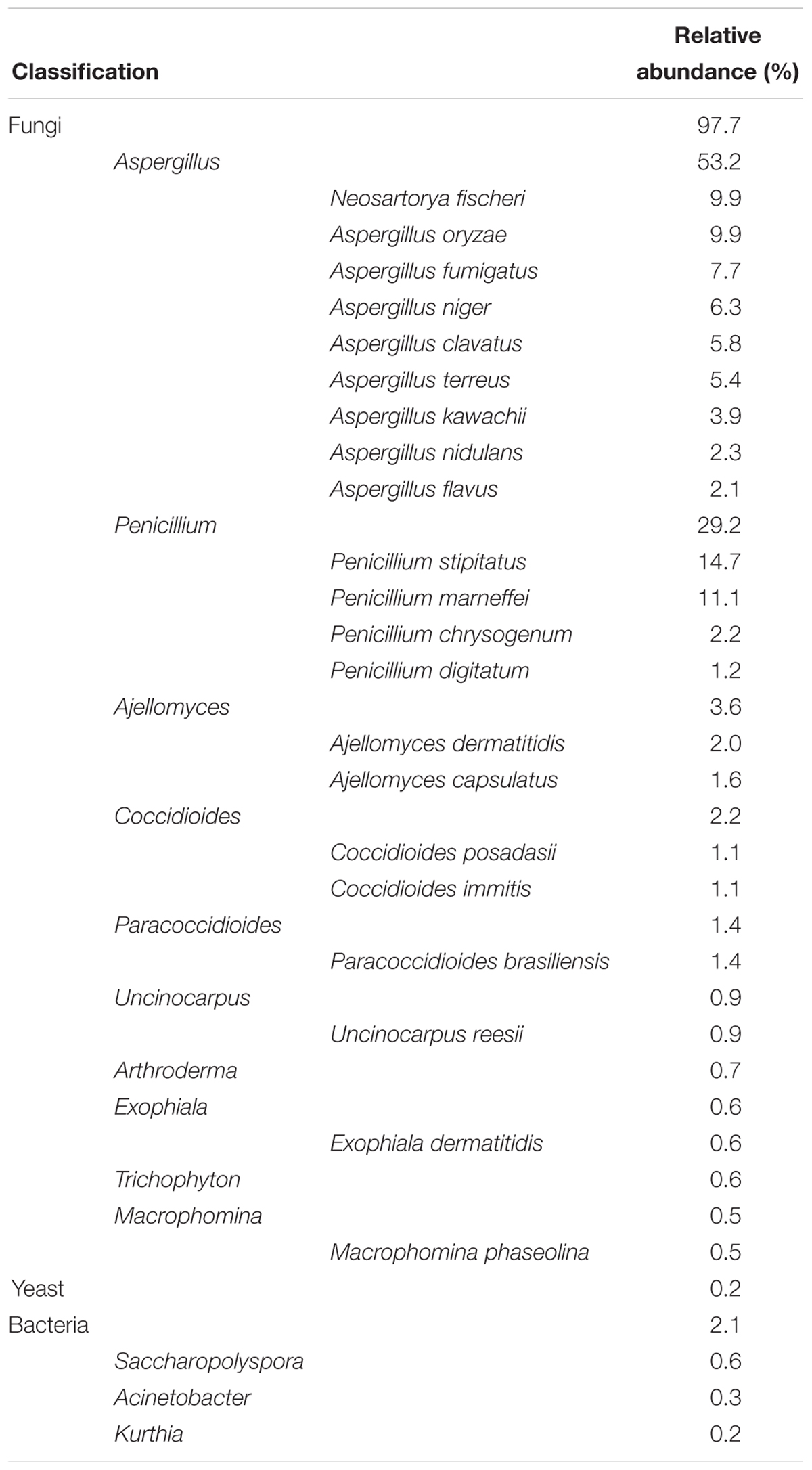
Table 1. Relative abundances of highly active fungal and bacterial taxa according to their designated gene numbers by the NR database.
Similar to the RNA extraction from NF daqu (Huang et al., 2017a), total RNA was extracted from JF daqu samples using borate buffer, cleaned with the RNeasy Midi Kit (Qiagen #75142, Venlo, Netherlands) and treated with DNase I (Fermentas, Waltham, MA, United States) according to the manufacturer’s protocols. The RNA integrity was evaluated by gel electrophoresis and RNA integrity number (RIN) was checked using an Agilent2100 Bioanalyzer (Santa Clara, CA, United States). RNA samples with RIN value greater than 7.0 and OD260/OD280 ratio greater than 1.8 were selected for deep sequencing.
Total RNA (approximately 20 μg) from J3 was used for the RNA sequencing. Prior to metatranscriptomic library construction, using a previously reported method (Huang et al., 2017a), mRNA was isolated using magnetic beads with Oligo (dT) for eukaryotes, and for prokaryotes, mRNA was obtained after removing ribosomal RNA. The isolated mRNA was first fragmented and then used as template for subsequent first- and second-strand cDNA synthesis with random primers. Short cDNA fragments were purified and resolved with EB buffer for end reparation and poly(A) addition. Thereafter, the short cDNA fragments were ligated to sequencing adapters and suitable sized cDNA fragments were purified as templates for polymerase chain reaction amplification. RNA sequencing of the library was performed using platform (Illumina, San Diego, CA, United States) at the Beijing Genomics Institute (BGI)- the HiSeqTM 2000 Shenzhen, China.
As for our previous metatranscriptomics assembly of NF daqu samples (N1–4), raw sequenced reads of J3 were first filtered by removing adaptors, low quality reads, and the rRNA sequences (Li et al., 2009). The clean reads of J3 were then de novo assembled using Trinity1 (Grabherr et al., 2011), by which unigene sequences were generated. To annotate the metatranscriptome, the unigene sequences were aligned using Blastx (version 2.5.0) with protein and nucleotide databases including Non-redundant protein (NR), Non-redundant nucleotide (NT), Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG), Clusters of Orthologous Groups (COG), and Gene Ontology (GO) (e-value < 10-5), and identified according to the highest similarity to known sequence. In cases of the non-alignment of unigenes against one of the listed databases, ESTScan was used to determine their coding directions. Thereafter, according to the standard codon usage, coding DNA sequences (CDSs) were translated into protein sequences. KEGG pathways were extracted from the KEGG web server2 (Kanehisa et al., 2017). WEGO software3 was used for GO classification (Ye et al., 2006). Carbohydrate-active enzymes (CAZymes) were retrieved from the Carbohydrate-Active Enzymes database (CAZy)4 (Lombard et al., 2014).
To compare the gene expression levels among J3 and NF daqu samples (N1–4), the predicted ORFs were combined after removing redundancy using cd-hit (Version 4.6.1)5 (Li and Godzik, 2006). Gene expression levels were calculated using the Reads Per Kilobase per Million mapped reads (RPKM) method (Mortazavi et al., 2008). DEGs among J3 and NF daqu samples were identified using a method based on the Poisson distribution (Audic and Claverie, 1997). DEGs between two samples were identified using p-value ≤ 0.05, Log2(RPKM ratio) ≥ 1, and false discovery rate (FDR) value ≤ 0.001 (Benjamini and Yekutieli, 2001). To analyze GO enrichment, all DEGs were mapped to terms of the GO database.
The raw and assembled metatranscriptomics data of J3 have been deposited to the GenBank database under accession numbers SRR7785758 and GGWC00000000, respectively.
After RNA sequencing of the J3 sample, 5.882 Gbp of raw data was generated, from which 5.663 Gbp of clean data was then obtained by filtering (Supplementary Table S1). These clean data were de novo assembled, from which 38,899 unigenes were identified with a total length of 46,187,298 nucleotides (nt) and N50 length of 2232 bp (Supplementary Table S2). As shown in Supplementary Figure S1, there were 3585 unigenes with sequence size > 3000 nt.
To annotate the unigenes of J3, blastx alignment against the protein and nucleotide databases of NR, NT, Swiss-Prot, KEGG COG and GO was performed; the results are shown in Supplementary Table S3. The CDSs that mapped to the protein database and were predicted by ESTscan numbered 30,615 and 1041, respectively. A total of 31,279 known unigenes were identified by blastx, among which 14,912 genes were annotated by COG classification. There were 25 classes in the COG classification with the largest number of unigenes being found solely in the class of “general function prediction” (15.1%; Supplementary Figure S2). In addition, 19,468 unigenes were also annotated by the GO database, which accounted for 50.1% of all the unigenes, with the annotations grouped into three categories (biological process; cellular component; and molecular function) (Supplementary Figure S3). “Metabolic processes,” “cell” and “catalytic activities” were dominant in the categories of biological processes, cellular components, and molecular functions, respectively.
Overall, 30,793 genes (e-value < 10-5) were annotated using the NR database (Supplementary Table S3), which is far higher than those by other databases; the composition of active bacterial and fungal taxa in J3 is presented in Table 1. Based on their gene numbers, the active fungal community was more prevalent than the bacterial community and accounted for 97.7% in J3. In the fungal component, Aspergillus and Penicillium were the pivotal genera with high relative abundances of 53.2 and 29.2%, respectively. In addition, the active yeast showed low relative abundances of 0.2% at this high temperature stage.
As shown in Figure 2, starch and sucrose metabolism had the highest number of unigenes in J3, and except for the citrate cycle (TCA cycle) and oxidative phosphorylation, all of the 30 most abundant KEGG pathways showed higher numbers of unigenes in J3 than those in the high temperature stage (N3) of NF daqu. Moreover, large differences were found between J3 and N3 in basic metabolisms (i.e., purine metabolism, RNA degradation, RNA transport, meiosis-yeast, MAPK signal pathway-yeast, cell cycle-yeast, spliceosome, and mRNA surveillance pathway), degradation of aromatic compounds (aminobenzoate degradation, naphthalene degradation, benzoate degradation and bisphenol degradation), starch and sucrose metabolism, and amino sugar and nucleotide sugar metabolism. Conversely, comparable numbers of unigenes were found between J3 and N3 in oxidative phosphorylation, glycolysis/gluconeogenesis, butanoate metabolism, and pyruvate metabolism. Additionally, metabolism of amino acids; i.e., tyrosine, glycine, serine, threonine, arginine, and proline, were also ranked in the top 30 of both J3 and N3.
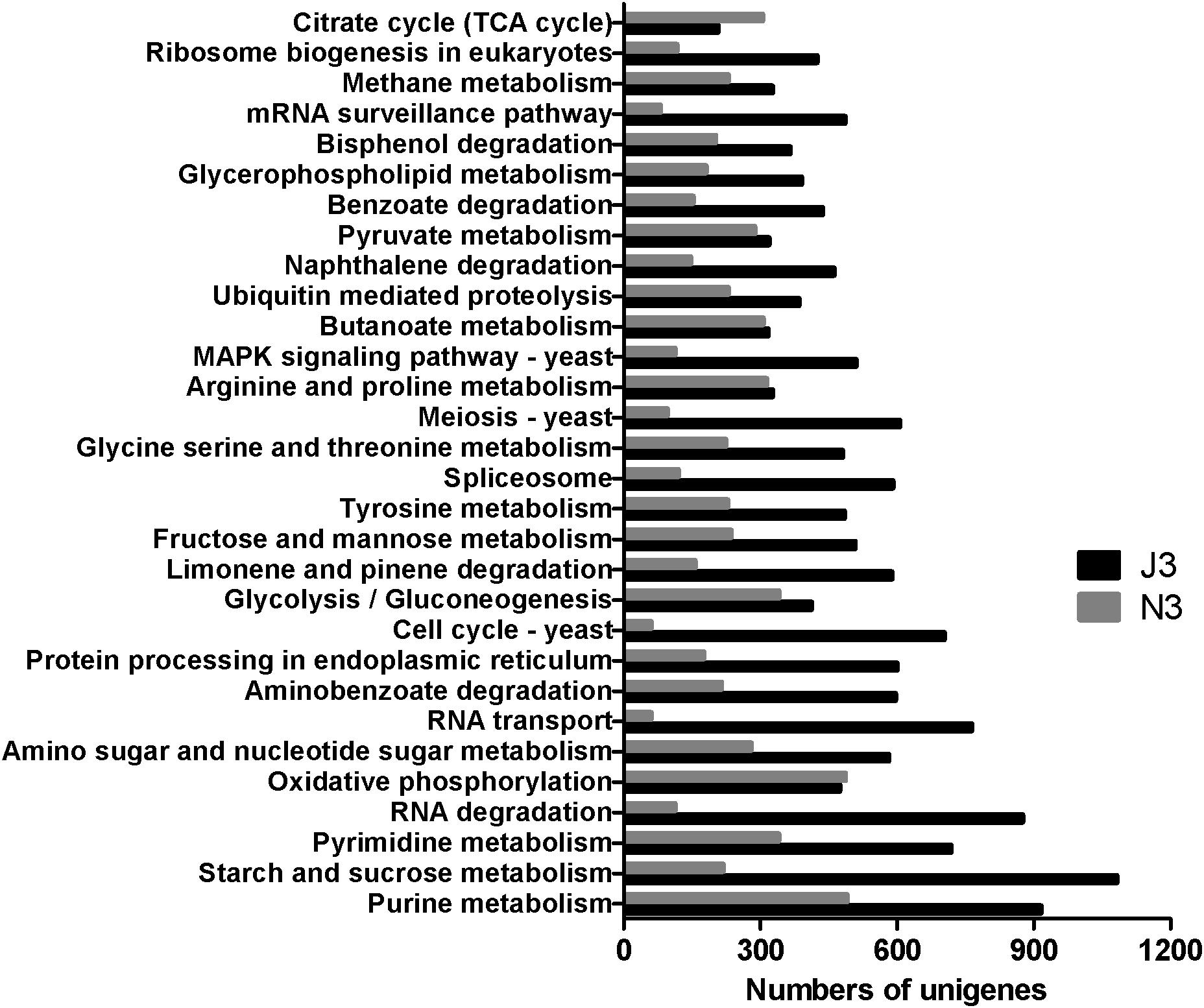
Figure 2. The 30 most abundant KEGG pathways in high temperature stage samples of J3 and N3. J3 was harvested after 8 days of JF daqu preparation and N3 was harvested after 9 days of NF daqu preparation. The temperatures of J3 and N3 were 70 and 62°C, respectively.
Screening for genes encoding putative CAZymes from the metatranscriptomic library of J3 identified glycoside hydrolases (GH) (119) and glycosyl transferases (GT) (91) as having higher numbers of unigenes than carbohydrate esterases (CE) (13) and carbohydrate-binding modules (CBM) (7) (Figure 3A). Additionally, the total number (230) of these CAZymes in J3 was lower than that (932) in the N3 sample of NF daqu (Huang et al., 2017a). The expression level for all CAZymes in J3 showed a total RPKM value of 460.8, which was markedly lower than that of 20789.9 in N3 (Supplementary Table S4). The CAZyme classes with relatively high expression levels in J3 comprised GH15 (28%), GH1 (22%), GH18 (15%), GT2 (10%), GH28 (6%), GT20 (6%), and GH79 (4%), which totally differed from those in N3 (Figure 3B and Supplementary Table S4). These major GH families may have activities of glucoamylase (GH15), beta-glucosidase (GH1), chitinase (GH18), cellulose synthase and chitin synthase (GT2), polygalacturonase (GH28), alpha, alpha-trehalose phosphate synthase (GT20), and β-glucuronidase (GH79).
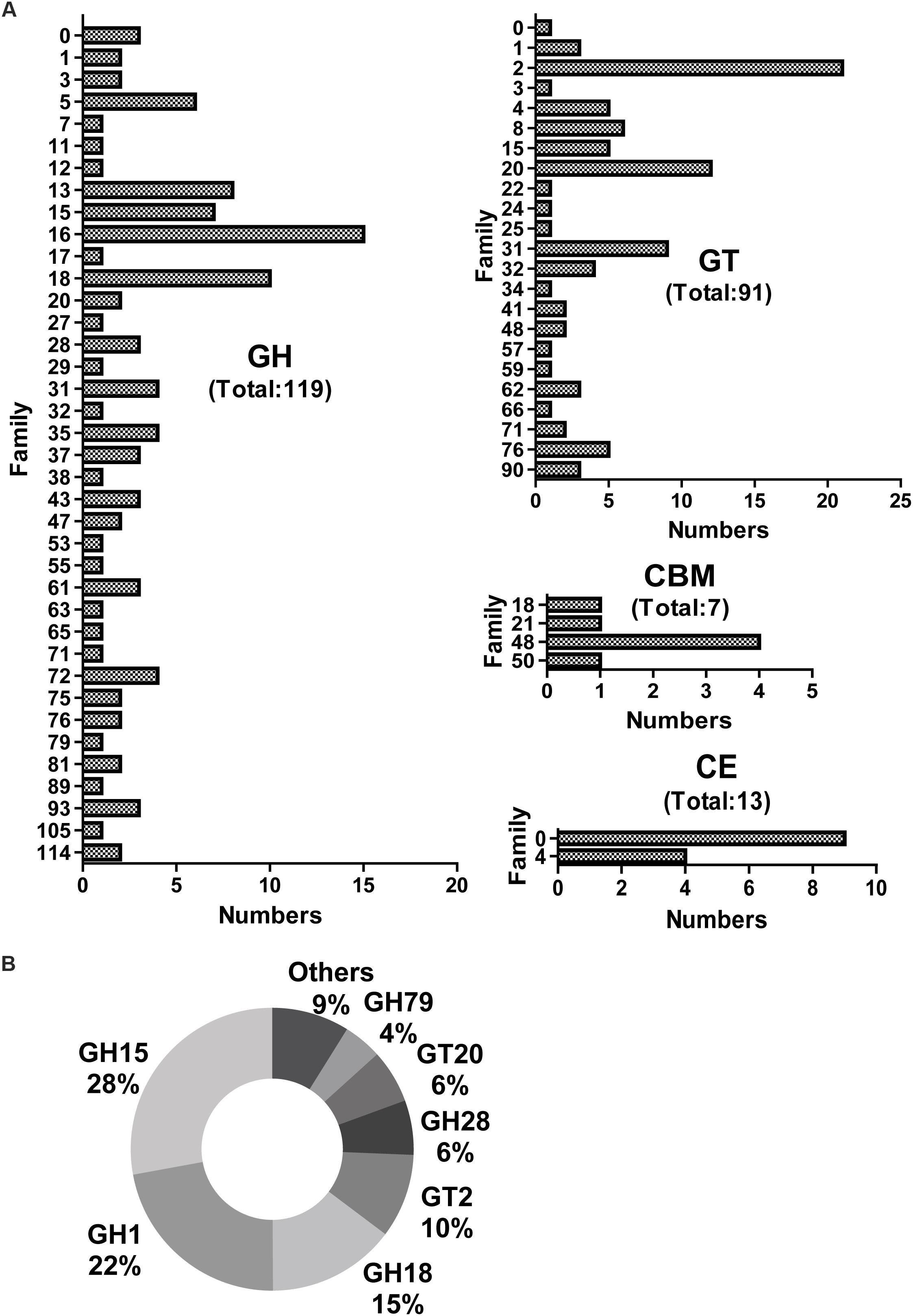
Figure 3. Numbers and expression levels of carbohydrate-active enzymes in J3. Matched unigenes of carbohydrate-active enzymes are shown with numbers (A) and expression levels (B). GH, glycoside hydrolase; GT, glycosyl transferase; CBM, carbohydrate-binding module; and CE, carbohydrate esterase.
Moreover, insoluble chromogenic AZCL polysaccharide assays at different reaction temperatures (35, 45, and 55°C), clearly detected only endo-β-1,3-1,4-glucanase, endo-1,4- β -D-galactanase, and rhamnogalacturonanase as exhibiting activity at the high temperature stage of J3 (Table 2), which to some extent was complementary to the metatranscriptic results. In comparison, a broad spectrum of CAZymes was detected in the initial sage (J1) and mature stage (J4), and four CAZymes were also found in the high temperature stage of J2. Notably, only one CAZyme of α-amylase with thermophilic activity was obviously present, showing higher activity at higher temperatures in J1, whereas more CAZymes were clearly found with thermophilic activities in J2, J3, and J4, such as α-amylase, endo-β-1,3-1,4-glucanase, endo-proteases, and endo-1,4- β -D-xylanase.
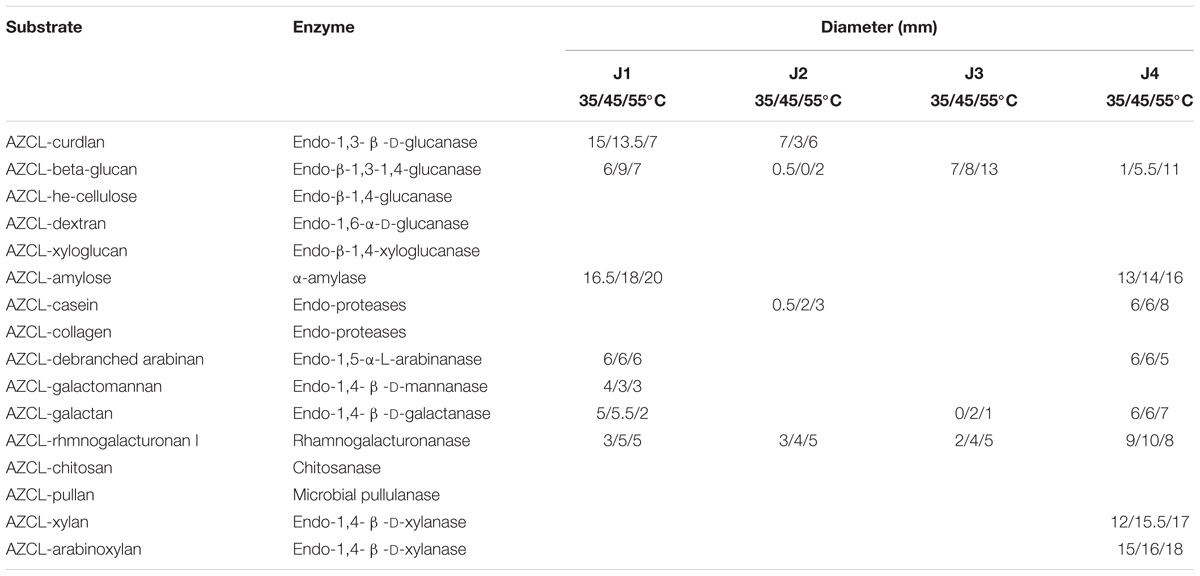
Table 2. Carbohydrate-active enzyme analysis of Jiang-flavor daqu.
The DEGs between the J3 and NF daqu samples were identified and a heatmap of hierarchical clustering of DEGs was constructed using log2(RPKM ratio) to visualize the respective patterns of DEGs. As shown in Figure 4, numerous DEGs (union) in J3, N2, N3, and N4 were clearly up-regulated with high log2(rations) values when compared with N1; thus, J3 together with N2–4 exhibit the largest differences in DEGs compared with N1. Alternatively, J3 presented the smallest differences in DEGs with N3. Similar results among J3 and NF daqu samples were also observed by analysis of hierarchical clustering of inter DEGs (Supplementary Figure S4). Furthermore, a comprehensive comparison performed between J3 and N3 identified a total of 14,149 unigenes as significant DEGs including 506 up- and 13,642 down-regulated genes (Supplementary Figure S5). In addition, for the GO functional classification (J3/N3), numerous DEGs were grouped into four dominant categories: “cellular processes,” “metabolic processes,” “binding,” and “catalytic activities” (Supplementary Figure S6).
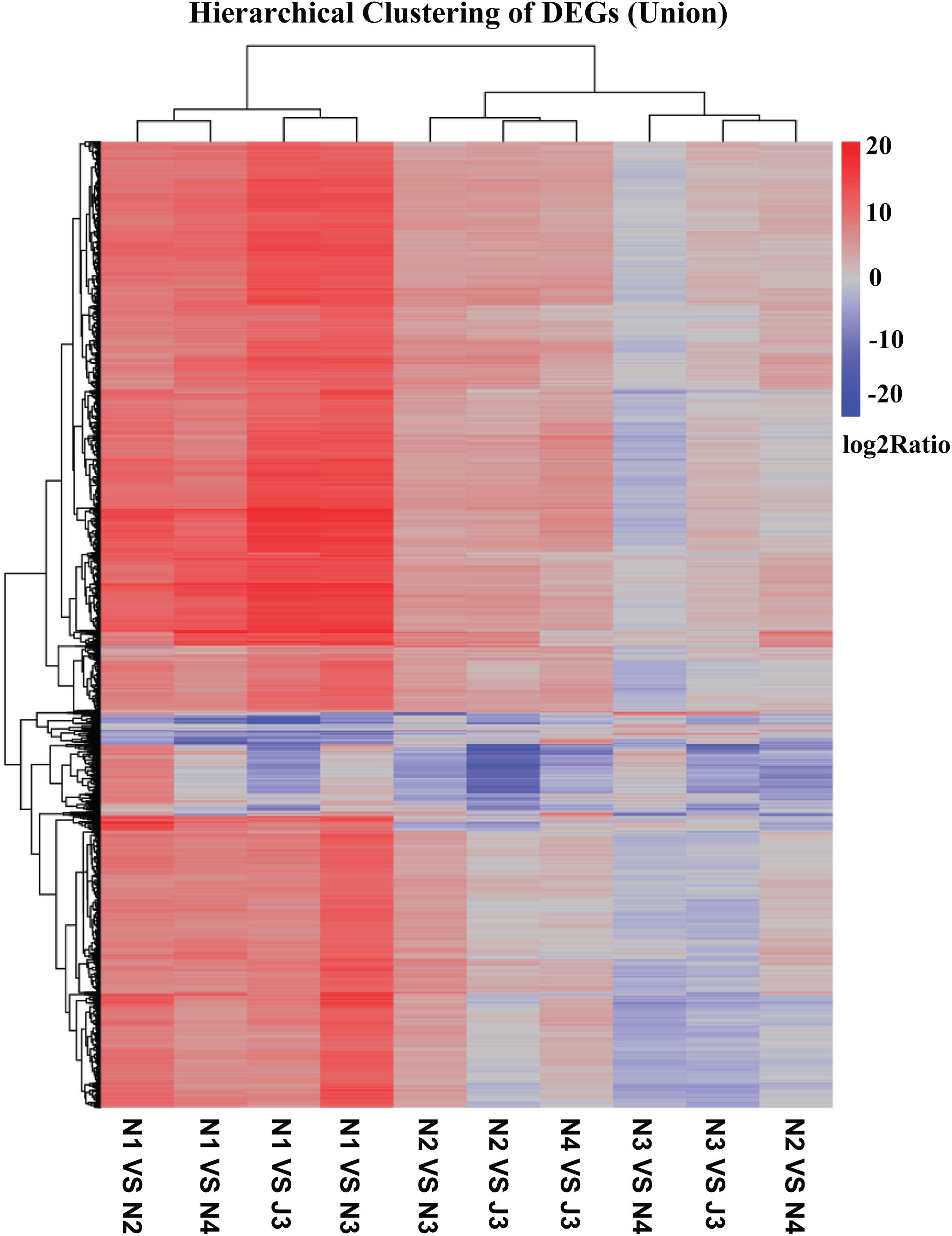
Figure 4. Hierarchical clustering of differentially expressed genes (DEGs) (Union) among J3 and NF samples (N1, N2, N3, and N4). DEGs between two samples were identified using p-value ≤ 0.05, Log2(RPKM ratio) ≥ 1, and false discovery rate (FDR) value ≤ 0.001.
For further functional comparison of DEGs between J3 and N3, metabolic pathways were analyzed based on the KEGG database. Moreover, several key carbohydrate and energy metabolisms that were associated with relatively high numbers of unigenes were selected for comparative analysis including starch and sucrose metabolism, glycolysis, pyruvate metabolism, and citrate cycle pathways. As shown in Figure 5, enzymes related to these selected pathways were mainly present, and a complete metabolic process of converting polymers into end-products was apparent in both J3 and N3. In addition, the majority of enzymes in these four key pathways exhibited lower expression levels in J3 than in N3, with the exception of e.g., aldehyde reductase (1.1.1.21), polygalacturonase (3.2.1.15), beta-fructofuranosidase (3.2.1.26), 4-alpha-glucanotransferase (2.4.1.25), 1,4-beta-cellobiosidase (3.2.1.91), and 2,3-bisphosphoglycerate-independent phosphoglycerate mutase (5.4.2.12) (Figure 5 and Supplementary Tables S5–S8).
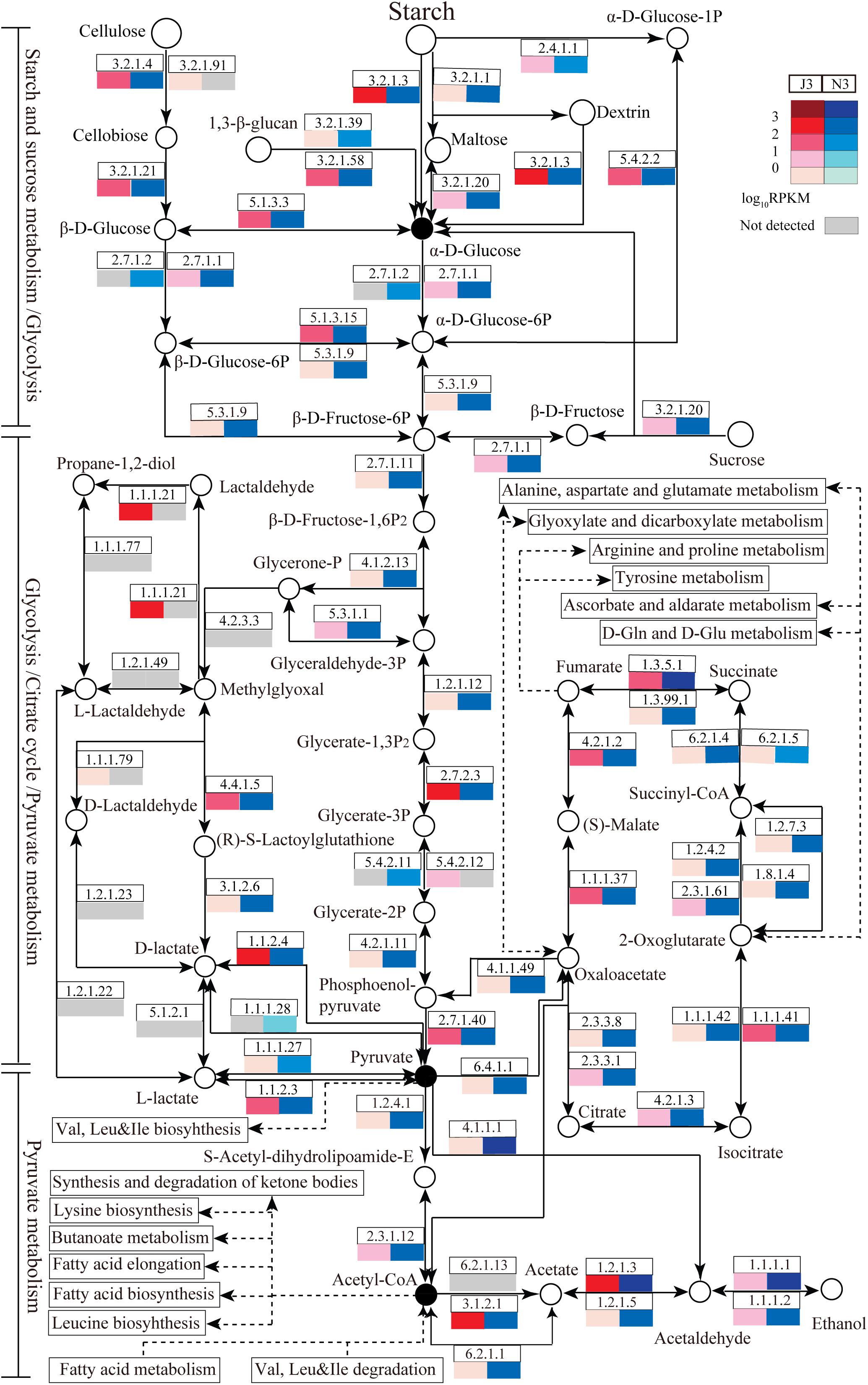
Figure 5. Relative abundances of enzymes related to carbohydrate and energy metabolisms in J3 and N3. Four abundant carbohydrate and energy metabolisms were analyzed including starch and sucrose metabolism, glycolysis, pyruvate metabolism, and the citrate cycle. In these pathways, enzymes with relatively high expression levels are partly presented by EC number and total RPKM. Relative expression [log10RPKM)] is shown in red for J3 and blue for N3. The key products are highlighted with black closed circles.
According to KEGG annotation, in starch and sucrose metabolism, glucoamylase (3.2.1.3), glucan 1,3-beta-glucosidase (3.2.1.58), endoglucanase (3.2.1.4), and beta-glucosidase (3.2.1.21) showed relatively high expression levels with RPKM values of 172.9, 70.1, 19.0, and 13.7, respectively, in J3; these are responsible for degrading polymers of starch, dextrin, 1,3-beta-glucan, and cellulose into glucose (Figure 5 and Supplementary Table S5). Moreover, phosphoglucomutase (5.4.2.2), which efficiently collaborates with starch phosphorylase (2.4.1.1) in converting starch into glucose-6P, also exhibited relatively high expression abundance with an RPKM value of 83.6 in J3. Additionally, most types of these enzymes were highly expressed by fungal species; e.g., Rasamsonia emersonii, Aspergillus fumigatus, Coccidioides immitis, and Aspergillus oryzae (Table 3). In comparison, some enzymes showed very low expression levels in J3, such as xylan 1,4-beta-xylosidase, alpha-amylase, and alpha-glucosidase, which to some extent was consistent with their low activities in AZCL polysaccharides assays of J3 (Table 2 and Supplementary Table S5).
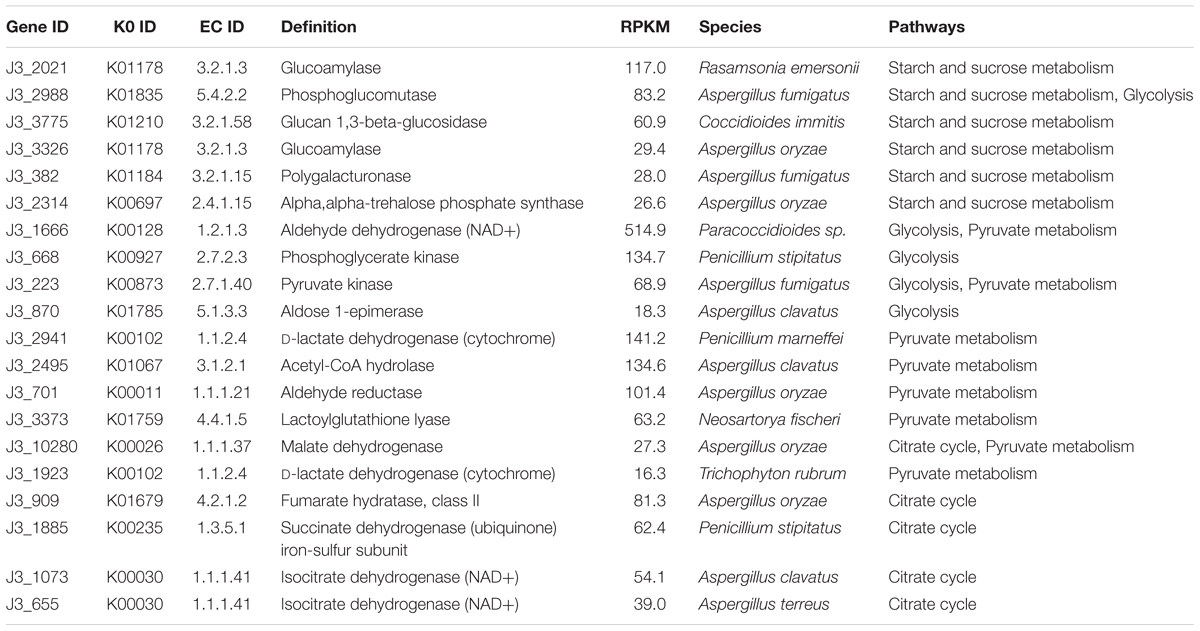
Table 3. The top 20 expressed enzymes in starch and sucrose metabolism, glycolysis, pyruvate metabolism, and the citrate cycle in J3.
In glycolysis and pyruvate metabolisms, J3 contained an integral serial of enzymes for converting glucose into the important product, pyruvate, which would then be reversibly converted to acetyl-coA under aerobic conditions (Figure 5). Hexokinase (2.7.1.1), 6-phosphofructokinase (2.7.1.11), and pyruvate kinase represent three key enzymes in glycolysis, although only pyruvate kinase (2.7.1.40) showed relatively high expression abundance (RPKM value of 75.5) in J3, which could irreversibly produce pyruvate from phosphenol-pyruvate (Figure 5 and Supplementary Tables S6, S7). Alternatively, pyruvate dehydrogenase E1 (1.2.4.1), pyruvate dehydrogenase E2 (2.3.1.12), and acetyl-CoA synthase (6.2.1.1) are responsible for producing acetyl-CoA from pyruvate and acetate, respectively; all of these showed relatively low RPKM values in J3. Moreover, aldehyde dehydrogenase (NAD+) (1.2.1.3), which reversibly produced acetate from acetaldehyde, had the highest relative expression level with an RPKM value of 515.0, and acetyl-CoA hydrolase (3.1.2.1), which irreversibly produced acetate from acetyl-CoA, was also highly expressed in glycolysis and pyruvate metabolisms of J3 (Figure 5 and Supplementary Tables S6, S7). Thus, acetaldehyde could be then converted to ethanol under anaerobic conditions by alcohol dehydrogenases (1.1.1.1 and 1.1.1.2) (Figure 5 and Supplementary Table S6), which were expressed at low levels by fungal species of A. fumigatus, C. immitis, Aspergillus terreus, Coccidioides posadasii, Penicillium marneffei, and Neosartorya fischeri in J3 (data not shown). Therefore, high concentration of acetate and low concentration of ethanol could be accumulated by collaborations of aldehyde dehydrogenase (NAD+), acetyl-CoA hydrolase, and alcohol dehydrogenases. In addition, aldehyde reductase (1.1.1.21), phosphoglycerate kinase (2.7.2.3), and lactoylglutathione lyase (4.4.1.5), which could produce lactaldehyde, phosphoglycerate, glucose, and lactoylglutathione, respectively, also showed relatively high expression levels and made large contributions in glycolysis and pyruvate metabolisms of J3. Highly expressed types of these enzymes also originated from fungal species, such as Paracoccidioides sp., Penicillium stipitatus, A. fumigatus, Aspergillus clavatus, Aspergillus oryzae, and N. fischeri (Table 3). Considering that pyruvate and acetyl-coA serve as important intermediates for Val, Leu, and Ile biosynthesis, fatty acid biosynthesis, butanoate metabolism, leucine biosynthesis, and the synthesis and degradation of ketone bodies (Figure 5), the J3 samples thus showed capacities for converting glucose to pivotal intermediates of pyruvate and acetyl-coA for fatty acids, amino acids, and carbohydrates, which would further make large contributions for generating specific flavor in JF daqu.
In conditions of insufficient oxygen, pyruvate can be reversibly converted to lactate by L-lactate dehydrogenase (1.1.1.27), D-lactate dehydrogenase (1.1.1.28), D-lactate dehydrogenase (cytochrome) (1.1.2.4), or L-lactate dehydrogenase (cytochrome) (1.1.2.3); in particular, D-lactate dehydrogenase (cytochrome) showed relatively high expression abundance with an RPKM value of 162.1 in J3 (Figure 5 and Supplementary Table S7). Members of D-lactate dehydrogenase (cytochrome) were specifically highly expressed by P. marneffei and Trichophyton rubrum (Table 3).
Almost all of the enzymes of the citrate cycle were present in the J3 sample (Figure 5). Among these, isocitrate dehydrogenase (NAD+) (1.1.1.41), fumarate hydratase (4.2.1.2), succinate dehydrogenase (1.3.5.1), and malate dehydrogenase (1.1.1.37) showed relatively high expression abundances with RPKM values of 94.1, 81.3, 63.0, and 37.8, respectively (Supplementary Table S8). Highly expressed members of these enzymes were mostly derived from fungal species, such as A. oryzae, T. stipitatus, A. clavatus, and A. terreus (Table 3). In addition, 2-oxoglutarate dehydrogenase E2 (2.3.1.61), citrate synthase (2.3.3.1), and aconitate hydratase 1 (4.2.1.3) were also clearly detected. Moreover, ATP citrate (pro-S)-lyase (2.3.3.8), phosphoenolpyruvate carboxykinase (ATP) (4.1.1.49), and pyruvate carboxylase (6.4.1.1), which are key enzymes that connect the citrate cycle and pyruvate metabolism, were still detected in J3. As shown in Figure 5, oxaloacetate, fumarate, and 2-oxoglutarate are pivotal intermediates for alanine, aspartate, and glutamate metabolism, arginine and proline metabolism, tyrosine metabolism, and D-Gln and D-Glu metabolism, which would then also contribute to specific flavor generation in JF daqu.
As large differences of unigene numbers were found between J3 and N3 with regard to the degradation of aromatic compounds (Figure 1), this study also focused on DEGs related to the degradation of aromatic compounds; i.e., aminobenzoate degradation (ABD), benzoate degradation (BD), bisphenol degradation (BPD), chlorobenzene degradation (CBD), ethylbenzene degradation (EBD), fluorobenzoate degradation (FBD), naphthalene degradation (ND), polycyclic aromatic hydrocarbon degradation (PD), styrene degradation (SD), toluene degradation (TD), and xylene degradation (XD) (Figure 6 and Supplementary Table S9). Notably, pathways in degradation of aromatic compounds are unnecessary for microbes, and many enzymes have not previously been identified in these pathways. As shown in Figure 6, among these pathways, benzoate degradation (BD) serves as the key pathway to connect most of other pathways via its pivotal products, benzoate, benzoyl-coA, catechol, and maleylacetate. A portion of enzymes related to these pathways was detected in J3 and N3, many of which showed lower expression levels in J3 than in N3. In particular, large numbers of enzymes were detected in J3 that were not identified in N3. Moreover, some of these, i.e., 2-deoxy-D-gluconate 3-dehydrogenase (1.1.1.125), 2-hydroxychromene-2-carboxylate isomerase (5.99.1.4), benzaldehyde dehydrogenase (NAD) (1.2.1.28), D-3-phosphoglycerate dehydrogenase (1.1.1.95), NADPH2:quinone reductase (1.6.5.5), L-iditol 2-dehydrogenase (1.1.1.14), 5-carboxymethyl-2-hydroxymuconate isomerase (5.3.3.10), and aldehyde reductase (1.1.1.21) clearly showed relatively high transcript abundances, with RPKM values ranging from 9.8 to 102.0, in pathways of BD, ND, BPD, ABD, EBD, XD, and TD, respectively (Supplementary Table S9 and Figure 6). Highly expressed members of these enzymes in J3 mostly originated from fungal species, e.g., N. fischeri, A. clavatus, Ajellomyces dermatitidis, Marssonina brunnea, Trichophyton equinum, Botryotinia fuckeliana, and A. oryzae (Table 4). Therefore, it was considered reasonable to postulate that trace aromatic derivatives would be differently produced between J3 and N3, some of which might be only produced in J3. In addition, both J3 and N3 could degrade aromatic compounds into important end-products, such as acetyl-coA, fumarate, acetoacetate, succinate, and glycolate, which serve as intermediates for the citrate cycle, propanoate metabolism, and glycoxylate and dicarboxylate metabolism (Figure 6).
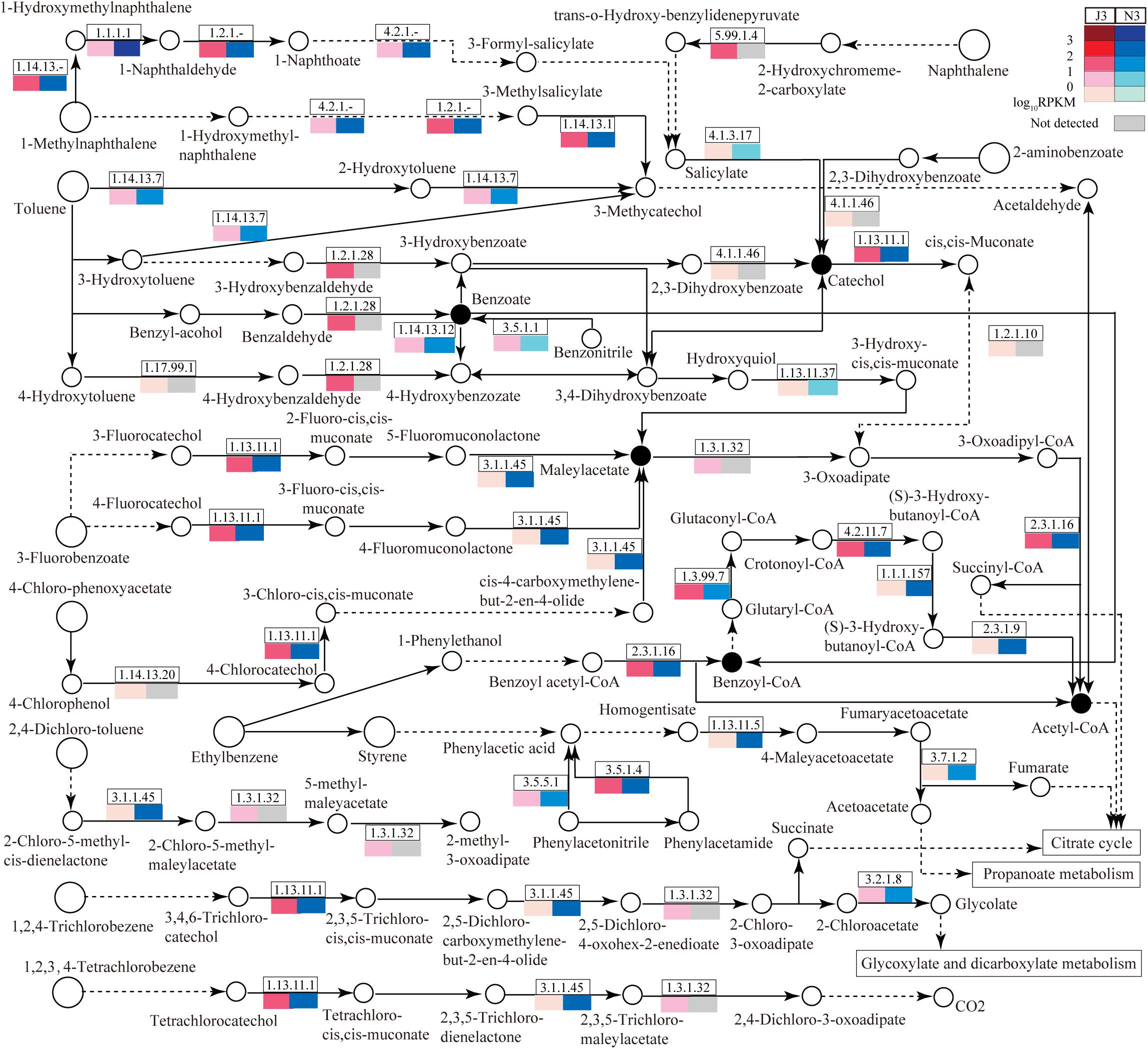
Figure 6. Relative abundances of enzymes related to the degradation of aromatic compounds in J3 and N3. A total of 11 abundant pathways associated with the degradation of aromatic compounds were analyzed: aminobenzoate degradation, benzoate degradation, fluorobenzoate degradation, chlorobenzene degradation, ethylbenzene degradation, naphthalene degradation, bisphenol degradation, styrene degradation, xylene degradation, polycyclic aromatic hydrocarbon degradation, and toluene degradation. In these pathways, only the portion of enzymes with relatively high expression levels is presented by EC number and total RPKM. Relative expression [log10RPKM)] is shown in red for J3 and blue for N3. The key products are highlighted with black closed circles.

Table 4. The top 20 expressed enzymes for the degradation of aromatic compounds in J3.
The daqus of Chinese JF and NF liquor, the most consumed liquors in China, undergo markedly different production processes that make large contributions to their special flavors. To ascertain the underlying factors, in comparison with our previous work of NF daqu, the present study comprehensively revealed the active microbial community and enzymes at the high temperature stage (J3) of JF daqu, and comparatively analyzed the active enzyme profiles at high temperature stages of JF and NF daqus. The active fungal community produced more diverse enzymes than those of the bacterial community, with Aspergillus and Penicillium representing the dominant genera at J3. This finding was complementary to the previous microbial diversity revealed for JF daqu by 16S rRNA and ITS sequencing, which indicated that the bacterial community was more diverse than the fungal community at J3 (Huang et al., 2017b). Meanwhile, low abundances of active yeast might be due to high temperature condition at J3, which may be well consistent with previous finding that yeast decreased quickly from J2 (55°C) to J3 (70°C) (Huang et al., 2017b). Additionally, the prevailing role of the active fungal community was also revealed in NF daqu samples by metatranscriptomics analysis (Huang et al., 2017a). Therefore, the present study further confirmed the suitability of metatranscriptomics for obtaining the active microbial community profiles in daqus.
JF daqu exhibited lower numbers and expression levels of CAZymes at the high temperature stage of J3 than those at the high temperature stage of N3 (Huang et al., 2017a). In addition, except for the initial stage of J1, most CAZymes were detected with lower activities and less diversities in the production process of JF daqu samples than those of NF daqu samples (Table 2) (Huang et al., 2017a). Lower activities and diversities of amylases (one kind of CAZymes) were similarly found in JF than in NF daqu via activities assay and protein electrophoresis (Liu et al., 2018). Thus, these findings might to some extent be consistent with the lower capacities of saccharification and liquefaction in JF daqu than those in NF daqu (Liu and Sun, 2018; Liu et al., 2018). Additionally, several thermostable CAZymes were detected in special stages of JF daqu samples, such as α-amylase in J1 and J4, endo-β-1,3-1,4-glucanase in J3 and J4, endo-proteases in J2 and J4, and endo-1,4- β -D-xylanase in J4, which suggests the feasibility of mining thermostable enzymes from special stages in the future.
Based on the functional annotation, starch and sucrose metabolism was the most abundant pathway in J3, which might imply that the microbial community has full capacity for degrading different polymers into glucose in J3. Upon comprehensive comparison between J3 and N3, most pathways showed higher diversities with more unigenes in J3 than in N3, which indicated more complicated metabolism for the microbial community in J3. Moreover, large differences of diversities were observed in basic metabolisms, degradation of aromatic compounds, starch and sucrose metabolism, and amino sugar and nucleotide sugar metabolism between J3 and N3, which further suggested that the microbial community of J3 might produce higher diversities of metabolites, some of which, such as phenol, benzaldehyde, and phenylethanol, might serve as precursors for aroma compounds (Fan et al., 2012; Wang et al., 2014; Xiao et al., 2016). In contrast, similar diversities were found in oxidative phosphorylation and glycolysis between J3 and N3, which indicated that the microbial communities released considerable bio-heat to maintain the high temperatures of 62 and 70°C for several days in NF and JF daqu, respectively (Huang et al., 2017b). In addition, butanoate metabolism and pyruvate metabolism were also similarly active between J3 and N3, suggesting that their intermediates, such as butanoate and acetate, represented important substrates for flavor compounds of e.g., butanol, acetic acid, butanoic acid, ethyl hexanoate, hexyl acetate, and isopentyl butanoate in JF and NF liquor (Fan et al., 2012; Wang et al., 2014; Xiao et al., 2016). Furthermore, six amino acid metabolisms were dominant in both J3 and N3, the products of which, i.e., amino acids, a-keto acids, and aldehydes, may serve as pre-substrates for important flavor precursors such as pyrazine, alcohol, and acids (Badrinarayanan and Sperry, 2012; Nashalian and Yaylayan, 2014; Scalone et al., 2015; Xu et al., 2017). Therefore, the microbial community was both active at the high temperature stages of JF and NF daqu for generating bio-heat (Huang et al., 2017a,b; Xiao et al., 2017) and releasing flavor precursors (Wu et al., 2009; Zheng et al., 2011), and JF daqu could provide larger diversities of flavor precursors than NF daqu from most of the active pathways, in particular from the degradation of aromatic compounds.
Similar DEG profiles were observed between the high temperature stages of J3 and N3; thus, detailed functional comparisons of DEGs were performed between these stages with regard to four key carbohydrate and energy metabolisms: starch and sucrose metabolism, glycolysis, pyruvate metabolism, and the citrate cycle, as their intermediates are essential for ethanol and flavor generation. The results showed that both J3 and N3 contained an intact process for converting polymers into glucose, pyruvate, acetyl-coA, and ethanol, indicating a complete system for saccharification, liquefaction, and fermentation. In general, the majority of enzymes related to these four key pathways showed lower expression levels in J3 than in N3, indicating lower activities for enzymes in J3 than in N3 to a degree that is consistent with the lower capacities in saccharification, liquefaction, and fermentation exhibited by high-temperature JF daqu than those by medium-temperature NF daqu (Liu and Sun, 2018; Liu et al., 2018). Low expression levels of enzymes might result from the inhibition caused by the high temperature (70°C) in J3. However, some enzymes were only detected in J3, albeit with relative low expression levels, indicating that a large number of minor intermediates would likely be specifically generated in J3. Notably, among enzymes related to saccharification and liquefaction in J3, glucoamylases were clearly active with high expression levels, indicating their collaborative roles along with high temperature in degrading starches, which would be spontaneously decomposed under high temperature, as well as suggesting a feasible way to mine thermostable glucoamylases from J3. The majority of enzymes related to saccharification and liquefaction in J3 were highly expressed by fungal species of R. emersonii, A. oryzae, A. fumigatus, and C. immitis, some of which have been found to secrete numerous carbohydrate-active enzymes and show high capacities toward degrading polymers, such as Aspergillus (Culleton et al., 2013; de Vries et al., 2017; Cologna et al., 2018) and R. emersonii (Hua et al., 2014; Martínez et al., 2016). In addition, J3 showed considerable potential for converting glucose to pivotal intermediates, such as acetate, ethanol, pyruvate, and acetyl-coA, which might then serve as direct or indirect substrates for JF flavor compounds including ethyl acetate, ethyl butanoate, ethyl propanoate, ethyl 2-hydroxypropanoate, ethyl 2-hydroxyhexanoate, acetic acid, 2-acetylpyridine, hexyl acetate, benzyl acetate ethyl, ethyl 3-methylbutanoate, ethyl benzeneacetate, and 3-methylbutyl acetate (Fan et al., 2012; Wang et al., 2014; Xiao et al., 2016; Gao et al., 2017). The highly expressed enzymes related to glycolysis and pyruvate metabolism were mostly derived from fungal species, some of which have been applied to the production of fermented foods and drugs, such as A. fumigatus (Qin et al., 2012; Wakefield et al., 2017), A. clavatus (Mo et al., 2008; Zutz et al., 2013; Li et al., 2017), and A. oryzae (Park et al., 2018; Son et al., 2018; Zhong et al., 2018). Furthermore, low concentration of ethanol might be generated by several fungi in J3, which to some extent agreed with the earlier finding that a small amount of ethanol could be directly produced by co-culture of fungi (Takano and Hoshino, 2012). Additionally, relatively high expression levels of D-lactate dehydrogenase (cytochrome) might indicate high concentration of lactate in J3, which may be consistent with the high level of lactate in the subsequent mature JF daqu (Wu et al., 2009). Moreover, intermediates of the citrate cycle also serve as pre-substrates for flavor compounds, and the highly expressed enzymes related to this pathway also originated from the fungal community, some of which have been applied to the saccharification and fermentation process of foods and drugs, including A. oryzae, A. clavatus, and A. terreus.
Numerous aromatic compounds, such as tannin, ferulic acid, and lignin have been identified in the materials of cereals, the degradation of which is strongly related to liquor flavor generation (Liu and Sun, 2018). Several laccases, feruloyl esterase and ferulic acid decarboxylase were detected with low expression levels from A. clavatus, C. posadasii, P. marneffei, A. terreus or Pseudomonas aeruginosa in J3 (data not shown), which might clearly confirm the degradations of ferulic acid and lignin during high temperature stage of JF daqu. Similarly, many aromatic compounds and phenols were identified in both JF and NF liquors (Wang et al., 2014; Xiao et al., 2016); consistent with this, in the present study some enzymes related to the degradation of aromatic compounds were also found to be expressed in JF and NF liquor starters, with most showing lower expression in the former. However, the remainder constituted those enzymes that were only detected (at low levels) in J3, indicating that trace aroma compounds were likely particularly associated with JF liquor flavor, such as ethyl benzeneacetate and benzaldehyde (Xiao et al., 2016). Highly expressed members of these enzymes were mostly derived from fungal species in J3, which appears consistent with the contributions of some fungi toward the degradation of aromatic compounds (Godoy et al., 2016; Sun et al., 2016; Vieira et al., 2018). Alternatively, enzymes expressed at low levels from bacteria may also substantively contribute to degradation of aromatic compounds (Pérez-Pantoja et al., 2015; Van der Waals et al., 2017). Therefore, both the fungal and bacterial communities appear to have an active role in degrading aromatic compounds in JF daqu (Bhattacharya et al., 2017; Wang et al., 2017; Kamyabi et al., 2018; Ma et al., 2018), especially in the high temperature and mature stage (Huang et al., 2017b).
In addition to the microbial community, temperature also makes large contributions to generate flavor compounds in JF daqu, such as pyrazines and their derivatives, which comprise pivotal impact aroma compounds of JF liquor (Zhu et al., 2007; Fan et al., 2012). In particular, their generation may be thermally induced from microbial metabolites by non-enzymatic browning via the Maillard reaction at 70°C in J3 (Richards et al., 2011; Nashalian and Yaylayan, 2014). Overall, JF liquor flavor thus appears to be determined by a highly complicated process and further analysis of the active microbial community, enzymes, and metabolites from the daqu preparation in addition to stacking fermentation and alcoholic fermentation processes are required to unravel the mystery of JF liquor flavor generation.
In the present study, fungi including Aspergillus and Penicillium, were identified as the most active microbial community members at the high temperature stage (J3: 70°C) of JF daqu by metatranscriptomics. Furthermore, the high temperature stage was found to not only lower the capacities of JF daqu toward saccharification and fermentation, but also enhance its ability in generating diverse minor flavor compounds, e.g., derivatives of aromatic compounds. Additionally, most of enzymes related to those capacities were highly expressed at 70°C by fungal genus of Aspergillus, Coccidioides, Paracoccidioides, Penicillium, and Rasamsonia. These exploratory findings shed light on our understanding of the JF baijiu fermentation system, in which the high temperature stage plays key roles in improving JF daqu by providing unique active microbiota and enzymes, and strongly contributing to the final distinctive aroma and taste of JF baijiu.
HZ, ZY, YF, YJ, LT, and KH designed the experiment. ZY performed the experiments and analyzed the data. ZY, YX, DL, and HL collected samples and communicated with the liquor factory. ZY and LC wrote the main manuscript. ZY, AD, YF, and HZ revised the manuscript. All authors revised and approved the final version of the manuscript.
This study was supported by the National Key Technology R&D Program of China (2015BAD15B01); the China Agriculture Research System (CARS-10-B22) the National Natural Science for Youth Foundation of China (21606218) the National Natural Science for General Foundation of China (31770395) the Key Deployment Project of the Chinese Academy of Sciences (ZDRW-ZS-2017-2-1) the Science and Technology Service Network Initiative (KFJ-STS-ZDTP-008); the Science & Technology Program of Sichuan Province (2017NZ0018 and 2017HH0077) and the Key Laboratory of Environmental and Applied Microbiology, Chengdu Institute of Biology, Chinese Academy of Sciences (KLCAS-2016-02 and KLCAS-2016-06).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Prof. Zhongyan Wang for providing valuable suggestions concerning daqu research.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00472/full#supplementary-material
Abu-Ali, G. S., Mehta, R. S., Lloyd-Price, J., Mallick, H., Branck, T., Ivey, K. L., et al. (2018). Metatranscriptome of human faecal microbial communities in a cohort of adult men. Nat. Microbiol. 3, 356–366. doi: 10.1038/s41564-017-0084-4
Audic, S., and Claverie, J.-M. (1997). The significance of digital gene expression profiles. Genome Res. 7, 986–995. doi: 10.1101/gr.7.10.986
Badrinarayanan, S., and Sperry, J. (2012). Pyrazine alkaloids via dimerization of amino acid-derived α-amino aldehydes: biomimetic synthesis of 2,5-diisopropylpyrazine, 2,5-bis(3-indolylmethyl)pyrazine and actinopolymorphol C. Org. Biomol. Chem. 10, 2126–2132. doi: 10.1039/c2ob06935k
Benjamini, Y., and Yekutieli, D. (2001). The control of the false discovery rate in multiple testing under dependency. Ann. Statist. 29, 1165–1188. doi: 10.1186/1471-2105-9-114
Bhattacharya, S., Das, A., Palaniswamy, M., and Angayarkanni, J. (2017). Degradation of benzo[α]pyrene by Pleurotus ostreatus PO-3 in the presence of defined fungal and bacterial co-cultures. J. Basic Microbiol. 57, 95–103. doi: 10.1002/jobm.201600479
Bokulich, N. A., Lewis, Z. T., Boundy-Mills, K., and Mills, D. A. (2016). A new perspective on microbial landscapes within food production. Curr. Opin. Biotechnol. 37, 182–189. doi: 10.1016/j.copbio.2015.12.008
Cologna, N. M. D., Gómez-Mendoza, D. P., Zanoelo, F. F., Giannesi, G. C., Guimarães, N. C. A., Moreira, L. R. S., et al. (2018). Exploring Trichoderma and Aspergillus secretomes: proteomics approaches for the identification of enzymes of biotechnological interest. Enzyme Microb. Technol. 109, 1–10. doi: 10.1016/j.enzmictec.2017.08.007
Culleton, H., Mckie, V., and De Vries, R. P. (2013). Physiological and molecular aspects of degradation of plant polysaccharides by fungi: what have we learned from Aspergillus? Biotechnol. J. 8, 884–894. doi: 10.1002/biot.201200382
de Vries, R. P., Riley, R., Wiebenga, A., Aguilar-Osorio, G., Amillis, S., Uchima, C. A., et al. (2017). Comparative genomics reveals high biological diversity and specific adaptations in the industrially and medically important fungal genus Aspergillus. Genome Biol. 18:28. doi: 10.1186/s13059-017-1151-0
Fan, W., Xu, Y., and Qian, M. C. (2012). “Identification of aroma compounds in Chinese “moutai” and “langjiu” liquors by normal phase liquid chromatography fractionation followed by gas chromatography/olfactometry,” in Flavor Chemistry of Wine and Other Alcoholic Beverages, eds M. C. Qian, and T. H. Shellhammer (Washington, DC: American Chemical Society), 303–338.
Fibri, D. L. N., and Frøst, M. B. (2019). Consumer perception of original and modernised traditional foods of Indonesia. Appetite 133, 61–69. doi: 10.1016/j.appet.2018.10.026
Gao, L., Liu, T., An, X., Zhang, J., Ma, X., and Cui, J. (2017). Analysis of volatile flavor compounds influencing Chinese-type soy sauces using GC–MS combined with HS-SPME and discrimination with electronic nose. J. Food Sci. Technol. 54, 130–143. doi: 10.1007/s13197-016-2444-0
Godoy, P., Reina, R., Calderon, A., Wittich, R. M., Garcia-Romera, I., and Aranda, E. (2016). Exploring the potential of fungi isolated from PAH-polluted soil as a source of xenobiotics-degrading fungi. Environ. Sci. Pollut. Res. Int. 23, 20985–20996. doi: 10.1007/s11356-016-7257-1
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29, 644–652. doi: 10.1186/1471-2105-12-S14-S2
Hua, H., Luo, H., Bai, Y., Wang, K., Niu, C., Huang, H., et al. (2014). A thermostable glucoamylase from Bispora sp. MEY-1 with stability over a broad pH range and significant starch hydrolysis capacity. PLoS One 9:e113581. doi: 10.1371/journal.pone.0113581
Huang, Y., Yi, Z., Jin, Y., Huang, M., He, K., Liu, D., et al. (2017a). Metatranscriptomics reveals the functions and enzyme profiles of the microbial community in Chinese nong-flavor liquor starter. Front. Microbiol. 8:1747. doi: 10.3389/fmicb.2017.01747
Huang, Y., Yi, Z., Jin, Y., Zhao, Y., He, K., Liu, D., et al. (2017b). New microbial resource: microbial diversity, function and dynamics in Chinese liquor starter. Sci. Rep. 7:14577. doi: 10.1038/s41598-017-14968-8
Jin, G., Zhu, Y., and Xu, Y. (2017). Mystery behind Chinese liquor fermentation. Trends Food Sci. Technol. 63, 18–28. doi: 10.1016/j.tifs.2017.02.016
Just, S., Mondot, S., Ecker, J., Wegner, K., Rath, E., Gau, L., et al. (2018). The gut microbiota drives the impact of bile acids and fat source in diet on mouse metabolism. Microbiome 6:134. doi: 10.1186/s40168-018-0510-8
Kamyabi, A., Nouri, H., and Moghimi, H. (2018). Characterization of pyrene degradation and metabolite identification by Basidioascus persicus and mineralization enhancement with bacterial-yeast co-culture. Ecotoxicol. Environ. Saf. 163, 471–477. doi: 10.1016/j.ecoenv.2018.07.098
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., and Morishima, K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. doi: 10.1093/nar/gkw1092
Kim, Y. S., Kim, M. C., Kwon, S. W., Kim, S. J., Park, I. C., Ka, J. O., et al. (2011). Analyses of bacterial communities in meju, a Korean traditional fermented soybean bricks, by cultivation-based and pyrosequencing methods. J. Microbiol. 49, 340–348. doi: 10.1007/s12275-011-0302-3
Li, P., Liang, H., Lin, W.-T., Feng, F., and Luo, L. (2015). Microbiota dynamics associated with environmental conditions and potential roles of cellulolytic communities in traditional Chinese cereal starter solid-state fermentation. Appl. Environ. Microbiol. 81, 5144–5156. doi: 10.1128/AEM.01325-15
Li, R., Yu, C., Li, Y., Lam, T. W., Yiu, S. M., Kristiansen, K., et al. (2009). SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967. doi: 10.1093/bioinformatics/btp336
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Li, W., Xiong, P., Zheng, W., Zhu, X., She, Z., Ding, W., et al. (2017). Identification and antifungal activity of compounds from the mangrove endophytic fungus Aspergillus clavatus R7. Mar. Drugs 15:E259. doi: 10.3390/md15080259
Liu, H., and Sun, B. (2018). Effect of fermentation processing on the flavor of Baijiu. J. Agric. Food Chem. 66, 5425–5432. doi: 10.1021/acs.jafc.8b00692
Liu, J., Chen, J., Fan, Y., Huang, X., and Han, B. (2018). Biochemical characterisation and dominance of different hydrolases in different types of Daqu – a Chinese industrial fermentation starter. J. Sci. Food Agric. 98, 113–121. doi: 10.1002/jsfa.8445
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., and Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. doi: 10.1093/nar/gkt1178
Ma, X.-K., Li, T.-T., Fam, H., Charles Peterson, E., Zhao, W.-W., Guo, W., et al. (2018). The influence of heavy metals on the bioremediation of polycyclic aromatic hydrocarbons in aquatic system by a bacterial–fungal consortium. Environ. Technol. 39, 2128–2137. doi: 10.1080/09593330.2017.1351492
Martínez, P. M., Appeldoorn, M. M., Gruppen, H., and Kabel, M. A. (2016). The two Rasamsonia emersoniiα-glucuronidases, ReGH67 and ReGH115, show a different mode-of-action towards glucuronoxylan and glucuronoxylo-oligosaccharides. Biotechnol. Biofuels 9:105. doi: 10.1186/s13068-016-0519-9
Mello, B. L., Alessi, A. M., Riaño-Pachón, D. M., Deazevedo, E. R., Guimarães, F. E. G., Espirito Santo, M. C., et al. (2017). Targeted metatranscriptomics of compost-derived consortia reveals a GH11 exerting an unusual exo-1,4-β-xylanase activity. Biotechnol. Biofuels 10:254. doi: 10.1186/s13068-017-0944-4
Mo, H., Zhu, Y., and Chen, Z. (2008). Microbial fermented tea – a potential source of natural food preservatives. Trends Food Sci. Technol. 19, 124–130. doi: 10.1016/j.tifs.2007.10.001
Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Nashalian, O., and Yaylayan, V. A. (2014). Thermally induced oxidative decarboxylation of copper complexes of amino acids and formation of strecker aldehyde. J. Agric. Food Chem. 62, 8518–8523. doi: 10.1021/jf502751n
Pandit, R. J., Hinsu, A. T., Patel, S. H., Jakhesara, S. J., Koringa, P. G., Bruno, F., et al. (2018). Microbiota composition, gene pool and its expression in Gir cattle (Bos indicus) rumen under different forage diets using metagenomic and metatranscriptomic approaches. Syst. Appl. Microbiol. 41, 374–385. doi: 10.1016/j.syapm.2018.02.002
Park, Y., Kim, D., Yang, I., Choi, B., Lee, J. W., Namkoong, S., et al. (2018). Decursin and Z-ligustilide in Angelica tenuissima root extract fermented by Aspergillus oryzae display anti-pigment activity in melanoma cells. J. Microbiol. Biotechnol. 28, 1061–1067. doi: 10.4014/jmb.1812.02044
Pérez-Pantoja, D., Leiva-Novoa, P., Donoso, R. A., Little, C., Godoy, M., Pieper, D. H., et al. (2015). Hierarchy of carbon source utilization in soil bacteria: hegemonic preference for benzoate in complex aromatic compound mixtures degraded by Cupriavidus pinatubonensis strain JMP134. Appl. Environ. Microbiol. 81, 3914–3924. doi: 10.1128/AEM.04207-14
Qin, J. H., Li, N., Tu, P. F., Ma, Z. Z., and Zhang, L. (2012). Change in tea polyphenol and purine alkaloid composition during solid-state fungal fermentation of postfermented tea. J. Agric. Food Chem. 60, 1213–1217. doi: 10.1021/jf204844g
Richards, G. J., Hill, J. P., Labuta, J., Wakayama, Y., Akada, M., and Ariga, K. (2011). Self-assembled pyrazinacene nanotubes. Phys. Chem. Chem. Phys. 13, 4868–4876. doi: 10.1039/c0cp02025g
Scalone, G. L. L., Cucu, T., De Kimpe, N., and De Meulenaer, B. (2015). Influence of free amino acids, oligopeptides, and polypeptides on the formation of pyrazines in maillard model systems. J. Agric. Food Chem. 63, 5364–5372. doi: 10.1021/acs.jafc.5b01129
Sha, S. P., Suryavanshi, M. V., Jani, K., Sharma, A., Shouche, Y., and Tamang, J. P. (2018). Diversity of yeasts and molds by culture-dependent and culture-independent methods for mycobiome surveillance of traditionally prepared dried starters for the production of indian alcoholic beverages. Front. Microbiol. 9:2237. doi: 10.3389/fmicb.2018.02237
Son, S. Y., Lee, S., Singh, D., Lee, N.-R., Lee, D.-Y., and Lee, C. H. (2018). Comprehensive secondary metabolite profiling toward delineating the solid and submerged-state fermentation of Aspergillus oryzae KCCM 12698. Front. Microbiol. 9:1076. doi: 10.3389/fmicb.2018.01076
Song, Z., Du, H., Zhang, Y., and Xu, Y. (2017). Unraveling core functional microbiota in traditional solid-state fermentation by high-throughput amplicons and metatranscriptomics sequencing. Front. Microbiol. 8:1294. doi: 10.3389/fmicb.2017.01294
Sun, S., Xie, S., Chen, H., Cheng, Y., Shi, Y., Qin, X., et al. (2016). Genomic and molecular mechanisms for efficient biodegradation of aromatic dye. J. Hazard. Mater. 302, 286–295. doi: 10.1016/j.jhazmat.2015.09.071
Takano, M., and Hoshino, K. (2012). Direct ethanol production from rice straw by coculture with two high-performing fungi. Front. Chem. Sci. Eng. 6:139. doi: 10.1007/s11705-012-1281-6
Tamang, J. P., Watanabe, K., and Holzapfel, W. H. (2016). Review: diversity of microorganisms in global fermented foods and beverages. Front. Microbiol. 7:377. doi: 10.3389/fmicb.2016.00377
Thanh, V. N., Mai, L. T., and Tuan, D. A. (2008). Microbial diversity of traditional Vietnamese alcohol fermentation starters (banh men) as determined by PCR-mediated DGGE. Int. J. Food Microbiol. 128, 268–273. doi: 10.1016/j.ijfoodmicro.2008.08.020
Van der Waals, M. J., Atashgahi, S., Da Rocha, U. N., Van Der Zaan, B. M., Smidt, H., and Gerritse, J. (2017). Benzene degradation in a denitrifying biofilm reactor: activity and microbial community composition. Appl. Microbiol. Biotechnol. 101, 5175–5188. doi: 10.1007/s00253-017-8214-8
Vieira, G. A. L., Magrini, M. J., Bonugli-Santos, R. C., Rodrigues, M. V. N., and Sette, L. D. (2018). Polycyclic aromatic hydrocarbons degradation by marine-derived basidiomycetes: optimization of the degradation process. Braz. J. Microbiol. 49, 749–756. doi: 10.1016/j.bjm.2018.04.007
Waché, Y., Do, T.-L., Do, T.-B.-H., Do, T.-Y., Haure, M., Ho, P.-H., et al. (2018). Prospects for food fermentation in South-East Asia, topics from the tropical fermentation and biotechnology network at the end of the AsiFood Erasmus+Project. Front. Microbiol. 9:2278. doi: 10.3389/fmicb.2018.02278
Wakefield, J., Hassan, H. M., Jaspars, M., Ebel, R., and Rateb, M. E. (2017). Dual induction of new microbial secondary metabolites by fungal bacterial co-cultivation. Front. Microbiol. 8:1284. doi: 10.3389/fmicb.2017.01284
Wang, H. Y., and Xu, Y. C. (2015). Effect of temperature on microbial composition of starter culture for Chinese light aroma style liquor fermentation. Lett. Appl. Microbiol. 60, 85–91. doi: 10.1111/lam.12344
Wang, P., Wu, Q., Jiang, X., Wang, Z., Tang, J., and Xu, Y. (2017). Bacillus licheniformis affects the microbial community and metabolic profile in the spontaneous fermentation of Daqu starter for Chinese liquor making. Int. J. Food Microbiol. 250, 59–67. doi: 10.1016/j.ijfoodmicro.2017.03.010
Wang, X., Fan, W., and Xu, Y. (2014). Comparison on aroma compounds in Chinese soy sauce and strong aroma type liquors by gas chromatography–olfactometry, chemical quantitative and odor activity values analysis. Eur. Food Res. Technol. 239, 813–825. doi: 10.1007/s00217-014-2275-z
Wu, X.-H., Zheng, X.-W., Han, B.-Z., Vervoort, J., and Nout, M. J. R. (2009). Characterization of Chinese liquor starter, “daqu”, by flavor type with 1H NMR-based nontargeted analysis. J. Agric. Food Chem. 57, 11354–11359. doi: 10.1021/jf902881p
Xia, Y., Yang, C., and Zhang, T. (2018). Microbial effects of part-stream low-frequency ultrasonic pretreatment on sludge anaerobic digestion as revealed by high-throughput sequencing-based metagenomics and metatranscriptomics. Biotechnol. Biofuels 11:47. doi: 10.1186/s13068-018-1042-y
Xiao, C., Lu, Z.-M., Zhang, X.-J., Wang, S.-T., Ao, L., Shen, C.-H., et al. (2017). Bio-heat is a key environmental driver shaping the microbial community of medium-temperature Daqu. Appl. Environ. Microbiol. 83:e01550-17. doi: 10.1128/AEM.01550-17
Xiao, Z., Yu, D., Niu, Y., Ma, N., and Zhu, J. (2016). Characterization of different aroma-types of chinese liquors based on their aroma profile by gas chromatography–mass spectrometry and sensory evaluation. Flavour Fragr. J. 31, 217–227. doi: 10.1002/ffj.3304
Xu, Y., Zhi, Y., Wu, Q., and Du, R. (2017). Zygosaccharomyces bailii is a potential producer of various flavor compounds in Chinese maotai-flavor liquor fermentation. Front. Microbiol. 8:2609. doi: 10.3389/fmicb.2017.02609
Yan, Z., Zheng, X.-W., Han, B.-Z., Han, J.-S., Robert Nout, M. J., and Chen, J.-Y. (2013). Monitoring the ecology of bacillus during daqu incubation, a fermentation starter, using culture-dependent and culture-independent methods. J. Microbiol. Biotechnol. 23, 614–622. doi: 10.4014/jmb.1211.11065
Ye, J., Fang, L., Zheng, H., Zhang, Y., Chen, J., Zhang, Z., et al. (2006). WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, W293–W297. doi: 10.1093/nar/gkl031
Yoshida, T., Nishimura, Y., Watai, H., Haruki, N., Morimoto, D., Kaneko, H., et al. (2018). Locality and diel cycling of viral production revealed by a 24 h time course cross-omics analysis in a coastal region of Japan. ISME J. 12, 1287–1295. doi: 10.1038/s41396-018-0052-x
Zheng, X.-W., and Han, B.-Z. (2016). Baijiu ( ), Chinese liquor: history, classification and manufacture. J. Ethn. Foods 3, 19–25. doi: 10.1016/j.jef.2016.03.001
), Chinese liquor: history, classification and manufacture. J. Ethn. Foods 3, 19–25. doi: 10.1016/j.jef.2016.03.001
Zheng, X.-W., Tabrizi, M. R., Nout, M. J. R., and Han, B.-Z. (2011). Daqu- a traditional Chinese liquor fermentation starter. J. Inst. Brew. 117, 82–90. doi: 10.1002/j.2050-0416.2011.tb00447.x
Zhong, Y., Lu, X., Xing, L., Ho, S. W. A., and Kwan, H. S. (2018). Genomic and transcriptomic comparison of Aspergillus oryzae strains: a case study in soy sauce koji fermentation. J. Ind. Microbiol. Biotechnol. 45, 839–853. doi: 10.1007/s10295-018-2059-8
Zhu, S., Lu, X., Ji, K., Guo, K., Li, Y., Wu, C., et al. (2007). Characterization of flavor compounds in Chinese liquor moutai by comprehensive two-dimensional gas chromatography/time-of-flight mass spectrometry. Anal. Chim. Acta 597, 340–348. doi: 10.1016/j.aca.2007.07.007
Keywords: flavor generation, Chinese baijiu, metatranscriptomics, Jiang-flavor daqu, saccharification, high temperature stage, degradation of aromatic compounds
Citation: Yi Z, Jin Y, Xiao Y, Chen L, Tan L, Du A, He K, Liu D, Luo H, Fang Y and Zhao H (2019) Unraveling the Contribution of High Temperature Stage to Jiang-Flavor Daqu, a Liquor Starter for Production of Chinese Jiang-Flavor Baijiu, With Special Reference to Metatranscriptomics. Front. Microbiol. 10:472. doi: 10.3389/fmicb.2019.00472
Received: 28 September 2018; Accepted: 22 February 2019;
Published: 12 March 2019.
Edited by:
Jyoti Prakash Tamang, Sikkim University, IndiaReviewed by:
Satyanarayana Tulasi, University of Delhi, IndiaCopyright © 2019 Yi, Jin, Xiao, Chen, Tan, Du, He, Liu, Luo, Fang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Fang, ZmFuZ3lhbmdAY2liLmFjLmNu Hai Zhao, emhhb2hhaUBjaWIuYWMuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.