 Sujan Timilsina1
Sujan Timilsina1 Juliana A. Pereira-Martin2
Juliana A. Pereira-Martin2 Gerald V. Minsavage1Fernanda Iruegas-Bocardo1Peter Abrahamian2
Gerald V. Minsavage1Fernanda Iruegas-Bocardo1Peter Abrahamian2 Neha Potnis3
Neha Potnis3 Bryan Kolaczkowski4
Bryan Kolaczkowski4 Gary E. Vallad2*Erica M. Goss1,5*
Gary E. Vallad2*Erica M. Goss1,5* Jeffrey B. Jones1*
Jeffrey B. Jones1*- 1Department of Plant Pathology, University of Florida, Gainesville, FL, United States
- 2Gulf Coast Research and Education Center, University of Florida, Gainesville, FL, United States
- 3Department of Entomology and Plant Pathology, Auburn University, Auburn, AL, United States
- 4Microbiology and Cell Science, University of Florida, Gainesville, FL, United States
- 5Emerging Pathogens Institute, University of Florida, Gainesville, FL, United States
Prior to the identification of Xanthomonas perforans associated with bacterial spot of tomato in 1991, X. euvesicatoria was the only known species in Florida. Currently, X. perforans is the Xanthomonas sp. associated with tomato in Florida. Changes in pathogenic race and sequence alleles over time signify shifts in the dominant X. perforans genotype in Florida. We previously reported recombination of X. perforans strains with closely related Xanthomonas species as a potential driving factor for X. perforans evolution. However, the extent of recombination across the X. perforans genomes was unknown. We used a core genome multilocus sequence analysis approach to identify conserved genes and evaluated recombination-associated evolution of these genes in X. perforans. A total of 1,356 genes were determined to be “core” genes conserved among the 58 X. perforans genomes used in the study. Our approach identified three genetic groups of X. perforans in Florida based on the principal component analysis (PCA) using core genes. Nucleotide variation in 241 genes defined these groups, that are referred as Phylogenetic-group Defining (PgD) genes. Furthermore, alleles of many of these PgD genes showed 100% sequence identity with X. euvesicatoria, suggesting that variation likely has been introduced by recombination at multiple locations throughout the bacterial chromosome. Site-specific recombinase genes along with plasmid mobilization and phage associated genes were observed at different frequencies in the three phylogenetic groups and were associated with clusters of recombinant genes. Our analysis of core genes revealed the extent, source, and mechanisms of recombination events that shaped the current population and genomic structure of X. perforans in Florida.
Introduction
Bacterial pathogens challenge the sustainability and economics of agricultural production. The most damaging bacterial plant pathogens combine rapid evolution with a tendency for emerging strains to spread quickly over long-distances (Carroll et al., 2014). Characterizing bacterial strains associated with disease outbreaks advances our understanding of changes in pathogen populations and geographic distribution of genetic variation as well as the potential to trace the source of outbreaks. Technological advancements in both sequencing and computational tools have facilitated translational research for bacterial disease management via epidemiological and resistance-based approaches in hosts ranging from humans to plants (Köser et al., 2012; Gétaz et al., 2018).
Evolutionary and epidemiological studies of bacterial populations use core genomes, pan-genomes, and intergenic regions to uncover patterns and processes of strain emergence and spread (Biek et al., 2015; McNally et al., 2016; Jibrin et al., 2018). The process of whole genome sequencing followed by gene-by-gene comparisons to identify core genes, which are present in all sampled genomes, expands MLSA (Multilocus Sequence Analysis) from a half-dozen to several hundred or even a thousand genes (Bialek-Davenet et al., 2014). This reproducible approach for phylogenetic comparisons is termed core genome MLSA (cgMLSA) (Maiden and Harrison, 2016; Ghanem et al., 2017; Moura et al., 2017).
The genus Xanthomonas is comprised of plant pathogenic bacteria affecting multiple plant hosts. Fresh market tomato production in Florida is severely affected by bacterial spot disease of tomato caused by Xanthomonas perforans (Jones et al., 2004; Horvath et al., 2012). Previous studies on X. perforans strains isolated from Florida have shown shifts in the bacterial population with regards to species, races, bactericide resistance, bacteriocin production, effector profiles, and phylogenetic groups (Timilsina et al., 2014; Schwartz et al., 2015; Abrahamian et al., 2018). Prior to the initial identification of X. perforans in 1991, only tomato race 1 (T1) strains of Xanthomonas euvesicatoria were reported on tomato in Florida (Horvath et al., 2012; Timilsina et al., 2016). The first X. perforans strains from Florida were identified as tomato race 3 (T3) strains (Jones, 2004; Timilsina et al., 2016). T3 strains carry the functional XopAF (avrXv3) and XopJ4 (avrXv4) effectors. In 1998, a tomato race 4 (T4) X. perforans strain was identified (Minsavage et al., 2003) that lacked a functional XopAF effector. Various surveys and independent isolations over the last two decades determined that T4 X. perforans has become the dominant pathogen causing bacterial spot on tomato in Florida (Horvath et al., 2012; Vallad et al., 2013). While selection for widespread copper tolerance in bacteria is expected due to the historical reliance on copper-based bactericides for the management of bacterial spot disease (Vallad et al., 2010), the drivers of tomato race change (in the absence of host resistance), host expansion, and introduction of novel effector genes are less obvious.
We previously identified at least two phylogenetic groups of X. perforans in strains isolated from Florida in 2006 and 2012 using MLSA of six housekeeping genes (Timilsina et al., 2014). Among the two groups, group 2 strains appeared recombinant based on the sequences of two housekeeping genes that were identical to X. euvesicatoria strain Xe85-10, isolated from pepper (Timilsina et al., 2014). Although X. perforans strains are regarded as tomato specific, a group 2 X. perforans strain, Xp2010, was isolated from pepper, and other group 2 strains from tomato were shown to cause disease on pepper (Timilsina et al., 2014; Schwartz et al., 2015). The phenotypic and genotypic changes in group 2 strains suggests that the genomic impact of recombination likely extends beyond the few genes we have previously reported (Jibrin et al., 2018).
Phylogenetic methods are commonly applied to the study of bacterial strain ancestry and diversification (Didelot and Falush, 2007). However, most phylogenetic analysis methods assume recombination is absent, and the presence of recombination in the history of a sample can cause incorrect phylogenies. For multilocus sequence analysis of bacterial populations, the tendency has been to remove recombination in order to correctly interpret ancestral relationships for the unrecombined portion of the genome, the “clonal frame” (Wicker et al., 2012; Croucher et al., 2014; Lu et al., 2016). However, considering the ubiquity and impact of recombination on bacterial genetic diversity and evolution, the effect of recombination on phylogenetic relationships should be considered (Didelot and Wilson, 2015; Mostowy et al., 2017). Horizontal gene transfer can expedite evolution and may influence host-specificity in bacteria (Ochman et al., 2000; Yan et al., 2008). Genetic transfer may result in trait convergence due to shared genes acquired by horizontal gene transfer, or lead to the formation of distinct lineages or phylogroups (McNally et al., 2016). Transduction via virus, transformation by donor DNA, and conjugation with the donor are the three mechanisms by which bacteria acquire genetic material (Ochman et al., 2000). The acquisition of genomic DNA can leave specific signals surrounding the introduced genes at the integration sites (Ochman et al., 2000). For example, genomic movement between bacterial species by transduction is limited by phage-host specificity and the events are mediated by mobile DNA vectors observed along with the translocated genomic DNA (Popa et al., 2017).
Our objectives were to determine the extent of recombination in X. perforans genomes from Florida strains, identify recombined genes that contribute to the observed population structure in Florida, and evaluate putative mechanisms of genetic transfer of recombined regions. Using a cgMLSA approach, our study provides insights into the extent of recombination and mechanisms of horizontal gene transfer affecting the core genes that constitute the majority of the genomic background of phylogenetically divergent X. perforans genomes. The presence of multiple recombination mechanism signals throughout the genome, affecting both core and pathogenicity associated genes, is consistent with high genome plasticity in X. perforans. We provide empirical evidence that recombination of core genes has defined the existing phylogenetic groups of X. perforans in Florida. The observed genomic patterns appear to be correlated with traits like host-specificity and overall pathogen fitness and indicate that recombination has an extraordinary impact on evolutionary processes in X. perforans.
Materials and Methods
Bacterial Strains, Genome Assembly, and Genome Similarity
The genomes of 58 X. perforans strains isolated from Florida in 1991, 2006, 2012/13, and 2015 were used in this study (Table 1). Draft whole genome sequences of 33 strains, including reference strain Xp91-118, were previously published (Potnis et al., 2011; Schwartz et al., 2015). The remaining 25 X. perforans strains collected in 2015 are also publicly available (Supplementary Table 1). The raw Illumina MiSeq 2x250 basepair reads were reassembled using Spades v.3.11 with read error correction and “--careful” switch (Bankevich et al., 2012). The assembled sequences were validated using filter-spades.py1 and Bowtie2 was used to align the assembled reads to identify inconsistencies (Langmead and Salzberg, 2012). Pilon (Walker et al., 2014) was used to remove the inconsistencies identified by Bowtie2. The assembled sequences were filtered to remove sections with coverage less than 2 and contig size less than 500 nucleotides. CheckM identified more than 99% genome completeness with less than 0.6% contamination per genome (Parks et al., 2015; Supplementary Table 1). The genomes were annotated using the IMG/JGI platform (Markowitz et al., 2013). Following assembly, pairwise Average Nucleotide Identity (ANI) based on blast was calculated using jSpecies v 1.2.1 (Richter and Rosselló-Móra, 2009).
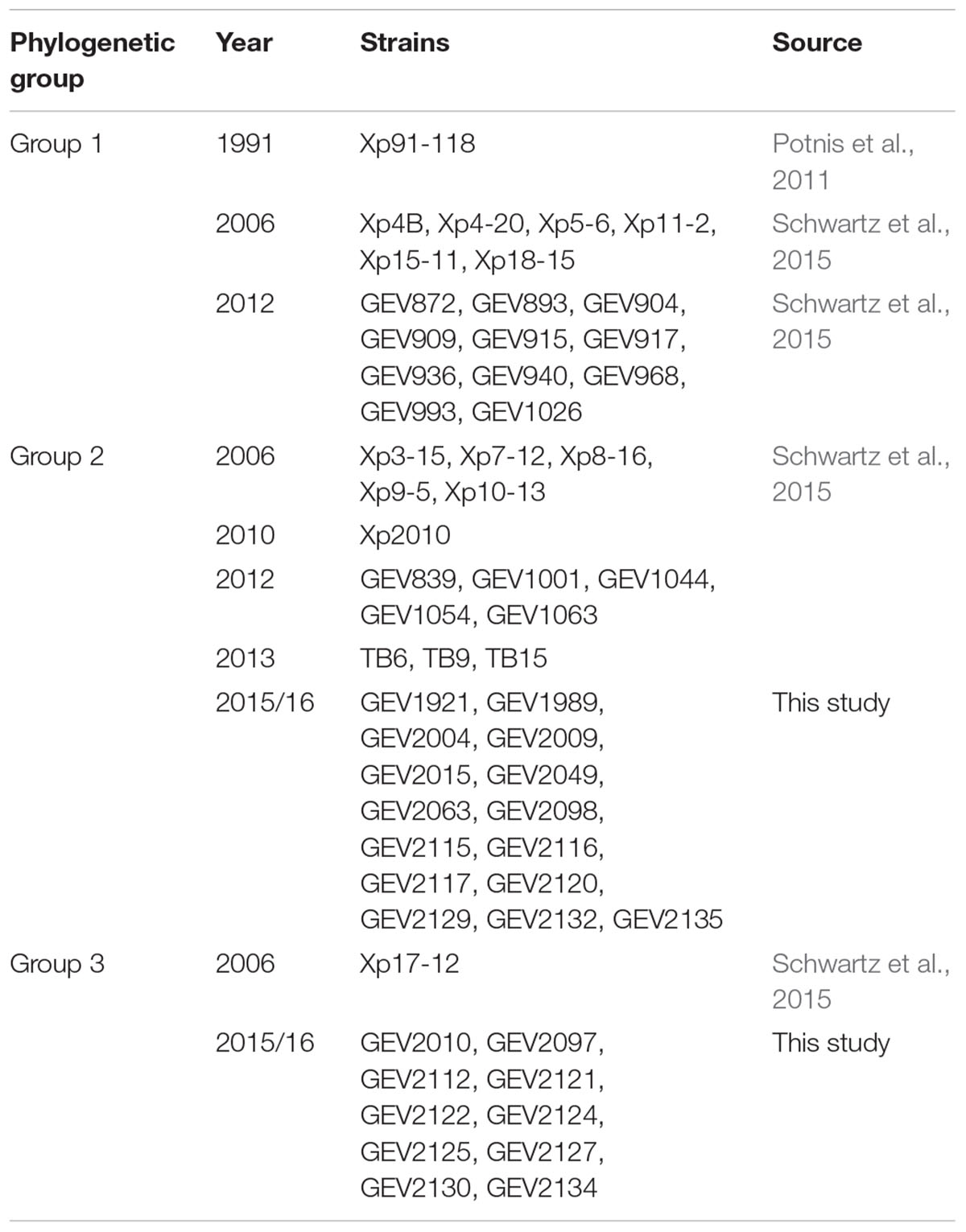
Table 1. List of strains used in this study.
Pan-Genome Size
For evaluation of the pan-genome of the 58 X. perforans strains, all genes were extracted from the 58 X. perforans strains using roary (Page et al., 2015) following gene annotation from prokka (Seemann, 2014). The method yielded a total of 7,245 genes. The pan-genome matrix of gene presence/absence in each genome was used as input for a rarefaction analysis to calculate the average number of genes added with each additional genome (Méric et al., 2014). The calculation was randomized by resampling 100 times. The Heaps law function was fitted to the data using the micropan package in R to the rarefaction curve (Tettelin et al., 2008; Snipen and Liliand, 2015). The Heaps law model estimates the parameter alpha. When alpha > 1, this suggests a closed pan genome and saturated sampling of the gene pool, while alpha < 1 suggests an open pan-genome.
Core Gene Identification and Alignment
The IMG/JGI annotated sequences were used to identify core genes among the 58 genomes. Nucleotide and amino acid sequences of annotated genes were used as input for core gene identification using get_homologues v.2.0.1.9 (Contreras-Moreira and Vinuesa, 2013). Genes present in at least 95% of the genomes and with 75% pairwise alignment coverage were retained. The genes were parsed using python scripts to strictly define core genes as genes present in 100% of the genomes with intact start and stop codons. This approach was taken to limit the core genes to those most likely to be functional, based on genome annotation, in all strains. Genes with multiple copies were also removed. A total of 1,356 genes met the above criteria. Nine genes annotated as functional by the get_homologues built-in annotation algorithm were not annotated by NCBI nor IMG/JGI, but were included in the analysis. The resulting nucleotide sequences of single copy core genes were individually aligned by MAFFT (Katoh and Standley, 2013) using a biopython script (Cock et al., 2009). Individual gene alignments were concatenated using sequence matrix software (Vaidya et al., 2011) to create a circa 1.09 megabases long sequence for each strain.
Sequence Typing and Gene Mapping
Individual core genes were sequence typed based on nucleotide sequence identity using a python script. Genes with identical sequences were assigned the same number, representing the sequence type. The process was repeated in a loop for all core genes and an output sequence type matrix was generated. This allowed quick comparison of core genes based on allelic variation. Invariable genes were stripped from the matrix to generate a heat map of allelic profiles using the ggplot2 package (Wickham, 2010) in R (R Core Team, 2013). The heat map was color coded to illustrate the allelic patterns for variable core genes, thus providing a genetic fingerprint.
The relative positions of the core genes were mapped based on the complete genome of X. perforans Xp91-118 (NCBI accession number: GCA_000192045.3). We used the collated nucleotide sequences of the core genes of Xp91-118 as queries to BLAST (Zhang et al., 2000) against the complete genome. The output was configured to list the start and end positions in the complete genome for all core genes, which were sorted by position using a python script. BRIG (BLAST Ring Image Generator) software v. 0.95 (Alikhan et al., 2011) was used to visualize the positions of individual core genes in Xp91-118.
Phylogenetic Analysis
Single gene evolution may be different from the evolution of the organism as a whole, particularly when there is horizontal gene transfer (Gogarten and Townsend, 2005). PhyML v.3.1 (Guindon et al., 2010) was used to construct maximum likelihood phylogenetic trees for single gene and concatenated core gene sequences. Nucleotide substitution models were estimated independently for individual genes and selected based on the log likelihood Akaike Information Criterion result calculated using jModelTest2 (Darriba et al., 2012). General time reversible model with gamma distributed rates and invariant sites (GTR+G+I) was identified as the best nucleotide substitution model for the concatenated sequence. Maximum likelihood trees were constructed with 500 bootstrap samples for both concatenated and single genes using the suggested substitution model. ClonalFrameML (Didelot and Wilson, 2015) was used to reconstruct maximum likelihood trees while accounting for recombination. ClonalFrameML calculates R/theta, nu, and 1/delta, which represent the relative rate of recombination to mutation, new polymorphisms introduced from recombination, and the inverse of average tract length of recombination (Didelot and Falush, 2007; Didelot and Wilson, 2015). The three parameters were calculated for all single gene trees and for the concatenated sequence tree (hereafter referred to as core genome tree). Additionally, genomic clustering observed in the phylogenetic trees was confirmed by principal component analysis (PCA). The sequence types of the core genes were used as input to conduct PCA using micropan package in R (Snipen and Liliand, 2015).
Detecting Genes Driving Phylogenic Relationships
Phylogenetic distances between the unrooted single gene trees and core genome tree, along with the sequence type matrix, were used to determine the genes influencing the core genome tree topology of the 58 strains of X. perforans. Congruency of single gene trees to the core genome tree was assessed using Robinson-Foulds (RF) symmetry. This index represents the distance between two phylogenetic trees by evaluating the number of nodes in a tree that are shared with a reference tree (Robinson and Foulds, 1981). We used the core genome tree as the reference tree. The RF symmetry values range between 0 and 1, such that 0 indicates identical tree topology and 1 indicates completely different tree topologies. For example, phylogenetic trees for genes that were identical in nucleotide sequence among all the 58 X. perforans strains did not share any nodes with the core genome tree and the RF value was 1. Alternatively, if any nodes in a single gene tree supported a node in the core genome tree, the resulting RF value was less than 1. RF symmetry was calculated using ETE3 Toolkit (Huerta-Cepas et al., 2016) for each single gene tree against the core genome tree to determine the genes that supported some part of the topology of the core genome tree. The sequence types of genes with RF symmetry < 1 were extracted. The variable genes that exhibited RF values < 1 and supported the phylogenetic grouping in the core genome topology are hereafter referred to as Phylogenetic group-Defining (PgD) genes. Maximum likelihood phylogenetic trees were constructed using concatenated sequences of 241 PgD and 1,115 non-PgD genes separately. The total tree length of the two phylogenetic trees were computed using Analysis of Phylogenetics and Evolution (ape) package in R (Paradis and Schliep, 2018) to confirm the role of PgD genes in phylogenetic grouping of X. perforans in the core genome tree.
Identifying Recombination Sources and Recombination Mechanisms
We used two methods to determine if PgD genes may have been horizontally transferred. The sequences of the PgD genes were compared to the NCBI sequence database to determine if alleles were shared with other closely related Xanthomonas species. We also calculated the relative impact of recombination to mutation on nucleotide substitution using ClonalFrameML. Clusters of genes identified as variable or PgD, particularly those with high recombination values, were identified and their gene neighborhoods and flanking regions were examined. Gene neighborhood regions from representatives of each phylogenetic group were aligned and examined for the presence of genes suggestive of prior transfer events, including features of plasmids, phages, and transposable elements (Chiu and Thomas, 2004). In addition to clusters of core genes, we confirmed the presence of these recombination associated signatures in neighborhood regions of effector genes that were previously suspected to be horizontal transferred (Timilsina et al., 2016).
Results
Xanthomonas perforans Pan-Genome
A total of 7,245 genes was identified in the pan-genome of 58 X. perforans strains and 2,866 genes were considered as core genes by roary (Supplementary Figure 1A). The Heaps law estimate, on the rarefaction curve of the number of new genes identified after randomly adding a genome, suggested an open pan-genome (Supplementary Figure 1B). The estimate for alpha was 0.813, suggesting that additional genes will be found upon sampling more X. perforans genomes.
Core Genome Phylogeny
The get_homologues pipeline identified 2,031 genes as core genes present in at least 95% of the sampled genomes. The variation in the core genes identified from roary and get_homologues is likely due to the two different annotation pipelines used to generate inputs for these programs. Roary used the annotation output from prokka whereas annotation based on Clusters of Orthologous Groups of proteins (COG) downloaded from IMG/JGI were used for the get_homologues platform for core gene extraction. We manually curated these to 1,356 core genes that were present and intact in all 58 X. perforans genomes (Supplementary Table 2). Nucleotide sequence comparison revealed that 783 genes were identical among all 58 genomes. At least two allele types were found in the remaining 573 genes (Supplementary Table 3).
The core genome phylogenetic analysis identified a third phylogenetic group in addition to the two previously described groups of X. perforans in Florida (Figure 1A). PCA of sequence types of the 1,356 core genes confirmed the three phylogenetic groups (Figure 2). Strain Xp17-12, which was previously considered to be within group 1 (Schwartz et al., 2015), clustered with 10 strains isolated in 2015/16 to form a separate phylogenetic clade that we refer as group 3. Among the 58 strains, 18 strains were designated group 1, 29 strains were designated group 2, and 11 strains were designated group 3. Group 1 is a heterogeneous group that includes 6 strains from 2006 and 11 from 2012 along with the reference tomato race 3 (T3) strain Xp91-118 from 1991. Strains from the 2015/16 season were in group 2 (15 strains) or group 3 (10 strains).
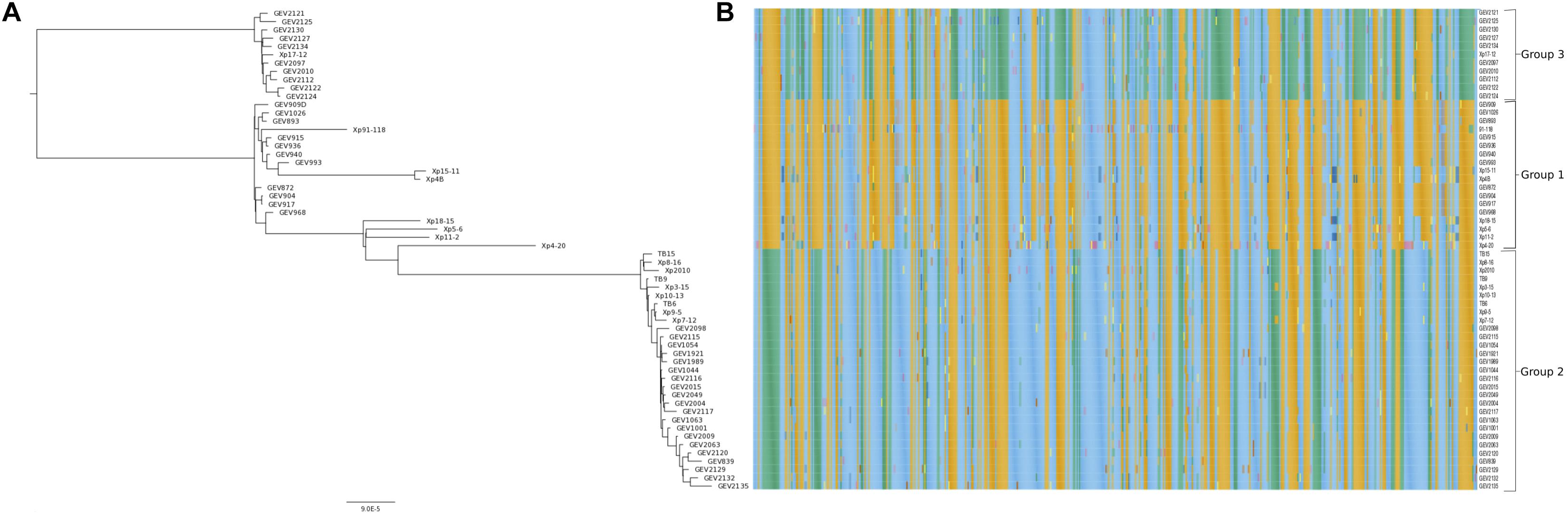
Figure 1. The core genome of Florida X. perforans strains shows three groups of strains. (A) Maximum likelihood phylogeny based on concatenated sequences of 1,356 core genes (∼1.3 Mb) and the branch topology were maintained regardless of the bootstrap support. (B) Heat map based on the allelic profile (sequence type) of variable genes (573) found within the core genes.
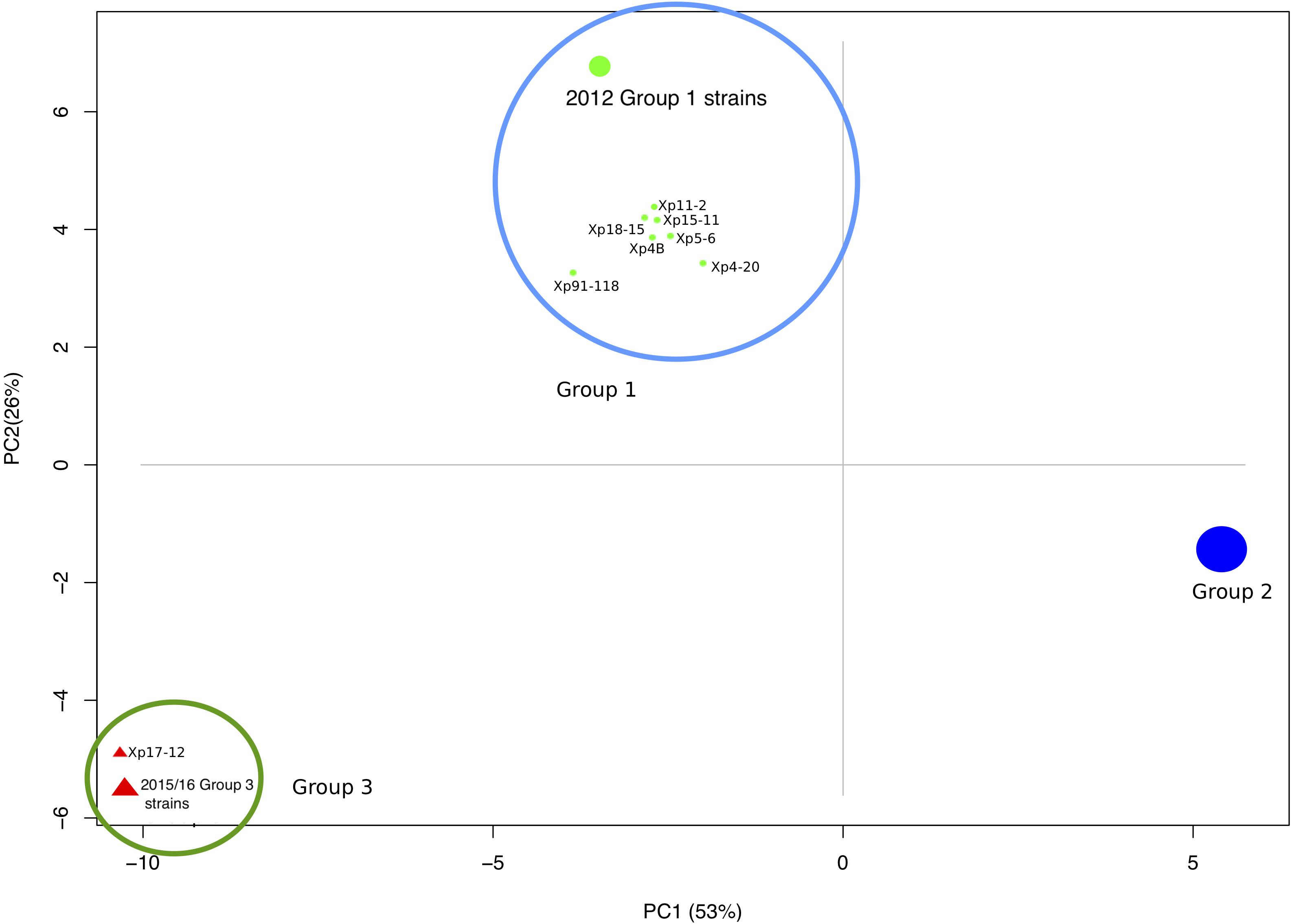
Figure 2. Principal component analysis showing three distinct clusters of X. perforans strains from Florida based on the sequence types of 1,356 conserved genes.
Within groups, the majority of strains shared more than 99.8% pairwise nucleotide sequence identity in the core genome (Supplementary Table 4). Between the groups, identity was reduced to 99.5%. The group 1 strains had relatively lower sequence identities of ∼99.5% in pairwise comparisons, and some strains had group 2 sequence types for several genes as observed in the heatmap and sequence type table (Figure 1B and Supplementary Table 3). Group 2 strains formed a monophyletic group with sequence identity above 99.7% among core genomes except for comparisons with Xp8-16 and Xp2010 (Figure 1A and Supplementary Table 4). Group 3 showed relatively low polymorphism with the majority of strains sharing core genome sequence identity above 99.9%. Average nucleotide identity based on BLAST using the whole genomes of these strains showed similar pairwise sequence identities to core genome comparisons (Supplementary Table 5).
Variable Core Genes by Phylogenetic Group
Core genes were distributed throughout the Xp91-118 genome (Figure 3). Among the 573 genes that had at least two allele types, referred to as variable genes, allelic variation was often between phylogenetic groups (Figure 1B). Sequences were generally monomorphic or had a single SNP at low frequency within phylogenetic groups. While only 783 genes were monomorphic across the 58 genomes, the number of genes with identical nucleotide sequences within groups were 1124, 1195, and 1239 for groups 1, 2, and 3, respectively.
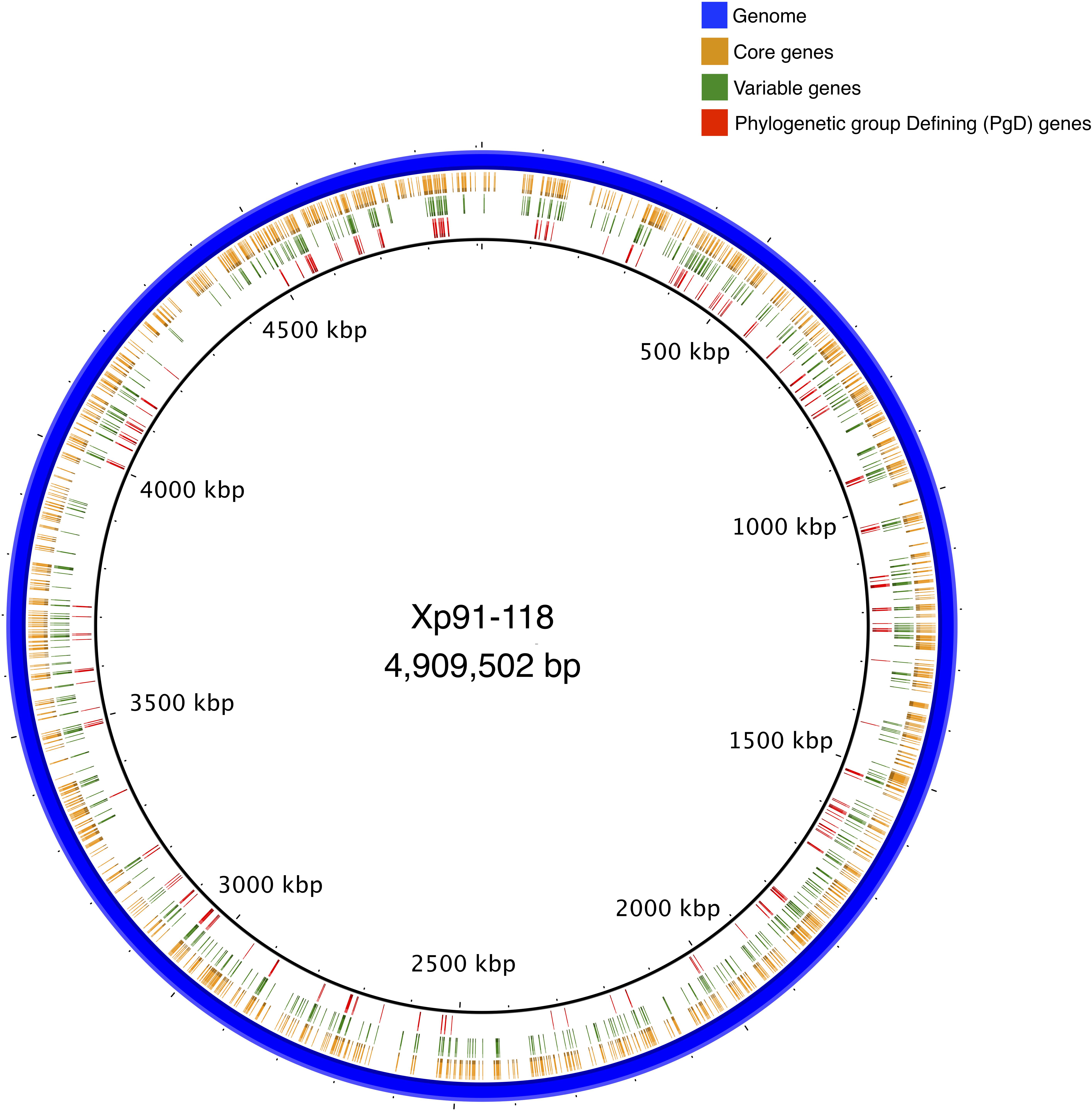
Figure 3. A circular representation of complete genome of Xp91-118 indicating relative positions of core, variable, and phylogenetic group defining (PgD) genes. Clusters of PgD suggest potential recombination hotspots.
Phylogenetic Group Defining Genes
We identified 241 core genes that supported one or more branches of the core genome tree topology that defined the phylogenetic groups (RF < 1). These genes are collectively distinguished as the phylogenetic group defining (PgD) genes as they drive the observed phylogenetic grouping of the 58 strains (Figure 1A and Supplementary Figure 2A). In particular, we observed that these PgD genes carried allele types that were often specific to phylogenetic groups (Figure 1B, Supplementary Table 3, and Supplementary Figure 2A). The annotations of genes identified as PgD are listed in Supplementary Table 2, among which 59 were annotated as hypothetical proteins and 2 as genes with domains of unknown functions. A total of 1,115 single gene trees did not support the core genome tree topology (RF = 1). These included 783 genes that were identical among all the X. perforans strains, plus 332 genes that had a variant allele type for at least one strain. The total length of tree based on 241 PgD genes was six times the length of the tree based on the remaining 1,115 core genes, signifying the larger contribution of PgD genes to strain variation (Supplementary Figure 2B). The non-PgD genes contribute to variation shared among a small numbers of strains.
Allele types distinguishing different groups were found in the PgD genes. Among the 241 PgD genes, 96 genes carried an allele specific to group 2 strains (different from the allele in group 1 and 3 strains), and 78 (81%) of those group 2 alleles were identical to X. euvesicatoria reference strain Xe85-10 (NCBI Accession no GCA_000009165.1). We found 142 genes with group 3-specific alleles, out of which 64 genes (45%) had alleles identical to X. euvesicatoria Xe85-10. An additional five PgD genes with group 3-specific alleles were identical to those of X. axonopodis pv. citrumelo strain F1 (NCBI Accession no. GCA_000225915.1), which included two hypothetical proteins, a protease modulator HflC, an anti-anti-sigma factor, and a type VI secretion system associated gene. Finally, group 3 alleles of two PgD genes were identical to those of X. perforans strain LH3 (NCBI Accession no. GCA_001908855.1), which was isolated from Mauritius in 2010 (Richard et al., 2017). These genes were N-acetyl-gamma-glutamyl-phosphate reductase (AQS75037.1) and aminoglycoside phosphotransferase (AQS78190.1). Therefore, LH3 is the only group 1 strain to contain these group 3 allele types. BLAST searches did not produce exact sequence matches to group 3 alleles for 71 genes. Unique allele types of PgD genes were distributed among group 1 strains. Group 1 strains isolated in 2006 carried specific allele types for 13 PgD genes that were identical to Xe85-10. Xp4-20 and Xp5-6 carried an additional 51and 19 unique allele types, respectively. The remaining four group 1 strains isolated in 2006 (Xp4B, Xp15-11, Xp11-2, and Xp18-15) had specific allele types for 15 additional genes. Group 1 strains isolated in 2012 were homogenous with 15 genes among the PgD genes identical to Xe85-10. Some of the allele types carried by group 1 strains collected in 2006 were identical to group 2 but different from the reference strain Xp91-118. Among all 241 PgD genes, we found three genes that each had three alleles that were specific to group: endopeptidase (AQS77891.1), TonB-dependent siderophore receptor (AQS78913.1), and septum formation protein Maf (AQS76051.1).
Mapping PgD genes to the complete genome of Xp91-118 identified the positions and proximity of these genes (Figure 3). For example, a ∼22 kb region between tryptophan-tRNA ligase (AQS77329.1) and catalase (AQS77307.1), encompassing 16 core genes (14 designated as PgD genes), exhibited diverged haplotypes specific to group 2 strains compared to group 1 and 3 strains. Similarly, an ∼8 kb region, between co-chaperone YbbN (AQS78328.1) and peptidyl-prolyl cis-trans isomerase (AQS78967.1) genes, exhibited a distinct haplotype in group 3 strains compared to the other two groups. The overall ratio of changes introduced by recombination relative to mutation in the concatenated core genome tree was estimated to be 16.75 by ClonalFrameML. These values ranged between 0.063 (AQS77927.1) and 184.159 (AQS77019.1) among the individual PgD gene trees (Supplementary Table 2).
Recombining Genes and Mechanism of Horizontal Gene Transfer
Genomic regions acquired via horizontal gene transfer may have signatures of integration associated with different modes of horizontal gene transfer (Ochman et al., 2000). We examined genomes representing each phylogenetic group for signals of recombination flanking clusters of PgD genes. We used two criteria to select genomic regions. First, we were interested in alleles of closely clustered PgD genes that were identical to Xe85-10 indicating gene transfer from X. euvesicatoria. Second, we focused on gene trees that exhibited higher ratios of recombination to mutation than the concatenated gene tree. Gene neighborhood comparisons around PgD genes showed the presence of multiple tRNAs, phage-associated and plasmid mobilization genes, along with site specific two-component system XerC and XerD, which were previously described to be associated with horizontal gene transfer (Aussel et al., 2002). For instance, group 1 strains isolated in 2012 have ∼9.6 kb of phage-associated genes between the tRNA-Leucine and tRNA-Glycine adjacent to the PgD genes AQS75151.1–AQS78555.1 (distinct alleles in group 2 and group 1 strains isolated in 2006). This unique ∼9.6 kb region found only in group 1 strains isolated in 2012 include AAA-domain containing protein, phage major capsid protein, phage portal protein, phage terminase-like protein with HTH domain, and hypothetical proteins. Nucleotide BLAST search in NCBI revealed 98% sequence similarity with only LH3 strain. The site-specific recombinase (XerD) gene was observed in all the group 1 strains between the tRNA-Leucine and phage associated genes, suggesting the integration of the unique genomic region was facilitated by bacteriophages (Figure 4A). In general, multiple copies of XerD, ranging from four to eight, were observed in all X. perforans genomes except for the 1991 reference strain, Xp91-118, which carries only one copy.
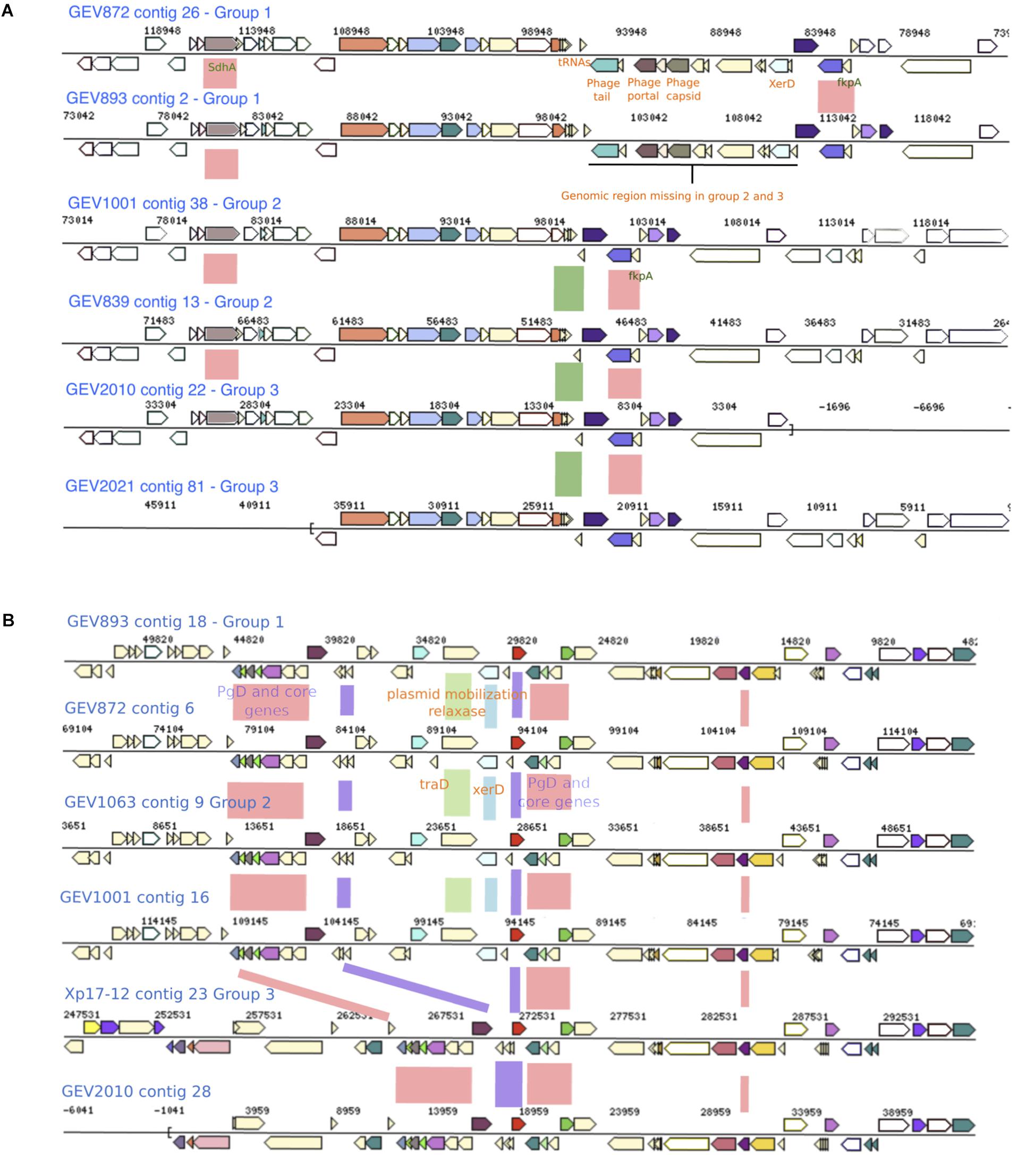
Figure 4. Genomic regions with genes exhibiting group specific alleles and recombination signals. (A) A genomic region present in group 1 strains, which contains phage associated genes and is flanked with tRNAs and XerD, is absent in Group 2 and 3 strains. Phylogenetic group defining (PgD) genes are indicated by pink boxes between the alignments with recombinant allele types in group 2 and group 1 2006 strains. The coordinates of this region in Xp91-118 are between 323182 and 325825 (B) Group 1 and 2 strains carry XerD, the site specific recombination gene, along with plasmid mobilization and Tra genes (green and blue boxes). This region is missing in Group 3 strains. PgD genes are indicated by pink and purple boxes with recombinant alleles in group 3. The coordinates of this region in Xp91-118 are 1604360-1620009.
Group 3 strains carried 71 PgD genes with unique alleles specific to the group. For example, group 3 alleles of six PgD genes between locus tags AQS76085.1–AQS76101.1 were distinct from the other two groups and were unique among sequences available in NCBI. In group 1 and 2, this genomic region is adjacent to XerD, mobile element protein (MobA), conjugal transfer protein (traD) and phage integrase family protein, suggesting integration via plasmid mobilization and site-specific recombination mechanisms (Figure 4B). The genomic region between the multiple tRNA sites and XerD present in group 1 and 2 included several unique hypothetical genes and DNA methyltransferase gene. Furthermore, the genetic variation in group 3 strains indicate the acquisition of novel genomic traits via recombination from multiple donors in addition to X. euvesicatoria.
Recombination Affecting Type III Effectors
Following the observation of at least two different mechanisms of genetic exchange that were associated with phage transfer and plasmid mobilization in core genes, we examined these signals throughout representative genomes. Interestingly, signatures of horizontal gene transfer were found surrounding effectors that show presence/absence polymorphism among strains and previously predicted to be acquired via recombination. Among the effectors found in bacterial spot causing X. perforans, avrXv4 (xopJ4) is found in all tomato pathogenic strains but not found in the strain, Xp2010, isolated from pepper. We found phage associated genes flanking XopJ4 (Figure 5). In Xp2010, both phage associated genes and the xopJ4 effector are absent. Similarly, the gene coding another XopJ family effector, avrBsT (XopJ2), found in the majority of tomato race 4 X. perforans, is flanked by genes for type IV secretion system proteins, conjugative transfer, chromosome partitioning protein, and hypothetical proteins (Supplementary Figure 3).
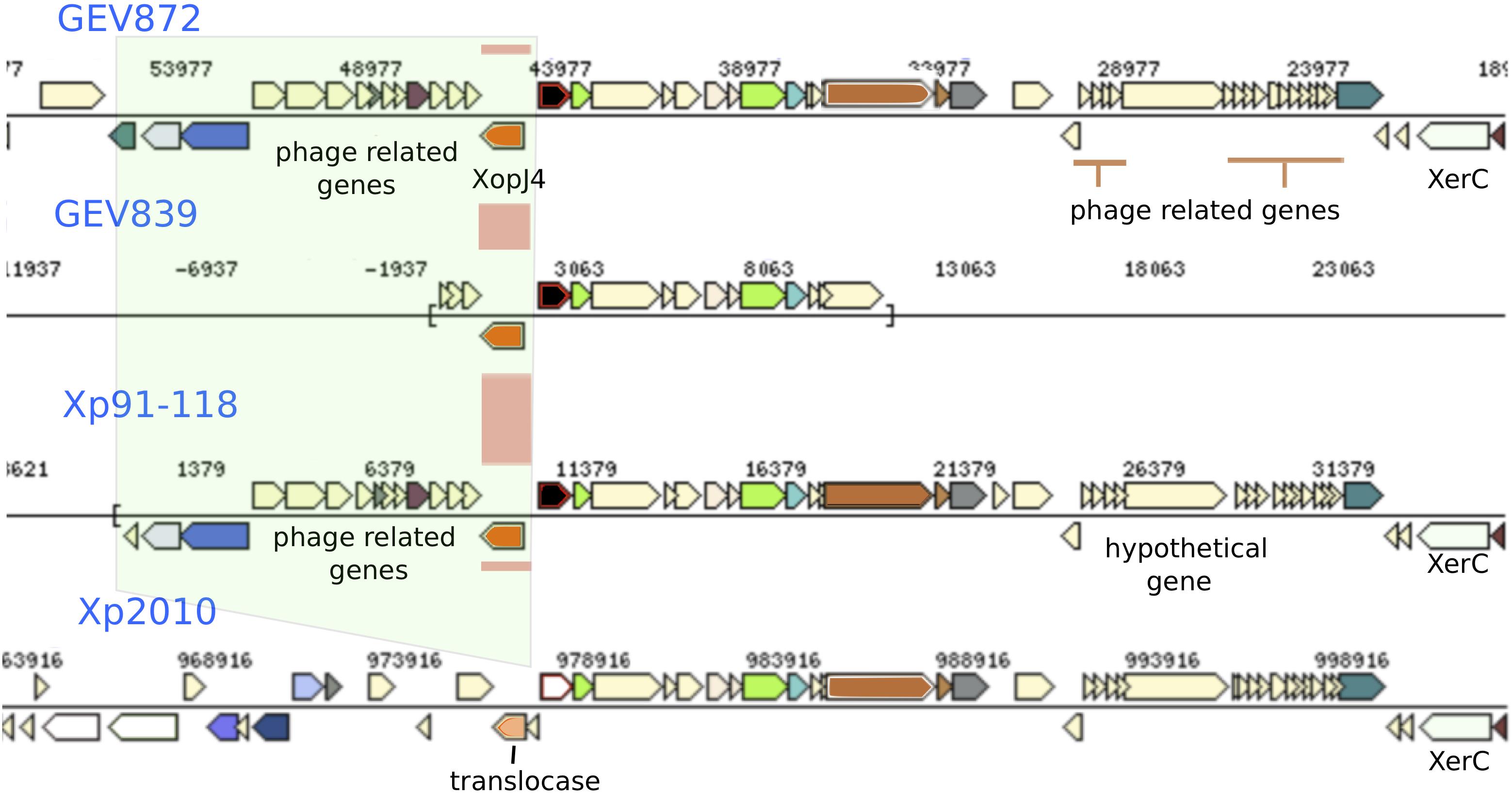
Figure 5. Recombination signals observed surrounding the gene for XopJ4 effector. The XopJ4 gene is flanked by phage associated genes in most strains, including T3 strains Xp91-118. In strain Xp2010, isolated from pepper, the effector gene and phage associated genes were replaced by a putative translocase.
Discussion
Using a cgMLSA approach, we determined that the X. perforans strains isolated in Florida at different times is defined by three groups of strains that are differentiated by hundreds of variable genes, the majority of which appear to be recombinant. All of the strains we analyzed were tomato race 4 X. perforans that were collected in the past two decades, except for the 1991 T3 reference strain, Xp91-118, and a pepper strain, Xp2010 (Schwartz et al., 2015). A high-resolution phylogeny of X. perforans strains was constructed as well as a core genome fingerprint, which shows allelic variation affecting genes throughout the chromosome. Core genome multilocus sequence analysis allows a holistic comparison of phylogenetic groups and their evolution while minimizing the individual differences between the strains (Maiden et al., 2013; Maiden and Harrison, 2016). Recombination was inferred to be widespread in core genes showing allelic variation with X. euvesicatoria strains as a major donor. Furthermore, we observed plasmid and phage associated site-specific recombination mechanisms surrounding clusters of putatively recombinant genes as well as genes that influence host-specificity and pathogen fitness. The open pan-genome further suggests high genomic plasticity in X. perforans. We hypothesize that these recombinant strains are epidemic lineages that have emerged in Florida tomato production from a highly recombinogenic source population.
We previously showed two major phylogenetic groups of X. perforans in Florida (Timilsina et al., 2014, 2016; Schwartz et al., 2015), but strains collected in the 2015/16 growing season revealed a third monophyletic group of 11 strains. Consistent with recent emergence, it was the most homogenous of the three groups as shown by pairwise nucleotide identity and PCA of sequence types. One strain from this group, Xp17-12, was isolated in 2006 and was previously described as an outlier within group 1 (Schwartz et al., 2015). The relative homogeneity among group 3 strains over time suggests that the 2015/16 group 3 strains were from the same source population. Group 1, which includes strains isolated in 1991, 2006 and 2012, is more heterogeneous than other groups (Figures 1, 2). Group 2 strains have been dominant in Florida since at least 2006 (Timilsina et al., 2016) but appear to be largely clonal (Figure 2). Three genes with alleles specific to each phylogenetic group were observed that could be used for assigning strains to groups to monitor their prevalence in Florida populations going forward. In general, we identified only a quarter of the total genes being shared among all X. perforans genomes, which suggests high genome plasticity in the species and is consistent with our finding of an open genome and is in agreement with another study of X. perforans (Jibrin et al., 2018).
We inferred recombination to have been the major source of genetic variation in the core genome. Horizontal inheritance of genes from multiple transfer events can obscure ancestral relationships among bacterial strains (Gogarten and Townsend, 2005). Consequently, recombinant loci are often removed from alignments prior to phylogenetic analysis. However, these loci also define the evolution of the recombinant strains (Didelot and Wilson, 2015). Phylogenetic reconstruction without the presence of these potentially recombined PgD genes significantly altered the observed phylogenetic relationships among the X. perforans groups (Supplementary Figure 2A). This variation, likely due to recombination from multiple donors, reinforces the necessity to include recombination in bacterial population studies. For group 2, most of the recombinant sequences appeared to have been acquired from X. euvesicatoria, which was displaced in Florida by X. perforans producing antagonistic bacteriocins against X. euvesicatoria (Hert et al., 2005; Timilsina et al., 2016). For group 3, X. euvesicatoria was one of multiple donors. The earliest strains of X. perforans isolated in Florida belonged to group 1, which had the least recombination signatures relative to the other two groups, as reflected by the copy numbers of site-specific recombinase XerCD genes. Correspondingly, the frequency of recombination observed with X. euvesicatoria and other closely related Xanthomonas was relatively low in these strains. Although, horizontal gene transfer is largely associated with acquisition of new traits, the tracts of homologous genes potentially acquired from closely related species shows the impact of recombination on the tempo and direction of X. perforans genome evolution. Recombination affecting the genetic background of a pathogen can affect niche adaptation, fitness, and microbial competition, and thus the establishment of recombinant strains (Alizon et al., 2009).
Our observations suggest horizontal gene transfer of long fragments of shared recombinant alleles through different genomic vectors. The horizontally introduced genomic fragments can be regulated by the carrying capacity of plasmid or phage vectors (Ochman et al., 2000; Bobay and Ochman, 2017). These two mechanisms of vector associated horizontal gene transfer were clearly visible in our X. perforans genomes. Phage and plasmid associated genes were present in the flanking regions of clusters of core genes showing evidence of recombination. Among the three modes of horizontal gene transfer, we were not able to directly identify genomic fragment acquisition via transformation. Several variable genes had variation attributed to recombination, but without any evidence of phage or plasmid associated genes in the flanking regions. These genes may have been transferred via transformation.
Phage mediated gene transfer appears to play a major role in influencing genomic diversity and evolution of X. perforans. We found phage genes throughout the genome in regions with potentially recombined core and effector genes. One such example is the XopJ family effector, avrXv4 (xopJ4). XopJ4 is a member of the XopJ effector family, which is similar to the YopJ, serine/threonine acetyltransferase superfamily (Szczesny et al., 2010). A previous study reported that xopJ4 was conserved in all X. perforans strains except for the Xp2010 strain that was isolated from pepper (Schwartz et al., 2015). The xopJ4 gene was located between phage associated genes. The whole genomic region including phage associated and XopJ4 genes is missing in the Xp2010 pepper strain. A similar XopJ family effector (98% amino acid identity) in X. citri pv. vignicola strain CFBP 7112 (Ruh et al., 2017) is also located between phage associated genes. Effector avrBsT is another XopJ family effector found in X. perforans that is generally associated with plasmids (Ciesiolka et al., 1999; Kay and Bonas, 2009; White et al., 2009). The avrBsT effector was not found in X. perforans until 1998 (Timilsina et al., 2016). The nucleotide sequence of avrBsT in X. perforans is identical to that in the more distantly related bacterial spot species, X. vesicatoria (Timilsina et al., 2016). An identical allele type of the plasmid-borne avrBsT gene in different Xanthomonas species, including X. perforans, suggests the gene is horizontally transferred across the genus (Timilsina et al., 2014, 2016; Jibrin et al., 2018). The majority of T4 X. perforans strains carry avrBsT and the gene has been found to provide a competitive advantage to bacterial strains in field conditions (Abrahamian et al., 2018). The variation created by mobile genetic elements in the core and accessory genes could influence host preference and pathogenicity of X. perforans strains.
Several PgD genes also showed evidence of plasmid associated horizontal gene transfer. An intriguing pattern that was evident in the genomes was the density of tRNAs and site-specific tyrosine recombinase genes flanking these recombined regions. The two-component site-specific tyrosine recombinase, XerC and XerD, catalyzes crossover and recombination at specific sites (Midonet and Barre, 2015). Site-specific recombination is characterized by cleavage of both DNA strands at two recombination sites that are later joined to new DNA partners without DNA degradation and phosphodiesters hydrolysis (Subramanya et al., 1997). The XerD recombinase works together with XerC, both of which belong to the λ-integrase family. XerD is reported to initiate recombination by strain exchange to form the Holliday junctions and that is reconstructed by XerC (Aussel et al., 2002; Crozat et al., 2015). In X. perforans genomes, the site-specific XerD gene was co-located with a plasmid mobilization and transfer (tra) gene where a ∼9.6-kb genomic region was present in group 1 genomes but absent in groups 2 and 3. Sequence comparison showed this region is specific to group 1 X. perforans strains. Along with XerCD genes, this genomic island was flanked by tRNAs (Figure 4A). The tRNAs serve as a gateway for integration of foreign DNA (Ochman et al., 2000; Williams, 2002). Boyd et al. (2009) reported that tRNA-Arg, -Leu, -Thr, and -Ser were commonly observed insertion sites. The genomic islands introduced by phage or plasmid in X. perforans seem to have specific attachment sites, facilitated by tRNAs and site-specific recombinase genes, that altered the core genomes and ultimately shaped the population of X. perforans in Florida. These specific sites serve as recombination hotspots in the bacterial genome.
Bacterial spot disease of tomato has posed a series of management challenges in Florida, including the introduction of X. perforans. In this study, we have begun to tease apart the genetic mechanisms driving population changes in X. perforans since its emergence in 1991, which appear primarily due to phage and plasmid-mediated horizontal gene transfer followed by integration into the chromosome. Our findings indicate rapid genomic evolution in the X. perforans population in Florida, which together with our previous findings of extensive recombination in strains from Nigeria and Italy (Jibrin et al., 2018), suggest a pathogen with a high probability of overcoming management practices, i.e., X. perforans poses a high “evolutionary risk” (McDonald and Linde, 2002). However, the clonal structure of the Florida population also indicates that a limited number of recombinant genotypes have been introduced to or have successfully established in Florida tomato production. If we could determine the population or populations that are highly recombinogenic, these populations could be specifically managed or movement out of these populations curtailed. For example, recombination events could be occurring in seed sources or other production regions that are not closely connected to Florida, thus exchanging few migrants. Efforts are also needed to understand why X. perforans readily recombines when other bacterial phytopathogens, including the bacterial spot pathogen X. gardneri, appear highly clonal (Schwartz et al., 2015; Timilsina et al., 2014).
Data Availability
The datasets generated for this study can be found in NCBI, PRJNA436012.
Author Contributions
ST, NP, GV, JJ, and EG conceived the project. PA collected additional bacterial strains and provided their genomes. JP-M and FI-B oversaw the genome assembly. GM and ST conducted the sequencing experiments. ST conducted all computational analyses and interpreted with the help of BK, EG, GV, and JJ. ST, EG, JJ, and GV wrote the final manuscript. All authors approved the final manuscript.
Funding
This research was supported in part by the United States Department of Agriculture, National Institute of Food and Agriculture under award number 2015-51181-24312. Publication of this article was funded in part by the University of Florida Open Access Publishing Fund.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00448/full#supplementary-material
Footnotes
References
Abrahamian, P., Timilsina, S., Minsavage, G. V., Goss, E. M., Jones, J. B., and Vallad, G. E. (2018). The type III effector AvrBsT enhances Xanthomonas perforans fitness in field-grown tomato. Phytopathology 108, 1355–1362. doi: 10.1094/PHYTO-02-18-0052-R
Alikhan, N.-F., Petty, N. K., Zakour, N. L. B., and Beatson, S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Alizon, S., Hurford, A., Mideo, N., and Van Baalen, M. (2009). Virulence evolution and the trade-off hypothesis: history, current state of affairs and the future. J. Evol. Biol. 22, 245–259. doi: 10.1111/j.1420-9101.2008.01658.x
Aussel, L., Barre, F.-X., Aroyo, M., Stasiak, A., Stasiak, A. Z., and Sherratt, D. (2002). FtsK is a DNA motor protein that activates chromosome dimer resolution by switching the catalytic state of the XerC and XerD recombinases. Cell 108, 195–205. doi: 10.1016/S0092-8674(02)00624-4
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bialek-Davenet, S., Criscuolo, A., Ailloud, F., Passet, V., Jones, L., Delannoy-Vieillard, A.-S., et al. (2014). Genomic definition of hypervirulent and multidrug-resistant Klebsiella pneumoniae clonal groups. Emerg. Infect. Dis. 20, 1812–1820. doi: 10.3201/eid2011.140206
Biek, R., Pybus, O. G., Lloyd-Smith, J. O., and Didelot, X. (2015). Measurably evolving pathogens in the genomic era. Trends Ecol. Evol. 30, 306–313. doi: 10.1016/j.tree.2015.03.009
Bobay, L. M., and Ochman, H. (2017). Impact of recombination on the base composition of bacteria and archaea. Mol. Biol. Evol. 34, 2627–2636. doi: 10.1093/molbev/msx189
Boyd, E. F., Almagro-Moreno, S., and Parent, M. A. (2009). Genomic islands are dynamic, ancient integrative elements in bacterial evolution. Trends Microbiol. 17, 47–53. doi: 10.1016/j.tim.2008.11.003
Carroll, S. P., Jørgensen, P. S., Kinnison, M. T., Bergstrom, C. T., Denison, R. F., Gluckman, P., et al. (2014). Applying evolutionary biology to address global challenges. Science 346:1245993. doi: 10.1126/science.1245993
Chiu, C.-M., and Thomas, C. M. (2004). Evidence for past integration of IncP-1 plasmids into bacterial chromosomes. FEMS Microbiol. Lett. 241, 163–169. doi: 10.1016/j.femsle.2004.10.016
Ciesiolka, L. D., Hwin, T., Gearlds, J. D., Minsavage, G. V., Saenz, R., Bravo, M., et al. (1999). Regulation of expression of avirulence gene avrRxv and identification of a family of host interaction factors by sequence analysis of avrBsT. Mol. Plant Microbe Interact. 12, 35–44. doi: 10.1094/MPMI.1999.12.1.35
Cock, P. J., Antao, T., Chang, J. T., Chapman, B. A., Cox, C. J., Dalke, A., et al. (2009). Biopython: freely available python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. doi: 10.1093/bioinformatics/btp163
Contreras-Moreira, B., and Vinuesa, P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701. doi: 10.1128/AEM.02411-13
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2014). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequence using Gubbins. Nucleic Acids Res. 43:e15. doi: 10.1093/nar/gku1196
Crozat, E., Fournes, F., Cornet, F., Hallet, B., and Rousseau, P. (2015). Resolution of multimeric forms of circular plasmids and chromosomes. Plasmids Biol. Impact Biotechnol. Discov. 2, 157–173. doi: 10.1128/microbiolspec.PLAS-0025-2014
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772. doi: 10.1038/nmeth.2109
Didelot, X., (2007). Inference of bacterial microevolution using multilocus sequence data. Genetics 175, 1251–1266. doi: 10.1534/genetics.106.063305
Didelot, X., and Wilson, D. J. (2015). ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput. Biol. 11:e1004041. doi: 10.1371/journal.pcbi.1004041
Gétaz, M.,Rezzonico, F., Smits, T. H. M., van der Wolf, J. M., and Pothier, J. F. (2018). Genome-based population structure analysis of the strawberry plant pathogen Xanthomonas fragariae reveals two distinct groups that evolved independently before its species description. Microb. Genomics 4. doi: 10.1099/mgen.0.000189
Ghanem, M., Wang, L., Zhang, Y., Edwards, S., Lu, A., Ley, D., et al. (2017). Core genome multilocus sequence typing (cgMLST): a standardized approach for molecular typing of Mycoplasma gallisepticum. J. Clin. Microbiol. 56:e01145-17. doi: 10.1128/JCM.01145-17
Gogarten, J. P., and Townsend, J. P. (2005). Horizontal gene transfer, genome innovation and evolution. Nat. Rev. Microbiol. 3, 679–687. doi: 10.1038/nrmicro1204
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hert, A. P., Roberts, P. D., Momol, M. T., Minsavage, G. V., Tudor-Nelson, S. M., and Jones, J. B. (2005). Relative importance of bacteriocin-like genes in antagonism of Xanthomonas perforans tomato race 3 to Xanthomonas euvesicatoria tomato race 1 strains. Appl. Environ. Microbiol. 71, 3581–3588. doi: 10.1128/AEM.71.7.3581-3588.2005
Horvath, D. M., Stall, R. E., Jones, J. B., Pauly, M. H., Vallad, G. E., Dahlbeck, D., et al. (2012). Transgenic resistance confers effective field level control of bacterial spot disease in tomato. PLoS One 7:e42036. doi: 10.1371/journal.pone.0042036
Huerta-Cepas, J., Serra, F., and Bork, P. (2016). ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 33, 1635–1638. doi: 10.1093/molbev/msw046
Jibrin, M. O., Potnis, N., Timilsina, S., Minsavage, G. V., Vallad, G. E., Roberts, P. D., et al. (2018). Genomic inference of recombination-mediated evolution in Xanthomonas euvesicatoria and X. perforans. Appl. Environ. Microbiol. 84:e00136-18. doi: 10.1128/AEM.00136-18
Jones, J. B., Lacy, G. H., Bouzar, H., Stall, R. E., and Schaad, N. W. (2004). Reclassification of the xanthomonads associated with bacterial spot disease of tomato and pepper. Syst. Appl. Microbiol. 27, 755–762. doi: 10.1078/0723202042369884
Jones, S. (2004). An overview of the basic helix-loop-helix proteins. Genome Biol. 5:226. doi: 10.1186/gb-2004-5-6-226
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kay, S., and Bonas, U. (2009). How Xanthomonas type III effectors manipulate the host plant. Curr. Opin. Microbiol. 12, 37–43. doi: 10.1016/j.mib.2008.12.006
Köser, C. U., Ellington, M. J., Cartwright, E. J. P., Gillespie, S. H., Brown, N. M., Farrington, M., et al. (2012). Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog. 8:e1002824. doi: 10.1371/journal.ppat.1002824
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357. doi: 10.1038/nmeth.1923
Lu, X., Zhou, H., Du, X., Liu, X., Xu, J., Cui, Z., et al. (2016). Population analysis of clinical and environmental Vibrio parahaemolyticus isolated from eastern provinces in China by removing the recombinant SNPs in the MLST loci. Infect. Genet. Evol. 45, 303–310. doi: 10.1016/j.meegid.2016.09.002
Maiden, M. C. J., and Harrison, O. B. (2016). Population and functional genomics of neisseria revealed with gene-by-gene approaches. J. Clin. Microbiol. 54, 1949–1955. doi: 10.1128/JCM.00301-16
Maiden, M. C. J., van Rensburg, M. J. J., Bray, J. E., Earle, S. G., Ford, S. A., Jolley, K. A., et al. (2013). MLST revisited: the gene-by-gene approach to bacterial genomics. Nat. Rev. Microbiol. 11, 728–736. doi: 10.1038/nrmicro3093
Markowitz, V. M., Chen, I.-M. A., Palaniappan, K., Chu, K., Szeto, E., Pillay, M., et al. (2013). IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 42, D560–D567. doi: 10.1093/nar/gkt963
McDonald, B. A., and Linde, C. (2002). Pathogen population genetics, evolutionary potential, and durable resistance. Annu. Rev. Phytopathol. 40, 349–379. doi: 10.1146/annurev.phyto.40.120501.101443
McNally, A., Oren, Y., Kelly, D., Pascoe, B., Dunn, S., Sreecharan, T., et al. (2016). Combined analysis of variation in core, accessory and regulatory genome regions provides a super-resolution view into the evolution of bacterial populations. PLoS Genet. 12:e1006280. doi: 10.1371/journal.pgen.1006280
Méric, G., Yahara, K., Mageiros, L., Pascoe, B., Maiden, M. C. J., Jolley, K. A., et al. (2014). A reference pan-genome approach to comparative bacterial genomics: identification of novel epidemiological markers in pathogenic campylobacter. PLoS One 9:e92798. doi: 10.1371/journal.pone.0092798
Midonet, C., and Barre, F.-X. (2015). Xer site-specific recombination: promoting vertical and horizontal transmission of genetic information. Mob. DNA III 2, 163–182. doi: 10.1128/microbiolspec.MDNA3-0056-2014
Minsavage, G. V., Balogh, B., Stall, R. E., and Jones, J. B. (2003). New tomato races of Xanthomonas campestris pv. vesicatoria associated with mutagenesis of tomato race 3 strains. Phytopatholoy 93:S62.
Mostowy, R., Croucher, N. J., Andam, C. P., Corander, J., Hanage, W. P., and Marttinen, P. (2017). Efficient inference of recent and ancestral recombination within bacterial populations. Mol. Biol. Evol. 34, 1167–1182. doi: 10.1093/molbev/msx066
Moura, A., Criscuolo, A., Pouseele, H., Maury, M. M., Leclercq, A., Tarr, C., et al. (2017). Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nat. Microbiol. 2:16185. doi: 10.1038/nmicrobiol.2016.185
Ochman, H., Lawrence, J. G., and Groisman, E. A. (2000). Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304. doi: 10.1038/35012500
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Paradis, E., and Schliep, K. (2018). ape 5.0: an environment for modern phylogenetic and evolutionary analyses in R. Bioinformatics 35, 526–528. doi: 10.1093/bioinformatics/bty633
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Popa, O., Landan, G., and Dagan, T. (2017). Phylogenomic networks reveal limited phylogenetic range of lateral gene transfer by transduction. ISME J. 11, 543–554. doi: 10.1038/ismej.2016.116
Potnis, N., Krasileva, K., Chow, V., Almeida, N. F., Patil, P. B., Ryan, R. P., et al. (2011). Comparative genomics reveals diversity among xanthomonads infecting tomato and pepper. BMC Genomics 12:146. doi: 10.1186/1471-2164-12-146
R Core Team (2013). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Richard, D., Boyer, C., Lefeuvre, P., Canteros, B. I., Beni-Madhu, S., Portier, P., et al. (2017). Complete genome sequences of six copper-resistant Xanthomonas strains causing bacterial spot of solanaceous plants, belonging to X. gardneri, X. euvesicatoria, and X. vesicatoria, using long-read technology. Genome Announc. 5:e1693-16.
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Robinson, D. F., and Foulds, L. R. (1981). Comparison of phylogenetic trees. Math. Biosci. 53, 131–147. doi: 10.1016/0025-5564(81)90043-2
Ruh, M., Briand, M., Bonneau, S., Jacques, M.-A., and Chen, N. W. G. (2017). First complete genome sequences of Xanthomonas citri pv. vignicola strains CFBP7111, CFBP7112, and CFBP7113 obtained using long-read technology. Genome Announc. 5:e813-17. doi: 10.1128/genomeA.00813-17
Schwartz, A. R., Potnis, N., Timilsina, S., Wilson, M., Patané, J., Martins, J. Jr., et al. (2015). Phylogenomics of Xanthomonas field strains infecting pepper and tomato reveals diversity in effector repertoires and identifies determinants of host specificity. Front. Microbiol. 6:535. doi: 10.3389/fmicb.2015.00535
Seemann, T. (2014). Proakka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Snipen, L., and Liliand, K. H. (2015). micropan: an R-package for microbial pan-genomics. BMC Bioinformatics 16:79. doi: 10.1186/s12859-015-0517-0
Subramanya, H. S., Arciszewska, L. K., Baker, R. A., Bird, L. E., Sherratt, D. J., and Wigley, D. B. (1997). Crystal structure of the site-specific recombinase, XerD. EMBO J. 16, 5178–5187. doi: 10.1093/emboj/16.17.5178
Szczesny, R., Büttner, D., Escolar, L., Schulze, S., Seiferth, A., and Bonas, U. (2010). Suppression of the AvrBs1-specific hypersensitive response by the YopJ effector homolog AvrBsT from Xanthomonas depends on a SNF1-related kinase. New Phytol. 187, 1058–1074. doi: 10.1111/j.1469-8137.2010.03346.x
Tettelin, H., Riley, D., Cattuto, C., and Medini, D. (2008). Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11, 472–477. doi: 10.1016/j.mib.2008.09.006
Timilsina, S., Abrahamian, P., Potnis, N., Minsavage, G. V., White, F. F., Staskawicz, B. J., et al. (2016). Analysis of sequenced genomes of Xanthomonas perforans identifies candidate targets for resistance breeding in tomato. Phytopathology 106, 1097–1104. doi: 10.1094/PHYTO-03-16-0119-FI
Timilsina, S., Jibrin, M. O., Potnis, N., Minsavage, G. V., Kebede, M., Schwartz, A., et al. (2014). Multilocus sequence analysis of xanthomonads causing bacterial spot of tomato and pepper reveals strains generated by recombination among species and recent global spread of Xanthomonas gardneri. Appl. Environ. Microbiol. 81, 1520–1529. doi: 10.1128/AEM.03000-14
Vaidya, G., Lohman, D. J., and Meier, R. (2011). SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27, 171–180. doi: 10.1111/j.1096-0031.2010.00329.x
Vallad, G. E., Pernezny, K. L., Balogh, B., Wen, A., Figueiredo, J. F. L., Jones, J. B., et al. (2010). Comparison of kasugamycin to traditional bactericides for the management of bacterial spot on tomato. HortScience 45, 1834–1840. doi: 10.21273/HORTSCI.45.12.1834
Vallad,S., Adkison, H., Potnis, N., Minsavage, G., Jones, J., et al. (2013). A recent survey of xanthomonads causing bacterial spot of tomato in florida provides insights into management strategies. Tomato Proc. 25.
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
White, F. F., Potnis, N., Jones, J. B., and Koebnik, R. (2009). The type III effectors of Xanthomonas. Mol. Plant Pathol. 10, 749–766. doi: 10.1111/j.1364-3703.2009.00590.x
Wicker, E., Lefeuvre, P., de Cambiaire, J.-C., Lemaire, C., Poussier, S., and Prior, P. (2012). Contrasting recombination patterns and demographic histories of the plant pathogen Ralstonia solanacearum inferred from MLSA. ISME J. 6, 961–974. doi: 10.1038/ismej.2011.160
Williams, K. P. (2002). Integration sites for genetic elements in prokaryotic tRNA and tmRNA genes: sublocation preference of integrase subfamilies. Nucleic Acids Res. 30, 866–875. doi: 10.1093/nar/30.4.866
Yan, S., Liu, H., Mohr, T. J., Jenrette, J., Chiodini, R., Zaccardelli, M., et al. (2008). Role of recombination in the evolution of the model plant pathogen Pseudomonas syringae pv. tomato DC3000, a very atypical tomato strain. Appl. Environ. Microbiol. 74, 3171–3181. doi: 10.1128/AEM.00180-08
Keywords: core genome multilocus sequence typing, bacterial evolution, recombination, horizontal gene transfer (HGT), Xanthomonas perforans, bacterial spot
Citation: Timilsina S, Pereira-Martin JA, Minsavage GV, Iruegas-Bocardo F, Abrahamian P, Potnis N, Kolaczkowski B, Vallad GE, Goss EM and Jones JB (2019) Multiple Recombination Events Drive the Current Genetic Structure of Xanthomonas perforans in Florida. Front. Microbiol. 10:448. doi: 10.3389/fmicb.2019.00448
Received: 30 November 2018; Accepted: 20 February 2019;
Published: 13 March 2019.
Edited by:
Dawn Arnold, University of the West of England, United KingdomReviewed by:
Prabhu B. Patil, Institute of Microbial Technology (CSIR), IndiaMarcus Michael Dillon, University of Toronto, Canada
Copyright © 2019 Timilsina, Pereira-Martin, Minsavage, Iruegas-Bocardo, Abrahamian, Potnis, Kolaczkowski, Vallad, Goss and Jones. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gary E. Vallad, Z3ZhbGxhZEB1ZmwuZWR1 Erica M. Goss, ZW1nb3NzQHVmbC5lZHU= Jeffrey B. Jones, amJqb25lc0B1ZmwuZWR1