 Yingxiu Cao1
Yingxiu Cao1 Mengyuan Song1
Mengyuan Song1 Feng Li1Congfa Li2Xue Lin2
Feng Li1Congfa Li2Xue Lin2 Yaru Chen1
Yaru Chen1 Yuanyuan Chen1
Yuanyuan Chen1 Jing Xu1
Jing Xu1 Qian Ding1
Qian Ding1 Hao Song1*
Hao Song1*- 1Key Laboratory of Systems Bioengineering (Ministry of Education), SynBio Research Platform, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), School of Chemical Engineering and Technology, Tianjin University, Tianjin, China
- 2College of Food Science and Technology, Hainan University, Haikou, China
Shewanella oneidensis MR-1 is a platform microorganism for understanding extracellular electron transfer (EET) with a fully sequenced and annotated genome. In comparison to other model microorganisms such as Escherichia coli, the available plasmid parts (such as promoters and replicons) are not sufficient to conveniently and quickly fine-tune the expression of multiple genes in S. oneidensis MR-1. Here, we constructed and characterized a plasmid toolkit that contains a set of expression vectors with a combination of promoters, replicons, antibiotic resistance genes, and an RK2 origin of transfer (oriT) cassette, in which each element can be easily changed by fixed restriction enzyme sites. The expression cassette is also compatible with BioBrick synthetic biology standards. Using green fluorescent protein (GFP) as a reporter, we tested and quantified the strength of promoters. The copy number of different replicons was also measured by real-time quantitative PCR. We further transformed two compatible plasmids with different antibiotic resistance genes into the recombinant S. oneidensis MR-1, enabling control over the expression of two different fluorescent proteins. This plasmid toolkit was further used for overexpression of the MtrCAB porin-c-type cytochrome complex in the S. oneidensis ΔmtrA strain. Tungsten trioxide (WO3) reduction and microbial fuel cell (MFC) assays revealed that the EET efficiency was improved most significantly when MtrCAB was expressed at a moderate level, thus demonstrating the utility of the plasmid toolkit in the EET regulation in S. oneidensis. The plasmid toolkit developed in this study is useful for rapid and convenient fine-tuning of gene expression and enhances the ability to genetically manipulate S. oneidensis MR-1.
Introduction
Shewanella oneidensis MR-1, a Gram-negative exoelectrogen, is regarded as an important model microorganism with extracellular electron transfer (EET) pathways for bio-electrochemical applications in environmental and energy fields, including bioelectricity generation and production of chemicals (Hou et al., 2009, 2011; Kouzuma et al., 2015; Shi et al., 2016; Kumar et al., 2017; Li et al., 2018b). After fully sequencing and annotating its genome (Venkateswaran et al., 1999; Heidelberg et al., 2002; Daraselia et al., 2003), a number of efforts have been made to engineer EET in S. oneidensis. For example, disruption of the putative cell surface polysaccharide biosynthesis gene SO3177 (Kouzuma et al., 2010) or overexpression of the c-type cytochrome gene mtrC (Bretschger et al., 2008) were undertaken to generate higher current output. With the advancement of synthetic biology, multiple genes were simultaneously manipulated to facilitate EET in Shewanella. Golitsch et al. (2013) used an arabinose-inducible promoter pBAD to upregulate the expression of an operon composed of the genes mtrF, mtrA, and mtrB, which had previously been deleted from the genome in S. oneidensis, showing that modulation of the operon expression levels is an efficient strategy in enhancing ferric iron reduction capacity and anode reduction performance. West et al. (2017) characterized a native inducible expression system induced by trimethylamine N-oxide (TMAO) in S. oneidensis to control EET, which was then successfully used to regulate EET by inducing mtrCAB gene expression with TMAO. The ability to induce this pathway was assessed by measuring iron reduction over time and by analyzing anodic current produced by cells grown in bioreactors. Yang et al. (2015) heterologously expressed a synthetic flavin biosynthesis pathway from Bacillus subtilis in the BioBrick compatible vector pYYDT, derived from the plasmid pHG101, to enhance the rate of EET in engineered S. oneidensis MR-1. By using this plasmid, Li et al. (2018a) assembled four genes that are mostly responsible for increasing NADH regeneration in S. oneidensis MR-1.
Improved genetic engineering strategies could provide additional opportunities to modulate the metabolism of microorganisms (Lin et al., 2018). Although manipulation of multiple genes has been employed to engineer S. oneidensis, the availability of gene expression tools is still not as abundant as other well-studied platform microorganisms, such as Escherichia coli or Saccharomyces cerevisiae (Keasling, 2012; Chae et al., 2017). In E. coli, a large number of expression and regulatory elements make the fine-tuning of gene expression quick and convenient. Researchers have combined various biological parts in E. coli, such as promoters with different strengths (e.g., pBAD, pTrc, pT5, pT7, placUV5, pLtetO-1, and double tac), replication origins (e.g., p15A, ColE, pBBR1, pSC101, and pBR322), antibiotic resistance markers [e.g., chloramphenicol (CmR), kanamycin (Kan), carbenicillin (Cb), ampicillin (Amp), and spectinomycin (Spect)], to fine-tune gene expression, which led to significant increases in the production of taxadiene (Ajikumar et al., 2010), n-butanol (Bond-Watts et al., 2011), L-tyrosine (Juminaga et al., 2012), and amorphadiene (Anthony et al., 2009). Heterologous expression of MtrCAB under the control of the T7 promoter in E. coli could create an artificial electrogenic cell (Jensen et al., 2010), but high MtrCAB expression in the presence of the Isopropyl β-D-Thiogalactoside (IPTG) inducer may impair EET efficiency in E. coli. Goldbeck et al. (2013) used E. coli with a more tunable induction system and a panel of constitutive promoters to express the MtrCAB pathway. They found that the efficiency of MtrC and MtrA synthesis decreased when controlled by strong promoters, leading to a significant decrease in EET efficiency. To improve the ability of manipulating S. oneidensis MR-1, more vectors were developed. Hajimorad and Gralnick (2016) demonstrated that different replication origins (e.g., p15A, pMB1, and pBBR1), selection markers (e.g., Kan and CmR), and an RK2 oriT cassette could be used in S. oneidensis MR-1, which provided a useful tool in engineering S. oneidensis. Continued development of synthetic biology parts, coupled with quantitative characterization, will enable more delicate or complicated engineering of S. oneidensis.
Here, we report the development of a synthetic plasmid toolkit for S. oneidensis MR-1. A number of available biological parts, such as promoters, replication origins, a conjugal transfer shuttle cassette, and antibiotic resistance genes were tested to design and construct easily available and compatible vectors for Shewanella (Figure 1). Firstly, we evaluated and characterized the strength of various promoters in E. coli and S. oneidensis MR-1 using the green fluorescent protein (GFP) as a reporter. Furthermore, we characterized the strength of inducible promoters by regulating inducer concentrations and induction times. Secondly, we quantified the copy numbers of different replicons in S. oneidensis MR-1 by real-time quantitative PCR (RT-qPCR) and found that copy number was directly correlated with the fluorescence intensity of GFP. Additionally, we simultaneously transformed two compatible plasmids into recombinant S. oneidensis MR-1, thus achieving fine control of the expression of two different fluorescent proteins [GFP and blue fluorescent protein (BFP)/superfolder cyan fluorescent protein (CFP)] through modulation of inducer concentration or the use of promoters with different strengths. Lastly, we took the porin-cytochrome complex MtrCAB for an example to demonstrate that EET efficiency can be fine-tuned in recombinant S. oneidensisΔmtrA strain by the plasmid toolkit we developed in this study. The plasmid toolkit developed in this study enabled rapid and convenient fine-tuning of gene expression with greater controllability and predictability, which will accelerate future genetic manipulations of S. oneidensis MR-1.
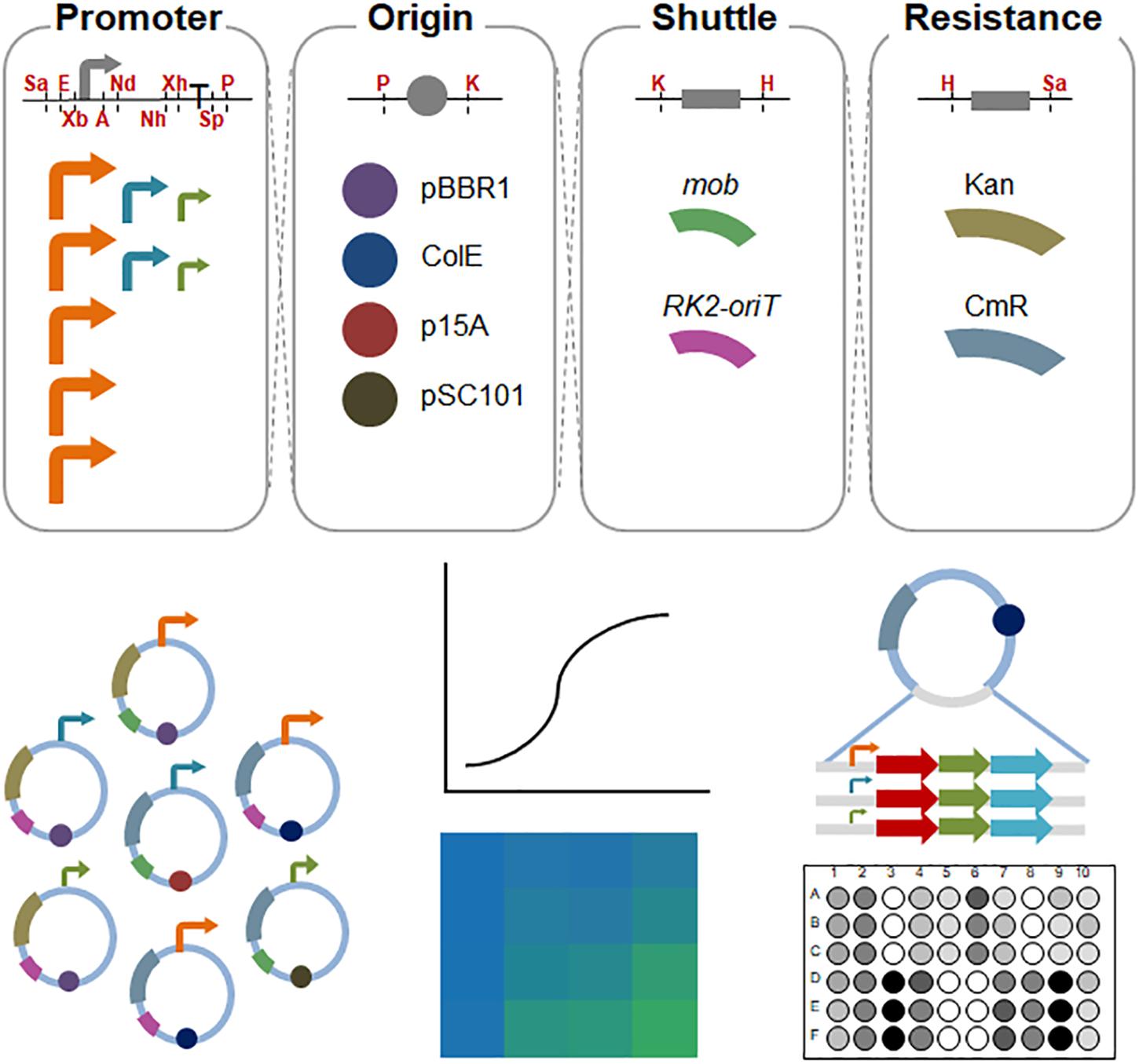
Figure 1. Schematic diagram of the assembled shuttle vectors in S. oneidensis MR-1. The cutting sites of the restriction enzymes are shown in red. Sa, SacI; E, EcoRI; Xb, XbaI; A, AvrII; Nd, NdeI; Nh, NheI; Xh, XhoI; Sp, SpeI; P, PstI; K, KpnI; H, HindIII. High-strength promoters (i.e., pBAD, pXyl, pCI, pJ23119, and pTac) are shown in orange; medium-strength promoters (pTet and pTrc∗) are shown in aqua; and low-strength promoters (placUV5 and pLlacO1) are shown in olive.
Materials and Methods
Strains and Growth Conditions
All bacterial strains used in this study are listed in Table 1. For genetic manipulation, E. coli Trans1-T1, BL21, MG1655, and WM3064 were cultivated in Luria-Bertani (LB) medium at 37°C. The wild-type and recombinant S. oneidensis MR-1 strains were cultured in LB medium at 30°C. Unless otherwise specified, genetic manipulation antibiotics were used at the following concentrations: kanamycin at 50 μg/ml and chloramphenicol at 34 μg/ml. The E. coli WM3064 culture was supplemented with 0.3 mM 2,6-Diaminopimelic acid (DAP).

Table 1. Strains and plasmids used in the study.
Plasmid Constructions
All the plasmids used in this study are listed in Table 1. Plasmid construction processes were performed in E. coli Trans1-T1 and E. coli BL21. The plasmid is composed of four elements: replication origin, antibiotic resistance gene, conjugal transfer shuttle cassette, and expression cassette, with fixed restriction enzyme sites between each part as shown in Figure 1 and Supplementary Figure S1. The sequences of each biological element and the corresponding restriction enzyme sites were synthesized by Genewiz (China), which are listed in Supplementary Table S2. Specifically, plasmid pHG12 was constructed from plasmid pHG11 (Cao et al., 2017) by site-directed mutagenesis of the NcoI restriction site and insertion of the SacI in front of EcoRI restriction site. The primers used are presented in Supplementary Table S1.
The sequences of green, blue and superfolder cyan fluorescent protein gens (gfp, bfp, and cfp) were codon-optimized and synthesized by Genewiz (China), which were listed in Supplementary Table S2. They were digested with NdeI/XhoI and inserted into the corresponding sites of the expression cassette in relative plasmid. The MtrCAB clusters were amplified from the S. oneidensis MR-1 genome using MtrCAB-F/MtrCAB-R primer pairs, listed in Supplementary Table S1, which were then digested with NdeI/XhoI and inserted into the corresponding sites of the plasmids carrying the promoter pBAD, pTet, and placUV5, respectively, resulting in three mtrCAB expression vectors.
Conjugation Assay
Transformed into S. oneidensis MR-1 or S. oneidensisΔmtrA strains (Cao et al., 2017), plasmids were firstly transformed into the plasmid donor cell E. coli WM3064 (a DAP auxotroph) and then transferred into S. oneidensis MR-1 or S. oneidensis ΔmtrA by conjugation. For the growth of E. coli WM3064 that has RP4 Tra function integrated into the chromosome, 0.3 mM 2,6-Diaminopimelic acid (DAP) was needed in the culture medium. In brief, 500 μl donor cells and 500 μl recipient cells were mixed and then the mixture was collected by centrifugation (5000 rpm for 10 min). Cells were suspended with 1 ml LB medium containing DAP and incubated for 1 h at 30°C. The appropriate volume of cells was next spread on LB plates with appropriate antibiotics and placed overnight in 30°C incubator to allow conjugation. To arrive at multiple-plasmid Shewanella strains, S. oneidensis MR-1 cells harboring a single-plasmid served as the recipient cell in the above steps.
Fluorescence Assay
Green, blue, and superfolder cyan fluorescent proteins (GFP, BFP, and CFP) were used to characterize the expression intensity. A total of 0.1 mM IPTG, 10 mM arabinose, 1 g/l xylose, or 100 ng/ml anhydrotetracycline hydrochloride (aTc) was used as the normal concentration for inducing the corresponding promoters. For concentration-response and time-response assays, GFP intensities in S. oneidensis MR-1 were tested with a series of inducer concentrations. IPTG, arabinose, and xylose were diluted to 10-3–104 μM, 10-2–105 μM, and 10-5–101 g/l using 10-fold serial dilutions, respectively. Additionally, aTc was used at the following concentrations (ng/ml): 25, 50, 100, 250, 500, 1000, 1500, 2000, and 2500. To simultaneously fine-tune the expression of multiple fluorescent proteins in S. oneidensis MR-1, we adjusted inducer concentrations for IPTG-inducible pTac or arabinose-inducible pBAD promoters. IPTG was used at the following concentrations (μM): 1, 12.5, 15, and 100. Arabinose was used at the following concentrations (mM): 0.1, 0.25, 0.5, and 1.
For fluorescence intensity assays, each strain was inoculated from a freshly transformed single colony on an LB agar plate into 2 ml LB medium as a seed culture. When cell accumulation reached stationary phase (assessed spectrophotometrically at OD600), 50 μl of seed culture was re-inoculated in test tubes containing 5 ml fresh LB medium supplemented with the appropriate antibiotic and the corresponding concentration of inducer. After 24 h of growth at 30°C with constant shaking (200 rpm), 200 μl suspensions from each test tube were centrifuged at 4000 rpm for 2 min to remove the supernatant, after which the culture was washed and resuspended in phosphate-buffered saline (PBS) and transferred to a 96-well polystyrene plate (black plate, clear bottom) (Corning Incorporated 3603, United States). Cell optical density and fluorescence intensity were detected by a SpectraMax M2 microplate reader. Optical density was measured at 600 nm. The excitation/emission wavelengths for GFP, BFP, and CFP were set at 485 nm/520 nm, 399 nm/456 nm, and 425 nm/475 nm, respectively. Assays were performed in triplicate, and untransformed E. coli MG1655 or S. oneidensis MR-1 strains were used as controls. Relative fluorescence was calculated as the fluorescence per OD600 for each strain subtracted from that of the untransformed E. coli MG1655 or untransformed S. oneidensis MR-1 control:
Determination of Replicon Copy Number by Real-Time Quantitative PCR (RT-qPCR)
Preparation of Template DNA for qPCR
Recombinant S. oneidensis MR-1 isolates harboring different plasmids (e.g., pHG13-pTrc∗-CoIE-GFP, pHG13-pTrc∗-p15A-GFP, pHG13-pTrc∗-pSC101-GFP, and pHG12-pTrc∗-GFP) were cultivated to exponential growth phase, after which total DNA was extracted using the Bacterial Genome DNA extraction kit (Kang Wei Century, China) according to the manufacturer’s instructions. Template DNA extracted from the recombinant S. oneidensis MR-1 was normalized to 2 ng/μl for consistency in the RT-qPCR assay (Lee et al., 2006).
Construction of Standards for RT-qPCR
The sequences of the four replicons (CoIE, p15A, pSC101, and pBBR1) were amplified using primers listed in Supplementary Table S1. The products were separately cloned into T vectors using a Puc-T TA cloning kit (Kang Wei Century, China). Each cloned plasmid was purified using the TIANquick Mini Purification kit (Tiangen, Beijing, China) and was used as a standard (Dhanasekaran et al., 2010). RT-qPCR was carried out using a real-time fluorescent quantitative PCR machine (Applied Biosystems) in white-walled PCR plates (96 wells). SYBR® Premix Ex TaqTM II (Tli RNaseH Plus) and ROXplus (TaKaRa) were used to make a Master Mix, which also contained TaKaRa Ex Taq HS, a dNTP mixture, Mg2+, Tli RNaseH, TB Green, and ROX Reference Dye. Reactions were prepared in a total volume of 18 μl, containing 10 μM of each standard RT-qPCR primer, 2× Master Mix, sterile water and 2 μl cDNA. The cycle conditions were set as follows: initial template denaturation at 95°C for 30 s, followed by 40 cycles of denaturation at 95°C for 5 s, and combined primer annealing/elongation at 60°C for 40 s, with the fluorescence data acquired at 60°C and analyzed using 7500 Software V2.0.4 (Applied Biosystems).
Construction of Standard Curves and Absolute Quantification
A 10-fold serial dilution series of each standard plasmid, ranging from 1 × 103 to 1 × 108 copies/μl, was used to construct the standard curves. The concentration of the plasmid was measured using a spectrophotometer (Thermo Scientific), and the corresponding copy number was calculated using the following equation (Whelan et al., 2003):
The threshold cycle (CT) values for each dilution were measured in triplicate by RT-qPCR with the CoIE-, p15A-, pSC101-, and pBBR1-sets to generate the corresponding standard curves. Standard curves were obtained by plotting the threshold cycle (CT) on the y-axis and the natural log of concentration (copies/μl) on the x-axis. CT, slope, PCR efficiency, correlation coefficient (R2), and percentage of variance in copy numbers were calculated using the default settings of 7500 Software V2.0.4 (Applied Biosystems). Absolute quantification of the copy numbers of target replicon genes were obtained by comparing the CT value of each template DNA sample to a standard curve (Yu et al., 2005).
Tungsten Trioxide Reduction Assay
This assay was performed according to Yuan et al. (2014). The process for the tungsten trioxide (WO3) nanorod assembly synthesis solution is as follows: Dilute 0.825 g of Na2WO4 ⋅ 2H2O and 0.290 g of NaCl in 20 ml of sterile water. Add 3 M HCl slowly under stirring until the pH reaches 2.0. This solution needs to be freshly prepared. The process for the sodium lactate minimal salt medium is as follows: Per liter, dissolve 2.02 g of sodium lactate, 5.85 g of NaCl, 11.91 g of HEPES, 0.3 g of NaOH, 1.498 g of NH4Cl, 0.097 g of KCl, 0.67 g of NaH2PO4 ⋅ 2H2O and 1 ml per liter trace mineral stack solution (containing per liter: 1.5 g of C6H9NO6, 30 g of MgSO4 ⋅ 7H2O, 5 g of MnSO4 ⋅ H2O, 10 g of NaCl, 1 g of FeSO4 ⋅ 7H2O, 1 g of CaCl2 ⋅ 2H2O, 1 g of CoCl2 ⋅ 6H2O, 1.3 g of ZnCl2, 0.1 g of CuSO4 ⋅ 5H2O, 0.1 g of AlK(SO4)2 ⋅ 12H2O, 0.1 g of H3BO3, 0.25 g of Na2MoO4 ⋅ 2H2O, 0.25 g of NiCl2 ⋅ 6H2O, and 0.25 g of Na2WO4 ⋅ 2H2O) in sterile water and autoclave the mixture immediately. For the WO3 nanorod assembly containing sodium lactate minimal salt suspension, the process is as follows: dissolve 5 g per liter of WO3 nanorod assembly in sodium lactate minimal salt suspension and autoclave the resulting suspension immediately.
Overexpression of the c-type cytochromes, MtrCAB, in engineered S. oneidensis ΔmtrA was tested under the control of placUV5, pTet, and pBAD promoters. The parental and recombinant strains were named as S-ΔmtrA, L-mtrCAB, M-mtrCAB, and H-mtrCAB, respectively, according to the expression level of the gene (i.e., L, low; M, medium; H, high). They were activated overnight in LB medium with or without chloramphenicol. Then, 50 μl from each bacterial culture suspension from a 2 ml LB medium seed culture (originating from a freshly transformed single colony on a LB agar plate) was used to inoculate a test tube containing 5 ml fresh LB medium supplemented with or without chloramphenicol. The inducers for each promoter (i.e., 0.1 mM IPTG for placUV5, 1,000 ng/ml aTc for pTet, and 10 mM arabinose for pBAD) were added to express mtrCAB when OD600 reached ∼0.5. After 24 h of induction, cells were collected by centrifugation at 4000 rpm at 4°C for 5 min, after which the pellets were resuspended in sterile sodium lactate minimal salt medium, with appropriate dilution to achieve an OD600 ∼0.8. Then, 100 μl of the above resuspended bacterial solution and 80 μl of 5 g/l sterile WO3 nanorod assembly containing sodium lactate minimal salt suspension were mixed completely before being added into a 96-well plate. After that, 80 μl of petrolatum oil was added into each well immediately to ensure anaerobic conditions for the bioelectrochromic reaction, and the plate was incubated at 30°C in an incubator. We observed conspicuous color development in the plate after 30 min, but maintained incubation for 6 h, after which we transferred the plate into a scanner. As the plate was being scanned, an opaque box was used to shield it from external light. Then, the average color intensity of each well was determined by analyzing their ‘Density (mean)’ of the area of interest in the scanned photograph with the ImageJ Software V.1.8.0. Culture medium without bacteria was used as a control to ensure that the WO3 electrochromic reaction was not caused by the culture medium. Triplicate experiments were performed for each strain.
MFC Setup and Electrochemical Analysis
Overnight S-ΔmtrA, L-mtrCAB, M-mtrCAB, and H-mtrCAB culture suspensions were inoculated into 100 ml fresh LB broth at 30°C with shaking (200 rpm). After around 10 h culture, the amplification-cultured cells were subsequently centrifuged and collected at 4°C and underwent 6000 rpm for 10 min to remove the supernatant. The concentrations of cell suspensions were adjusted to the same level (OD600 ≈ 0.5), and the suspensions were dispersed into the anode chambers. In this MFC, S-ΔmtrA was used as a control. Whenever needed, 34 μg/ml chloramphenicol was added in anolytes to ensure consistent culture condition. For recombinant S. oneidensisΔmtrA strain harboring the plasmid under the control of the inducible promoter, the medium was supplemented with 0.1 mM IPTG for promoter placUV5, 1,000 ng/ml aTc for pTet, or 10 mM arabinose for pBAD. All MFCs were incubated in a 30°C incubator and each group was tripled for parallel experiments. Dual-chamber MFCs with a working volume of 110 mL separated by the Nafion 117 membrane (DuPont Inc., United States) were applied in this study. Carbon cloth was used as the electrodes for both anode (2.5 cm × 2.5 cm, i.e., the geometric area is 6.25 cm2) and cathode (2.5 cm × 3 cm). The anolyte consisted of M9 buffer (Na2HPO4, 6 g/l; KH2PO4, 3 g/l; NaCl, 0.5 g/l; NH4Cl, 1 g/l; MgSO4, 1 mM; CaCl2, 0.1 mM), supplemented with 18 mM lactate and 5% (v/v) LB broth. The catholyte was made of 50 mM K3[Fe (CN)6] in 50 mM KH2PO4 and 50 mM K2HPO4 solution. To measure the voltage generation, a 2 kΩ external resistor was connected into the external circuit of MFCs, and the output voltages were recorded across the external loading resistor with a digital multimeter (DT9205A). Linear sweep voltammetry (LSV) analysis with a scan rate (0.1 mV/s) was conducted on a two-electrode mode, where the anode performed as the working electrode and the cathode as the reference as well as the counter electrode to obtain the polarization curves to estimate the maximum power density (the potential decreased from the open circuit potential (OCP) to around -0.3 V). Power density (P) was calculated as P = V (output voltage) × I (current density). Both I and P were normalized to the projected area of the anode surface.
Results and Discussion
Characterization of Promoters in S. oneidensis MR-1
In this study, we designed a number of shuttle vectors, in which each element could be constructed or replaced using fixed restriction enzyme sites (Figure 1 and Supplementary Figure S1). Firstly, we characterized 9 promoters in S. oneidensis MR-1, which included pBAD, pLlacO1, placUV5, pTet, pXyl, pCI, pTac, pJ23119, and pTrc∗. These promoters could be classified into two categories: constitutive promoters (e.g., pCI, pJ23119, and pTrc∗) and inducible promoters (e.g., IPTG-inducible pTac, IPTG-inducible pLlacO1, IPTG-inducible placUV5, arabinose-inducible pBAD, xylose-inducible pXyl, and aTc-inducible pTet). The strength of these promoters was characterized using GFP as a reporter in E. coli MG1655 and S. oneidensis MR-1. 0.1 mM IPTG, 10 mM arabinose, 1 g/l xylose, or 100 ng/ml aTc was used to induce the corresponding promoters (Figure 2). In E. coli MG1655, the relative fluorescence showed a ladder-like distribution, which ranged from about 1,000 to more than 20,000 caused by the different expression strengths of the promoters (from low to high: pBAD, pLlacO1, placUV5, pTet, pXyl, pCI, pTac, pJ23119, and pTrc∗). However, the relative fluorescence intensities revealed only three expression levels in S. oneidensis MR-1. The pLlacO1 and placUV5 promoters showed low expression levels (1,000 ∼ 2,000); pTet and pTrc∗ showed medium expression levels (∼20,000); pBAD, pXyl, pCI, pJ23119, and pTac showed substantially higher expression levels (∼40,000). The highest fluorescence level was 40-fold higher than the lowest fluorescence level for S. oneidensis MR-1. Furthermore, the highest fluorescence intensity observed in S. oneidensis MR-1 was ∼2-fold higher than that in E. coli.
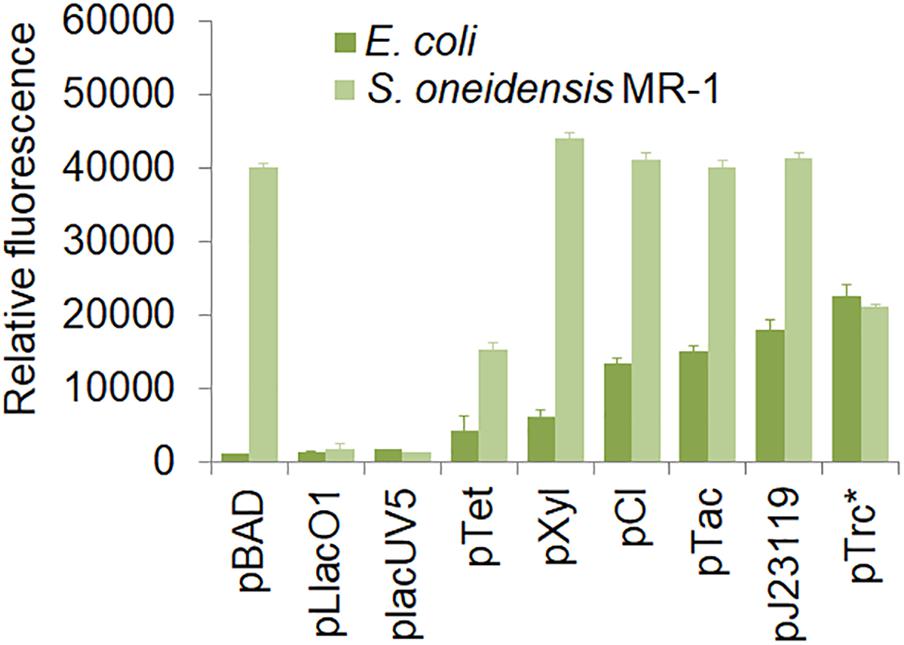
Figure 2. Characterization of various promoters in S. oneidensis MR-1. E. coli MG155 and S. oneidensis MR-1 harbored a single plasmid containing the pBAD, pLlacO1, placUV5, pTet, pXyl, pCI, pTac, pJ23119, and pTrc∗(no repressor lacO) promoters, respectively. The error bars (mean ± SD) were derived from triplicate experiments for each strain.
To further assess the levels of gene expression in S. oneidensis MR-1, the inducible promoters were characterized by adjusting inducer concentration and induction time (Figure 3). The distribution of relative fluorescence intensities was narrow in the early stage of induction but became broader with increasing inducer concentrations and longer induction times. With increasing inducer concentrations, GFP expression increased until a plateau level was reached for each promoter. All promoters resulted in a characteristic sigmoidal “S”-shaped curve. For all inducible promoters, the higher expression levels were achieved at the normal inducer concentrations. Almost full induction of the pTac, pLlacO1, and placUV5 promoters occurred at IPTG concentrations of ∼0.1, ∼1, and ∼1 mM, respectively (Figures 3A–C). pBAD and pXyl were fully induced at an arabinose or a xylose concentration of about 10 mM or 1 g/l, respectively (Figures 3D,E). The pTet promoter resulted in a higher expression at an aTc concentration of about 100 ng/ml (Figure 3F). The IPTG-inducible pTac, pLlacO1, and placUV5 promoters and the arabinose-inducible pBAD promoter could tightly control gene expression, since GFP fluorescence was basically not observed at low inducer concentrations. This feature is particularly important for the expression of genes whose products may be toxic, such that expression can occur at very low levels. In contrast, we found that the xylose-inducible pXyl and the aTc-inducible pTet promoters were susceptible to leaky expression. In addition, the inducible promoters were not only regulated by inducer concentration, but were also influenced by induction time. At the beginning, the fluorescence intensities were enhanced with longer induction times, but GFP fluorescence eventually reached a higher and more stable level by 24 h. The relative fluorescence intensities of all promoters at various inducer concentrations were compared after 24 h of incubation (Figure 3G), allowing for easy selection of promoters and inducer concentrations depending on the experimental needs.
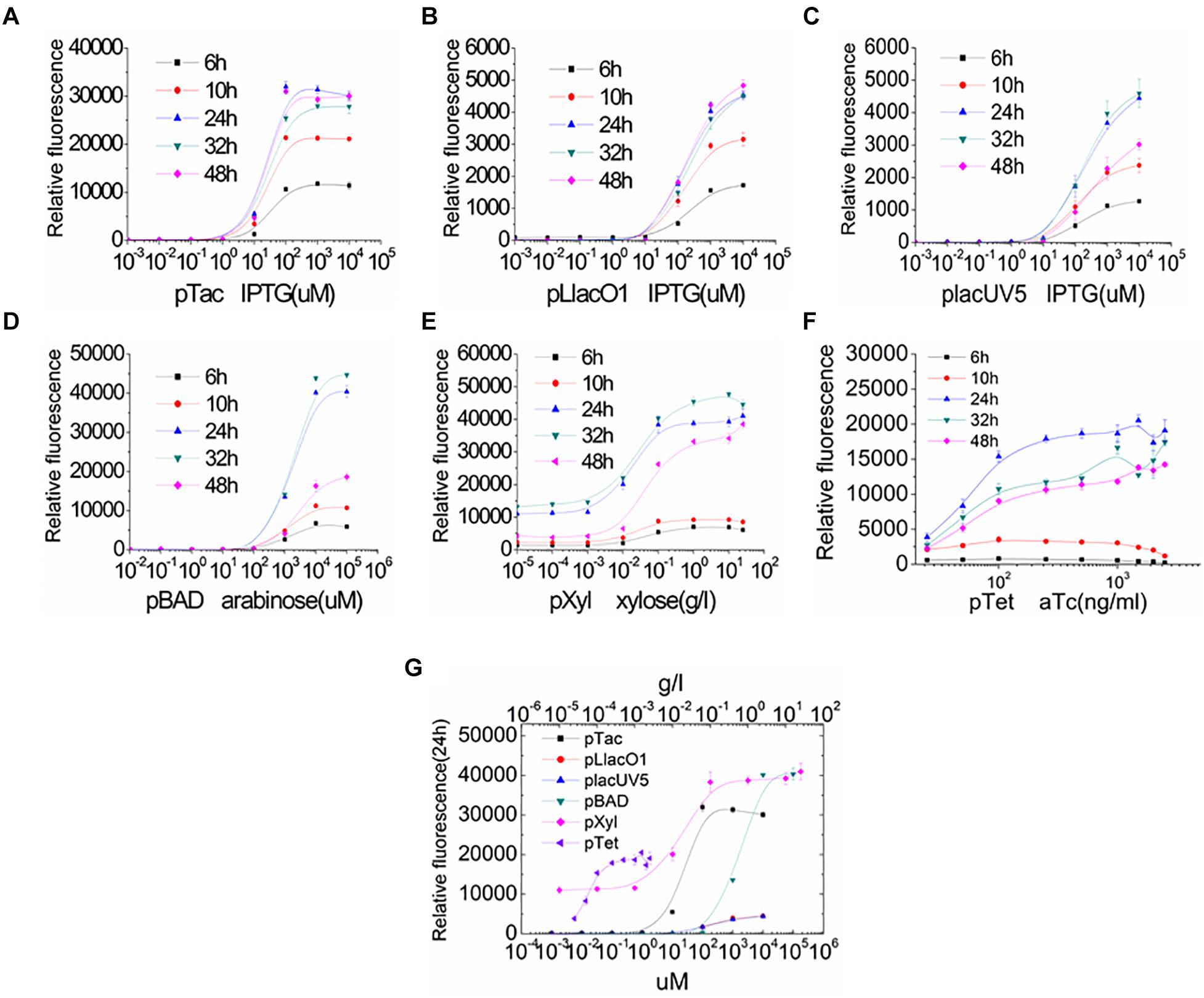
Figure 3. Characterization of the inducible promoters by regulating inducer concentration and induction time in S. oneidensis MR-1. (A–F) GFP expression under the control of inducible promoters was measured at 6, 10, 24, 32, and 48 h after induction with a series of different inducer concentrations. (A–C) IPTG-inducible pTac, pLlacO1, and placUV5; (D) arabinose-inducible pBAD; (E) xylose-inducible pXyl; (F) aTc-inducible pTet). (G) GFP expression was measured at 24 h after induction with different inducer concentrations. The error bars (mean ± SD) were derived from triplicate experiments for each strain.
It is worth mentioning that for expression cassettes, we designed and constructed biological parts that conformed to BioBrick standards (Canton et al., 2008; Shetty et al., 2008). BioBrick vectors feature four isocaudomer pairs (AvrII, NdeI, XbaI, and SpeI), can form the monocistron or polycistron, and support the modular assembly of numbers of molecular components and multigene pathways, collectively contributing to convenient and rapid gene combination and plasmid construction (Supplementary Figure S2).
Characterization of Antibiotic Resistance Genes, Replication Origins, and Coexistence of Two Plasmids in S. oneidensis
For fine-tuning the expression of multiple genes, genes should be expressed from two separate plasmids. As such, knowledge of compatible replication origins, different antibiotic resistances, and the shuttle component involved in conjugation and transformation in S. oneidensis MR-1 are required.
We first assessed the ability of S. oneidensis MR-1 to resist different antibiotics to identify suitable resistance markers for maintaining multiple plasmids. Sensitivities of the wild-type S. oneidensis MR-1 to three antibiotics (kanamycin at 50 or 100 μg/ml, chloramphenicol at 15 or 34 μg/ml, and ampicillin at 100 μg/ml) were tested in LB broths and on LB agar plates (Supplementary Figure S3). We found that the wild-type S. oneidensis MR-1 could grow on media containing ampicillin, indicating natural resistance to this antibiotic. Based on antibiotic sensitivity tests, two different genes conferring resistance to chloramphenicol and kanamycin were selected. Next, we addressed the use of the shuttle element oriT, an alternative to the mob gene, since transformation of S. oneidensis MR-1 relies on conjugation. Bacterial conjugation machinery is composed of RK2-oriT modules containing the traJ gene (Smillie et al., 2010), which is needed within a plasmid to enable its uptake by the recipient cell through conjugation. We introduced the RK2-oriT cassette, which is smaller than the mob gene, into the vector backbone and demonstrated successful transformation from E. coli to S. oneidensis MR-1. Using this efficient transfer system, longer genes can be accommodated and expressed in the vectors.
In order to be compatible with the commonly used vector pHG101, we used the chloramphenicol resistance (CmR) gene and the shuttle oriT gene to test various replication origins. We chose ColE, p15A, and pSC101, which are commonly used in E. coli, and repB, which was used as a replication origin in the shuttle vector pSXM33 from Shewanella xiamenensis BC01 (Zhou and Ng, 2016). We determined that shuttle vectors carrying ColE, p15A, or pSC101 could be efficiently imported into S. oneidensis MR-1. However, S. oneidensis MR-1 carrying repB could not grow due to failed transformation into E. coli and S. oneidensis MR-1. When considering and assessing replication origins, plasmid copy number is a key parameter. Hajimorad and Gralnick (2016) showed that different replication origins (p15A, pMB1, and pBBR1) could be maintained in Shewanella by counting colony forming unites (CFU). Here, we determined the copy number of different replicons using real-time quantitative PCR (RT-qPCR). As depicted in Figure 4A, the results indicated that the CoIE replication origin had a copy number of 54, which was higher than the other replication origins: pSC101 (40), p15A (33), and pBBR1 (23). Meanwhile, we also measured the corresponding fluorescence of strains carrying these replication origins (Figure 4B), demonstrating that fluorescence was in accordance with copy number.
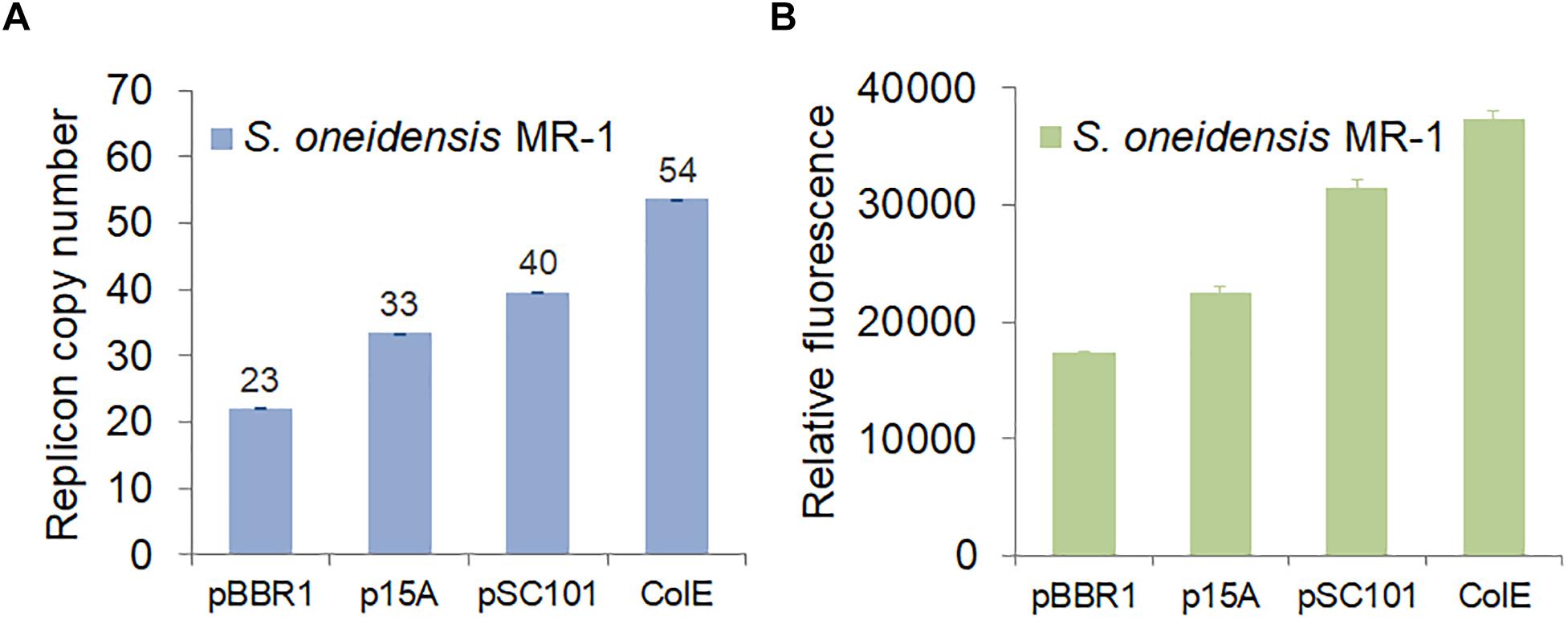
Figure 4. Characterization of replication origins in S. oneidensis MR-1. (A) Replicon copy numbers of different replication origins in S. oneidensis MR-1 determined by RT-qPCR. (B) GFP expression of various replication origins in S. oneidensis MR-1. S. oneidensis MR-1 harbored a single plasmid carrying pBBR1, p15A, pSC101, or ColE replication origin. In each case, the gfp gene was expressed from the same promoter, pTrc∗ (no repressor lacO). The error bars (mean ± SD) were derived from triplicate experiments for each strain.
While screening the replication origins, we also tested whether the origins ColE, p15A, and pSC101 were compatible with the commonly used replication origin, pBBR1, in S. oneidensis MR-1. We co-transformed S. oneidensis MR-1 with two vectors carrying different replication origins: pBBR1/ColE, pBBR1/p15A, and pBBR1/pSC101. Plasmids carrying the pBBR1 origin confer kanamycin resistance, while plasmids carrying the ColE, p15A, or pSC101 origin confer chloramphenicol resistance. Transfer and maintenance of either a single plasmid or two plasmids (containing two replication origins) in S. oneidensis MR-1 were verified by colony PCR (Figure 5) using primers listed in Supplementary Table S1. The expected lengths of PCR products were 840 bp, 1,145 bp, 1,132 bp, and 2,535 bp for pBBR1, CoIE, p15A, and pSC101, respectively. Agarose gel electrophoresis analysis confirmed band sizes, as shown from Lane 1 to Lane 4 in Figure 5. Lanes 5–10 depict three groups of two-plasmid containing replication origins: pBBR1/ColE (Lanes 5 and 6), pBBR1/p15A (Lanes 7 and 8), and pBBR1/pSC101 (Lanes 9 and 10). The results revealed compatibility of ColE/pBBR1, p15A/pBBR1, and pSC101/pBBR1 as well as successful co-transformation and maintenance of multiple plasmids in S. oneidensis MR-1.
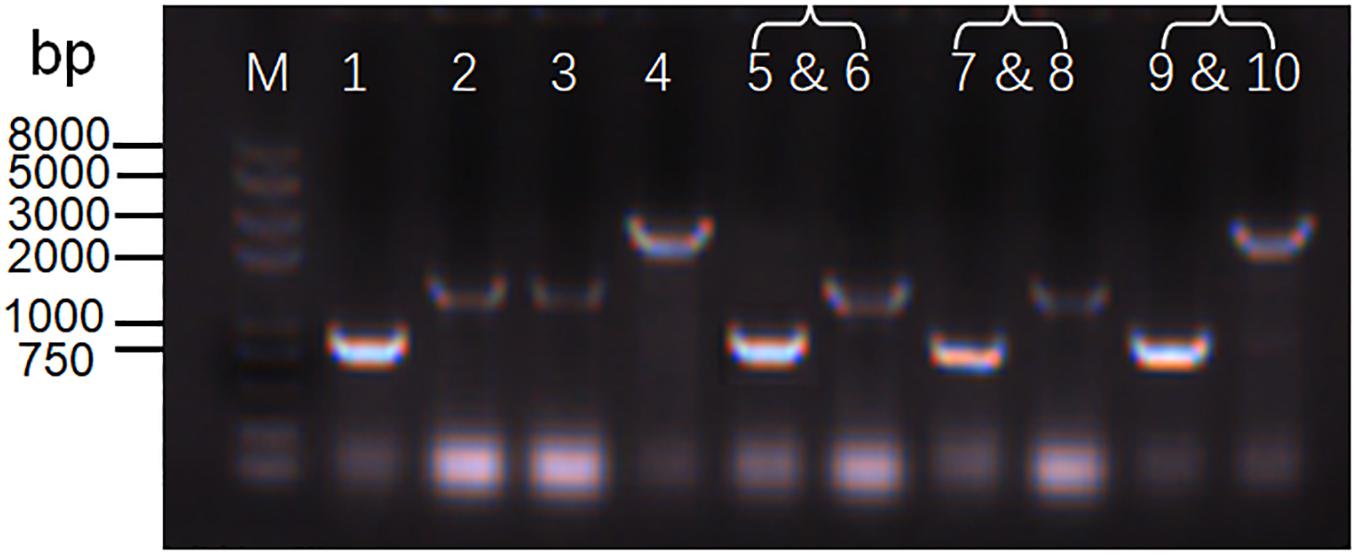
Figure 5. Determination of the coexistence of two plasmids in S. oneidensis MR-1. Agarose gel electrophoresis analysis of replication origins from a single plasmid or two plasmids in S. oneidensis MR-1. M: Trans2K Plus ll DNA Marker. Lanes 1–4: a single plasmid containing replication origin pBBR1 (Lane 1: 840 bp), ColE (Lane 2: 1,145 bp), p15A (Lane 3: 1,132 bp), and pSC101 (Lane 4: 2,535 bp). Lanes 5–10: three groups of two plasmids containing replication origins pBBR1/ColE (Lanes 5 and 6), pBBR1/p15A (Lanes 7 and Lane 8), and pBBR1/pSC101 (Lanes 9 and 10).
Fine-Tuning the Expression of Multiple Fluorescent Proteins in S. oneidensis
The above results demonstrated the utility of the developed plasmid toolkit, which enabled precise control of gene expression using different strength promoters, regulating the inducer concentration or induction time for inducible promoters, and using various replication origins with different copy numbers. Fine-tuning gene expression can be achieved by adopting suitable elements with different levels of expression according to the actual experimental needs. The next step was to test the ability of the modular plasmids to modulate the simultaneous expression of multiple genes in S. oneidensis MR-1. For this purpose, three fluorescent proteins (i.e., GFP, BFP, and CFP) were chosen and expressed in separated plasmids.
Two manipulation strategies were applied. Firstly, two different fluorescent genes were expressed using different types of promoters (e.g., IPTG-inducible or arabinose-inducible) with two-gene expression fine-tuned by adjusting the inducer concentrations. GFP was expressed using vectors with the IPTG-inducible promoter pTac, the pBBR1 replication origin, and the kanamycin resistance (Kan) gene. The following IPTG concentrations were assessed: 1, 12.5, 15, and 100 μM. BFP was expressed using vectors with the arabinose-inducible promoter pBAD, the ColE replication origin, and the chloramphenicol resistance (CmR) gene. The following arabinose concentrations were used: 0.1, 0.25, 0.5, and 1 mM. As shown in Figure 6A and Supplementary Figure S4, higher GFP fluorescence was observed for higher concentrations of IPTG and lower concentrations of arabinose. Likewise, higher BFP fluorescence was observed for higher concentrations of arabinose and lower concentrations of IPTG. Furthermore, similar results were obtained when BFP was replaced with CFP (Figure 6B). The only difference was that CFP expression level was relatively low (Supplementary Figure S5). In all, these results demonstrated that the expression of different genes in S. oneidensis MR-1 could be obtained by adjusting inducer concentrations for the inducible promoters.
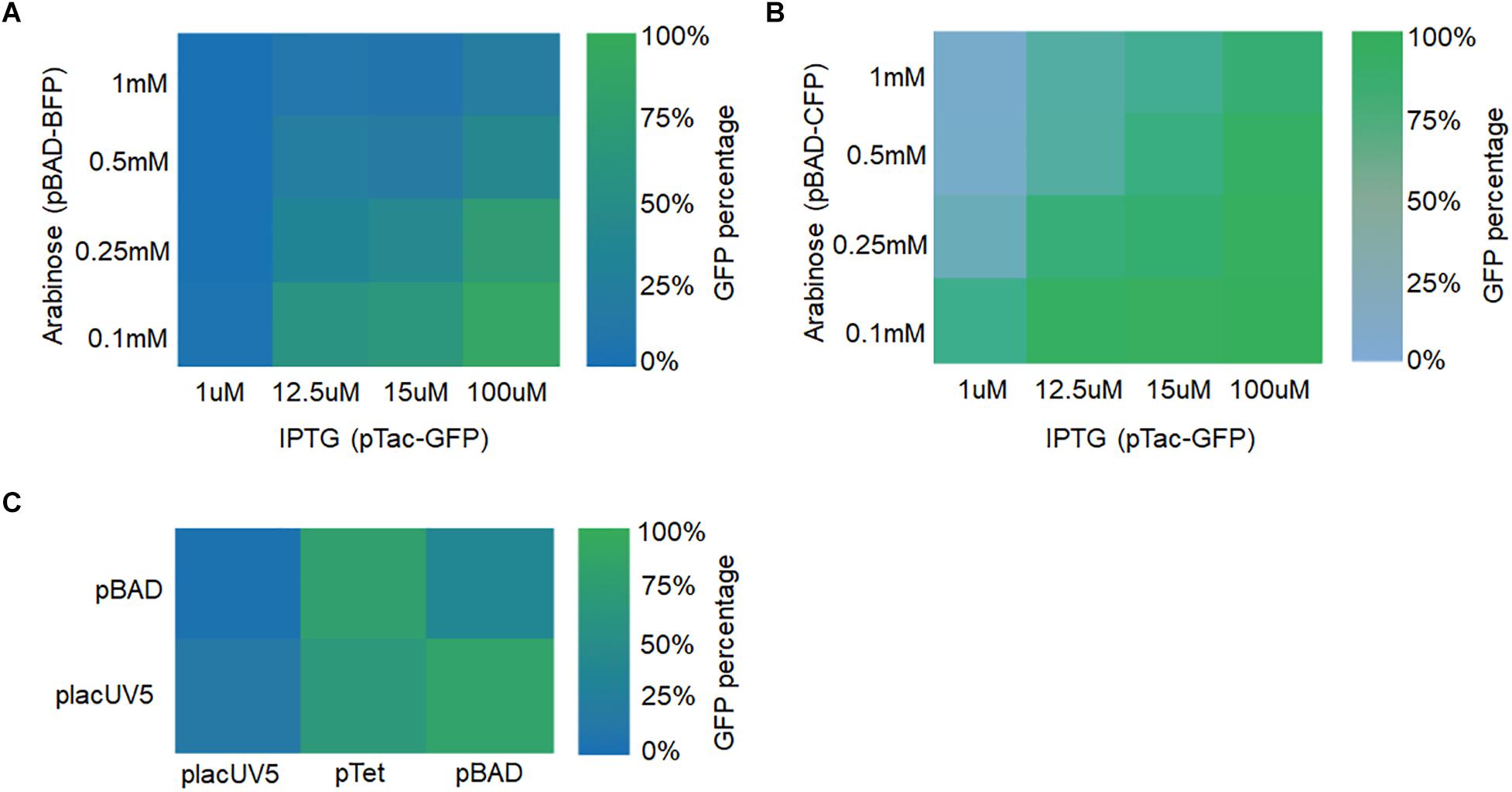
Figure 6. Fine-tuning multiple fluorescent protein expression in S. oneidensis MR-1 harboring two plasmids with replicons pBBR1 and CoIE. (A) GFP and BFP expression were regulated by the IPTG-inducible pTac promoter (IPTG concentrations: 1, 12.5, 15, and 100 μM) and the arabinose-inducible pBAD promoter (arabinose concentrations: 0.1, 0.25, 0.5, and 1 mM), respectively. (B) GFP and CFP expression were adjusted by the promoters pTac and pBAD at the above concentrations, respectively. (C) GFP and BFP expression were under the control of placUV5, pTet, or pBAD, and placUV5 or pBAD, respectively. The error bars (mean ± SD) were derived from triplicate experiments for each strain.
Secondly, fine-tuning of gene expression was achieved by utilizing different strength promoters (low, medium, and high) at their normal induction concentrations. GFP was expressed under the control of placUV5, pTet, and pBAD promoters on a plasmid containing the pBBR1 replication origin and the kanamycin resistance (Kan) gene. BFP was expressed under the control of placUV5 and pBAD promoters on a separate plasmid containing the ColE replication origin and the chloramphenicol resistance (CmR) gene. We co-transformed the two plasmid types into S. oneidensis MR-1. As depicted in Figure 6C and Supplementary Figure S6, when the GFP was expressed under the control of the high-strength pBAD promoter and BFP was expressed under the control of the low-strength placUV5 promoter, high GFP and low BFP were detected. Similarly, when the BFP was expressed under the control of the high-strength pBAD promoter and GFP was expressed under the control of the low-strength placUV5 promoter, low GFP and high BFP were detected. In all, different levels of gene expression could also be achieved by using promoters of different strengths. Overall, the plasmid toolkit developed here can be used to delicately fine-tune the simultaneous expression of multiple genes using different strategies in S. oneidensis.
Fine-Tuning MtrCAB Expression in the S. oneidensis ΔmtrA Strain
The EET pathway of S. oneidensis MR-1 is comprised of c-type cytochromes that shuttle electrons from oxidizing enzymes in the cytoplasm and inner membrane to the outside of the cell during anaerobic incubation (Schuetz et al., 2009; Dan et al., 2010; Gralnick and Newman, 2010; Ross et al., 2012). The c-type cytochrome MtrCAB complex is regarded as the primary route of EET and a potentially minimal set. In E. coli, heterologous expression and regulation of the genes in the Mtr pathway (Gescher et al., 2010; Jensen et al., 2010; Goldbeck et al., 2013) suggest that too much expression of MtrCAB could cause pleotropic impairments in the host organism and diminish EET efficiency. Thus, MtrCAB expression should be delicately fine-tuned to improve EET capacity.
To illustrate whether the plasmid toolkit could be applied in EET manipulation in S. oneidensis, the mtrCAB was taken as an example to be overexpressed. The mtrCAB gene cluster was cloned from the S. oneidensis genome and overexpressed in the S. oneidensis ΔmtrA strain under the control of placUV5, pTet, and pBAD promoters, which displayed low, medium, and high levels of gene expression, respectively. The parental and recombinant strains were named as S-ΔmtrA, L-mtrCAB, M-mtrCAB, and H-mtrCAB, respectively. Firstly, we performed a tungsten trioxide (WO3) reduction assay (Yuan et al., 2014) to assess the fine-tuning effect of mtrCAB on EET. Culture medium without bacteria was used as a control to eliminate disturbances caused by abiotic factors. EET from S-ΔmtrA, L-mtrCAB, M-mtrCAB, or H-mtrCAB to the WO3 nanorod assembly probe was accompanied by a bioelectrochromic reaction causing conspicuous color development in the 96-well plate, which enabled us to evaluate EET either qualitatively with the naked eye or quantitatively through image analysis (Figure 7A). For the latter, the color intensity of each well was determined by analyzing the ‘Density (mean)’ (Wang et al., 2009; Tatro et al., 2013) of the area of interest with ImageJ Software V.1.8.0 (Figure 7B). The results showed that the EET capacity of MtrCAB under the control of the low-, medium-, and high-strength promoters was increased by 22.3, 133.5, and 65.3% over that of the parental strain, suggesting that overexpression of MtrCAB at a medium level improved EET most significantly in the S. oneidensis ΔmtrA strain.
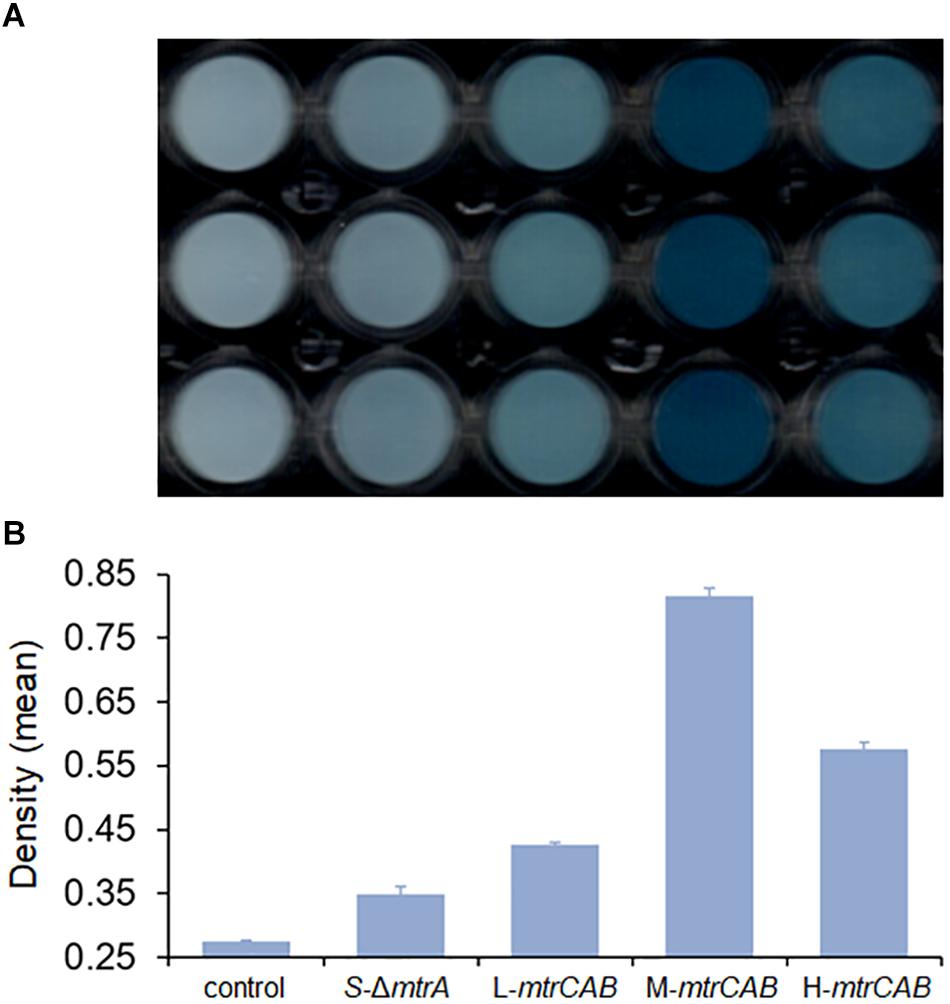
Figure 7. Fine-tuning MtrCAB expression to affect EET efficiency determined by WO3 reduction assay. (A) Color development of the WO3 reduction assay. Culture medium without bacteria was used as a control to eliminate disturbances caused by abiotic factors. The mtrCAB gene was overexpressed in the S. oneidensis ΔmtrA strain under the control of promoters placUV5, pTet, and pBAD, which were named as S-ΔmtrA, L-mtrCAB, M-mtrCAB, and H-mtrCAB, respectively, according to their level of expression (L, low; M, medium; H, high). (B) ‘Density (mean)’ analysis of each strain from the WO3 reduction assay using ImageJ Software V.1.8.0. The error bars (mean ± SD) were derived from triplicate experiments for each strain.
To more quantitatively investigate EET efficiency upon fine-tuning the overexpression of MtrCAB, bio-electrochemical analyses were conducted in microbial fuel cells (MFCs). The parental and engineered strains were inoculated into the anodic chamber of MFCs, respectively, with a 2 kΩ external resistor (Figure 8). Experimental results showed that MtrCAB expression in the mutant S. oneidensis could increase the voltage output, with significant differences among the recombinant strains. The maximum voltage increased 140.3, 595.8, and 443.2%, when the mtrCAB complex was expressed at low, medium and high levels, respectively, compared to the parental S. oneidensisΔmtrA strain (Figure 8A). Bioelectrochemical analyses were further conducted to study EET efficiency of these recombinant strains in MFCs (Figure 8B). Polarization discharge curves and power density output curves were obtained by linear sweep voltammetry (LSV) with a 0.1 mV/s scan rate. The dropping slope of the polarization curve obtained from M-mtrCAB (shown in the blue curve) was smaller than that of the other two recombinant strains (L-mtrCAB shown in red; and H-mtrCAB, shown in magenta), implying that the internal charge transfer resistance of the MFC inoculated with M-mtrCAB was relatively smaller. Furthermore, the power density output curves showed that the M-mtrCAB strain obtained a maximum power density of ∼17.0 mW/m2, which was ∼1.8, ∼3.1, and ∼4.5 times higher than that of H-mtrCAB, L-mtrCAB, and S-ΔmtrA, respectively (Figure 8B). These MFC results demonstrated that EET efficiency was improved most significantly when mtrCAB was expressed using a moderate promoter in the mutant S. oneidensis, which was consistent with the WO3 assay analyses. These results validate that the plasmid toolkit we developed could indeed be applied to manipulate EET and metabolism in S. oneidensis.
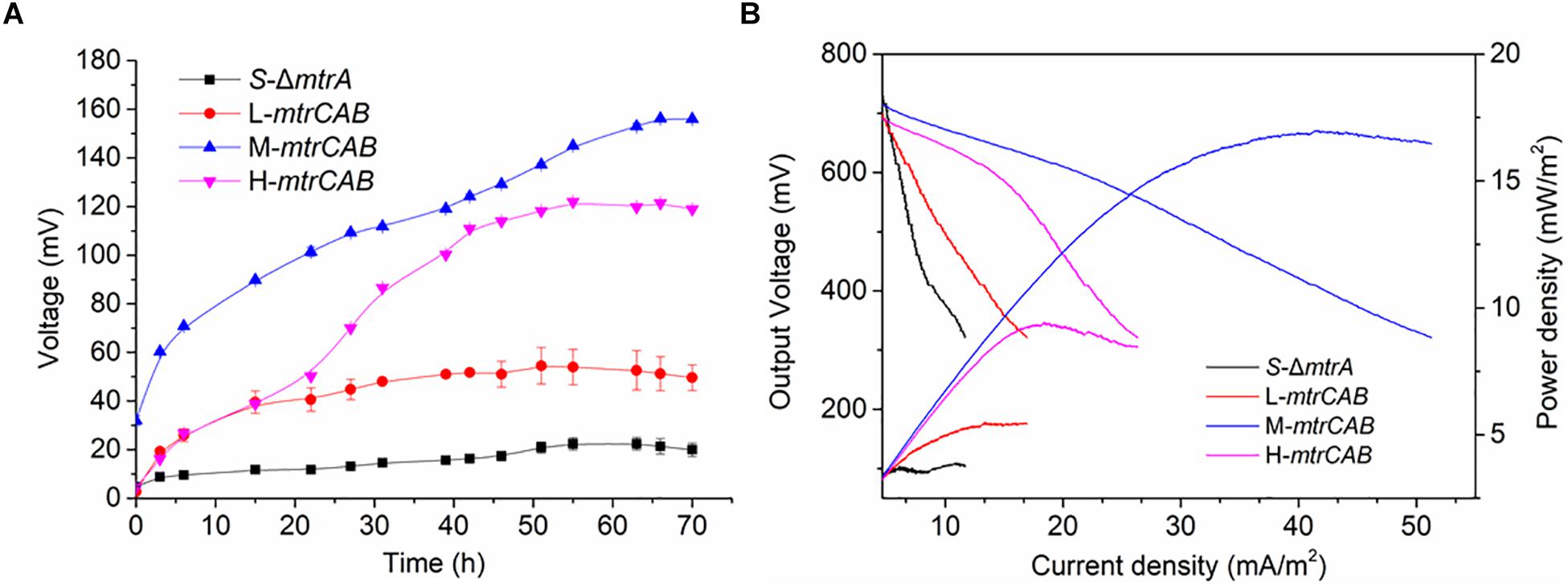
Figure 8. Fine-tuning MtrCAB expression to affect EET efficiency determined by MFC assay. (A) Voltage output of the S-ΔmtrA, L-mtrCAB, M-mtrCAB, and H-mtrCAB strains in MFCs. (B) MFC polarization curves and power density output curves obtained by linear sweep voltammetry (LSV) with a scan rate of 0.1 mV/s. The error bars (mean ± SD) were derived from triplicate experiments for each strain.
Conclusion
We constructed and characterized a synthetic plasmid toolkit for S. oneidensis MR-1, which contains a set of shuttle vectors with a combination of promoters, replication origins, antibiotic resistance genes, and a shuttle component. We quantified the strength of promoters and the copy number of different replicons. We further transformed two plasmids conferring different antibiotic resistances into recombinant S. oneidensis MR-1 strains, enabling the expression of multiple genes (e.g., fluorescent reporters) to be fine-tuned. The synthetic plasmid toolkit was then successfully used to modulate the expression of MtrCAB and achieve improved EET efficiency. Moderate overexpression level of MtrCAB improved the EET efficiency most significantly in the S. oneidensis ΔmtrA strain. The plasmid toolkit developed in this study allows researchers to rapidly and conveniently fine-tune gene expressions with greater controllability and predictability, thus accelerating the development and application of genetic manipulations in S. oneidensis.
Data Availability
This manuscript contains previously unpublished data. The name of the repository and accession number are not available.
Author Contributions
Y-XC designed the experiments. M-YS and Y-XC performed all the experiments. Y-RC, Y-YC, JX, and QD performed a part of MFC experiments. FL, C-FL, and XL helped with revising the manuscript. M-YS, Y-XC, and HS wrote the manuscript.
Funding
This research was supported by the National Natural Science Foundation of China (21621004 and 21506153).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00410/full#supplementary-material
References
Ajikumar, P. K., Xiao, W. H., Tyo, K. E., Wang, Y., Simeon, F., Leonard, E., et al. (2010). Isoprenoid pathway optimization for Taxol precursor overproduction in Escherichia coli. Science 330, 70–74. doi: 10.1126/science.1191652
Anthony, J. R., Anthony, L. C., Nowroozi, F., Kwon, G., Newman, J. D., and Keasling, J. D. (2009). Optimization of the mevalonate-based isoprenoid biosynthetic pathway in Escherichia coli for production of the anti-malarial drug precursor amorpha-4,11-diene. Metab. Eng. 11, 13–19. doi: 10.1016/j.ymben.2008.07.007
Bond-Watts, B. B., Bellerose, R. J., and Chang, M. C. Y. (2011). Enzyme mechanism as a kinetic control element for designing synthetic biofuel pathways. Nat. Chem. Biol. 7, 222–227. doi: 10.1038/nchembio.537
Bretschger, O., Obraztsova, A., Sturm, C. A., Chang, I. S., Gorby, Y. A., Reed, S. B., et al. (2008). Current production and metal oxide reduction by Shewanella oneidensis MR-1 wild type and mutants (vol 73, pg 7003, 2007). Appl. Environ. Microbiol. 74, 553–562. doi: 10.1128/Aem.02560-07
Canton, B., Labno, A., and Endy, D. (2008). Refinement and standardization of synthetic biological parts and devices. Nat. Biotechnol. 26, 787–793. doi: 10.1038/nbt1413
Cao, Y., Li, X., Li, F., and Song, H. (2017). CRISPRi-sRNA: transcriptional-translational regulation of extracellular electron transfer in Shewanella oneidensis. ACS Synth. Biol. 6, 1679–1690. doi: 10.1021/acssynbio.6b00374
Chae, T. U., Choi, S. Y., Kim, J. W., Ko, Y. S., and Lee, S. Y. (2017). Recent advances in systems metabolic engineering tools and strategies. Curr. Opin. Biotechnol. 47, 67–82. doi: 10.1016/j.copbio.2017.06.007
Dan, C., Baron, D. B., Bond, D. R., and Gralnick, J. A. (2010). The Mtr respiratory pathway is essential for reducing flavins and electrodes in Shewanella oneidensis. J. Bacteriol. 192, 467–474. doi: 10.1128/JB.00925-09
Daraselia, N., Dernovoy, D., Tian, Y., Borodovsky, M., Tatusov, R., and Tatusova, T. (2003). Reannotation of Shewanella oneidensis genome. OMICS 7, 171–175. doi: 10.1089/153623103322246566
Dhanasekaran, S., Doherty, T. M., Kenneth, J., and Group, T. B. T. S. (2010). Comparison of different standards for real-time PCR-based absolute quantification. J. Immunol. Methods 354, 34–39. doi: 10.1016/j.jim.2010.01.004
Gescher, J. S., Cordova, C. D., and Spormann, A. M. (2010). Dissimilatory iron reduction in Escherichia coli: identification of CymA of Shewanella oneidensis and NapC of E. coli as ferric reductases. Mol. Microbiol. 68, 706–719. doi: 10.1111/j.1365-2958.2008.06183.x
Goldbeck, C. P., Jensen, H. M., TerAvest, M. A., Beedle, N., Appling, Y., Hepler, M., et al. (2013). Tuning promoter strengths for improved synthesis and function of electron conduits in Escherichia coli. ACS Synth. Biol. 2, 150–159. doi: 10.1021/sb300119v
Golitsch, F., Bucking, C., and Gescher, J. (2013). Proof of principle for an engineered microbial biosensor based on Shewanella oneidensis outer membrane protein complexes. Biosens. Bioelectron. 47, 285–291. doi: 10.1016/j.bios.2013.03.010
Gralnick, J. A., and Newman, D. K. (2010). Extracellular respiration. Mol. Microbiol. 65, 1–11. doi: 10.1111/j.1365-2958.2007.05778.x
Hajimorad, M., and Gralnick, J. A. (2016). Towards enabling engineered microbial-electronic systems: RK2-based conjugal transfer system for Shewanella synthetic biology. Electron. Lett. 52, 426–428. doi: 10.1049/el.2015.3226
Heidelberg, J. F., Paulsen, I. T., Nelson, K. E., Gaidos, E. J., Nelson, W. C., Read, T. D., et al. (2002). Genome sequence of the dissimilatory metal ion-reducing bacterium Shewanella oneidensis. Nat. Biotechnol. 20, 1118–1123. doi: 10.1038/nbt749
Hou, H. J., Li, L., Cho, Y., de Figueiredo, P., and Han, A. (2009). Microfabricated microbial fuel cell arrays reveal electrochemically active microbes. PLoS One 4:e6570. doi: 10.1371/journal.pone.0006570
Hou, H. J., Li, L., de Figueiredo, P., and Han, A. (2011). Air-cathode microbial fuel cell array: a device for identifying and characterizing electrochemically active microbes. Biosens. Bioelectron. 26, 2680–2684. doi: 10.1016/j.bios.2010.06.037
Jensen, H. M., Albers, A. E., Malley, K. R., Londer, Y. Y., Cohen, B. E., Helms, B. A., et al. (2010). Engineering of a synthetic electron conduit in living cells. Proc. Natl. Acad. Sci. U.S.A. 107, 19213–19218. doi: 10.1073/pnas.1009645107
Juminaga, D., Baidoo, E. E., Redding-Johanson, A. M., Batth, T. S., Burd, H., Mukhopadhyay, A., et al. (2012). Modular engineering of L-tyrosine production in Escherichia coli. Appl. Environ. Microbiol. 78, 89–98. doi: 10.1128/AEM.06017-11
Keasling, J. D. (2012). Synthetic biology and the development of tools for metabolic engineering. Metab. Eng. 14, 189–195. doi: 10.1016/j.ymben.2012.01.004
Kouzuma, A., Kasai, T., Hirose, A., and Watanabe, K. (2015). Catabolic and regulatory systems in Shewanella oneidensis MR-1 involved in electricity generation in microbial fuel cells. Front. Microbiol. 6:609. doi: 10.3389/fmicb.2015.00609
Kouzuma, A., Meng, X. Y., Kimura, N., Hashimoto, K., and Watanabe, K. (2010). Disruption of the putative cell surface polysaccharide biosynthesis gene SO3177 in Shewanella oneidensis MR-1 enhances adhesion to electrodes and current generation in microbial fuel cells. Appl. Environ. Microbiol. 76, 4151–4157. doi: 10.1128/Aem.00117-10
Kumar, A., Hsu, L. H. H., Kavanagh, P., Barriere, F., Lens, P. N. L., Lapinsonniere, L., et al. (2017). The ins and outs of microorganism-electrode electron transfer reactions. Nat. Rev. Chem. 1, 0024–0082. doi: 10.1038/s41570-017-0024
Lee, C., Kim, J., Shin, S. G., and Hwang, S. (2006). Absolute and relative QPCR quantification of plasmid copy number in Escherichia coli. J. Biotechnol. 123, 273–280. doi: 10.1016/j.jbiotec.2005.11.014
Li, F., Li, Y., Sun, L., Chen, X., An, X., Yin, C., et al. (2018a). Modular engineering intracellular NADH regeneration boosts extracellular electron transfer of Shewanella oneidensis MR-1. ACS Synth. Biol. 7, 885–895. doi: 10.1021/acssynbio.7b00390
Li, F., Yin, C., Sun, L., Li, Y., Guo, X., and Song, H. (2018b). Synthetic Klebsiella pneumoniae-Shewanella oneidensis consortium enables glycerol-fed high-performance microbial fuel cells. Biotechnol. J. 13:e1700491. doi: 10.1002/biot.201700491
Lin, T., Ding, W. Q., Sun, L. M., Wang, L., Liu, C. G., and Song, H. (2018). Engineered Shewanella oneidensis-reduced graphene oxide biohybrid with enhanced biosynthesis and transport of flavins enabled a highest bioelectricity output in microbial fuel cells. Nano Energy 50, 639–648. doi: 10.1016/j.nanoen.2018.05.072
Ross, D. E., Flynn, J. M., Baron, D. B., Gralnick, J. A., and Bond, D. R. (2012). Towards electrosynthesis in Shewanella: energetics of reversing the mtr pathway for reductive metabolism. PLoS One 6:e16649. doi: 10.1371/journal.pone.0016649
Schuetz, B., Schicklberger, M., Kuermann, J., Spormann, A., and Gescher, J. (2009). Periplasmic electron transfer via the c-Type cytochromes MtrA and FccA of Shewanella oneidensis MR-1. Appl. Environ. Microbiol. 75, 7789–7796. doi: 10.1128/AEM.01834-09
Shetty, R. P., Endy, D., and Knight, T. F. Jr. (2008). Engineering BioBrick vectors from BioBrick parts. J. Biol. Eng. 2:5. doi: 10.1186/1754-1611-2-5
Shi, L., Dong, H. L., Reguera, G., Beyenal, H., Lu, A. H., Liu, J., et al. (2016). Extracellular electron transfer mechanisms between microorganisms and minerals. Nat. Rev. Microbiol. 14, 651–662. doi: 10.1038/nrmicro.2016.93
Smillie, C., Garcillan-Barcia, M. P., Francia, M. V., Rocha, E. P. C., and de la Cruz, F. (2010). Mobility of plasmids. Microbiol. Mol. Biol. Rev. 74, 434–452. doi: 10.1128/Mmbr.00020-10
Tatro, E. T., Risbrough, V., Soontornniyomkij, B., Young, J., Shumaker-Armstrong, S., Jeste, D. V., et al. (2013). Short-term recognition memory correlates with regional CNS expression of microRNA-138 in mice. Am. J. Geriatr. Psychiatry 21, 461–473. doi: 10.1016/j.jagp.2012.09.005
Venkateswaran, K., Moser, D. P., Dollhopf, M. E., Lies, D. P., Saffarini, D. A., MacGregor, B. J., et al. (1999). Polyphasic taxonomy of the genus Shewanella and description of Shewanella oneidensis sp. nov. Int. J. Syst. Bacteriol. 49, 705–724. doi: 10.1099/00207713-49-2-705
Wang, C. J., Zhou, Z. G., Holmqvist, A., Zhang, H., Li, Y., Adell, G., et al. (2009). Survivin expression quantified by image pro-plus compared with visual assessment. Appl. Immunohistochem. Mol. Morphol. 17, 530–535. doi: 10.1097/Pai.0b013e3181a13bf2
West, E. A., Jain, A., and Gralnick, J. A. (2017). Engineering a native inducible expression system in Shewanella oneidensis to control extracellular electron transfer. ACS Synth. Biol. 6, 1627–1634. doi: 10.1021/acssynbio.6b00349
Whelan, J. A., Russell, N. B., and Whelan, M. A. (2003). A method for the absolute quantification of cDNA using real-time PCR. J. Immunol. Methods 278, 261–269. doi: 10.1016/s0022-1759(03)00223-0
Yang, Y., Ding, Y., Hu, Y., Cao, B., Rice, S. A., Kjelleberg, S., et al. (2015). Enhancing bidirectional electron transfer of Shewanella oneidensis by a synthetic flavin pathway. ACS Synth. Biol. 4, 815–823. doi: 10.1021/sb500331x
Yu, Y., Lee, C., Kim, J., and Hwang, S. (2005). Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol. Bioeng. 89, 670–679. doi: 10.1002/bit.20347
Yuan, S. J., Li, W. W., Cheng, Y. Y., He, H., Chen, J. J., Tong, Z. H., et al. (2014). A plate-based electrochromic approach for the high-throughput detection of electrochemically active bacteria. Nat. Protoc. 9, 112–119. doi: 10.1038/nprot.2013.173
Keywords: Shewanella oneidensis MR-1, plasmid toolkit, BioBrick, c-type cytochrome, fine-tuning, synthetic biology
Citation: Cao Y-X, Song M-Y, Li F, Li C-F, Lin X, Chen Y-R, Chen Y-Y, Xu J, Ding Q and Song H (2019) A Synthetic Plasmid Toolkit for Shewanella oneidensis MR-1. Front. Microbiol. 10:410. doi: 10.3389/fmicb.2019.00410
Received: 23 November 2018; Accepted: 18 February 2019;
Published: 08 March 2019.
Edited by:
Pascal E. Saikaly, King Abdullah University of Science and Technology, Saudi ArabiaReviewed by:
Jeffrey A. Gralnick, University of Minnesota Twin Cities, United StatesKrishna Prasad Katuri, King Abdullah University of Science and Technology, Saudi Arabia
Copyright © 2019 Cao, Song, Li, Li, Lin, Chen, Chen, Xu, Ding and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Song, aHNvbmdAdGp1LmVkdS5jbg==