 Ratree Takhampunya1*
Ratree Takhampunya1* Achareeya Korkusol1Chalermpol Pongpichit2Komsan Yodin2Artharee Rungrojn1
Achareeya Korkusol1Chalermpol Pongpichit2Komsan Yodin2Artharee Rungrojn1 Nitima Chanarat1Sommai Promsathaporn1Taweesak Monkanna1Sasikanya Thaloengsok1Bousaraporn Tippayachai1Naruemon Kumfao2Allen L. Richards3Silas A. Davidson1
Nitima Chanarat1Sommai Promsathaporn1Taweesak Monkanna1Sasikanya Thaloengsok1Bousaraporn Tippayachai1Naruemon Kumfao2Allen L. Richards3Silas A. Davidson1- 1Department of Entomology, US Army Medical Directorate of the Armed Forces Research Institute of Medical Sciences (USAMD-AFRIMS), Bangkok, Thailand
- 2Bo Kluea Hospital, Nan, Thailand
- 3Viral and Rickettsial Diseases Department, Naval Medical Research Center, Silver Spring, MD, United States
In this study, we used a metagenomic approach to analyze bacterial communities from diverse populations (humans, animals, and vectors) to investigate the role of these microorganisms as causative agents of disease in human and animal populations. Wild rodents and ectoparasites were collected from 2014 to 2018 in Nan province, Thailand where scrub typhus is highly endemic. Samples from undifferentiated febrile illness (UFI) patients were obtained from a local hospital. A total of 200 UFI patient samples were obtained and 309 rodents and 420 pools of ectoparasites were collected from rodents (n = 285) and domestic animals (n = 135). The bacterial 16S rRNA gene was amplified and sequenced with the Illumina. Real-time PCR and Sanger sequencing were used to confirm the next-generation sequencing (NGS) results and to characterize pathogen species. Several pathogens were detected by NGS in all populations studied and the most common pathogens identified included Bartonella spp., Rickettsia spp., Leptospira spp., and Orientia tsutsugamushi. Interestingly, Anaplasma spp. was detected in patient, rodent and tick populations, although they were not previously known to cause human disease from this region. Candidatus Neoehrlichia, Neorickettsia spp., Borrelia spp., and Ehrlichia spp. were detected in rodents and their associated ectoparasites. The same O. tsutsugamushi genotypes were shared among UFI patients, rodents, and chiggers in a single district indicating that the chiggers found on rodents were also likely responsible for transmitting to people. Serological testing using immunofluorescence assays in UFI samples showed high prevalence (IgM/IgG) of Rickettsia and Orientia pathogens, most notably among samples collected during September–November. Additionally, a higher number of seropositive samples belonged to patients in the working age population (20–60 years old). The results presented in this study demonstrate that the increased risk of human infection or exposure to chiggers and their associated pathogen (O. tsutsugamushi) resulted in part from two important factors; working age group and seasons for rice cultivation and harvesting. Evidence of pathogen exposure was shown to occur as there was seropositivity (IgG) in UFI patients for bartonellosis as well as for anaplasmosis. Using a metagenomic approach, this study demonstrated the circulation and transmission of several pathogens in the environment, some of which are known causative agents of illness in human populations.
Introduction
Most public health surveillance systems and laboratories rely on serological and molecular assays that were developed to detect specific pathogens. However, conventional laboratory assays are often ineffective at detecting all causative agents of disease. Studies have shown that 40% of gastroenteritis cases (Finkbeiner et al., 2008) and as many as 60% of encephalitis cases (Ambrose et al., 2011) went undetected by conventional laboratory testing. Pathogens can go undetected if they are novel or are not known to previously occur in an area. There are many examples of the emergence of novel pathogens or reemergence of known organisms in new places where the available surveillance systems were inadequate, such as occurred with outbreaks of H7N9 influenza (Gao et al., 2013), Middle East respiratory syndrome coronavirus (MERS-CoV) (van Boheemen et al., 2012; Kindler et al., 2013), and the severe acute respiratory syndrome (SARS) outbreak in 2003 (Wang and Jolly, 2004).
Conventional diagnostic tests used by most reference laboratories require culture, microscopy, serology, and polymerase chain reaction (PCR). Such tools are useful for pathogen detection but only if culture conditions, test sensitivity, and primers are compatible and suitable for the microbial target. Other molecular approaches can be used to capture a wider range of pathogenic species such as multiplex PCR that targets highly conserved DNA regions or multiplex assays that target many of the most common pathogens known to cause similar symptoms. However, it is worth noting that even when multiplex assays are used, pathogens not included in the multiplexing may go undetected. The use of 16S rDNA was first proposed by Woese and Fox (1977) and Woese et al. (1990) as a tool for the molecular identification and characterization of microorganisms. The 16S rDNA gene is highly conserved among prokaryotes and some parts of its sequence are hypervariable between species, which makes it an ideal marker for species identification and for understanding evolutionary relationships (Gill et al., 2006; Sogin et al., 2006; Dethlefsen et al., 2008; McInerney et al., 2008; Tringe and Hugenholtz, 2008; Sunagawa et al., 2009). Metagenomics allows for comparisons of genetic material from multiple samples. One of the most common metagenomic approaches is deep amplicon sequencing (DAS), which employs universal primer to amplify parts of the 16S rRNA gene from specimens. A major benefit of metagenomics is the simultaneous detection of all microorganisms in clinical samples without prior knowledge of their identities. In addition, metagenomics has the potential to detect rare and novel pathogens. Current surveillance assays are limited in their ability to detect the emergence of novel pathogens or ones not previously known to be present in a given region. Metagenomic approaches can fulfill such gaps by identifying unknown etiological agents and assisting in the development of a new test for pathogen detection (Miller et al., 2013; Mokili et al., 2013; Wan et al., 2013).
Metagenomic approaches are especially suitable for zoonotic diseases. It is estimated that more than 60% of human pathogens are of animal origin (Taylor et al., 2001). Rodents are major reservoirs that account for a wide range of emerging zoonotic diseases in humans and livestock (Jones et al., 2008; Meerburg et al., 2009). Co-infection of multiple pathogens within individual rodents is frequently observed and the interaction between pathogens can have significant effects (Cox, 2001). Such co-infections can cause rodents to be more or less susceptible to other microparasites (Tadin et al., 2012). Generally, multiple infections in wildlife can increase disease severity in a host (Lello et al., 2005), affecting the survival and reproduction of animal hosts (Davidar and Morton, 2006; Holmstad et al., 2008). Disease surveillance in rodents and other wildlife can provide important information for public health preparedness. Surveillance can also be used to measure biodiversity and disease emergence which are both directly linked to the stability of ecosystems (Keesing et al., 2010; Grogan et al., 2014). Metagenomic approaches combined with NGS can be powerful tools to disentangle complex patterns of pathogen transmission among ectoparasites, animal reservoirs, and humans. For example, NGS has been used to perform blood meal analysis to determine the wide-range of animals that vectors feed on and possible reservoirs (Alcaide et al., 2009). NGS has also been useful in finding unexpected pathogens not normally associated with particular vectors (Vayssier-Taussat et al., 2013) and has been used to show the genetic diversity of bacteria that are specific to certain animal hosts and vectors (Pierlé et al., 2014; Swei et al., 2015). Such information can be used to correlate infections in people with important vectors and reservoir hosts.
In this study, metagenomics and NGS technology were used to characterize human (patients with undifferentiated febrile illness (UFI)), reservoir host (rodents and small mammals), and ectoparasite (chiggers, ticks, fleas, and lice) populations for bacterial pathogens. All samples were collected from Nan province in northern Thailand. Since all samples were from the same sites, bacteria could be compared from different populations to determine potential vectors and reservoirs. Nan province is highly endemic for scrub typhus, caused by the agent Orientia tsutsugamushi, and one of the major goals of this study was to determine the etiology and transmission dynamics of scrub typhus in the area. Another goal was to identify other bacterial pathogens that were under-reported or not previously known from this region. NGS results were verified by conventional methods such as real-time PCR, PCR, and DNA sequencing to confirm the pathogenic potential of detected bacteria and to better characterize those important pathogens to the species level. In addition, serological tests were performed to determine the seroprevalence and the history of human exposure to the pathogens detected by the NGS approach. The in-depth characterization of bacteria performed in this study from humans, animal hosts, and ectoparasites allowed us to determine the transmission dynamics of pathogens and identify several new and previously unreported pathogens from this area.
Materials and Methods
Ethics Statement
Rodents were trapped according to the institutional animal collection protocol entitled “Field Sampling of Small Mammal (Orders: Erinaceomorpha, Soricomorpha, Scandentia, Macroscelidea, and Rodentia) Populations to Support Zoonotic Disease Surveillance and Ectoparasite Collection” (PN# 12–06), reviewed and approved by the USAMC-AFRIMS Institutional Animal Care and Use Committee (IACUC). All sampling procedures and experimental manipulations were reviewed and approved as part of the animal collection protocol (PN# 12–06). Research was conducted in compliance with the Animal Welfare Act and other federal statutes and regulations related to animals and experiments involving animals, and adhered to principles outlined in the “Guide for the Care and Use of Laboratory Animals,” NRC Publication, 2011 edition.
Undifferentiated Febrile Illness (UFI) Patients
A total of 200 individual UFI patients’ coded specimens were received from Bo Kluea hospital, Nan province, Thailand. Samples were from outpatients and inpatients presenting to Bo Kluea hospital with UFI and suspicion of scrub typhus infection during February–November 2017. Residual whole blood and serum samples from routine laboratory testing were coded and sent to the Department of Entomology to be tested for the possible causative agent of UFI as well as for scrub typhus or murine typhus infection, caused by Rickettsia typhi. The protocol was determined on February 01, 2016 by WRAIR Human Subjects Protection Branch (HSPB) to be research not involving human subjects for this investigation, since the work described herein involves the use of existing, coded specimens wherein investigators will not receive associated identifiable data, the project did not require a review by the Institutional Review Board (IRB) and 45 CFR 46 and 32 CFR 219 does not apply.
Study Locations
Rodents and ectoparasites were collected during the wet (June–September) and dry (November–April) seasons in Bo Kluea, Mae Charim, and Phu Phiang districts of Nan province, Thailand in 2014–2018 (Figure 1 and Table 1). Ectoparasites were also collected from domesticated mammals (dogs, cats, and cattle). All study sites were on private land, and permission was obtained from each of the owners to conduct research on their land. None of the field studies involved endangered or protected species. Rodents were captured using live traps baited with bananas, palm fruit, or dried fish, and were collected from orchards, palm and rubber plantations, cultivated rice-fields, grassland areas, edges of dense forest, stream margins, and around dwellings. Traps were set for 3–5 nights and were checked early in the morning. Captured rodents were removed from the traps, euthanized using carbon dioxide, and processed immediately at the site of collection. Blood, serum, and tissue samples (liver, spleen, kidney, and lung) were collected and stored on dry ice. Ears were removed and stored in 70% ethanol for chigger collection. All tissues were then transported to the AFRIMS laboratory for further processing. All rodents were later identified to the species level as described previously (Muul, 1979).
Figure 1. Map of collection sites; Bo Kluea, Mae Charim, and Phu Phiang Districts, Nan province, Thailand.
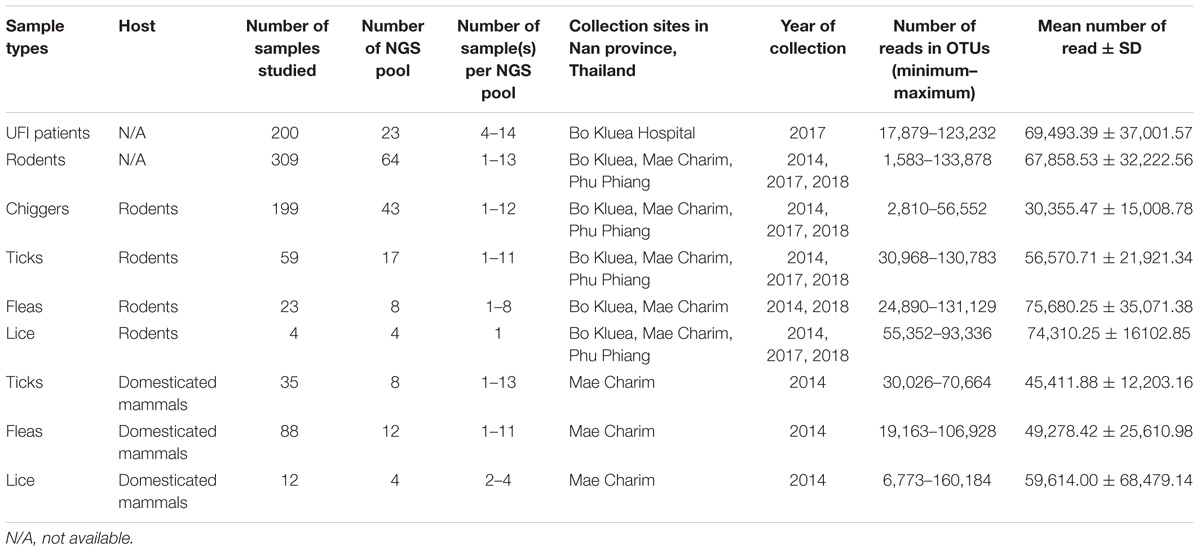
Table 1. Summary of sample description, sample size, pooling, and NGS coverage.
Genomic DNA Extraction
UFI Whole Blood
Genomic DNA was extracted from whole blood samples by automated extraction machine, a QIAsymphony® SP instrument (Qiagen, Hombrechtikon, Switzerland) with QIAsymphony® DNA Mini Kit (Qiagen, Germany). For each patient, 250 μl of whole blood was used for the DNA extraction with DNA Blood 200 DSP protocol. DNA was eluted in 50 μl and stored at −20°C until use. Ultrapure DNA/RNA-free distilled water was also included in every extraction procedure as an extraction control.
Rodent Tissue
Spleen and kidney tissues from each rodent were cut into pieces (∼3 mm in diameter) and added to 230 μl of ATL Tissue Lysis Buffer and 20 μl of Proteinase K solution (20 mg/ml), then incubated at 55°C for 1 h or until the tissues were homogenized. A total volume of 250 μl homogenized solution was then used for DNA extraction on the QIAsymphony® SP with QIAsymphony® DNA Mini Kit and Tissue HC 200 DSP protocol. The DNA was eluted in 200 μl and stored at −20°C until use. Ultrapure DNA/RNA-free distilled water was also included as an extraction control.
Ectoparasite Morphological Identification and DNA Extraction
Ectoparasites (chiggers, ticks, fleas, and lice) collected from rodents and small mammals were morphologically identified and pooled by genus, the host species they were collected from, and ectoparasitic stage. Chiggers were identified to genus level using a taxonomic key (Nadchatram, 1974). Other ectoparasites (fleas, ticks, and lice) were identified morphologically (Hopkins and Miriam, 1953; Tanskull and Inlao, 1989; Durden and Musser, 1994) and pooled by the host species they were collected from, type, stage, and gender of ectoparasites. Each pool was subjected to genomic DNA extraction using a modified protocol of QIAamp DNA Mini Kit (Qiagen). Briefly, ectoparasites in 180 μl of ATL buffer were punctured with a fine needle under a stereomicroscope to release the tissue from the hard chitin exoskeleton prior to adding 20 μl of Proteinase K solution (20 mg/ml). Samples were then incubated at 55°C for 1 h or until the ectoparasites were homogenized. A volume of 200 μl of AL buffer was added to the sample and the sample mixed and incubated at 70°C for 10 min. Then 100 μl of absolute ethanol was added to precipitate DNA. The solution was transferred to a QIAamp DNA column then centrifuged at 8,000 rpm for 1 min. The supernatant was discarded. DNA was washed twice with 500 μl of AW1 and AW2, respectively. The DNA was eluted at 50 μl of AE buffer and stored at −20°C until used. Ultrapure DNA/RNA-free distilled water was also included as an extraction control.
Amplification of Bacterial 16S DNA
Following DNA extraction of patient whole blood and rodent tissue, bacterial-specific 16S rDNA (V3–V4, a 550 bp fragment) was amplified in three replicates using the universal bacterial primer set; 16S amplicon PCR Forward primer (TCGTCGGCA GCGTCAGATGTGTATAAGAGACAG CCTACGGGNGGCW GCAG) and Reverse primer (GTCTCGTGGGCTCGGAG ATGTGTATAAGAGACAG GACTACHVGGGTATCTAATCC) (gene- specific sequences are underlined). PCRs were performed in a 20-μl volume containing 5 μl (1–100 ng/μL) of DNA template, 400 nM each primer, 200 μM dNTPs, 1.5 mM MgCl2, 1× PCR buffer, and 0.4 U of iProof High-Fidelity DNA Polymerase (Bio-Rad, Hercules, CA, United States). Amplification was performed using a T100 DNA thermal cycler (Bio-Rad) under the following conditions: initial denaturation at 98°C for 3 min; 40 cycles of 98°C for 10 s, 60°C for 20 s, and 72°C for 30 s; and a final extension at 72°C for 5 min.
For DNA from all ectoparasites, a fragment of 16S rDNA (V1–V6) region (1,016 bp) was amplified in triplicate in a first-round PCR using primers; 27F-Y (5′-GAGTTTGATCCTGGCTYAG-3′), 1061R (5′-CRRCACGAGCTGACGAC-3′) (Ong et al., 2013), and 2.5 μM MidBlocker oligonucleotide to inhibit 16S Candidatus Midichloria mitochondrii amplification (Gofton et al., 2015b). The reaction was performed in a 20-μl volume containing 3 μl of ectoparasite DNA, 400 nM each primer, 200 μM dNTPs, 1.5 mM MgCl2, 1× PCR buffer, and 0.4 U of iProof High-Fidelity DNA Polymerase (Bio-Rad). Amplification was performed using a T100 DNA thermal cycler (Bio-Rad) under the following conditions: initial denaturation at 98°C for 3 min; 40 cycles of 98°C for 10 s, 60°C for 20 s, and 72°C for 30 s; and a final extension at 72°C for 5 min. The second amplification was performed as described above for human and rodent samples. Negative control PCR reactions were included in every experimental run using Ultrapure DNA/RNA-free distilled water in place of DNA template. PCR reactions were also performed with eluates from mock DNA extractions.
Three PCR products from each sample were pooled and cleaned using AMPure magnetic bead-based purification system (Beckman Coulter, United Kingdom) following the manufacturer’s instructions. Purified PCR products were eluted and quantified using the Quant-iT PicoGreen dsDNA assay (Invitrogen Life Technologies, MA) according to the manufacturer’s protocol. Each purified PCR was normalized and then pooled again with other purified PCR products from other samples by; (i) gender and age group for UFI patients, (ii) season of collection, location (sub-district/district), and rodent genera/species for rodents and rodent chiggers, and (iii) the host type they were collected from, genus/species and stages of ectoparasites for all other ectoparasites (ticks, fleas, and lice) collected from rodents and domesticated mammals (dogs and cows). Additional details on sample pooling for NGS are provided in Supplementary Table S2.
Library Preparation and High Throughput Sequencing
Indexing Samples
The dual indices and Illumina sequencing adapters were attached to pooled, purified PCR products using the Nextera XT Index Kit following the manufacturer’s protocol (Illumina). Index control reaction: combination of index primers that were not used with samples, was also included with PCR grade water as template. The number of reads recovered from these particular index combinations should be used to filter the cross-contaminations between indexed PCR primers and to identify errors in an Illumina sample sheet.
Library Clean Up, Normalization and Pooling
The final products were cleaned using Agencourt AMPure XP beads. The purity of the libraries was checked on the QIAxcel Advanced System (Qiagen) with a QIAxcel DNA High Resolution Cartridge. Purified amplicon libraries were quantified using the Qubit dsDNA HS Assay Kit (Invitrogen). DNA concentration was calculated and normalized to reach 4.0 nM for each library. Five microliters of DNA from each library were pooled (each NGS pool had 29–78 DNA libraries) for a NGS run (1–5 runs in total). Pooled libraries were denatured and diluted to a final concentration of 8 pM with a 10% PhiX (Illumina) control. Sequencing was performed using the MiSeq Reagent Kit V3 on the Illumina MiSeq System.
Data Analysis
The sequence reads generated by the 16S rRNA on MiSeq sequencers were processed on the CLC Genomics workbench v 11.0.1 (Qiagen, Aarhus A/S1). High-throughput sequences were imported into CLC Genomics Workbench according to quality scores of Illumina pipeline 1.8. In order to achieve the highest quality sequences for clustering, paired reads were merged in CLC microbial genomics module v3.0 using default settings (mismatch cost = 1; minimum score = 40; gap cost = 4 and maximum unaligned end mismatch = 5). Primer sequences were trimmed from merged reads using parameters (trim using quality scores = 0.01, trim ambiguous nucleotides = 2, and discard read length shorter than 150 bp). Samples were removed from analysis if the number of reads was less than 100 or less than 25% from the median (the median number of reads across all samples).of minimum read from the median. Chimeric sequences were detected and removed. Filtered sequences were clustered into operational taxonomic units (OTUs) according to a threshold of 97% sequence identity. All such processes were performed using CLC microbial genomics module v3.0. Reference OTU data used in the present study were downloaded from the Greengenes database V13.8 (DeSantis et al., 2006) and SILVA 16S V132 (Quast et al., 2013). Alpha rarefaction curve plots were generated among samples using CLC Microbial Genomics Module v3.0 with default parameter settings (minimum depth = 1, maximum depth = 100,000 and number of point = 20).
Pathogen Characterization by PCR Amplification and Sanger Sequencing
Real-time PCR and PCR assays were performed on positive NGS pools to confirm the detection of pathogen and the taxonomic species assignment generated by NGS analysis. The detail of assays and target gene(s) for selected pathogens was provided as online Supplementary Data (Supplementary Table S1) (Norman et al., 1995; Barbour et al., 1996; Horinouchi et al., 1996; Schwaiger et al., 2001; Scoles et al., 2001; Smythe et al., 2002; Park et al., 2004; Klee et al., 2006; Chmielewski et al., 2009; Colborn et al., 2010; Ganoza et al., 2010; Billeter et al., 2011; Parola et al., 2011; Diaz et al., 2012; Lalzar et al., 2012; Shakya et al., 2013; Gofton et al., 2015a; Pereira et al., 2018). For all real-time PCR, the reaction consisted of 1X Platinum quantitative PCR SuperMix-UDG (Invitrogen) using standard real-time PCR conditions with primer/probe concentrations and annealing temperatures as indicated in Supplementary Table S1. For conventional PCR, the assay was carried out in a 50 μl reaction volume containing 0.5 U of iProof High-Fidelity DNA Polymerase, 200 μM dNTPs, MgCl2 and primer concentration as indicated (Supplementary Table S1). The PCR conditions consisted of 98°C for 3 min, followed by 40 cycles of 98°C for 10 s, annealing temperature for 30 s, and 72°C for 45 s.
Estimating the Prevalence of Each Pathogen in Samples Studied
After performing NGS analysis of pooled samples, all samples in each NGS-positive pool for potential pathogenic bacteria were individually tested by their respective confirmatory assays using either real-time PCR or conventional PCR as indicated in Supplementary Table S1. Any positive signal was then confirmed by DNA sequencing by the Sanger method for species characterization. The prevalence rate for each pathogen was calculated based on the number of positive samples verified by confirmatory assays in the total number of samples studied. For some pathogens including O. tsutsugamushi and Bartonella spp., all samples were screened as routine tests. Therefore, the prevalence rate was calculated based on the number of combined positive samples detected by the NGS analysis then confirmatory assays and routine screening tests.
DNA Sequence and Phylogenetic Analysis
PCR amplicons were purified using the QIAquick® PCR Purification Kit (Qiagen) according to the manufacturer’s instructions. The PCR products were cycle-sequenced using an ABI BigDyeTM Terminator v3.1 Cycle Sequencing Kit, ethanol precipitated, and run on a SeqStudio Genetic Analyzer (Applied Biosystems Thermo Fisher, Thailand). Sequences of each sample and pathogen were assembled using SequencherTM ver. 5.1 (Gene Codes Corp., Ann Arbor, MI, United States). The pathogen sequences were aligned with reference sequences retrieved from the GenBank database using the MUSCLE codon alignment algorithm (Edgar, 2004). A maximum likelihood phylogenetic tree was then constructed from bacterial target gene(s) (Supplementary Table S1) using the best fit model of nucleotide substitution with bootstrapping (1000 replicates) in MEGA 6 (Tamura et al., 2013).
Serological Tests
Immunofluorescence Assay (IFA)
Scrub typhus and typhus group and spotted fever group of rickettsial diseases IFA tests were used to detect group-specific IgM antibodies against scrub typhus orientiae, and murine typhus and the spotted fever group of rickettsiae. The Rickettsia Screen IFA IgM Antibody Kit was used following the manufacturers’ instruction (Fuller Laboratories, Fullerton, CA, United States). The assay is intended for the simultaneous detection and semi-quantitation of IgM human antibody to both typhus group (TG) and spotted fever group (SFG) rickettsiae. An O. tsutsugamushi IFA IgM Antibody Kit (Fuller) included 4 strains (Boryong, Gilliam, Karp, and Kato) in one well. Positive reaction appears as bright staining (at least 1+) of positive control cut-off level in any of the four antigens areas. Bartonella henselae and Anaplasma phagocytophilum IFA tests were conducted for the detection of human IgG antibodies from serum using commercial kits [B. henselae IFA Human IgG Antibody Kit, A. phagocytophilum (HGA) IFA IgG Antibody Kit, Fuller]. Serum screening dilutions for B. henselae and A. phagocytophilum were 1:64 and 1:80, respectively.
In-house B. quintana IFA assay was prepared by CDC, Fort Collins (CO, United States) for testing the presence of human IgG against B. quintana. The protocol has been published previously (Iralu et al., 2006; Myint et al., 2011). Briefly, the human serum (1:32 dilution) was added to a slide fixed with B. quintana antigen, prepared by infecting Vero E6 cells with the bacteria. The slide was then incubated in a moist chamber at 35°C for 30 min, washed with PBS for 10 min, rinsed with distilled water, and air dried. Anti-human fluorescein isothiocyanate-labeled IgG conjugate was added to the slide which was processed as before. The slide was then mounted and read on a fluorescent microscope. All positive samples were then serially diluted in PBS, and an IFA-endpoint titer was determined using the same procedure; the end cut-off value for B. quintana was a titer greater than 1:200.
Enzyme-Linked Immunosorbent Assay (ELISA)
The determination of serological reactivity to O. tsutsugamushi 56-kDa recombinant protein was performed by in-house ELISA as previously described (Jiang et al., 2004). The ELISA plates were coated with 4 recombinants of O. tsutsugamushi 56-kDa protein from Karp, Gilliam, Kato, and TA763 genotypes. Patient sera were diluted at 1:100 with PBS for screening procedure. Samples considered positive (>0.5 OD) were further titered to determine their endpoint. The titer procedure was performed by diluting the positive sera by a factor of 4 (1:100, 1:400, 1:1,600, and 1:6,400) and tested again with the same procedure. If the sample had a total absorbance for all 4 dilutions of 1.00 or greater for the net OD, then the sample was considered reactive and the titer value was the inverse of the highest dilution with the OD of 0.2 or greater.
Enzyme-linked immunosorbent assay for the detection of IgG class antibody against R. typhi and spotted fever group Rickettsia in human serum was performed using commercial kits (R. typhi EIA IgG Antibody Kit, Spotted Fever Rickettsia IgG EIA Antibody Kit, Fuller). The kits utilized a group-specific lipopolysaccharide (rLPS) antigen extracted from spotted fever group Rickettsia species and a species-specific protein (rOmpB) purified from R. typhi.
Statistical Analysis and Data Visualization
All statistical analyses (linear regression and two-way ANOVA tests) were performed using GraphPad Prism version 5.04 for Windows (GraphPad Software, San Diego, CA, United States2). Some graphical illustrations presented in this study were performed in the R environment for statistical computing (Wickham, 2009). A nucleotide distance matrix was generated using “DNADist DNA Distance Matrix” in BioEdit (Hall, 1999). Maps used in this study were created by QGIS software (QGIS Development Team, 2009. QGIS Geographic Information System. Open Source Geospatial Foundation3).
Results
Sample Collection and NGS Procedure
Samples included in this study were from UFI patients (n = 200), rodents and small mammals (n = 309), rodent-associated ectoparasites (chiggers = 199 pools, ticks = 59 pools, fleas = 23 pools, and lice = 4 pools), and ectoparasites collected from larger animals including dogs, cats, and cattle (ticks = 35 pools, fleas = 88 pools and lice = 12 pools) (Table 1). Samples were collected mainly from Bo Kluea and from nearby districts of Nan province (Figure 1), Thailand. UFI samples were from inpatients and outpatients visiting the hospital with symptoms similar to scrub typhus infection or fever of unknown origin throughout the year 2017. The sampling of rodents and ectoparasites took place twice each year (wet and dry seasons) in 2014 and 2017, and only once in 2018 (dry season). Each sample was amplified in triplicate reactions to minimize PCR bias (Acinas et al., 2005; Sipos et al., 2007; Aird et al., 2011) and PCR products from the three reactions were pooled for each sample and purified before pooling with other samples for library preparation before NGS. All totaled, 929 samples were pooled according to their sample type, area of collection, season of collection, and host species into 183 NGS pools (Table 2). After NGS quality control procedures, 13,225,584 16S sequences from the field-collected samples and 38 control samples (6 extraction controls, 25 PCR controls, and 7 index controls) were used for analysis. From the 38 control samples, 153 bacterial genera were detected and then subtracted from the field samples’ 16S sequence dataset before conducting downstream analysis. These bacterial genera were considered to be contaminants from molecular reagents, the environment (water), or from cross-contamination during sample processing and between NGS runs (Salter et al., 2014). The number of pass-filtered raw reads per NGS pool ranged from 24,939 to 627,297, with the highest read (1,625,690) belonging to an ectoparasite pool collected from domesticated mammals (mean ±SD = 294,518 ± 152,314). The number of reads per NGS pool used in OTU assignment ranged from 1,583 to 160,184 (mean ±SD = 110,802 ± 34,095) (Table 1 and Figure 2A). The majority of samples with low numbers of reads were from chigger samples. Overall, 7.8% of OTUs (1,032,389 reads) were unclassifiable at the phylum level. Rarefaction curves demonstrated that sequence data for all samples approached completeness as indicated by the curve plateaus (Figure 2B). These data suggest that most bacterial profiles of all samples studied were nearly complete.
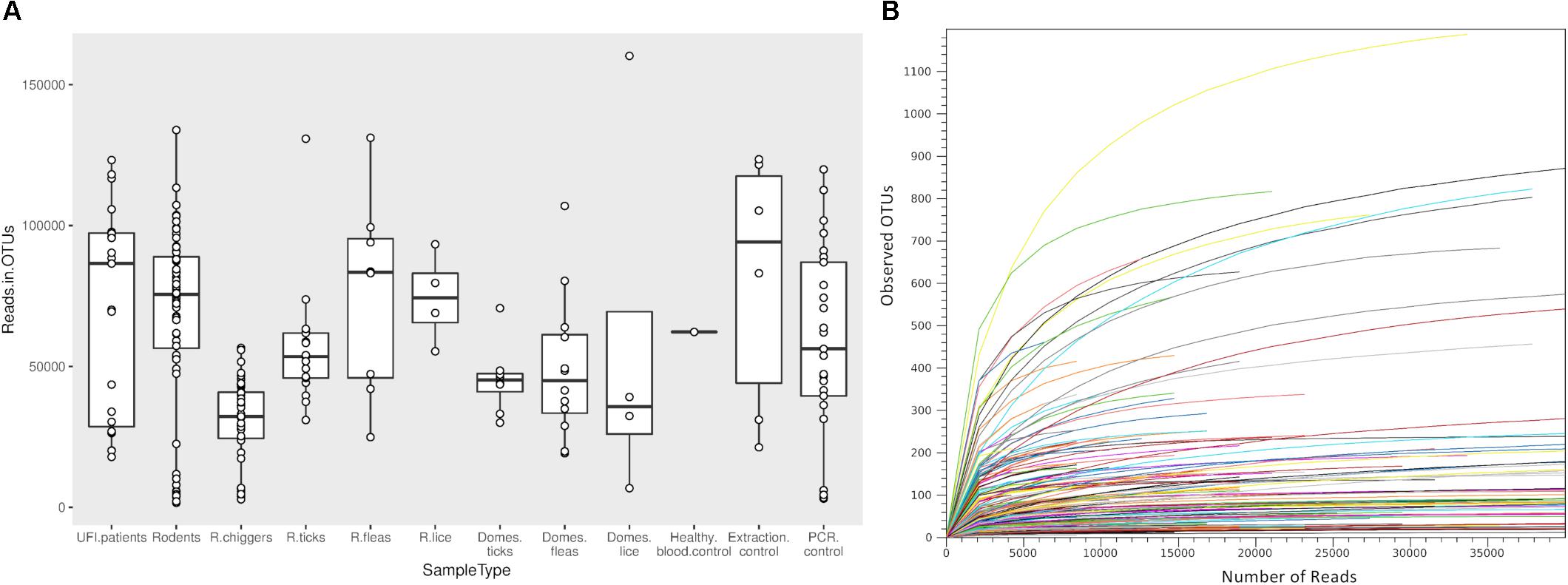
Figure 2. Box plot showing the distribution of number of reads from each NGS pool used for OTU assignment in this study (A). Rarefaction curves for all samples included in this study (B). The curves show the number of taxonomic units (OTUs) as a function of the number of sequences, indicating the sampling completeness. R, rodents; Domes, domesticated mammals.
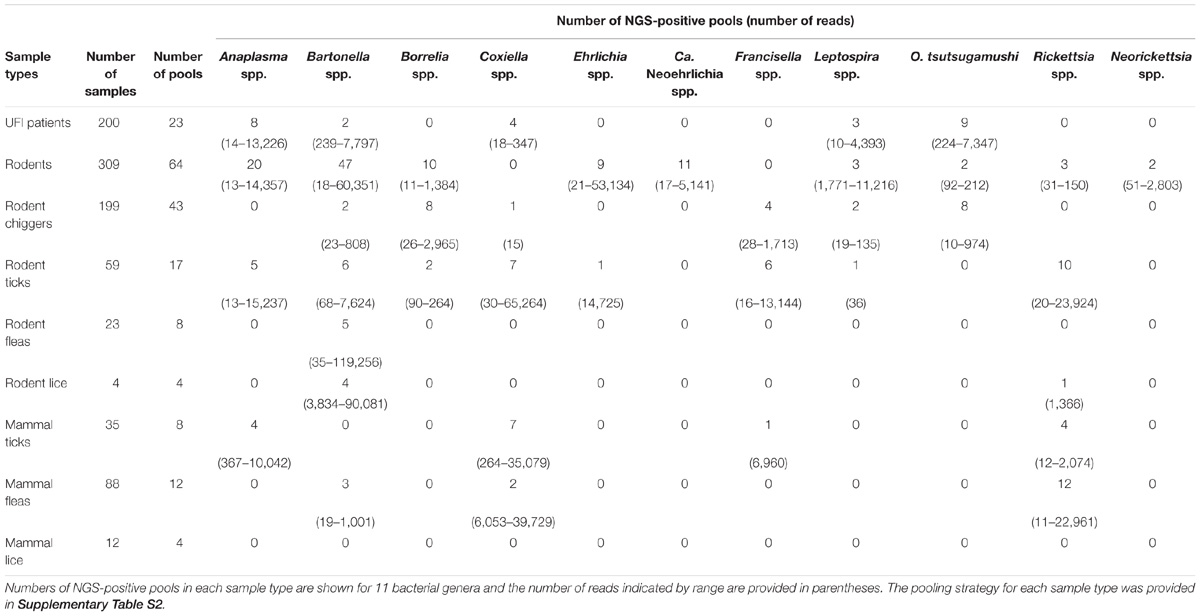
Table 2. Eleven pathogenic bacterial genera detected in samples.
Microbial Profiling in Human, Rodent, and Vector Populations
The classification of OTUs from each sample were made against Greengenes reference databases, with SILVA reference databases as a secondary database, and the similarity confidence threshold for each taxonomic level was set at 0.97 in CLC microbial genomic module. There were 19 recorded phyla found among all sample types and the ten most abundant phyla were seen in all sample types (Proteobacteria, Firmicutes, Actinobacteria, Spirochaetes, Tenericutes, Bacteroidetes, [Thermi], Planctomycetes, Fusobacteria, Cyanobacteria) (Figure 3A). The most prevalent phylum in all sample types was Proteobacteria and it was most abundant in ectoparasites from rodents (88.0%), domesticated mammals (83.0%), and UFI patients (70.0%) (Figure 3A). In rodent populations, the phyla Proteobacteria, Tenericutes, and Firmicutes were most abundant (38.0, 31.0, and 27.0%, respectively). In chiggers, the highest abundance was of Actinobacteria (43.0%), followed by Firmicutes (32.0%), and Proteobacteria (9.0%). Other interesting phyla include Spirochaetes found in UFI patients (5%), chiggers (5%), and rodents (1%); and two phyla, Thermi and Plantomycetes, that were found most commonly in UFI patients and chigger samples, respectively.
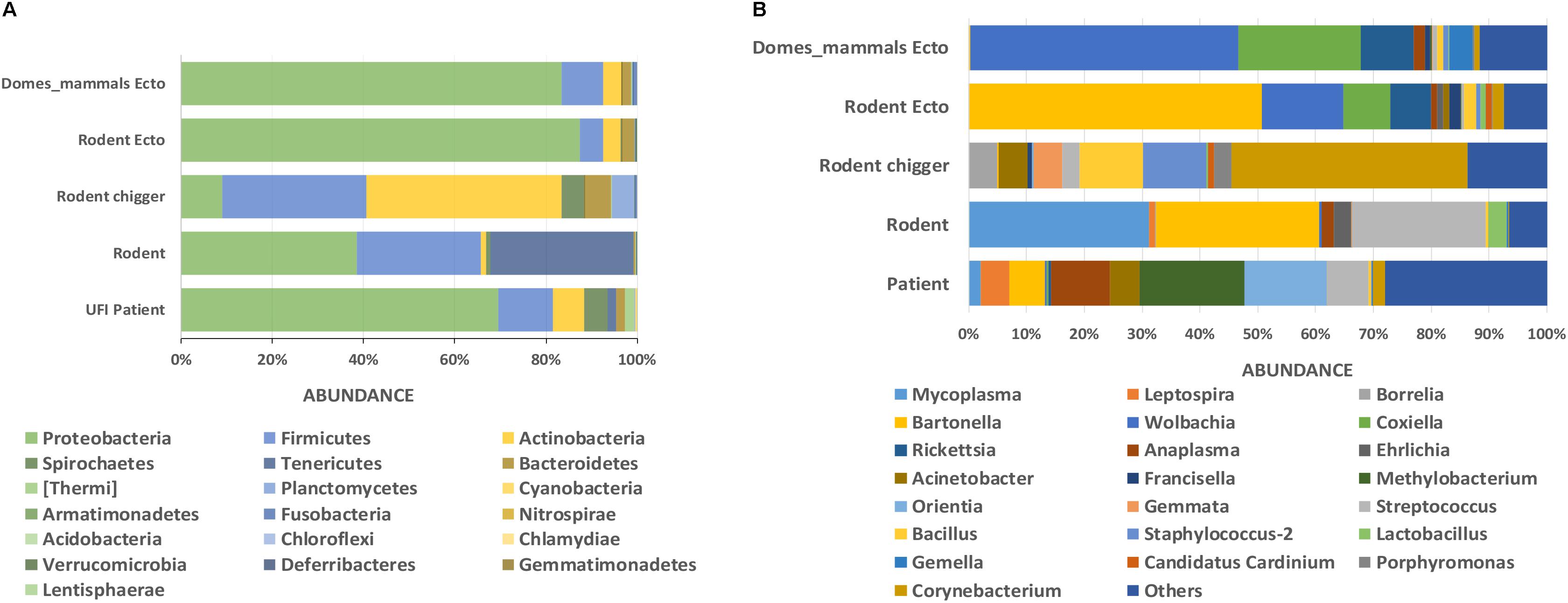
Figure 3. Taxonomic diversity and relative abundance at the phylum (A) and genus (B) level of bacterial community in UFI patients, rodents, chiggers, and ectoparasites collected from rodents, and ectoparasites collected from domesticated mammals. Phyla were identified on the basis of a confidence threshold cutoff of 77%, and genera on a confidence threshold cutoff of >90% using the Green Genes reference database. The percent relative abundances are of the total number of OTUs. Color legend for each phylum (A) or genus (B) was indicated below the bar graph.
At the genus level, as many as 651 genera were found among sample populations with the greatest diversity of taxa belonging to the phylum Proteobacteria (n = 254 genera), followed by Actinobacteria (n = 152), Firmicutes (n = 122), and Bacteroidetes (n = 57). The most abundant reads belonged to genera in Proteobacteria from UFI patients (Methylobacterium = 18%, Orientia = 14%, Anaplasma = 10%), rodent associated ectoparasites (Bartonella = 50%, Wolbachia = 14%, Coxiella = 8%, and Rickettsia = 7%), and ectoparasites collected from domesticated mammals (Wolbachia = 46%, Coxiella = 21%, and Rickettsia = 9%) (Figure 3B). In rodent samples, the most abundant bacterial genera were distributed among three major phyla; Tenericutes (Mycoplasma = 31%), Proteobacteria (Bartonella = 28%), and Firmicutes (Streptococcus = 23%). With the chigger samples, the majority of reads belonged to Corynebacterium spp. in the Actinobacteria phylum (41%), followed by Bacillus spp. and Staphylococcus spp. in the Firmicutes phylum (both with 11% abundance).
Bacterial Endosymbionts in Ectoparasites From Rodents and Domesticated Mammals
Wolbachia spp. was found mainly in mammal fleas (n = 10 NGS pools) with one pool each in rodents and ticks, lice, and fleas collected from rodents. The data of endosymbionts detected in NGS pools were provided as online Supplementary Data (Supplementary Figure S1). Few reads were detected in UFI patients (273 reads). This may have been due to cross-contamination during the sample preparation process. Coxiella endosymbionts were equally found in ticks collected from rodents and domesticated mammals. Few (n = 2) were associated with Francisella spp., and they were suspected as endosymbionts since they tested negative by a confirmatory assay (qPCR and PCR). Only one NGS pool of chiggers was found to carry a Coxiella endosymbiont. Francisella endosymbiont was mainly found in chiggers and ticks collected from rodents but one pool was also found in ticks from a dog. Candidatus Cardinium was mostly detected in chiggers; however, it was also detected in one pool of rodent fleas and in one pool of rodent ticks. Rickettsia spp. were mainly detected in ticks and lice from rodents and fleas and ticks from domesticated mammals. However, some Rickettsia were pathogenic species as confirmed by real-time PCR and DNA sequencing. This phenomenon was mostly observed in Rickettsia from flea pools (mostly from domesticated mammals) where Rickettsia could be identified to species and excluded from being identified as endosymbiont bacteria. Therefore, Rickettsia endosymbiont was not discussed here. When considering only known and confirmed endosymbionts (Wolbachia, Coxiella, and Ca. Cardinium) found in vectors, there was usually one or two predominant endosymbionts harbored by each vector type.
Detection of Pathogenic Bacteria in all Populations by NGS Results and Prevalence Rate After Verification by Confirmatory Assays
After NGS analysis, eleven potential pathogenic bacteria were detected among samples. Table 2 shows details about positive NGS pools and the number of reads indicated by range for each potential pathogen detected among sample populations. Bartonella spp. were the most prevalent and detectable bacteria among samples studied and the highest infection rate was found in rodent population (47/64 NGS pools). Bartonella spp. were found in most sample types with the exception of ticks and lice from domesticated mammals (69/183 NGS pools). The second most prevalent bacteria was Anaplasma spp. (37/183 NGS pools) which was found in UFI patients (8/23), rodents (20/64) and rodent-associated ticks (5/17), and ticks collected from domesticated mammals (4/8). Rickettsia spp. were also found in high prevalence as well (30/183), mostly in pools of fleas collected from domesticated mammals (12/12), 10 out of 17 pools of rodent ticks, 4/8 pools of ticks from domesticated mammals, 3/64 pools of rodents, and 1/4 pools of rodent lice. Borrelia spp., Coxiella spp., and Orientia spp. were equally detected; however, the distribution among populations was less similar. Borrelia spp. was found among rodents and its ectoparasites (chiggers and ticks), while Orientia spp. was detected among UFI patients, rodents, and chigger populations. Coxiella spp. were detected in a wide range of populations such as UFI patients, chiggers and ticks collected from rodents, and ticks and fleas collected from domesticated mammals. Other less prevalent pathogens such as Ehrlichia spp. (Rodents and their ticks), Candidatus Neoehrlichia spp. (rodents), Francisella spp. (chiggers and ticks from rodents, ticks from domesticated mammals), Leptospira spp. (UFI patients, rodents and their associated chiggers and ticks), and Neorickettsia spp. (rodents) were also detected in 2–11 out of 183 pools. Rodents carried the most diverse and wide-range of potential pathogenic bacteria and as many as 9 genera were detected. Likewise, ticks collected from rodents had the greatest pathogenic bacterial diversity of any vectors sampled (8 genera).
In this study, all samples present in each positive NGS pool were individually tested by confirmatory assays using either real-time PCR or conventional PCR (Supplementary Table S1). All amplified products were sequenced by the Sanger method. The correlation between the number of NGS-positive pools and the number of positive pools verified by confirmatory assays was determined using linear regression analysis (Figure 4). The results from both assays were positively correlated with R2 = 0.8968 (95% confidence interval = 0.7440–0.9368) or R2 = 0.6004 (95% confidence interval = 0.3950–0.7024) when the far point was removed. Prevalence rates for each pathogen were calculated based on NGS results verified by confirmatory assays or a combination of both NGS results and routine screening tests as mentioned earlier. Table 3 shows the prevalence rate for each pathogen detected among samples. A high prevalence of Bartonella spp. was seen in rodents, rodent fleas, and rodent lice populations (41.1, 65.2, and 75.0%, respectively). The prevalence of Rickettsia spp. was highest in fleas collected from domesticated mammals, mostly from dogs (84.1%), followed by rodent lice (25.0%), and rodent ticks (6.8%). Coxiella spp. was detected at highest prevalence in ticks collected from rodents and domesticated mammals (32.2 and 71.4%), but later was identified as a Coxiella endosymbiont (Table 4). Other highly pathogenic species known to cause disease in humans and animals were detected among vectors, rodents, and UFI patient samples, although at low prevalence. These included O. tsutsugamushi, Anaplasma spp., Borrelia spp., and Leptospira spp.
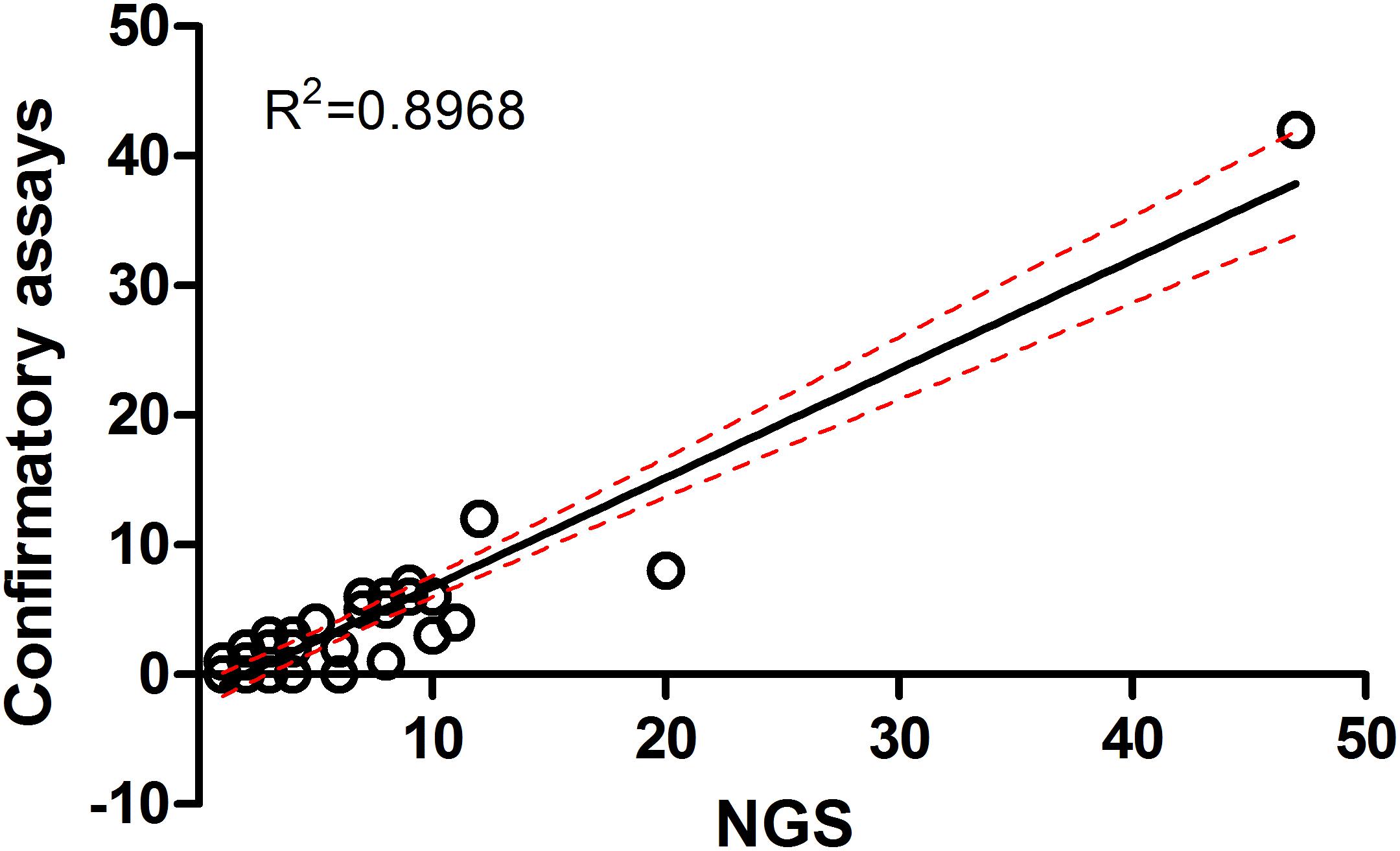
Figure 4. Correlation of metagenomic sequencing with confirmatory assays using qPCR and PCR methods for detection of bacterial pathogens in a variety of samples.
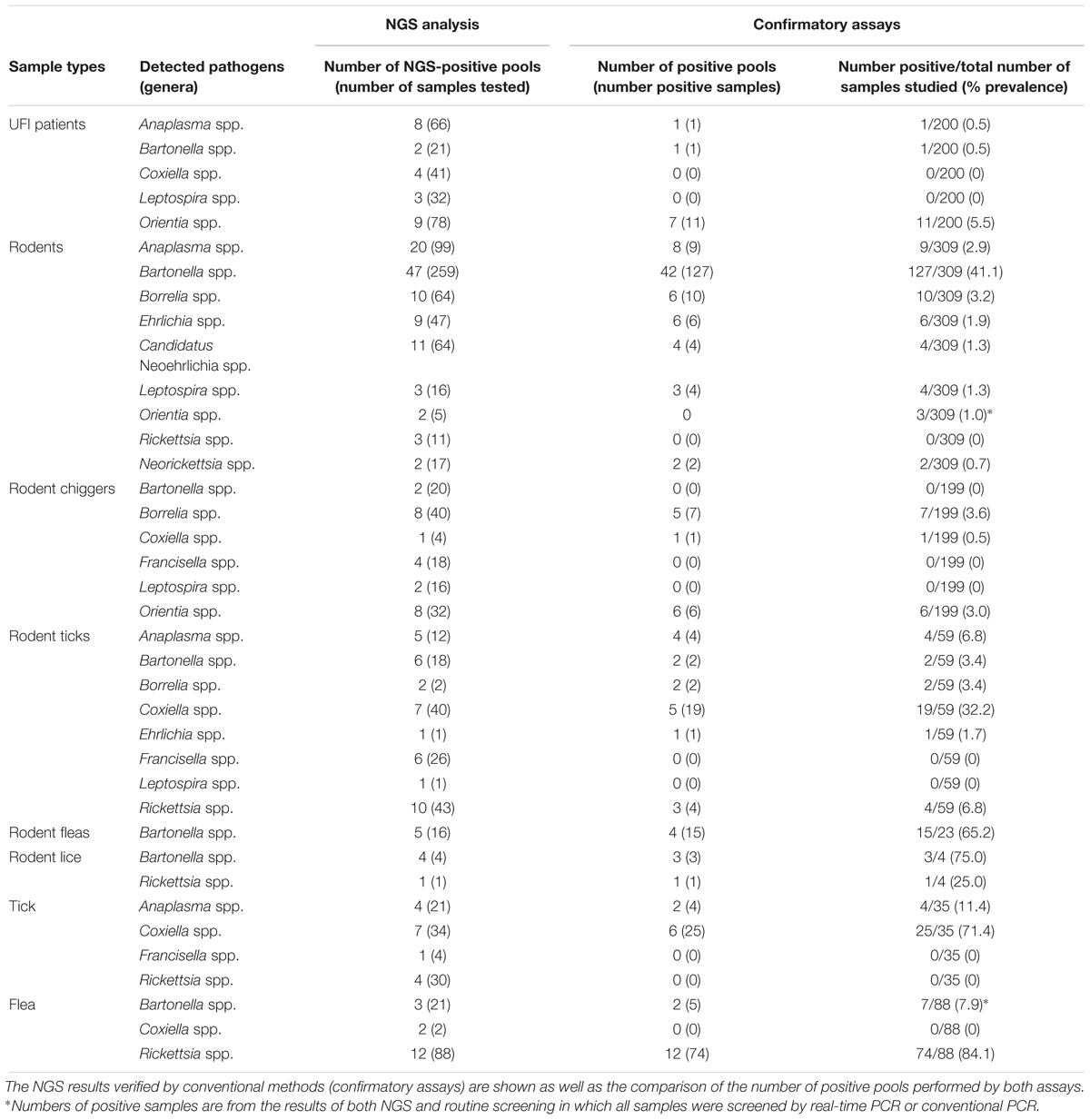
Table 3. Prevalence of pathogenic bacteria detected in sample populations.
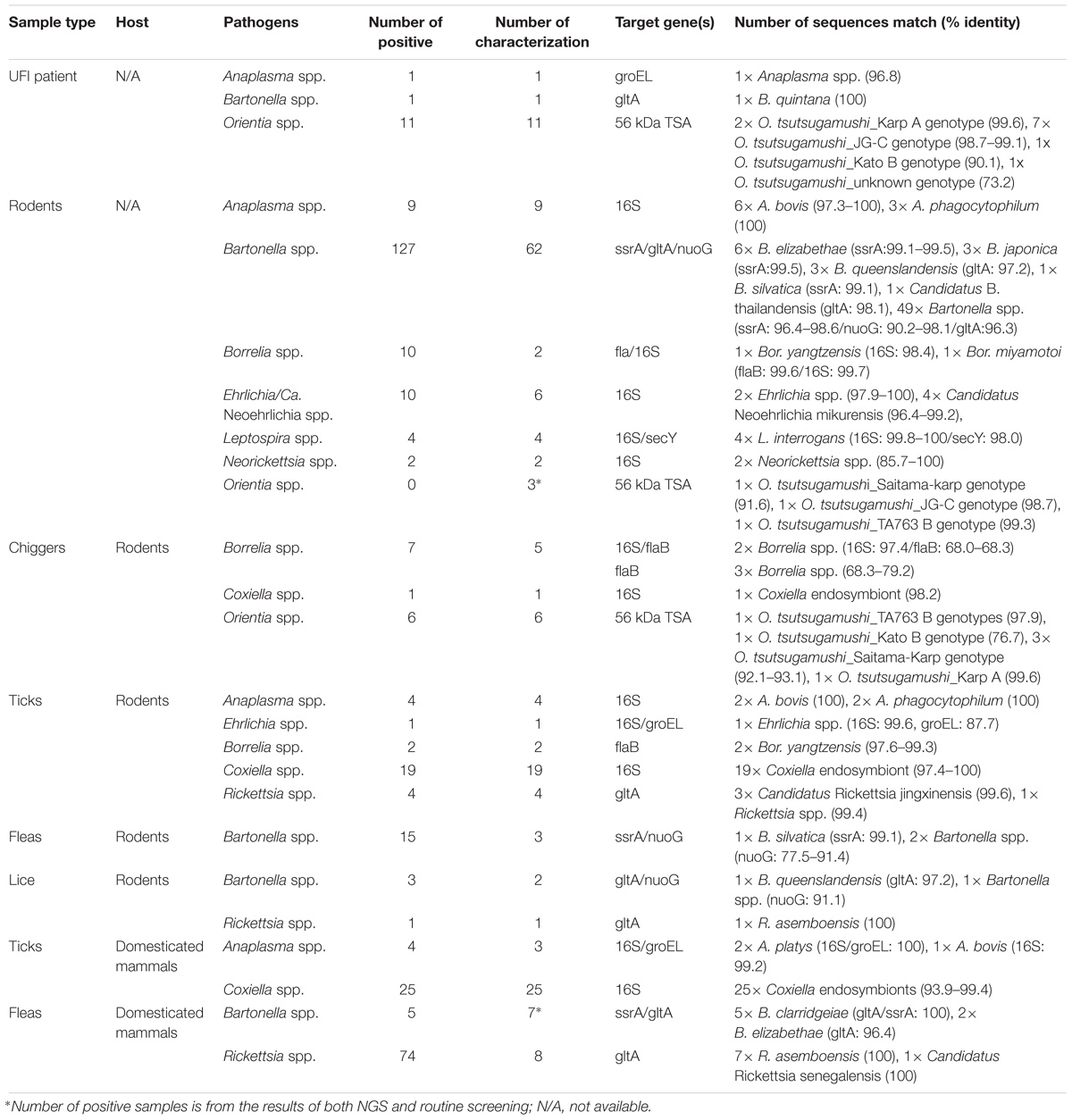
Table 4. Pathogen characterization by DNA sequence and phylogenetic analyses.
Orientia tsutsugamushi was found among UFI patients (11/200, 5.5%), chiggers (6/199, 3.0%) (a well-known scrub typhus vector), and rodents (3/309, 1.0%). Anaplasma spp. were also found in one UFI patient (1/200, 0.5%), rodents (9/309, 2.9%) and ticks collected from rodents (4/59, 6.8%) and domesticated mammals (4/35, 11.4%), while Borrelia spp. were found only in rodents and their associated ticks and chiggers with prevalence rates ranging from 3.2 to 3.6%. Other bacteria such as Leptospira spp., Ehrlichia spp., Candidatus Neoehrlichia spp., and Neorickettsia spp. were found in rodents and their associated ticks with prevalence rates in the range of 0.7–1.7%.
Characterization of Bacterial Species by DNA Sequence (Sanger Sequencing) and Phylogenetic Analyses
Several bacterial species were identified based on their highest similarity to reference sequences (GenBank database) as shown in Table 4, as well as their identity (%) corresponding to highly matched reference sequences as indicated in parenthesis after each pathogen. Only some important pathogenic bacteria are discussed here. O. tsutsugamushi and Leptospira spp. are well-known pathogenic bacteria endemic to Thailand and were frequently detected in the samples studied. O. tsutsugamushi was detected among UFI patients, rodents, and chiggers and it was observed that some populations shared the same genotypes as demonstrated by the phylogenetic analysis present in Figure 5. Although L. interrogans was confirmed to be present in rodent population, the pathogenic status of Leptospira spp. (NGS reads in the range 36–4,393) belonging to other populations could not be verified (Tables 3, 4). B. quintana, the causative agent of trench fever, was detected in one UFI patient but not in other populations. However, other common rodent-associated Bartonella species were found in rodents and their associated fleas and lice. Interestingly, B. clarridgeiae, a possible causative agent of cat-scratch disease, was found in 2 pools of fleas (Ctenocephalides felis) collected from domesticated mammals (dogs). Human granulocytic anaplasmosis (A. phagocytophilum) was also detected in rodents and their associated Ixodes ticks. Other anaplasmosis causative agents, A. bovis and A. platys were also detected from rodents and their ticks, and ticks of domesticated mammals. Interestingly, one UFI patient was positive for Anaplasma spp. with 96.8% identity to Uncultured Anaplasma spp. detected in a tick from China (GenBank accession No. KF728361.1). Even though its identity to pathogenic species, A. phagocytophilum and A. bovis, was 92.3–92.7%, the phylogenetic relationship of its groEL sequence with these pathogenic species was relatively closer than other known species in the tree (online Supplementary Data). Borrelia miyamotoi was detected in one rodent (Niviventer spp.) with 99.6% identity to reference sequence of flagellin and 16S rRNA genes. Borrelia yangtzensis, a newly recognized B. valaisiana-like strain, was also found in one rodent and 2 tick pools collected from rodents (Ixodes spp.). Borrelia spp. detected in chiggers were sequenced and the results showed that only 2 sequences (16S rRNA gene) were highly similar to Borrelia species (97.4%) which were not grouped in any of 2 Borrelia groups; relapsing fever and Lyme. All five flaB sequences had very low identity to Borrelia species (68.0–79.2%) and were very distantly related to the genus of Borrelia as demonstrated by the phylogenetic tree analysis. The phylogenetic trees for most of pathogens detected were provided as online Supplementary Data.
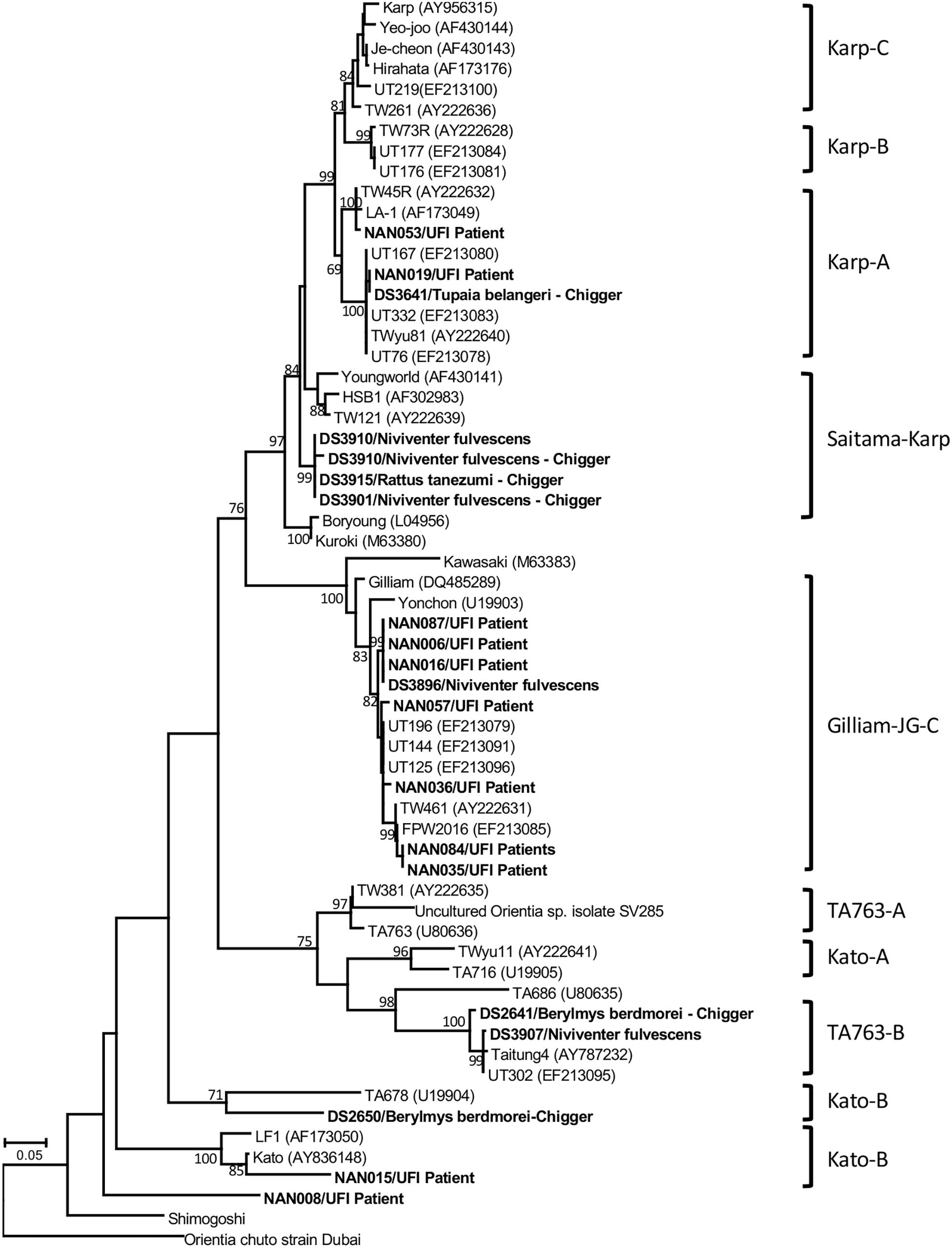
Figure 5. Phylogenetic tree analysis of O. tsutsugamushi genotypes detected among UFI patients, rodents, and chiggers in Nan province, Thailand (indicated in bold letters). A maximum likelihood tree was constructed based on 56-kDa type-specific antigen gene sequences using the GTR+G model of nucleotide substitution in the MEGA 6 program with bootstrapping (1000 replicates).
Seroprevalence in UFI Patients for Selected Pathogens With Highest Prevalence in Rodent and Vector Populations
The seroprevalence of the most prevalent pathogenic bacteria was determined in UFI patient sera (n = 200). IFA and ELISA assays were performed to examine the presence of IgM or IgG antibodies and to measure the titer of IgG antibody in UFI patients (Table 5). The results of IFA assays testing for IgM antibodies against scrub typhus, murine typhus and spotted fever group Rickettsia (SFGR) showed relatively high numbers of patients with past infection. Patients with IgM antibody against murine typhus had the highest number accounting for 33.0% (66/200), followed by SFGR (28.0%, 56/200), and scrub typhus (20.0%, 40/200). However, when the same set of sera (n = 200) were tested for their IgG titer, scrub typhus seems to dominate the other two diseases with 154 patients having higher titers (1,600 and >6,400), compared to SFGR (n = 26) or murine typhus (n = 0) (Table 5 and Figure 6A). Cross-reactivity between scrub typhus and rickettsiosis was determined using positive controls from commercial kits and the results showed no cross-reactivity was found among these pathogens for IgG and IgM.
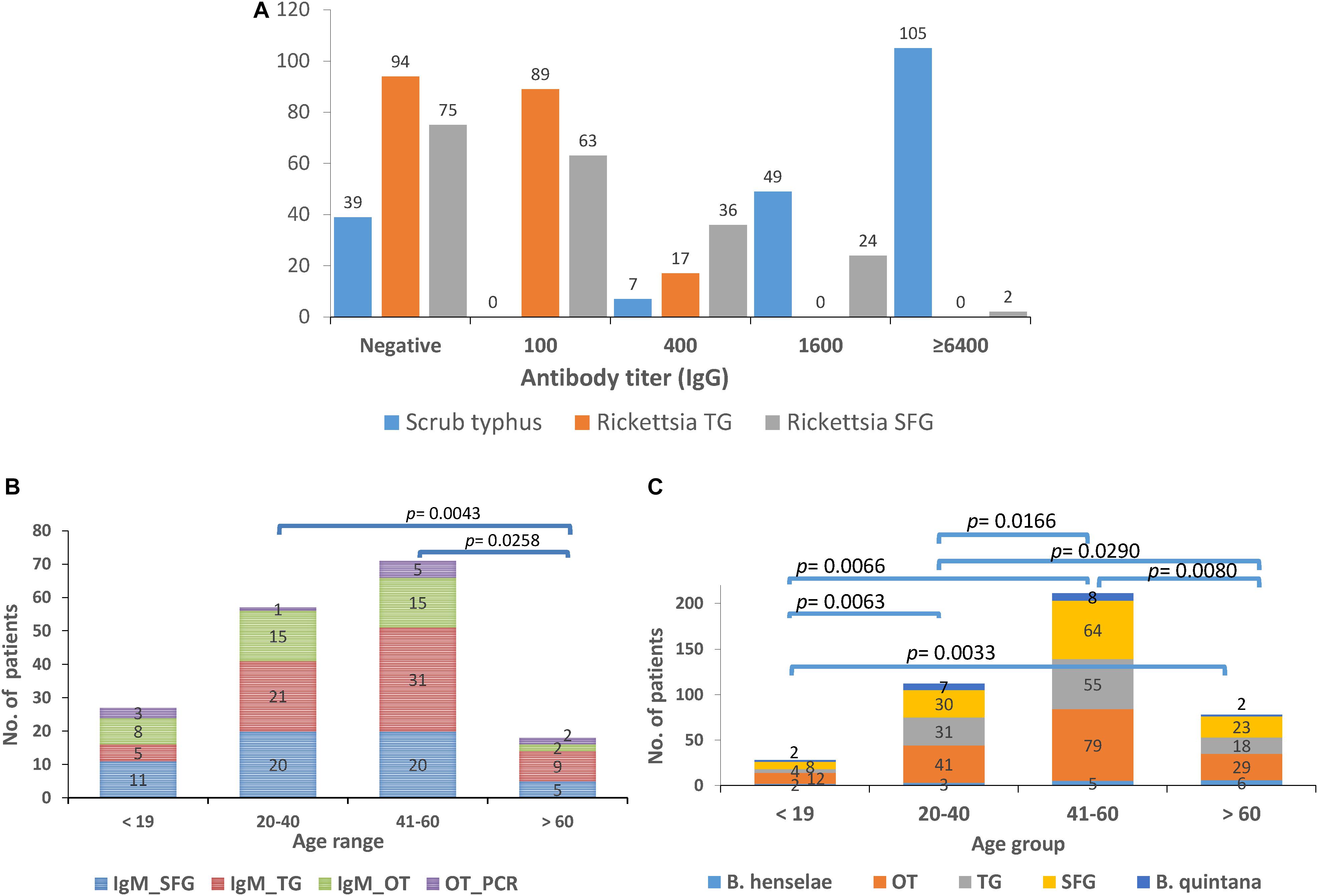
Figure 6. Seroprevalence (IgM and IgG) of scrub typhus [O. tsutsugamushi (OT)], and rickettsiosis in UFI patients from Bo Kluea hospital, Nan province. The IgG titers for scrub typhus and rickettsiosis [typhus group (TG) and spotted fever group (SFG)] are shown (A) as well as the seroprevalence of IgM (B) and IgG (C) antibodies among age groups (<19, 20–40, 41–60, >60 years old).
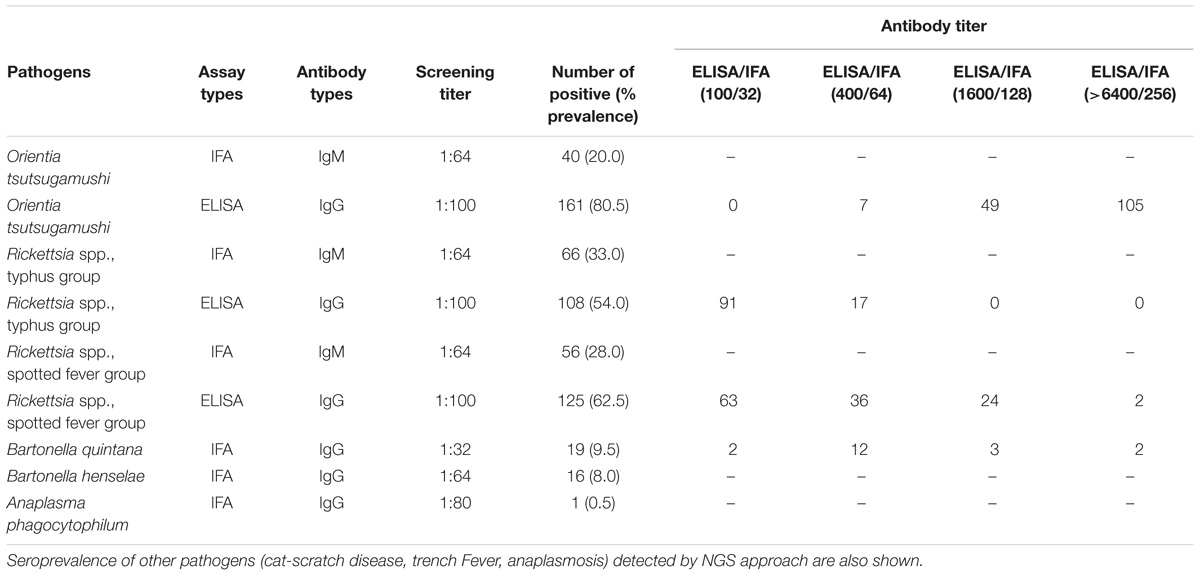
Table 5. Seroprevalence of scrub typhus, Rickettsia typhus group, and Rickettsia spotted fever group in UFI patients, Nan province, Thailand.
Patients were grouped into four age groups; <19, 20–40, 41–60, and >60 years old, and by sex; male and female, to determine how seroprevalence to rickettsiosis and scrub typhus differed among the groups. The data showed higher seroprevalence (both IgM and IgG) of the two diseases in two age groups (20–40 and 41–60 years old) compared to the other two age groups (<19 and >60) with statistical significance (Figures 6B,C). However, the overall prevalence was not different between male and female patients. In addition, lower seroprevalence (IgG) was observed for other pathogens such as B. quintana (9.5%, 19/200), B. henselae (8.0%, 16/200), and A. phagocytophilum (0.5%, 1/200) (Table 5).
Circulation of Pathogenic Bacteria Among Human, Reservoir Host, and Vector Populations
A Venn diagram illustrates the shared bacterial species among samples (Figure 7A). Samples were grouped into four groups [UFI patients, rodents, rodent-associated ectoparasites (including chiggers)], and ectoparasites collected from domesticated mammals. The diagram illustrates the sharing of O. tsutsugamushi genotypes among UFI patients, chiggers, and rodent populations. Coordinates of all positive samples for O. tsutsugamushi were mapped and it was shown that almost all positive samples clustered together in Bo Kluea district (Figure 7B). A. bovis was another pathogen found circulating among animal reservoirs and ticks. The pathogen was detected in Rattus rats and Tupaia glis (common treeshrew) as well as in Rhipicephalus sanguineus and Haemaphysalis bandictota ticks where both tick species were known to share common hosts (Bandicota indica and Rattus rats) (Tanskul et al., 1983). Other pathogens such as A. phagocytophilum, Borrelia yanzensis, and various O. tsutsugamushi genotypes were shared between rodents and their associated vectors and are shown in Figure 7A. Pathogens solely detected in each population are also indicated in the figure, especially Borrelia miyamotoi, B. clarridgeiae, and Leptospirosis interrogans which are known to cause infections in humans. A. platys was found from R. sanguineus ticks collected from a dog and is the causative agent of canine ehrlichiosis.
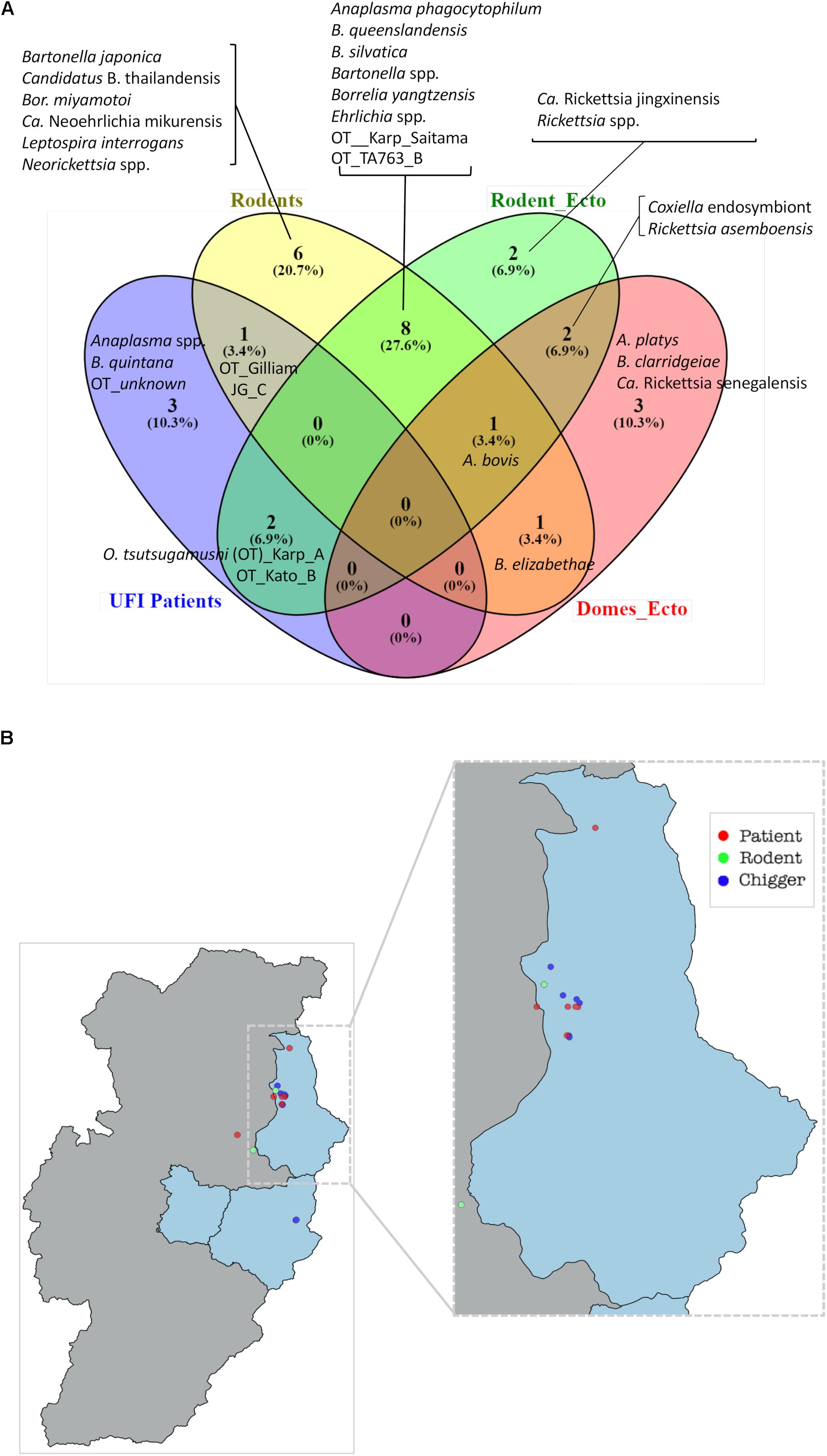
Figure 7. Venn diagram (Oliveros et al., 2007–2015) indicates the bacteria species shared between populations or unique to each of them (A). Bacterial species were identified on the basis of DNA sequence and phylogenetic analyses of their target genes (Supplementary Table S1). Rodent_ecto = ectoparasites (chiggers, ticks, fleas, and lice) collected from rodents, Domes_ecto = ectoparasites collected from domesticated mammals. Map of O. tsutsugamushi-positive samples in Bo Kluea district, Nan (B). Each dot represents only positive samples found among UFI patients (red), chigger pools (blue), and rodent populations (green).
The effect of environmental factors on the prevalence and transmission of scrub typhus among populations studied was also evaluated. Rainfall (mm) and temperature in 2017 from Nan province was acquired from the Thai Meteorological Department4. The rainy season started from late April through September corresponding to increased rainfall (mm) recorded during this period of the year (Figure 8A). The temperature was relatively constant throughout the year except a slight decrease at the end and the beginning of the year (October–March). The chigger index (no. of chigger/number of hosts collected) and the O. tsutsugamushi infection rates in rodents and chiggers were examined each month (Figure 8B). The chigger index slightly increased at the beginning of the year and peaked around June–September. O. tsutsugamushi in chigger could be found almost every month and the highest infection rates were in March. On the other hand, the number of UFI patients with IgM antibody against scrub typhus slowly increased from April to October and peaked at the end through the beginning of the year (November–February) (Figure 8C). O. tsutsugamushi was also detected by PCR from patient whole blood samples but the number did not correlate well with the number of patients having IgM seroactivity. Interestingly, the number of patients with rickettsiosis IgM increased sharply from April to September and this high number continued through the rest of the year. Likewise, the same pattern was observed with their corresponding IgGs although the number of patients with scrub typhus IgG and its titer seemed to be higher than that observed for rickettsiosis (TG and SFG) (Figure 8C and Table 5).
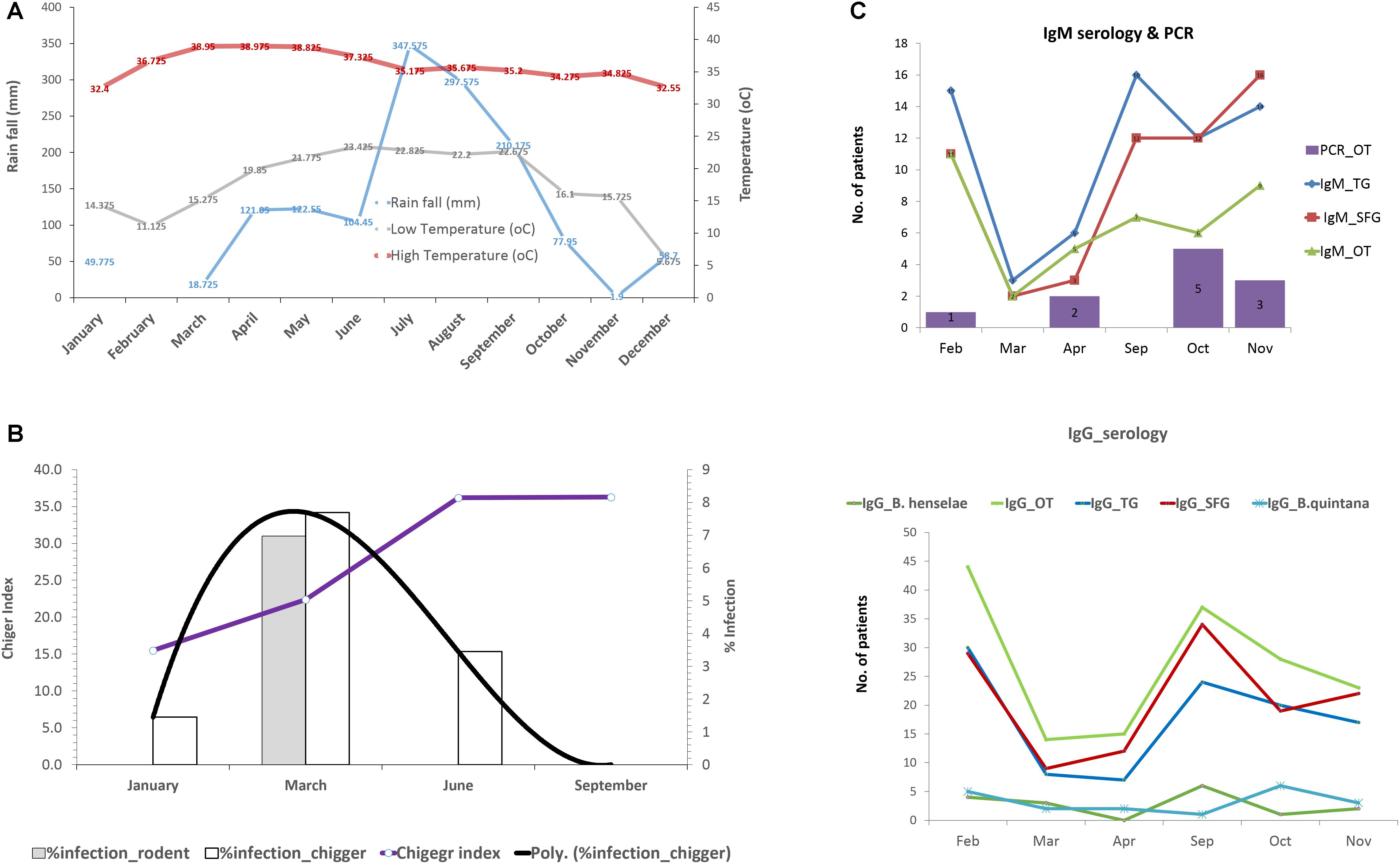
Figure 8. Rainfall (mm) and temperature (°C) during the year 2017 in Nan province (A). Infection rate of O. tsutsugamushi in rodents and chigger pools as well as the chigger index recovered from rodents during wet (April–September) and dry (October–March) seasons (B). Seroprevalence (IgM and IgG) of scrub typhus, rickettsiosis, and O. tsutsugamushi detection in UFI patients were plotted according to the time of collection from Bo Kluea hospital, Nan (C).
Discussion
Scrub typhus is a major public health problem in Nan province with the highest cases of scrub typhus infection (152.64 per 100,000 population) reported to Bureau of Epidemiology, Department of Disease Control, the Ministry of Public Health (MoPH), Thailand in 20175. Samples analyzed in this study included all factors/populations that are involved in disease transmission. Most samples from UFI patients were collected from Bo Kluea hospital in 2017, while rodents and ectoparasites were collected twice a year (dry and wet seasons) from 2014 until the beginning of 2018 at field collection sites in Mae Charim, Phu Phiang, and Bo Kluea districts.
In this study, extraction and PCR controls were included in each NGS run to exclude bacteria genera commonly found in molecular reagents, water, and other environments (Tanner et al., 1998; Goodrich et al., 2014). Several common bacterial genera were found in controls similar to previous studies (Salter et al., 2014; Razzauti et al., 2015). Some potential zoonotic and pathogenic bacteria were also detected in controls such as Bartonella spp., Leptospira spp., and Rickettsia spp. albeit at relatively low numbers of reads. Therefore, we applied cut-off values (number of reads detected in controls) for those bacteria detected in controls and applied these numbers to all samples in our study. Contamination likely came from cross-contamination during sample processing and carry-over between sequencing runs (Swei et al., 2013). The NGS technique has many benefits over conventional tests since it does not require prior knowledge of the target pathogens which conventional tests most often rely upon. However, there is no standardized protocol for all laboratories and contamination from reagents and the environment can complicate the analysis. Therefore, conventional methods were also employed as confirmatory assays in this study. The 16S sequence can only discriminate pathogens to the genus level with confidence. However, one genus may consist of multiple species, some of which may not be pathogenic to humans or animals. Therefore, it is necessary to further identify the bacteria genera detected by NGS to the species level using conventional PCR or Sanger sequencing.
Several bacterial genera are saprophytes and commensals or can be found as contaminants in reagents and the environment. Therefore, they are considered non-pathogenic bacteria and were not considered in our analysis (Razzauti et al., 2015). A list of bacteria commonly detected in reagents and laboratory contamination was previously published by Salter et al. (2014). In this study, most of the bacterial genera found in sample populations were commensals or saprophytes such as Methylobacterium in UFI patients, Mycoplasma and Streptococcus in rodents, and Corynebacterium in chiggers. These bacteria comprised 18–41% of the total OTU reads in each population. Detection of bacterial DNA in human blood was not unexpected since healthy blood donors also contain bacterial DNA such as Proteobacteria (>80%), Actinobacteria, Firmicutes, and Bacteroidetes (Paisse et al., 2016) which is similar to what we found in UFI patients. Bacterial endosymbionts were highly abundant in ectoparasites such as Wolbachia (46% in fleas from dogs) or Coxiella endosymbionts in ticks from rodents and dogs. What we found in this study is that fleas and ticks collected from domesticated mammals harbored one predominant endosymbiont such as Wolbachia in fleas (Ctenocephalides felis) and Coxiella endosymbiont in R. sanguineus ticks collected from dogs. Chiggers and ticks collected from rodents had two predominant endosymbionts such as Haemaphysalis ticks which carried both Coxiella and Francisella endosymbionts, while chiggers had both Candidatus Cardinium and Francisella endosymbionts. However, since ectoparasites were pooled before the NGS procedure, the number of endosymbionts or co-infection of endosymbionts in single vectors could not be determined.
In this study, a high prevalence from NGS results was observed for few bacterial genera; however, some genera could not be verified with conventional assays such as real-time PCR, PCR, or DNA sequencing. The main reason was likely that the number of reads was too low and below the limit of detection for the conventional test, or they could have been non-pathogenic strains or species and were not picked up by confirmatory assays. For example, Leptospira spp. comprise both pathogenic, intermediate, and saprophytic (non-pathogenic) species that can be introduced as contaminants from the environment into samples. Here Leptospira spp. were only detected in the rodent population, although NGS analysis showed reads were also detected in UFI patients, chiggers, and ticks at lower levels. However, testing with confirmatory assay resulted in no signal or PCR product using a genus-based assay (Ahmed et al., 2009) with primer sets targeting house-keeping genes such as gyrB or SecY (Slack et al., 2006; Victoria et al., 2008). In some cases, PCR assays targeting Leptospira 16S rRNA genes showed some positive bands for UFI patients but the product size was shorter than expected and DNA sequences from these products matched only human DNA (100%). Rickettsia spp. and Francisella spp. could not be verified in some sample types such as ticks, chiggers, and rodents. These could be endosymbiont bacteria which our assays could not detect (Wright et al., 2011; Takhampunya et al., 2017).
Originally, the confirmatory assays did not verify the NGS results (2 pools) of O. tsutsugamushi detected in rodents. However, since scrub typhus detection has been run in our lab as part of routine surveillance assays, three O. tsutsugamushi-positive rodents were verified by a routine real-time PCR test (Table 4). Similarly, four Bartonella species detected in fleas from domesticated mammals were also verified by a routine real-time PCR test and they were included in Table 4. NGS seems to have less sensitivity than the conventional method. In support of this observation, a previous study has compared MiSeq and RNA-seq, and found that MiSeq cannot detect bacteria at a value lower than 4% prevalence in the population and thus RNA-seq is better in terms of sensitivity (Razzauti et al., 2015). In all likelihood, this is due to differences in sequencing depth for each of the techniques used. The detection of B. quintana in one UFI patient and B. clarridgeiae in Ctenocephalides felis fleas provides significant evidence that Trench fever and cat-scratch disease-causing bacteria are present in the study area. The seroprevalence data (IgG) of B. quintana and B. henselae also confirmed previous human exposure to these bacteria. Although we detected A. phagocytophilum (anaplasmosis) in rodents and ticks, only one UFI patient was seropositive (IgG) to anaplasmosis. Further characterization of Anaplasma species detected in the UFI patient is required to identify this pathogenic species causing human infection. Given the fact that a few Anaplasma species were detected from rodents and ticks in this study area such as A. phagocytophilum, A. bovis, and A. platys, knowing what species caused infection in humans would lead to a better understanding of the transmission dynamics among the vector, host, and reservoir enable to and to better understand its transmission in the area and reservoir host and the vector involved. Other bovine and canine ehrlichiosis were also detected in ticks. Interestingly, Bor. miyamotoi and Bor. yangtzensis were detected in rodents and Ixodes ticks which marks the first detection of these human pathogenic species in Thailand. More research and surveillance is needed to further characterize their prevalence and distribution in the country. Borrelia spp. detected in chiggers could be some other unidentified bacteria since their sequence identity to most Borrelia species were quite low based on flaB gene sequences (64.0–70.2%), while the percent identity among all reference sequences used in the alignment ranges from 69.6 to100%. While the 16S rRNA sequences of two chigger pools were 97.4% identical to some unknown Borrelia species and Candidatus Borrelia africana (Accession No. KT364339), additional analysis such as multilocus sequence typing (MLST) should be performed in order to determine whether Borrelia spp. detected in chiggers are new Borrelia species or some other bacterial genus. Since some sample types included in this study were not collected across all years of sampling (2014, 2017, and 2018) such as ectoparasites collected from domesticated mammals (2014) and UFI patients (2017), the observed pathogens reported here might not represent the true picture of pathogens shared among the vectors, reservoirs, and hosts in Bo Kluea district, Nan province. In this study, co-infection between Anaplasma spp. (A. phagocytophilum, A. bovis) and Bartonella spp. was observed in Rattus and Bandicota rats (2/309, 0.65%). It is unfortunate that the co-infection/co-occurrence patterns in ectoparasites could not be examined in this study due to our pooling procedure for ectoparasites which was performed immediately after they were collected from animal hosts.
Seroprevalence (IgM and IgG) for scrub typhus and rickettsiosis in UFI patients confirmed that the two diseases are highly endemic to the region, especially for scrub typhus. O. tsutsugamushi was present in all related samples studied and human exposure was clearly observed with high prevalence and titers (n = 154 with 1600, >6400 titers). Although human rickettsiosis was not detected in rodents or vectors, the levels of IgM and IgG seroprevalence for TGR and SFGR indicate the circulation of these pathogens in the area as well. Some pathogens were detected in animals and vectors but not in humans; however, seroprevalence (IgG) of the pathogens in patients indicated previous exposure in humans, such as B. henselae. It is worth noting that serological differentiation between B. henselae and B. quintana IgG antibody might not be possible since there could be some cross-reactivity between the two species. With O. tsutsugamushi, when age and sex of patients were considered, prevalence was significantly higher in 20–40 and 41–60 year-old groups, which shows the working age population having increased risk of contracting the diseases. The incidence of scrub typhus infections in humans seems to occur at higher rates during the rainy season corresponding to the time when local people start rice/corn cultivation and continues throughout the year until the harvesting season ends in October/November as the high seroprevalence in working age groups (20–60 years old) strongly supports this speculation. Moreover, the infection rate of O. tsutsugamushi in rodents and chiggers was highest in March just before the rainy season, followed by the increase of chigger indexes (number of chiggers per host) possibly leading to increased potential for disease transmission.
The discovery of certain bacterial pathogens was expected based on previous surveillance data and reported cases from the Ministry of Public Health. Additionally, other unexpected pathogens such as Anaplasma spp. and B. quintana (an agent causing Trench fever) were detected among UFI patients as well as Bor. miyamotoi in rodent populations. However, to date in-depth analyses as to how, when and where transmission occurs are lacking. Human, animal, and vector interactions play a major role in disease transmission and form a dynamic transmission cycle. Pathogens can spread from animal-to-animal or animal-to-human by several modes of transmission. Probably the most important method of transmission occurs during feeding by parasitic arthropods. This study employed NGS and metagenomics to characterize bacterial pathogens and understand their transmission in animal, human, and vector populations. Several pathogens were detected in rodent and vector populations indicating the complex ecology of bacterial pathogens and their reservoir hosts and vectors in the area close to where human activities occur which increase the risk of human–animal interface. The most apparent example is scrub typhus where O. tsutsugamushi was found in UFI patients, chiggers, and rodent populations. These data clearly illustrate the complex picture of pathogen transmission from animal reservoir hosts to humans via arthropod vectors. Local public health officials can effectively use the data to assist in understanding the seasonality of diseases such as scrub typhus and the populations most at risk. Information can be shared locally and preventive measures, such as repellents, can be used when appropriate. From this study, multiple bacterial pathogens known to cause human diseases in other locations were identified for the first time in Nan province. Such information is useful for local medical providers as they try to diagnose and treat patients with undifferentiated fevers. Finally, the data presented in this study effectively illustrate the utility of metagenomics in future epidemiological surveys involving multiple types of samples.
Author Contributions
RT and AK conceived and designed the experiments. AR, NC, SP, TM, ST, and BT performed the experiments. CP, KY, and NK were the hospital staff involved in this study. RT, AK, and SD analyzed the data. RT, SD, and AK wrote the manuscript. ALR provided reagents and reviewed the manuscript.
Funding
This work was funded by the Armed Forces Health Surveillance Branch, Global Emerging Infections Surveillance and Response System (AFHSB-GEIS), Silver Spring, Maryland, United States under study #P0081_18_AF_04.
Disclaimer
Material has been reviewed by the Walter Reed Army Institute of Research. There is no objection to its presentation and/or publication. The opinions or assertions contained herein are the private views of the author, and are not to be construed as official, or as reflecting true views of the Department of the Army or the Department of Defense. Research was conducted under an approved animal use protocol in an AAALACi accredited facility in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adheres to principles stated in the “Guide for the Care and Use of Laboratory Animals”, NRC Publication, 2011 edition. The investigators have adhered to the policies for protection of human subjects as prescribed in Army Regulations 70–25.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the staff (laboratory technicians and physicians) at Bo Kluea Hospital, Nan province, for their excellent collaboration during this study. The authors are grateful to MK for providing materials and reagents for serological tests and to Dr. Brian P. Evans for assisting with the editing of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00319/full#supplementary-material
Footnotes
- ^ http://www.clcbio.com
- ^ www.graphpad.com
- ^ https://www.qgis.org/en/site/
- ^ http://climate.tmd.go.th/content/category/17
- ^ http://www.boe.moph.go.th/boedb/surdata/index.php
References
Acinas, S. G., Sarma-Rupavtarm, R., Klepac-Ceraj, V., and Polz, M. F. (2005). PCR-induced sequence artifacts and bias: insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl. Environ. Microbiol. 71, 8966–8969. doi: 10.1128/AEM.71.12.8966-8969.2005
Ahmed, A., Engelberts, M. F., Boer, K. R., Ahmed, N., and Hartskeerl, R. A. (2009). Development and validation of a real-time PCR for detection of pathogenic leptospira species in clinical materials. PLoS One 4:e7093. doi: 10.1371/journal.pone.0007093
Aird, D., Ross, M. G., Chen, W. S., Danielsson, M., Fennell, T., Russ, C., et al. (2011). Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 12:R18. doi: 10.1186/gb-2011-12-2-r18
Alcaide, M., Rico, C., Ruiz, S., Soriguer, R., Muñoz, J., and Figuerola, J. (2009). Disentangling vector-borne transmission networks: a universal DNA barcoding method to identify vertebrate hosts from arthropod bloodmeals. PLoS One 4:e7092. doi: 10.1371/journal.pone.0007092
Ambrose, H. E., Granerod, J., Clewley, J. P., Davies, N. W., Keir, G., Cunningham, R., et al. (2011). Diagnostic strategy used to establish etiologies of encephalitis in a prospective cohort of patients in England. J. Clin. Microbiol. 49, 3576–3583. doi: 10.1128/JCM.00862-11
Barbour, A. G., Maupin, G. O., Teltow, G. J., Carter, C. J., and Piesman, J. (1996). Identification of an uncultivable Borrelia species in the hard tick Amblyomma americanum: possible agent of a Lyme disease-like illness. J. Infect. Dis. 173, 403–409. doi: 10.1093/infdis/173.2.403
Billeter, S. A., Gundi, V. A., Rood, M. P., and Kosoy, M. Y. (2011). Molecular detection and identification of Bartonella species in Xenopsylla cheopis fleas (Siphonaptera: Pulicidae) collected from Rattus norvegicus rats in Los Angeles, California. Appl. Environ. Microbiol. 77, 7850–7852. doi: 10.1128/AEM.06012-11
Chmielewski, T., Podsiadly, E., Karbowiak, G., and Tylewska-Wierzbanowska, S. (2009). Rickettsia spp. in ticks. Poland Emerg. Infect. Dis. 15, 486–488. doi: 10.3201/eid1503.080711
Colborn, J. M., Kosoy, M. Y., Motin, V. L., Telepnev, M. V., Valbuena, G., Myint, K. S., et al. (2010). Improved detection of Bartonella DNA in mammalian hosts and arthropod vectors by real-time PCR using the NADH dehydrogenase gamma subunit (nuoG). J. Clin. Microbiol. 48, 4630–4633. doi: 10.1128/JCM.00470-10
Cox, F. E. (2001). Concomitant infections, parasites and immune responses. Parasitology 122(Suppl.), S23–S38. doi: 10.1017/S003118200001698X
Davidar, P., and Morton, E. S. (2006). Are multiple infections more severe for purple martins than single infections? Auk 123, 141–147. doi: 10.1642/0004-8038(2006)123[0141:AMIMSF]2.0.CO;2
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
Dethlefsen, L., Huse, S., Sogin, M. L., and Relman, D. A. (2008). The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 6:e0280. doi: 10.1371/journal.pbio.0060280
Diaz, M. H., Bai, Y., Malania, L., Winchell, J. M., and Kosoy, M. Y. (2012). Development of a novel genus-specific real-time PCR assay for detection and differentiation of Bartonella species and genotypes. J. Clin. Microbiol. 50, 1645–1649. doi: 10.1128/JCM.06621-11
Durden, L. A., and Musser, G. G. (1994). The sucking lice (Insecta, Anoplura) of the world—a taxonomic checklistwith records of mammalian hosts and geographical distributions. Am. Mus. Nat. Hist. 218, 1–90.
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Finkbeiner, S. R., Allred, A. F., Tarr, P. I., Klein, E. J., Kirkwood, C. D., and Wang, D. (2008). Metagenomic analysis of human diarrhea: viral detection and discovery. PLoS Pathog. 4:e1000011. doi: 10.1371/journal.ppat.1000011
Ganoza, C. A., Matthias, M. A., Saito, M., Cespedes, M., Gotuzzo, E., and Vinetz, J. M. (2010). Asymptomatic renal colonization of humans in the peruvian amazon by leptospira. PLoS Negl. Trop. Dis. 4:e0612. doi: 10.1371/journal.pntd.0000612
Gao, R., Cao, B., Hu, Y., Feng, Z., Wang, D., Hu, W., et al. (2013). Human infection with a novel avian-origin influenza A (H7N9) virus. N. Engl. J. Med. 368, 1888–1897. doi: 10.1056/NEJMoa1304459
Gill, S. R., Pop, M., Deboy, R. T., Eckburg, P. B., Turnbaugh, P. J., Samuel, B. S., et al. (2006). Metagenomic analysis of the human distal gut microbiome. Science 312, 1355–1359. doi: 10.1126/science.1124234
Gofton, A. W., Doggett, S., Ratchford, A., Oskam, C. L., Paparini, A., Ryan, U., et al. (2015a). Bacterial profiling reveals novel “Ca. Neoehrlichia”, Ehrlichia, and Anaplasma Species in Australian Human-Biting Ticks. PLoS One 10:e0145449. doi: 10.1371/journal.pone.0145449
Gofton, A. W., Oskam, C. L., Lo, N., Beninati, T., Wei, H., Mccarl, V., et al. (2015b). Inhibition of the endosymbiont “Candidatus Midichloria mitochondrii” during 16S rRNA gene profiling reveals potential pathogens in Ixodes ticks from Australia. Parasit. Vectors 8:345. doi: 10.1186/s13071-015-0958-3
Goodrich, J. K., Di Rienzi, S. C., Poole, A. C., Koren, O., Walters, W. A., Caporaso, J. G., et al. (2014). Conducting a microbiome study. Cell 158, 250–262. doi: 10.1016/j.cell.2014.06.037
Grogan, L. F., Berger, L., Rose, K., Grillo, V., Cashins, S. D., and Skerratt, L. F. (2014). Surveillance for emerging biodiversity diseases of wildlife. PLoS Pathog. 10:e1004015. doi: 10.1371/journal.ppat.1004015
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98.
Holmstad, P. R., Jensen, K. H., and Skorping, A. (2008). Ectoparasite intensities are correlated with endoparasite infection loads in willow ptarmigan. Oikos 117, 515–520. doi: 10.1111/j.0030-1299.2008.16219.x
Hopkins, G. H. E., and Miriam, E. (1953). An Illustrated Catalogue of the Rothschild Collection of Fleas (Siphonaptera) in the British Museum (Natural History) with Keys and Short Descriptions for the Identification of Families, Genera, Species and Subspecies, Vol. I. London: Tungidae and Pulicidae.
Horinouchi, H., Murai, K., Okayama, A., Nagatomo, Y., Tachibana, N., and Tsubouchi, H. (1996). Genotypic identification of Rickettsia tsutsugamushi by restriction fragment length polymorphism analysis of DNA amplified by the polymerase chain reaction. Am. J. Trop. Med. Hyg. 54, 647–651. doi: 10.4269/ajtmh.1996.54.647
Iralu, J., Bai, Y., Crook, L., Tempest, B., Simpson, G., McKenzie, T., et al. (2006). Rodent-associated bartonella febrile illness, southwestern united states. Emerg. Infect. Dis. 12, 1081–1086. doi: 10.3201/eid1207.040397
Jiang, J., Chan, T. C., Temenak, J. J., Dasch, G. A., Ching, W. M., and Richards, A. L. (2004). Development of a quantitative real-time polymerase chain reaction assay specific for orientia tsutsugamushi. Am. J. Trop. Med. Hyg. 70, 351–356. doi: 10.4269/ajtmh.2004.70.351
Jones, K. E., Patel, N. G., Levy, M. A., Storeygard, A., Balk, D., Gittleman, J. L., et al. (2008). Global trends in emerging infectious diseases. Nature 451, 990–993. doi: 10.1038/nature06536
Keesing, F., Belden, L. K., Daszak, P., Dobson, A., Harvell, C. D., Holt, R. D., et al. (2010). Impacts of biodiversity on the emergence and transmission of infectious diseases. Nature 468, 647–652. doi: 10.1038/nature09575
Kindler, E., Jonsdottir, H. R., Muth, D., Hamming, O. J., Hartmann, R., Rodriguez, R., et al. (2013). Efficient replication of the novel human betacoronavirus EMC on primary human epithelium highlights its zoonotic potential. mBio 4:e00611–12. doi: 10.1128/mBio.00611-12
Klee, S. R., Tyczka, J., Ellerbrok, H., Franz, T., Linke, S., Baljer, G., et al. (2006). Highly sensitive real-time PCR for specific detection and quantification of Coxiella burnetii. BMC Microbiol. 6:2. doi: 10.1186/1471-2180-6-2
Lalzar, I., Harrus, S., Mumcuoglu, K. Y., and Gottlieb, Y. (2012). Composition and seasonal variation of rhipicephalus turanicus and Rhipicephalus sanguineus bacterial communities. Appl. Environ. Microbiol. 78, 4110–4116. doi: 10.1128/AEM.00323-12
Lello, J., Boag, B., and Hudson, P. J. (2005). The effect of single and concomitant pathogen infections on condition and fecundity of the wild rabbit (Oryctolagus cuniculus). Int. J. Parasitol. 35, 1509–1515. doi: 10.1016/j.ijpara.2005.06.002
McInerney, J. O., Cotton, J. A., and Pisani, D. (2008). The prokaryotic tree of life: past, present. and future?. Trends Ecol. Evol. 23, 276–281. doi: 10.1016/j.tree.2008.01.008
Meerburg, B. G., Singleton, G. R., and Kijlstra, A. (2009). Rodent-borne diseases and their risks for public health. Crit. Rev. Microbiol. 35, 221–270. doi: 10.1080/10408410902989837
Miller, R. R., Montoya, V., Gardy, J. L., Patrick, D. M., and Tang, P. (2013). Metagenomics for pathogen detection in public health. Genome Med. 5:81. doi: 10.1186/gm485
Mokili, J. L., Dutilh, B. E., Lim, Y. W., Schneider, B. S., Taylor, T., Haynes, M. R., et al. (2013). Identification of a novel human papillomavirus by metagenomic analysis of samples from patients with febrile respiratory illness. PLoS One 8:e58404. doi: 10.1371/journal.pone.0058404
Muul, I. (1979). Lekagul, boonsong, and jeffrey a. mcneely. mammals of thailand. association for the conservation of wildlife∗, Bangkok, Thailand, li + 758 pp., 1977. Price $50.00. J. Mammal. 60, 241–242. doi: 10.2307/1379792
Myint, K. S. A., Gibbons, R. V., Iverson, J., Shrestha, S. K., Pavlin, J. A., Mongkolsirichaikul, D., et al. (2011). Serological response to Bartonella species in febrile patients from Nepal. Trans. R. Soc. Trop. Med. Hyg. 105, 740–742. doi: 10.1016/j.trstmh.2011.08.002
Nadchatram, M. D. A. (1974). A Pictorial Key to the Subfamilies, Genera and Subgenera of SoutheastAsian Chiggers (Acari, Prostigmata, Trombiculidae). Kuala Lumpur: Institute for Medical Research.
Norman, A. F., Regnery, R., Jameson, P., Greene, C., and Krause, D. C. (1995). Differentiation ofBartonella-like isolates at the species level by PCR-restriction fragment length polymorphism in the citrate synthase gene. J. Clin. Microbiol. 33, 1797–1803.
Oliveros, J. C. (2007–2015). Venny. An interactive Tool for Comparing Lists with Venn’s Diagrams. Available at: http://bioinfogp.cnb.csic.es/tools/venny/index.html
Ong, S. H., Kukkillaya, V. U., Wilm, A., Lay, C., Ho, E. X., Low, L., et al. (2013). Species identification and profiling of complex microbial communities using shotgun Illumina sequencing of 16S rRNA amplicon sequences. PLoS One 8:e060811. doi: 10.1371/journal.pone.0060811
Paisse, S., Valle, C., Servant, F., Courtney, M., Burcelin, R., Amar, J., et al. (2016). Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion 56, 1138–1147. doi: 10.1111/trf.13477
Park, H. S., Lee, J. H., Jeong, E. J., Koh, S. E., Park, T. K., Jang, W. J., et al. (2004). Evaluation of groEL gene analysis for identification of Borrelia burgdorferi sensu lato. J. Clin. Microbiol. 42, 1270–1273. doi: 10.1128/JCM.42.3.1270-1273.2004
Parola, P., Diatta, G., Socolovschi, C., Mediannikov, O., Tall, A., Bassene, H., et al. (2011). Tick-borne relapsing fever borreliosis, rural senegal. Emerg. Infect. Dis. 17, 883–885. doi: 10.3201/eid1705.100573
Pereira, A., Parreira, R., Cotao, A. J., Nunes, M., Vieira, M. L., Azevedo, F., et al. (2018). Tick-borne bacteria and protozoa detected in ticks collected from domestic animals and wildlife in central and southern Portugal. Ticks Tick Borne Dis. 9, 225–234. doi: 10.1016/j.ttbdis.2017.09.008
Pierlé, S. A., Rosshandler, I. I., Kerudin, A. A., Sambono, J., Lew-Tabor, A., Rolls, P., et al. (2014). Genetic diversity of tick-borne rickettsial pathogens; insights gained from distant strains. Pathogens 3, 57–72. doi: 10.3390/pathogens3010057
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Razzauti, M., Galan, M., Bernard, M., Maman, S., Klopp, C., Charbonnel, N., et al. (2015). A Comparison between transcriptome sequencing and 16S metagenomics for detection of bacterial pathogens in wildlife. PLoS Negl. Trop. Dis. 9:e0003929. doi: 10.1371/journal.pntd.0003929
Salter, S. J., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffatt, M. F., et al. (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12:87. doi: 10.1186/s12915-014-0087-z
Schwaiger, M., Peter, O., and Cassinotti, P. (2001). Routine diagnosis of Borrelia burgdorferi (sensu lato) infections using a real-time PCR assay. Clin. Microbiol. Infect. 7, 461–469. doi: 10.1046/j.1198-743x.2001.00282.x
Scoles, G. A., Papero, M., Beati, L., and Fish, D. (2001). A relapsing fever group spirochete transmitted by Ixodes scapularis ticks. Vector Borne Zoonotic Dis. 1, 21–34. doi: 10.1089/153036601750137624
Shakya, M., Quince, C., Campbell, J. H., Yang, Z. K., Schadt, C. W., and Podar, M. (2013). Comparative metagenomic and rRNA microbial diversity characterization using archaeal and bacterial synthetic communities. Environ. Microbiol. 15, 1882–1899. doi: 10.1111/1462-2920.12086
Sipos, R., Székely, A. J., Palatinszky, M., Révész, S., Márialigeti, K., and Nikolausz, M. (2007). Effect of primer mismatch, annealing temperature and PCR cycle number on 16S rRNA gene-targetting bacterial community analysis. FEMS Microbiol. Ecol. 60, 341–350. doi: 10.1111/j.1574-6941.2007.00283.x
Slack, A. T., Symonds, M. L., Dohnt, M. F., and Smythe, L. D. (2006). Identification of pathogenic leptospira species by conventional or real-time PCR and sequencing of the DNA gyrase subunit B encoding gene. BMC Microbiol. 6:95. doi: 10.1186/1471-2180-6-95
Smythe, L. D., Smith, I. L., Smith, G. A., Dohnt, M. F., Symonds, M. L., Barnett, L. J., et al. (2002). A quantitative PCR (TaqMan) assay for pathogenic Leptospira spp. BMC Infect. Dis. 2:13. doi: 10.1186/1471-2334-2-13
Sogin, M. L., Morrison, H. G., Huber, J. A., Mark Welch, D., Huse, S. M., Neal, P. R., et al. (2006). Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. U.S.A. 103, 12115–12120. doi: 10.1073/pnas.0605127103
Sunagawa, S., Desantis, T. Z., Piceno, Y. M., Brodie, E. L., Desalvo, M. K., Voolstra, C. R., et al. (2009). Bacterial diversity and white plague disease-associated community changes in the caribbean coral Montastraea faveolata. ISME J. 3, 512–521. doi: 10.1038/ismej.2008.131
Swei, A., Bowie, V. C., and Bowie, R. C. (2015). Comparative genetic diversity in lyme disease bacteria from northern california ticks and their vertebrate hosts. Ticks Tick Borne Dis. 6, 414–423. doi: 10.1016/j.ttbdis.2015.03.011
Swei, A., Russell, B. J., Naccache, S. N., Kabre, B., Veeraraghavan, N., Pilgard, M. A., et al. (2013). The genome sequence of lone star virus, a highly divergent bunyavirus found in the Amblyomma americanum tick. PLoS One 8:e62083. doi: 10.1371/journal.pone.0062083
Tadin, A., Turk, N., Korva, M., Margaletic, J., Beck, R., Vucelja, M., et al. (2012). Multiple co-infections of rodents with hantaviruses, Leptospira, and Babesia in Croatia. Vector Borne Zoonotic Dis. 12, 388–392. doi: 10.1089/vbz.2011.0632
Takhampunya, R., Kim, H. C., Chong, S. T., Korkusol, A., Tippayachai, B., Davidson, S. A., et al. (2017). Francisella-Like endosymbiont detected in haemaphysalis ticks (Acari: Ixodidae) from the republic of korea. J. Med. Entomol. 54, 1735–1742. doi: 10.1093/jme/tjx123
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tanner, M. A., Goebel, B. M., Dojka, M. A., and Pace, N. R. (1998). Specific ribosomal DNA sequences from diverse environmental settings correlate with experimental contaminants. Appl. Environ. Microbiol. 64, 3110–3113.
Tanskull, P., and Inlao, I. (1989). Keys to the adult ticks of haemaphysalis koch, 1844, in Thailand with notes on changes in taxonomy (Acari: Ixodoidea: Ixodidae). J. Med. Entomol. 26, 573–601. doi: 10.1093/jmedent/26.6.573
Tanskul, P., Stark, H. E., and Inlao, I. (1983). A checklist of ticks of Thailand (Acari: Metastigmata: Ixodoidea). J. Med. Entomol. 20, 330–341. doi: 10.1093/jmedent/20.3.330
Taylor, L. H., Latham, S. M., and Woolhouse, M. E. (2001). Risk factors for human disease emergence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356, 983–989. doi: 10.1098/rstb.2001.0888
Tringe, S. G., and Hugenholtz, P. (2008). A renaissance for the pioneering 16S rRNA gene. Curr. Opin. Microbiol. 11, 442–446. doi: 10.1016/j.mib.2008.09.011
van Boheemen, S., De Graaf, M., Lauber, C., Bestebroer, T. M., Raj, V. S., Zaki, A. M., et al. (2012). Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3:e473–12. doi: 10.1128/mBio.00473-12
Vayssier-Taussat, M., Moutailler, S., Michelet, L., Devillers, E., Bonnet, S., Cheval, J., et al. (2013). Next generation sequencing uncovers unexpected bacterial pathogens in ticks in western Europe. PLoS One 8:e081439. doi: 10.1371/journal.pone.0081439
Victoria, B., Ahmed, A., Zuerner, R. L., Ahmed, N., Bulach, D. M., Quinteiro, J., et al. (2008). Conservation of the S10-spc-alpha locus within otherwise highly plastic genomes provides phylogenetic insight into the genus Leptospira. PLoS One 3:e2752. doi: 10.1371/journal.pone.0002752
Wan, X. F., Barnett, J. L., Cunningham, F., Chen, S., Yang, G., Nash, S., et al. (2013). Detection of African swine fever virus-like sequences in ponds in the Mississippi Delta through metagenomic sequencing. Virus Genes 46, 441–446. doi: 10.1007/s11262-013-0878-2
Wang, M. D., and Jolly, A. M. (2004). Changing virulence of the SARS virus: the epidemiological evidence. Bull. World Health Organ. 82, 547–548.
Wickham, H. (2009). ggplot2: Elegant Graphics for Data Analysis. New York, NY: Springer-Verlag. doi: 10.1007/978-0-387-98141-3
Woese, C. R., and Fox, G. E. (1977). Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc. Natl. Acad. Sci. U.S.A. 74, 5088–5090. doi: 10.1073/pnas.74.11.5088
Keywords: metagenomic, bacterial community, disease epidemiology, disease transmission, scrub typhus, undifferentiated febrile illness
Citation: Takhampunya R, Korkusol A, Pongpichit C, Yodin K, Rungrojn A, Chanarat N, Promsathaporn S, Monkanna T, Thaloengsok S, Tippayachai B, Kumfao N, Richards AL and Davidson SA (2019) Metagenomic Approach to Characterizing Disease Epidemiology in a Disease-Endemic Environment in Northern Thailand. Front. Microbiol. 10:319. doi: 10.3389/fmicb.2019.00319
Received: 25 November 2018; Accepted: 06 February 2019;
Published: 26 February 2019.
Edited by:
Michael Kosoy, Centers for Disease Control and Prevention (CDC), United StatesReviewed by:
Clifton McKee, Colorado State University, United StatesSara Moutailler, Agence Nationale de Sécurité Sanitaire de l’Alimentation, de l’Environnement et du Travail (ANSES), France
Copyright © 2019 Takhampunya, Korkusol, Pongpichit, Yodin, Rungrojn, Chanarat, Promsathaporn, Monkanna, Thaloengsok, Tippayachai, Kumfao, Richards and Davidson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ratree Takhampunya, cmF0cmVldC5mc25AYWZyaW1zLm9yZw==