
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 27 February 2019
Sec. Infectious Agents and Disease
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.00082
This article is part of the Research Topic New epidemiology of Staphylococcus Aureus Complex from the Tropics View all 6 articles
 Ana Maria Nunes Botelho1
Ana Maria Nunes Botelho1 Maiana Oliveira Cerqueira e Costa2
Maiana Oliveira Cerqueira e Costa2 Ahmed M. Moustafa3
Ahmed M. Moustafa3 Cristiana Ossaille Beltrame1
Cristiana Ossaille Beltrame1 Fabienne Antunes Ferreira1
Fabienne Antunes Ferreira1 Marina Farrel Côrtes1
Marina Farrel Côrtes1 Bruno Souza Scramignon Costa1Deborah Nascimento Santos Silva1Paula Terra Bandeira1Nicholas Costa Barroso Lima2
Bruno Souza Scramignon Costa1Deborah Nascimento Santos Silva1Paula Terra Bandeira1Nicholas Costa Barroso Lima2 Rangel Celso Souza2
Rangel Celso Souza2 Luiz Gonzaga Paula de Almeida2
Luiz Gonzaga Paula de Almeida2 Ana Tereza Ribeiro Vasconcelos2Apurva Narechania4Chanelle Ryan3Kelsey O’Brien3
Ana Tereza Ribeiro Vasconcelos2Apurva Narechania4Chanelle Ryan3Kelsey O’Brien3 Sergios-Orestis Kolokotronis4,5
Sergios-Orestis Kolokotronis4,5 Paul J. Planet3,4*
Paul J. Planet3,4* Marisa Fabiana Nicolás2*
Marisa Fabiana Nicolás2* Agnes Marie Sá Figueiredo1*
Agnes Marie Sá Figueiredo1*The global spread of specific clones of methicillin-resistant Staphylococcus aureus (MRSA) has become a major public health problem, and understanding the dynamics of geographical spread requires worldwide surveillance. Over the past 20 years, the ST239 lineage of MRSA has been recognized as an emerging clone across the globe, with detailed studies focusing on isolates from Europe and Asia. Less is known about this lineage in South America, and, particularly, Brazil where it was the predominant lineage of MRSA in the early 1990s to 2000s. To gain a better understanding about the introduction and spread of ST239 MRSA in Brazil we undertook a comparative phylogenomic analysis of ST239 genomes, adding seven completed, closed Brazilian genomes. Brazilian ST239 isolates grouped in a subtree with those from South American, and Western, romance-language-speaking, European countries, here designated the South American clade. After an initial worldwide radiation in the 1960s and 1970s, we estimate that ST239 began to spread in South America and Brazil in approximately 1988. This clone demonstrates specific genomic changes that are suggestive of local divergence and adaptational change including agrC single-nucleotide polymorphisms variants, and a distinct pattern of virulence-associated genes (mainly the presence of the chp and the absence of sea and sasX). A survey of a geographically and chronologically diverse set of 100 Brazilian ST239 isolates identified this virulence genotype as the predominant pattern in Brazil, and uncovered an unexpectedly high prevalence of agr-dysfunction (30%). ST239 isolates from Brazil also appear to have undergone transposon (IS256) insertions in or near global regulatory genes (agr and mgr) that likely led to rapid reprogramming of bacterial traits. In general, the overall pattern observed in phylogenomic analyses of ST239 is of a rapid initial global radiation, with subsequent local spread and adaptation in multiple different geographic locations. Most ST239 isolates harbor the ardA gene, which we show here to have in vivo anti-restriction activity. We hypothesize that this gene may have improved the ability of this lineage to acquire multiple resistance genes and distinct virulence-associated genes in each local context. The allopatric divergence pattern of ST239 also may suggest strong selective pressures for specific traits in different geographical locations.
Staphylococcus aureus is a challenging human pathogen capable of causing a range of hospital (HA) and community-associated (CA) infections, varying from local and uncomplicated skin/soft tissue infections to more severe illnesses such as necrotizing fasciitis, pneumonia, bacteremia, osteomyelitis, and endocarditis. This extraordinary pathogenic potential is generally attributed to a plethora of well-known virulence-associated genes, and a relatively high rate of genomic plasticity that results in the frequent acquisition of new genes (Figueiredo and Ferreira, 2014). MRSA remains one of the leading causes of hospital infections worldwide, despite aggressive infection control measures (World Health Organisation [WHO], 2017). Interestingly, like CA-MRSA strains, the global diversity of HA-MRSA is represented by just a few, mostly clonal, epidemic lineages that appear to have adapted to specific challenges of the hospital environment (Hsu et al., 2015). In many cases, the specific factors that enhance the fitness of HA-MRSA strains are unknown, but it is likely that environmental and host challenges as well as inter-strain competition affect selection (Figueiredo and Ferreira, 2014). The mechanisms that lead to the spread of organisms over large geographical areas, even though they manifest mostly in hospitals, are also obscure, betraying a more complex epidemiology that includes large-scale waves of replacement of one predominant clone by another (Figueiredo and Ferreira, 2014; Hsu et al., 2015; Planet et al., 2017).
One of the most successful HA-MRSA lineages is the multilocus sequence type (ST) 239 that carries the staphylococcal cassette chromosome type III (SCCmecIII). ST239-SCCmecIII appears to have originated as a naturally occurring “hybrid,” from a large recombination event involving S. aureus ancestors of two major lineages, CC8 and CC30, which contribute about 80% and 20% of its genome, respectively (Robinson and Enright, 2004). The ST239-SCCmecIII MRSA lineage has been associated with outbreaks in healthcare settings on all populated continents, and members of this lineage are generally characterized by their ability to cause serious, disseminated infections (Holden et al., 2010). Additionally, these bacteria present high-level resistance to many types of available antimicrobial drugs. Glycopeptide antibiotics and some more recent anti-MRSA-targeted antibiotics remain effective, but heteroresistance to vancomycin (hVISA) has recently been detected in ST239 isolates from Australia (van Hal et al., 2011).
Several studies have analyzed the genomic epidemiology and phylogeny of ST239 MRSA from different geographic regions (Harris et al., 2010; Yamamoto et al., 2012; Wang et al., 2014; Baines et al., 2015; Hsu et al., 2015; Arias et al., 2017). These studies focused mostly on isolates from Europe, Asia, and Australia; although a handful of ST239 strains from Latin America have been noted to form their own clade. Within this clade is a ST239 lineage referred to as the Brazilian epidemic clone (BEC) that was first reported in 1992 (Teixeira et al., 1995). For over 20 years this clone has been one of the most common MRSA isolates collected in Brazilian hospitals (Figueiredo and Ferreira, 2014). To gain a better understanding of this lineage we completed, closed, and annotated genomes for seven ST239 isolates of Brazilian MRSA (ST239-BRC). We used publicly available ST239 genome sequences to assess the origin and phylogeography of these isolates, and to generate a more complete picture of the spread and local adaptation of ST239.
Seven representatives of the ST239-SCCmecIII lineage obtained from clinical samples in Brazil, including agr-functional and -dysfunctional isolates, were selected for whole-genome sequencing. These MRSA isolates were collected from patients presenting with local or invasive hospital-associated (HA) infections, admitted at different public general hospitals; and from colonization cases involving patients enrolled in a home-care system, from 1993 to 2001, as indicated in Table 1. The strains BMB9393, HC1335, and HC1340 were isolated in Rio de Janeiro, RJ (southeast region of Brazil), GV69, GV51, and GV88 in Teresina, PI (Northeast region), and Be62 in Belém, PA (North region). The distance from Rio de Janeiro to Belem is about 2,464 km and from Belém to Teresina approximately 752 km. The genomes of seven of these Brazilian ST239 MRSA were completely closed and referred collectively as ST239-BRC.
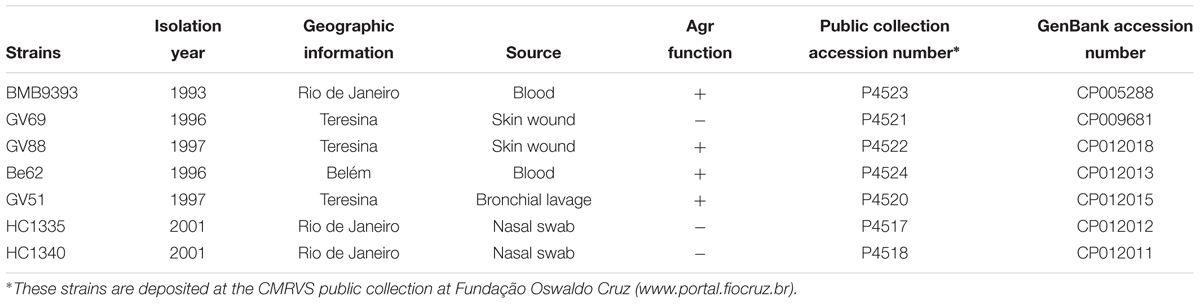
Table 1. Clinical source, public collection and GenBank accession numbers of the ST239 MRSA strains from Brazil sequenced.
Additionally, a collection of 100 hospital MRSA isolates previously genotyped as ST239-SCCmecIII (Teixeira et al., 1995) from different geographic regions of Brazil was used in experimental assays. We refer to this collection as ST239-BR100. These isolates were collected from sites of infection (n = 62) or from nasal colonization (n = 38), and only one isolate per patient was included. The main characteristics of these isolates are listed in Supplementary Table S1.
For detailed comparative genomic analyses, we used two other representative genomic subsets (ST239-INTC, non-ST239-INTC). First we defined an international subset of completed and annotated ST239 genomes from GenBank (ST239-INTC). These genomes were from the following isolates: (i) TW20 (GenBank Accession Number: FN433596), isolated in 2003 from a 2-year MRSA outbreak in an intensive care unit (ICU), in London (Holden et al., 2010); (ii) JKD6008 (Acc: CP002120), a vancomycin-intermediate S. aureus (VISA) strain obtained in 2003 from blood after 42 day-vancomycin treatment, in New Zealand (Howden et al., 2010); (iii) T0131 (Acc: CP002643), isolated in 2006 from an 87-year-old in China (Li et al., 2011); (iv) Z172 (Acc: CP006838), a VISA isolate obtained in 2010 from blood of an elderly patient in an ICU in Taiwan (Chen et al., 2013) and (v) XN108 (Acc: CP007447), also a VISA isolate collected from a burned patient with a wound infection, in China (Zhang et al., 2014). We also used another comparative group of well-described, international, complete HA-MRSA genomes from outside of the ST239 lineage (non-ST239-INTC), which included the following genomes: (i) MRSA252 (Acc: BX571856), a ST36-SCCmecII isolate, representative of the EMRSA-16 clone (Holden et al., 2004); (ii) Mu50 (Acc:: BA0000 17), a ST5-SCCmecII isolate, representative of the USA100 clone (Kuroda et al., 2001); (iii) CA-347 (Acc: CP006044), a ST45-SCCmecIV isolate, representative of the USA600 clone (Stegger et al., 2013); (iv) H-EMRSA-15 (Acc: CP007659), a ST22-SCCmecIV isolate, representative of the EMRSA-15 clone (Sabirova et al., 2014), and (v) 2395 (Acc: CP007499), a ST8-SCCmecIV isolate, representative of the USA500 clone (Benson et al., 2014). The characteristics of ST239-INTC and non-ST239-INTC are summarized in Table 2.
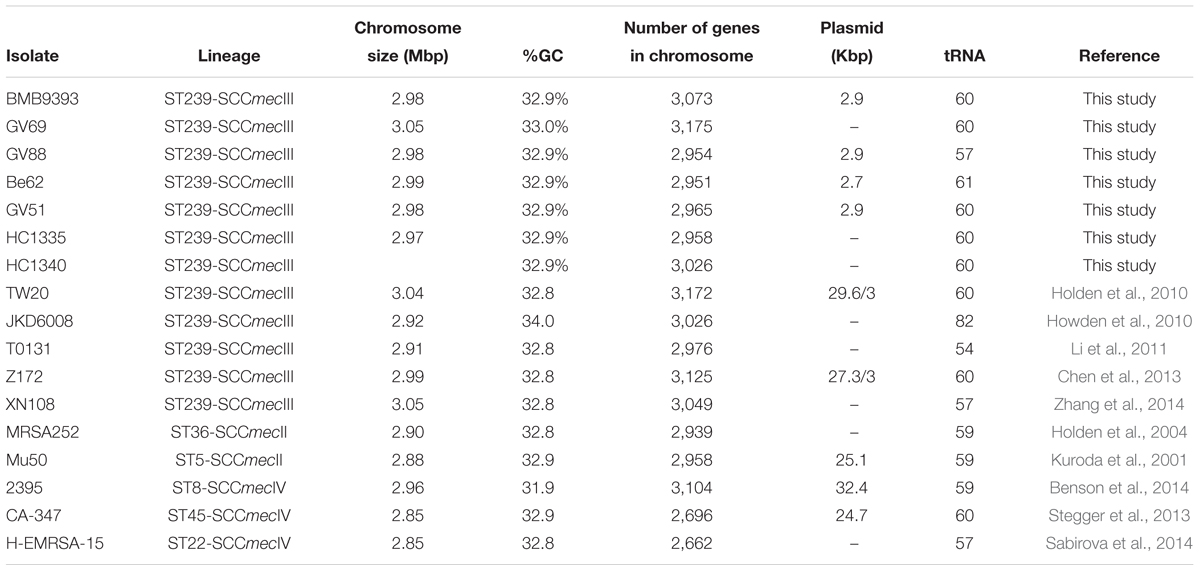
Table 2. General characteristics of the closed genomes of the HA–MRSA strains from ST239 and non-ST239 lineages.
Besides the seven genomes sequenced for this work (ST239-BRC) and the five completely closed genomes available on NCBI (ST239-INTC), a total of 171 ST239 and 7 single-locus variant (SLV) raw genome sequences deposited in the GenBank1, along with those used in the phylogenetic studies published by Castillo-Ramírez et al. (2012) and Harris et al. (2010) deposited in the EMBL-Bank (Acc: ERA000102) were also used in this study for phylogenetic analyses, altogether totaling 190 genomes. Only genomes of strains with reported isolation date were considered for the molecular clock analysis (n = 167 genomes). The accession numbers for these assemblies are listed in Supplementary Table S2. For comparative genomic studies, the genome of the Brazilian strain BMB9393 was used as reference, in order to easily highlight the differences between Brazilian and international ST239 genomes. The strain TW20 that belongs to the ST239 lineage and has been used as reference in genomic studies of ST239 MRSA was chosen as the reference for phylogenetic analyses.
DNA from the seven ST239-BRC MRSA isolates used was obtained by phenol extraction and ethanol precipitation (Sambrook et al., 1989). The concentration and purity of the DNA were assessed using a Qubit® 2.0 fluorometer (Invitrogen, Eugene, OR, United States). A total of 5 μg genomic DNA was used to prepare a paired-end library. The genome sequencing was performed using a 454 GS FLX Titanium (3-kb paired-end library) approach (Roche Diagnostics Corporation, Indianapolis, IN, United States). The assembly was accomplished using Newbler v2.6 (Roche) (Margulies et al., 2005) and Celera Assembler v6.1 (Myers et al., 2000). Gaps within scaffolds resulting from repetitive sequences were resolved by in silico gap filling. The complete genome assembly was accomplished using a combination of Newbler v 2.6 (Roche Inc.) and GapFiller (Boetzer and Pirovano, 2012). The consensus sequences assembled into contigs and scaffolds were aligned against a database of complete genomes from the Staphylococcus genus to determine the orientation and order of the contigs and scaffolds (Supplementary Figure S1). The gaps, intra- and inter-scaffolds, were filled by individual assemblies of the reads falling in both termini of a given gap (Supplementary Figure S1). This was accomplished by selecting reads that formed the end of contigs adjacent to each gap stretches. Those reads were assembled separately with Newbler. Contiguous sequences generated by this approach that were able to complete the gap and anchor on the two adjacent contigs were added to the sequence, thus closing the gap. As a last step of gap closure, the remaining gaps were closed using the GapFiller program.
Genome annotation was performed using the System for Automated Bacterial Integrated Annotation (SABIA) (Almeida et al., 2004). This software uses an automated annotation pipeline, where each open-reading frame (ORF) is submitted for comparison with several databases (NCBI-nr, KEGG, InterPro, and UniProtKB/Swiss-Prot), and the results are made available on the screen for assessment by expert users. All possible ORFs were predicted by Glimmer (Delcher et al., 2007) and GeneMark (Besemer and Borodovsky, 2005) and the transfer RNAs (tRNAs) by tRNAscan-SE (Schattner et al., 2005). The identification of bona fide ORFs and their probable functions included similarity searches using both nucleotide and amino acid sequences by Basic Local Alignment Search Tool (BLAST) against KEGG, NCBI-nr, and UniProtKB/Swiss-Prot databases, as well as identification of protein domains and important sites using InterPro (Mitchell et al., 2015).
Chromosomal architecture and genome organization were initially analyzed using ProgressiveMauve (Darling et al., 2010), with default parameters. The inversions found in the GV69 genome (ST239-BRC), using BMB9393 genome (ST239-BRC) as reference, were confirmed by a PCR-based assay using specific primers listed in Supplementary Table S3. Thus, to confirm the position of each locally collinear block (LCB) in the GV69 genome, primers were designed using the genome of GV69 based on contiguous sequences of adjacent LCBs. The rearrangement found in silico was confirmed when the PCR resulted in the expected amplicon size. The MicroScope platform (Vallenet et al., 2009, 2013) was used for determination of the RGPs and unique/shared gene content identification. RGPs were defined as DNA segments over 5 kbp that were possibly related to events of horizontal exchanges. Additional features for identification of RGPs included G + C% deviation, compositional biases, presence of prophage genes, synteny breaks, and proximity to tRNAs. For these analyses, the BMB9393 chromosome was set as the reference and RGPs were mapped in ST239-BRC, ST239-INTC, and non-ST239-INTC genomes. Each identified RGP was manually inspected based on the genomic context and conserved genes located at the flanking regions. Other genomic analyses were performed using the comparative genomic tools available in SABIA platform (Almeida et al., 2004). The Geneious software (Biomatters Ltd., Auckland, New Zealand) was also used for genome alignment and visualization of the regions analyzed. Putative bacteriophages were detected using the phage search tool, PHAST (Zhou et al., 2011). The set of virulence genes (virulome) and staphylococcal pathogenicity island (SaPI) and other genomic islands (GIs) were detected using VirulenceFinder 1.5 (minimum length = 80%, threshold id = 98%) and manual inspection using Uniprot/Swissprot, BLAST, and literature review (Joensen et al., 2014). The EasyFig software (Sullivan et al., 2011) was used for creating linear comparative figures of multiple genomic loci, and the BLASTn atlas was generated with GVIEW, applying as parameters identity >80% and e-values <10-10 (Petkau et al., 2010).
We used PCR to search for important genomic features in the whole collection of ST239-BR100 isolates. The primers used for each assay are listed in Supplementary Table S3.
Besides the genomes sequenced in this study (ST239-BRC) and the international representative genomes used (ST239-INTC), ST239 genomes were downloaded from NCBI or from EMBL (n = 190) and were used for whole-genome alignment with reference to the S. aureus TW20 genome, using the NUCmer and show-snps utilities of MUMmer2. All regions from the reference genome annotated as MGEs were excluded as well as the approximately 20% of the genome thought to be derived from CC30 (as in Holden et al., 2010; Supplementary Table S4). We also applied a mask that excluded repetitive sequences from the reference genome that were >80% identical over at least 100 nucleotides to other genomic loci, based on pairwise MegaBLAST-based analysis. Phylogenetic reconstruction was carried out by using the alignment of polymorphic sites in IQ-TREE v1.6.2 (Nguyen et al., 2015). One BIONJ (Gascuel, 1997) and 100 parsimony trees were used as starting trees for the maximum-likelihood (ML) tree search. The substitution model implemented was the general time-reversible (GTR) (Lanave et al., 1984) and empirical base frequencies, among-site rate heterogeneity was modeled according to the free-rate model (Yang, 1995; Soubrier et al., 2012), and ascertainment bias for variable-only sites was corrected (Lewis, 2001). Internode branch support was evaluated with the ultrafast bootstrap approximation method (UFBoot) and a maximum of 1000 replicates (Minh et al., 2013), using a convergence criterion, as well as with the Shimodaira–Hasegawa–aLRT branch test (Guindon et al., 2010) and 1,000 replicates.
To estimate the time that elapsed since the genomic divergence of the isolates in our dataset, we employed a Bayesian phylogenetic framework implemented in BEAST v2.5.1 (Drummond et al., 2012). For this analysis, genomes without collection date report were excluded, leading to a total of 167 genomes included. In addition to the above masked regions that were potentially mobile (Supplementary Table S4), we sought to exclude any regions with evidence of recombination. For this analysis, we rebuilt the genome matrix using simulated paired-end reads that were generated for the 167 assembled genomes that have dates using wgsim3 in the haplotype mode without introducing errors, mutations, or indels. Read mapping to TW20 reference genome and variants calling were performed using Snippy v.4.3.34. The regions in TW20 excluded in the previous step were masked in this analysis as well. A whole genome alignment, including SNPs and invariant sites, produced by Snippy was used to infer an initial phylogenetic tree in RAxML v8.2.4 (Stamatakis, 2014) using the GTR substitution model (Lanave et al., 1984) accounting for among-site rate heterogeneity using the Γ distribution and four rate categories (Yang, 1995) for 100 individual searches with maximum parsimony random-addition starting trees. Node support was evaluated with 100 nonparametric bootstrap pseudoreplicates (Felsenstein, 1985). The initial ML newick tree and the whole-genome alignment were used as input for ClonalFrameML (Didelot and Wilson, 2015) to infer recombination using 100 pseudo-bootstrap replicates. The maskrc-svg script5 was then used to mask recombinant regions from the original whole-genome alignment. A SNP alignment was then called from the resultant alignment, from maskrc-svg, using the SNP-sites tool (Page et al., 2016).
The SNP alignment was then used to estimate divergence times in BEAST v2.5.1 (Bouckaert et al., 2014). In order to gauge the potential for temporal signal of our dataset, we used a regression of root-to-tip genetic distance vs. isolation time as a diagnostic tool, as implemented in TempEst v1.5.1 (Rambaut et al., 2016). A positive correlation between genetic divergence and isolation time was observed (plot not shown; R2 = 0.6413), indicating suitability of this dataset for downstream molecular clock analysis in BEAST. The Hasegawa–Kishino–Yano nucleotide substitution model was used (Hasegawa et al., 1985) with among-site rate heterogeneity modeled with the Γ distribution and four discrete rate categories (Yang, 1995), and ascertainment bias for variable-only sites was corrected by factoring in the number of invariable sites based on fully sequenced MRSA genomes. We implemented an uncorrelated lognormal relaxed clock model with a random starting tree, a Bayesian skyline coalescent (Drummond, 2005), and a uniform prior probability distribution of 10-4–10-8 substitutions/site/year. The chain length was set at 200 million MCMC steps with a 1,000-step thinning. The sampled parameters and their effective sample size (ESS > 200) were inspected in Tracer v1.66. LogCombiner was then used to resample posterior distribution of the trees file at a lower frequency (10,000 steps). We also implemented a strict clock in BEAST with a random starting tree, a Bayesian skyline coalescent (Drummond, 2005), and a uniform prior probability distribution of 10-4-10-8 substitutions/site/year. The chain length was set at 50 million MCMC steps with a 5,000-step thinning. Two independent runs were combined post convergence, following inspections of the sampled parameters and their ESS. The chronograms were plotted on the basis of the maximum clade credibility tree using the TreeAnnotator program from the BEAST package and were visualized in FigTree v1.4.37. The trees and divergence times produced by the strict and relaxed clock techniques were largely indistinguishable.
The detection of Agr activity in the collection of one hundred (ST239-BR100) isolates was performed using MALDI–TOF–MS according to a previously described method (Josten et al., 2014). Briefly, individual colonies were subjected to MALDI-TOF-MS for detection of a peak at m/z 2415 representing the phenol soluble modulin PSM-mec, an Agr-regulated gene harbored on some types of SCCmec, including SCCmecIII, which is carried by the ST239 lineage.
Strains BMB9393 and HC1335 were also tested using a real-time quantitative reverse transcription-polymerase chain reaction (real-time qRT-PCR) for the RNAIII transcript with expression of the 16S rRNA gene as a control. For RNA preparations, bacterial cells grown in BHI broth (18 h; 37°C; 250 rpm) were collected in the stationary phase. Total RNA was prepared using RNeasy Mini kit (Qiagen) and quantified using a Nanodrop Lite Spectrophotometer (Thermo Scientific, ThermoFisher Scientific). RNA integrity was analyzed by gel electrophoresis. Real-time qRT-PCR was performed using the Power SYBR Green RNA-to-CTTM 1-Step Kit (Applied Biosystems, ThermoFisher Scientific) as recommended. The run was performed in a StepOne Real-Time PCR System (Applied Biosystems, ThermoFisher Scientific) and analyzed using the StepOne Software v2.2 (Applied Biosystems, ThermoFisher Scientific). The cycling conditions for all primers were performed as follows: 48°C for 30 min (cDNA preparation); 95°C for 10 min; 35 cycles of 95°C for 30 s, 55°C for 45 s, and 72°C for 45 s (cDNA amplification cycles). At least three biological replicates were run with four technical replicates each. Primers used are listed in Supplementary Table S3.
Genomic analysis showed an IS256-related element upstream mgrA ORF in the genome of BMB9393 strain (ST230-BRC). The frequency of this insertion in our collection of 100 ST239-BR isolates was assayed using primers designed based on the BMB9393 genome sequence, with the forward primer in the IS256 sequence and the reverse in the mgrA sequence. Likewise, the IS256 insertion found disrupting agrC ORF in the genome of HC1335 (ST239-BRC) was also searched for in the whole ST239-BR collection using a PCR-based approach, with forward primer in the agrC sequence and the reverse in the IS256 sequence (based on the HC1335 genome sequence). Amplicons with the expected sizes were considered positive for the correspondent insertion.
We mined agrC coding sequences from all assembled genomes and were condensed into 15 unique haplotypes. Those were then aligned by coding frame using MAFFT v7.407 (Katoh and Standley, 2013), and first checked for recombination breakpoints using the GARD algorithm (Kosakovsky Pond et al., 2006) on the DataMonkey web server8. We then employed several tests for positive or diversifying selection at different levels on this dataset. First, we examined whether or not over the length of the gene and across the phylogeny there had been at least one codon site that experienced diversifying selection on a branch. This was done using Branch-Site Unrestricted Statistical Test for Episodic Diversification (BUSTED) as a gene-wide test by testing all branches across the tree (Murrell et al., 2015). We then used different methods to investigate the potential for diversifying selection having acted on individual codon sites and, separately, on branches of the phylogeny. We employed two sitewise methods: Mixed Effects Model of Evolution (MEME) (Murrell et al., 2012) and Fast Unconstrained Bayesian AppRoximation (FUBAR) (Murrell et al., 2013), asking whether the intensity of natural selection has been relaxed or increased along internal branches of the phylogeny using the RELAX framework (Wertheim et al., 2015).
The ardA sequence was amplified using the primers ardA-fwd and ardA-rev (Supplementary Table S3) and genomic DNA from S. aureus BMB9393 as template. The ardA gene was initially cloned into the p-GEM T easy vector (Promega) and subcloned into the expression vector pCN40 using BamHI and EcoRI restriction sites (Charpentier et al., 2004). The recombinant plasmid pCN40A (pCN40:PblaZ-ardA) or empty pCN40 were transformed by electroporation into DC10B competent cells (Monk et al., 2012). The recombinant plasmids (pCN40A) and pCN40 were obtained (QIAfilter Plasmid Midi Kit; Qiagen), and transformed into RN4220 by electroporation (Monk et al., 2012), yielding the clones 42P40E (RN4220; empty pCN40) and 42P40A (RN4220; pCN40A). Transformants were confirmed by DNA sequencing and the expression of ardA in 42P40E was detected using real-time qRT-qPCR.
The natural S. aureus plasmid pBMB (Costa et al., 2013) and the shuttle-vector pLI50 (Addgen) were obtained from S. aureus BMB9393 and Escherichia coli DC10B strains, respectively. 42P40E or 42P40A competent cells were submitted to electroporation with 0.1 mg/μl of pBMB and 10 mg/μl of pLI50. Chloramphenicol-resistant colonies derived from 42P40A or 42P40E were measured as colony forming units (CFU)/ml.
We used Student’s t-test (α = 0.05) where noted. In addition to the selection tests noted above, a Poisson distribution was used to determine the probability of a nucleotide change in agrC occurring at the same rate observed for other ST239 S. aureus virulence regulator genes. To determine the most representative genetic pattern of a clade, defined by presence/absence of the loci (sasX, sae, chp, and agrC clade-specific SNPs), we used the Grubbs’ test. Subsequently, a χ2-test for independence was used to test whether the main genetic pattern of a clade and its SLVs were associated with a specific clade. We also used a χ2-test to correlate these genetic patterns and broad geographic location (Eastern Mediterranean, Asia, South America). Because there are many sequenced genomes from Tunisia and Turkey, we randomly selected only three representative genomes of each of these two countries.
To understand genomic diversity of our sequenced genomes (ST239-BRC), we did a detailed comparison to an international set of five completely closed ST239 genomes from different continents available in GenBank (ST239-INTC). We also defined a set of five other non-ST239, closed, and completed, S. aureus genome representatives from internationally disseminated lineages (non-ST239-INTC) commonly associated with hospital infections, including ST5, ST8, ST22, ST36, and ST45. The results of our analysis are shown in Figure 1.
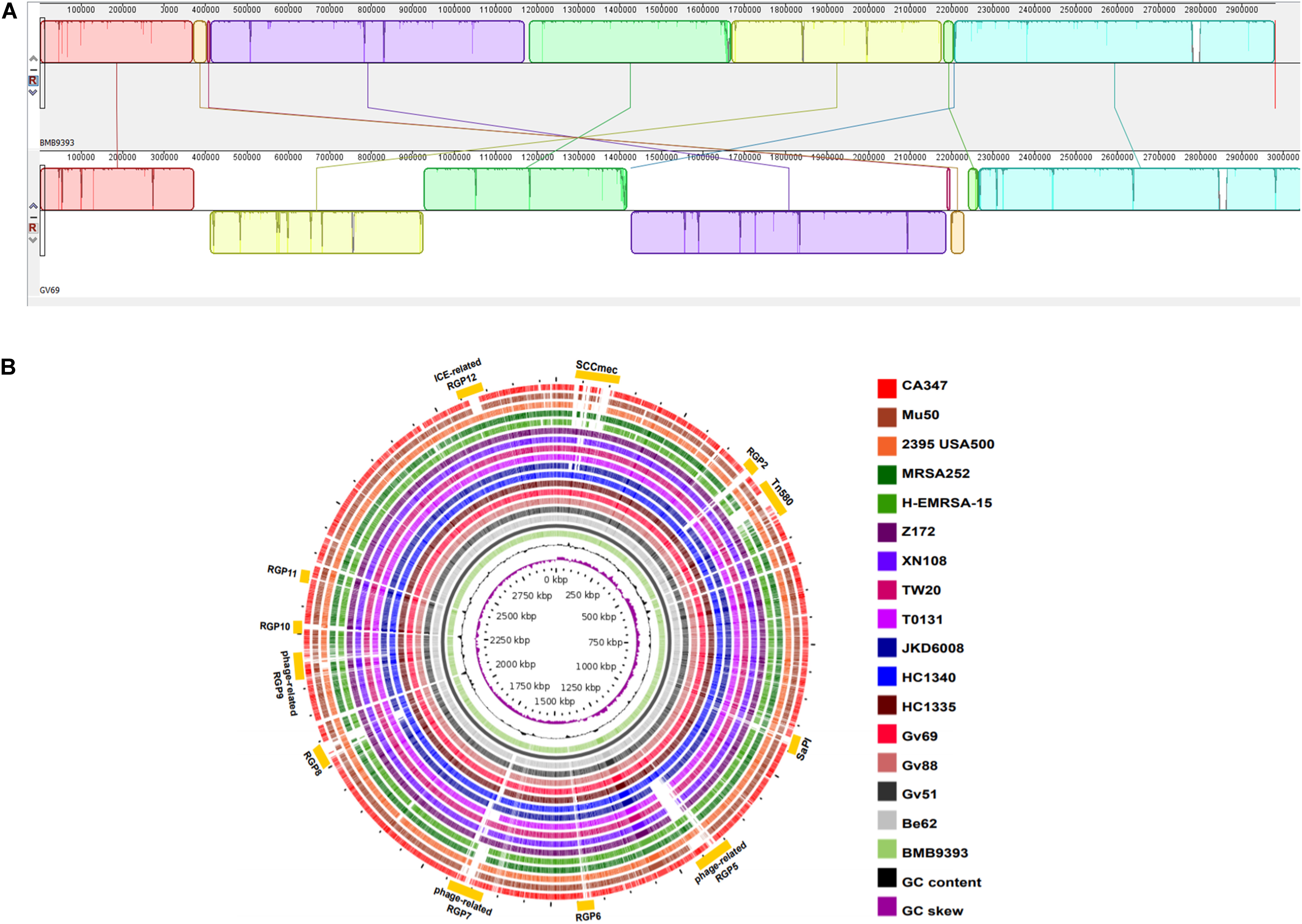
Figure 1. Genomic organization. (A) Genome alignments of ST239-BRC strains BMB9393 and GV69 highlighting the distinct syntenic patterns among them. The genome of the strain BMB9393 (ST239-BRC) was used as reference. (B) The innermost ring (purple) plots the G+C skew of the reference, followed by its G+C content (black). The third ring represents the reference chromosome of strain BMB9393 and its coordinates. The following colored rings depict BLASTN (>80% identity and e-values < 10-10) comparisons obtained by GVIEW, between the chromosome of BMB9393 and those of ST239-BRC strains (Be62, Gv51, Gv88, Gv69, HC1335, HC1340), ST239-INTC (JKD6008, T0131, TW20, XN108, Z172), and non-ST239-INTC (H-EMRSA-15, MRSA252, 2395, Mu50, CA-347). The outermost, interspaced rings (in dark yellow) represent the localization of the predicted RGPs in the BMB9393 chromosome, and the labels of each region as listed in Table 3.
The main characteristics of the completely sequenced and closed genomes of seven MRSA isolates from Brazil (ST239-BRC) are described in Table 2. Whole-genome alignments revealed a large genomic inversion in the GV69 genome compared to other ST239-BRC genomes (Figure 1A). This inversion was confirmed by PCR amplification of the expected products (see the section “Materials and Methods”). Additional chromosomal rearrangements were detected in comparison with ST239-INTC genomes (Figure 1B), as represented by breaks in the synteny between 10 genomic LCBs (Figure 1B).
Comparison of ST239-BRC, ST239-INTC, and non-ST239-INTC genomes performed using the SABIA platform (Almeida et al., 2004) revealed 1,991 genes in the ST239-SCCmecIII core genome, defined here as the genes common to all ST239 completely closed genomes. When compared with the non-ST239-INTc genomes, only 11 ORFs were exclusively found among the ST239-BRC genomes studied. These unique ORFs are predicted to encode five distinct hypothetical proteins (BMB9393 SABB_01759, BMB9393 SABB_01754, BMB9393 SABB_01753, BMB9393 SABB_02921, and BMB9393 SABB_01926), a putative transposase (BMB9393 SABB_00433), the cassette chromosome recombinase A (BMB9393 SABB_01756), the ribosome-binding factor A (BMB9393 SABB_00236), an ATP-binding protein (BMB9393 SABB_02230), a putative phi PVL-like protein (BMB9393 SABB_04160), and a superantigen-like protein (BMB9393 SABB_02209).
A total of 82 ORFs were exclusively present in the ST239-BRC set compared with the ST239-INTC genomes (Supplementary Table S5): 19 (23.2%) encode hypothetical proteins, 40 (48.8%) encode putative proteins related to bacteriophages and other MGEs) and the remaining 23 (28%) ORFs have diverse predicted biological functions including some putative and known virulence-related genes, such as the gene for the chemotaxis inhibitory protein (chp; BMB9393 SABB_02361). A total of 59 ORFs (Supplementary Table S5) were exclusively found in ST239-INTC genomes: 33 (55.9%) encode hypothetical proteins, 8 (13.6%) encode predicted proteins related to bacteriophages and other MGEs, and 18 (30.5%) encode putative proteins with different biological functions including the genes for known virulence-factors, such as enterotoxin A (Sea; TW20 SATW20_19410) and staphylococcal enterotoxin-like Q (SelQ; TW20 SATW20_08910). Most of these exclusive, or diagnostic, genes were found within RGPs, which will be discussed in the sections below.
The S. aureus autolysin gene atl (BMB9393 SABB_01019), in all ST239-BRC genomes, has a deletion of 243 bp in the coding region (Supplementary Figure S2A). PCR-based screening detected this deletion in 84% of the collection ST239-BR100 clinical isolates. The truncated atl gene was significantly more prevalent in the group of ST239-BR100 isolates that was collected from infection cases compared with that of colonization cases (p < 0.05), although this difference was relatively small (Figure 2A). It is possible that the absence of functional atl might be beneficial for immune evasion by reducing the amount of immune stimulation that is due to bacterial cell lysis (Humann and Lenz, 2009). It is possible that this could contribute to higher levels of invasive disease associated with these isolates.
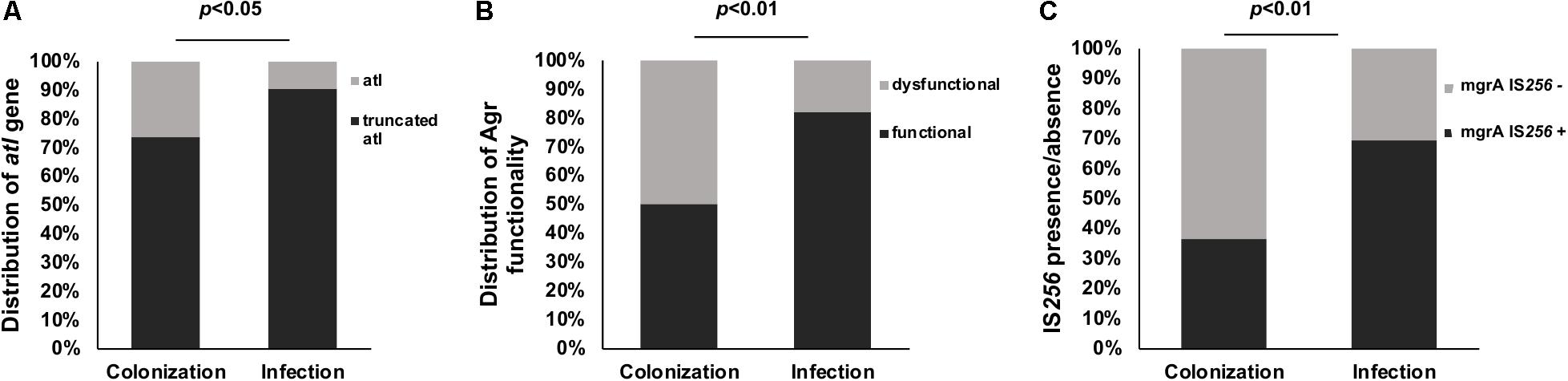
Figure 2. Differences in isolates collected from colonization and infection sites. (A) Percentage of truncated (black) and not truncated (gray) atl gene (PCR detected) in hundred ST239 isolates from Brazil collected from colonization and infection cases. (B) Distribution of functional (black) and dysfunctional (gray) Agr among 100 ST239 isolates obtained from colonization (n = 38) and infection cases (n = 62) in Brazil. (C) Presence (black) and absence (gray) of IS256 insertion in the mgrA gene in 100 ST239 MRSA isolates from Brazil collected from colonization and infection cases. Note a significantly increased number of IS256 insertion among infection cases compared with colonization cases (p-values calculated by Fisher’s exact).
We identified 12 RGPs in Brazilian isolates through our comparative approach (Figure 1B). These RGPs contain multiple signatures of genomic plasticity and MGEs including sequences related to transposable elements, phage-structures, insertion sites for integrases or recombinases, as well as adjacent tRNA for insertions sites of GIs, as summarized in Table 3. The amount of DNA located in the RGPs accounted for at least 429.5 kbp or 14.4% of the chromosome of the BMB9393 reference genome. Another feature of these RGPs is that they have GC content typically equivalent to the chromosomal DNA, suggesting that HGT events have predominantly involved S. aureus or other bacteria with low GC content (Lindsay, 2008). The main characteristics of the most prominent RGPs are discussed below, with additional descriptions provided in Supplementary Table S6.
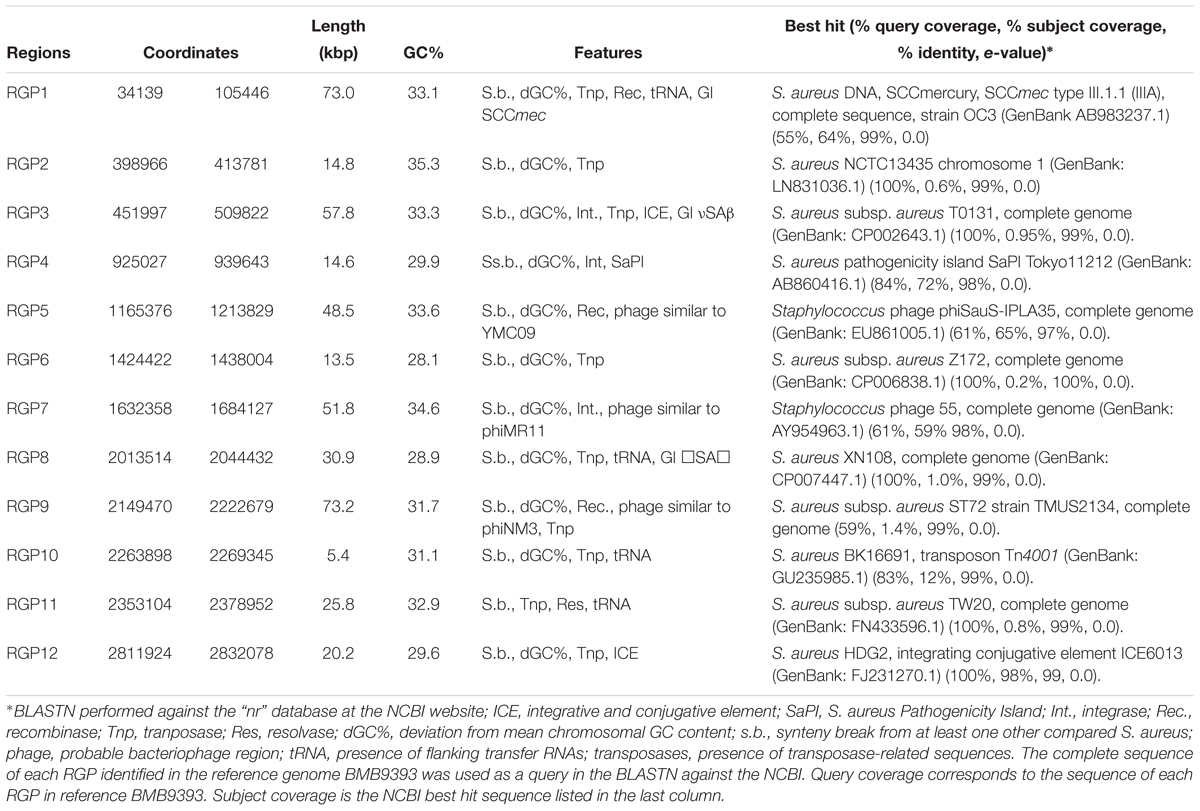
Table 3. RGPs identified comparing ST239 and non-ST239-closed genomes using the chromosome of the Brazilian strain BMB9393 as reference.
This region in ST239-BRC genomes carries two SCC elements, the SCCmercury (also known as SCCHg) and SCCmec. All ST239 closed genomes (ST239C) shared a SCCmec type III, although ST239-BRC and ST239-INTC display different architectures for this element. The ST239-BRC genomes carry SCCmecIII.1.1.2 (named SCCmecIIIA), an approximately 28.0 kbp cassette, with 15-bp att direct-repeat sequences (attL, attR); however, this cassette is markedly distinct from TW20 (a representative of ST239 from the United Kingdom), which has SCCmecIII.1.1.1 (approx. 35.3 kbp), as shown in Supplementary Figure S2B. The SCCmecIII.1.1.1 (named SCCmecIII) carries four copies of the IS431 with the same orientation of those found in SCCmecIII.1.1.2, suggesting that the SCCmecIII.1.1.2 originated from a recombination event occurring between two IS431 copies (Ito et al., 2001; Supplementary Figure S2B).
In the 5′-portion of this RGP (Supplementary Figure S2C), there is an integrative and conjugative element (ICE) with similarity to Tn5801, while another segment corresponds to a GI of approximately 27.6 kbp known as νSAα. This GI, which is conserved among all ST239 genomes studied, carries a gene encoding a truncated transposase (BMB9393 SABB_02229) that shows high similarity to that of the IS256 family; hsdM (BMB9393 SABB_05215) and hsdS (BMB9393 SABB_05408) genes encoding a restriction/modification system; a cluster of 10 ssl (staphylococcal superantigen-like) ORFs (BMB9393 SABB_02212 to SABB_02203) and putative ORFs encoding several lipoproteins (lpl) (BMB9393 SABB_02197 to SABB_05337). These two clusters are considered markers of the νSAα island (Baba et al., 2008).
In addition to tetM, this RGP also contains genes associated with conjugation, regulation, excision/integration systems, and a gene encoding the anti-restriction protein ArdA (BMB9393 SABB_02743). RGP3 showed sequence divergence for the genomes of the non-ST239-INTC (strains CA-347, H-EMRSA-15, and MRSA252) but it was highly conserved among all our comparison ST239 genomes.
A PCR screen detected the ardA gene, which is present in the genome of all ST239 comparison genomes, and in the majority (95/100) of isolates from our Brazilian ST239-BR100 collection. The ardA gene is also found in other non-ST239 HA-MRSA genomes [e.g., MU50 (ST5-SCCmecII)], and in other bacterial pathogens, such as E. coli (McMahon et al., 2009). It is important to note that typical CA-MRSA strains, such as USA300 and USA1100, do not harbor this gene, according to BLAST searches in public databases (data not shown). In E. coli, the acquisition of ardA has a significant impact on gene exchange due to the ability of the ArdA protein to bind and inactivate type I restriction modification (RM) enzymes, which, in turn, results in increased dissemination of MGEs among these bacteria. ArdA from S. aureus is active against the EcoKI type I RM system of E. coli K12 (McMahon et al., 2009), but this has not been demonstrated in S. aureus. To test the function of ArdA in S. aureus, we cloned the ardA gene into the S. aureus expression vector pCN40 and transformed it into the ardA-negative, ST8, strain RN4220 (42P40A). The ability to acquire exogenous DNA from both E. coli and S. aureus of a different lineage (ST5) increased significantly (p < 0.001) in 42P40A in comparison with 42P40E (RN4220 with empty vector pCN40) (Figure 3).
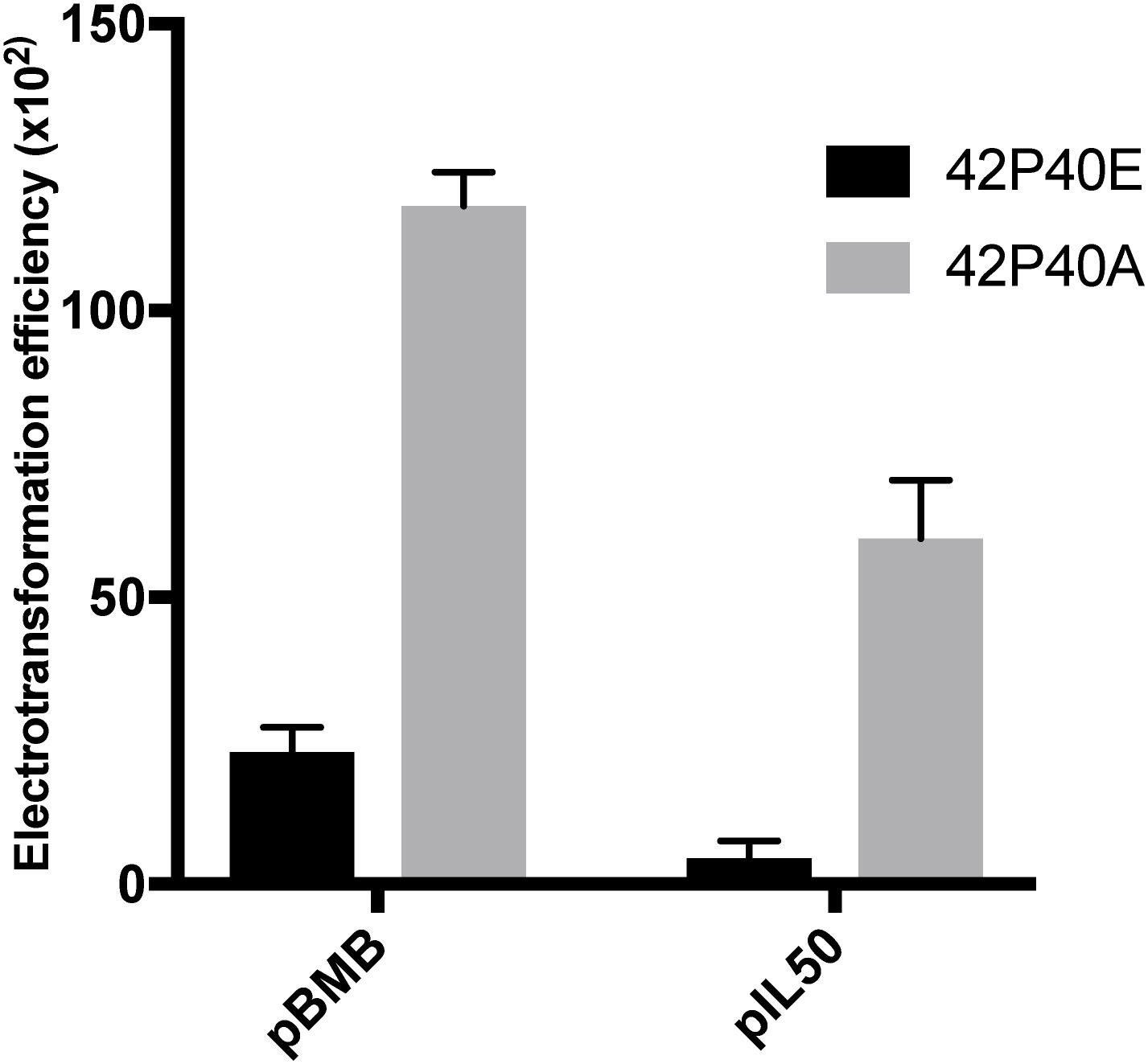
Figure 3. Competence. Electrotransformation efficiency (CFU/μg DNA) of 42P40A (black) and 42P40E (gray) clones after electroporation with plasmids recovered from S. aureus (pBMB) or E. coli (pLI50) (p < 0.001, two-way ANOVA).
Recent studies have shown that the high-level multi-resistant ST239 isolates are adapted to the hospital environment (Alp et al., 2009; Xiao et al., 2013). The global dissemination of a bacterial clone is likely to be dependent on specific mechanisms for transmissibility, bacterial adaptability, and fitness (Amaral et al., 2005; Li et al., 2012). Thus, it is reasonable to posit that the presence of the ardA gene may represent a substantial evolutionary event in the adaptation to the nosocomial niche, assisting not only in the acquisition of resistance determinants but also of genes associated with virulence/fitness.
RGP4 is associated with a unique S. aureus pathogenicity island (SaPI, Supplementary Figure S2D). SaPIs belong to a large family of phage-inducible chromosomal islands. This SaPI – composed of 14.6 kbp, carries 25 ORFs and is highly similar to SaPITokyo11212. The identified integration site, att, belongs to the att/int IV group (Novick et al., 2010), with the core sequence TTATTTAGCAGGAATAA, which includes the insertion site and an intact ORF for an integrase gene (BMB9393 SABB_00863). Despite the fact that RGP4 and SaPI1 share a homologous integrase, and are located at the same site att in the metQ gene, they show few similar segments, even in the rep/oriT sequences (Supplementary Figure S2D). SaPIs can be composed of modular units from other SaPIs (Novick and Subedi, 2007). Indeed, when RGP4 and SaPI2 were compared (Acc: EF010993) there was greater overall similarity than with SaPI1, specifically in the region related to the modules of DNA replication and SaPI packaging.
Interestingly, modular conservation could be related to the fact that SaPI2 is very similar to SaPI1028 (Subedi et al., 2007), for which the “auxiliary” phage is PT1028, which itself is highly similar to the phage found in RGP4. It is important to emphasize that in the ST239-INTC genomes analyzed, the phages related with PT1028 harbor enterotoxin-like genes, such as selK and selQ, which are absent in all ST239-BRC. In fact, the ST239-BRC genomes do not carry any of the known enterotoxin or enterotoxin-like genes, clearly indicating that – despite the role of some enterotoxins in S. aureus virulence in animal models (Stach et al., 2014) – the acquisition of these superantigen genes is not crucial for the pathogenesis of severe nosocomial infections including bloodstream infections and other disseminated diseases. On the other hand, the gene encoding the SAP domain-containing protein is lost in all non-ST239-INTc strains studied here. This observation is in agreement with the assumption that most SaPIs show an association with specific MRSA lineages instead of widespread distribution among different lineages (Lindsay, 2008).
This 30.9 kbp region shows similar features to GIs, such as the presence of a putative truncated transposase, hsdS (BMB9393 SABB_06193) and hsdM (BMB9393 SABB_06194) encoding an RM system, and tRNAs flanking both ends. This GI has been classified as νSAβ type I in ST239 genomes (Baba et al., 2008) and carries the entire serine–protease operon splABCDEF (BMB9393 SABB_01932 to SABB_01937) and the gene cluster encoding staphylococcal lantibiotics (BMB9393 SABB_01939 to SABB_05404). In general, S. aureus extracellular proteases are considered important virulence factors. For instance, a S. aureus mutant derived from USA300 LAC – for which all 10 protease genes were deleted including spl proteases – showed lower mortality rates in mice compared with the isogenic wild-type strain (Kolar et al., 2013).
νSAβ is another example of a GI that is widely dispersed in S. aureus and not strictly associated with particular lineages, demonstrating that its acquisition may have important adaptive benefits for S. aureus strains. Among the strains analyzed, other than JKD6008, only CA347 (USA600) and H-EMRSA-15 do not carry this GI (Supplementary Figure S2E).
This RGP is 73.2 kbp and corresponds to a bacteriophage-related plasticity region that shows high similarity (BLASTp ≥ 70%) with ϕNM3 (Acc:NC_008617). This region is highly conserved among all ST239-BRC genomes (Supplementary Figure S2F) and is known to be a β-hemolysin converting prophage. RGP9 can also carry the immune evasion cluster (IEC) (Van Wamel et al., 2006), which has also been associated with S. aureus host-adaptation and virulence mechanisms (Van Wamel et al., 2006). The previously described S. aureus IEC carries genes encoding the chemotaxis-inhibitory protein (CHIPS), staphylococcal complement inhibitor (SCIN), staphylokinase (Sak), and the staphylococcal enterotoxin A (Sea). ST239-BRc genomes have genes for all of these proteins except Sea. We could not detect the sea gene in any ST239-BR100 isolates from our collection.
While the chp gene is found in all ST239-BRC strains, it is not found in any of the ST239-INTC genomes analyzed, raising questions about the specific importance of this gene in the microevolution of Brazilian ST239 (Supplementary Figure S2F). Bae et al. (2006) showed a significant reduction in the ability of S. aureus strain Newman to replicate in a murine liver abscess model when ϕNM3, harboring chp, was cured. The product of the chp gene (BMB9393 SABB_02361), CHIPS, specifically binds to C5a and formylated peptide receptors inhibiting human neutrophils and monocytes chemotaxis (de Haas et al., 2004; Postma et al., 2004). We detected the chp gene by PCR in all Brazilian isolates from our ST239-BR100 collection (100%). It is intriguing that in GV69, the IEC cluster including chp are carried by a different phage (similar to tp310-3), which may indicate a strong pressure for the acquisition of this gene cluster in Brazilian ST239. Notably, GV69 also does not have the sea gene similar to other Brazilian ST239. It is important to note that chp is also present in other successful HA-MRSA lineages including ST36 (MRSA252; representative of the EMRSA-16), ST22 (H-EMRSA-15; representative of the EMRSA-16), and ST45 (CA-347; representative of the USA600), within a phage related to ϕNM3. Both scn and sak are present in all ST239C genomes.
In general, the set of virulence genes in ST239 genomes is well conserved (Supplementary Table S7). The ST239-BRC and ST239-INTC genomes harbor a plethora of surface proteins named microbial surface components recognizing adhesive matrix molecules (MSCRAMMs), which are required for the establishment of infections. A notable exception is the absence of genes encoding fibronectin-binding protein B (fnbB) and clumping factor A (clfA) in T0131 (ST239-INTC) and the absence of clumping factor B (clfB) in T0131 and XN108 (ST239-INTC). These genes are found in all ST239-BRC genomes (Supplementary Table S7).
Until now, few studies have reported the tst gene in ST239 isolates (Iwao et al., 2012; Kong et al., 2016), and most of the enterotoxin and enterotoxin-like genes found in the other HA-MRSA lineages were absent in the ST239 genomes we studied. In fact, the only enterotoxin genes found in any ST239 genomes were selK, selQ, and sea, none of which are found in ST239-BRC. The reason why it would be an advantage for ST239 isolates to be devoid of enterotoxin genes is unclear, but it is possible that loss of these genes may lead to less cytotoxicity and greater persistence (Das et al., 2016).
The arginine catabolic mobile element (ACME) is a putative virulence determinant, integrated into the chromosome at the same site as SCCmec (Diep et al., 2006; Barbier et al., 2011). While there are no reports of ST239 strains carrying ACME I, there are a few reports of ST239 isolates carrying ACME II (Espedido et al., 2012; Hsu et al., 2015). Using a BLAST-based search, we did not find the ACME-encoded arcA gene in any of 190 genome sequences analyzed, probably due to the fact that the geographic regions from which ACME+ ST239 strains reported are under-represented in this set of genomes. Nonetheless, ACME does not seem to be important to the biology of ST239 overall.
A unique, conserved cluster consisting of 11 ORFs for putative staphylococcal exotoxin-like genes (set or ssl) was found in the genomes of ST239-BRC and ST239-INTC. Among the other non-ST239-INTC genomes, only USA500 carries a ssl gene cluster similar to ST239. Like ST239, USA500 is a member of CC8 and it is possible that this region of ssl genes is associated with CC8 evolution (Aguiar-Alves et al., 2006). The role of some ssl gene products in impairing neutrophil chemotaxis and complement activation has been reported (Bestebroer et al., 2010; Koymans et al., 2016).
ST239 genomes carry a number of genes conserved in all S. aureus, encoding exoproteins likely important for bacterial dissemination in host tissues, such as phospholipases, proteases, hyaluronidase, coagulase, among others. The ST239C genomes are also equipped with several hemolysin-encoded genes including hla, hld, hlgABC, and the leukocidins lukD and lukE. Interestingly, while JKD6008 has a gene for LukE it lacks the lukD gene. As generally observed for canonical hospital strains, the ST239C genomes analyzed do not carry the genes encoding the Panton–Valentine leukocidin (Figueiredo and Ferreira, 2014).
The agr operon encodes the main S. aureus quorum sensing system (QSS) (Novick et al., 2000), but at least one Brazilian isolate (HC1335) has a transposable element similar to IS256 disrupting the agrC coding sequence (Botelho et al., 2016), an insertion that is expected to inactivate the entire Agr system including multiple virulence determinants, since agrC encodes the Agr autoinducer peptide (AIP) receptor. A similar IS256 insertion was also found in the Chinese ST239 isolate T0131, located 36 nt upstream of the insertion site observed for HC1335 (Supplementary Figure S2G).
Agr dysfunction was assayed in the entire collection of ST239-BR100 isolates using a mass spectrometry approach to detect PSM-mec, an Agr-regulated gene. Thirty percent of isolates (n = 30) had decreased Agr activity. Most of the Agr-dysfunctional isolates were from colonization cases (p < 0.01) (Figure 2B). Among these 30 Agr-dysfunctional isolates, the IS256 insertion in agrC was found by PCR in four isolates (13.3%) suggesting multiple possible inactivation mechanisms. The high prevalence of Agr dysfunction may seem counterintuitive given the prominent role of the Agr system in pathogenicity and virulence (Ferreira et al., 2013), but it is possible that in the specific context of the hospital where patients are debilitated and/or immunocompromised, Agr dysfunction may provide a fitness advantage (Paulander et al., 2012; Baines et al., 2015). However, one important caveat of this analysis is that multiple studies have documented within-patient heterogeneity in Agr dysfunction, and future work should incorporate multiple isolates from the same sample or paired isolates from the same individual.
Point mutations inactivating Agr QSS have predominately been described in the agrA or agrC genes (Traber and Novick, 2006; Shopsin et al., 2010). To investigate mutational evolution in these genes, we aligned the agrC genes of all 190 genomes used for our phylogenetic analysis. This analysis revealed 180 SNPs in the agrC genes distributed in 135 genomes, corresponding to 9.0 × 10-4 SNPs/nucleotide. The average number of SNPs in several other important global gene regulators (agrA, agrB, mgrA, rot, saeRS, sarA, sarR, sarS, sarX, sarZ, and sigB) was significantly lower (2.1 × 10-5 SNPs/nucleotide) (Supplementary Figure S3). The 180 SNPs in agrC genes were located in 10 positions in the agrC sequence. Eight SNPs were nonsynonymous mutations. Three nonsynonymous SNPs resulted in amino acid changes, S6R, I311T, and A343T that were conserved in 60, 61, and 40 of the 190 ST239 genomes studied, respectively (Supplementary Figure S3), suggesting that these changes are not strongly detrimental. To test whether substitutions in the agrC gene occur at a significantly different frequency compared to the other virulence regulators studied, we used a statistical test based on Poisson probability distribution. The probability that agrC mutation rate was the same or lower compared to other genes was extremely low (p = 9.4 × 10-12) suggesting that this gene may be undergoing more rapid mutational change, a hallmark of possible positive or diversifying selection. No recombination breakpoints were found in agrC. Scans for evidence of the action of diversifying selection using two methods (FUBAR and MEME) were in disagreement about selection at codon site 6 (S6R change). FUBAR found evidence for codon site 6 (Prob[dN > dS] = 0.908, Bayes factor = 12.254), whereas MEME did not (P = 0.2). Across the phylogeny, the test for stringency of selection yielded a selection intensity parameter k = 1.74 that is usually indicative of an intensification of selection strength (k > 1), but it was not significant (p = 0.264).
In a recent study that identified two separate clades of ST239 MRSA in Australia, clade 2 also showed agr-dysfunction (Baines et al., 2015), which is thought to be due to mutations located upstream of agr. In two of the three Agr-dysfunctional ST239-BRc genomes, the mutation was not found in the agr operon.
Several studies have demonstrated the importance of IS256 in MRSA evolution. Benson et al. (2014) reported an insertion of IS256 upstream of the repressor of toxins gene, rot, in USA500-related strains, leading to a hypervirulent variant of this lineage. Frisch et al. (2018) showed that strains with multiple copies of IS256 not only grouped together in phylogenetic analysis, but also displayed higher levels of antibiotic resistance. It is notable that in this study the high-level multi-resistant ST239 also showed high copy numbers of IS256 (12–24 insertions in the ST239-BRC genomes).
It is noteworthy that a copy of an IS256-related element was also found inserted at -210 bp of the mgrA gene, encoding another global regulatory protein, in the genomes of BMB9393, GV88, Be62, GV51, and HC1335 (ST239-BRC) (Supplementary Figure S4). This IS256 insertion was exclusively found among the Brazilian isolates. It is absent in the genomes of international ST239 strains. mgrA is a negative regulator of autolysis and consequently of biofilm development (Ingavale et al., 2003; Gupta et al., 2015). The expression of mgrA also has a positive effect on agr transcription (Ingavale et al., 2003), which is coordinated by the P1 (more active in stationary phase of growth) and P2 promoters (more active in logarithmic phase) (Ingavale et al., 2003; Gupta et al., 2015). The IS256 element in the ST239-BRC genomes was inserted between the P1 and P2 promoters (Supplementary Figure S4). It is notable that agr-RNAIII is a key molecule in the stabilization of the 5′-UTR mRNA transcribed under the action of P2mgrA. Gupta et al. (2015) demonstrated that two adjacent regions transcribed from the upstream P2mgrA, located between -300 and -200 bp, form a stable complex with two regions of agr-RNAIII near its 5′ and 3′-ends. It was suggested that agr exerts its regulatory function through MgrA, which could act as an intermediate controller. According to this model, one might expect that the IS256 insertion near the mgrA gene at -210 bp impairs RNAIII-based stability of P2 transcript, thus decreasing the level of mgrA transcripts under the coordination of P2mgrA promoter.
The entire collection of ST239-BR100 isolates was assessed for the IS256 insertion in the mgrA gene using PCR-based detection. Fifty-seven (57%) of the total isolates carry the IS256 insertion between the mgrA promoters. Notably, 43 out of these 57 (75.4%) isolates were from infection-associated cases (p < 0.01) (Figure 2C). It has been shown that inactivation of mgrA resulted in increased autolytic activity and decreased transcription of several global regulators, including Agr (Luong et al., 2006). Other studies showed that MgrA regulates, directly or indirectly, a total of 355 genes, up-regulating 175 (including genes encoding exoproteins) and down-regulates the remaining 180 genes, among which are genes encoding surface proteins (Ingavale et al., 2005; Luong et al., 2006). Furthermore, the deletion of mgrA decreased virulence in an animal model (Jonsson et al., 2008). It is conceivable that the IS256 insertion found in 57% of ST239-BR isolates would not completely abolish mgrA transcription, because this insertion occurred in a position (-210 bp) that would be predicted to affect only P2 transcripts without disturbing P1 regulatory region, located at -28 to +16, which is the binding site for the non-coding RNA, RsaA (Romilly et al., 2014).
Maximum-likelihood (Supplementary Tables S2, 10 and Supplementary Figure S5) trees were constructed based on core genome SNPs using 190 complete genomes of ST239 and ST239-like SLVs of both draft and completely closed chromosomes that were available on NCBI during this study-period. Genomes used in the phylogenetic studies by Harris et al. (2010) and Castillo-Ramírez et al. (2012) were also included in this study. Trees were constructed both including and excluding the recombined region thought to be derived from CC30 (corresponding to positions 2848181-3043210 and 1-427978 on the TW20 genome; data not shown), but we used datasets excluding this region for our final analyses. Trees were rooted using outgroups from ST8.
The tree architecture showed three primary ST239 clades referred to here as clades I (n = 77 genomes), II (n = 33), and III (n = 80), that are roughly equivalent to the primary groupings defined in Castillo-Ramírez et al. (2012) (n = 165). Both our trees and previously published trees showed a similar branch length distribution with multiple deeply divergent lineages and short branches near the root of the tree possibly suggesting deep and rapid radiation. These deep branches show no consistent geographic location, and they are not restricted to Europe, so it remains unclear where the ST239 lineage originated. This branching pattern also may cause some instability in the placement of early branching taxa in the major early bifurcations. Like previous studies, our tree also showed a similar pattern of three well-defined subclades that had strong geographical affinities and partitioned with long internal branches suggesting local adaptation. For standardization purposes, these subclades were designated as in Harris et al. (2010) and Castillo-Ramírez et al. (2012) as the Asian, South American, and Turkish subclades. All ST239-BRC genomes were found in the main clade II and grouped with genomes mostly from other Latin American and romance language-speaking European countries (South American clade; n = 27). ST239-INTC genomes were located in the two other main clades (I and III). Closed genomes of TW20 from the United Kingdom, Z172 from Taiwan, and XN108 from China clustered in clade I, in the Asian subclade (n = 63). In clade I, there is a paraphyletic group (n = 13) composed largely of strains from Europe (e.g., P32 from Poland, H160 from Hungary, HSA10 from Portugal among others). The ST239-INTC genomes from Australia (JKD6008) and from China (T0131) clustered with the remaining 80 genomes in clade III, in which the derived subclade is represented mostly by Turkish isolates (Turkish subclade; n = 70).
For estimation of divergence times using Bayesian estimation (Figure 4), the genomes included were only those for which the isolation date of each isolate was reported (n = 167). The resulting Bayesian phylogenetic tree topology, calibrated by incorporating the temporal information of the isolation date and relaxed (Figure 4 and Supplementary Table S8) and strict (Supplementary Figure S7 and Supplementary Table S9) evolutionary clocks, suggests a global initial radiation of ST239 isolates in the 1960s and 1970s. The root of the tree is found at approximately 1962–1966. We estimate that the Asian clade originated in 1976 (HPD 95%: 1972–1980), the South American clade in 1988 (HPD 95%: 1986–1990), and the Turkish clade in 1981 (HPD 95%: 1976–1986). Even more recent intercontinental spread can be inferred based on the branching patterns within each clade. For example, European strains TW20 and D71 from the United Kingdom and Germany, respectively, clustered within the Asian clade; ES26 and M278 from Spain and Portugal, respectively, within the South American clade; and strains T0131 and TN79 from China in the Turkish clade. The intercontinental spread of ST239 isolates has also been documented by Harris et al. (2010) and Gray et al. (2011) among others. Our molecular dating techniques estimate that the expansion of the Brazilian strains began in 1988, coincident with the introduction of the South American clade. Indeed, the first report of ST239 in Brazil was published in 1995 using MRSA isolates collected in the 1992–1994 period (Teixeira et al., 1995). This data are in accordance with previous analyses which estimate that the ST239 introduction into South America occurred in the beginning of the 1990s (Smyth et al., 2010; Castillo-Ramírez et al., 2012). At that time, 87% of the total MRSA isolates detected in multiple Brazilian hospitals from the South to the North of the country belonged to the ST239 lineage, and all of them displayed the same or very similar PFGE patterns, suggesting a very recent clonal spread (Teixeira et al., 1995).
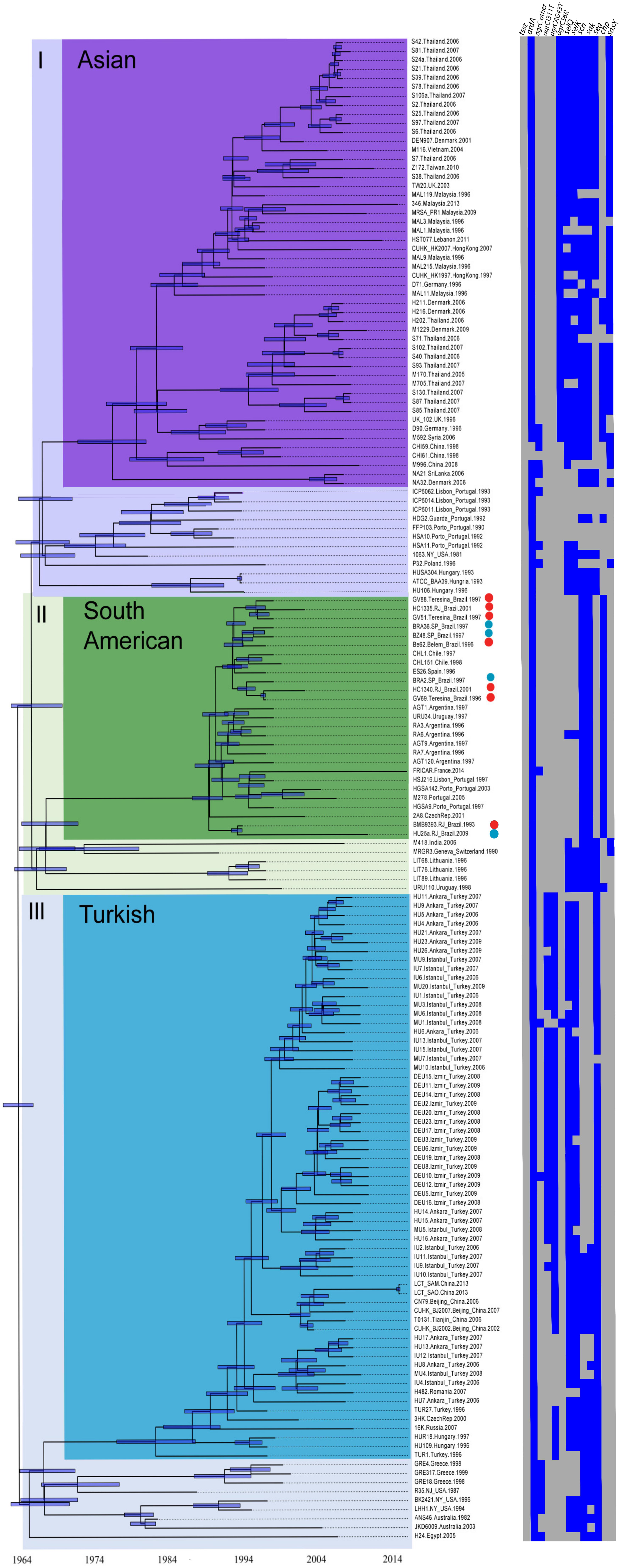
Figure 4. Time-calibrated phylogenetic tree. Maximum clade credibility tree estimated using a log normal-relaxed clock (see the section “Materials and Methods”) from the genomic SNPs revealing three major ST239 clades (I, II, and III), which encompass subclades named as following previous classification: Asian (purple), South American (green), Turkish (light blue). Heat map shows the presence (blue) and absence (gray) of tsst, ardA, selQ, selK, scn, sak, sea, chp, sasX, chp, sea, and the main SNPs in agrC associated with each clade.
Local BLAST was performed to compare all 190 ST239 genomes in relation to virulence-associated genes that diverged or were not homogeneously present in the virulome of the ST239C genomes. Our analyses revealed that several genes are associated with specific clades on the phylogenetic trees. The chp gene is present in 28 of 33 (81.8%) genomes of clade II (where the South American subclade is located). There is only one genome (URU110; Uruguay-1998; clade II) among the 190 genomes that carries both chp and sea genes, and all others carrying chp lack sea (81.8%, n = 27). Only five genomes in clade II lack chp and carry sea (M418, India-2006; MRGR3 from Switzerland-1990; LIT68, LIT89, and LIT76 from Lithuania-1996). These genomes together with URU110 are found in a paraphyletic group in a basal position of clade II. Among all 190 genomes analyzed, only 17 do not carry chp or sea (Supplementary Table S2).
Clade I is composed of a total of 77 genomes, and the great majority, 75.6% (n = 59), carry the sasX gene encoding a LPXTG motif protein. All of these group in the Asian subclade, except for one genome (P32 from Poland) located in a basal position in this clade. SasX promotes bacterial aggregation, enhances nasal colonization and immune evasion, and plays a role in lung infection and abscess formation in animal models (Li et al., 2012). Outside the clade I, only two other ST239 genomes carry sasX, M418 (India, 2006), and MRGR3 (Switzerland, 1990). Both of these group at the base of clade II. Thus, it is possible that the common ancestor (dated to approximately 1966, 95% HPD 1963–1970) of the Asian and South American subclades carried sasX but, after local diversification, only the Asian clade conserved this gene.
There are only four genomes (5.1%) in the Asian subclade (M592, Syria-2006; D90, Germany-1996; United Kingdom 102, UK-1996; and M170, Thailand-2005) where the chp gene shows 100% identity with the chp sequence found in the South American subclade (chp-1 allele). In addition, in clade I, there are an additional 11 genomes (S40, S85, S87, S93, S102, S130, H202, M705, from Thailand-2007; H211, H216, from Denmark-2006; HDG2, from Portugal-1992) in which the chp gene displays up to four non-synonymous SNPs compared to the reference (variant chp-2). The genomes with chp-2 are all found in the Asian subclade except for the Portuguese strain HDG2 that is located in a basal position in this subclade (Supplementary Table S2). Most Asian-subclade chp-2 positive strains also carry sasX genes (12/15), highlighting the acquisition of sasX as an important divergent evolutionary event for the Asian subclade. Many of the genomes grouped in clade I also carry sea (60.3%, n = 47). The genome of the strain OC3 (Russia, 2007–2009) is the only genome in this clade to harbor the tst gene (Supplementary Table S2). For all 80 genomes in clade III (where the Turkish subclade is located), both sasX and chp are absent, and sea is present in all but one genome (MU7) from Turkey-2007 (97.5%, n = 78) (Figure 4).
While agr-dysfunction was detected in 30% of the ST239 isolates from the Brazilian collection, the large majority of the genomes found in clade II show no variants in agrC (n = 32, 97%). Only one genome (RA3, Argentina-1996) shows any variation, a substitution of glutamic acid (E) for lysine (K) at position 175. None of the genomes found in the two other main clades share this point mutation.
Variants in agrC in the other two main clades (I and III) are also well conserved. The nonsynonymous variant A343T (n = 58) and I311T (n = 61) in the AgrC protein are found in 74.4 and 78.2% of the genomes grouped in the clade III. Another AgrC nonsynonymous variant S6R (n = 39) is specifically detected among many strains from clade I (48.1%), although one strain (MU1) from Turkey (clade III) also has this substitution. The agrC variants reported here are distinct from positions reported in other studies involving non-ST239 MRSA lineages (Geisinger et al., 2009; Shopsin et al., 2010).
Grouping genomes by their virulence gene patterns, based on absence or presence of sea, chp, and sasX genes and agrC SNPs (Figure 4), we used the Grubbs’ test statistic to detect outliers and find a predominant virulence profile for each phylogenetic group (Supplementary Figure S6). Accordingly, the predominant pattern of each group was clade I: sasX+, chp-, sea+, agrC S6R+, agrC I311T-, agrC A343T-; clade II: sasX-, chp+, sea-, agrC S6R-, agrC I311T-, agrC A343T-; and clade III: sasX-, chp-, sea+, agrC S6R-, agrC I311T+, agrC A343T+; Each of these patterns and corresponding SLVs were strongly associated (χ2-square, p < 0.0001; Supplementary Figure S6) with each of the three main clades and, independently, with broad geographical location of isolation. Gene patterns from a geographic origin were significantly independent from those from another region (p < 0.0001). This pattern strongly suggests the geographic (allopatric) influence on the diversification of ST239 MRSA. The selective pressures that may have driven this diversification remain unknown. It is possible that pathogen–immune system and pathogen–microbiome interactions play a role, in addition to broad pressures related to medical practices and socio-cultural influences (Georgiades and Raoult, 2011).
Our genomic and phylogenetic analysis of the ST239 lineage revealed specific patterns of local geographic divergence involving well-known virulence factors. The genomes grouped with the Brazilian ST239 isolates show a specific virulence pattern represented by the presence of chp-1, lack of sasX and sea (both of which are commonly observed in genomes grouped in the Asian subclade). In addition, we also found that the ardA gene was present and conserved in all 190 ST239 genomes analyzed, and confirmed ArdA-antirestriction activity in S. aureus, resulting in high-frequency plasmid-borne acquisition of resistance genes. We posit that the ardA gene played a role in the acquisition of high-level antimicrobial resistance and the plethora of virulence genes observed in this successful HA-MRSA lineage. Further we posit that the presence of this gene might have facilitated the pattern of local diversification seen in each of the major ST239 subclades, allowing each to rapidly adapt to its local context.
Mobile genetic elements such as phages and GIs have clearly played an important role in the evolution of virulence of this MRSA lineage. The insertion of transposable elements (derived from IS256) in, or close to, the important global regulatory genes such as agr and mgrA, may also function as an additional strategy for MRSA evolution, representing a rapid mechanism for global reprogramming of virulence attributes (Benson et al., 2014). Mutations driving agr dysfunction, represented by point mutation or IS256 insertion in the agrC gene, and also mutations upstream of the agr locus may be instances of convergent functional evolution, that may have decreased toxicity, and increased colonization and persistence (Le and Otto, 2015; Edmiston et al., 2016), balancing the increased fitness costs of antimicrobial resistance or other nosocomial adaptations.
Finally, much of the diversification we identified in the three major ST239 subclades appears to have occurred in the 1970–1980s, when MRSA emerged causing hospital outbreaks in Europe, United States, and Australia (Aires de Sousa et al., 1998). To more fully understand the process of virulence gene diversification it is possible that targeted efforts on banked culture or tissue samples may more fully reveal the process of bacterial adaptation. Further studies might also be focused on uncovering the selective processes that favored one set of virulence-associated genes over another in each of the three major areas of diversification, and also in understanding evolution in other geographical locations or countries that have been so far neglected. It is clear that broad taxon sampling is critical to unraveling diversity patterns and processes that drive global radiations, allopatric diversification, or multigeographic shifts in pathogens.
AB carried out most of the experiments in this study, manual annotation, in addition to bioinformatics analyses using Local BLAST and Geneious. AMNB wrote the draft manuscript. MC and MN manual annotation, genome architecture, and RGP analysis. FF, MC, BC, and PB manual annotation. DS and CB manual annotation, ardA molecular cloning and transformation experiments with ardA-positive and -negative isogenic clones. RS, LA, and AV sequencing experiments, genome assembly, run automatic annotation, and other bioinformatics analyses. AN, AM, PP, and KO’B phylogenetic analysis. S-OK, AM, and PP phylogenetic analysis, divergence times estimation, recombination scan and analysis, and natural selection scan. AF responsible for the study design. PP and AF wrote the final version of the manuscript. All others contributed to and reviewed the final version of the manuscript.
This work was supported in part by grants from the Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), #E-26/010.001764/2014, E-26/201.147/2014, and E-26/202.803/2017, Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), #303067/2015-2. AB and PB has a fellowship from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) – Brasil, Finance Code 001. PP was supported by NIH 1K08AI101005.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00082/full#supplementary-material
FIGURE SI | An illustration of the assembly and gap-closing strategies. Gaps intra- and inter-scaffolds were resolved using an hybrid assembly strategy with the Newbler and GapFiller programs, with local assemblies in gap regions using Newbler.
FIGURE S2 | Regions of Genetic Plasticity. Graphical representation of linear comparative alignments. Arrows represent predicted genes and their respective transcription strands (forward/reverse). Vertical red segments connecting regions represent direct conserved segments (BLASTN > 70%). Plots above each region indicate the corresponding % G+C content. Figures were generated using Easyfig program. (A) Chromosome region harboring the atl gene in BMB9393 (ST239 closed genome; Brazil) and TW20 (ST239 closed genome from United Kingdom). atl is highlighted in green. (B) SCCmec type III for BMB9393 (ST239 closed genome; Brazil) and TW20 (ST239 closed genome; United Kingdom) sequences. Genes coding for the recombinases are represented in blue; mecA and mecRI, in dark and light pink, respectively; and IS437 elements, in green. (C) RGP3 context for MU50 (non-ST239 closed genome, Japan), BMB9393 (ST239 closed genome; Brazil) and TW20 (ST239 closed genome; United Kingdom) sequences. The ardA gene, which encodes for an antirestriction protein, is represented in yellow. Strain BMB9393 lacks genes for an integrase and a hypothetical protein. (D) RGP4 carrying a unique SaPI. (top) BMB9393 (ST239 closed genome; Brazil) and SaPM sequences, (bottom) BMB9393 and SaPI2 sequences. In light pink, gene encoding an integrase; in light green, gene encoding an excisionase; in brown, pri genes; in dark green, rep gene; in dark blue, p/fgene; in light blue, operon required to DNA packaging; in dark pink, gene encoding a recombinase protein. (E) RGP8 carrying the vSAp genomic island using the following genomic sequences: JKD6008 (ST239 closed genome; Australia), BMB9393 (ST239 closed genome; Brazil) and MRSA252 (non-ST239 closed genome; United Kingdom). In green, hsdS and hsdM; in pink, the gene cluster encoding for serine proteases; in yellow, the gene cluster encoding for staphylococcal lantibiotics. (F) RGP9 carrying a bacteriophage related to PhiNM3 in the genomes of BMB9393 (ST239 close genome from Brazil; ST239-BRC) and TW20 (ST239 closed genome from United Kingdom; ST239-INTC). Arrows represent predicted ORFs and their respective transcription strands (forward/reverse). In light blue, gene encoding a prophage integrase; in blue, hlb encoding a trucated p-hemolysin; in yellow, chp gene encoding a chemotaxis inhibitory protein (exclusively found in ST239-BRC); in green and pink, sen and sak genes encoding the staphylococccal complement inhibitory protein SCIN and staphylokinase; and in purple sea encoding staphylococcal enterotoxin A (absent in BMB9393 and present in TW20). (G) agr operon for the genome of the strains BMB9393 (ST239 closed genome from Brazil; ST239-BRC), HC1335 (ST239-BRC) and T0131 (ST239 closed genome from China; ST239-INTC). Arrows in blue hid (delta-toxin); agrB] agrD; agrC (blue); agrA and the transposase (yellow).
FIGURE S3 | Substitutions in agrC vs. other global gene regulatiors. (A) SNP allele frequency found in pools of DNA from the genomes of 190 ST239 deposited om the Genbank for the regulators of agrC (blue circle), agrA, rngrA, rot, sacRS, sarA, sarR, sarS, sarXy sarZ, and sigB (other colored cirlces.) The horizontal line represents the average frequency. The probability P = 9.4x×lOA-12 is the chance of a SNP in agrC to occur at the same mean frequency observed for the other regulators using Poisson distribution. (B) Graphical representation shows the amino acid change and its position in the AgrC protein. In parenthesis is the number of genomes that show the correspondent SNP. From the 10 types of SNP substitution found, eight resulted in nonsynonymous changes.
FIGURE S4 | IS256 insertion at mgrA locus. Schematic representation of the rngrA gene regulator showing the position of the insertion of the IS256 between P2 (log-phase promoter) and PI (stationary-phase promoter).
FIGURE S5 | Maximum likelihood tree. Patterns within the three clades are denoted by a darker shade of the parent clade color: Asian pattern as purple, South American pattern as green, and Turkish pattern as blue. See methods for tree construction parameters. Red circles denote an isolate from this study while blue circles denote an isolate obtained from GenBank.
FIGURE S6 | Virulence gene patterns in clades. Distribution of the predominant pattern and its one-locus variants (clade specific patterns) among the genomes clustered in the Type I, Type II and Type III clades. Turkish predominant pattern (sosX1, chp-, sea+, agrC S6R-, agrC I311T+ agrC A343T+), South American predominant pattern (sasX, chp+, sea1, agrC S6R∼, agrC I311T, ogrC A343T), Asian predominant pattern sasX+, chp-, sea+, agrC S6R+, agrC I311T, agrC A343T). Other patterns comprises all patterns that differed by two or more loci from the specific clade pattern. The distribution of the clade specific patterns was extremely significant associated with the tree topology and geographic location of the strains. ∗∗∗ = (p < 0.0001).
FIGURE S7 | A Bayesian phylogenetic tree based on core genome SNPs yields three distinct ST239 patterns (named based on predominant locations of strains): Turkish (light blue), South American (green) and Asian (purple). The Bayesian tree was constructed using r 190 and 167 complete genomes of ST239 and ST239-like single locus variants (SLVs) of both draft and completely closed chromosomes (obtained from NCBI) as well as genomes used in the phylogenetic studies by Harris et al). A heat map shows the presence (grey)/absence (blue) of sasX, chp, sea and main SNP mutation in agrC associated with each clade.
TABLE S1 | Main characteristics of the ST239-BR100 strains.
TABLE S2 | List of genomes used on ML analysis and their main characteristics.
TABLE S3 | Primers used on PCR assays in this work.
TABLE S4 | Sequences that were potentially mobile excluded from SNP dataset.
TABLE S5 | Exclusive clusters comparing ST239-BRC and ST239-INTC.
TABLE S6 | Description of RGPs main features.
TABLE S7 | Virulence set of ST239-BRC, ST239-INTC and non-ST239-INTC.
TABLE S8 | BEAST uncorrelated, lognormal tree.
TABLE S9 | BEAST strict clock tree.
TABLE S10 | Maximum likelihood tree.
CA-MRSA, community-acquired methicillin-resistant Staphylococcus aureus; CC, clonal complex; HA-MRSA, hospital-acquired methicillin-resistant Staphylococcus aureus; IS, insertion sequence; MALDI–TOF–MS, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry; MGE, mobile genetic element; MRSA, methicillin-resistant Staphylococcus aureus; RGP, region of genomic plasticity; SCCmec, staphylococcal cassette chromosome mec; SET, staphylococcal enterotoxin-like proteins; SNP, single-nucleotide polymorphism; SSL, staphylococcal superantigen-like proteins; ST, sequence typing; TE, transposase element.
Aguiar-Alves, F., Medeiros, F., Fernandes, O., Gudziki Pereira, R. M., Perdreau-Remington, F., and Riley, L. W. (2006). New Staphylococcus aureus genotyping method based on exotoxin (set) genes. J. Clin. Microbiol. 44, 2728–2732. doi: 10.1128/JCM.00376-06
Aires de Sousa, M., Sanches, I. S., Ferro, M. L., Vaz, M. J., Saraiva, Z., Tendeiro, T., et al. (1998). Intercontinental spread of a multidrug-resistant methicillin-resistant Staphylococcus aureus clone. J. Clin. Microbiol. 36, 2590–2596.
Almeida, L. G. P., Paixão, R., Souza, R. C., da Costa, G. C., Barrientos, F. J. A., Trindade dos Santos, M., et al. (2004). A system for automated bacterial (genome) integrated annotation - SABIA. Bioinformatics 20, 2832–2833. doi: 10.1093/bioinformatics/bth273
Alp, E., Klaassen, C. H. W., Doganay, M., Altoparlak, U., Aydin, K., Engin, A., et al. (2009). MRSA genotypes in Turkey: persistence over 10 years of a single clone of ST239. J. Infect. 58, 433–438. doi: 10.1016/j.jinf.2009.04.006
Amaral, M. M., Coelho, L. R., Flores, R. P., Souza, R. R., Silva-Carvalho, M. C., Teixeira, L. A., et al. (2005). The predominant variant of the Brazilian epidemic clonal complex of methicillin-resistant Staphylococcus aureus has an enhanced ability to produce biofilm and to adhere to and invade airway epithelial cells. J. Infect. Dis. 192, 801–810. doi: 10.1086/432515
Arias, C. A., Reyes, J., Carvajal, L. P., Rincon, S., Diaz, L., Panesso, D., et al. (2017). A prospective cohort multicenter study of molecular epidemiology and phylogenomics of Staphylococcus aureus bacteremia in nine Latin American countries. Antimicrob. Agents Chemother. 61:e00816-17. doi: 10.1128/AAC.00816-17
Baba, T., Bae, T., Schneewind, O., Takeuchi, F., and Hiramatsu, K. (2008). Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J. Bacteriol. 190, 300–310. doi: 10.1128/JB.01000-07
Bae, T., Baba, T., Hiramatsu, K., and Schneewind, O. (2006). Prophages of Staphylococcus aureus Newman and their contribution to virulence. Mol. Microbiol. 62, 1035–1047. doi: 10.1111/j.1365-2958.2006.05441.x
Baines, S. L., Holt, K. E., Schultz, M. B., Seemann, T., Howden, B. O., Jensen, S. O., et al. (2015). Convergent adaptation in the dominant global hospital clone ST239 of methicillin-resistant Staphylococcus aureus. mBio 6:e00080-15. doi: 10.1128/mBio.00080-15
Barbier, F., Lebeaux, D., Hernandez, D., Delannoy, A. S., Caro, V., and Francois, P. (2011). High prevalence of the arginine catabolic mobile element in carriage isolates of methicillin-resistant Staphylococcus epidermidis. J. Antimicrob. Chemother. 66, 29–36. doi: 10.1093/jac/dkq410
Benson, M. A., Ohneck, E. A., Ryan, C., Alonzo, F., Smith, H., Narechania, A., et al. (2014). Evolution of hypervirulence by a MRSA clone through acquisition of a transposable element. Mol. Microbiol. 93, 664–681. doi: 10.1111/mmi.12682
Besemer, J., and Borodovsky, M. (2005). GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 33, W451–W454. doi: 10.1093/nar/gki487
Bestebroer, J., Aerts, P. C., Rooijakkers, S. H. M., Pandey, M. K., Köhl, J., Van Strijp, J. A. G., et al. (2010). Functional basis for complement evasion by staphylococcal superantigen-like 7. Cell. Microbiol. 12, 1506–1516. doi: 10.1111/j.1462-5822.2010.01486.x
Boetzer, M., and Pirovano, W. (2012). Toward almost closed genomes with GapFiller. Genome Biol. 13:R56. doi: 10.1186/gb-2012-13-6-r56
Botelho, A. M. N., Costa, M. O. C., Beltrame, C. O., Ferreira, F. A., Lima, N. C. B., Costa, B. S. S., et al. (2016). Complete genome sequence of the MRSA isolate HC1335 from ST239 lineage displaying a truncated AgrC histidine kinase receptor. Genome Biol. Evol. 8, 3187–3192. doi: 10.1093/gbe/evw225
Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu, C.-H., Xie, D., et al. (2014). BEAST 2: a software platform for bayesian evolutionary analysis. PLoS Comput. Biol. 10:e1003537. doi: 10.1371/journal.pcbi.1003537
Castillo-Ramírez, S., Corander, J., Marttinen, P., Aldeljawi, M., Hanage, W. P., Westh, H., et al. (2012). Phylogeographic variation in recombination rates within a global clone of methicillin-resistant Staphylococcus aureus. Genome Biol. 13:R126. doi: 10.1186/gb-2012-13-12-r126
Charpentier, E., Anton, A. I., Barry, P., Alfonso, B., Fang, Y., and Novick, R. P. (2004). Novel Cassette-based shuttle vector system for Gram-positive bacteria. Appl. Environ. Microbiol. 70, 6076–6085. doi: 10.1128/AEM.70.10.6076-6085.2004
Chen, F.-J., Lauderdale, T.-L., Wang, L.-S., and Huang, I. (2013). Complete genome sequence of Staphylococcus aureus Z172, a vancomycin-intermediate and daptomycin-nonsusceptible methicillin-resistant strain isolated in Taiwan. Genome Announc. 1:e01011-13. doi: 10.1128/genomeA.01011-13
Costa, M. O. C., Beltrame, C. O., Ferreira, F. A., Botelho, A. M. N., Lima, N. C. B., Souza, R. C., et al. (2013). Complete genome sequence of a variant of the methicillin-resistant Staphylococcus aureus ST239 Lineage, Strain BMB9393, displaying superior ability to accumulate ica-independent biofilm. Genome Announc. 1:e00576-13. doi: 10.1128/genomeA.00576-13
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
Das, S., Lindemann, C., Young, B. C., Muller, J., Österreich, B., Ternette, N., et al. (2016). Natural mutations in a Staphylococcus aureus virulence regulator attenuate cytotoxicity but permit bacteremia and abscess formation. Proc. Natl. Acad. Sci. U.S.A. 113, E3101–E3110. doi: 10.1073/pnas.1520255113
de Haas, C. J. C., Veldkamp, K. E., Peschel, A., Weerkamp, F., Van Wamel, W. J. B., Heezius, E. C. J. M., et al. (2004). Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory Agent. J. Exp. Med. 199, 687–695. doi: 10.1084/jem.20031636
Delcher, A. L., Bratke, K. A., Powers, E. C., and Salzberg, S. L. (2007). Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679. doi: 10.1093/bioinformatics/btm009
Didelot, X., and Wilson, D. J. (2015). ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput. Biol. 11:e1004041. doi: 10.1371/journal.pcbi.1004041
Diep, B. A., Gill, S. R., Chang, R. F., Phan, T. H., Chen, J. H., Davidson, M. G., et al. (2006). Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367, 731–739. doi: 10.1016/S0140-6736(06)68231-7
Drummond, A. J. (2005). Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22, 1185–1192. doi: 10.1093/molbev/msi103
Drummond, A. J., Suchard, M. A., Xie, D., and Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973. doi: 10.1093/molbev/mss075
Edmiston, C. E., Ledeboer, N. A., Buchan, B. W., Spencer, M., Seabrook, G. R., and Leaper, D. (2016). Is staphylococcal screening and suppression an effective interventional strategy for reduction of surgical site infection? Surg. Infect. 17, 158–166. doi: 10.1089/sur.2015.257
Espedido, B. A., Steen, J. A., Barbagiannakos, T., Mercer, J., Paterson, D. L., Grimmond, S. M., et al. (2012). Carriage of an ACME II variant may have contributed to methicillin-resistant Staphylococcus aureus sequence type 239-Like strain replacement in Liverpool hospital, Sydney, Australia. Antimicrob. Agents Chemother. 56, 3380–3383. doi: 10.1128/AAC.00013-12
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Ferreira, F. A., Souza, R. R., de Sousa Moraes, B., de Amorim Ferreira, A. M., Américo, M. A., Fracalanzza, S. E. L., et al. (2013). Impact of agr dysfunction on virulence profiles and infections associated with a novel methicillin-resistant Staphylococcus aureus (MRSA) variant of the lineage ST1-SCCmec IV. BMC Microbiol. 13:93. doi: 10.1186/1471-2180-13-93
Figueiredo, A. M. S., and Ferreira, F. A. (2014). The multifaceted resources and microevolution of the successful human and animal pathogen methicillin-resistant Staphylococcus aureus. Mem. Inst. Oswaldo Cruz 109, 265–278. doi: 10.1590/0074-0276140016
Frisch, M. B., Castillo-Ramírez, S., Petit, R. A., Farley, M. M., Ray, S. M., Albrecht, V. S., et al. (2018). Invasive methicillin-resistant Staphylococcus aureus USA500 strains from the U.S. Emerging infections program constitute three geographically distinct lineages. mSphere 3:e00571-17. doi: 10.1128/mSphere.00571-17
Gascuel, O. (1997). BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14, 685–695. doi: 10.1093/oxfordjournals.molbev.a025808
Geisinger, E., Muir, T. W., and Novick, R. P. (2009). agr receptor mutants reveal distinct modes of inhibition by staphylococcal autoinducing peptides. Proc. Natl. Acad. Sci. U.S.A. 106, 1216–1221. doi: 10.1073/pnas.0807760106
Georgiades, K., and Raoult, D. (2011). Defining pathogenic bacterial species in the genomic era. Front. Microbiol. 1:151. doi: 10.3389/fmicb.2010.00151
Gray, R. R., Tatem, A. J., Johnson, J. A., Alekseyenko, A. V., Pybus, O. G., Suchard, M. A., et al. (2011). Testing spatiotemporal hypothesis of bacterial evolution using methicillin-resistant Staphylococcus aureus ST239 genome-wide data within a bayesian framework. Mol. Biol. Evol. 28, 1593–1603. doi: 10.1093/molbev/msq319
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Gupta, R. K., Luong, T. T., and Lee, C. Y. (2015). RNAIII of the Staphylococcus aureus agr system activates global regulator MgrA by stabilizing mRNA. Proc. Natl. Acad. Sci. U.S.A. 112, 14036–14041. doi: 10.1073/pnas.1509251112
Harris, S. R., Feil, E. J., Holden, M. T. G., Quail, M. A., Nickerson, E. K., Chantratita, N., et al. (2010). Evolution of MRSA during hospital transmission and intercontinental spread. Science 327, 469–474. doi: 10.1126/science.1182395
Hasegawa, M., Kishino, H., and Yano, T. (1985). Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22, 160–174. doi: 10.1007/BF02101694
Holden, M. T. G., Feil, E. J., Lindsay, J. A., Peacock, S. J., Day, N. P. J., Enright, M. C., et al. (2004). Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc. Natl. Acad. Sci. U.S.A. 101, 9786–9791. doi: 10.1073/pnas.0402521101
Holden, M. T. G., Lindsay, J. A., Corton, C., Quail, M. A., Cockfield, J. D., Pathak, S., et al. (2010). Genome sequence of a recently emerged, highly transmissible, multi-antibiotic- and antiseptic-resistant variant of methicillin-resistant Staphylococcus aureus, sequence type 239 (TW). J. Bacteriol. 192, 888–892. doi: 10.1128/JB.01255-09
Howden, B. P., Seemann, T., Harrison, P. F., McEvoy, C. R., Stanton, J. A. L., Rand, C. J., et al. (2010). Complete genome sequence of Staphylococcus aureus strain JKD6008, an ST239 clone of methicillin-resistant Staphylococcus aureus with intermediate-level vancomycin resistance. J. Bacteriol. 192, 5848–5849. doi: 10.1128/JB.00951-10
Hsu, L.-Y., Harris, S. R., Chlebowicz, M. A., Lindsay, J. A., Koh, T.-H., Krishnan, P., et al. (2015). Evolutionary dynamics of methicillin-resistant Staphylococcus aureus within a healthcare system. Genome Biol. 16:81. doi: 10.1186/s13059-015-0643-z
Humann, J., and Lenz, L. L. (2009). Bacterial peptidoglycan-degrading enzymes and their impact on host muropeptide detection. J. Innate Immun. 1, 88–97. doi: 10.1159/000181181
Ingavale, S., Van Wamel, W., Luong, T. T., Lee, C. Y., and Cheung, A. L. (2005). Rat/MgrA, a regulator of autolysis, is a regulator of virulence genes in Staphylococcus aureus. Infect. Immun. 73, 1423–1431. doi: 10.1128/IAI.73.3.1423-1431.2005
Ingavale, S. S., Van Wamel, W., and Cheung, A. L. (2003). Characterization of RAT, an autolysis regulator in Staphylococcus aureus. Mol. Microbiol. 48, 1451–1466. doi: 10.1046/j.1365-2958.2003.03503.x
Ito, T., Katayama, Y., Asada, K., Mori, N., Tsutsumimoto, K., Tiensasitorn, C., et al. (2001). Structural comparison of three types of staphylococcal cassette chromosome mec integrated in the chromosome in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 45, 1323–1336. doi: 10.1128/AAC.45.5.1323-1336.2001
Iwao, Y., Khokhlova, O. E., Takano, T., Hung, W., Isobe, H., Peryanova, O. V., et al. (2012). Fatal pneumonia in HIV-infected patients from a novel ST239 methicillin-resistant Staphylococcus aureus carrying the toxic shock syndrome toxin-1 gene in Krasnoyarsk, Siberian Russia. Jpn. J. Infect. Dis. 65, 184–186.
Joensen, K. G., Scheutz, F., Lund, O., Hasman, H., Kaas, R. S., Nielsen, E. M., et al. (2014). Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 52, 1501–1510. doi: 10.1128/JCM.03617-13
Jonsson, I.-M., Lindholm, C., Luong, T. T., Lee, C. Y., and Tarkowski, A. (2008). mgrA regulates staphylococcal virulence important for induction and progression of septic arthritis and sepsis. Microbes Infect. 10, 1229–1235. doi: 10.1016/j.micinf.2008.07.026
Josten, M., Dischinger, J., Szekat, C., Reif, M., Al-Sabti, N., Sahl, H.-G., et al. (2014). Identification of agr-positive methicillin-resistant Staphylococcus aureus harbouring the class A mec complex by MALDI-TOF mass spectrometry. Int. J. Med. Microbiol. 304, 1018–1023. doi: 10.1016/j.ijmm.2014.07.005
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kolar, S. L., Ibarra, J. A., Rivera, F. E., Mootz, J. M., Davenport, J. E., Stevens, S. M., et al. (2013). Extracellular proteases are key mediators of Staphylococcus aureus virulence via the global modulation of virulence-determinant stability. Microbiologyopen 2, 18–34. doi: 10.1002/mbo3.55
Kong, Z., Zhao, P., Liu, H., Yu, X., Qin, Y., Su, Z., et al. (2016). Whole-Genome sequencing for the investigation of a hospital outbreak of MRSA in China. PLoS One 11:e0149844. doi: 10.1371/journal.pone.0149844
Kosakovsky Pond, S. L., Posada, D., Gravenor, M. B., Woelk, C. H., and Frost, S. D. W. (2006). Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 23, 1891–1901. doi: 10.1093/molbev/msl051
Koymans, K., Bisschop, A., Vughs, M., van Kessel, K., de Haas, C., and van Strijp, J. (2016). Staphylococcal superantigen-like protein 1 and 5 (SSL1 & SSL5) limit neutrophil chemotaxis and migration through MMP-inhibition. Int. J. Mol. Sci. 17:1072. doi: 10.3390/ijms17071072
Kuroda, M., Ohta, T., Uchiyama, I., Baba, T., Yuzawa, H., Kobayashi, I., et al. (2001). Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357, 1225–1240. doi: 10.1016/S0140-6736(00)04403-2
Lanave, C., Preparata, G., Saccone, C., and Serio, G. (1984). A new method for calculating evolutionary substitution rates. J. Mol. Evol. 20, 86–93. doi: 10.1007/BF02101990
Le, K. Y., and Otto, M. (2015). Quorum-sensing regulation in staphylococci — an overview. Front. Microbiol. 6:1174. doi: 10.3389/fmicb.2015.01174
Lewis, P. O. (2001). A likelihood approach to estimating phylogeny from discrete morphological character data. Syst. Biol. 50, 913–925. doi: 10.1080/106351501753462876
Li, M., Du, X., Villaruz, A. E., Diep, B. A., Wang, D., Song, Y., et al. (2012). MRSA epidemic linked to a quickly spreading colonization and virulence determinant. Nat. Med. 18, 816–819. doi: 10.1038/nm.2692
Li, Y., Cao, B., Zhang, Y., Zhou, J., Yang, B., and Wang, L. (2011). Complete genome sequence of Staphylococcus aureus T0131, an ST239-MRSA-SCCmec type III clone isolated in China. J. Bacteriol. 193, 3411–3412. doi: 10.1128/JB.05135-11
Lindsay, J. (ed.) (2008). “S. aureus evolution: lineages and mobile genetic elements (MGEs),” in Staphylococcus: Molecular Genetics, (Norfolk: Caister Academic Press), 45–70.
Luong, T. T., Dunman, P. M., Murphy, E., Projan, S. J., and Lee, C. Y. (2006). Transcription profiling of the mgrA regulon in Staphylococcus aureus. J. Bacteriol. 188, 1899–1910. doi: 10.1128/JB.188.5.1899-1910.2006
Margulies, M., Egholm, M., Altman, W. E., Attiya, S., Bader, J. S., Bemben, L. A., et al. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380. doi: 10.1038/nature04726
McMahon, S. A., Roberts, G. A., Johnson, K. A., Cooper, L. P., Liu, H., White, J. H., et al. (2009). Extensive DNA mimicry by the ArdA anti-restriction protein and its role in the spread of antibiotic resistance. Nucleic Acids Res. 37, 4887–4897. doi: 10.1093/nar/gkp478
Minh, B. Q., Nguyen, M. A. T., and von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi: 10.1093/molbev/mst024
Mitchell, A., Chang, H., Daugherty, L., Fraser, M., Hunter, S., Lopez, R., et al. (2015). The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43, D213–D221. doi: 10.1093/nar/gku1243
Monk, I. R., Shah, I. M., Xu, M., Tan, M.-W., and Foster, T. J. (2012). Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. mBio 3:e00277-11. doi: 10.1128/mBio.00277-11
Murrell, B., Moola, S., Mabona, A., Weighill, T., Sheward, D., Kosakovsky Pond, S. L., et al. (2013). FUBAR: a Fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 30, 1196–1205. doi: 10.1093/molbev/mst030
Murrell, B., Weaver, S., Smith, M. D., Wertheim, J. O., Murrell, S., Aylward, A., et al. (2015). Gene-wide identification of episodic selection. Mol. Biol. Evol. 32, 1365–1371. doi: 10.1093/molbev/msv035
Murrell, B., Wertheim, J. O., Moola, S., Weighill, T., Scheffler, K., and Kosakovsky Pond, S. L. (2012). Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 8:e1002764. doi: 10.1371/journal.pgen.1002764
Myers, E. W., Sutton, G. G., Delcher, A. L., Dew, I. M., Fasulo, D. P., Flanigan, M. J., et al. (2000). A whole-genome assembly of Drosophila. Science 287, 2196–2204. doi: 10.1126/science.287.5461.2196
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Novick, R., Ross, H., Figueiredo, A., and Abramochkin, G. (2000). Activation and Inhibition of the staphylococcal agr system. Science 287:391. doi: 10.1126/science.287.5452.391a
Novick, R. P., Christie, G. E., and Penadés, J. R. (2010). The phage-related chromosomal islands of Gram-positive bacteria. Nat. Rev. Microbiol. 8, 541–551. doi: 10.1038/nrmicro2393
Novick, R. P., and Subedi, A. (2007). The SaPIs: mobile pathogenicity islands of Staphylococcus. Chem. Immunol. Allergy 93, 42–57. doi: 10.1159/0000100857
Page, A. J., Delaney, A. J., Taylor, B., Seemann, T., Harris, S. R., and Soares, J. (2016). SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genomics 2:e000056. doi: 10.1099/mgen.0.000056
Paulander, W., Nissen Varming, A., Baek, K. T., Haaber, J., Frees, D., and Ingmer, H. (2012). Antibiotic-mediated selection of quorum-sensing-negative Staphylococcus aureus. mBio 3:e00459-12. doi: 10.1128/mBio.00459-12
Petkau, A., Stuart-Edwards, M., Stothard, P., and Van Domselaar, G. (2010). Interactive microbial genome visualization with GView. Bioinformatics 26, 3125–3126. doi: 10.1093/bioinformatics/btq588
Planet, P. J., Narechania, A., Chen, L., Mathema, B., Boundy, S., Archer, G., et al. (2017). Architecture of a species: phylogenomics of Staphylococcus aureus. Trends Microbiol. 25, 153–166. doi: 10.1016/j.tim.2016.09.009
Postma, B., Poppelier, M. J., van Galen, J. C., Prossnitz, E. R., van Strijp, J. A. G., de Haas, C. J. C., et al. (2004). Chemotaxis inhibitory protein of Staphylococcus aureus binds specifically to the C5a and formylated peptide receptor. J. Immunol. 172, 6994–7001. doi: 10.4049/jimmunol.172.11.6994
Rambaut, A., Lam, T. T., Max Carvalho, L., and Pybus, O. G. (2016). Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2:vew007. doi: 10.1093/ve/vew007
Robinson, D. A., and Enright, M. C. (2004). Evolution of Staphylococcus aureus by large chromosomal replacements. J. Bacteriol. 186, 1060–1064. doi: 10.1128/JB.186.4.1060-1064.2004
Romilly, C., Lays, C., Tomasini, A., Caldelari, I., Benito, Y., Hammann, P., et al. (2014). A Non-coding RNA promotes bacterial persistence and decreases virulence by regulating a regulator in Staphylococcus aureus. PLoS Pathog. 10:e1003979. doi: 10.1371/journal.ppat.1003979
Sabirova, J., Xavier, B. B., Hernalsteens, J. P., De Greve, H., Malhotra-Kumar, S., and Goossens, H. (2014). Complete genome sequences of two prolific biofilm-forming Staphylococcus aureus isolates belonging to USA300 and EMRSA-15. Genome Announc. 2:e00610-14. doi: 10.1128/genomeA.00610-14.Copyright
Sambrook, J., Fritch, E., and Maniatis, T. (1989). “Commonly used techniques in molecular cloning,” in Molecular Cloning: A Laboratory Manual, eds J. Sambrook, E. Fritch, and T. Maniatis (New York, NY: Cold Spring Harbor Laboratory Press), E3–E4.
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 33, W686–W689. doi: 10.1093/nar/gki366
Shopsin, B., Eaton, C., Wasserman, G. X. A., Mathema, B., Adhikari, R. X. P., Agolory, S., et al. (2010). Mutations in agr do not persist in natural populations of methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 202, 1593–1599. doi: 10.1086/656915
Smyth, D. S., McDougal, L. K., Gran, F. W., Manoharan, A., Enright, M. C., Song, J.-H., et al. (2010). Population structure of a hybrid clonal group of methicillin-resistant Staphylococcus aureus, ST239-MRSA-III. PLoS One 5:e8582. doi: 10.1371/journal.pone.0008582
Soubrier, J., Steel, M., Lee, M. S. Y., Der Sarkissian, C., Guindon, S., Ho, S. Y. W., et al. (2012). The influence of rate heterogeneity among sites on the time dependence of molecular rates. Mol. Biol. Evol. 29, 3345–3358. doi: 10.1093/molbev/mss140
Stach, C. S., Herrera, A., and Schlievert, P. M. (2014). Staphylococcal superantigens interact with multiple host receptors to cause serious diseases. Immunol. Res. 59, 177–181. doi: 10.1007/s12026-014-8539-7
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stegger, M., Driebe, E. M., Roe, C., Lemmer, D., Bowers, J. R., Engelthaler, D. M., et al. (2013). Genome sequence of Staphylococcus aureus strain CA-347, a USA600 methicillin-resistant isolate. Genome Announc. 1:e00517-13. doi: 10.1128/genomeA.00517-13
Subedi, A., Ubeda, C., Adhikari, R. P., Penadés, J. R., and Novick, R. P. (2007). Sequence analysis reveals genetic exchanges and intraspecific spread of SaPl2, pathogenicity island involved in menstrual toxic shock. Microbiology 153, 3235–3245. doi: 10.1099/mic.0.2007/006932-0
Sullivan, M. J., Petty, N. K., and Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. doi: 10.1093/bioinformatics/btr039
Teixeira, L. A., Resende, C. A., Ormonde, L. R., Rosenbaum, R., Figueiredo, A. M. S., De Lencastre, H., et al. (1995). Geographic spread of epidemic multiresistant Staphylococcus aureus clone in Brazil. J. Clin. Microbiol. 33, 2400–2404.
Traber, K., and Novick, R. (2006). A slipped-mispairing mutation in AgrA of laboratory strains and clinical isolates results in delayed activation of agr and failure to translate δ- and α-haemolysins. Mol. Microbiol. 59, 1519–1530. doi: 10.1111/j.1365-2958.2006.04986.x
Vallenet, D., Belda, E., Calteau, A., Cruveiller, S., Engelen, S., Lajus, A., et al. (2013). MicroScope–an integrated microbial resource for the curation and comparative analysis of genomic and metabolic data. Nucleic Acids Res. 41, D636–D647. doi: 10.1093/nar/gks1194
Vallenet, D., Engelen, S., Mornico, D., Cruveiller, S., Fleury, L., Lajus, A., et al. (2009). MicroScope: a platform for microbial genome annotation and comparative genomics. Database 2009:ba021. doi: 10.1093/database/bap021
van Hal, S. J., Jones, M., Gosbell, I. B., and Paterson, D. L. (2011). Vancomycin heteroresistance is associated with reduced mortality in ST239 methicillin-resistant Staphylococcus aureus blood stream infections. PLoS One 6:e21217. doi: 10.1371/journal.pone.0021217
Van Wamel, W. J. B., Rooijakkers, S. H. M., Ruyken, M., Van Kessel, K. P. M., and Van Strijp, J. A. G. (2006). The innate immune modulators staphylococcal complement inhibitor and chemotaxis inhibitory protein of Staphylococcus aureus are located on β-hemolysin-converting bacteriophages. J. Bacteriol. 188, 1310–1315. doi: 10.1128/JB.188.4.1310-1315.2006
Wang, Z., Zhou, H., Wang, H., Chen, H., Leung, K. K., Tsui, S., et al. (2014). Comparative genomics of methicillin-resistant Staphylococcus aureus ST239: distinct geographical variants in Beijing and Hong Kong. BMC Genomics 15:529. doi: 10.1186/1471-2164-15-529
Wertheim, J. O., Murrell, B., Smith, M. D., Kosakovsky Pond, S. L., and Scheffler, K. (2015). RELAX: detecting relaxed selection in a phylogenetic framework. Mol. Biol. Evol. 32, 820–832. doi: 10.1093/molbev/msu400
World Health Organisation [WHO] (2017). Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. Geneva: WHO.
Xiao, M., Wang, H., Zhao, Y., Mao, L.-L., Brown, M., Yu, Y.-S., et al. (2013). National surveillance of methicillin-resistant Staphylococcus aureus in China highlights a still-evolving epidemiology with 15 novel emerging multilocus sequence Types. J. Clin. Microbiol. 51, 3638–3644. doi: 10.1128/JCM.01375-13
Yamamoto, T., Takano, T., Higuchi, W., Iwao, Y., Singur, O., Reva, I., et al. (2012). Comparative genomics and drug resistance of a geographic variant of ST239 methicillin-resistant Staphylococcus aureus emerged in Russia. PLoS One 7:e29187. doi: 10.1371/journal.pone.0029187
Yang, Z. (1995). A space-time process model for the evolution of DNA sequences. Genetics 139, 993–1005.
Zhang, X., Xu, X., Yuan, W., Hu, Q., Shang, W., Hu, X., et al. (2014). Complete genome sequence of Staphylococcus aureus XN108, an ST239-MRSA-SCCmec III strain with intermediate vancomycin resistance isolated in Mainland China. Genome Announc. 2:e00449-14. doi: 10.1128/genomeA.00449-14
Keywords: methicillin-resistant Staphylococcus aureus, ST239-SCCmecIII, Brazilian epidemic clone, Brazilian/Hungarian clone, comparative genomics
Citation: Botelho AMN, Cerqueira e Costa MO, Moustafa AM, Beltrame CO, Ferreira FA, Côrtes MF, Costa BSS, Silva DNS, Bandeira PT, Lima NCB, Souza RC, Almeida LGdP, Vasconcelos ATR, Narechania A, Ryan C, O’Brien K, Kolokotronis S-O, Planet PJ, Nicolás MF and Figueiredo AMS (2019) Local Diversification of Methicillin- Resistant Staphylococcus aureus ST239 in South America After Its Rapid Worldwide Dissemination. Front. Microbiol. 10:82. doi: 10.3389/fmicb.2019.00082
Received: 18 August 2018; Accepted: 16 January 2019;
Published: 27 February 2019.
Edited by:
Steven Tong, The Peter Doherty Institute for Infection and Immunity, AustraliaReviewed by:
Santiago Castillo Ramírez, National Autonomous University of Mexico, MexicoCopyright © 2019 Botelho, Cerqueira e Costa, Moustafa, Beltrame, Ferreira, Côrtes, Costa, Silva, Bandeira, Lima, Souza, Almeida, Vasconcelos, Narechania, Ryan, O’Brien, Kolokotronis, Planet, Nicolás and Figueiredo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul J. Planet, cGxhbmV0cEBlbWFpbC5jaG9wLmVkdQ== Marisa Fabiana Nicolás, bWFyaXNhQGxuY2MuYnI= Agnes Marie Sá Figueiredo, YWduZXNAbWljcm8udWZyai5icg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.