 Océane Sorel
Océane Sorel Benjamin G. Dewals
Benjamin G. Dewals
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol., 09 January 2019
Sec. Virology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.03315
This article is part of the Research TopicHerpesvirus LatencyView all 14 articles
Gammaherpesviruses are important pathogens that establish latent infection in their natural host for lifelong persistence. During latency, the viral genome persists in the nucleus of infected cells as a circular episomal element while the viral gene expression program is restricted to non-coding RNAs and a few latency proteins. Among these, the genome maintenance protein (GMP) is part of the small subset of genes expressed in latently infected cells. Despite sharing little peptidic sequence similarity, gammaherpesvirus GMPs have conserved functions playing essential roles in latent infection. Among these functions, GMPs have acquired an intriguing capacity to evade the cytotoxic T cell response through self-limitation of MHC class I-restricted antigen presentation, further ensuring virus persistence in the infected host. In this review, we provide an updated overview of the main functions of gammaherpesvirus GMPs during latency with an emphasis on their immune evasion properties.
Herpesviruses are enveloped double-stranded DNA viruses that are in general responsible for persistent infections in a large number of animal species. In 2008, the International Committee on Taxonomy of Viruses (ICTV) created the order Herpesvirales comprising three families: the family Malacoherpesviridae composed of viruses infecting molluscs such as oysters, the family Alloherpesviridae composed of viruses infecting fish species and amphibians, and the predominantly studied family Herpesviridae that includes viruses of mammals and birds, itself classified into the three subfamilies Alpha-, Beta-, and Gammaherpesvirinae. A hallmark of all herpesviruses is their unique capacity to induce lifelong infection through establishing and maintaining latent infection. The definition of herpesvirus latency involves: (i) the presence of the viral genome in the nucleus of the infected cell (either as an episome or integrated in cellular chromosomes), (ii) reduced viral gene expression together with the absence of virion production, and (iii) the ability of latently infected cells to reactivate lytic viral replication either in vivo and/or in vitro (Lieberman, 2016). In addition, latent lifelong infection requires evasion mechanisms from the host immune response. Most alphaherpesviruses such as herpes simplex virus (HSV-1 or human alphaherpesvirus 1 – HHV-1) establish latency in non-dividing sensory neurons through maintenance of a quiescent episomal genome and expression of viral transcripts, in the absence of viral protein detection (Roizman and Whitley, 2013). In betaherpesviruses, myeloid cells such as macrophages are the main target of latent infection but the latency mechanisms in this subfamily have yet to be fully deciphered (Goodrum, 2016; Collins-McMillen et al., 2018). Gammaherpesviruses essentially establish latency in either B or T lymphocytes, although some species such as bovine gammaherpesvirus 4 (BoHV-4) seem to infect cells of the monocyte/macrophage lineage (Machiels et al., 2011, 2013). The mechanisms regulating latency establishment, maintenance of such a dormant infection in actively dividing cells, and how gammaherpesviruses escape the immune system of the infected host have been thoroughly studied (Stevenson and Efstathiou, 2005; Blake, 2010; Barton et al., 2011; White et al., 2012; Lieberman, 2013; Schuren et al., 2016; Dong et al., 2017; Ueda, 2018).
Most significant advances in the understanding of herpesvirus latency mechanisms have been identified in gammaherpesviruses, which can probably be explained by the fact that one major latency protein, named the “genome maintenance protein” (GMP): (i) is encoded by the genome of all described gammaherpesvirus species, (ii) is expressed during latent infection, (iii) regulates the maintenance of viral episomes in actively dividing lymphocytes through tethering the viral genome to cellular chromosomes, and (iv) evades immune detection (Verma et al., 2007; Frappier, 2015). The main objective of this review is to briefly summarize the importance of gammaherpesvirus infections and how GMPs maintain viral episomes in infected lymphocytes, before focusing on a more detailed description of the mechanisms mediated by GMPs to escape immune surveillance, in particular CD8+ cytotoxic T cells (CTLs), during latent infection.
Based on genomic and biological characteristics, gammaherpesviruses have been classified into four genera: the Lymphocryptovirus genus, the Rhadinovirus genus, the Percavirus genus and the Macavirus genus (Davison et al., 2009). Lymphocryptoviruses mainly infect human and non-human primates, and include one of the two gammaherpesviruses infecting humans: Epstein-Barr virus (EBV or human gammaherpesvirus 4 – HHV-4) (Jha et al., 2016). Rhadinoviruses also infect human and non-human primates, and include the second human gammaherpesvirus, Kaposi’s sarcoma-associated herpesvirus (KSHV or human gammaherpesvirus 8 – HHV-8) (Li et al., 2017). In addition to KSHV and viruses infecting Old World primates such as macaques, gorillas and chimpanzees, some rhadinoviruses also infect New World monkeys. Two examples are saimiriine gammaherpesvirus 2 (or SaHV-2) also known as herpesvirus saimiri (HVS); and ateline gammaherpesvirus 3, which infects spider monkeys (Damania and Desrosiers, 2001). In addition to viruses infecting primates, rhadinoviruses also include a number of viral species infecting other mammalians (Davison et al., 2009). For instance, murid gammaherpesvirus 4 (MuHV-4), also referred to as murine gammaherpesvirus 68 (MHV-68) is a natural pathogen of the yellow-necked field mouse (Ehlers et al., 2007) and is largely used in the laboratory mouse (Mus musculus). Furthermore, Bovine herpesvirus 4 (BoHV-4) infection is prevalent in cows while this virus is thought to have originally evolved in another Bovinae, the African buffalo (Syncerus caffer) (Dewals et al., 2005). Macaviruses are viruses infecting ruminants and are mainly associated with a lymphoproliferative disease named malignant catarrhal fever (MCF). Among these, alcelaphine gammaherpesvirus 1 (AlHV-1) and ovine gammaherpesvirus 2 (OvHV-2) naturally infect wildebeest (Connochaetes taurinus) and sheep (Ovis aries). They are responsible for wildebeest-derived and sheep-associated MCF in ruminants, respectively. The genus Percavirus is less well-defined and includes virus species infecting horses or mustelids.
The importance of latent infection by gammaherpesviruses is evident, both in term of lifelong persistence and related clinical diseases. The majority of epidemiological and clinical data comes from human gammaherpesviruses, although some veterinary viruses also induce latency-associated malignancies. EBV infects >90% of the human population, with seroconversion occurring early during childhood (Henle et al., 1969; Andersson, 2000). Whereas EBV latent infection is mostly asymptomatic, a number of clinical manifestations are associated with EBV infection. Beside infectious mononucleosis when primary infection occurs during adolescence (Callan et al., 1996), EBV is further associated with malignancies including Burkitt’s and Hodgkin’s lymphomas and other types of cancers (Rezk et al., 2018). In addition, EBV infection has been positively correlated with multiple sclerosis (MS). Nonetheless, the exact mechanisms and the causative role of EBV in MS induction remain open for investigation (Levin et al., 2010; Munger et al., 2011; Pender, 2011; Pakpoor et al., 2013; Moreno et al., 2018). The prevalence of KSHV is more variable and ranges from 5 to 50% depending on regions across the world. Like EBV, KSHV infection is in general asymptomatic but can be responsible for severe malignancies such as Kaposi’s sarcoma in immunocompromised patients but also other cancers such as multicentric Castlemans’s disease or primary effusion lymphoma (Li et al., 2017). Just like EBV and KSHV, animal gammaherpesviruses are also extensively studied, either for their veterinary importance or used as experimental models to study the biology of gammaherpesvirus infection in vivo. The former category includes MCF-inducing AlHV-1 that is responsible for the induction of a deadly peripheral T cell lymphoma-like disease in cattle caused by latently infected CD8+ T lymphocytes (Dewals et al., 2008, 2011; Dewals and Vanderplasschen, 2011; Palmeira et al., 2013). MuHV-4 and SaHV-2 are two examples of animal gammaherpesviruses of the latter category. Whereas the functions of gammaherpesvirus GMPs have been extensively investigated for EBV and KSHV in cell culture in vitro, animal gammaherpesviruses have provided important insights on the role of GMPs during in vivo infection.
After primary infection of target cells, gammaherpesviruses enter the lytic cycle that leads to production of viral particles and cell death. However, and depending on the infected cell type, the latent phase of the infection is in general rapidly established and is accompanied with the production of latency transcripts, including GMP. All sequenced gammaherpesviruses encode a predicted GMP (Table 1). In lymphocryptoviruses, GMPs are encoded by open reading frame (ORF) BKRF1 and are named according to Epstein-Barr virus (EBV) nuclear antigen 1 (EBNA-1). The GMPs in Rhadinovirus, Percavirus, and Macavirus genera are encoded by ORF73 and can be named after KSHV latency-associated nuclear antigen 1 (LANA-1). GMPs are DNA-binding proteins able to bind sequences within the viral genome while at the same time interacting with cell chromosome-associated proteins, to ensure partitioning to daughter cells during mitosis. Early studies have already demonstrated that GMPs, such as EBNA-1 and LANA-1, are essential for episome persistence (Ballestas et al., 1999; Ballestas and Kaye, 2001; Sears et al., 2003). Similar data have been generated for SaHV-2 and MuHV-4 for which it has been demonstrated that the gene ORF73 is required for efficient establishment of latency (Fowler et al., 2003; Calderwood et al., 2005). Likewise, deletion from the BoHV-4 genome of ORF73 impaired viral persistence in a macrophage cell line in vitro and in vivo in the rabbit model (Thirion et al., 2010). In addition, the ORF73-encoded protein of strain H26-95 of macacine gammaherpesvirus 5 was shown to bind to the viral episome and to be essential for establishment of latency (DeWire and Damania, 2005; Wen et al., 2009). The deletion of ORF73 from the genome of AlHV-1 also rendered AlHV-1 unable to persist and induce MCF in vivo, whereas impairment of its expression did not affect viral lytic replication (Palmeira et al., 2013). To enable partitioning in proliferating cells and avoid losing the episomal genomes in the cytoplasm, GMPs bind viral episomes to host chromosomes. Tethering of viral episomes to host DNA is accomplished by the ability of GMPs to simultaneously bind to several chromosome-associated proteins, other cellular components of the mitotic apparatus and specific viral DNA sequences (Yates et al., 1984; Cotter and Robertson, 1999; Shire et al., 1999; Cruickshank et al., 2000; Wu et al., 2000; Ballestas and Kaye, 2001; Cotter et al., 2001; Kapoor and Frappier, 2003; Verma and Robertson, 2003; Barbera et al., 2004, 2006; Calderwood et al., 2004; Waldmann et al., 2004; Kapoor et al., 2005; You et al., 2006; Kelley-Clarke et al., 2007; Habison et al., 2012; Verma et al., 2013; Gupta et al., 2016).
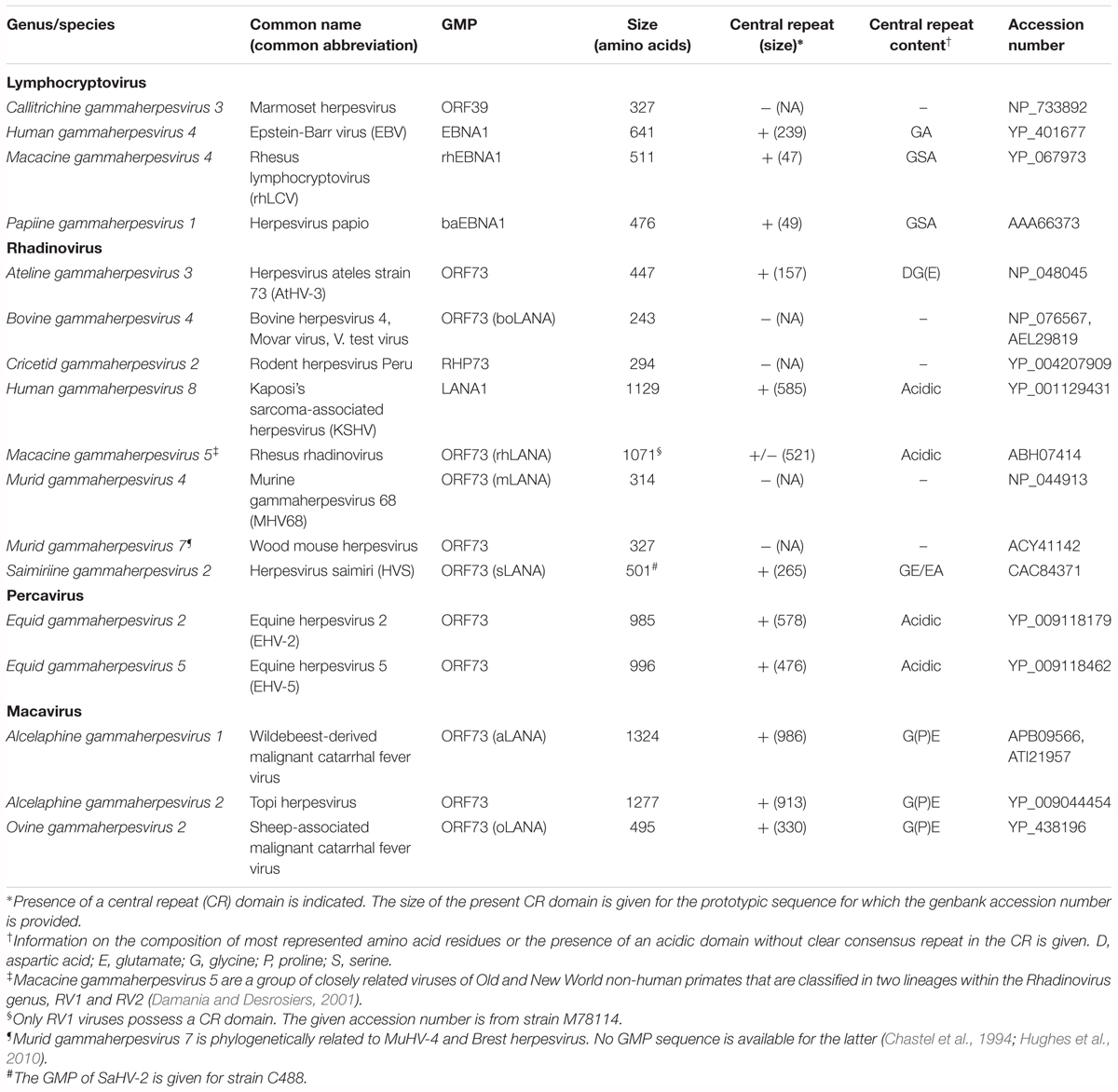
Table 1. Gammaherpesvirus genome maintenance proteins based on functional evidence and/or sequence prediction.
Genome maintenance proteins are nuclear proteins with very little sequence similarity among gammaherpesviruses, even though GMPs within one genus show higher sequence similarity. The C-terminal region shows the highest degree of sequence similarity and is involved in DNA-binding (Tellam et al., 2012). Regarding protein primary structure, most GMPs contain a central repeat (CR) domain composed of repeated amino acid motives that are divergent between gammaherpesvirus species. Interestingly, the size of the different GMPs greatly varies due to the presence of the CR domain (Figure 1). However, such variation in size does not seem to alter its role in maintaining the viral genome and the CR domain appears to be dispensable for genome maintenance properties with some gammaherpesviruses being devoid of a CR domain, such as MuHV-4 or BoHV-4 (Lomonte et al., 1995; Bennett et al., 2005). In addition to genome-maintenance functions, studies performed essentially on EBV EBNA-1 and KSHV LANA-1 have revealed additional roles for GMPs during latency and latency-associated diseases. GMPs are involved in initiating viral DNA replication during latency to generate sufficient copies of viral episomes prior to cell division (Wysokenski and Yates, 1989; Harrison et al., 1994; Yates et al., 2000; Ballestas and Kaye, 2001; Cotter et al., 2001; Stedman et al., 2004; Wong and Wilson, 2005; Verma et al., 2006; Lu et al., 2012), modulating viral gene expression to promote latency and repress reactivation (Gahn and Sugden, 1995; Evans et al., 1996; Schafer et al., 2003; Lu et al., 2006; Sivachandran et al., 2012), promoting tumorigenesis (Radkov et al., 2000; Humme et al., 2003; Altmann et al., 2006; Cai et al., 2006, 2012), and evading the immune system.
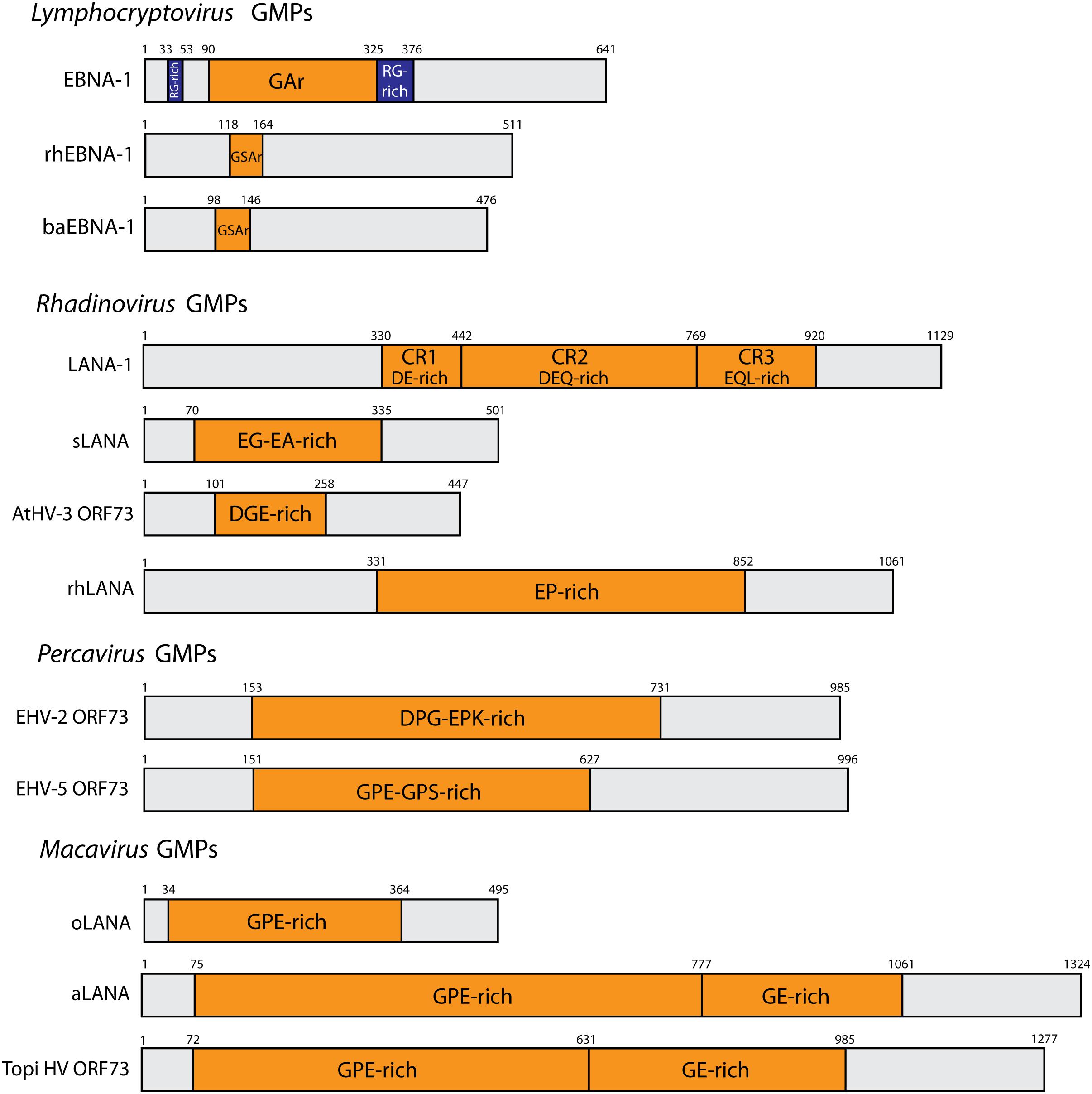
Figure 1. Schematic representation of representative gammaherpesvirus GMPs. N- and C- terminal domains are separated by a central amino acid repeat domain (CR), highlighted in orange. Repeat residues are indicated. The RG-rich regions of EBNA-1 are depicted in blue. Genus Lymphocryptovirus: EBNA-1 (EBV, strain B95.8), rhEBNA1 (rhLCV, strain LCL8664) and baEBNA-1 (baLCV, strain S594). Genus Rhadinovirus: AtHV3 ORF73 (strain 73), LANA-1 (KSHV, strain JK-18), sLANA (SaHV-2, strain C488), rhLANA (M78114). Genus Percavirus: EHV-2 ORF73 (strain 86/87), EHV-5 (strain 2-141/67). Genus Macavirus: oLANA (OvHV-2, strain BJ1035), and aLANA (AlHV-1, strain C500).
To ensure lifelong latency, gammaherpesviruses must maintain their genomes in dividing cells and remain undetected by virus-specific CD8+ CTLs. Thus, the GMPs must overcome the dilemma of efficiently maintaining viral episomes within infected cells while, at the same time, evading immune surveillance. Viral proteins are expressed endogenously within cells and are thus degraded by the proteasome into antigenic peptides before being translocated from the cytosol to the endoplasmic reticulum (ER) and loaded on major histocompatibility class I (MHC class I) molecules to form MHC-I-peptide (MHCp) complexes. MHCp complexes are exported to the cell surface for recognition by CD8+ cytotoxic T lymphocytes (CTLs) (Blum et al., 2013) (Figure 2).
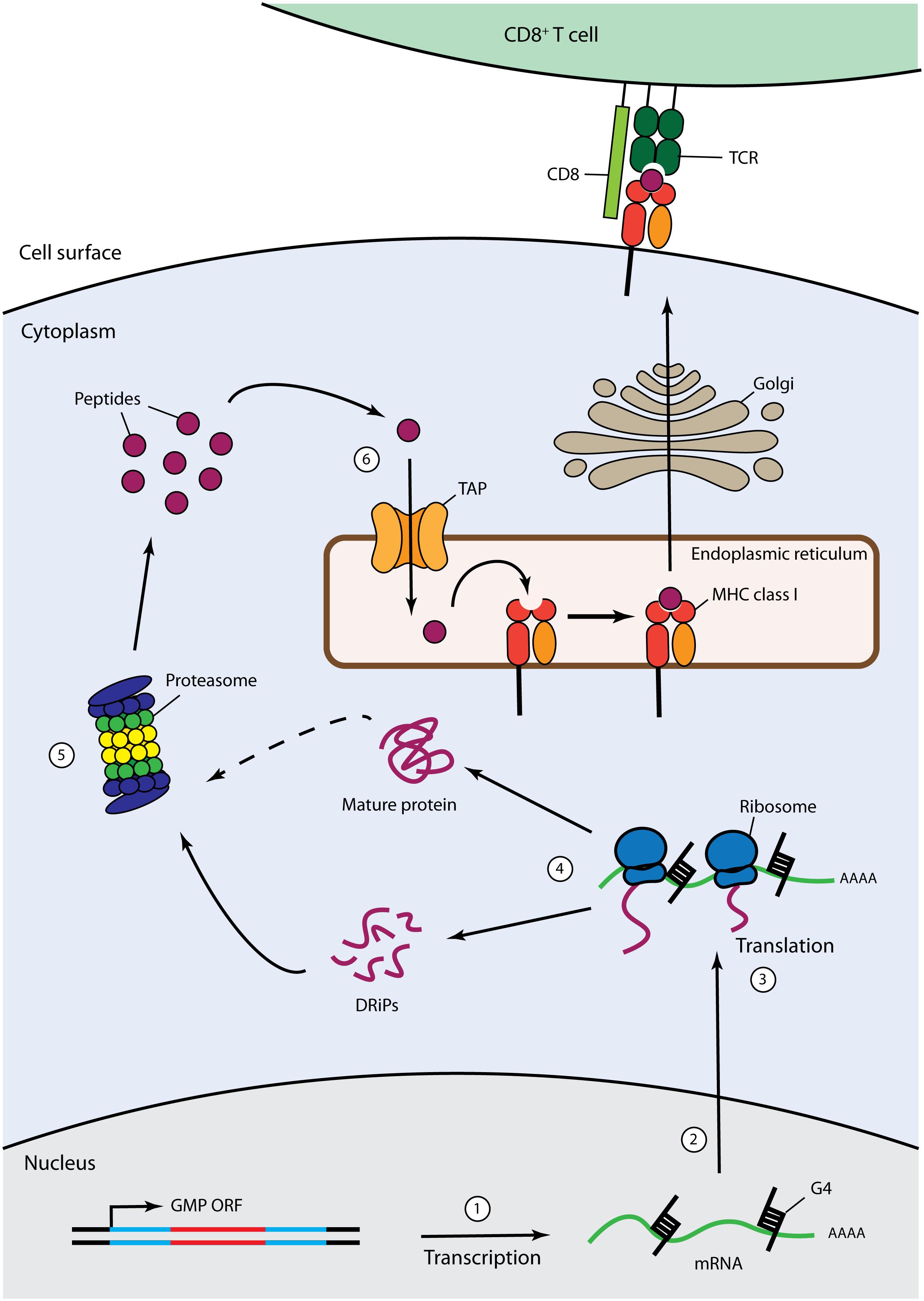
Figure 2. Cis-acting immune evasion of MHC Class I antigen presentation of gammaherpesvirus GMPs. The MHC class I antigen presentation pathway is depicted with described GMP-mediated cis-acting immune evasion mechanisms. Cytoplasmic endogenously expressed viral proteins are degraded by the proteasome into antigenic peptides that are then translocated from the cytosol to the endoplasmic reticulum (ER) through the transporter for antigen processing (TAP). Then, antigenic peptides are loaded on MHC class I molecules to form MHC-I-peptides complexes that are subsequently exported to the cell surface through the Golgi apparatus for recognition by CD8+ cytotoxic T lymphocytes. GMPs have been demonstrated to inhibit this process through various mechanisms: (1) sLANA and aLANA were shown to decrease their own steady-state RNA levels; (2) EBNA-1 can inhibit pre-mRNA processing of the primary EBNA-1 transcript; (3) structural constraints, such as G-quadruplexes (G4), contained in aLANA and EBNA-1 mRNA rather than protein sequences can regulate self-translation; (4) EBNA-1, aLANA, LANA-1, sLANA, and mLANA are able to induce retardation of self-translation; (5) EBNA-1, mLANA, and LANA-1 were shown to be protected from proteasomal degradation; (6) LANA-1 was reported to hold an inhibitory effect prior to translocation of its own cytoplasmic peptides into the ER.
The main source of viral antigens was previously thought to uniquely arise from the turnover of mature proteins. However, more recent studies highlighted an alternate hypothesis regarding the origin of MHC class I-restricted viral peptides, pointing to a major role of defective ribosomal products (DRiPs) as the main source of antigenic peptides during viral infection (Yewdell et al., 1996; Yewdell, 2011). DRiPs are translational products derived from prematurely terminated or misfolded polypeptides. Prioritizing DRiPs as the main source of antigenic peptides is believed to provide opportunities for the immune system to rapidly detect an active viral infection and thus optimize immune surveillance (Anton and Yewdell, 2014; Wei and Yewdell, 2018). Nevertheless, data supporting this hypothesis are still limited and further studies are necessary to exactly quantify whether MHC class I antigen presentation can be attributed to DRiPs of newly synthesized proteins. Indeed, the MHC class I-presented peptides seem to come from both short-lived and stable mature proteins depending on the origin of proteins and the biological status of the cell environment (Rock et al., 2014, 2016). Although autophagy is in some key aspects involved in the induction of adaptive immunity and control of some viral infections (Paludan et al., 2005), it appears that gammaherpesviruses have rather evolved to develop strategies to co-opt autophagy for viral benefit, during the lytic cycle but also during latency. However, GMPs do not seem to be directly involved in such mechanisms [see recent review (Lussignol and Esclatine, 2017)].
Gammaherpesviruses have evolved to acquire different strategies to escape the immune response. These mechanisms have been extensively reviewed in the past (Stevenson and Efstathiou, 2005; Means et al., 2007; Blake, 2010; Feng et al., 2013; Hu and Usherwood, 2014; Ressing et al., 2015; Sorel and Dewals, 2016; Zuo et al., 2017). The significant downregulation of viral gene expression during latent infection contributes to immune evasion, with viral gene expression being restricted to non-coding RNAs and latency proteins, including GMPs. GMPs must indeed be expressed in infected cells due to their key functions during long-term latency but at the same time need to remain hidden from the immune system. In order to evade CTL recognition of latently infected cells, all studied gammaherpesvirus GMPs have evolved to put into place immune evasion mechanisms consisting of the inhibition of their own presentation in the context of MHC class I on the cell surface, a mechanism that has been termed “cis-acting immune evasion” (Figure 2). Pioneer work came from studying EBV GMP where the CR domain could directly be involved in self-inhibition of antigen presentation (Levitskaya et al., 1995). Although most GMPs encoded by gammaherpesviruses contain a CR domain and are generally involved in the described cis-acting immune evasion mechanism, GMPs encoded by MuHV-4 (mLANA) or BoHV-4 (boLANA) do not have a CR domain (Lomonte et al., 1995; Bennett et al., 2005). While no data are available for immune evasion mechanism by boLANA, mLANA was able to inhibit self-presentation in MHC class I despite its lack of a CR domain (Bennett et al., 2005). Intriguingly, despite the conserved functions of gammaherpesviruses GMPs, the peptidic sequence in their repeat regions differs to a great extent from one ortholog to another. In contrast, comparative mRNA sequence analysis revealed that the internal repeat regions of GMP mRNAs displayed high nucleotide sequence similarities (Tellam et al., 2012). The results detailed in the next sections suggest that GMPs have evolved to adopt various strategies depending on the viral species, in order to achieve the ultimate goal consisting of the inhibition of their own presentation by MHC class I.
Epstein-Barr virus EBNA-1 is one of the most studied GMPs with regards to its different functional aspects, from genome maintenance (Yates et al., 1984, 1985), to cis-acting immune evasion. Early studies have demonstrated that EBNA-1 is able to prevent MHC class I-restricted self-peptide presentation in cis to CTLs through a mechanism involving its CR domain (GAr) (Levitskaya et al., 1995). The GAr domain is a region rich in glycine (G) and alanine (A) residues, which corresponds to 239 aa in strain B95.8 of EBV (Baer et al., 1984). Although its size can vary based on the strain, all isolates contain a GAr region (Falk et al., 1995). In order to decipher the immune evasion mechanism driving this effect, several studies have shown that the GAr domain provides increased stability to the EBNA-1 protein by inhibiting proteasome-mediated degradation (Levitskaya et al., 1997; Sharipo et al., 1998; Heessen et al., 2002, 2003; Hoyt et al., 2006; Coppotelli et al., 2011). In these studies, increased CTL responses could be induced with EBNA-1ΔGAr recombinant proteins, where the GAr was removed. Using conventional chromium-51 cytotoxicity assays, MHC-tetramer stains and/or peptide restimulation, it appeared that removal of the GAr domain resulted in increased presentation of a model T-cell epitope inserted into the GMP backbone but also increased presentation of T-cell epitopes of EBNA-1 itself, including the HLA-B∗35:01-restricted CTL epitope (HPVGEADYFEY). Despite the potential self-protection of EBNA-1 from MHC class I antigen presentation, several studies found that EBNA-1-specific CTLs exist in EBV-seropositive patients (Blake et al., 1997, 2000; Subklewe et al., 1999; Sim et al., 2013). Whereas inhibition of proteasome degradation was initially suggested, other reports found that GAr would more likely self-inhibit protein translation efficiency to reduce presentation of CTL epitopes from EBNA-1. These latter observations led to the conclusion that the GAr domain could inhibit the production of translation-dependent DRiPs. However, it remains unresolved whether control of the production of DRiPs during de novo translation is the only mechanism explaining EBNA-1 cis-acting immune evasion. Indeed, additional data contradicted the direct implication of the GAr domain in protecting EBNA-1 from proteasome degradation and increased protein stability (Yin et al., 2003; Lee et al., 2004; Tellam et al., 2004, 2007a; Voo et al., 2004; Daskalogianni et al., 2008).
What is clear from the studies investigating cis-acting immune evasion of EBNA-1 is that the GAr-mediated self-inhibition of antigen presentation to CTLs is not absolute. Indeed, although reduced in presence of GAr, EBNA-1 can be immunogenic and lead to the development of EBNA-1-specific CTLs in humans (Blake et al., 1997, 2000; Subklewe et al., 1999; Sim et al., 2013), but also in a mouse model where EBNA-1 was transduced using an adenovirus expression vector (Tellam et al., 2014). Recent advances have put forward a hypothesis to explain how the GAr domain is able to reduce self-translation efficiency. Studies which investigated mRNA translation of EBNA-1 suggested that the nascent GAr peptide alone was able to delay the assembly of the translation initiation complex mRNA, therefore reducing mRNA translation (Apcher et al., 2009, 2010). However, more recent reports suggested that EBNA-1 mRNA structure itself rather than the GAr peptidic sequence could regulate EBNA-1 protein translation (Tellam et al., 2008). Further findings supported this hypothesis by demonstrating that mRNA sequence could regulate the level of self-synthesis and antigen presentation of EBNA-1 in vitro through the GAr domain (Tellam et al., 2012). These authors highlighted the fact that the GAr domain, but also most GMP CR domains, display a nucleotidic sequence bias with enrichment of purines that is associated with reduced efficiency of protein translation. The role of purine-rich domains was demonstrated when replacement of the third base position of codons by pyrimidines led to increased translation of the protein and CTL activation (Tellam et al., 2008, 2014). Moreover, generating frameshifts in the EBNA-1 GAr internal repeat sequence to create alternate peptidic repeats had no effect on the cis-acting immune evasion (Tellam et al., 2012). Indeed, EBNA-1 frameshift mutants expressing GQE-rich or GRS-rich repeats could inhibit the presentation of a linked model epitope with an efficacy similar to native EBNA-1. Shortly after, the same group highlighted a key role played by clusters of unusual structural elements within the EBNA-1 mRNA sequence, named G-quadruplexes (G4), in the modulation of protein synthesis (Murat et al., 2014). G4 are secondary structures of nucleic acids that form within G-rich DNA or RNA sequences (Murat and Tellam, 2015). Four guanine bases can associate through hydrogen bonding to form a guanine tetrad and two or more guanine tetrads can stack on top of each other to constitute a G4 structure (Metifiot et al., 2014). Globally, these structures are present in telomeres, promoters, and gene bodies where they perform important regulatory roles in diverse biological processes including replication, transcription and translation (Rhodes and Lipps, 2015). Bioinformatics analysis of the EBNA-1 mRNA sequence revealed the presence of multiple putative G4 structures within the GAr domain (Murat et al., 2014). These authors further demonstrated that destabilization of G4 structures using antisense oligonucleotides led to an increase of EBNA-1 mRNA translation (Murat et al., 2014). To go further, as mentioned above, a modification of codon usage to reduce the purine bias in GAr resulted in reverted in vivo MHC class I epitope presentation and early priming of CD8+ T cells (Tellam et al., 2014). The mechanism underlying this effect was suggested to be determined by a capacity of G4 structures present in GAr to alter the association of ribosomes with EBNA-1 mRNA by inducing premature termination and/or ribosome stalling, therefore impeding protein translation (Murat et al., 2014). Nonetheless, whether G4 structures are present in all gammaherpesvirus GMPs and involved in self-inhibition of protein translation for immune evasion, needs to be further elucidated. Interestingly, the generation of memory T cell response was not affected by the codon-modification within the GAr domain (Tellam et al., 2014). These results were of high interest as they reported that promoting CTL priming against EBNA-1 through impairment of the GAr-dependent cis-acting immune evasion mechanisms could result in a more rapid CTL response and the establishment of efficient immune memory. In addition to translation regulation, previous studies have also established that EBNA-1 could act at the transcriptional level through inhibition of pre-mRNA processing of the primary EBNA-1 transcript (Yoshioka et al., 2008).
Studies on the GMPs encoded by baboon lymphocryptovirus (baLCV) and rhesus lymphocryptovirus (rhLCV), namely baEBNA-1 and rhEBNA-1, respectively, provided conflicting data. Indeed, the first results suggested that both the ba- and rhGSAr domains were not able to prevent MHC class I restricted peptide presentation in cis (Blake et al., 1999), whereas a second study showed that rhEBNA-1-specific CTLs expanded in vitro from rhLCV-infected animals failed to recognize endogenously expressed rhEBNA-1 (Fogg et al., 2005). A more recent study provided data supporting the hypothesis whereby rhEBNA-1 and baEBNA-1 proteins do not possess cis-acting immune evasion properties (Tellam et al., 2007b). Both proteins were translated at a higher rate than EBV EBNA-1 with no effect of deletion of the GSAr domains on translation rates and the rhGSAr domain could not avoid cis-acting presentation of a model epitope. A potential explanation for the adversarial result regarding the lack of recognition of rhEBNA-1 by the rhEBNA-1-specific CTLs reported in a study by Fogg and collaborators, could be that the specific clones isolated were of low affinity against the GMP (Blake, 2010).
LANA-1 is also able to act in cis to inhibit MHC class I-restricted epitope presentation to CTLs through involvement of the CR domain (Zaldumbide et al., 2007). LANA-1 is subdivided into three domains based on the peptidic sequence, with imperfect repeats: CR1 (aa 330–442) is a DE-rich region, CR2 (aa 442–768) is a DEQ-rich region, and CR3 (aa 769–920) consists of an EQL-rich region (Figure 1). Interestingly, the size of the CR domain varies between different KSHV strains or isolates (Gao et al., 1999). While a junctional domain between LANA-1 CR2 and CR3 has been mapped to contribute to retardation of translation and inhibition of proteasomal degradation of LANA-1 (Kwun et al., 2007), neither the CR2 nor CR3 domains were found to be involved in the inhibition of peptide presentation (Kwun et al., 2011). These data suggested that, in contrast to EBNA-1, the mechanism combining protection of LANA-1 from proteasomal degradation and reduction in the DRiPs generation level is not sufficient to block peptide presentation on MHC class I. Another notifiable difference with EBNA-1 is that the retardation of LANA-1 translation seems to be due to CR amino acid sequence rather than to the nucleotide level. Indeed, the introduction of a stop codon between CR2 and CR3 resulted in increased translation (Kwun et al., 2007). This observation is of importance considering the high degree (about 50% for CR1 and CR2, and about 70% for CR3) of similarity between EBNA-1 and LANA-1 in terms of nucleotidic sequence (Tellam et al., 2012). Conversely, CR1 has been implicated in LANA-1 cis-acting immune evasion through an apparent inhibitory effect prior to translocation of cytoplasmic peptides into the ER (Kwun et al., 2011). Here, fusion of a signal peptide to LANA-1 led to efficient processing of the protein for MHC Class I presentation. However, the presence or absence of CR1 had no effect on protein translation or proteasomal degradation. Using interferon-dependent induction of proteasomal degradation and proteasome inhibitor MG132, Kwun and collaborators further highlighted that LANA-1 is processed for MHC I presentation through the canonical proteasome pathway with little contribution of autophagy (Kwun et al., 2007, 2011). Thus, LANA-1 seems to have evolved to adopt immune evasion mechanisms that differ from EBNA-1, despite having conserved nucleotide sequence similarities. Although G4 structures have been involved in KSHV DNA replication and episomal persistence (Madireddy et al., 2016), it remains unappreciated whether G4 structures are present in LANA-1 or not, as demonstrated for EBNA-1. EBNA-1-specific CD8+ CTLs could be identified in patients (Blake et al., 1997, 2000; Subklewe et al., 1999; Sim et al., 2013). Likewise, several studies have identified LANA-1-specific CD8+ T-cell responses in KSHV seropositive subjects, highlighting the premise that the self-protection of GMPs against MHC class I-restricted epitope presentation is not absolute (Brander et al., 2002; Woodberry et al., 2005; Bihl et al., 2007; Lepone et al., 2010). Nonetheless, to our knowledge, no LANA-1-specific T cell clone is available to be tested in vitro.
Infection of squirrel monkeys with SaHV-2 results in asymptomatic latency in T lymphocytes. However, co-species transmission to New World non-human primates can lead to the development of acute T-cell lymphomas (Fickenscher and Fleckenstein, 2001). Interestingly, SaHV-2 can induce transformation of human and rabbit T lymphocytes (Fleckenstein and Ensser, 2004). The transforming capability of SaHV-2 has been identified to be mainly driven by two viral proteins termed Stp and Tip (Fleckenstein and Ensser, 2007). SaHV-2 infection of T lymphocytes is associated with episomal maintenance in absence of production of viral particles and the GMP encoded by SaHV-2 (sLANA) has been demonstrated to be essential for episomal maintenance (Calderwood et al., 2005). The CR domain of sLANA varies between strains, with a EG-rich domain of 15 aa in strain A11 or of 111 aa in strain C488; and a EA-rich region of 147 and 132 aa in both strains, respectively. One study thoroughly investigated the role of sLANA in evading CTL recognition (Gao et al., 2009). In this study, Gao and collaborators demonstrated that sLANA could reduce the MHC class I presentation of the linked-OT1 epitope SIINFEKL (Gao et al., 2009). The authors further observed that the steady-state levels of sLANA protein were reduced due to the presence of the CR domain rich in EG and EA residues, an observation that could be explained by a decrease in the steady-state levels of sLANA mRNA. Unexpectedly, the CR domain was not responsible for an increased turnover of sLANA mRNA but for a better stability over time of sLANA mRNAs compared to constructs deleted of the EG-EA repeat. Moreover, the authors showed that neither protein stability nor the efficiency of protein translation were influenced by the CR region, which revealed significant differences compared to both EBNA-1 and LANA-1. Finally, a single copy of the motif EEAEEAEEE, which is present multiple times in the EA-rich domain of two strains of SaHV-2, was shown to be sufficient to inhibit MHC class I-restricted antigen presentation when fused in frame with the sequence of the heterologous ovalbumin protein (Gao et al., 2009). The mechanism underlying this effect was suggested to be due to both stabilization of mRNA and repression of self-transcription. Thus, the presence of the EA-rich region could potentially influence the total amount of translated sLANA and protein synthesis efficiency, which in turn could potentially reduce the generation of DRiPs although this aspect has not been directly addressed.
MuHV-4 infects and persists in the laboratory mouse. Following primary infection, usually experimental intranasal or intra-tracheal infection, MuHV-4 replicates in epithelial cells and macrophages before reaching secondary lymphoid organs where the virus is maintained as episomal genomes in memory B lymphocytes (Barton et al., 2011; Gillet et al., 2015). MuHV-4 GMP (mLANA) has been demonstrated to be essential for episome maintenance in vitro but also in vivo as shown using recombinant strains of MuHV-4 impaired for the expression of mLANA (Fowler et al., 2003; Forrest et al., 2007; Habison et al., 2012). Among gammaherpesvirus GMPs, mLANA is probably one of the most intriguing proteins. Indeed, despite a lack of CR domain, mLANA has retained the ability of its orthologs to act in cis to self-inhibit MHC class I antigen presentation through a region mapped to amino acids 170–220 of mLANA (Bennett et al., 2005). This region was shown to be able to decrease the steady-state levels of mLANA protein while at the same time contributing to enhancing protein stability and protection from proteasomal degradation (Bennett et al., 2005), similar to EBNA-1. However, the exact mechanisms of action have not been fully deciphered and it remains unclear how protein translation efficiency and potential G4 structures could be involved. MuHV-4 provides an invaluable model to study gammaherpesvirus infection in vivo, including viral pathogenesis and latency (Barton et al., 2011). Using recombinant strains of MuHV-4 expressing model T-cell epitopes in tandem under the control of ORF73, Bennett and collaborators used an internal ribosome entry site following the ORF73 coding sequence to bypass the cis-acting evasion of ORF73 and to force expression of a tandem sequence of three T-cell epitopes in vivo. By doing so, latently-infected cells could express, in trans, CTL epitopes encoded by the ORF73 mRNA. Using this recombinant virus, infected mice showed a critical MHC class I-restricted and CTL-dependent reduction in viral latency, demonstrating that trans-acting immune evasion could not inhibit peptide presentation to CTLs during latency but rather indirectly suggests that cis-acting evasion by the GMP is critical for normal establishment of long-term latency in vivo. This study was the first to tackle the immune evasion mechanisms of GMPs in vivo. Interestingly, three recent studies used chimeric MuHV-4 recombinant viruses where mLANA was replaced by functional KSHV LANA-1 (Gupta et al., 2017; Habison et al., 2017; Pires de Miranda et al., 2018). These studies demonstrated the ability of LANA-1-expressing chimeric MuHV-4 to be maintained and establish latency in vivo, although at lower levels compared to wildtype MuHV-4. These studies are encouraging for future prospects to further investigate the role of immune evasion mechanisms in vivo that are directly mediated by LANA-1 or even other related GMPs.
Numerous clinical syndromes have been identified in equid species in association with EHV-2 or EHV-5 infection. However, true evidence for causal implication in the described diseases remain elusive (Marenzoni et al., 2015), with the exception of pulmonary fibrosis induced by EHV-5 infection (Williams et al., 2013). The full sequence of only two members of the Percavirus genus has been recently obtained. Although originally thought to not express a GMP (Telford et al., 1995), a recent report identified ORF73-encoding GMPs in two strains of equid gammaherpesvirus 2 (EHV-2), including the initially sequenced strain, and one strain of equid gammaherpesvirus 5 (EHV-5) (Wilkie et al., 2015). Whereas strain 86/67 of EHV-2 expresses a GMP of 985 amino acids, strain G9/92 expresses a 949-aa GMP. Strain 2-141/67 of EHV-5 encodes a GMP of 996 residues. Although GMPs of EHV-2 and EHV-5 contain a predicted CR domain, no data is currently available on a potential cis-acting immune evasion. More studies need be performed to uncover the role of EHV-2 and EHV-5 GMPs in infection of equids with these viruses.
All sequenced macaviruses encode a GMP but only the role of AlHV-1 GMP (aLANA) has recently been investigated experimentally (Palmeira et al., 2013; Sorel et al., 2017). AlHV-1 infects and persists in wildebeest asymptomatically and one can assume that the entire population of free-ranging wildebeest are infected. However, upon reactivation events, AlHV-1 can be transmitted to a range of phylogenetically related ruminant species, like cattle. In these susceptible species, AlHV-1 induces MCF that ultimately leads to the death of the infected animal. In both wildebeest and cattle, AlHV-1 establishes latency but results either in true quiescent/latent infection (in wildebeest) or latency-associated lymphoproliferation of CD8+ T lymphocytes (in cattle). During AlHV-1-associated MCF, aLANA is highly expressed (Palmeira et al., 2013). Thus, an adaptive immune response is likely induced against the protein, potentially including CD8+ CTLs that are specific to aLANA-derived antigenic peptides. However, if such a putative response exists, it fails to be protective as MCF-susceptible animals ultimately develop MCF and die upon infection. In our recent study, aLANA was shown to have acquired cis-acting immune evasion properties similarly to its orthologs (Sorel et al., 2017). In particular, this immune evasion mechanism was shown to be mediated through the CR domain of aLANA that is rich in G and E residues (termed GE). Importantly, the inhibitory properties of GE could be transferred to a heterologous protein such as enhanced green fluorescent protein, which is consistent with the data obtained with the EA-rich domain of sLANA (Gao et al., 2009). Mutant constructs expressing aLANA deleted for the GE-rich domain exhibited similar protein and mRNA turnover suggesting that GE inhibits proteasome-dependent antigen presentation through a mechanism that does not involve protein or mRNA degradation processes (Sorel et al., 2017). Although these data are consistent with the results obtained with sLANA (Gao et al., 2009), the internal region of several GMPs, including LANA-1 and EBNA-1, as well as the amino acid region 170–220 of mLANA were shown to mediate decreased protein turnover (Levitskaya et al., 1997; Bennett et al., 2005; Kwun et al., 2007). However, the CR2CR3 region of LANA-1 that was mapped to inhibit proteasomal degradation was, however, not found to be involved in the self-inhibition of antigen presentation (Kwun et al., 2011). Although the mechanisms involved in immune evasion are not necessarily shared by all gammaherpesvirus GMPs, these data are nonetheless strengthened by another study that revealed that the half-life of a polypeptide does not determine antigen presentation (Apcher et al., 2010). Thus, it can be suggested that protection of GMPs from proteasomal degradation might not be sufficient to block antigen presentation. The lack of aLANA GE resulted in increased protein expression levels due to a combination of enhanced translation efficiency and increased steady-state RNA levels (Sorel et al., 2017), which resulted in increased proteasome-dependent processing of aLANA for MHC-I presentation. This mechanism was, however, independent of autophagy, as treatments with the autophagy inducer rapamycin, or autophagy inhibitors chloroquine or 3-methyladenine, did not affect peptide presentation by MHC-I. Thus, these results suggested that the GE-rich domain could inhibit self-antigen presentation through regulation of both protein translation and RNA transcription levels, leading as a consequence to a decrease in DRiPs generation. Several related gammaherpesviruses were shown to have acquired mechanisms that lead to reduced steady-state protein levels of their respective GMPs (Tellam et al., 2007a; Yoshioka et al., 2008; Gao et al., 2009), resulting in a potential reduction of DRiPs production. Thus, targeting pathways leading to DRiPs production seems to be a valuable mechanism to ensure episome persistence during latency while avoiding detection by the immune system. Furthermore, replacing the native GE-rich region of aLANA by a synthetic codon-modified sequence, in order to reduce the purine bias in the mRNA sequence without modifying the protein sequence, similar to EBNA-1 GAr (Tellam et al., 2014), led to significantly enhanced antigen presentation and increased activation of antigen-specific CTLs in a mouse model of DNA immunization (Sorel et al., 2017). These results were suggestive of potential constraints, such as G4 structures, within native GE mRNA structure that could limit antigen presentation in a similar manner as EBNA-1 (Tellam et al., 2008; Murat et al., 2014). Then, mRNA constraints contained in the GE-rich domain of aLANA, rather than peptidic sequence, is likely responsible for CTL immune evasion. However, the GE-mediated cis-limitation of MHC class I antigen presentation of aLANA was further shown to be dispensable for the induction of MCF in the experimental rabbit model (Sorel et al., 2017). Indeed, a recombinant virus expressing a GE-deleted form of aLANA could induce MCF in rabbits in a similar manner to a wild type virus expressing aLANA. Although the viral-specific CTL response could not be monitored to determine the role of the GE-rich domain in the efficient priming of CTLs by aLANA in vivo, it clearly appears that aLANA-mediated cis-acting immune evasion is not determinant during MCF. While the mechanisms explaining this finding in the context of MCF remain to be identified, these results suggest that the immune evasion functions of aLANA are more likely to play a role in the context of lifelong infection of the natural host of AlHV-1, the wildebeest.
All sequenced gammaherpesviruses encode a GMP that tethers viral genomes to the cellular chromosomes, ensuring even segregation of viral episomes in daughter cells during cell division (Blake, 2010). Besides their role in viral persistence, GMPs can also modify the cellular environment to promote cell immortalization and tumorigenesis in gammaherpesvirus-induced malignancies. Because of their essential roles in gammaherpesvirus latency, GMPs need to be expressed while remaining hidden from immune surveillance in the infected host. Evasion mechanisms of the cytotoxic T cell response through self-limitation of MHC class I antigen presentation constitute unique properties developed by GMPs to ensure gammaherpesvirus long-term persistence. Importantly, more questions need to be addressed for a complete understanding of how GMPs successfully achieve both viral persistence and escape of the CTL response. Such future understanding is of interest to develop potential treatments to target and efficiently disrupt latency. Among these questions, we could ask whether the presence of G4 structures does represent a major and common mechanism in gammaherpesviruses to control the production of DRiPs from nascent GMP proteins during latency? Moreover, how important is the cis-acting immune evasion during lymphoproliferation, a hallmark of gammaherpesvirus-associated malignancies? Indeed, a recombinant strain of AlHV-1 expressing a mutated aLANA unable to self-inhibit protein processing for presentation by MHC class I was, however, fully able to induce normal MCF. Whether this observation after AlHV-1 infection represents a general rule or just an exception is unknown. Thus, it makes no doubt that understanding the degree of involvement of GMP cis-acting immune evasion during gammaherpesvirus latency will be determined depending on our understanding of latency mechanisms themselves and, for instance, how distinct are silent latency in healthy individuals and latency-dependent lymphoproliferative diseases. In other words, would self-inhibition of antigen presentation by GMP represent the essential mechanism to avoid CTL recognition during gammaherpesvirus-induced lymphoproliferation? We are eager to uncover future investigations that will clarify these questions.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was funded through F.R.S.-FNRS grant #MIS-F.4501.15.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
BD is a Research Associate of the F.R.S.-FNRS (Belgium). The authors are grateful to Léa Morvan for careful reading of the manuscript.
Altmann, M., Pich, D., Ruiss, R., Wang, J., Sugden, B., and Hammerschmidt, W. (2006). Transcriptional activation by EBV nuclear antigen 1 is essential for the expression of EBV’s transforming genes. Proc. Natl. Acad. Sci. U.S.A. 103, 14188–14193. doi: 10.1073/pnas.0605985103
Andersson, J. (2000). An overview of epstein-barr virus: from discovery to future directions for treatment and prevention. Herpes 7, 76–82.
Anton, L. C., and Yewdell, J. W. (2014). Translating DRiPs: MHC class I immunosurveillance of pathogens and tumors. J. Leukoc. Biol. 95, 551–562. doi: 10.1189/jlb.1113599
Apcher, S., Daskalogianni, C., Manoury, B., and Fahraeus, R. (2010). Epstein Barr virus-encoded EBNA1 interference with MHC class I antigen presentation reveals a close correlation between mRNA translation initiation and antigen presentation. PLoS Pathog. 6:e1001151. doi: 10.1371/journal.ppat.1001151
Apcher, S., Komarova, A., Daskalogianni, C., Yin, Y., Malbert-Colas, L., and Fåhraeus, R. (2009). mRNA translation regulation by the Gly-Ala repeat of Epstein-Barr virus nuclear antigen 1. J. Virol. 83, 1289–1298. doi: 10.1128/jvi.01369-08
Baer, R., Bankier, A. T., Biggin, M. D., Deininger, P. L., Farrell, P. J., Gibson, T. J., et al. (1984). DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 310, 207–211. doi: 10.1038/310207a0
Ballestas, M. E., Chatis, P. A., and Kaye, K. M. (1999). Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284, 641–644. doi: 10.1126/science.284.5414.641
Ballestas, M. E., and Kaye, K. M. (2001). Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through cis-acting terminal repeat (TR) sequence and specifically binds TR DNA. J. Virol. 75, 3250–3258. doi: 10.1128/JVI.75.7.3250-3258.2001
Barbera, A. J., Ballestas, M. E., and Kaye, K. M. (2004). The Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen 1 N terminus is essential for chromosome association, DNA replication, and episome persistence. J. Virol. 78, 294–301. doi: 10.1128/JVI.78.1.294-301.2004
Barbera, A. J., Chodaparambil, J. V., Kelley-Clarke, B., Joukov, V., Walter, J. C., Luger, K., et al. (2006). The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science 311, 856–861. doi: 10.1126/science.1120541
Barton, E., Mandal, P., and Speck, S. H. (2011). Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu. Rev. Immunol. 29, 351–397. doi: 10.1146/annurev-immunol-072710-081639
Bennett, N. J., May, J. S., and Stevenson, P. G. (2005). Gamma-herpesvirus latency requires T cell evasion during episome maintenance. PLoS Biol. 3:e120. doi: 10.1371/journal.pbio.0030120
Bihl, F., Narayan, M., Chisholm, J. V. III, Henry, L. M., Suscovich, T. J., Brown, E. E., et al. (2007). Lytic and latent antigens of the human gammaherpesviruses Kaposi’s sarcoma-associated herpesvirus and Epstein-Barr virus induce T-cell responses with similar functional properties and memory phenotypes. J. Virol. 81, 4904–4908. doi: 10.1128/jvi.02509-06
Blake, N. (2010). Immune evasion by gammaherpesvirus genome maintenance proteins. J. Gen. Virol. 91(Pt 4), 829–846. doi: 10.1099/vir.0.018242-0
Blake, N., Haigh, T., Shaka’a, G., Croom-Carter, D., and Rickinson, A. (2000). The importance of exogenous antigen in priming the human CD8+ T cell response: lessons from the EBV nuclear antigen EBNA1. J. Immunol. 165, 7078–7087. doi: 10.4049/jimmunol.165.12.7078
Blake, N., Lee, S., Redchenko, I., Thomas, W., Steven, N., Leese, A., et al. (1997). Human CD8+ T cell responses to EBV EBNA1: HLA class I presentation of the (Gly-Ala)-containing protein requires exogenous processing. Immunity 7, 791–802. doi: 10.1016/S1074-7613(00)80397-0
Blake, N. W., Moghaddam, A., Rao, P., Kaur, A., Glickman, R., Cho, Y. G., et al. (1999). Inhibition of antigen presentation by the glycine/alanine repeat domain is not conserved in simian homologues of Epstein-Barr virus nuclear antigen 1. J. Virol. 73, 7381–7389.
Blum, J. S., Wearsch, P. A., and Cresswell, P. (2013). Pathways of antigen processing. Annu. Rev. Immunol. 31, 443–473. doi: 10.1146/annurev-immunol-032712-095910
Brander, C., Raje, N., O’Connor, P. G., Davies, F., Davis, J., Chauhan, D., et al. (2002). Absence of biologically important Kaposi sarcoma-associated herpesvirus gene products and virus-specific cellular immune responses in multiple myeloma. Blood 100, 698–700. doi: 10.1182/blood.V100.2.698
Cai, Q., Xiao, B., Si, H., Cervini, A., Gao, J., Lu, J., et al. (2012). Kaposi’s sarcoma herpesvirus upregulates Aurora A expression to promote p53 phosphorylation and ubiquitylation. PLoS Pathog. 8:e1002566. doi: 10.1371/journal.ppat.1002566
Cai, Q. L., Knight, J. S., Verma, S. C., Zald, P., and Robertson, E. S. (2006). EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 tumor suppressors. PLoS Pathog. 2:e116. doi: 10.1371/journal.ppat.0020116
Calderwood, M., White, R. E., Griffiths, R. A., and Whitehouse, A. (2005). Open reading frame 73 is required for herpesvirus saimiri A11-S4 episomal persistence. J. Gen. Virol. 86(Pt 10), 2703–2708. doi: 10.1099/vir.0.81230-0
Calderwood, M. A., Hall, K. T., Matthews, D. A., and Whitehouse, A. (2004). The herpesvirus saimiri ORF73 gene product interacts with host-cell mitotic chromosomes and self-associates via its C terminus. J. Gen. Virol. 85(Pt 1), 147–153. doi: 10.1099/vir.0.19437-0
Callan, M. F., Steven, N., Krausa, P., Wilson, J. D., Moss, P. A., Gillespie, G. M., et al. (1996). Large clonal expansions of CD8+ T cells in acute infectious mononucleosis. Nat. Med. 2, 906–911. doi: 10.1038/nm0896-906
Chastel, C., Beaucournu, J. P., Chastel, O., Legrand, M. C., and Le Goff, F. (1994). A herpesvirus from an European shrew (Crocidura russula). Acta Virol. 38:309.
Collins-McMillen, D., Buehler, J., Peppenelli, M., and Goodrum, F. (2018). Molecular determinants and the regulation of human cytomegalovirus latency and reactivation. Viruses 10:E444. doi: 10.3390/v10080444
Coppotelli, G., Mughal, N., Marescotti, D., and Masucci, M. G. (2011). High avidity binding to DNA protects ubiquitylated substrates from proteasomal degradation. J. Biol. Chem. 286, 19565–19575. doi: 10.1074/jbc.M111.224782
Cotter, M. A. II, and Robertson, E. S. (1999). The latency-associated nuclear antigen tethers the Kaposi’s sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 264, 254–264. doi: 10.1006/viro.1999.9999
Cotter, M. A. II, Subramanian, C., and Robertson, E. S. (2001). The Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen binds to specific sequences at the left end of the viral genome through its carboxy-terminus. Virology 291, 241–259. doi: 10.1006/viro.2001.1202
Cruickshank, J., Shire, K., Davidson, A. R., Edwards, A. M., and Frappier, L. (2000). Two domains of the epstein-barr virus origin DNA-binding protein, EBNA1, orchestrate sequence-specific DNA binding. J. Biol. Chem. 275, 22273–22277. doi: 10.1074/jbc.M001414200
Damania, B., and Desrosiers, R. C. (2001). Simian homologues of human herpesvirus 8. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356, 535–543. doi: 10.1098/rstb.2000.0782
Daskalogianni, C., Apcher, S., Candeias, M. M., Naski, N., Calvo, F., and Fahraeus, R. (2008). Gly-Ala repeats induce position- and substrate-specific regulation of 26 S proteasome-dependent partial processing. J. Biol. Chem. 283, 30090–30100. doi: 10.1074/jbc.M803290200
Davison, A. J., Eberle, R., Ehlers, B., Hayward, G. S., McGeoch, D. J., Minson, A. C., et al. (2009). The order Herpesvirales. Arch. Virol. 154, 171–177. doi: 10.1007/s00705-008-0278-4
Dewals, B., Boudry, C., Farnir, F., Drion, P. V., and Vanderplasschen, A. (2008). Malignant catarrhal fever induced by alcelaphine herpesvirus 1 is associated with proliferation of CD8+ T cells supporting a latent infection. PLoS One 3:e1627. doi: 10.1371/journal.pone.0001627
Dewals, B., Gillet, L., Gerdes, T., Taracha, E. L., Thiry, E., and Vanderplasschen, A. (2005). Antibodies against bovine herpesvirus 4 are highly prevalent in wild African buffaloes throughout eastern and southern Africa. Vet. Microbiol. 110, 209–220. doi: 10.1016/j.vetmic.2005.08.006
Dewals, B., Myster, F., Palmeira, L., Gillet, L., Ackermann, M., and Vanderplasschen, A. (2011). Ex vivo bioluminescence detection of alcelaphine herpesvirus 1 infection during malignant catarrhal fever. J. Virol. 85, 6941–6954. doi: 10.1128/JVI.00286-11
Dewals, B. G., and Vanderplasschen, A. (2011). Malignant catarrhal fever induced by Alcelaphine herpesvirus 1 is characterized by an expansion of activated CD3+CD8+CD4- T cells expressing a cytotoxic phenotype in both lymphoid and non-lymphoid tissues. Vet. Res. 42:95. doi: 10.1186/1297-9716-42-95
DeWire, S. M., and Damania, B. (2005). The latency-associated nuclear antigen of rhesus monkey rhadinovirus inhibits viral replication through repression of Orf50/Rta transcriptional activation. J. Virol. 79, 3127–3138. doi: 10.1128/JVI.79.5.3127-3138.2005
Dong, S., Forrest, J. C., and Liang, X. (2017). Murine gammaherpesvirus 68: a small animal model for gammaherpesvirus-associated diseases. Adv. Exp. Med. Biol. 1018, 225–236. doi: 10.1007/978-981-10-5765-6_14
Ehlers, B., Kuchler, J., Yasmum, N., Dural, G., Voigt, S., Schmidt-Chanasit, J., et al. (2007). Identification of novel rodent herpesviruses, including the first gammaherpesvirus of Mus musculus. J. Virol. 81, 8091–8100. doi: 10.1128/jvi.00255-07
Evans, T. J., Farrell, P. J., and Swaminathan, S. (1996). Molecular genetic analysis of Epstein-Barr virus Cp promoter function. J. Virol. 70, 1695–1705.
Falk, K., Gratama, J. W., Rowe, M., Zou, J. Z., Khanim, F., Young, L. S., et al. (1995). The role of repetitive DNA sequences in the size variation of Epstein-Barr virus (EBV) nuclear antigens, and the identification of different EBV isolates using RFLP and PCR analysis. J. Gen. Virol. 76(Pt 4), 779–790. doi: 10.1099/0022-1317-76-4-779
Feng, P., Moses, A., and Fruh, K. (2013). Evasion of adaptive and innate immune response mechanisms by gamma-herpesviruses. Curr. Opin. Virol. 3, 285–295. doi: 10.1016/j.coviro.2013.05.011
Fickenscher, H., and Fleckenstein, B. (2001). Herpesvirus saimiri. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356, 545–567. doi: 10.1098/rstb.2000.0780
Fleckenstein, B., and Ensser, A. (2004). Herpesvirus saimiri transformation of human T lymphocytes. Curr. Protoc. Immunol. 63, 7.21.1–7.21.11. doi: 10.1002/0471142735.im0721s63
Fleckenstein, B., and Ensser, A. (2007). “Gammaherpesviruses of new world primates,” in Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis, eds A. Arvin, G. Campadelli-Fiume, E. Mocarski, P. S. Moore, B. Roizman, R. Whitley, et al. (Cambridge: Cambridge University Press).
Fogg, M. H., Kaur, A., Cho, Y. G., and Wang, F. (2005). The CD8+ T-cell response to an Epstein-Barr virus-related gammaherpesvirus infecting rhesus macaques provides evidence for immune evasion by the EBNA-1 homologue. J. Virol. 79, 12681–12691. doi: 10.1128/jvi.79.20.12681-12691.2005
Forrest, J. C., Paden, C. R., Allen, R. D. III, Collins, J., and Speck, S. H. (2007). ORF73-null murine gammaherpesvirus 68 reveals roles for mLANA and p53 in virus replication. J. Virol. 81, 11957–11971. doi: 10.1128/JVI.00111-07
Fowler, P., Marques, S., Simas, J. P., and Efstathiou, S. (2003). ORF73 of murine herpesvirus-68 is critical for the establishment and maintenance of latency. J. Gen. Virol. 84(Pt 12), 3405–3416. doi: 10.1099/vir.0.19594-0
Frappier, L. (2015). EBNA1. Curr. Top. Microbiol. Immunol. 391, 3–34. doi: 10.1007/978-3-319-22834-1_1
Gahn, T. A., and Sugden, B. (1995). An EBNA-1-dependent enhancer acts from a distance of 10 kilobase pairs to increase expression of the Epstein-Barr virus LMP gene. J. Virol. 69, 2633–2636.
Gao, J., Coulson, J. M., Whitehouse, A., and Blake, N. (2009). Reduction in RNA levels rather than retardation of translation is responsible for the inhibition of major histocompatibility complex class I antigen presentation by the glutamic acid-rich repeat of Herpesvirus saimiri open reading frame 73. J. Virol. 83, 273–282. doi: 10.1128/JVI.01532-08
Gao, S. J., Zhang, Y. J., Deng, J. H., Rabkin, C. S., Flore, O., and Jenson, H. B. (1999). Molecular polymorphism of Kaposi’s sarcoma-associated herpesvirus (Human herpesvirus 8) latent nuclear antigen: evidence for a large repertoire of viral genotypes and dual infection with different viral genotypes. J. Infect. Dis. 180, 1466–1476. doi: 10.1086/315098
Gillet, L., Frederico, B., and Stevenson, P. G. (2015). Host entry by gamma-herpesviruses–lessons from animal viruses? Curr. Opin. Virol. 15, 34–40. doi: 10.1016/j.coviro.2015.07.007
Goodrum, F. (2016). Human cytomegalovirus latency: approaching the gordian knot. Annu. Rev. Virol. 3, 333–357. doi: 10.1146/annurev-virology-110615-042422
Gupta, A., Oldenburg, D. G., Salinas, E., White, D. W., and Forrest, J. C. (2017). Murine gammaherpesvirus 68 expressing kaposi sarcoma-associated herpesvirus latency-associated nuclear antigen (LANA) reveals both functional conservation and divergence in LANA homologs. J. Virol. 91:e00992-17. doi: 10.1128/JVI.00992-17
Gupta, N., Thakker, S., and Verma, S. C. (2016). KSHV encoded LANA recruits nucleosome assembly protein NAP1L1 for regulating viral DNA replication and transcription. Sci. Rep. 6:32633. doi: 10.1038/srep32633
Habison, A. C., Beauchemin, C., Simas, J. P., Usherwood, E. J., and Kaye, K. M. (2012). Murine gammaherpesvirus 68 LANA acts on terminal repeat DNA to mediate episome persistence. J. Virol. 86, 11863–11876. doi: 10.1128/JVI.01656-12
Habison, A. C., de Miranda, M. P., Beauchemin, C., Tan, M., Cerqueira, S. A., Correia, B., et al. (2017). Cross-species conservation of episome maintenance provides a basis for in vivo investigation of Kaposi’s sarcoma herpesvirus LANA. PLoS Pathog. 13:e1006555. doi: 10.1371/journal.ppat.1006555
Harrison, S., Fisenne, K., and Hearing, J. (1994). Sequence requirements of the Epstein-Barr virus latent origin of DNA replication. J. Virol. 68, 1913–1925.
Heessen, S., Dantuma, N. P., Tessarz, P., Jellne, M., and Masucci, M. G. (2003). Inhibition of ubiquitin/proteasome-dependent proteolysis in Saccharomyces cerevisiae by a Gly-Ala repeat. FEBS Lett. 555, 397–404. doi: 10.1016/S0014-5793(03)01296-1
Heessen, S., Leonchiks, A., Issaeva, N., Sharipo, A., Selivanova, G., Masucci, M. G., et al. (2002). Functional p53 chimeras containing the Epstein-Barr virus Gly-Ala repeat are protected from Mdm2- and HPV-E6-induced proteolysis. Proc. Natl. Acad. Sci. U.S.A. 99, 1532–1537. doi: 10.1073/pnas.022306499
Henle, G., Henle, W., Clifford, P., Diehl, V., Kafuko, G. W., Kirya, B. G., et al. (1969). Antibodies to Epstein-Barr virus in Burkitt’s lymphoma and control groups. J. Natl. Cancer Inst. 43, 1147–1157.
Hoyt, M. A., Zich, J., Takeuchi, J., Zhang, M., Govaerts, C., and Coffino, P. (2006). Glycine-alanine repeats impair proper substrate unfolding by the proteasome. EMBO J. 25, 1720–1729. doi: 10.1038/sj.emboj.7601058
Hu, Z., and Usherwood, E. J. (2014). Immune escape of gamma-herpesviruses from adaptive immunity. Rev. Med. Virol. 24, 365–378. doi: 10.1002/rmv.1791
Hughes, D. J., Kipar, A., Milligan, S. G., Cunningham, C., Sanders, M., Quail, M. A., et al. (2010). Characterization of a novel wood mouse virus related to murid herpesvirus 4. J. Gen. Virol. 91(Pt 4), 867–879. doi: 10.1099/vir.0.017327-0
Humme, S., Reisbach, G., Feederle, R., Delecluse, H. J., Bousset, K., Hammerschmidt, W., et al. (2003). The EBV nuclear antigen 1 (EBNA1) enhances B cell immortalization several thousandfold. Proc. Natl. Acad. Sci. U.S.A. 100, 10989–10994. doi: 10.1073/pnas.1832776100
Jha, H. C., Pei, Y., and Robertson, E. S. (2016). Epstein-Barr Virus: diseases linked to infection and transformation. Front. Microbiol. 7:1602. doi: 10.3389/fmicb.2016.01602
Kapoor, P., and Frappier, L. (2003). EBNA1 partitions Epstein-Barr virus plasmids in yeast cells by attaching to human EBNA1-binding protein 2 on mitotic chromosomes. J. Virol. 77, 6946–6956. doi: 10.1128/jvi.77.12.6946-6956.2003
Kapoor, P., Lavoie, B. D., and Frappier, L. (2005). EBP2 plays a key role in Epstein-Barr virus mitotic segregation and is regulated by aurora family kinases. Mol. Cell. Biol. 25, 4934–4945. doi: 10.1128/mcb.25.12.4934-4945.2005
Kelley-Clarke, B., Ballestas, M. E., Komatsu, T., and Kaye, K. M. (2007). Kaposi’s sarcoma herpesvirus C-terminal LANA concentrates at pericentromeric and peri-telomeric regions of a subset of mitotic chromosomes. Virology 357, 149–157. doi: 10.1016/j.virol.2006.07.052
Kwun, H. J., da Silva, S. R., Qin, H., Ferris, R. L., Tan, R., Chang, Y., et al. (2011). The central repeat domain 1 of Kaposi’s sarcoma-associated herpesvirus (KSHV) latency associated-nuclear antigen 1 (LANA1) prevents cis MHC class I peptide presentation. Virology 412, 357–365. doi: 10.1016/j.virol.2011.01.026
Kwun, H. J., da Silva, S. R., Shah, I. M., Blake, N., Moore, P. S., and Chang, Y. (2007). Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mimics Epstein-Barr virus EBNA1 immune evasion through central repeat domain effects on protein processing. J. Virol. 81, 8225–8235. doi: 10.1128/JVI.00411-07
Lee, S. P., Brooks, J. M., Al-Jarrah, H., Thomas, W. A., Haigh, T. A., Taylor, G. S., et al. (2004). CD8 T cell recognition of endogenously expressed epstein-barr virus nuclear antigen 1. J. Exp. Med. 199, 1409–1420. doi: 10.1084/jem.20040121
Lepone, L., Rappocciolo, G., Knowlton, E., Jais, M., Piazza, P., Jenkins, F. J., et al. (2010). Monofunctional and polyfunctional CD8+ T cell responses to human herpesvirus 8 lytic and latency proteins. Clin. Vaccine Immunol. 17, 1507–1516. doi: 10.1128/CVI.00189-10
Levin, L. I., Munger, K. L., O’Reilly, E. J., Falk, K. I., and Ascherio, A. (2010). Primary infection with the Epstein-Barr virus and risk of multiple sclerosis. Ann. Neurol. 67, 824–830. doi: 10.1002/ana.21978
Levitskaya, J., Coram, M., Levitsky, V., Imreh, S., Steigerwald-Mullen, P. M., Klein, G., et al. (1995). Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature 375, 685–688. doi: 10.1038/375685a0
Levitskaya, J., Sharipo, A., Leonchiks, A., Ciechanover, A., and Masucci, M. G. (1997). Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. U.S.A. 94, 12616–12621. doi: 10.1073/pnas.94.23.12616
Li, S., Bai, L., Dong, J., Sun, R., and Lan, K. (2017). Kaposi’s sarcoma-associated herpesvirus: epidemiology and molecular biology. Adv. Exp. Med. Biol. 1018, 91–127. doi: 10.1007/978-981-10-5765-6_7
Lieberman, P. M. (2013). Keeping it quiet: chromatin control of gammaherpesvirus latency. Nat. Rev. Microbiol. 11, 863–875. doi: 10.1038/nrmicro3135
Lieberman, P. M. (2016). Epigenetics and genetics of viral latency. Cell Host Microbe 19, 619–628. doi: 10.1016/j.chom.2016.04.008
Lomonte, P., Bublot, M., van Santen, V., Keil, G. M., Pastoret, P. P., and Thiry, E. (1995). Analysis of bovine herpesvirus 4 genomic regions located outside the conserved gammaherpesvirus gene blocks. J. Gen. Virol. 76(Pt 7), 1835–1841. doi: 10.1099/0022-1317-76-7-1835
Lu, F., Day, L., Gao, S. J., and Lieberman, P. M. (2006). Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi’s sarcoma-associated herpesvirus lytic transcription. J. Virol. 80, 5273–5282. doi: 10.1128/JVI.02541-05
Lu, F., Tsai, K., Chen, H. S., Wikramasinghe, P., Davuluri, R. V., Showe, L., et al. (2012). Identification of host-chromosome binding sites and candidate gene targets for Kaposi’s sarcoma-associated herpesvirus LANA. J. Virol. 86, 5752–5762. doi: 10.1128/JVI.07216-11
Lussignol, M., and Esclatine, A. (2017). Herpesvirus and autophagy: “all right, everybody be cool, this is a robbery!” Viruses 9:E372. doi: 10.3390/v9120372
Machiels, B., Lete, C., de Fays, K., Mast, J., Dewals, B., Stevenson, P. G., et al. (2011). The bovine herpesvirus 4 Bo10 gene encodes a nonessential viral envelope protein that regulates viral tropism through both positive and negative effects. J. Virol. 85, 1011–1024. doi: 10.1128/JVI.01092-10
Machiels, B., Stevenson, P. G., Vanderplasschen, A., and Gillet, L. (2013). A gammaherpesvirus uses alternative splicing to regulate its tropism and its sensitivity to neutralization. PLoS Pathog. 9:e1003753. doi: 10.1371/journal.ppat.1003753
Madireddy, A., Purushothaman, P., Loosbroock, C. P., Robertson, E. S., Schildkraut, C. L., and Verma, S. C. (2016). G-quadruplex-interacting compounds alter latent DNA replication and episomal persistence of KSHV. Nucleic Acids Res. 44, 3675–3694. doi: 10.1093/nar/gkw038
Marenzoni, M. L., Stefanetti, V., Danzetta, M. L., and Timoney, P. J. (2015). Gammaherpesvirus infections in equids: a review. Vet. Med. 6, 91–101. doi: 10.2147/VMRR.S39473
Means, R. E., Lang, S. M., and Jung, J. U. (2007). “Human gammaherpesvirus immune evasion strategies,” in Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis, eds A. Arvin, G. Campadelli-Fiume, E. Mocarski, P. S. Moore, B. Roizman, R. Whitley, et al. (Cambridge: Cambridge University Press).
Metifiot, M., Amrane, S., Litvak, S., and Andreola, M. L. (2014). G-quadruplexes in viruses: function and potential therapeutic applications. Nucleic Acids Res. 42, 12352–12366. doi: 10.1093/nar/gku999
Moreno, M. A., Or-Geva, N., Aftab, B. T., Khanna, R., Croze, E., Steinman, L., et al. (2018). Molecular signature of Epstein-Barr virus infection in MS brain lesions. Neurol. Neuroimmunol. Neuroinflamm. 5:e466. doi: 10.1212/NXI.0000000000000466
Munger, K. L., Levin, L. I., O’Reilly, E. J., Falk, K. I., and Ascherio, A. (2011). Anti-Epstein-Barr virus antibodies as serological markers of multiple sclerosis: a prospective study among United States military personnel. Mult. Scler. 17, 1185–1193. doi: 10.1177/1352458511408991
Murat, P., and Tellam, J. (2015). Effects of messenger RNA structure and other translational control mechanisms on major histocompatibility complex-I mediated antigen presentation. Wiley Interdiscip. Rev. RNA 6, 157–171. doi: 10.1002/wrna.1262
Murat, P., Zhong, J., Lekieffre, L., Cowieson, N. P., Clancy, J. L., Preiss, T., et al. (2014). G-quadruplexes regulate Epstein-Barr virus-encoded nuclear antigen 1 mRNA translation. Nat. Chem. Biol. 10, 358–364. doi: 10.1038/nchembio.1479
Pakpoor, J., Disanto, G., Gerber, J. E., Dobson, R., Meier, U. C., Giovannoni, G., et al. (2013). The risk of developing multiple sclerosis in individuals seronegative for Epstein-Barr virus: a meta-analysis. Mult. Scler. 19, 162–166. doi: 10.1177/1352458512449682
Palmeira, L., Sorel, O., Van Campe, W., Boudry, C., Roels, S., Myster, F., et al. (2013). An essential role for gamma-herpesvirus latency-associated nuclear antigen homolog in an acute lymphoproliferative disease of cattle. Proc. Natl. Acad. Sci. U.S.A. 110, E1933–E1942. doi: 10.1073/pnas.1216531110
Paludan, C., Schmid, D., Landthaler, M., Vockerodt, M., Kube, D., Tuschl, T., et al. (2005). Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 307, 593–596. doi: 10.1126/science.1104904
Pender, M. P. (2011). The essential role of Epstein-Barr virus in the pathogenesis of multiple sclerosis. Neuroscientist 17, 351–367. doi: 10.1177/1073858410381531
Pires de Miranda, M., Quendera, A. P., McVey, C. E., Kaye, K. M., and Simas, J. P. (2018). In vivo persistence of chimeric virus after substitution of the Kaposi’s sarcoma-associated herpesvirus LANA DNA binding domain with that of murid herpesvirus 4. J. Virol. 92:e01251-18. doi: 10.1128/JVI.01251-18
Radkov, S. A., Kellam, P., and Boshoff, C. (2000). The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 6, 1121–1127. doi: 10.1038/80459
Ressing, M. E., van Gent, M., Gram, A. M., Hooykaas, M. J., Piersma, S. J., and Wiertz, E. J. (2015). Immune evasion by Epstein-Barr virus. Curr. Top. Microbiol. Immunol. 391, 355–381. doi: 10.1007/978-3-319-22834-1_12
Rezk, S. A., Zhao, X., and Weiss, L. M. (2018). Epstein-Barr virus (EBV)-associated lymphoid proliferations, a 2018 update. Hum. Pathol. 79, 18–41. doi: 10.1016/j.humpath.2018.05.020
Rhodes, D., and Lipps, H. J. (2015). G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 43, 8627–8637. doi: 10.1093/nar/gkv862
Rock, K. L., Farfan-Arribas, D. J., Colbert, J. D., and Goldberg, A. L. (2014). Re-examining class-I presentation and the DRiP hypothesis. Trends Immunol. 35, 144–152. doi: 10.1016/j.it.2014.01.002
Rock, K. L., Reits, E., and Neefjes, J. (2016). Present yourself! By MHC Class I and MHC Class II molecules. Trends Immunol. 37, 724–737. doi: 10.1016/j.it.2016.08.010
Roizman, B., and Whitley, R. J. (2013). An inquiry into the molecular basis of HSV latency and reactivation. Annu. Rev. Microbiol. 67, 355–374. doi: 10.1146/annurev-micro-092412-155654
Schafer, A., Lengenfelder, D., Grillhosl, C., Wieser, C., Fleckenstein, B., and Ensser, A. (2003). The latency-associated nuclear antigen homolog of Herpesvirus saimiri inhibits lytic virus replication. J. Virol. 77, 5911–5925. doi: 10.1128/JVI.77.10.5911-5925.2003
Schuren, A. B., Costa, A. I., and Wiertz, E. J. (2016). Recent advances in viral evasion of the MHC Class I processing pathway. Curr. Opin. Immunol. 40, 43–50. doi: 10.1016/j.coi.2016.02.007
Sears, J., Kolman, J., Wahl, G. M., and Aiyar, A. (2003). Metaphase chromosome tethering is necessary for the DNA synthesis and maintenance of oriP plasmids but is insufficient for transcription activation by Epstein-Barr nuclear antigen 1. J. Virol. 77, 11767–11780. doi: 10.1128/JVI.77.21.11767-11780.2003
Sharipo, A., Imreh, M., Leonchiks, A., Imreh, S., and Masucci, M. G. (1998). A minimal glycine-alanine repeat prevents the interaction of ubiquitinated I kappaB alpha with the proteasome: a new mechanism for selective inhibition of proteolysis. Nat. Med. 4, 939–944. doi: 10.1038/nm0898-939
Shire, K., Ceccarelli, D. F., Avolio-Hunter, T. M., and Frappier, L. (1999). EBP2, a human protein that interacts with sequences of the Epstein-Barr virus nuclear antigen 1 important for plasmid maintenance. J. Virol. 73, 2587–2595.
Sim, A. C., Too, C. T., Oo, M. Z., Lai, J., Eio, M. Y., Song, Z., et al. (2013). Defining the expression hierarchy of latent T-cell epitopes in Epstein-Barr virus infection with TCR-like antibodies. Sci. Rep. 3:3232. doi: 10.1038/srep03232
Sivachandran, N., Wang, X., and Frappier, L. (2012). Functions of the Epstein-Barr virus EBNA1 protein in viral reactivation and lytic infection. J. Virol. 86, 6146–6158. doi: 10.1128/jvi.00013-12
Sorel, O., Chen, T., Myster, F., Javaux, J., Vanderplasschen, A., and Dewals, B. G. (2017). Macavirus latency-associated protein evades immune detection through regulation of protein synthesis in cis depending upon its glycin/glutamate-rich domain. PLoS Pathog. 13:e1006691. doi: 10.1371/journal.ppat.1006691
Sorel, O., and Dewals, B. G. (2016). MicroRNAs in large herpesvirus DNA genomes: recent advances. Biomol. Concepts 7, 229–239. doi: 10.1515/bmc-2016-0017
Stedman, W., Deng, Z., Lu, F., and Lieberman, P. M. (2004). ORC, MCM, and histone hyperacetylation at the Kaposi’s sarcoma-associated herpesvirus latent replication origin. J. Virol. 78, 12566–12575. doi: 10.1128/JVI.78.22.12566-12575.2004
Stevenson, P. G., and Efstathiou, S. (2005). Immune mechanisms in murine gammaherpesvirus-68 infection. Viral Immunol. 18, 445–456. doi: 10.1089/vim.2005.18.445
Subklewe, M., Chahroudi, A., Bickham, K., Larsson, M., Kurilla, M. G., Bhardwaj, N., et al. (1999). Presentation of epstein-barr virus latency antigens to CD8(+), interferon-gamma-secreting, T lymphocytes. Eur. J. Immunol. 29, 3995–4001. doi: 10.1002/(SICI)1521-4141(199912)29:12<3995::AID-IMMU3995>3.0.CO;2-E
Telford, E. A., Watson, M. S., Aird, H. C., Perry, J., and Davison, A. J. (1995). The DNA sequence of equine herpesvirus 2. J. Mol. Biol. 249, 520–528. doi: 10.1006/jmbi.1995.0314
Tellam, J., Connolly, G., Green, K. J., Miles, J. J., Moss, D. J., Burrows, S. R., et al. (2004). Endogenous presentation of CD8+ T cell epitopes from Epstein-Barr virus-encoded nuclear antigen 1. J. Exp. Med. 199, 1421–1431. doi: 10.1084/jem.20040191
Tellam, J., Fogg, M. H., Rist, M., Connolly, G., Tscharke, D., Webb, N., et al. (2007a). Influence of translation efficiency of homologous viral proteins on the endogenous presentation of CD8+ T cell epitopes. J. Exp. Med. 204, 525–532. doi: 10.1084/jem.20062508
Tellam, J., Rist, M., Connolly, G., Webb, N., Fazou, C., Wang, F., et al. (2007b). Translation efficiency of EBNA1 encoded by lymphocryptoviruses influences endogenous presentation of CD8+ T cell epitopes. Eur. J. Immunol. 37, 328–337. doi: 10.1002/eji.200636153
Tellam, J., Smith, C., Rist, M., Webb, N., Cooper, L., Vuocolo, T., et al. (2008). Regulation of protein translation through mRNA structure influences MHC class I loading and T cell recognition. Proc. Natl. Acad. Sci. U.S.A. 105, 9319–9324. doi: 10.1073/pnas.0801968105
Tellam, J. T., Lekieffre, L., Zhong, J., Lynn, D. J., and Khanna, R. (2012). Messenger RNA sequence rather than protein sequence determines the level of self-synthesis and antigen presentation of the EBV-encoded antigen, EBNA1. PLoS Pathog. 8:e1003112. doi: 10.1371/journal.ppat.1003112
Tellam, J. T., Zhong, J., Lekieffre, L., Bhat, P., Martinez, M., Croft, N. P., et al. (2014). mRNA structural constraints on EBNA1 synthesis impact on in vivo antigen presentation and early priming of CD8+ T cells. PLoS Pathog. 10:e1004423. doi: 10.1371/journal.ppat.1004423
Thirion, M., Machiels, B., Farnir, F., Donofrio, G., Gillet, L., Dewals, B., et al. (2010). Bovine herpesvirus 4 ORF73 is dispensable for virus growth in vitro, but is essential for virus persistence in vivo. J. Gen. Virol. 91(Pt 10), 2574–2584. doi: 10.1099/vir.0.023192-0
Ueda, K. (2018). KSHV genome replication and maintenance in latency. Adv. Exp. Med. Biol. 1045, 299–320. doi: 10.1007/978-981-10-7230-7_14
Verma, S. C., Cai, Q., Kreider, E., Lu, J., and Robertson, E. S. (2013). Comprehensive analysis of LANA interacting proteins essential for viral genome tethering and persistence. PLoS One 8:e74662. doi: 10.1371/journal.pone.0074662
Verma, S. C., Choudhuri, T., Kaul, R., and Robertson, E. S. (2006). Latency-associated nuclear antigen (LANA) of Kaposi’s sarcoma-associated herpesvirus interacts with origin recognition complexes at the LANA binding sequence within the terminal repeats. J. Virol. 80, 2243–2256. doi: 10.1128/JVI.80.5.2243-2256.2006
Verma, S. C., Lan, K., and Robertson, E. (2007). Structure and function of latency-associated nuclear antigen. Curr. Top. Microbiol. Immunol. 312, 101–136. doi: 10.1007/978-3-540-34344-8_4
Verma, S. C., and Robertson, E. S. (2003). ORF73 of Herpesvirus saimiri strain C488 tethers the viral genome to metaphase chromosomes and binds to cis-acting DNA sequences in the terminal repeats. J. Virol. 77, 12494–12506. doi: 10.1128/JVI.77.23.12494-12506.2003
Voo, K. S., Fu, T., Wang, H. Y., Tellam, J., Heslop, H. E., Brenner, M. K., et al. (2004). Evidence for the presentation of major histocompatibility complex class I-restricted Epstein-Barr virus nuclear antigen 1 peptides to CD8+ T lymphocytes. J. Exp. Med. 199, 459–470. doi: 10.1084/jem.20031219
Waldmann, T., Scholten, I., Kappes, F., Hu, H. G., and Knippers, R. (2004). The DEK protein–an abundant and ubiquitous constituent of mammalian chromatin. Gene 343, 1–9. doi: 10.1016/j.gene.2004.08.029
Wei, J., and Yewdell, J. W. (2018). Flu DRiPs in MHC Class I immunosurveillance. Virol. Sin. doi: 10.1007/s12250-018-0061-y [Epub ahead of print].
Wen, K. W., Dittmer, D. P., and Damania, B. (2009). Disruption of LANA in rhesus rhadinovirus generates a highly lytic recombinant virus. J. Virol. 83, 9786–9802. doi: 10.1128/JVI.00704-09
White, D. W., Suzanne Beard, R., and Barton, E. S. (2012). Immune modulation during latent herpesvirus infection. Immunol. Rev. 245, 189–208. doi: 10.1111/j.1600-065X.2011.01074.x
Wilkie, G. S., Kerr, K., Stewart, J. P., Studdert, M. J., and Davison, A. J. (2015). Genome sequences of equid herpesviruses 2 and 5. Genome Announc. 3:e00119-15. doi: 10.1128/genomeA.00119-15
Williams, K. J., Robinson, N. E., Lim, A., Brandenberger, C., Maes, R., Behan, A., et al. (2013). Experimental induction of pulmonary fibrosis in horses with the gammaherpesvirus equine herpesvirus 5. PLoS One 8:e77754. doi: 10.1371/journal.pone.0077754
Wong, L. Y., and Wilson, A. C. (2005). Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen induces a strong bend on binding to terminal repeat DNA. J. Virol. 79, 13829–13836. doi: 10.1128/JVI.79.21.13829-13836.2005
Woodberry, T., Suscovich, T. J., Henry, L. M., Martin, J. N., Dollard, S., O’Connor, P. G., et al. (2005). Impact of Kaposi sarcoma-associated herpesvirus (KSHV) burden and HIV coinfection on the detection of T cell responses to KSHV ORF73 and ORF65 proteins. J. Infect. Dis. 192, 622–629. doi: 10.1086/432103
Wu, H., Ceccarelli, D. F., and Frappier, L. (2000). The DNA segregation mechanism of Epstein-Barr virus nuclear antigen 1. EMBO Rep. 1, 140–144. doi: 10.1038/sj.embor.embor611
Wysokenski, D. A., and Yates, J. L. (1989). Multiple EBNA1-binding sites are required to form an EBNA1-dependent enhancer and to activate a minimal replicative origin within oriP of Epstein-Barr virus. J. Virol. 63, 2657–2666.
Yates, J., Warren, N., Reisman, D., and Sugden, B. (1984). A cis-acting element from the Epstein-Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. Proc. Natl. Acad. Sci. U.S.A. 81, 3806–3810. doi: 10.1073/pnas.81.12.3806
Yates, J. L., Camiolo, S. M., and Bashaw, J. M. (2000). The minimal replicator of Epstein-Barr virus oriP. J. Virol. 74, 4512–4522. doi: 10.1128/JVI.74.10.4512-4522.2000
Yates, J. L., Warren, N., and Sugden, B. (1985). Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature 313, 812–815. doi: 10.1038/313812a0
Yewdell, J. W. (2011). DRiPs solidify: progress in understanding endogenous MHC class I antigen processing. Trends Immunol. 32, 548–558. doi: 10.1016/j.it.2011.08.001
Yewdell, J. W., Anton, L. C., and Bennink, J. R. (1996). Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? J. Immunol. 157, 1823–1826.
Yin, Y., Manoury, B., and Fahraeus, R. (2003). Self-inhibition of synthesis and antigen presentation by Epstein-Barr virus-encoded EBNA1. Science 301, 1371–1374. doi: 10.1126/science.1088902
Yoshioka, M., Crum, M. M., and Sample, J. T. (2008). Autorepression of Epstein-Barr virus nuclear antigen 1 expression by inhibition of pre-mRNA processing. J. Virol. 82, 1679–1687. doi: 10.1128/jvi.02142-07
You, J., Srinivasan, V., Denis, G. V., Harrington, W. J. Jr., Ballestas, M. E., Kaye, K. M., et al. (2006). Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen interacts with bromodomain protein Brd4 on host mitotic chromosomes. J. Virol. 80, 8909–8919. doi: 10.1128/JVI.00502-06
Zaldumbide, A., Ossevoort, M., Wiertz, E. J., and Hoeben, R. C. (2007). In cis inhibition of antigen processing by the latency-associated nuclear antigen I of Kaposi sarcoma herpes virus. Mol. Immunol. 44, 1352–1360. doi: 10.1016/j.molimm.2006.05.012
Keywords: herpesvirus, viral latency, genome maintenance protein, immune evasion, antigen presentation, viral proteins
Citation: Sorel O and Dewals BG (2019) The Critical Role of Genome Maintenance Proteins in Immune Evasion During Gammaherpesvirus Latency. Front. Microbiol. 9:3315. doi: 10.3389/fmicb.2018.03315
Received: 28 September 2018; Accepted: 20 December 2018;
Published: 09 January 2019.
Edited by:
Michael Nevels, University of St Andrews, United KingdomReviewed by:
Neil Blake, University of Liverpool, United KingdomCopyright © 2019 Sorel and Dewals. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benjamin G. Dewals, YmdkZXdhbHNAdWxpZWdlLmJl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.